Dernière mise à jour le 03/08/2026
FIBRYGA 1 g, poudre et solvant pour solution injectable/pour perfusion
Indications thérapeutiques
Classe pharmacothérapeutique : antihémorragiques, fibrinogène humain, code ATC : B02BB01
Qu’est-ce que FIBRYGA, poudre et solvant pour solution injectable/pour perfusion ?
FIBRYGA contient du fibrinogène humain, une protéine importante pour la coagulation du sang. Un manque en fibrinogène fait que le sang ne coagule pas aussi bien qu'il ne le devrait, ce qui augmente la tendance aux saignements. Le remplacement du fibrinogène humain par FIBRYGA corrigera le défaut de coagulation.
Dans quel cas FIBRYGA, poudre et solvant pour solution injectable/pour perfusion est-il utilisé ?
FIBRYGA est utilisé pour :
· le traitement des épisodes de saignement et la prophylaxie en cas d’intervention chirurgicale chez des patients présentant une hypo- ou afibrinogènémie congénitale, avec une tendance aux saignements ;
· la supplémentation en fibrinogène chez les patients présentant un saignement grave non contrôlé, accompagné d’un déficit acquis en fibrinogène pendant une intervention chirurgicale.
Présentations
> 1 flacon en verre de 1 g - 1 flacon en verre de 50 mL avec 1 dispositif de transfert nextaro
Code CIP : 34009 302 111 6 8
Déclaration de commercialisation : 11/01/2021
Cette présentation est agréée aux collectivités
Service médical rendu (SMR)
Les libellés affichés ci-dessous ne sont que des résumés ou extraits issus des avis rendus par la Commission de la Transparence. Seul l'avis complet de la Commission de la Transparence fait référence.
Cet avis est consultable à partir du lien `Avis du jj/mm/aaaa` ou encore sur demande auprès de la HAS (plus d'informations dans l'aide). Les avis et synthèses d'avis contiennent un paragraphe sur la place du médicament dans la stratégie thérapeutique.
| Valeur du SMR | Avis | Motif de l'évaluation | Résumé de l'avis |
|---|---|---|---|
| Important | Avis du 07/04/2021 | Inscription (CT) | Le service médical rendu par FIBRYGA (fibrinogène humain) est important dans les hypo-, dys- ou afibrinogénémies constitutionnelles et dans l'hypofibrinogénémie acquise au cours d’une intervention chirurgicale. |
Amélioration du service médical rendu (ASMR)
Les libellés affichés ci-dessous ne sont que des résumés ou extraits issus des avis rendus par la Commission de la Transparence. Seul l'avis complet de la Commission de la Transparence fait référence.
Cet avis est consultable à partir du lien `Avis du jj/mm/aaaa` ou encore sur demande auprés de la HAS (plus d'informations dans l'aide). Les avis et synthéses d'avis contiennent un paragraphe sur la place du médicament dans la stratégie thérapeutique.
| Valeur de l'ASMR | Avis | Motif de l'évaluation | Résumé de l'avis |
|---|---|---|---|
| V (Inexistant) | Avis du 07/04/2021 | Inscription (CT) | En l’absence de démonstration d’un bénéfice en termes d’efficacité ou de tolérance en comparaison à un autre concentré de fibrinogène, la Commission considère que FIBRYGA (fibrinogène humain) n’apporte pas d’amélioration du service médical rendu (ASMR V) par rapport aux autres concentrés de fibrinogène disponibles dans le traitement des saignements et la prophylaxie périopératoire chez des patients présentant une hypo- ou afibrinogénémie congénitale, avec une tendance aux saignements. En l’absence de démonstration d’un bénéfice en termes d’efficacité ou de tolérance en comparaison au concentré de fibrinogène CLOTTAFACT, la Commission considère que FIBRYGA n’apporte pas d’amélioration du service médical rendu (ASMR V) par rapport au concentré de fibrinogène CLOTTAFACT en tant que traitement complémentaire dans la prise en charge d’une hémorragie sévère incontrôlée chez les patients présentant une hypo fibrinogénémie acquise au cours d’une intervention chirurgicale. |
Autres informations
- Titulaire de l'autorisation : OCTAPHARMA France
- Conditions de prescription et de délivrance :
- liste I
- prescription hospitalière
- Statut de l'autorisation : Valide
- Type de procédure : Procédure de reconnaissance mutuelle
- Code CIS : 6 976 812 3
ANSM - Mis à jour le : 16/07/2024
FIBRYGA 1 g, poudre et solvant pour solution injectable/pour perfusion
2. COMPOSITION QUALITATIVE ET QUANTITATIVE
Fibrinogène humain
Chaque flacon de FIBRYGA contient 1 g de fibrinogène humain. FIBRYGA contient approximativement 20 mg/mL de fibrinogène humain après reconstitution avec 50 mL d'eau pour préparations injectables.
Le contenu en facteurs de coagulation est déterminé conformément à la Pharmacopée européenne pour le fibrinogène humain.
Produit à partir du plasma de donneurs humains.
Excipient(s) à effet notoire : jusqu'à 132 mg (5,8 mmol) de sodium par flacon.
Pour la liste complète des excipients, voir rubrique 6.1.
Poudre et solvant pour solution injectable/pour perfusion.
La poudre est hygroscopique, a également l’aspect d’un amas friable, et de couleur blanche ou jaune pâle.
Le solvant est un liquide transparent et incolore.
4.1. Indications thérapeutiques
Traitement complémentaire dans la prise en charge d’une hémorragie sévère incontrôlée chez les patients présentant une hypofibrinogènémie acquise au cours d’une intervention chirurgicale.
4.2. Posologie et mode d'administration
Posologie
La dose et la durée du traitement substitutif dépendent de la sévérité des troubles, de la localisation, de l'étendue du saignement, et de l'état clinique du patient.
La concentration (fonctionnelle) en fibrinogène doit être déterminée afin de calculer la posologie individuelle. Le nombre et la fréquence des injections doivent être adaptés à chaque patient en fonction du dosage régulier de la concentration plasmatique en fibrinogène et du suivi continu de l'état clinique du patient, et des autres traitements de substitution utilisés.
En cas d'intervention chirurgicale majeure, une surveillance étroite du traitement de substitution par des tests de coagulation est essentielle.
1. Prophylaxie chez des patients présentant une hypo- ou afibrinogènémie congénitale avec une tendance connue aux saignements.
Pour prévenir un saignement excessif au cours de procédures chirurgicales, un traitement prophylactique est recommandé pour augmenter la concentration en fibrinogène jusqu'à 1 g/l puis de la maintenir à ce niveau jusqu'à l'obtention de l'hémostase, et au-dessus de 0,5 g/l jusqu'à guérison complète des plaies.
En cas de procédure chirurgicale ou de traitement d'un épisode de saignement, la dose doit être calculée comme suit :
Dose (mg/kg de poids corporel) = [Concentration cible (g/L) - concentration mesurée (g/L)]
0,018 (g/L par mg/kg de poids corporel)
La posologie suivante (dose et fréquence des injections) doit être adaptée en fonction de l'état clinique du patient et des résultats des tests de laboratoire.
La demi-vie biologique du fibrinogène est de 3 à 4 jours. Ainsi, en l'absence de dégradation, un traitement répété avec le fibrinogène humain n'est habituellement pas nécessaire. Compte tenu de l'accumulation survenant en cas d'administration répétée dans un but prophylactique, la dose et la fréquence doivent être déterminées en fonction des objectifs thérapeutiques du médecin pour chaque patient.
Population pédiatrique
En cas de procédure chirurgicale ou de traitement d’un épisode de saignement, la dose pour les adolescents doit être calculée selon la formule indiquée ci‑dessus pour les adultes, tandis que la dose pour les enfants âgés de moins de 12 ans doit être calculée comme suit :
Dose (mg/kg de poids corporel) = [Concentration cible (g/L) - concentration mesurée (g/L)]
0,014 (g/L par mg/kg de poids corporel)
La posologie doit être adaptée en fonction de l’état clinique du patient et des résultats des tests de laboratoire.
Patients âgés
Les études cliniques menées avec FIBRYGA n'ont pas inclus de patients âgés de 65 ans et plus pour fournir des données probantes et concluantes permettant de savoir si ces patients répondent différemment au traitement par rapport à des patients plus jeunes.
2. Traitement des saignements
Saignements chez des patients présentant une hypo- ou afibrinogènémie congénitale
Les épisodes de saignement doivent être traités en utilisant les formules ci-dessus adaptées aux adultes/adolescents ou aux enfants, respectivement de manière à atteindre la concentration plasmatique cible de 1 g/l recommandée en fibrinogène. Ce taux doit être maintenu jusqu'à ce qu'une hémostase durable soit obtenue.
Saignements chez des patients présentant un déficit acquis en fibrinogène
Adultes
Une dose de 1-2 g est généralement administrée initialement, suivie de perfusions autant que nécessaire. En cas d’hémorragie sévère, par exemple au cours d’une intervention chirurgicale majeure, de grandes quantités (4-8 g) de fibrinogène peuvent être nécessaires.
Population pédiatrique
La posologie doit être déterminée en fonction du poids corporel et des besoins cliniques mais se situe généralement entre 20 et 30 mg/kg.
Mode d’administration
Perfusion ou injection intraveineuse.
FIBRYGA doit être administré lentement par voie intraveineuse à la vitesse maximale recommandée de 5 ml/min chez les patients présentant une hypo- ou afibrinogènémie congénitale. La vitesse maximale recommandée est de 10 ml/min chez les patients présentant un déficit acquis en fibrinogène.
Pour des instructions concernant la reconstitution du médicament avant administration, voir la rubrique 6.6.
Hypersensibilité à la substance active ou à l’un des excipients mentionnés à la rubrique 6.1
4.4. Mises en garde spéciales et précautions d'emploi
Pour améliorer la traçabilité des médicaments biologiques, le nom et le numéro de lot du produit administré doivent être clairement consignés.
Thrombo-embolie
Il existe un risque de thrombose chez les patients présentant un déficit congénital ou acquis, traités avec du fibrinogène humain, en particulier à fortes doses ou à répétition. Les patients recevant du fibrinogène humain doivent être étroitement surveillés afin de détecter des signes ou symptômes de thrombose.
Le bénéfice potentiel du traitement par du fibrinogène humain devra être soigneusement évalué en fonction du risque de complications thrombo-emboliques chez les patients ayant des antécédents de maladie coronarienne ou d'infarctus du myocarde, les patients ayant une maladie hépatique, les patients en période périopératoire ou postopératoire, chez les nouveau-nés ou chez les patients à risque d'événements thrombo-emboliques ou de coagulation intravasculaire disséminée. Il est nécessaire de faire preuve de prudence et d'un suivi attentif.
L'hypofibrinogénémie acquise est associée à de faibles concentrations plasmatiques de tous les facteurs de coagulation (pas seulement le fibrinogène) et d'inhibiteurs. Il convient donc d’envisager un traitement avec des produits sanguins contenant des facteurs de coagulation. Une surveillance minutieuse de la coagulation est nécessaire.
Réactions allergiques ou de type anaphylactique
L'injection/perfusion doit être arrêtée immédiatement en cas de survenue de réactions allergiques ou de type anaphylactique. En cas de choc anaphylactique, le traitement médical standard en ces circonstances doit être instauré.
Teneur en sodium
Ce médicament contient jusqu'à 132 mg de sodium par flacon, ce qui équivaut à 6,6 % de l’apport alimentaire quotidien maximal recommandé par l’OMS de 2 g de sodium pour un adulte. Cela doit être pris en compte chez les patients suivant un régime contrôlé en sodium.
Sécurité vis-à-vis des virus
Les mesures classiques de prévention des infections liées à l’utilisation de médicaments préparés à partir de sang ou de plasma humain incluent la sélection des donneurs, le dépistage dans chaque don et pool de plasma de marqueurs spécifiques d’infection et l’inclusion d’étapes de fabrication pour l'inactivation/élimination efficace des virus. Malgré cela, lorsque des médicaments préparés à partir de sang ou de plasma humain sont administrés, la possibilité de transmettre des agents infectieux ne peut pas être totalement écartée. Cela concerne également des virus inconnus ou émergents et d’autres agents pathogènes.
Les mesures prises sont jugées efficaces contre les virus enveloppés, tels que le VIH, le VHB et le VHC, et contre les virus non-enveloppés tels que le VHA. Les mesures prises peuvent n'avoir qu'une valeur limitée contre les virus non-enveloppés, tels que le parvovirus B19. L'infection par le parvovirus B19 peut être grave chez la femme enceinte (infection du fœtus) et chez les personnes ayant une immunodéficience ou une érythropoïèse augmentée (p. ex., anémie hémolytique).
Une vaccination appropriée (hépatite A et B) doit être envisagée pour les patients qui reçoivent régulièrement/de façon répétée des produits dérivés du plasma humain.
Immunogénicité
Des réactions immunologiques ont été observées dans le cas de traitements de substitution avec des facteurs de coagulation dans les autres déficits congénitaux, mais il n'existe pas actuellement de données avec les concentrés de fibrinogène.
4.5. Interactions avec d'autres médicaments et autres formes d'interactions
Aucune interaction entre les produits à base de fibrinogène humain et un autre médicament n'est connue.
4.6. Fertilité, grossesse et allaitement
Grossesse
La sécurité d’utilisation de FIBRYGA chez les femmes enceintes n’a pas été établie au cours d’études cliniques contrôlées. Les données cliniques sur les produits à base de fibrinogène dans le traitement de complications obstétricales laissent penser qu’aucun effet délétère sur le déroulement de la grossesse ou sur la santé du fœtus ou du nouveau-né ne devrait être attendu. Aucune étude sur la reproduction animale n’a été menée avec FIBRYGA (voir rubrique 5.3). Dans la mesure où la substance active est d'origine humaine, elle est catabolisée de la même manière que les propres protéines des patients. Ces composants physiologiques du sang humain ne devraient pas induire d'effets indésirables sur la reproduction ou sur le fœtus.
Le bénéfice de FIBRYGA au cours de la grossesse doit être évalué en tenant compte de la disponibilité des données provenant de l’expérience clinique sur les concentrés en fibrinogène, mais également de l’absence de données issues d’études cliniques contrôlées.
Allaitement
On ignore si FIBRYGA est excrété dans le lait humain. Toutefois, en raison de la nature de la substance, aucun effet sur le nouveau-né/nourrisson allaité n’est attendu.
Ainsi, la décision quant à l’utilisation de FIBRYGA au cours de l’allaitement doit tenir compte du bénéfice de l’allaitement pour l’enfant et du bénéfice du traitement pour la mère.
Fertilité
Aucune donnée n'est disponible sur la fertilité.
4.7. Effets sur l'aptitude à conduire des véhicules et à utiliser des machines
FIBRYGA n'a aucune influence sur l'aptitude à conduire des véhicules et à utiliser des machines.
Résumé du profil de sécurité
Il n'existe pas de données solides sur la fréquence des effets indésirables liés à ce produit au cours des études cliniques.
Les effets indésirables suivants ont été décrits au cours des études cliniques : fièvre, éruption cutanée liée au médicament, phlébite et thrombose.
Les effets indésirables suivants ont été décrits pour FIBRYGA et d'autres concentrés de fibrinogène :
|
Classe de systèmes d’organes MedDRA usuelle |
Effets indésirables |
Fréquence* |
|
Affections du système immunitaire : |
Réactions allergiques ou de type anaphylactique Réactions cutanées |
Inconnue |
|
Affections vasculaires : |
Épisodes thromboemboliques (y compris infarctus du myocarde et embolie pulmonaire) (voir rubrique 4.4) Thrombophlébite |
Inconnue |
|
Troubles généraux et anomalies au site d’administration : |
Élévation de la température corporelle (pyrexie) |
Inconnue |
*Fréquence inconnue car elle n’a pas pu être déterminée à partir des données disponibles. Une légère pyrexie et une réaction cutanée se sont produites une fois au cours des études cliniques. Les réactions allergiques ou de type anaphylactique, des épisodes thromboemboliques (y compris infarctus du myocarde et embolie pulmonaire) et une thrombophlébite sont des effets propres à la classe.
Pour la sécurité envers des agents transmissibles, voir rubrique 4.4.
Population pédiatrique
Vingt‑six patients, âgés de 1 an à moins de 18 ans, ont été inclus dans l'analyse de sécurité effectuée dans le déficit congénital en fibrinogène, parmi lesquels : 12 adolescents âgés de 12 ans à moins de 18 ans, 8 enfants âgés de 6 ans à moins de 12 ans et 6 enfants âgés de 1 an à moins de 6 ans.
Le profil global de sécurité des enfants et des adolescents n'est pas différent de celui des adultes.
Il n’existe aucune donnée relative à l’utilisation de FIBRYGA chez les patients pédiatriques présentant un déficit acquis en fibrinogène.
Déclaration des effets indésirables suspectés
La déclaration des effets indésirables suspectés après autorisation du médicament est importante. Elle permet une surveillance continue du rapport bénéfice/risque du médicament. Les professionnels de santé déclarent tout effet indésirable suspecté via le système national de déclaration : Agence nationale de sécurité du médicament et des produits de santé (ANSM) et réseau des Centres Régionaux de Pharmacovigilance - Site internet : https://signalement.social-sante.gouv.fr/.
Une surveillance régulière du taux de fibrinogène plasmatique est recommandée pour éviter un surdosage (voir rubrique 4.2).
En cas de surdosage, le risque de survenue de complications thromboemboliques est accru.
5. PROPRIETES PHARMACOLOGIQUES
5.1. Propriétés pharmacodynamiques
Classe pharmacothérapeutique : antihémorragiques, fibrinogène, code ATC : B02BB01
Le fibrinogène humain (Facteur I de la coagulation), en présence de thrombine, de Facteur XIII activé (FXIIIa) de la coagulation, et d’ions calciums, est converti en réseau de fibrine hémostatique tridimensionnel, élastique et stable.
L'administration de fibrinogène humain induit une augmentation du taux de fibrinogène plasmatique et peut corriger temporairement le défaut de coagulation chez les patients porteurs d'un déficit en fibrinogène.
Une étude pharmacocinétique de phase 2, croisée à deux bras en dose unique, ouverte, prospective, randomisée et contrôlée, chez 22 patients présentant un déficit congénital en fibrinogène (afibrinogènémie) (voir rubrique 5.2) a également évalué l’efficacité hémostatique grâce au critère substitutif de la fermeté maximale du caillot (FMC) (FORMA-01). Le FMC a été déterminé par thromboélastométrie (ROTEM). Le FMC a été déterminé pour chaque patient avant (valeur à l’état basal) et une heure après l'administration d'une dose unique de FIBRYGA. Les valeurs du FMC après l'administration de FIBRYGA sont significativement plus élevées que les valeurs à l’état basal (voir le tableau ci-dessous).
Tableau 1 : Fermeté Maximum du Caillot (FMC) [mm] (population en ITT) ; n = 22
|
Point temporel |
Moyenne ± Ecart-type |
Médiane (étendue) |
|
Avant la perfusion |
0 ± 0 |
0 (0 - 0) |
|
1 heure après la perfusion |
9,7 ± 3,0 |
10,0 (4,0 - 16,0) |
|
Variation moyenne (analyse principale)* |
9,7 ± 3,0 |
10,0 (4,0 - 16,0) |
FMC= Fermeté maximum du caillot ; ITT = intention de traiter.
*P < 0,0001 (intervalle de confiance de 95 % : 8,37 à 10,99)
Une étude prospective, ouverte, multicentrique, non contrôlée de phase 3 (FORMA-02) a été menée chez 25 patients âgés de 12 à 54 ans (6 adolescents, 19 adultes) présentant un déficit congénital en fibrinogène (afibrinogénémie et hypofibrinogénémie). Le traitement de 89 épisodes de saignement et 12 procédures chirurgicales a été analysé durant cette étude. Il y a eu un changement significatif de la FMC mesurée par ROTEM et des concentrations plasmatiques en fibrinogène, par rapport aux valeurs de référence.
La dose médiane de FIBRYGA par perfusion pour le traitement des épisodes de saignement était de 57,5 mg/kg et la dose totale médiane était de 59,4 mg/kg. La dose totale médiane de FIBRYGA par intervention chirurgicale était de 85,8 mg/kg. Pour 98,9 % des épisodes de saignement traités et 100 % des interventions chirurgicales, l’efficacité hémostatique globale a été évaluée en succès (bonne ou excellente efficacité) par le comité indépendant d’arbitrage utilisant un système de notation objectif.
Une autre étude prospective, ouverte, multicentrique, non contrôlée de phase 3 (FORMA‑04) a été menée chez 14 enfants âgés de 1 à 10 ans (6 enfants de moins de 6 ans et 8 enfants âgés de 6 ans à moins de 12 ans) présentant un déficit congénital en fibrinogène (afibrinogénémie et hypofibrinogénémie). Le traitement de 10 épisodes de saignement et 3 procédures chirurgicales, ainsi qu’une évaluation de la pharmacocinétique d’une dose unique ont été évalués dans cette étude. Il y a eu un changement significatif de la FMC mesurée par ROTEM et des concentrations plasmatiques en fibrinogène par rapport aux valeurs de référence. La dose médiane de FIBRYGA par perfusion pour le traitement des épisodes de saignement était de 70,2 mg/kg et la dose totale médiane était de 73,9 mg/kg. La dose totale médiane de FIBRYGA par intervention chirurgicale était de 108 mg/kg. Pour 100 % des épisodes de saignement traités et des interventions chirurgicales, l’efficacité hémostatique globale a été évaluée en succès (bonne ou excellente efficacité) par le comité indépendant d’arbitrage utilisant un système de notation objectif.
L’étude prospective randomisée et contrôlée FORMA-05 a étudié l’efficacité hémostatique et la sécurité de FIBRYGA par rapport au cryoprécipité comme source de supplémentation en fibrinogène chez des patients présentant un déficit acquis en fibrinogène lors d’une intervention chirurgicale de réduction tumorale pour le pseudomyxome péritonéal étendu abdominal. L’étude des données conformément au protocole a porté sur 43 patients adultes (PP), 21 patients traités par FIBRYGA et 22 patients traités avec le cryoprécipité. En peropératoire, une supplémentation en fibrinogène a été réalisée préventivement (à savoir après 60-90 minutes d’intervention chirurgicale, lors d’une hémorragie sévère, mais avant une perte supérieure à 2 Litres de sang) avec des doses de 4 gr de FIBRYGA ou de 2 mélanges de 5 unités de cryoprécipité, répétés si nécessaire. Au cours des 7,8 ± 1,7 heures de chirurgie, 6,5 ± 3 g de FIBRYGA (89 ± 39 mg/kg de poids corporel) et 4,1 ± 2,2 pools de 5 unités de cryoprécipité ont été utilisés, respectivement. Une médiane de 1 unité et 0,5 unité de CGR ont été administrées en peropératoire aux patients traités par FIBRYGA et par cryoprécipité, avec une médiane de 0 unité de CGR au cours des 24 premières heures postopératoires dans les deux groupes (voir le tableau ci-dessous). Aucun Plasma Frais Congelé ou Concentré Plaquettaire n’a été transfusé au cours de l’étude. Un traitement hémostatique basé sur la supplémentation en fibrinogène a été jugé réussi pour 100 % des interventions chirurgicales dans les deux groupes par le comité indépendant d’arbitrage utilisant un système de notation objectif.
Tableau 2 : Transfusion de CGR* [unités] en peropératoire et au cours des 24 premières heures en postopératoire (population PP)
|
Intervalle de temps |
Groupe FIBRYGA (n=21) Médiane (étendue) |
Groupe de cryoprécipité (n=22) Médiane (étendue) |
|
Peropératoire |
1 (0-4) |
0,5 (0-5) |
|
24 premières heures en postopératoire |
0 (0-2) |
0 (0-2) |
CGR = concentrés de Globules Rouges; PP = Per Protocole.
*pas de transfusion d’autres produits sanguins allogéniques comme les plasmas frais congelés ou concentrés plaquettaires.
Population pédiatrique
Dans le déficit congénital en fibrinogène, FIBRYGA a été administré à 20 patients âgés de 1 an à moins de 18 ans (6 adolescents âgés de 12 ans à moins de 18 ans, 8 enfants âgés de 6 ans à moins de 12 ans et 6 enfants âgés de 1 an à moins de 6 ans) dans le cadre de 2 études cliniques (FORMA‑02 et FORMA‑04). Selon un comité indépendant d’arbitrage, l’efficacité hémostatique a été jugée en succès pour la totalité des épisodes de saignement traités (10 épisodes de saignement chez les adolescents, 5 chez les enfants âgés de 6 ans à moins de 12 ans et 5 chez les enfants âgés de 1 an à moins de 6 ans). L’efficacité hémostatique a également été jugée en succès en prophylaxie pour les 4 interventions chirurgicales pratiquées chez ces patients (1 chez un adolescent et 3 chez des enfants âgés de 1 an à moins de 6 ans).
5.2. Propriétés pharmacocinétiques
Une étude de phase 2 croisée à deux bras ouverte, prospective, randomisée et contrôlée menée chez 22 patients présentant un déficit congénital en fibrinogène (afibrinogènémie), âgés de 12 à 53 ans (6 adolescents, 16 adultes), a comparé chez les mêmes patients les propriétés pharmacodynamiques d'une dose unique de FIBRYGA à celles d'un autre concentré de fibrinogène commercialisé (FORMA‑01). Chaque patient a reçu une dose intraveineuse unique de 70 mg/kg de FIBRYGA et du produit comparatif. Des échantillons de sang ont été prélevés pour déterminer l'activité du fibrinogène avant l'administration et jusqu'à 14 jours après la perfusion. Les paramètres pharmacocinétiques de FIBRYGA dans l'analyse per protocole (PP) (n = 21) sont résumés dans le tableau ci-dessous.
Tableau 3 : Paramètres pharmacocinétiques (n = 21) de l'activité du fibrinogène (population PP*)
|
Paramètre |
Moyenne ± ÉT |
Écart |
|
Demi-vie (heure) |
75,9 ± 23,8 |
40,0 à 157,0 |
|
Cmax (mg/dL) |
139,0 ± 36,9 |
83,0 à 216,0 |
|
ASCnorm pour une dose de 70 mg/kg (mg*h/mL) |
113,7 ± 31,5 |
59,7 à 175,5 |
|
Clairance (mL/h/kg) |
0,67 ± 0,2 |
0,4 à 1,2 |
|
Temps moyen résiduel (h) |
106,3 ± 30,9 |
58,7 à 205,5 |
|
Volume de distribution à la phase de plateau (mL/kg) |
70,2 ± 29,9 |
36,9 à 149,1 |
* Un patient exclu de la population PP parce qu'ayant reçu < 90 % de la dose prévue de FIBRYGA et du comparateur.
Cmax = concentration plasmatique maximum ; ASCnorm = aire sous la courbe normalisée pour la dose administrée ; ÉT = écart-type
La récupération incrémentielle in vivo (IVR) a été déterminée à partir des taux obtenus jusqu'à 4 heures après la perfusion. L'IVR incrémentiel médian était une augmentation de 1,8 mg/dL (écart : 1,08 à 2,62 mg/dL) par mg/kg. L'IVR médian indique qu'une dose de 70 mg/kg augmentera la concentration plasmatique du fibrinogène du patient d'environ 125 mg/dL.
Pharmacocinétique dans des populations particulières
Aucune différence statistiquement pertinente de l'activité du fibrinogène n'a été observée entre les participants masculins et féminins à l'étude.
Population pédiatrique
Les données de pharmacocinétique chez les adolescents âgés de 12 ans à moins de 18 ans proviennent de l’étude FORMA‑02. Dans l’analyse PP, une légère différence de valeur de la demi-vie a été observée entre les adolescents (n = 5) et les adultes (n = 16), avec respectivement 72,8 ± 16,5 heures et 76,9 ± 26,1 heures. La clairance était pratiquement identique dans les deux groupes d’âge, avec des valeurs de 0,68 ± 0,18 mL/h/kg et de 0,66 ± 0,21 mL/h/kg, respectivement.
Les propriétés pharmacocinétiques de FIBRYGA ont ensuite été évaluées dans l’étude FORMA‑04 chez 13 enfants âgés de moins de 12 ans présentant un déficit congénital en fibrinogène (afibrinogénémie). Chaque patient a reçu une dose intraveineuse unique de 70 mg/kg de FIBRYGA. Les paramètres pharmacocinétiques de FIBRYGA sont résumés dans le tableau ci‑dessous. La récupération incrémentielle in vivo médiane correspondait à une augmentation de 1,4 mg/dL (intervalle : 1,3 à 2,1 mg/dL) par mg/kg.
Tableau 4 : Paramètres pharmacocinétiques (n = 13) pour l’activité du fibrinogène
|
Paramètre |
Moyenne ± ÉT |
Intervalle |
|
Demi-vie (heure) |
63,3 ± 12,0 |
45,6 à 91,6 |
|
Cmax (mg/dL) |
107,2 ± 16,8 |
93,0 à 154,0 |
|
ASCnorm pour une dose de 70 mg/kg (mg*h/mL)* |
92,0 ± 20,0 |
69,7 à 134,2 |
|
Clairance (mL/h/kg)* |
0,8 ± 0,2 |
0,5 à 1,0 |
|
Temps moyen résiduel (h)* |
88,0 ± 16,8 |
63,6 à 126,7 |
|
Volume de distribution à l’état d’équilibre (mL/kg)* |
67,6 ± 7,1 |
52,8 à 76,8 |
* Paramètres calculés pour 10 des 13 patients en raison d’un nombre insuffisant de valeurs quantifiables chez 3 patients.
Cmax = concentration plasmatique maximale ; ASCnorm = aire sous la courbe normalisée pour la dose administrée ; ÉT = écart-type
5.3. Données de sécurité préclinique
Chlorhydrate de L-arginine
Glycine
Chlorure de sodium
Citrate de sodium dihydraté
Solvant
Eau pour préparations injectables
Ce médicament ne doit pas être mélangé avec d’autres médicaments.
3 ans.
La stabilité physicochimique en cours d'utilisation de la solution reconstituée pendant 24 heures à température ambiante (max. : 25°C) a été démontrée. D'un point de vue microbiologique, le produit doit être utilisé immédiatement après reconstitution. S'il n'est pas utilisé immédiatement, les délais et conditions de conservation en cours d'utilisation sont de la responsabilité de l'utilisateur. La solution reconstituée ne doit pas être congelée ou conservée au réfrigérateur. Les flacons partiellement utilisés doivent être éliminés.
6.4. Précautions particulières de conservation
Conserver à une température ne dépassant pas 25°C. Ne pas congeler. Conserver le flacon dans l’emballage d’origine à l’abri de la lumière.
Pour les conditions de conservation du médicament après reconstitution, voir la rubrique 6.3.
6.5. Nature et contenu de l'emballage extérieur
Chaque emballage contient :
· 1 g de fibrinogène humain dans un flacon en verre incolore de 100 ml, type II de la Ph. Eur., scellé avec bouchon pour perfusion (caoutchouc de bromobutyle) et une capsule à opercule en aluminium
· 50 ml de solvant (eau pour préparations injectables) dans un flacon en verre incolore de 50 ml, type II de la Ph. Eur., scellé avec bouchon pour perfusion (caoutchouc d’halobutyle) et une capsule à opercule en aluminium
· 1 dispositif de transfert nextaro
6.6. Précautions particulières d’élimination et de manipulation
Instructions générales
La solution reconstituée doit être presque incolore et légèrement opalescente. Ne pas utiliser les solutions troubles ou présentant des dépôts.
· FIBRYGA est à usage unique. Aucun des éléments contenus dans la boite ne doit être réutilisé.
· Pour des raisons de sécurité microbiologique, la solution doit être administrée immédiatement après reconstitution. La stabilité physico-chimique de la solution reconstituée a été démontrée pendant 24 heures à température ambiante (25 °C max.). Après reconstitution, ne pas mettre la solution de FIBRYGA au réfrigérateur et ne pas la congeler.
Reconstitution
|
|||||
|
2. Retirer les opercules amovibles du flacon de poudre (FIBRYGA) et du flacon de solvant afin d'exposer la partie centrale du bouchon pour la perfusion. Nettoyer les bouchons en caoutchouc avec un tampon alcoolisé et laisser sécher les bouchons. |
|
||||
|
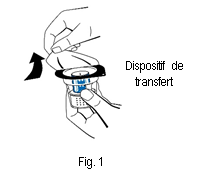
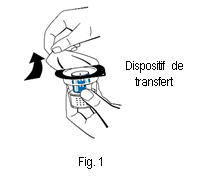
3. Ouvrir l'emballage du dispositif de transfert (nextaro) en détachant la pellicule du couvercle (fig. 1). Laisser le dispositif de transfert dans la coque transparente de l'emballage afin de maintenir sa stérilité. Ne pas toucher le perforateur.
|
|
||||
|
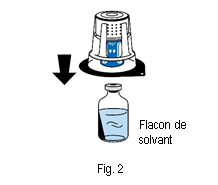
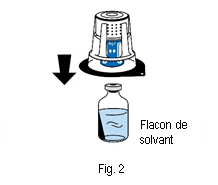
4. Placer le flacon de solvant sur une surface plane et propre et le tenir fermement. Sans retirer la coque de l’emballage, placer la partie bleue du dispositif de transfert sur le haut du flacon de solvant. Appuyer fermement, tout droit, jusqu’à ce que le dispositif se bloque en place (fig. 2). Ne pas tourner pendant la fixation. Remarque :
|
|
||||
|
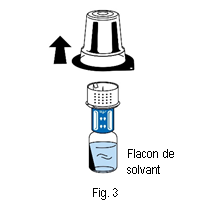
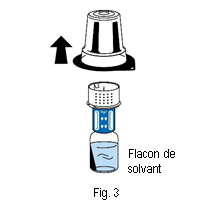
5. Tenir le flacon de solvant et retirer délicatement la coque de l’emballage du dispositif de transfert (nextaro) en la tirant tout droit vers le haut. Veiller à ce que le dispositif de transfert reste fermement fixé sur le flacon de solvant (fig. 3).
|
|
||||
|
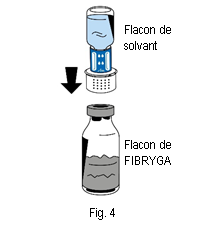
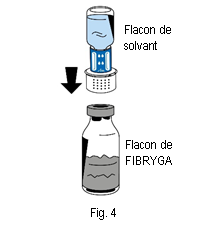
6. Placer le flacon de poudre (FIBRYGA) sur une surface plane et propre et le tenir fermement. Prendre le flacon de solvant avec le dispositif de transfert fixé dessus et le retourner. Placer la partie blanche du connecteur du dispositif de transfert sur le haut du flacon de poudre (FIBRYGA) et appuyer fermement jusqu’à ce qu’il se bloque en place (fig. 4). Ne pas tourner pendant la fixation. Le solvant s’écoulera automatiquement dans le flacon de poudre (FIBRYGA).
|
|
||||
|
7. En laissant le flacon de solvant fixé, faire tournoyer doucement le flacon de FIBRYGA jusqu’à ce que la poudre soit entièrement dissoute. Ne pas secouer le flacon afin d’éviter la formation de mousse. La poudre devrait être complètement dissoute au bout de 5 minutes environ. La dissolution de la poudre ne doit pas prendre plus de 20 minutes. Si la poudre n’est pas dissoute au bout de 20 minutes, le produit doit être éliminé.
|
|||||
|
8. Dans les rares cas où des amas flottants de poudre non reconstituée continueraient à flotter pendant le transfert du solvant, ou en cas de prolongation inattendue du temps nécessaire à la reconstitution, il est possible d’accélérer le processus de dissolution au moyen d’une agitation horizontale plus vigoureuse du flacon. |
|||||
|
|
||||
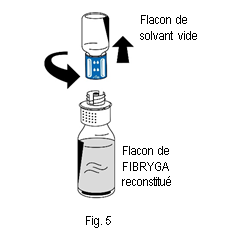
|
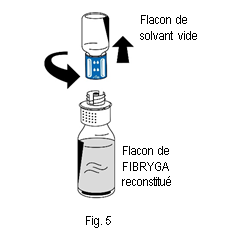
10. Éliminer le flacon de solvant vide ainsi que la partie bleue du dispositif de transfert.
|
||||
Administration
|
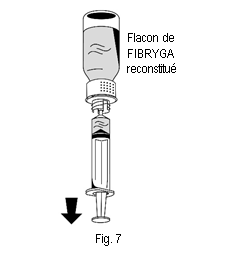
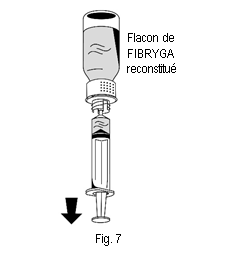
2. Retourner le flacon de FIBRYGA et aspirer la solution dans la seringue (fig. 7). |
|||||
|
|
|
||||
|
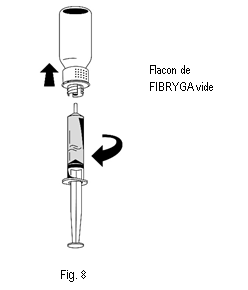
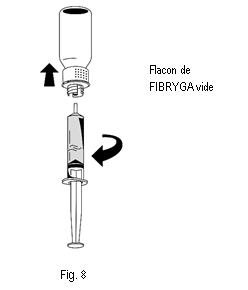
3. Une fois la solution transférée, tenir fermement le cylindre de la seringue (en laissant le piston de la seringue dirigé vers le bas) et détacher la seringue du dispositif de transfert (fig. 8). |
|||||
|
|
|||||
|
4. Éliminer la partie blanche du dispositif de transfert ainsi que le flacon vide de FIBRYGA. |
|||||
|
||||||
Un dispositif standard pour perfusion est recommandé pour l'administration intraveineuse de la solution reconstituée à température ambiante.
Tout médicament non utilisé et les déchets doivent être éliminés conformément à la réglementation locale en vigueur.
7. TITULAIRE DE L’AUTORISATION DE MISE SUR LE MARCHE
62 BIS AVENUE ANDRE MORIZET
92100 BOULOGNE BILLANCOURT
8. NUMERO(S) D’AUTORISATION DE MISE SUR LE MARCHE
· 34009 302 111 6 8 : 1 flacon de 1 g de fibrinogène humain + 1 flacon de 50 ml de solvant (eau pour préparations injectables).
9. DATE DE PREMIERE AUTORISATION/DE RENOUVELLEMENT DE L’AUTORISATION
[à compléter ultérieurement par le titulaire]
10. DATE DE MISE A JOUR DU TEXTE
[à compléter ultérieurement par le titulaire]
Sans objet.
12. INSTRUCTIONS POUR LA PREPARATION DES RADIOPHARMACEUTIQUES
Liste I
Médicament soumis à prescription hospitalière.
ANSM - Mis à jour le : 16/07/2024
Poudre et solvant pour solution injectable/pour perfusion
Fibrinogène humain
· Gardez cette notice. Vous pourriez avoir besoin de la relire.
· Si vous avez d’autres questions, interrogez votre médecin ou votre pharmacien.
· Ce médicament vous a été personnellement prescrit. Ne le donnez pas à d’autres personnes. Il pourrait leur être nocif, même si les signes de leur maladie sont identiques aux vôtres.
· Si vous ressentez un quelconque effet indésirable, parlez-en à votre médecin ou votre pharmacien. Ceci s’applique aussi à tout effet indésirable qui ne serait pas mentionné dans cette notice. Voir rubrique 4.
1. Qu'est-ce que FIBRYGA, poudre et solvant pour solution injectable/pour perfusion et dans quels cas est-il utilisé ?
2. Quelles sont les informations à connaître avant d'utiliser FIBRYGA, poudre et solvant pour solution injectable/pour perfusion ?
3. Comment utiliser FIBRYGA, poudre et solvant pour solution injectable/pour perfusion ?
4. Quels sont les effets indésirables éventuels ?
5. Comment conserver FIBRYGA, poudre et solvant pour solution injectable/pour perfusion ?
6. Contenu de l’emballage et autres informations.
1. QU’EST-CE QUE FIBRYGA, poudre et solvant pour solution injectable/pour perfusion ET DANS QUELS CAS EST-IL UTILISE ?
Classe pharmacothérapeutique : antihémorragiques, fibrinogène humain, code ATC : B02BB01
Qu’est-ce que FIBRYGA, poudre et solvant pour solution injectable/pour perfusion ?
FIBRYGA contient du fibrinogène humain, une protéine importante pour la coagulation du sang. Un manque en fibrinogène fait que le sang ne coagule pas aussi bien qu'il ne le devrait, ce qui augmente la tendance aux saignements. Le remplacement du fibrinogène humain par FIBRYGA corrigera le défaut de coagulation.
Dans quel cas FIBRYGA, poudre et solvant pour solution injectable/pour perfusion est-il utilisé ?
FIBRYGA est utilisé pour :
· le traitement des épisodes de saignement et la prophylaxie en cas d’intervention chirurgicale chez des patients présentant une hypo- ou afibrinogènémie congénitale, avec une tendance aux saignements ;
· la supplémentation en fibrinogène chez les patients présentant un saignement grave non contrôlé, accompagné d’un déficit acquis en fibrinogène pendant une intervention chirurgicale.
2. QUELLES SONT LES INFORMATIONS A CONNAITRE AVANT D’UTILISER FIBRYGA, poudre et solvant pour solution injectable/pour perfusion ?
N’utilisez JAMAIS FIBRYGA, poudre et solvant pour solution injectable/pour perfusion :
· si vous êtes allergique au fibrinogène humain ou à l’un des autres composants contenus dans ce médicament (mentionnés dans la rubrique 6).
· si vous avez présenté dans le passé des réactions allergiques à FIBRYGA.
Veuillez informer votre médecin si vous êtes allergique à un médicament.
Avertissements et précautions
Adressez-vous à votre médecin ou pharmacien avant d’utiliser FIBRYGA.
Risque de caillots dans les vaisseaux sanguins
Votre médecin doit évaluer les avantages de ce médicament au regard du risque de caillot dans les vaisseaux sanguins, en particulier si :
· vous avez reçu une forte dose ou des doses répétées de ce médicament,
· vous avez eu une crise cardiaque (antécédents d'atteinte des artères coronaires ou d'infarctus du myocarde),
· vous avez une maladie du foie (hépatique),
· vous venez de subir une intervention chirurgicale (patients en période postopératoire),
· vous allez subir une intervention chirurgicale (patients en période préopératoire),
· chez les nouveau-nés,
· vous êtes susceptible de développer des caillots sanguins ou des problèmes de coagulation dans les vaisseaux sanguins (patients à risque d'événements thromboemboliques ou de coagulation intravasculaire disséminée).
Votre médecin peut vous demander de subir des tests supplémentaires de coagulation du sang afin de contrôler le risque.
Réactions allergiques ou de type anaphylactique
Tout médicament, tel que FIBRYGA, préparé à partir de sang humain (qui contient des protéines) et qui est injecté dans une veine (administré par voie intraveineuse) peut entraîner des réactions allergiques. Si vous avez présenté dans le passé des réactions allergiques à FIBRYGA, votre médecin vous indiquera si un médicament antiallergique est nécessaire.
Votre médecin vous expliquera quels sont les signes d'alerte d'une réaction allergique ou d'une réaction de type anaphylactique.
Veuillez être attentifs aux signes précoces de réactions allergiques (hypersensibilité), notamment :
· urticaire
· éruption cutanée
· sensation d'oppression dans la poitrine
· respiration sifflante
· pression artérielle basse
· ou anaphylaxie (quand n'importe lequel ou tous les symptômes précédents se développent rapidement et sont intenses).
· Si cela survient, l'injection/la perfusion de FIBRYGA doit être arrêtée immédiatement.
Sécurité vis-à-vis des virus
Lorsque des médicaments sont fabriqués à partir de sang ou de plasmas humains, certaines mesures sont prises pour éviter que des infections soient transmises aux patients. Elles incluent :
· une sélection soigneuse des donneurs de sang et de plasma pour assurer que les donneurs susceptibles d’être porteurs d’infections sont exclus,
· la recherche de la présence de virus/marqueurs d’infections dans chaque don et pool de plasma,
· l'inclusion d'étapes d’inactivation et d’élimination des virus dans le traitement du sang et du plasma.
Malgré ces mesures, lorsque des médicaments préparés à partir de sang ou de plasma humain sont administrés, la possibilité de transmettre une infection ne peut pas être totalement écartée. Cela concerne également des virus inconnus ou émergents ou d’autres types de pathogènes.
Les mesures prises sont jugées efficaces contre les virus enveloppés, tels que le virus de l’immunodéficience humaine (VIH), le virus de l’hépatite B et le virus de l’hépatite C, et contre les virus non enveloppés tels que le virus de l’hépatite A. Les mesures prises peuvent n'avoir qu'une efficacité limitée contre les virus non enveloppés, tels que le parvovirus B19.
L'infection par le parvovirus B19 peut être grave chez la femme enceinte (infection de l'enfant à naître) et chez les personnes ayant un système immunitaire déprimé ou présentant certaines formes d'anémie (par exemple, une drépanocytose ou destruction anormale des globules rouges).
Chaque fois que vous recevez une dose de FIBRYGA, il est fortement recommandé que le nom et le numéro de lot du produit soient notés afin de conserver une traçabilité des lots utilisés.
Votre médecin pourrait vous recommander d'envisager une vaccination contre l'hépatite A et l'hépatite B si vous recevez régulièrement/de façon répétée des produits à base de fibrinogène d'origine plasmatique.
Enfants et adolescents
Il n’y a aucun avertissement et aucune mise en garde spécifique ou supplémentaire pour les enfants et les adolescents.
Autres médicaments et FIBRYGA, poudre et solvant pour solution injectable/pour perfusion
Informez votre médecin ou pharmacien si vous utilisez, avez récemment utilisé ou pourriez utiliser tout autre médicament.
FIBRYGA ne doit pas être mélangé avec d'autres médicaments hormis ceux cités dans la rubrique « Les informations suivantes sont destinées exclusivement aux professionnels de la santé / Reconstitution ».
Grossesse, allaitement et fertilité
Si vous êtes enceinte ou que vous allaitez, si vous pensez être enceinte ou planifiez une grossesse, demandez conseil à votre médecin ou pharmacien avant de prendre ce médicament. Ce produit ne doit être pris pendant la grossesse ou l'allaitement qu'après avoir demandé l'avis de votre médecin ou pharmacien.
Conduite de véhicules et utilisation de machines
FIBRYGA n'a aucune influence sur l'aptitude à conduire des véhicules et à utiliser des machines.
FIBRYGA, poudre et solvant pour solution injectable/pour perfusion contient du sodium.
Ce médicament contient jusqu'à 132 mg de sodium (composant principal du sel de cuisine/table) dans chaque flacon. Cela équivaut à 6,6 % de l’apport alimentaire quotidien maximal recommandé de sodium pour un adulte. Cela doit être pris en compte chez les patients suivant un régime contrôlé en sodium.
3. COMMENT utiliser FIBRYGA, poudre et solvant pour solution injectable/pour perfusion ?
FIBRYGA est administré par perfusion intraveineuse (perfusion dans une veine) par un professionnel de la santé.
La dose et la posologie dépendent de :
· votre poids
· la sévérité de votre maladie
· la localisation du saignement ou
· la nature de votre opération et
· votre état de santé
Utilisation chez les enfants et les adolescents
L'administration de FIBRYGA chez les enfants et adolescents (voie intraveineuse) n'est pas différente de celle des adultes.
Si vous avez utilisé plus de FIBRYGA, poudre et solvant pour solution injectable/pour perfusion que vous n’auriez dû
Pour éviter le risque de surdosage, votre médecin effectuera régulièrement des analyses de sang pour doser votre taux de fibrinogène.
En cas de surdosage, il peut y avoir une augmentation du risque de caillots anormaux dans vos vaisseaux sanguins.
Si vous avez d’autres questions sur l’utilisation de ce médicament, demandez plus d'information à votre médecin ou à votre pharmacien.
Si vous avez utilisé ou trop pris de FIBRYGA, prenez immédiatement contact avec votre médecin ou votre pharmacien.
Mode d’administration
Ce médicament doit être injecté ou perfusé dans les veines après reconstitution avec le solvant fourni. Si vous avez d’autres questions sur l’utilisation de ce produit, demandez plus d'information à votre médecin ou à votre pharmacien.
4. QUELS SONT LES EFFETS INDESIRABLES EVENTUELS ?
Veuillez communiquer immédiatement avec votre médecin :
· si l'un des effets indésirables survient,
· si vous constatez un effet indésirable qui ne serait pas mentionné dans cette notice.
Les effets indésirables suivants ont été décrits pour FIBRYGA et d'autres produits à base de fibrinogène (la fréquence des effets indésirables cités n'est pas connue) :
· Réactions allergiques ou de type anaphylactique : réactions cutanées telles que rash ou rougeur cutanée (voir rubrique 2 « Avertissements et précautions »)
· Affections cardiovasculaires : inflammation des veines et formation de caillots sanguins (voir rubrique 2 « Avertissements et précautions »)
· Élévation de la température corporelle
Si vous ressentez l’un des symptômes ci-dessus, contactez votre médecin le plus tôt possible.
Déclaration des effets secondaires
Si vous ressentez un quelconque effet indésirable, parlez-en à votre médecin ou votre pharmacien. Ceci s’applique aussi à tout effet indésirable qui ne serait pas mentionné dans cette notice. Vous pouvez également déclarer les effets indésirables directement via le système national de déclaration : Agence nationale de sécurité du médicament et des produits de santé (ANSM) et réseau des Centres Régionaux de Pharmacovigilance - Site internet: https://signalement.social-sante.gouv.fr/.
En signalant les effets indésirables, vous contribuez à fournir davantage d’informations sur la sécurité du médicament.
5. COMMENT CONSERVER FIBRYGA, poudre et solvant pour solution injectable/pour perfusion ?
Tenir hors de la vue et de la portée des enfants.
N’utilisez pas ce médicament après la date de péremption indiquée sur l’étiquette et l’emballage. La date de péremption fait référence au dernier jour de ce mois.
Conserver à une température ne dépassant pas 25 °C. Ne pas congeler. Conserver le flacon dans l’emballage d’origine à l’abri de la lumière.
La poudre doit être dissoute immédiatement avant l'injection/la perfusion. La stabilité de la solution reconstituée pendant 24 heures à température ambiante a été démontrée (maximum 25°C). Néanmoins, afin de prévenir toute contamination, la solution doit être utilisée immédiatement et en une seule fois. Le produit reconstitué ne doit pas être conservé au réfrigérateur ou au congélateur.
Ne jetez aucun médicament au tout-à-l’égout ni avec les ordures ménagères. Demandez à votre pharmacien d’éliminer les médicaments que vous n’utilisez plus. Ces mesures contribuent à protéger l’environnement.
6. CONTENU DE L’EMBALLAGE ET AUTRES INFORMATIONS
Ce que contient FIBRYGA, poudre et solvant pour solution injectable/pour perfusion
· La substance active est du fibrinogène humain.
FIBRYGA contient 1 g de fibrinogène humain par flacon ou 20 mg/ml de fibrinogène humain après reconstitution avec le solvant fourni (50 ml d'eau pour préparations injectables).
· Les autres ingrédients sont le chlorhydrate de L-arginine, la glycine, le chlorure de sodium et le citrate de sodium dihydraté.
FIBRYGA est présenté sous la forme d’une poudre et d’un solvant pour solution injectable/pour perfusion et est disponible en flacons en verre.
La poudre hygroscopique, ayant également l’aspect d’un solide friable, est de couleur blanche ou jaune pâle.
Le solvant est un liquide transparent et incolore.
La solution reconstituée est presque incolore et légèrement opalescente.
FIBRYGA est vendu dans une boîte contenant:
· 1 flacon de poudre pour solution injectable/pour perfusion
· 1 flacon de solvant (eau pour préparations injectables)
· 1 dispositif de transfert nextaro
Titulaire de l’autorisation de mise sur le marché
62 BIS AVENUE ANDRE MORIZET
92100 BOULOGNE BILLANCOURT
Exploitant de l’autorisation de mise sur le marché
OCTAPHARMA FRANCE
62 BIS AVENUE ANDRE MORIZET
92100 BOULOGNE BILLANCOURT
OCTAPHARMA PHARMAZEUTIKA PRODUKTIONSGES.M.B.H.
OBERLAAER STRASSE 235
1100 VIENNE
AUTRICHE
ou
OCTAPHARMA AB
LARS FORSSELLS GATA 23
112 75 STOCKHOLM
SUÈDE
Noms du médicament dans les Etats membres de l'Espace Economique Européen
Ce médicament est autorisé dans les Etats membres de l'Espace Economique Européen sous les noms suivants : Conformément à la réglementation en vigueur.
[À compléter ultérieurement par le titulaire]
La dernière date à laquelle cette notice a été révisée est :
[à compléter ultérieurement par le titulaire]
Les informations suivantes sont destinées exclusivement aux professionnels de santé :
Posologie
La dose et la durée du traitement substitutif dépendent de la sévérité des troubles, de la localisation, de l'étendue du saignement, et de l'état clinique du patient. La concentration (fonctionnelle) en fibrinogène doit être déterminée afin de calculer la posologie individuelle. Le nombre et la fréquence des injections doivent être adaptés à chaque patient en fonction du dosage régulier de la concentration plasmatique en fibrinogène et du suivi continu de l'état clinique du patient, et des autres traitements de substitution utilisés. En cas d'intervention chirurgicale majeure, une surveillance étroite du traitement de substitution par des tests de coagulation est essentielle.
1. Prophylaxie chez des patients présentant une hypo- ou afibrinogènémie congénitale avec une tendance connue aux saignements.
Pour prévenir un saignement excessif au cours de procédures chirurgicales, un traitement prophylactique est recommandé pour augmenter la concentration en fibrinogène jusqu'à 1 g/l puis de la maintenir à ce niveau jusqu'à l'obtention de l'hémostase, et au-dessus de 0,5 g/l jusqu'à guérison complète des plaies.
En cas de procédure chirurgicale ou de traitement d'un épisode de saignement, la dose doit être calculée comme suit :
Dose (mg/kg de poids corporel) = [Concentration cible (g/L) - concentration mesurée (g/L)]
0,018 (g/L par mg/kg de poids corporel)
La posologie suivante (dose et fréquence des injections) doit être adaptée en fonction de l'état clinique du patient et des résultats des tests de laboratoire.
La demi-vie biologique du fibrinogène est de 3 à 4 jours. Ainsi, en l'absence de dégradation, un traitement répété avec le fibrinogène humain n'est habituellement pas nécessaire. Compte tenu de l'accumulation survenant en cas d'administration répétée dans un but prophylactique, la dose et la fréquence doivent être déterminées en fonction des objectifs thérapeutiques du médecin pour chaque patient.
Population pédiatrique
En cas de procédure chirurgicale ou de traitement d’un épisode de saignement, la dose pour les adolescents doit être calculée selon la formule indiquée ci‑dessus pour les adultes, tandis que la dose pour les enfants âgés de moins de 12 ans doit être calculée comme suit :
Dose (mg/kg de poids corporel) = [Concentration cible (g/L) - concentration mesurée (g/L)]
0,014 (g/L par mg/kg de poids corporel)
La posologie doit être adaptée en fonction de l’état clinique du patient et des résultats des tests de laboratoire.
Patients âgés
Les études cliniques menées avec FIBRYGA n'ont pas inclus de patients âgés de 65 ans et plus pour fournir des données probantes et concluantes permettant de savoir si ces patients répondent différemment au traitement par rapport à des patients plus jeunes.
2. Traitement des saignements
Saignements chez des patients présentant une hypo- ou afibrinogènémie congénitale
Les épisodes de saignement doivent être traités en utilisant les formules ci-dessus adaptées aux adultes/adolescents ou aux enfants, respectivement, de manière à atteindre la concentration plasmatique cible de 1 g/l recommandée en fibrinogène. Ce taux doit être maintenu jusqu'à ce qu'une hémostase durable soit obtenue.
Saignements chez des patients présentant un déficit acquis en fibrinogène
Adultes
Une dose de 1-2 g est généralement administrée initialement, suivie de perfusions autant que nécessaire. En cas d’hémorragie sévère, par exemple au cours d’une intervention chirurgicale majeure, de grandes quantités (4-8 g) de fibrinogène peuvent être nécessaires.
Population pédiatrique
La posologie doit être déterminée en fonction du poids corporel et des besoins cliniques mais se situe généralement entre 20 et 30 mg/kg.
Instructions pour la préparation et l’administration
Instructions générales
La solution reconstituée doit être presque incolore et légèrement opalescente. Ne pas utiliser les solutions troubles ou présentant des dépôts.
· FIBRYGA est à usage unique. Aucun des éléments contenus dans la boite ne doit être réutilisé.
· Pour des raisons de sécurité microbiologique, la solution doit être administrée immédiatement après reconstitution. La stabilité physico-chimique de la solution reconstituée a été démontrée pendant 24 heures à température ambiante (25 °C max.). Après reconstitution, ne pas mettre la solution de FIBRYGA au réfrigérateur et ne pas la congeler.
Reconstitution
|
1. Veiller à ce que le flacon de poudre (FIBRYGA) et le flacon de solvant soient à température ambiante. Cette température doit être maintenue pendant la reconstitution. Si un bain-marie est utilisé pour le réchauffement, faire le nécessaire pour que l’eau n’entre pas en contact avec les bouchons en caoutchouc ou les opercules amovibles des récipients. La température du bain-marie ne doit pas dépasser 37 °C. |
|
|||||
|
2. Retirer les opercules amovibles du flacon de poudre (FIBRYGA) et du flacon de solvant afin d'exposer la partie centrale du bouchon pour la perfusion. Nettoyer les bouchons en caoutchouc avec un tampon alcoolisé et laisser sécher les bouchons. |
|
|||||
|
3. Ouvrir l'emballage du dispositif de transfert (nextaro) en détachant la pellicule du couvercle (fig. 1). Laisser le dispositif de transfert dans la coque transparente de l'emballage afin de maintenir sa stérilité. Ne pas toucher le perforateur.
|
|
|
||||
|
4. Placer le flacon de solvant sur une surface plane et propre et le tenir fermement. Sans retirer la coque de l’emballage, placer la partie bleue du dispositif de transfert sur le haut du flacon de solvant. Appuyer fermement, tout droit, jusqu’à ce que le dispositif se bloque en place (fig. 2). Ne pas tourner pendant la fixation. Remarque :
|
|
|
||||
|
5. Tenir le flacon de solvant et retirer délicatement la coque de l’emballage du dispositif de transfert (nextaro) en la tirant tout droit vers le haut. Veiller à ce que le dispositif de transfert reste fermement fixé sur le flacon de solvant (fig. 3).
|
|
|
||||
|
6. Placer le flacon de poudre (FIBRYGA) sur une surface plane et propre et le tenir fermement. Prendre le flacon de solvant avec le dispositif de transfert fixé dessus et le retourner. Placer la partie blanche du connecteur du dispositif de transfert sur le haut du flacon de poudre (FIBRYGA) et appuyer fermement jusqu’à ce qu’il se bloque en place (fig. 4). Ne pas tourner pendant la fixation. Le solvant s’écoulera automatiquement dans le flacon de poudre (FIBRYGA). |
|
|
||||
|
7. En laissant le flacon de solvant fixé, faire tournoyer doucement le flacon de FIBRYGA jusqu’à ce que la poudre soit entièrement dissoute. Ne pas secouer le flacon afin d’éviter la formation de mousse. La poudre devrait être complètement dissoute au bout de 5 minutes environ. La dissolution de la poudre ne doit pas prendre plus de 20 minutes. Si la poudre n’est pas dissoute au bout de 20 minutes, le produit doit être éliminé. |
|
|||||
|
8. Dans les rares cas où des amas flottants de poudre non reconstituée continueraient à flotter pendant le transfert du solvant, ou en cas de prolongation inattendue du temps nécessaire à la reconstitution, il est possible d’accélérer le processus de dissolution au moyen d’une agitation horizontale plus vigoureuse du flacon. |
|
|||||
|
|
|
|||||
|
9. Une fois la reconstitution terminée, dévisser le dispositif de transfert (partie bleue) en tournant dans le sens contraire des aiguilles d’une montre de façon à séparer les deux parties (fig. 5). Ne pas toucher le connecteur Luer lock sur la partie blanche du dispositif de transfert. |
|
|||||
|
10. Éliminer le flacon de solvant vide ainsi que la partie bleue du dispositif de transfert. |
||||||
|
Administration |
||||||
|
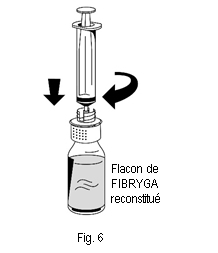
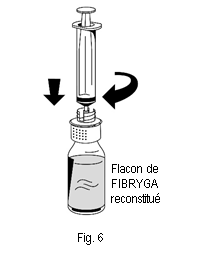
1. Raccorder délicatement la seringue au connecteur Luer lock de la partie blanche du dispositif de transfert (fig. 6). 2. Retourner le flacon de FIBRYGA et aspirer la solution dans la seringue (fig. 7). |
||||||
|
|
|
|||||
|
3. Une fois la solution transférée, tenir fermement le cylindre de la seringue (en laissant le piston de la seringue dirigé vers le bas) et détacher la seringue du dispositif de transfert (fig. 8). |
||||||
|
|
||||||
|
4. Éliminer la partie blanche du dispositif de transfert ainsi que le flacon vide de FIBRYGA. |
||||||
Un dispositifstandard pour perfusion est recommandé pour l'administration intraveineuse de la solution reconstituée à température ambiante.
Tout médicament non utilisé etles déchets doivent être éliminés conformément à la réglementation locale en vigueur.
Mode d’administration
Perfusion ou injection intraveineuse.
FIBRYGA doit être administré lentement par voie intraveineuse à la vitesse maximale recommandée de 5 ml/min chez les patients présentant une hypo- ou afibrinogènémie congénitale. La vitesse maximale recommandée est de 10 ml/min chez les patients présentant un déficit acquis en fibrinogène.
Incompatibilités
Ce médicament ne doit pas être mélangé avec d’autres médicaments.