Base de données publique
des médicaments
Visiter [medicaments.gouv.fr] ![Visiter [medicaments.gouv.fr]](/img/icone_lien.png "Visiter [medicaments.gouv.fr] - nouvelle fenêtre")



FULVESTRANT ZENTIVA 250 mg, solution injectable en seringue préremplie - Résumé des caractéristiques du produit |
 |
|
ANSM - Mis à jour le : 11/02/2021
FULVESTRANT ZENTIVA 250 mg, solution injectable en seringue préremplie
2. COMPOSITION QUALITATIVE ET QUANTITATIVE 
Pour une seringue préremplie de 5 mL.
Excipients à effet notoire (par 5 mL)
Ethanol (96 %) (alcool): 500 mg
Alcool benzylique (E1519): 500 mg
Benzoate de benzyle : 750 mg
Pour la liste complète des excipients, voir rubrique 6.1.
Solution injectable en seringue préremplie.
Solution visqueuse, limpide, incolore à jaune, exempte de particules visibles.
4.1. Indications thérapeutiques
· en monothérapie dans le traitement du cancer du sein, localement avancé ou métastatique, positif pour les récepteurs aux estrogènes, chez les femmes ménopausées :
o non traitées précédemment par une hormonothérapie, ou
o avec une récidive pendant ou après un traitement adjuvant par un anti-estrogène ou une progression de la maladie sous traitement par anti-estrogène.
· en association avec le palbociclib dans le traitement du cancer du sein localement avancé ou métastatique, positif pour les récepteurs hormonaux (RH), négatif pour le récepteur 2 du facteur de croissance épidermique humain (HER2), chez les femmes ayant été traitées antérieurement par hormonothérapie (voir rubrique 5.1).
Chez les femmes en pré- ou périménopause, le traitement en association avec le palbociclib doit être associé à un agoniste de l’hormone de libération de la lutéinostimuline (LHRH).
4.2. Posologie et mode d'administration
Femmes adultes (y compris les personnes âgées)
La dose recommandée est de 500 mg une fois par mois, avec une dose supplémentaire de 500 mg deux semaines après la dose initiale.
Lorsque le fulvestrant est utilisé en association avec le palbociclib, veuillez également vous référer au Résumé des Caractéristiques du Produit du palbociclib.
Avant le début et pendant toute la durée du traitement associant le fulvestrant et le palbociclib, les femmes en pré/périménopause doivent être traitées par des agonistes de la LHRH, conformément à la pratique clinique locale.
Populations particulières
Insuffisance rénale
Aucun ajustement posologique n’est recommandé chez les patientes atteintes d’une insuffisance rénale légère à modérée (clairance de la créatinine ≥ 30 mL/min). La sécurité et l’efficacité n’ont pas été évaluées chez les patientes atteintes d’insuffisance rénale sévère (clairance de la créatinine < 30 mL/min) et, en conséquence, la prudence est recommandée chez ces patientes (voir rubrique 4.4).
Insuffisance hépatique
Aucun ajustement posologique n’est recommandé chez les patientes atteintes d’une insuffisance hépatique légère à modérée. Cependant, comme l’exposition au fulvestrant peut être augmentée chez ces patientes, le fulvestrant doit être utilisé avec précaution. Il n’y a pas de données chez les patientes atteintes d’insuffisance hépatique sévère (voir rubriques 4.3, 4.4 et 5.2).
Population pédiatrique
La sécurité et l’efficacité du fulvestrant chez les enfants âgés de moins de 18 ans n’ont pas été établies. Les données actuellement disponibles sont décrites aux rubriques 5.1 et 5.2, mais aucune recommandation sur la posologie ne peut être donnée.
Mode d’administration
Le fulvestrant doit être administré en deux injections consécutives de 5 mL par injection intramusculaire lente (1–2 minutes/injection), une dans chaque fesse (zone du fessier).
Il convient de faire preuve de prudence lors de l’injection de fulvestrant au niveau du site dorso-fessier en raison de la proximité du nerf sciatique sous-jacent.
Pour des instructions détaillées sur l’administration, voir rubrique 6.6.
Hypersensibilité à la substance active ou à l’un des excipients mentionnés à la rubrique 6.1.
Grossesse et allaitement (voir rubrique 4.6).
Insuffisance hépatique sévère (voir rubriques 4.4 et 5.2).
4.4. Mises en garde spéciales et précautions d'emploi
Le fulvestrant doit être utilisé avec prudence chez les patientes atteintes d’insuffisance rénale sévère (clairance de la créatinine inférieure à 30 mL/min).
En raison de la voie d’administration intramusculaire, le fulvestrant doit être administré avec prudence en cas de diathèse hémorragique, de thrombopénie ou chez les patientes traitées par des anticoagulants.
Des événements thromboemboliques sont fréquemment observés chez les patientes atteintes de cancer du sein à un stade avancé et ont été rapportés avec le fulvestrant dans les études cliniques (voir rubrique 4.8). Ceci doit être pris en compte lorsque le fulvestrant est prescrit à des patientes à risque.
Des événements liés au site d'injection comprenant sciatiques, névralgies, douleurs neuropathiques et neuropathies périphériques ont été rapportés lors de l’injection de fulvestrant. Des précautions doivent être prises lors de l'administration de fulvestrant au site d'injection dorso-fessier en raison de la proximité du nerf sciatique sous-jacent (voir rubriques 4.2 et 4.8).
Il n’y a pas de données sur les effets à long terme du fulvestrant sur les os. Etant donné le mécanisme d’action du fulvestrant, il existe un risque potentiel d’ostéoporose.
L’efficacité et la sécurité du fulvestrant (en monothérapie ou en association avec le palbociclib) n’ont pas été étudiées chez les patientes présentant une maladie viscérale grave.
Lorsque le fulvestrant est utilisé en association avec le palbociclib, veuillez également vous référer au Résumé des Caractéristiques du Produit du palbociclib.
Interférence avec des tests de dosage d’estradiol par anticorps
En raison de la similarité structurale du fulvestrant et de l'estradiol, le fulvestrant peut interférer avec des dosages d’estradiol par anticorps et peut entraîner une fausse augmentation du taux d'estradiol.
Ethanol
Ce médicament contient 10 % p/v d’éthanol (alcool), c'est-à-dire jusqu’à 500 mg par injection, ce qui équivaut à 10 mL de bière ou 4 ml de vin. Cela peut être nocif pour les personnes souffrant d’alcoolisme, et doit être pris en considération chez les groupes à haut risque comme les patientes présentant une affection hépatique ou une épilepsie.
Alcool benzylique
Ce médicament contient comme excipient de l’alcool benzylique qui peut provoquer des réactions allergiques.
Population pédiatrique
L’utilisation du fulvestrant n’est pas recommandée chez les enfants et les adolescentes car la sécurité et l’efficacité n’ont pas été établies dans ce groupe de patients (voir rubrique 5.1).
4.5. Interactions avec d'autres médicaments et autres formes d'interactions
Aucun ajustement posologique n’est donc nécessaire chez les patientes recevant, de façon concomitante, du fulvestrant et des inhibiteurs ou inducteurs du CYP3A4.
4.6. Fertilité, grossesse et allaitement
Femmes en âge de procréer
Les patientes en âge de procréer doivent utiliser une contraception efficace pendant le traitement par FULVESTRANT ZENTIVA et pendant 2 ans après l’administration de la dernière dose.
Grossesse
Le fulvestrant est contre-indiqué lors de la grossesse (voir rubrique 4.3). Il a été démontré que le fulvestrant traverse le placenta après une injection intramusculaire unique chez la rate et la lapine. Des études chez l’animal ont montré une toxicité sur les fonctions de reproduction, y compris une augmentation de l’incidence des anomalies et des morts fœtales (voir rubrique 5.3). En cas de grossesse survenant lors du traitement par fulvestrant, la patiente devra être avertie du risque potentiel pour le fœtus et du risque potentiel de fausse couche.
L’allaitement doit être interrompu pendant le traitement par fulvestrant. Le fulvestrant est excrété dans le lait de rates qui allaitent. Il n’y a pas de données sur l’excrétion du fulvestrant dans le lait maternel. Compte tenu du risque potentiel d’effets indésirables graves du fulvestrant pour le nourrisson allaité, l’utilisation pendant l’allaitement est contre-indiquée (voir rubrique 4.3).
Fertilité
Les effets du fulvestrant sur la fertilité dans l’espèce humaine n’ont pas été étudiés.
4.7. Effets sur l'aptitude à conduire des véhicules et à utiliser des machines
Résumé du profil de sécurité
Monothérapie
Cette rubrique fournit des informations basées sur tous les effets indésirables observés lors des études cliniques, des études post-commercialisation ou des déclarations spontanées. Sur l’ensemble des données combinées relatives au fulvestrant en monothérapie, les effets indésirables les plus fréquemment rapportés étaient : réactions au site d’injection, asthénie, nausées, et augmentation des enzymes hépatiques (ALAT, ASAT, phosphatases alcalines).
Dans le tableau 1, les catégories de fréquence des effets indésirables (EI) ci-après ont été calculées sur la base du groupe traité par fulvestrant 500 mg à partir des analyses de tolérance combinées des études qui ont comparé le fulvestrant 500 mg au fulvestrant 250 mg [CONFIRM (Etude D6997C00002), FINDER 1 (Etude D6997C00004), FINDER 2 (Etude D6997C00006), et NEWEST (Etude D6997C00003)] ou de FALCON (Etude D699BC00001) seule qui a comparé le fulvestrant 500 mg à l’anastrozole 1 mg. Lorsque les fréquences sont différentes entre l’analyse de tolérance combinée et FALCON, la fréquence la plus élevée est présentée. Les fréquences dans le tableau 1 ont été établies à partir de tous les effets indésirables rapportés, indépendamment de l’évaluation de la causalité par l’investigateur. La durée médiane du traitement par fulvestrant 500 mg sur l’ensemble des données combinées (y compris les études mentionnées ci-dessus et FALCON) était de 6,5 mois.
Liste des effets indésirables sous forme de tableau
Les effets indésirables listés ci-dessous sont classés par fréquence et par classe de systèmes d’organes (SOC). Les groupes de fréquence sont définis selon la convention suivante : très fréquent (≥ 1/10), fréquent (≥ 1/100, < 1/10), peu fréquent (≥ 1/1 000, < 1/100). Au sein de chaque groupe de fréquence, les effets indésirables sont présentés par ordre décroissant de gravité.
Tableau 1 Effets indésirables rapportés chez les patientes traitées par fulvestrant en monothérapie
|
Effets indésirables par classe de systèmes d’organes et fréquence |
||
|
Infections et infestations |
Fréquent |
Infections des voies urinaires |
|
Affections hématologiques et du système lymphatique |
Fréquent |
Numération plaquettaire réduitee |
|
Affections du système immunitaire |
Très fréquent |
Réactions d’hypersensibilitée |
|
Peu fréquent |
Réactions anaphylactiques |
|
|
Troubles du métabolisme et de la nutrition |
Fréquent |
Anorexiea |
|
Affections du système nerveux |
Fréquent |
Céphalées |
|
Affections vasculaires |
Très fréquent |
Bouffées de chaleure |
|
Fréquent |
Thrombo-embolies veineusesa |
|
|
Affections gastro-intestinales |
Très fréquent |
Nausées |
|
Fréquent |
Vomissements, diarrhées |
|
|
Affections hépatobiliaires |
Très fréquent |
Enzymes hépatiques élevées (ALAT, ASAT, phosphatases alcalines)a |
|
Fréquent |
Bilirubine élevéea |
|
|
Peu fréquent |
Insuffisance hépatiquec,f, hépatitef, gamma-GT élevéef |
|
|
Affections de la peau et du tissu sous-cutané |
Très fréquent |
Rashe |
|
Affections musculo-squelettiques et systémiques |
Très fréquent |
Douleurs articulaires et musculo- squelettiquesd |
|
Fréquent |
Dorsalgiesa |
|
|
Affections des organes de reproduction et du sein |
Fréquent |
Hémorragie vaginalee |
|
Peu fréquent |
Moniliase vaginalef, leucorrhéef, |
|
|
Troubles généraux et anomalies au site d’administration |
Très fréquent |
Asthéniea, réactions au site d’injectionb |
|
Fréquent |
Neuropathie périphériquee, sciatiquee |
|
|
Peu fréquent |
Hémorragie au site d’injectionf, hématome au site d’injectionf, névralgiec,f |
|
a Inclut des effets indésirables pour lesquels l’étendue exacte de la contribution du fulvestrant ne peut être évaluée en raison de la maladie sous-jacente.
b Le terme réactions au site d’injection n’inclut pas les termes hémorragies au site d’injection, hématomes au site d’injection, sciatique, névralgie et neuropathie périphérique.
c L’événement n’a pas été observé dans les études cliniques majeures (CONFIRM, FINDER 1, FINDER 2, NEWEST). La fréquence a été calculée en utilisant la limite supérieure de l’intervalle de confiance à 95 % pour l’estimation de la valeur. Elle est calculée à 3/560 (où 560 est le nombre de patientes des études cliniques majeures), ce qui correspond à la catégorie de fréquence « peu fréquent ».
d Inclut : arthralgies et, moins fréquemment, douleurs musculo-squelettiques, myalgies et douleurs dans les extrémités.
e La catégorie de fréquence diffère entre les données de tolérance combinées et FALCON.
f Cet effet indésirable n’a pas été observé dans FALCON.
Description des effets indésirables sélectionnés
Les descriptions incluses ci-dessous sont basées sur la population d’analyse de la tolérance composée de 228 patientes ayant respectivement reçu au moins une (1) dose de fulvestrant et de 232 patientes ayant reçu au moins une (1) dose d’anastrozole, dans l’étude de phase III FALCON.
Douleurs articulaires et musculo-squelettiques
Dans l’étude FALCON, le nombre de patientes ayant rapporté un effet indésirable de douleurs articulaires et musculo-squelettiques a été de 65 (31,2 %) et de 48 (24,1 %) respectivement dans les bras fulvestrant et anastrozole. Sur les 65 patientes du bras fulvestrant, 40 % (26/65) ont rapporté des douleurs articulaires et musculo-squelettiques au cours du premier mois de traitement et 66,2 % (43/65) au cours des 3 premiers mois de traitement. Aucune patiente n’a rapporté d’événements de grade CTCAE ≥ 3 ou ayant nécessité une réduction de la dose, une interruption du traitement ou l’arrêt du traitement en raison de ces effets indésirables.
Traitement en association avec le palbociclib
Le profil de sécurité global du fulvestrant lorsqu'il est utilisé en association avec le palbociclib est basé sur les données de 517 patientes atteintes d’un cancer du sein avancé ou métastatique RH-positif, HER2-négatif dans l'étude randomisée PALOMA3 (voir rubrique 5.1). Les effets indésirables les plus fréquents (≥ 20 %) tous grades confondus rapportés chez les patientes recevant du fulvestrant en association avec le palbociclib étaient la neutropénie, la leucopénie, les infections, la fatigue, les nausées, l'anémie, la stomatite, la diarrhée, la thrombopénie et les vomissements. Les effets indésirables de grade ≥ 3 les plus fréquents (≥ 2 %) étaient la neutropénie, la leucopénie, l'anémie, les infections, l'augmentation du taux d’ASAT, la thrombopénie et la fatigue.
Le tableau 2 rapporte les effets indésirables de PALOMA3.
La durée médiane de l'exposition au fulvestrant était de 11,2 mois dans le bras fulvestrant + palbociclib et de 4,8 mois dans le bras fulvestrant + placebo. La durée médiane de l'exposition au palbociclib dans le bras fulvestrant + palbociclib était de 10,8 mois.
Tableau 2 Effets indésirables basés sur l’étude PALOMA3 (N = 517)
|
Classe de systèmes d’organes Fréquence Terme préférentiela |
Fulvestrant + palbociclib (N = 345) |
Fulvestrant + placebo (N = 172) |
|||
|
Tous grades n (%) |
Grade ≥ 3 n (%) |
Tous grades n (%) |
Grade ≥ 3 n (%) |
||
|
Infections et infestations |
|||||
|
Très fréquent |
|
|
|
|
|
|
Infectionsb |
188 (54,5) |
19 (5,5) |
60 (34,9) |
6 (3,5) |
|
|
Affections hématologiques et du système lymphatique |
|||||
|
Très frequent |
|
|
|
|
|
|
Neutropéniec |
290 (84,1) |
240 (69,6) |
6 (3,5) |
0 |
|
|
Leucopénied |
207 (60,0) |
132 (38,3) |
9 (5,2) |
1 (0,6) |
|
|
Anémiee |
109 (31,6) |
15 (4,3) |
24 (14,0) |
4 (2,3) |
|
|
Thrombopénief |
88 (25,5) |
10 (2,9) |
0 |
0 |
|
|
Peu frequent |
|
|
|
|
|
|
Neutropénie fébrile |
3 (0,9) |
3 (0,9) |
0 |
1 (0,6) |
|
|
Troubles du métabolisme et de la nutrition |
|||||
|
Très fréquent |
|
|
|
|
|
|
Diminution de l’appétit |
60 (17,4) |
4 (1,2) |
18 (10,5) |
1 (0,6) |
|
|
Affections du système nerveux |
|||||
|
Fréquent |
|
|
|
|
|
|
Dysgueusie |
27 (7,8) |
0 |
6 (3,5) |
0 |
|
|
Affections oculaires |
|||||
|
Fréquent |
|
|
|
|
|
|
Augmentation de la sécrétion lacrymale |
25 (7,2) |
0 |
2 (1,2) |
0 |
|
|
Vision trouble |
24 (7,0) |
0 |
3 (1,7) |
0 |
|
|
Sécheresse oculaire |
15 (4,3) |
0 |
3 (1,7) |
0 |
|
|
Affections respiratoires, thoraciques et médiastinales |
|||||
|
Fréquent |
|
|
|
|
|
|
Epistaxis |
25 (7,2) |
0 |
4 (2,3) |
0 |
|
|
Affections gastro-intestinales |
|||||
|
Très fréquent |
|
|
|
|
|
|
Nausées |
124 (35,9) |
2 (0,6) |
53 (30,8) |
1 (0,6) |
|
|
Stomatiteg |
104 (30,1) |
3 (0,9) |
24 (14,0) |
0 |
|
|
Diarrhée |
94 (27,2) |
0 |
35 (20,3) |
2 (1,2) |
|
|
Vomissements |
75 (21,7) |
2 (0,6) |
28 (16,3) |
1 (0,6) |
|
|
Affections de la peau et du tissu sous-cutané |
|||||
|
Très fréquent |
|
|
|
|
|
|
Alopécie |
67 (19,4) |
NA |
11 (6,4) |
NA |
|
|
Rashh |
63 (18,3) |
3 (0,9) |
10 (5,8) |
0 |
|
|
Fréquent |
|
|
|
|
|
|
Sécheresse cutanée |
28 (8,1) |
0 |
3 (1,7) |
0 |
|
|
Troubles généraux et anomalies au site d’administration |
|||||
|
Très fréquent |
|
|
|
|
|
|
Fatigue |
152 (44,1) |
9 (2,6) |
54 (31,4) |
2 (1,2) |
|
|
Fièvre |
47 (13,6) |
1 (0,3) |
10 (5,8) |
0 |
|
|
Fréquent |
|
|
|
|
|
|
Asthénie |
27 (7,8) |
1 (0,3) |
13 (7,6) |
2 (1,2) |
|
|
Investigations |
|||||
|
Très fréquent |
|
|
|
|
|
|
Augmentation des ASAT |
40 (11,6) |
11 (3,2) |
13 (7,6) |
4 (2,3) |
|
|
Fréquent |
|
|
|
|
|
|
Augmentation des ALAT |
30 (8,7) |
7 (2,0) |
10 (5,8) |
1 (0,6) |
|
ALAT = alanine aminotransférase ; ASAT = aspartate aminotransférase ; N/n = nombre de patientes ; NA = non applicable
a Les termes préférentiels (TP) sont répertoriés selon le dictionnaire MedDRA 17.1.
b Le terme infections inclut tous les TP de la classe de systèmes d’organes Infections et infestations.
c Le terme neutropénie inclut les TP suivants : neutropénie, neutrophiles diminués.
d Le terme leucopénie inclut les TP suivants : leucopénie, globules blancs diminués.
e Le terme anémie inclut les TP suivants : anémie, hémoglobine diminuée, hématocrite diminuée.
f Le terme thrombopénie inclut les TP suivants : thrombopénie, numération plaquettaire diminuée.
g Le terme stomatite inclut les TP suivants : stomatite aphteuse, chéilite, glossite, glossodynie, ulcération buccale, inflammation muqueuse, douleur buccale, gêne oropharyngée, douleur oropharyngée, stomatite.
h Le terme rash inclut les TP suivants : rash, rash maculopapuleux, rash prurigineux, rash érythémateux, rash papuleux, dermatite, dermatite acnéiforme, éruption cutanée toxique.
Description des effets indésirables sélectionnés
Neutropénie
Chez les patientes recevant du fulvestrant en association avec le palbociclib dans l'étude PALOMA3, une neutropénie tous grades confondus a été rapportée chez 290 patientes (84,1 %), une neutropénie de grade 3 étant rapportée chez 200 patientes (58,0 %) et une neutropénie de grade 4 étant rapportée chez 40 patientes (11,6 %). Dans le bras fulvestrant + placebo (n = 172), une neutropénie tous grades confondus a été rapportée chez 6 patientes (3,5 %). Aucun cas de neutropénie de grade 3 et 4 n’a été rapporté dans le bras fulvestrant + placebo.
Chez les patientes recevant du fulvestrant en association avec le palbociclib, le délai médian jusqu’au premier épisode de neutropénie tous grades confondus était de 15 jours (entre 13 et 512 jours) et la durée moyenne de neutropénie de grade ≥ 3 était de 16 jours. Une neutropénie fébrile a été rapportée chez 3 des patientes (0,9 %) recevant du fulvestrant en association avec le palbociclib.
Déclaration des effets indésirables suspectés
La déclaration des effets indésirables suspectés après autorisation du médicament est importante. Elle permet une surveillance continue du rapport bénéfice/risque du médicament. Les professionnels de santé déclarent tout effet indésirable suspecté via le système national de déclaration : Agence nationale de sécurité du médicament et des produits de santé (ANSM) et réseau des Centres Régionaux de Pharmacovigilance - Site internet : www.signalement-sante.gouv.fr.
Des cas isolés de surdosage ont été rapportés avec le fulvestrant chez l’être humain. En cas de surdosage, un traitement symptomatique et de soutien est recommandé. Au cours des études menées chez l’animal, aucun effet autre que ceux liés directement ou indirectement à l’activité anti-estrogène n’a été mis en évidence à des doses plus élevées de fulvestrant (voir rubrique 5.3).
5. PROPRIETES PHARMACOLOGIQUES
5.1. Propriétés pharmacodynamiques
Classe pharmacothérapeutique : Thérapeutique endocrine, Anti-estrogènes, Code ATC : L02BA03.
Mécanisme d’action et effets pharmacodynamiques
Le fulvestrant est un antagoniste compétitif des récepteurs aux estrogènes (RE) avec une affinité comparable à l’estradiol. Le fulvestrant bloque les actions trophiques des estrogènes sans posséder une quelconque activité agoniste partielle (de type estrogène). Son mécanisme d’action est associé à une diminution des taux d’expression de la protéine du récepteur aux estrogènes. Des études cliniques menées chez des patientes ménopausées présentant un cancer primaire du sein ont montré que le fulvestrant diminuait significativement l’expression de la protéine RE dans les tumeurs RE positives par comparaison au placebo. Une diminution significative de l’expression des récepteurs à la progestérone a été également observée, en corrélation avec l’absence d’effet estrogénique intrinsèque. Il a été également montré que le fulvestrant 500 mg diminue l’expression de la protéine RE et la prolifération du marqueur Ki67, d’une manière plus importante que le fulvestrant 250 mg en traitement néo-adjuvant des tumeurs mammaires après la ménopause.
Efficacité et sécurité cliniques dans le cancer du sein à un stade avancé
Monothérapie
Une étude clinique de phase III a été réalisée chez 736 femmes ménopausées atteintes d’un cancer du sein à un stade avancé, dont la maladie a récidivé pendant ou après une hormonothérapie adjuvante ou dont la maladie a progressé suite à une hormonothérapie. L’étude a inclus 423 patientes dont la maladie a récidivé ou progressé sous traitement par anti-estrogène (sous-groupe AE) et 313 patientes dont la maladie a récidivé ou progressé sous traitement par inhibiteur de l’aromatase (sous-groupe IA). Cette étude a comparé l’efficacité et la tolérance du fulvestrant 500 mg (n = 362) au fulvestrant 250 mg (n = 374). Le critère principal était la survie sans progression (SSP), les critères secondaires clés d’efficacité incluaient le taux de réponse objective (TRO), le taux de bénéfice clinique (TBC) et la survie globale (SG). Les résultats d’efficacité de l’étude CONFIRM sont résumés dans le tableau 3.
Tableau 3 Résumé des résultats du critère principal d’efficacité (SSP) et des critères secondaires clés d’efficacité de l’étude CONFIRM
|
Variable |
Méthode de calcul ; comparaison des traitements |
Fulvestrant 500 mg (N = 362) |
Fulvestrant 250 mg (N = 374) |
Comparaison entre les groupes |
||
|
Rapport de risque |
IC à 95 % |
Valeur de p |
||||
|
SSP |
Médiane en mois de K-M ; |
|
|
|
|
|
|
Population globale |
6,5 |
5,5 |
0,80 |
0,68 - 0,94 |
0,006 |
|
|
-Sous-groupe AE (n = 423) |
8,6 |
5,8 |
0,76 |
0,62 - 0,94 |
0,013 |
|
|
-Sous-groupe IA (n = 313)a |
5,4 |
4,1 |
0,85 |
0,67 - 1,08 |
0,195 |
|
|
SGb |
Médiane en mois de K-M ; |
|
|
|
|
|
|
Population globale |
26,4 |
22,3 |
0,81 |
0,69 - 0,96 |
0,016c |
|
|
-Sous-groupe AE (n = 423) |
30,6 |
23,9 |
0,79 |
0,63 - 0,99 |
0,038c |
|
|
-Sous-groupe IA (n = 313)a |
24,1 |
20,8 |
0,86 |
0,67 - 1,11 |
0,241c |
|
|
Variable |
Méthode de calcul ; |
Fulvestrant 500 mg (N = 362) |
Fulvestrant 250 mg (N = 374) |
Comparaison entre les groupes |
||
|
|
|
|
|
Différence absolue en % |
IC à 95 % |
|
|
TROd |
% de patientes avec une RO ; |
|
|
|
|
|
|
Population globale |
13,8 |
14,6 |
-0,8 |
-5,8 – 6,3 |
||
|
-Sous-groupe AE (n = 296) |
18,1 |
19,1 |
-1,0 |
-8,2 – 9,3 |
||
|
-Sous-groupe IA (n = 205)a |
7,3 |
8,3 |
-1,0 |
-5,5 – 9,8 |
||
|
TBCe |
% de patientes avec un BC ; |
|
|
|
|
|
|
Population globale |
45,6 |
39,6 |
6,0 |
-1,1 – 13,3 |
||
|
-Sous-groupe AE (n = 423) |
52,4 |
45,1 |
7,3 |
-2,2 – 16,6 |
||
|
-Sous-groupe IA (n = 313)a |
36,2 |
32,3 |
3,9 |
-6,1 – 15,2 |
||
a Le fulvestrant est indiqué chez les patientes dont la maladie a récidivé ou progressé sous traitement par anti-estrogène. Les résultats du sous-groupe IA ne peuvent faire l’objet d’une conclusion.
b La SG est présentée pour les analyses finales de survie à 75 % de maturité.
c Valeur de p sans ajustements pour la multiplicité des tests entre les analyses initiales de la survie globale à 50 % de maturité et les analyses actualisées de survie à 75 % de maturité.
d Le TRO a été calculé chez les patientes qui étaient évaluables à l’inclusion (c.-à-d., celles avec une maladie mesurable à l’inclusion : 240 patientes dans le groupe fulvestrant 500 mg et 261 patientes dans le groupe fulvestrant 250 mg).
e Patientes dont la meilleure réponse objective est soit une réponse complète, soit une réponse partielle, soit une stabilité de la maladie ≥ 24 semaines.
SSP : survie sans progression ; TRO : taux de réponse objective ; RO : réponse objective ; TBC : taux de bénéfice clinique ; BC : bénéfice clinique ; SG : survie globale ; K-M : Kaplan-Meier ; IC : intervalle de confiance ; IA : inhibiteur de l’aromatase ; AE : anti-estrogène.
Une étude multicentrique de phase III, randomisée, en double aveugle, à double placebo, évaluant le fulvestrant 500 mg versus l’anastrozole 1 mg, a été conduite chez des femmes ménopausées atteintes d’un cancer du sein localement avancé ou métastatique, avec des RE positifs et/ou des RP positifs, non traitées précédemment par hormonothérapie. Au total, 462 patientes ont été randomisées de manière séquentielle selon un rapport 1/1 pour recevoir soit le fulvestrant 500 mg soit l’anastrozole 1 mg.
La randomisation a été stratifiée en fonction du stade de la maladie (localement avancée ou métastatique), de la chimiothérapie antérieure pour une maladie avancée et des signes mesurables de la maladie.
Le critère principal d’efficacité de l’étude était la survie sans progression (SSP) évaluée par l’investigateur selon les critères RECIST 1.1 (Response Evaluation Criteria in Solid Tumors). Les critères secondaires clés d’efficacité incluaient la survie globale (SG) et le taux de réponse objective (TRO).
L’âge médian des patientes incluses dans cette étude était de 63 ans (de 36 à 90 ans). La majorité des patientes (87,0 %) présentaient une maladie métastatique à l’inclusion. Cinquante-cinq pour cent (55,0 %) des patientes présentaient des métastases viscérales à l’inclusion. Au total, 17,1 % des patientes avaient reçu une chimiothérapie antérieure pour une maladie avancée ; 84,2 % des patientes présentaient une maladie mesurable.
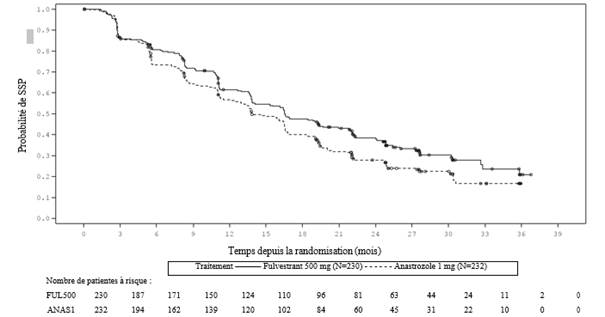
Des résultats cohérents ont été observés dans la majorité des sous-groupes de patientes pré-spécifiés. Pour le sous-groupe de patientes avec une maladie limitée à des métastases non viscérales (n = 208), le HR a été de 0,592 (IC à 95 % : 0,419–0,837) dans le bras fulvestrant comparé au bras anastrozole. Pour le sous-groupe de patientes avec des métastases viscérales (n = 254), le HR a été de 0,993 (IC à 95 % : 0,740–1,331) dans le bras fulvestrant comparé au bras anastrozole. Les résultats d’efficacité de l’étude FALCON sont présentés dans le tableau 4 et la figure 1.
Tableau 4 Résumé des résultats du critère principal d’efficacité (SSP) et des critères secondaires clés d’efficacité (évaluation par l’investigateur, population en intention de traiter) — étude FALCON
|
|
Fulvestrant 500 mg (N = 230) |
Anastrozole 1 mg (N = 232) |
|
Survie sans progression |
||
|
Nombre d’événements de SSP (%) |
143 (62,2 %) |
166 (71,6 %) |
|
Rapport de risque pour la SSP (IC à 95 %) et valeur de p |
HR 0,797 (0,637 – 0,999) |
|
|
p = 0,0486 |
||
|
Médiane de SSP [mois (IC à 95 %)] |
16,6 (13,8 ; 21,0) |
13,8 (12,0 ; 16,6) |
|
Nombre d’événements de SG* |
67 (29,1 %) |
75 (32,3 %) |
|
Rapport de risque pour la SG (IC à 95 %) et valeur de p |
HR 0,875 (0,629 – 1,217) |
|
|
p = 0,4277 |
||
|
TRO** |
89 (46,1 %) |
88 (44,9 %) |
|
Odds ratio pour le TRO (IC à 95 %) et valeur de p |
OR 1,074 (0,716 – 1,614) |
|
|
p = 0,7290 |
||
|
DDR médiane (mois) |
20,0 |
13,2 |
|
TBC |
180 (78,3 %) |
172 (74,1 %) |
|
Odds ratio pour le TBC (IC à 95 %) et valeur de p |
OR 1,253 (0,815 – 1,932) |
|
|
p = 0,3045 |
||
* (31 % de maturité) — analyse de la SG non finale
** pour les patientes avec une maladie mesurable
Figure 1 Courbe de Kaplan-Meier de la survie sans progression (évaluation par l’investigateur, population en intention de traiter) — étude FALCON
Deux études cliniques de phase III ont été réalisées chez 851 femmes ménopausées atteintes d’un cancer du sein à un stade avancé, dont la maladie a récidivé pendant ou après une hormonothérapie adjuvante ou dont la maladie a progressé suite à une hormonothérapie. Soixante-dix-sept pour cent (77 %) de la population de l’étude présentait un cancer du sein avec des récepteurs aux estrogènes positifs. Ces études ont comparé la tolérance et l’efficacité d’une administration mensuelle du fulvestrant 250 mg à celles de l’administration quotidienne de 1 mg d’anastrozole (inhibiteur de l’aromatase). Le fulvestrant à la dose mensuelle de 250 mg s’est montré, dans son ensemble, au moins aussi efficace que l’anastrozole en termes de survie sans progression, de réponse objective, et de temps jusqu’au décès. Aucune différence statistiquement significative n’a été observée sur ces critères d’évaluation entre les deux groupes de traitement.
Le critère d’évaluation principal était la survie sans progression. L’analyse combinée des deux études montre que 83 % des patientes du groupe fulvestrant ont vu leur maladie progresser contre 85 % des patientes du groupe anastrozole. L’analyse combinée des deux études montrait que le rapport de risque du fulvestrant 250 mg par rapport à l’anastrozole pour la survie sans progression était de 0,95 (IC à 95 % 0,82 à 1,10). Le taux de réponse objective était de 19,2 % dans le groupe fulvestrant 250 mg, comparé à 16,5 % dans le groupe anastrozole. Le délai médian jusqu’au décès a été de 27,4 mois pour les patientes traitées par le fulvestrant et de 27,6 mois pour les patientes traitées par l’anastrozole. Le rapport de risque du fulvestrant 250 mg par rapport à l’anastrozole pour le temps jusqu’au décès était de 1,01 (IC à 95 % 0,86 à 1,19).
Traitement en association avec le palbociclib
Une étude multicentrique de phase III, internationale, randomisée, en double aveugle, en groupes parallèles, évaluant fulvestrant 500 mg plus palbociclib 125 mg versus fulvestrant 500 mg plus placebo, a été conduite chez des femmes atteintes d’un cancer du sein localement avancé, RH positif, HER2 négatif, non éligible à une résection chirurgicale ou à une radiothérapie à visée curative, ou d’un cancer du sein métastatique, quel que soit leur statut ménopausique, dont la maladie a progressé après une hormonothérapie antérieure dans le cadre d’un traitement (néo) adjuvant ou métastatique.
Au total, 521 femmes en pré/périménopause et ménopausées dont la maladie avait progressé durant l’hormonothérapie adjuvante ou dans les 12 mois suivant l’arrêt de l’hormonothérapie adjuvante, durant une hormonothérapie antérieure pour une maladie avancée ou au cours du mois suivant une hormonothérapie antérieure pour une maladie avancée, ont été randomisées selon un rapport de 2/1 pour recevoir l’association fulvestrant plus palbociclib ou fulvestrant plus placebo, et stratifiées selon la sensibilité documentée à une hormonothérapie antérieure, le statut ménopausique à l’entrée dans l’étude (pré/périménopause versus ménopause), et la présence de métastases viscérales. Les femmes en pré/périménopause ont reçu la goséréline en tant qu’agoniste de la LHRH. Les patientes présentant une extension viscérale, symptomatique, avancée/métastatique, et qui risquaient des complications mettant en jeu leur pronostic vital à court terme (incluant les patientes avec épanchement massif non contrôlé [pleural, péricardique, péritonéal], lymphangite pulmonaire et atteinte hépatique supérieure à 50 %), n’étaient pas éligibles à l’inclusion dans l’étude.
Les patientes ont poursuivi le traitement attribué jusqu’à progression objective de la maladie, détérioration des symptômes, toxicité inacceptable, décès, ou retrait du consentement, en fonction de la survenue du premier événement.
Un cross-over entre les bras de traitement n’était pas autorisé.
Les patientes étaient bien appariées selon les caractéristiques démographiques et pronostiques à l’entrée dans l’étude entre le bras fulvestrant plus palbociclib et le bras fulvestrant plus placebo. L’âge médian des patientes incluses dans cette étude était de 57 ans (de 29 à 88 ans). Dans chacun des bras de traitement, la majorité des patientes étaient d’origine caucasienne, présentaient une sensibilité documentée à une hormonothérapie antérieure et étaient ménopausées.
Environ 20 % des patientes étaient en pré/périménopause. Toutes les patientes avaient reçu un traitement systémique antérieur et la plupart des patientes de chacun des bras de traitement avaient reçu une chimiothérapie préalable pour leur diagnostic initial. Plus de la moitié d’entre elles (62 %) présentaient un indice de performance ECOG de 0 ; 60 % présentaient des métastases viscérales et 60 % avaient reçu plusieurs traitements hormonaux antérieurs pour leur diagnostic initial.
Le critère d’évaluation principal de l’étude a été la SSP évaluée par l’investigateur selon les critères RECIST 1.1. Des analyses complémentaires sur la SSP ont été réalisées sur la base d’une revue radiologique centralisée indépendante. Les critères d’évaluation secondaires ont été les suivants : TRO, TBC, survie globale (SG), tolérance, et délai avant détérioration (TTD) du critère composite de la douleur.
L’étude a atteint son critère d’évaluation principal sur l’allongement de la SSP évaluée par l’investigateur lors de l’analyse intermédiaire réalisée sur 82 % des événements de SSP prévus ; les résultats ont dépassé la limite d’efficacité pré-spécifiée de Haybittle-Peto (α = 0,00135), démontrant un allongement statistiquement significatif de la SSP et un effet cliniquement significatif du traitement. Des données d’efficacité actualisées sont présentées dans le tableau 5.
Après une période de suivi médiane de 45 mois, l’analyse finale de la SG a été effectuée sur la base de 310 événements (60 % des patientes randomisées). Une différence de 6,9 mois a été observée au niveau de la SG médiane dans le bras palbociclib plus fulvestrant par rapport au bras placebo plus fulvestrant ; ce résultat n’était pas statistiquement significatif au seuil de significativité prédéfini de 0,0235 (test unilatéral). Dans le bras placebo plus fulvestrant, 15,5 % des patientes randomisées ont reçu du palbociclib et d’autres inhibiteurs des CDK comme traitements ultérieurs post-progression.
Les résultats de la SSP évaluée par l’investigateur et les données de SG finales provenant de l’étude PALOMA3 sont présentés dans le tableau 5. Les courbes de Kaplan-Meier correspondantes sont représentées aux figures 2 et 3, respectivement.
Tableau 5 Résultats d’efficacité — étude PALOMA3 (évaluation par l’investigateur, population en intention de traiter)
|
|
Analyse actualisée (au 23 octobre 2015) |
|
|
Fulvestrant plus palbociclib (N = 347) |
Fulvestrant plus placebo (N = 174) |
|
|
Survie sans progression |
|
|
|
Médiane [mois (IC à 95 %)] |
11,2 (9,5 ; 12,9) |
4,6 (3,5 ; 5,6) |
|
Rapport de risque (IC à 95 %) et valeur de p |
0,497 (0,398 ; 0,620), p < 0,000001 |
|
|
Critères d’évaluation secondaires* |
||
|
RO [% (IC à 95 %)] |
26,2 (21,7 ; 31,2) |
13,8 (9,0 ; 19,8) |
|
RO (maladie measurable) [% (IC à 95 %)] |
33,7 (28,1 ; 39,7) |
17,4 (11,5 ; 24,8) |
|
TBC [% (IC à 95 %)] |
68,0 (62,8 ; 72,9) |
39,7 (32,3 ; 47,3) |
|
Survie globale (SG) finale (au 13 avril 2018) |
||
|
Nombre d’événements (%) |
201 (57,9) |
109 (62,6) |
|
Médiane [mois (IC à 95%)] |
34,9 (28,8 ; 40,0) |
28,0 (23,6 ; 34,6) |
|
Rapport de risque (IC à 95 %) et valeur de p† |
0,814 (0,644 ; 1,029) p = 0,0429† |
|
TBC = taux de bénéfice clinique ; IC = intervalle de confiance ; N = nombre de patientes ; RO = réponse objective ;
Les résultats du critère d’évaluation secondaire reposent sur les réponses confirmées et non confirmées selon les critères RECIST 1.1.
* Non statistiquement significatif.
† Valeur de p unilatérale basée sur un test du log-rank stratifié par la présence de métastases viscérales et la sensibilité à l’hormonothérapie antérieure par randomisation.
Figure 2 Courbe de Kaplan-Meier de la survie sans progression (évaluation par l’investigateur, population en intention de traiter) — étude PALOMA3 (au 23 octobre 2015)
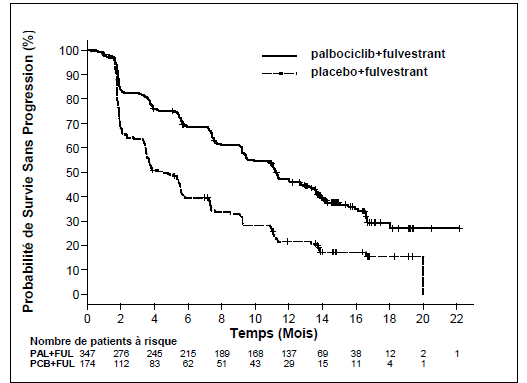
FUL = fulvestrant ; PAL = palbociclib ; PCB = placebo.
Dans le bras fulvestran plus palbociclib, une réduction du risque de progression de la maladie ou de décès a été observée dans tous les sous-groupes de patientes définis par les facteurs de stratification et les caractéristiques initiales. Cela a été mis en évidence chez les femmes en pré/périménopause (HR de 0,46 [IC à 95 % : 0,28 – 0,75]) et les femmes ménopausées (HR de 0,52 [IC à 95 % : 0,40 – 0,66]), les patientes présentant des métastases viscérales (HR de 0,50 [IC à 95 % : 0,38 – 0,65]) et les patientes présentant des métastases non viscérales (HR de 0,48 [IC à 95 % : 0,33 – 0,71]). Un bénéfice a également pu être observé quel que soit le nombre de lignes de traitement antérieur dans le contexte métastatique : que ce soit 0 (HR de 0,59 [IC à 95 % : 0,37 – 0,93]), 1 (HR de 0,46 [IC à 95 % : 0,32 – 0,64]), 2 (HR de 0,48 [IC à 95 % : 0,30 – 0,76]), ou ≥ 3 lignes (HR de 0,59 [IC à 95 % : 0,28 – 1,22]).
Figure 3 Courbe de Kaplan-Meier de la survie globale (population en intention de traiter) — Etude PALOMA3 (au 13 avril 2018)
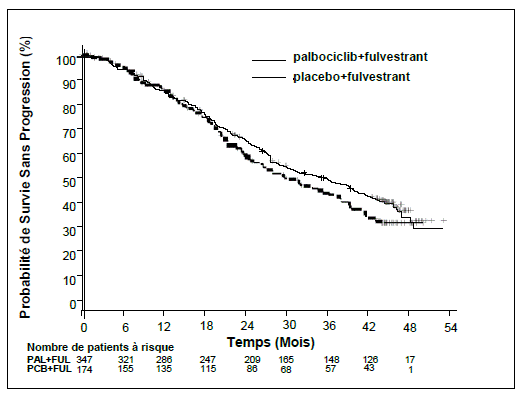
FUL = fulvestrant ; PAL = palbociclib ; PCB = placebo.
Des paramètres d’efficacité additionnels (RO et TRT) évalués dans des sous-groupes de patientes avec ou sans maladie viscérale sont présentés au tableau 6.
Tableau 6 Résultats d’efficacité de l’étude PALOMA3 dans la maladie viscérale et non viscérale (population en intention de traiter)
|
|
Maladie viscérale |
Maladie non viscérale |
||
|
|
Fulvestrant plus palbociclib (N = 206) |
Fulvestrant plus placebo (N = 105) |
Fulvestrant plus palbociclib (N = 141) |
Fulvestrant plus placebo (N = 69) |
|
RO [% (IC à 95 %)] |
35,0 |
13,3 |
13,5 |
14,5 |
|
(28,5 ; 41,9) |
(7,5 ; 21,4) |
(8,3 ; 20,2) |
(7,2 ; 25,0) |
|
|
TRT*, Médiane [mois (intervalle)] |
3,8 |
5,4 |
3,7 |
3,6 |
|
(3,5 ; 16,7) |
(3,5 ; 16,7) |
(1,9 ; 13,7) |
(3,4 ; 3,7) |
|
* Critères d’évaluation de la réponse sur la base des réponses confirmées et non confirmées.
N = nombre de patientes ; IC = intervalle de confiance ; RO = réponse objective ; TRT = temps jusqu’à obtention de la première réponse tumorale.
Les symptômes rapportés par les patientes ont été évalués à l’aide du questionnaire sur la qualité de vie de l’European Organization for Research and Treatment of Cancer (EORTC) (QLQ)-C30 et de son module spécifique sur le cancer du sein (EORTC QLQ-BR23). Au total, 335 patientes du bras fulvestrant plus palbociclib et 166 patientes du bras fulvestrant plus placebo ont rempli le questionnaire à l’inclusion et au cours d’au moins une visite après l’inclusion dans l’étude.
Le délai avant détérioration était prédéfini comme étant le délai entre l’inclusion et la première survenue d’une augmentation ≥ 10 points des scores en termes de symptômes douloureux. L’ajout du palbociclib au fulvestrant a apporté un bénéfice sur les symptômes en allongeant significativement le délai avant détérioration de la douleur en comparaison avec l’association de fulvestrant plus placebo (médiane de 8,0 mois versus 2,8 mois ; HR de 0,64 [IC à 95 % : 0,49 – 0,85] ; p < 0,001).
Effets sur l’endomètre post-ménopausique
Les données pré-cliniques ne suggèrent pas un effet stimulant du fulvestrant sur l’endomètre post-ménopausique (voir rubrique 5.3). Une étude de 2 semaines réalisée chez des volontaires saines ménopausées traitées par 20 microgrammes/jour d’éthinylestradiol a montré qu’un pré-traitement par 250 mg de fulvestrant réduisait significativement la stimulation de l’endomètre post-ménopausique comparé à un pré-traitement par placebo, évaluée par ultrasons de l’épaisseur de l’endomètre.
Un traitement néo-adjuvant jusqu’à 16 semaines chez des patientes ayant un cancer du sein traitées par fulvestrant 500 mg ou fulvestrant 250 mg n’a pas entraîné des modifications cliniquement significatives de l’épaisseur de l’endomètre, indiquant une absence d’effet agoniste. Il n’y a aucune preuve d’effets indésirables sur l’endomètre chez les patientes étudiées atteintes d’un cancer du sein. Aucune donnée n’est disponible sur la morphologie de l’endomètre.
Dans deux études à court terme (1 et 12 semaines) portant sur des patientes préménopausées souffrant de pathologies gynécologiques bénignes, aucune différence significative au niveau de l’épaisseur de l’endomètre (mesures par ultra-sons) n’a été observée entre les groupes fulvestrant et placebo.
Effets sur l’os
Il n’y a pas de données sur les effets à long terme du fulvestrant sur l’os. Un traitement néo-adjuvant jusqu’à 16 semaines chez des patientes ayant un cancer du sein traitées par fulvestrant 500 mg ou fulvestrant 250 mg n’a pas entraîné des modifications cliniquement significatives au niveau des marqueurs sériques du remodelage osseux.
Population pédiatrique
Le fulvestrant n’est pas indiqué pour une utilisation chez les enfants. L’Agence européenne des médicaments a différé l’obligation de soumettre les résultats d’études réalisées avec le fulvestrant dans plusieurs sous-groupes de la population pédiatrique ayant un cancer du sein (voir rubrique 4.2 pour les informations concernant l’usage pédiatrique).
Une étude en ouvert de phase II a étudié la tolérance, l’efficacité et la pharmacocinétique du fulvestrant chez 30 filles âgées de 1 à 8 ans présentant une puberté précoce progressive associée à un syndrome de McCune Albright (SMA). Les patientes pédiatriques ont reçu une dose intramusculaire mensuelle de 4 mg/kg de fulvestrant. Cette étude de 12 mois a évalué plusieurs critères du SMA et a montré une réduction de la fréquence des saignements vaginaux et un ralentissement du taux d’augmentation de l’âge osseux. Les concentrations minimales à l’état d’équilibre du fulvestrant chez l’enfant dans cette étude étaient alignées avec celles observées chez les adultes (voir rubrique 5.2). Aucune nouvelle donnée concernant la tolérance n’a été rapportée au cours de cette étude à faible effectif, mais les données à 5 ans ne sont pas encore disponibles.
5.2. Propriétés pharmacocinétiques
Après injection intramusculaire de fulvestrant à action prolongée, le fulvestrant est absorbé lentement et la concentration plasmatique maximale (Cmax) est atteinte au bout de 5 jours environ. L’administration de fulvestrant 500 mg aboutit à des niveaux d’exposition proches de (ou équivalant à) l’état d’équilibre dans le premier mois de l’administration (moyenne [CV] : ASC 475 [33,4 %] ng.jours/mL, Cmax 25,1 [35,3 %] ng/mL, Cmin 16,3 [25,9 %] ng/mL, respectivement). A l’état d’équilibre, les concentrations plasmatiques du fulvestrant se situent dans une fenêtre relativement étroite et présentent jusqu’à environ un facteur 3 entre les concentrations maximales et les concentrations minimales observées. Après injection intramusculaire, les concentrations plasmatiques augmentent proportionnellement à la dose injectée, pour des doses allant de 50 à 500 mg.
Distribution
La distribution du fulvestrant est rapide et importante. Le volume apparent de distribution élevé à l’état d’équilibre (Vdss) d’environ 3 à 5 L/kg suggère une forte distribution extravasculaire.
Le fulvestrant est fortement lié (99 %) aux protéines plasmatiques, principalement aux lipoprotéines de très basse densité (VLDL), de basse densité (LDL) et de haute densité (HDL). Aucune étude d’interactions par compétition au niveau des sites de fixation protéique n’a été menée. Le rôle éventuel des globulines se fixant aux hormones sexuelles (SHBG) n’a pas été déterminé.
Biotransformation
Le métabolisme du fulvestrant n’a pas été pleinement étudié mais il implique divers processus éventuels de biotransformation analogues à ceux des stéroïdes endogènes. Les métabolites identifiés (incluant les métabolites 17-cétone, sulfone, 3-sulfate, 3- et 17-glucuronide) présentent une activité inférieure ou similaire à celle du fulvestrant dans les modèles d’études de l’activité anti-estrogénique. Les études réalisées avec des préparations de foie humain et des enzymes humaines recombinantes montrent que, parmi les iso-enzymes du cytochrome P450, seul le CYP3A4 intervient dans l’oxydation du fulvestrant, alors que des mécanismes autres que la voie P450 semblent prédominants in vivo. Les données obtenues in vitro suggèrent que le fulvestrant n’inhibe pas les isoenzymes du cytochrome P450.
Elimination
Le fulvestrant est principalement éliminé sous forme métabolisée. L’élimination se fait principalement dans les fèces, moins de 1 % de la dose étant éliminé dans les urines. La clairance du fulvestrant est élevée, 11 ± 1,7 mL/min/kg, suggérant un taux d’extraction hépatique élevé. La demi-vie terminale (t1/2) après administration intramusculaire dépend du taux d’absorption et a été estimée à 50 jours.
Populations particulières
L’analyse de pharmacocinétique de population effectuée sur les données des études de phase III n’a détecté aucune modification du profil pharmacocinétique du fulvestrant en fonction de l’âge (33 à 89 ans), du poids (40 à 127 kg) ou de l’origine ethnique des patientes.
Insuffisance rénale
Chez les patientes présentant une insuffisance rénale légère à modérée, aucune modification cliniquement significative des paramètres pharmacocinétiques du fulvestrant n’a été observée.
Insuffisance hépatique
La pharmacocinétique du fulvestrant a été évaluée en dose unique dans une étude clinique conduite chez des femmes atteintes d’une insuffisance hépatique légère à modérée (scores de Child-Pugh A et B). Une dose élevée d’une formulation injectable intramusculaire, de courte durée d’action, a été utilisée. L’ASC des femmes présentant une insuffisance hépatique a été jusqu’à environ 2,5 fois supérieure à celle des sujets sains. Chez les patientes recevant du fulvestrant, une augmentation de l’exposition de cette amplitude devrait être bien tolérée. Les femmes présentant une insuffisance hépatique sévère (score de Child-Pugh C) n’ont pas été évaluées.
Population pédiatrique
La pharmacocinétique du fulvestrant a été évaluée dans une étude clinique chez 30 filles présentant une puberté précoce progressive associée à un syndrome de McCune-Albright (voir rubrique 5.1). Les patientes pédiatriques étaient âgées de 1 à 8 ans et ont reçu, par voie intramusculaire, une dose mensuelle de 4 mg/kg de fulvestrant. La moyenne géométrique (écart-type) des concentrations minimales à l’état d’équilibre (Cmin,ss) et l’aire sous la courbe à l’état d’équilibre (ASCss) étaient respectivement de 4,2 (0,9) ng/mL et 3 680 (1 020) ng.h/mL. Bien que les données recueillies soient limitées, les concentrations minimales à l’état d’équilibre du fulvestrant chez les enfants semblent être alignées avec celles observées chez les adultes.
5.3. Données de sécurité préclinique
La toxicité aiguë du fulvestrant est faible.
Le fulvestrant et d’autres formulations de fulvestrant ont été bien tolérés dans les espèces animales étudiées lors des études à doses multiples. Des réactions locales au site d’injection, incluant myosite et granulome ont été attribuées aux excipients, mais la sévérité de la myosite chez le lapin a été plus importante dans le groupe fulvestrant que dans le groupe contrôle (solution saline). Lors des études de toxicité par administration intramusculaire réitérée menées chez le rat et le chien, la plupart des effets observés, particulièrement les effets sur les organes reproducteurs femelles mais aussi sur les organes hormono-sensibles des deux sexes, ont pu être attribués à l’activité anti-estrogénique du fulvestrant. Une artérite au niveau de différents tissus a été observée chez plusieurs chiens après une administration chronique (12 mois).
Dans des études chez des chiens, après administration orale ou intraveineuse, des effets sur le système cardio-vasculaire ont été observés : léger allongement du segment S-T de l’électrocardiogramme (voie orale) et pause sinusale chez un chien (voie intraveineuse). Ces effets sont apparus pour des expositions supérieures à celles utilisées chez des patientes (Cmax > 15 fois) et sont considérés comme peu significatifs en matière de sécurité d’emploi pour l’espèce humaine aux doses cliniques.
Le fulvestrant n’a montré aucun potentiel génotoxique.
Les effets constatés, à des doses similaires aux doses cliniques, sur la reproduction et sur le développement embryonnaire et fœtal sont la conséquence de l’activité anti-estrogène du fulvestrant. Chez les rats, une diminution réversible de la fertilité des femelles, une diminution de la survie embryonnaire, une dystocie et une augmentation de la fréquence des anomalies fœtales, y compris de la courbure du tarse, ont été observées. Chez des lapines ayant reçu du fulvestrant, la gestation n’a pu être maintenue. Une augmentation du poids du placenta et des pertes post-implantatoires des fœtus ont été observées. Chez les lapines, il y a eu une augmentation de l’incidence des modifications fœtales (bascule en arrière du bassin et de la vertèbre présacrée 27).
Une étude de cancérogénicité de deux ans chez les rats (administration intramusculaire de fulvestrant) a mis en évidence une augmentation de la fréquence des tumeurs ovariennes bénignes des cellules de la granulosa chez les rats femelles pour de fortes doses de 10 mg/rat/15 jours, et une augmentation des tumeurs des cellules testiculaires de Leydig chez les mâles. Lors d’une étude de cancérogénicité de deux ans chez la souris (administration orale quotidienne), il y a eu une augmentation de l’incidence des tumeurs du stroma et des cordons sexuels de l’ovaire (à la fois bénignes et malignes) à des doses de 150 et 500 mg/kg/jour. A la dose sans effet pour ces événements, les niveaux d’exposition systémique (ASC) étaient, chez les rats, approximativement 1,5 fois les niveaux d’exposition attendus chez la femelle et 0,8 fois chez le mâle et, chez les souris, approximativement 0,8 fois les niveaux d’exposition attendus à la fois chez le mâle et chez la femelle. L’induction de telles tumeurs est cohérente avec les modifications pharmacologiques endocriniennes des taux de gonadotropine provoquées par les anti-estrogènes chez des animaux en activité hormonale. De ce fait, ces résultats ne sont pas considérés comme significatifs dans le cadre de l’utilisation du fulvestrant chez des femmes ménopausées souffrant d’un cancer du sein au stade avancé.
Evaluation du risque environnemental (ERE)
Les études sur l’évaluation du risque environnemental ont montré que le fulvestrant présente un potentiel de toxicité pour l’environnement aquatique (voir rubrique 6.6).
Ethanol (96 %), alcool benzylique (E1519), benzoate de benzyle, huile de ricin raffinée.
6.4. Précautions particulières de conservation
A conserver et transporter réfrigéré (entre 2 °C et 8 °C).
Les excursions de température en dehors de 2 °C–8 °C doivent être limitées. La conservation à des températures supérieures à 30 °C est exclue, et la durée de conservation à une température moyenne pour le produit inférieure à 25 °C (mais supérieure à 2 °C–8 °C) ne doit pas dépasser 28 jours. Après des excursions de température, le produit doit être replacé immédiatement dans les conditions de conservation recommandées (à conserver et transporter réfrigéré entre 2 °C et 8 °C). Les excursions de température ont un effet cumulatif sur la qualité du produit et la durée de 28 jours ne doit pas être dépassée au cours de la durée de conservation de 2 ans de fulvestrant (voir rubrique 6.3). Une exposition à des températures inférieures à 2 °C n’endommagera pas le produit sous réserve de ne pas le conserver en dessous de -20 °C.
Conserver la seringue préremplie dans l’emballage d’origine à l’abri de la lumière.
6.5. Nature et contenu de l'emballage extérieur
La présentation en seringue préremplie se compose de :
Une seringue préremplie en verre transparent de type I, avec tige de piston en polystyrène et bouchon de piston en élastomère, munie d’un embout rigide en plastique, contenant 5 mL de solution de fulvestrant pour injection. Une aiguille protégée (BD SafetyGlide) à connecter au corps de la seringue est également fournie.
Ou
Deux seringues préremplies en verre transparent de type I, avec tige de piston en polystyrène et bouchon de piston en élastomère, munies d’un embout rigide en plastique, contenant chacune 5 mL de solution de fulvestrant pour injection. Deux aiguilles protégées (BD SafetyGlide) à connecter à chaque corps de la seringue sont également fournies.
Ou
2 x deux seringues préremplies en verre transparent de type I, avec tige de piston en polystyrène et bouchon de piston en élastomère, munies d'un embout rigide en plastique, contenant chacune 5 mL de solution de fulvestrant pour injection. Deux aiguilles protégées (BD SafetyGlide) à connecter à chaque corps de la seringue sont également fournies, pour chaque emballage.
Ou
Quatre seringues préremplies en verre transparent de type I avec tige de piston en polystyrène et bouchon de piston en élastomère, munies d'un embout rigide en plastique, contenant chacune 5 ml de solution de fulvestrant pour injection. Quatre aiguilles protégées (BD SafetyGlide) à connecter à chaque corps de la seringue sont également fournies.
Ou
Six seringues préremplies en verre transparent de type I, avec tige de piston en polystyrène et bouchon de piston en élastomère, munies d’un embout rigide en plastique, contenant chacune 5 mL de solution de fulvestrant pour injection. Six aiguilles protégées (BD SafetyGlide) à connecter à chaque corps de la seringue sont également fournies.
Toutes les présentations peuvent ne pas être commercialisées.
6.6. Précautions particulières d’élimination et de manipulation
Instructions pour l’administration
Administrer l'injection selon les recommandations locales pour réaliser des injections intramusculaires de grand volume.
REMARQUE : en raison de la proximité du nerf sciatique sous-jacent, des précautions doivent être prises en cas d'administration de fulvestrant au site d'injection dorso-fessier (voir rubrique 4.4).
Avertissement — Ne jamais stériliser à l’autoclave l’aiguille protégée (aiguille hypodermique protégée BD SafetyGlideTM) avant utilisation. Pendant toute la durée de l’utilisation et de la procédure d’élimination de l’aiguille, les mains doivent toujours rester derrière l’aiguille.
Pour chacune des deux seringues :
· Retirez le corps de la seringue en verre du plateau-support et vérifiez qu’il n’a pas été endommagé.
· Retirez l’emballage extérieur de l’aiguille protégée (SafetyGlide).
· Les solutions à usage parentéral doivent être contrôlées visuellement avant administration afin de détecter la présence de particules ou un changement de coloration.
· Tenez la seringue à la verticale sur la partie nervurée (C). Avec l'autre main, saisissez le capuchon (A) et tournez soigneusement l’embout en plastique rigide dans le sens inverse des aiguilles d’une montre (voir Figure 1) :
Figure 1
· Retirez le capuchon (A) droit vers le haut. Pour maintenir la stérilité, ne pas toucher l’embout de la seringue (B) (voir figure 2).
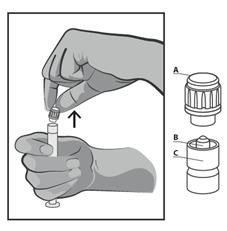
Figure 2
· Fixez l’aiguille protégée sur la connexion Luer Lock et tournez fermement jusqu’à fixation (voir figure 3).
· Vérifiez que l'aiguille est verrouillée sur la connexion Luer avant de bouger hors du plan vertical.
· Débloquez le protège-aiguille en tirant d’un coup sec afin de ne pas en endommager la pointe de l’aiguille.
· Transportez la seringue remplie au point d’administration.
· Retirez le manchon d’aiguille.
· Chassez les bulles de la seringue.
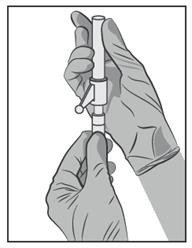
Figure 3
· Injectez lentement en intramusculaire (1–2 minutes/injection) dans le muscle fessier (zone du fessier). Pour plus de facilité, le biseau de l’aiguille est orienté du côté du bras du levier (voir figure 4).
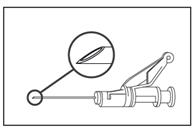
Figure 4
· Après injection, enclenchez immédiatement le dispositif de protection de l’aiguille en poussant à fond le bras du levier avec le doigt (voir figure 5).
REMARQUE : tenez la seringue à distance de vous-même et d’autrui pour enclencher la protection. Le clic doit se faire entendre et vous devez vérifier visuellement que l’aiguille a bien été complètement recouverte.
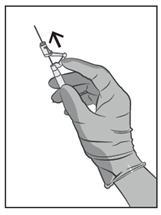
Figure 5
Elimination
Les seringues préremplies sont exclusivement à usage unique.
Ce médicament peut présenter un risque pour l’environnement aquatique. Tout médicament non utilisé ou déchet doit être éliminé conformément à la réglementation en vigueur (voir rubrique 5.3).
7. TITULAIRE DE L’AUTORISATION DE MISE SUR LE MARCHE
35 RUE DU VAL DE MARNE
75013 PARIS
8. NUMERO(S) D’AUTORISATION DE MISE SUR LE MARCHE
· 34009 302 137 7 3 : 5 mL de solution en seringue préremplie (verre), boîte de 2 seringues + 2 aiguilles.
· 34009 550 759 9 8 : 5 mL de solution en seringue préremplie (verre), boîte de 6 seringues + 6 aiguilles.
· 34009 302 235 7 4 : 5 mL de solution en seringue préremplie (verre), boîte de 2 x 2 seringues + 2 x 2 aiguilles.
· 34009 550 759 9 8 : 5 mL de solution en seringue préremplie (verre), boîte de 4 seringues + 4 aiguilles.
9. DATE DE PREMIERE AUTORISATION/DE RENOUVELLEMENT DE L’AUTORISATION
[à compléter ultérieurement par le titulaire]
Date de première autorisation:{JJ mois AAAA}
10. DATE DE MISE A JOUR DU TEXTE
[à compléter ultérieurement par le titulaire]
{JJ mois AAAA}
Sans objet.
12. INSTRUCTIONS POUR LA PREPARATION DES RADIOPHARMACEUTIQUES
Liste I
|
|
| Plan du site | Accessibilité | Contact | Téléchargement | Declaration de confidentialité | Service-Public.fr | Legifrance | Gouvernement.fr |