Dernière mise à jour le 01/06/2026
TOBRAMYCINE ZENTIVA 300 mg/5 mL, solution pour inhalation par nébuliseur
Indications thérapeutiques
Classe pharmacothérapeutique : Aminosides antibiotiques - code ATC : J01GB01.
Qu’est-ce que TOBRAMYCINE ZENTIVA ?
TOBRAMYCINE ZENTIVA contient de la tobramycine. Il s’agit d’un antibiotique de la famille des aminosides.
Dans quels cas TOBRAMYCINE ZENTIVA est-il utilisé ?
TOBRAMYCINE ZENTIVA est utilisé chez les patients âgés de 6 ans et plus atteints de mucoviscidose pour traiter les infections respiratoires dues à une bactérie appelée Pseudomonas aeruginosa.
TOBRAMYCINE ZENTIVA combat l’infection provoquée par la bactérie Pseudomonas dans vos poumons et vous aide à mieux respirer.
Comment fonctionne TOBRAMYCINE ZENTIVA ?
Lorsque vous inhalez TOBRAMYCINE ZENTIVA, l’antibiotique arrive directement dans vos poumons pour lutter contre la bactérie responsable de l’infection. Pour obtenir les meilleurs résultats de ce traitement, prenez ce médicament comme cela est expliqué dans cette notice.
Qu’est-ce que Pseudomonas aeruginosa ?
C’est une bactérie très fréquente qui infecte presque tous les patients atteints de mucoviscidose à un moment ou un autre de leur vie. Chez certaines personnes, cette infection ne se développe que très tardivement, tandis que chez d’autres, elle se développe très jeune.
C’est l’une des bactéries les plus nocives pour les patients atteints de mucoviscidose. Si l’infection n’est pas contrôlée correctement, elle continuera à altérer vos poumons entraînant des difficultés respiratoires supplémentaires.
TOBRAMYCINE ZENTIVA tue la bactérie qui provoque les infections pulmonaires. L’infection peut être contrôlée avec succès si elle est traitée suffisamment tôt.
Présentations
> 56 ampoules unidoses polyéthylène basse densité (PEBD) surpochées de 5 mL
Code CIP : 34009 301 777 3 0
Déclaration de commercialisation : 15/02/2021
Cette présentation est agréée aux collectivités
- Prix hors honoraire de dispensation : 1482,68 €
- Honoraire de dispensation : 1,02 €
- Prix honoraire compris : 1483,70 €
- Taux de remboursement :65%
Service médical rendu (SMR)
Les libellés affichés ci-dessous ne sont que des résumés ou extraits issus des avis rendus par la Commission de la Transparence. Seul l'avis complet de la Commission de la Transparence fait référence.
Cet avis est consultable à partir du lien `Avis du jj/mm/aaaa` ou encore sur demande auprès de la HAS (plus d'informations dans l'aide). Les avis et synthèses d'avis contiennent un paragraphe sur la place du médicament dans la stratégie thérapeutique.
| Valeur du SMR | Avis | Motif de l'évaluation | Résumé de l'avis |
|---|---|---|---|
| Important | Avis du 19/02/2020 | Inscription (CT) | Le service médical rendu par la spécialité TOBRAMYCINE ALTAN est important dans les indications de l’AMM. |
Amélioration du service médical rendu (ASMR)
Les libellés affichés ci-dessous ne sont que des résumés ou extraits issus des avis rendus par la Commission de la Transparence. Seul l'avis complet de la Commission de la Transparence fait référence.
Cet avis est consultable à partir du lien `Avis du jj/mm/aaaa` ou encore sur demande auprés de la HAS (plus d'informations dans l'aide). Les avis et synthéses d'avis contiennent un paragraphe sur la place du médicament dans la stratégie thérapeutique.
| Valeur de l'ASMR | Avis | Motif de l'évaluation | Résumé de l'avis |
|---|---|---|---|
| V (Inexistant) | Avis du 19/02/2020 | Inscription (CT) | La spécialité TOBRAMYCINE ALTAN n’apporte pas d’amélioration du service médical rendu (ASMR V) par rapport aux autres spécialités à base de tobramycine par voie inhalée déjà disponibles. |
Autres informations
- Titulaire de l'autorisation : ZENTIVA France
- Conditions de prescription et de délivrance :
- liste I
- prescription initiale hospitalière semestrielle
- renouvellement non restreint
- Statut de l'autorisation : Valide
- Type de procédure : Procédure décentralisée
- Code CIS : 6 947 245 5
ANSM - Mis à jour le : 10/11/2025
TOBRAMYCINE ZENTIVA 300 mg/5 mL, solution pour inhalation par nébuliseur
2. COMPOSITION QUALITATIVE ET QUANTITATIVE
Pour une ampoule à dose-unique de 5 mL.
Excipient(s) à effet notoire : sodium : 4,43 mg/ampoule.
Pour la liste complète des excipients, voir rubrique 6.1.
Solution pour inhalation par nébuliseur.
Solution claire, légèrement jaune.
4.1. Indications thérapeutiques
Il convient de tenir compte des recommandations officielles concernant l’utilisation appropriée des antibactériens.
4.2. Posologie et mode d'administration
TOBRAMYCINE ZENTIVA s’administre par inhalation et non par voie parentérale.
La dose recommandée chez les adultes et les enfants est une ampoule deux fois par jour pendant 28 jours. L'intervalle entre les doses doit être le plus proche possible de 12 heures et ne pas être inférieur à 6 heures. Après 28 jours de traitement, le patient doit interrompre TOBRAMYCINE ZENTIVA pendant les 28 jours suivants. Chaque cycle de 28 jours de traitement actif doit être suivi de 28 jours d'interruption du traitement.
Le dosage n'a pas à être ajusté en fonction du poids. Tous les patients reçoivent une ampoule de TOBRAMYCINE ZENTIVA (300 mg de tobramycine) deux fois par jour.
Les essais cliniques contrôlés, réalisés sur une période de 6 mois selon le schéma d'administration du TOBRAMYCINE ZENTIVA, ont montré que l'amélioration de la fonction pulmonaire est maintenue au-dessus de l'état de base pendant les périodes de 28 jours sans traitement.
Schéma d'administration de la tobramycine au cours des essais cliniques contrôlés
|
Cycle 1 |
Cycle 2 |
Cycle 3 |
|||
|
28 jours |
28 jours |
28 jours |
28 jours |
28 jours |
28 jours |
|
Tobramycine 300 mg deux fois par jour en plus des soins habituels |
Soins habituels |
Tobramycine 300 mg deux fois par jour en plus des soins habituels |
Soins habituels |
Tobramycine 300 mg deux fois par jour en plus des soins habituels |
Soins habituels |
La tolérance et l'efficacité dans le traitement au long cours des infections pulmonaires chroniques dues à Pseudomonas aeruginosa ont été évaluées au cours d'essais contrôlés en ouvert sur une période de 96 semaines (12 cycles), elles n'ont cependant pas été étudiées chez les patients âgés de moins de 6 ans, les patients ayant un volume expiratoire maximum-seconde (VEMS) < 25 % ou > 75 %, ou les patients infectés par Burkholderia cepacia.
Le traitement doit être initié par un médecin expérimenté dans le domaine de la mucoviscidose. Le traitement par TOBRAMYCINE ZENTIVA doit être poursuivi de manière cyclique aussi longtemps que le médecin considère qu'il existe un bénéfice clinique pour le patient. En cas de détérioration évidente de la fonction pulmonaire, on envisagera un traitement anti-pseudomonal additionnel. Les essais cliniques ont montré qu'une résistance in vitro à la tobramycine ne signifiait pas forcément une absence de bénéfice clinique pour le patient.
Populations particulières
Patients âgés
Les données disponibles dans cette population sont insuffisantes pour recommander ou non une adaptation posologique.
Insuffisance rénale
Aucune donnée n’est disponible dans cette population pour recommander ou non une adaptation posologique de TOBRAMYCINE ZENTIVA. Voir également les informations sur la néphrotoxicité en rubrique 4.4 et sur l’élimination en rubrique 5.2.
Insuffisance hépatique
Aucune étude n’a été conduite chez les patients présentant une insuffisance hépatique. La tobramycine n’étant pas métabolisée au niveau du foie, l’insuffisance hépatique ne devrait pas avoir d’effet sur l’exposition à la tobramycine.
Patients ayant eu une transplantation d’organe
Il n'existe pas de données adéquates sur l'utilisation du TOBRAMYCINE ZENTIVA chez les patients transplantés d’organe.
Population pédiatrique
La sécurité et l’efficacité de TOBRAMYCINE ZENTIVA chez les enfants âgés de moins de 6 ans n’ont pas encore été établies. Les données actuellement disponibles sont décrites à la rubrique 5.1 mais aucune recommandation sur la posologie ne peut être donnée.
Mode d’administration
L'ampoule doit être vidée dans le nébuliseur et administrée par inhalation en 15 minutes environ, à l'aide d'un nébuliseur à main réutilisable PARI LC PLUS, équipé d'un compresseur approprié. Les compresseurs appropriés sont ceux qui, fixés sur un nébuliseur PARI LC PLUS, fournissent un débit de 4-6 L/min et/ou une contre-pression de 110-217 kPa. Pour l'utilisation et l'entretien du nébuliseur et du compresseur, suivre les instructions du fabricant.
TOBRAMYCINE ZENTIVA doit être inhalé par le patient en position assise ou debout en respirant normalement au travers de la pièce buccale du nébuliseur. Des pinces nasales peuvent aider le patient à respirer par la bouche. Le patient doit continuer sa kinésithérapie respiratoire habituelle. L'usage de bronchodilatateurs appropriés doit être poursuivi tant que cela paraît nécessaire. Lorsqu'un patient reçoit plusieurs traitements respiratoires différents, il est recommandé d'observer l'ordre suivant : bronchodilatateur, kinésithérapie respiratoire, autres médicaments en inhalation et enfin TOBRAMYCINE ZENTIVA 300 mg/5 mL.
Dose quotidienne maximale tolérée
La dose quotidienne maximale tolérée de TOBRAMYCINE ZENTIVA n'a pas été établie.
Hypersensibilité à la substance active ou à l’un des excipients mentionnés à la rubrique 6.1.
4.4. Mises en garde spéciales et précautions d'emploi
Mises en garde spéciales
Pour tout renseignement sur la grossesse et l'allaitement, voir rubrique 4.6.
TOBRAMYCINE ZENTIVA doit être administré avec précaution en cas de troubles rénaux, auditifs, vestibulaires ou neuromusculaires connus ou suspectés ou d'hémoptysie sévère active.
Les concentrations sériques de tobramycine doivent être surveillées chez les patients présentant des troubles auditifs ou rénaux connus ou suspectés. En cas de survenue d'une oto- ou d’une néphrotoxicité chez un patient traité par TOBRAMYCINE ZENTIVA, le traitement par la tobramycine doit être interrompu jusqu'à ce que la concentration sérique soit inférieure à 2 µg/mL.
Les concentrations sériques de tobramycine doivent être surveillées chez les patients recevant un traitement parentéral concomitant par aminosides (ou autres médicaments pouvant modifier l'excrétion rénale). Ces patients doivent faire l'objet d'une surveillance clinique appropriée.
La concentration sérique de la tobramycine doit être uniquement contrôlée à partir d'une ponction veineuse et non à partir d'une piqûre du doigt. Une contamination de la peau des doigts avec de la tobramycine peut entraîner une fausse augmentation des concentrations sériques mesurées de ce médicament. Cette contamination ne peut être complètement évitée par un lavage des mains précédant le test.
Bronchospasme
L'inhalation de médicaments est susceptible de provoquer un bronchospasme, des cas ont été rapportés avec la tobramycine nébulisée. La première dose de TOBRAMYCINE ZENTIVA doit être administrée sous surveillance avec utilisation d'un bronchodilatateur avant la nébulisation si cela fait partie du traitement habituel du patient. Le volume expiratoire maximum-seconde (VEMS) sera mesuré avant et après la nébulisation. En cas d'apparition d'un bronchospasme induit par le traitement chez un patient ne recevant pas de bronchodilatateur, répéter le test à un autre moment en utilisant un bronchodilatateur. L'apparition d'un bronchospasme au cours d'un traitement par bronchodilatateur peut être le signe d'une réaction allergique. Si une réaction allergique est suspectée, le traitement par TOBRAMYCINE ZENTIVA doit être interrompu. Tout bronchospasme doit faire l'objet d'un traitement médical approprié.
Troubles neuromusculaires
TOBRAMYCINE ZENTIVA doit être utilisé avec de grandes précautions chez les patients présentant des troubles neuromusculaires connus ou suspectés tels que la maladie de Parkinson ou d'autres pathologies caractérisées par une myasthénie (y compris la myasthénie grave), puisque les aminosides peuvent aggraver la faiblesse musculaire en raison de leur effet curarisant sur les fonctions neuromusculaires.
Néphrotoxicité
Bien que les aminosides par voie parentérale soient connus pour leur néphrotoxicité, les essais cliniques réalisés avec la tobramycine n'ont révélé aucun effet de ce type.
Le produit doit être utilisé avec précaution chez les patients ayant des troubles connus ou suspectés de la fonction rénale et les concentrations sériques de tobramycine doivent être surveillées. Les patients souffrant d'insuffisance rénale sévère avec une créatinine sérique > 2mg/dL (176,8 μmol/L), n'ont pas été inclus dans les essais cliniques.
La pratique clinique actuelle suggère d'évaluer la fonction rénale initiale. Les taux d'urée et de créatinine doivent ensuite être mesurés au bout de 6 cycles complets de traitement par tobramycine (180 jours de traitement par aminosides nébulisés).
Voir également ci-dessus « Surveillance des concentrations sériques de tobramycine ».
Ototoxicité
Une ototoxicité, se manifestant à la fois sous forme d'une toxicité auditive et d’une toxicité vestibulaire, a été rapportée lors de l'administration d'aminosides par voie parentérale. La toxicité vestibulaire peut se manifester par des vertiges, une ataxie ou des étourdissements.
Les essais cliniques contrôlés réalisés avec la tobramycine n'ont révélé aucun cas d’ototoxicité, les patients n'ayant pas rapporté de perte auditive et les résultats des examens audiométriques étant restés normaux. Des essais menés en ouvert et l'expérience après commercialisation ont montré que des pertes auditives avaient été observées chez des patients ayant bénéficié d’un traitement par aminosides intraveineux prolongé antérieur ou concomitant à la tobramycine. Les patients avec perte auditive ont souvent rapporté des acouphènes. Les médecins doivent tenir compte des risques de toxicités vestibulaire et cochléaire potentielles induits par les aminosides et mettre en œuvre une évaluation appropriée de la fonction auditive pendant toute la durée du traitement par TOBRAMYCINE ZENTIVA. Chez les patients plus exposés en raison d'un traitement systémique préalable et prolongé par aminosides, il peut être nécessaire d'envisager un examen de l'audition avant de commencer le traitement par TOBRAMYCINE ZENTIVA. L'apparition d'acouphènes doit faire l'objet d'une attention particulière étant donné qu'ils sont un prodrome d'ototoxicité.
TOBRAMYCINE ZENTIVA doit être prescrit avec précaution chez les patients présentant des troubles auditifs ou vestibulaires connus ou suspectés. Les médecins doivent envisager un examen auditif chez les patients ayant manifesté des signes de troubles auditifs, ou chez les patients ayant un risque élevé de présenter ces troubles.
Si un patient se plaint d'acouphènes ou de perte auditive pendant le traitement par aminosides, le médecin devra envisager un examen de l'audition.
Il existe un risque accru d’ototoxicité chez les patients présentant des mutations de l’ADN mitochondrial (en particulier une substitution du nucléotide 1555 A par G dans le gène de l’ARNr 12S), même si les taux sériques d’aminoside restent dans la plage recommandée pendant le traitement. D’autres approches thérapeutiques doivent être envisagées chez ces patients.
Chez les patients présentant des antécédents maternels de mutations pertinentes ou de surdité induite par les aminosides, d’autres traitements ou des tests génétiques doivent être envisagés avec l’administration.
Voir également ci-dessus « Surveillance des concentrations sériques de tobramycine ».
Hémoptysie
L'inhalation de solutions nébulisées est susceptible d'induire une toux réflexe. L'utilisation de TROBRAMYCINE ZENTIVA chez les patients présentant une hémoptysie sévère active ne doit être envisagée que si les bénéfices du traitement sont plus importants que les risques de déclencher une nouvelle hémorragie.
Résistance microbienne
Au cours des essais cliniques, à partir d'isolats de P. aeruginosa, il a été montré une augmentation de la concentration minimale inhibitrice des aminosides pour quelques patients sous tobramycine. Il existe un risque théorique que les patients traités par tobramycine inhalée, développent des souches de P. aeruginosa résistantes à la tobramycine intraveineuse (voir rubrique 5.1).
4.5. Interactions avec d'autres médicaments et autres formes d'interactions
L'administration concomitante et/ou séquentielle de la tobramycine avec d'autres médicaments pouvant entraîner des effets neurotoxiques, néphrotoxiques ou ototoxiques est à éviter. Certains diurétiques sont susceptibles d'accroître la toxicité des aminosides par modification des concentrations d'antibiotiques dans le sérum et les tissus. TOBRAMYCINE ZENTIVA ne doit pas être administré de façon concomitante avec l’acide éthacrynique, le furosémide, l'urée ou le mannitol en intraveineux.
D'autres médicaments sont susceptibles d'augmenter le potentiel toxique des aminosides administrés par voie parentérale :
· Amphotéricine B, céfalotine, cyclosporine, tacrolimus, polymyxines (risque accru de néphrotoxicité),
· Dérivés du platine (risque accru de néphrotoxicité et d'ototoxicité),
· Anticholinestérases, toxine botulinique (effets neuromusculaires).
Lors des essais cliniques, il a été montré que le profil des effets indésirables était similaire à celui du groupe contrôle chez les patients traités de façon concomitante par la tobramycine et par dornase alfa, β-agonistes, corticostéroïdes par inhalation et autres antibiotiques anti-Pseudomonas par voie orale ou parentérale.
4.6. Fertilité, grossesse et allaitement
TOBRAMYCINE ZENTIVA ne doit pas être administré pendant la grossesse ni au cours de l'allaitement sauf si le bénéfice escompté pour la mère est supérieur au risque potentiel pour le fœtus, le nouveau-né ou le nourrisson.
Grossesse
Il n'y a pas de données suffisantes concernant l'utilisation de la tobramycine administrée par inhalation chez la femme enceinte. Les études chez l'animal n'ont mis en évidence aucun effet tératogène de la tobramycine (voir rubrique 5.3. Données de sécurité précliniques). Cependant, les aminosides peuvent être néfastes pour le fœtus (par ex. surdité congénitale) lorsque des concentrations systémiques élevées sont atteintes chez la femme enceinte. En cas d'utilisation de TOBRAMYCINE ZENTIVA pendant la grossesse, ou si une grossesse survient en cours de traitement par TOBRAMYCINE ZENTIVA, la patiente doit être informée des risques encourus par le fœtus.
La tobramycine administrée par voie systémique est excrétée dans le lait maternel. Il n’y a pas de données suffisantes pour déterminer si l’administration de TOBRAMYCINE ZENTIVA entraîne des concentrations sériques suffisamment élevées pour être détectées dans le lait maternel. En raison de l'ototoxicité et de la néphrotoxicité de la tobramycine chez les nourrissons, il faudra soit arrêter l'allaitement soit interrompre le traitement par TOBRAMYCINE ZENTIVA.
Fertilité
Aucun effet sur la fertilité des mâles ou des femelles n'a été observé dans les études chez l'animal après administration sous-cutanée (voir rubrique 5.3).
4.7. Effets sur l'aptitude à conduire des véhicules et à utiliser des machines
Deux études cliniques de 24 semaines, randomisées, en double aveugle, contrôlées contre placebo, en groupes parallèles, ont été conduites avec la tobramycine chez 520 patients âgés de 6 à 63 ans atteints de mucoviscidose.
Les événements indésirables les plus fréquemment rapportés (≥ 10 %) avec la tobramycine dans les études contrôlées contre placebo ont été : toux, pharyngite, toux productive, asthénie, rhinite, dyspnée, fièvre, troubles pulmonaires, céphalées, douleur thoracique, expectoration teintée, hémoptysie, anorexie, exploration fonctionnelle respiratoire diminuée, asthme, vomissements, douleur abdominale, dysphonie, nausées et perte de poids.
Chez les patients recevant le placebo, la plupart des événements ont été rapportés à des fréquences supérieures ou égales à celles observées chez les patients traités par la tobramycine. La dysphonie et les acouphènes sont les seuls effets indésirables rapportés par un nombre significativement supérieur de patients traités par la tobramycine ; (groupe tobramycine 12,8 % vs. placebo 6,5 %) et (groupe tobramycine 3,1 % vs. placebo 0 %) respectivement. Ces épisodes d'acouphènes ont été transitoires et ont disparu sans qu'il ait été nécessaire d'interrompre le traitement par la tobramycine sans perte définitive d'audition constatée sur audiogramme. Les risques d'acouphènes n'ont pas augmenté au cours des cycles d'administration par la tobramycine (voir rubrique 4.4 Ototoxicité).
Tableau de synthèse des effets indésirables
Dans les études à 24 semaines contrôlées contre placebo et leurs extensions en ouvert sous traitement actif, 313, 264 et 120 patients au total ont été traités par la tobramycine pendant respectivement 48, 72 et 96 semaines.
Le Tableau 1 montre la fréquence des effets indésirables survenus sous traitement en fonction des critères suivants : rapportés avec une incidence ≥ 2 % chez les patients traités par la tobramycine, survenus à une fréquence plus élevée dans le groupe tobramycine et évalués comme imputables au médicament chez ≥ 1 % des patients.
Les effets indésirables survenus au cours des essais cliniques sont listés par classe de système organe selon MedDRA. Au sein de chaque classe de système organe, les effets indésirables sont classés suivant un ordre décroissant de fréquence. Au sein de chaque groupe de fréquence, les effets indésirables sont classés suivant un ordre décroissant de gravité. De plus, pour chaque effet indésirable, la catégorie de fréquence correspondante est également présentée selon la convention suivante (CIOMS III) : très fréquent (≥ 1/10) ; fréquent (≥ 1/100 ; < 1/10) ; peu fréquent (≥ 1/1 000 ; < 1/100) ; rare (≥ 1/10 000 ; < 1/1 000) ; très rare (< 1/10 000), y compris les cas isolés.
Tableau 1 Effets indésirables au cours des essais cliniques
|
Classe de système d’organe |
Très fréquent |
Fréquent |
|
Affections respiratoires, thoraciques et médiastinales |
Troubles pulmonaires Rhinitie Dysphonie Expectoration teintée |
|
|
Troubles généraux et anomalies au site d'administration |
|
Malaise |
|
Investigations |
Exploration fonctionnelle respiratoire diminuée |
|
|
Affections de l'oreille et du labyrinthe |
|
Acouphènes |
|
Affections musculo-squelettiques et systémiques |
|
Myalgies |
|
Infections et infestations |
|
Laryngite |
A mesure que la durée d'exposition à la tobramycine augmentait au cours des deux études d'extension en ouvert, l'incidence de la toux productive et de la diminution de la fonction pulmonaire semblaient augmenter ; toutefois l'incidence de la dysphonie semblait diminuer. Dans l'ensemble, l'incidence des événements indésirables relatifs aux classes de système organe (selon MedDRA) suivantes a diminué avec l'augmentation de l'exposition à la tobramycine : affections respiratoires, thoraciques et médiastinales, affections gastro-intestinales et troubles généraux et anomalies au site d'administration.
Effets indésirables issus des notifications spontanées
Les effets indésirables notifiés spontanément, présentés ci-dessous, ont été notifiés de manière volontaire et il n'est pas toujours possible d'établir avec fiabilité leur fréquence ou la relation de causalité avec l'exposition au médicament.
Affections de l'oreille et du labyrinthe
Perte auditive
Affections de la peau et du tissu sous-cutané
Hypersensibilité, prurit, urticaire, éruption cutanée
Affections du système nerveux
Aphonie, dysgueusie
Affections respiratoires, thoraciques et médiastinales
Bronchospasme, douleurs oropharyngées
Les essais menés en ouvert et l'expérience après commercialisation ont montré que des pertes auditives avaient été observées chez des patients ayant bénéficié de traitement par aminosides intraveineux prolongé antérieur ou concomitant à la tobramycine (voir rubrique 4.4). Des réactions d'hypersensibilité, d'ototoxicité et de néphrotoxicité ont été associées à l'administration par voie parentérale d'aminosides (voir rubriques 4.3 et 4.4).
Déclaration des effets indésirables suspectés
La déclaration des effets indésirables suspectés après autorisation du médicament est importante. Elle permet une surveillance continue du rapport bénéfice/risque du médicament. Les professionnels de santé déclarent tout effet indésirable suspecté via le système national de déclaration : Agence nationale de sécurité du médicament et des produits de santé (ANSM) et réseau des Centres Régionaux de Pharmacovigilance - Site internet : https://signalement.social-sante.gouv.fr
La biodisponibilité systémique de la tobramycine est faible après administration par inhalation. L'enrouement prononcé peut être un des symptômes du surdosage par aérosol.
En cas d'ingestion accidentelle de TOBRAMYCINE ZENTIVA, les risques de toxicité sont peu probables, la tobramycine n'étant que faiblement absorbée au niveau d'un tractus gastro-intestinal non pathologique.
En cas d'administration par inadvertance de TOBRAMYCINE ZENTIVA par voie intraveineuse, les signes et les symptômes d'un surdosage parentéral de tobramycine possibles peuvent survenir : étourdissements, acouphènes, vertiges, perte de l'acuité auditive, détresse respiratoire et/ou blocage neuromusculaire et altération de la fonction rénale.
En cas d'intoxication aiguë, il faut interrompre immédiatement le traitement par TOBRAMYCINE ZENTIVA et évaluer la fonction rénale. Le dosage des concentrations sériques de tobramycine peut être utile pour contrôler l'état du patient lors d'un surdosage.
En cas de surdosage, il faut tenir compte des interactions médicamenteuses possibles, susceptibles de modifier l'élimination de TOBRAMYCINE ZENTIVA ou d'autres médicaments.
5. PROPRIETES PHARMACOLOGIQUES
5.1. Propriétés pharmacodynamiques
Classe pharmacothérapeutique : Aminosides antibiotiques, code ATC : J01GB01.
Mécanisme d’action
La tobramycine est un antibiotique de la famille des aminosides produit à partir du Streptomyces tenebrarius. Elle agit principalement en bloquant la synthèse des protéines, altérant ainsi la perméabilité de la membrane cellulaire, entraînant la rupture progressive de l'enveloppe cellulaire puis éventuellement la mort de la cellule. Elle possède une action bactéricide à des concentrations égales ou légèrement supérieures aux concentrations inhibitrices.
Limites de sensibilité
Les limites de sensibilité déterminées pour l'administration parentérale de tobramycine ne conviennent pas pour l'administration par aérosol du médicament.
Les expectorations des patients atteints de mucoviscidose présentent une action inhibitrice sur l'activité biologique locale des aminosides nébulisés. De ce fait, les concentrations de tobramycine en aérosol dans les expectorations doivent être respectivement 10 et 25 fois supérieures aux CMI (concentrations minimales inhibitrices) pour arrêter la croissance de P. aeruginosa et permettre une activité bactéricide. Au cours des essais cliniques contrôlés, 97 % des patients recevant la tobramycine présentaient dans les expectorations des concentrations égales à 10 fois la CMI la plus élevée de P. aeruginosa obtenue et 95 % présentaient des concentrations supérieures à 25 fois la CMI la plus élevée. Pour la majorité des patients dont les souches cultivées présentaient des valeurs de CMI supérieures à la limite de sensibilité parentérale, un bénéfice clinique a tout de même été obtenu.
Sensibilité
En l'absence de limites de sensibilité conventionnelles pour l'administration par nébulisation, il faut être prudent lors de la définition de la sensibilité ou insensibilité de l'organisme à la tobramycine par nébulisation. Les études cliniques avec la tobramycine ont toutefois révélé qu’un examen microbiologique indiquant une résistance in vitro à la substance n’est pas nécessairement synonyme d’une absence de bénéfice clinique pour le patient.
La majorité des patients ayant au début du traitement par la tobramycine, une CMI < 128 μg/mL à la tobramycine vis-à-vis de P. aeruginosa ont eu une amélioration de leur fonction pulmonaire après traitement. Les patients ayant au début du traitement par la tobramycine, une CMI ≥ 128 μg/mL vis-à-vis de P. aeruginosa ont une probabilité moindre de répondre au traitement. Cependant, au cours des essais contrôlés versus placebo, 7 patients sur 13 (54 %) ayant acquis sous traitement des souches avec une CMI ≥ 128 μg/mL, ont eu une amélioration de leur fonction pulmonaire après traitement.
Sur toute la durée des études d'extension de 96 semaines, la CMI50 de la tobramycine vis-à-vis de P. aeruginosa a augmenté de 1 à 2 µg/mL et la CMI90 a augmenté de 8 à 32 µg/mL.
Le spectre établi à partir des données in vitro et/ou cliniques pour les bactéries impliquées dans les infections pulmonaires au cours de la mucoviscidose est le suivant :
|
Espèces sensibles |
Pseudomonas aeruginosa Haemophilus influenzae Staphylococcus aureus |
|
Espèces résistantes |
Burkholderia cepacia Stenotrophomonas maltophilia Alcaligenes xylosoxidans |
Les essais cliniques avec la tobramycine ont mis en évidence une faible mais nette augmentation des Concentrations Minimales Inhibitrices de la tobramycine, l'amikacine et la gentamicine sur les souches de P. aeruginosa. Chaque période supplémentaire de 6 mois de traitement a montré une augmentation similaire à celle observée durant les 6 mois de l'étude contrôlée.
Le principal mécanisme de résistance aux aminosides constaté sur les souches de P. aeruginosa sur des patients atteints de mucoviscidose et souffrant d'infection chronique est l'imperméabilité, qui se traduit par une perte de sensibilité à tous les aminosides. On a observé également sur les souches de P. aeruginosa provenant de patients mucoviscidosiques, une résistance aux aminosides caractérisée par un retour à la sensibilité en l'absence de l'antibiotique.
Autres informations
Il n'a pas été démontré que les patients traités par tobramycine pendant 18 mois avaient un risque accru d'infection par B. cepacia, S. maltophilia ou A. xylosoxidans, par rapport aux patients ne recevant pas la tobramycine. Des espèces d'Aspergillus ont été plus fréquemment retrouvées dans les expectorations des patients sous tobramycine ; cependant, des conséquences cliniques telles que l'Aspergillose Bronchopulmonaire Allergique (ABPA) ont été rarement rapportées et avec une fréquence similaire à celle du groupe contrôle.
Les données cliniques de sécurité et d’efficacité disponibles sont insuffisantes chez les enfants de moins de 6 ans.
Dans une étude en ouvert, non comparative, 88 patients atteints de mucoviscidose (37 patients âgés de 6 mois à 6 ans, 41 patients âgés de 6 à 18 ans et 10 patients âgés de plus de 18 ans) avec primo-colonisation (infection non chronique) à P. aeruginosa ont été traités pendant 28 jours avec la tobramycine. Après 28 jours, les patients ont été randomisés selon un ratio 1:1 soit pour arrêter le traitement (n = 45) soit pour 28 jours supplémentaires de traitement (n = 43).
Le critère principal était le temps médian de récidive de l’infection à P. aeruginosa (quelle que soit la souche) qui a été respectivement de 26,1 et 25,8 mois pour 28 jours et 56 jours de traitement. Il a été constaté que 93 % et 92 % des patients ne présentaient plus d’infection à P.aeruginosa un mois après la fin du traitement après 28 jours et 56 jours de traitement, respectivement. L’utilisation de la tobramycine selon un schéma posologique de plus de 28 jours de traitement continu, n’est pas approuvée.
Dans une étude clinique en double aveugle, randomisée, contrôlée contre placebo, 51 patients âgés de 3 mois à moins de 7 ans ayant un diagnostic confirmé de mucovicidose et une primo-colonisation à P. aeruginosa (définie comme ayant pour la première fois une culture positive ou bien ayant une première culture positive après au moins 1 an sans culture positive) ont été traités par de la Tobramycine 300 mg/5 mL ou par le placebo, les deux étant inhalés à l’aide d’un nébuliseur (PARI LC Plus®) deux fois par jour pendant 28 jours. Les patients traités par un traitement à visée anti-pseudomonale l’année précédente ont été exclus. Au total, 26 patients ont été randomisés dans le groupe traité par la Tobramycine et 25 dans le groupe placebo. Le critère principal était la proportion de patients sans aucune souche de P. aeruginosa à la culture de prélèvement d’expectoration/de gorge réalisée après l’achèvement d’une période de 28 jours de traitement et était de 84,6 % (22 patients sur 26) pour le groupe traité par la Tobramycine et 24 % (6 patients sur 25) pour le groupe placebo (p< 0,001). La fréquence, le type et la sévérité des effets indésirables observés chez l’enfant de moins de 7 ans étaient en ligne avec le profil de tolérance connu de la Tobramycine. L’utilisation de Tobramycine n’est pas indiquée chez l’enfant de moins de 6 ans (voir rubrique 4.2 Posologie et mode d’administration).
Efficacité clinique
Deux études cliniques de 24 semaines ayant un plan identique, randomisées, en double aveugle, contrôlées contre placebo, en groupes parallèles (Etude 1 et Etude 2), ont été conduites chez des patients atteints de mucoviscidose par P. aeruginosa afin d'étayer la demande initiale d'autorisation de mise sur le marché du produit, qui a été accordée en 1999. Ces études ont inclus 520 patients avec un VEMS à la sélection compris entre 25 % et 75 % de la valeur normale théorique. Les patients âgés de moins de 6 ans, ceux avec un taux de créatinine > 2 mg/dl à la sélection et ceux infectés par Burkholderia cepacia ont été exclus. Dans ces études cliniques, 258 patients ont reçu un traitement par tobramycine en ambulatoire à l'aide d'un nébuliseur à main réutilisable PARI LC PLUS, équipé d'un compresseur DeVilbiss® Pulmo-Aide®.
Dans chaque étude, les patients traités par tobramycine ont présenté une amélioration significative de la fonction pulmonaire et une réduction significative du nombre d'unités formant colonie (UFC) de P. aeruginosa dans les expectorations au cours des périodes sous traitement. Le VEMS moyen est resté supérieur à la valeur initiale au cours des périodes sans traitement de 28 jours, même s'il y a eu la plupart du temps une certaine inversion. La densité bactérienne dans les expectorations est revenue à la valeur initiale au cours des périodes sans traitement. Les réductions de la densité bactérienne dans les expectorations ont été plus faibles lors de chaque cycle successif.
Les patients traités par tobramycine ont en moyenne présenté moins de jours d'hospitalisation et nécessité moins de jours de traitement antibiotique à visée anti-pseudomonale par voie parentérale que les patients sous placebo.
Sur les 464 patients ayant terminé l'une ou l'autre des deux études 1 et 2 en double aveugle de 24 semaines, 396 patients ont participé aux extensions en ouvert de ces études. Au total, 313, 264 et 120 patients ont été traités par tobramycine pendant respectivement 48, 72 et 96 semaines. Le taux de diminution de la fonction pulmonaire a été significativement plus faible après l'initiation du traitement par tobramycine que celui observé chez les patients recevant le placebo au cours de la période de traitement randomisé en double aveugle. La pente de diminution de la fonction pulmonaire estimée au moyen d'un modèle de régression a été de -6,52 % au cours du traitement en aveugle par placebo et de - 2,53 % au cours du traitement par tobramycine (p = 0,0001).
5.2. Propriétés pharmacocinétiques
La tobramycine est une molécule polaire cationique qui ne traverse pas facilement la membrane épithéliale. L’exposition systémique à la tobramycine après inhalation de la tobramycine devrait résulter de l’absorption pulmonaire de la fraction de dose délivrée aux poumons étant donné que la tobramycine n’est pas absorbée de manière appréciable lorsqu’elle est administrée par voie orale. La biodisponibilité de la tobramycine peut varier en raison des différences interindividuelles selon les performances du nébuliseur utilisé et selon l’atteinte respiratoire.
Concentrations dans les expectorations :
10 minutes après l'inhalation de la première dose de 300 mg de tobramycine, la concentration moyenne de tobramycine dans les expectorations était de 1 237 μg/g (de 35 à 7 414 μg/g). La tobramycine ne s'accumule pas dans les expectorations ; après 20 semaines de traitement par tobramycine, la concentration moyenne de tobramycine dans les expectorations, 10 minutes après l'inhalation était de 1 154 μg/g (de 39 à 8 085 μg/g). Une forte variabilité des concentrations de tobramycine dans les expectorations a été observée. Deux heures après l'inhalation, les concentrations dans les expectorations avaient diminué, passant à environ 14 % des taux de tobramycine relevés 10 minutes après l'inhalation.
Concentrations sériques :
La concentration sérique moyenne de tobramycine 1 heure après l'inhalation d'une dose unique de 300 mg de tobramycine chez les patients atteints de mucoviscidose était de 0,95 μg/mL (de indétectable à 3,62 μg/mL). Après 20 semaines de traitement par tobramycine, la concentration moyenne sérique de tobramycine 1 heure après l'administration était de 1,05 μg/mL (de indétectable à 3,41 μg/mL). Pour comparaison, le pic de concentration après administration intraveineuse ou intramusculaire d’une dose unique de 1,5 à 2 mg/kg de tobramycine varie généralement entre 4 et 12 µg/mL.
Distribution
Suite à l’administration de la tobramycine, elle se concentre principalement dans les voies respiratoires. Le taux de liaison de la tobramycine aux protéines plasmatiques est inférieur à 10 %.
Biotransformation
La tobramycine n’est pas métabolisée et est principalement excrétée sous forme inchangée dans les urines.
Élimination
L'élimination de la tobramycine administrée par inhalation n'a pas été étudiée.
Après administration par voie intraveineuse, la tobramycine est principalement éliminée par filtration glomérulaire sous forme inchangée. La demi-vie d’élimination apparente de la tobramycine sérique après inhalation d’une dose unique de 300 mg de tobramycine a été de 3 heures chez les patients atteints de mucoviscidose. La fonction rénale devrait modifier l’exposition à la tobramycine. Cependant, les données ne sont pas disponibles, les patients ayant une créatininémie supérieure ou égale à 2 mg/dL (176,8 µmol/L) ou un taux d'urée sanguine supérieur ou égal à 40 mg/dL n’ayant pas été inclus dans les études cliniques.
Il est probable que la tobramycine non absorbée après administration par inhalation soit éliminée principalement dans les expectorations.
5.3. Données de sécurité préclinique
Sur la base des études de pharmacologie, de toxicité en administration répétée, de génotoxicité ou de toxicité sur la reproduction, les principaux risques chez l'homme mis en évidence par les données précliniques sont une toxicité rénale et une ototoxicité. Au cours des études de toxicité par administration réitérée, les organes cibles de la toxicité sont le rein et les fonctions vestibulaires et cochléaire. En général, cette toxicité apparaît pour des taux systémiques de tobramycine plus élevés que ceux pouvant être atteints par inhalation à la dose recommandée.
Les études de carcinogénicité avec la tobramycine inhalée n'ont mis en évidence aucune augmentation de l'incidence d'aucun type de tumeur. La tobramycine n'a pas montré de potentiel génotoxique dans une batterie de tests de génotoxicité.
Aucun essai de toxicologie sur la reproduction n'a été effectué avec la tobramycine en inhalation, mais l'administration sous-cutanée de tobramycine à des doses de 100 mg/kg/jour chez le rat et à la dose maximale tolérée de 20 mg/kg/jour chez le lapin pendant l'organogenèse ne s'est pas révélée tératogène. La tératogénicité ne peut pas être évaluée à des doses parentérales supérieures (supérieures ou égales à 40 mg/kg/jour) chez le lapin car ces doses induisent une toxicité maternelle et des avortements. L’ototoxicité n’a pas été évaluée chez la progéniture lors des études non cliniques de toxicité sur la reproduction avec la tobramycine. Sur la base des données disponibles chez l'animal, un risque de toxicité (par exemple : ototoxicité) ne peut être exclu lors de l'exposition prénatale au médicament.
L'administration sous-cutanée de doses allant jusqu’à 100 mg/kg de tobramycine n'a pas affecté le comportement d'accouplement ou altéré la fécondité chez des rats mâles ou femelles.
5 ans.
À usage unique. L'intégralité du contenu de l'ampoule doit être utilisée immédiatement après ouverture (voir rubrique 6.6).
Toute solution restante après ouverture doit être éliminée.
6.4. Précautions particulières de conservation
À conserver au réfrigérateur (entre 2°C et 8°C).
Ne pas congeler.
À conserver dans l’emballage d’origine, à l’abri de la lumière.
La solution TOBRAMYCINE ZENTIVA est normalement légèrement jaune, cependant, des variations de couleur peuvent être observées. Cela n'indique pas de perte de l'activité si le produit a été conservé conformément aux recommandations.
6.5. Nature et contenu de l'emballage extérieur
TOBRAMYCINE ZENTIVA est conditionné dans des ampoules unidoses en polyéthylène de basse densité (PEBD) de 5 ml. Chaque boîte contient 56, 112 ou 168 ampoules réparties respectivement en 4, 8 ou 12 poches en film métallisé scellées. Elles contiennent chacune 7 ampoules sur un plateau en plastique.
Toutes les présentations peuvent ne pas être commercialisées.
6.6. Précautions particulières d’élimination et de manipulation
TOBRAMYCINE ZENTIVA est une préparation aqueuse stérile, apyrogène pour usage unique. Ce médicament ne contenant pas de conservateur, l'intégralité du contenu de l'ampoule doit être utilisée immédiatement après ouverture et toute solution non utilisée éliminée. Les ampoules ouvertes ne doivent jamais être conservées pour être réutilisées.
Tout médicament non utilisé ou déchet doit être éliminé conformément à la réglementation en vigueur.
7. TITULAIRE DE L’AUTORISATION DE MISE SUR LE MARCHE
35 RUE DU VAL DE MARNE
75013 PARIS
8. NUMERO(S) D’AUTORISATION DE MISE SUR LE MARCHE
· 34009 301 777 3 0 : Solution pour inhalation par nébuliseur en ampoule (PEBD) de 5 mL ; boîte de 56.
· 34009 550 647 6 3 : Solution pour inhalation par nébuliseur en ampoule (PEBD) de 5 mL ; boîte de 112.
· 34009 550 647 8 7 : Solution pour inhalation par nébuliseur en ampoule (PEBD) de 5 mL ; boîte de 168.
9. DATE DE PREMIERE AUTORISATION/DE RENOUVELLEMENT DE L’AUTORISATION
[à compléter ultérieurement par le titulaire]
10. DATE DE MISE A JOUR DU TEXTE
[à compléter ultérieurement par le titulaire]
Sans objet.
12. INSTRUCTIONS POUR LA PREPARATION DES RADIOPHARMACEUTIQUES
Liste I.
Médicament soumis à prescription initiale hospitalière semestrielle. Renouvellement non restreint.
ANSM - Mis à jour le : 10/11/2025
TOBRAMYCINE ZENTIVA 300 mg/5 mL, solution pour inhalation par nébuliseur
Tobramycine
· Gardez cette notice. Vous pourriez avoir besoin de la relire.
· Si vous avez d’autres questions, interrogez votre médecin ou votre pharmacien.
· Ce médicament vous a été personnellement prescrit. Ne le donnez pas à d’autres personnes. Il pourrait leur être nocif, même si les signes de leur maladie sont identiques aux vôtres.
· Si vous ressentez un quelconque effet indésirable, parlez-en à votre médecin ou votre pharmacien. Ceci s’applique aussi à tout effet indésirable qui ne serait pas mentionné dans cette notice. Voir rubrique 4.
1. Qu'est-ce que TOBRAMYCINE ZENTIVA 300 mg/5 mL, solution pour inhalation par nébuliseur et dans quels cas est-il utilisé ?
2. Quelles sont les informations à connaître avant d'utiliser TOBRAMYCINE ZENTIVA 300 mg/5 mL, solution pour inhalation par nébuliseur ?
3. Comment utiliser TOBRAMYCINE ZENTIVA 300 mg/5 mL, solution pour inhalation par nébuliseur ?
4. Quels sont les effets indésirables éventuels ?
5. Comment conserver TOBRAMYCINE ZENTIVA 300 mg/5 mL, solution pour inhalation par nébuliseur ?
6. Contenu de l’emballage et autres informations.
1. QU’EST-CE QUE TOBRAMYCINE ZENTIVA 300 mg/5 mL, solution pour inhalation par nébuliseur ET DANS QUELS CAS EST-IL UTILISE ?
Classe pharmacothérapeutique : Aminosides antibiotiques - code ATC : J01GB01.
Qu’est-ce que TOBRAMYCINE ZENTIVA ?
TOBRAMYCINE ZENTIVA contient de la tobramycine. Il s’agit d’un antibiotique de la famille des aminosides.
Dans quels cas TOBRAMYCINE ZENTIVA est-il utilisé ?
TOBRAMYCINE ZENTIVA est utilisé chez les patients âgés de 6 ans et plus atteints de mucoviscidose pour traiter les infections respiratoires dues à une bactérie appelée Pseudomonas aeruginosa.
TOBRAMYCINE ZENTIVA combat l’infection provoquée par la bactérie Pseudomonas dans vos poumons et vous aide à mieux respirer.
Comment fonctionne TOBRAMYCINE ZENTIVA ?
Lorsque vous inhalez TOBRAMYCINE ZENTIVA, l’antibiotique arrive directement dans vos poumons pour lutter contre la bactérie responsable de l’infection. Pour obtenir les meilleurs résultats de ce traitement, prenez ce médicament comme cela est expliqué dans cette notice.
Qu’est-ce que Pseudomonas aeruginosa ?
C’est une bactérie très fréquente qui infecte presque tous les patients atteints de mucoviscidose à un moment ou un autre de leur vie. Chez certaines personnes, cette infection ne se développe que très tardivement, tandis que chez d’autres, elle se développe très jeune.
C’est l’une des bactéries les plus nocives pour les patients atteints de mucoviscidose. Si l’infection n’est pas contrôlée correctement, elle continuera à altérer vos poumons entraînant des difficultés respiratoires supplémentaires.
TOBRAMYCINE ZENTIVA tue la bactérie qui provoque les infections pulmonaires. L’infection peut être contrôlée avec succès si elle est traitée suffisamment tôt.
2. QUELLES SONT LES INFORMATIONS A CONNAITRE AVANT D’UTILISER TOBRAMYCINE ZENTIVA 300 mg/5 mL, solution pour inhalation par nébuliseur ?
N’utilisez jamais TOBRAMYCINE ZENTIVA 300 mg/5 mL, solution pour inhalation par nébuliseur :
· si vous êtes allergique à la tobramycine, à un antibiotique de la famille des aminosides ou à l’un des autres composants contenus dans ce médicament (mentionnés dans la rubrique 6).
Si vous êtes dans l’une de ces situations, ne prenez pas ce médicament et parlez-en à votre médecin.
Si vous pensez que vous pouvez être allergique, demandez conseil à votre médecin.
Avertissements et précautions
Adressez-vous à votre médecin avant d’utiliser TOBRAMYCINE ZENTIVA 300 mg/5 mL, solution pour inhalation par nébuliseur, si vous présentez ou avez déjà présenté un des troubles suivants :
· Problèmes auditifs (notamment bourdonnements d’oreilles et sensations vertigineuses),
· Problèmes de reins,
· Difficultés respiratoires inhabituelles avec une respiration sifflante ou une toux, et une oppression dans la poitrine,
· Sang dans les expectorations (sécrétions émises en toussant),
· Faiblesse musculaire qui dure ou s’aggrave avec le temps, symptôme principalement lié à une maladie telle que la myasthénie ou la maladie de Parkinson.
Si vous êtes dans l’une de ces situations, parlez-en à votre médecin avant de prendre TOBRAMYCINE ZENTIVA.
Si vous ou les membres de votre famille maternelle souffrez d’une mutation mitochondriale (maladie génétique) ou d’une perte d’audition due à des antibiotiques, il est conseillé d’en informer votre médecin ou votre pharmacien avant de prendre ce médicament ; certaines mutations mitochondriales pourrait augmenter le risque de perte d’audition avec ce produit. Votre médecin peut vous recommander un test génétique avant l’administration de TOBRAMYCINE ZENTIVA.
Les médicaments inhalés peuvent entraîner une oppression thoracique et une respiration sifflante et ceci peut arriver avec TOBRAMYCINE ZENTIVA. Votre médecin vous surveillera lors de votre première prise de TOBRAMYCINE ZENTIVA et vérifiera votre fonction pulmonaire avant et après l’administration. Si vous n’en utilisez pas déjà, votre médecin peut vous demander d’utiliser un bronchodilatateur (par exemple salbutamol) avant de prendre TOBRAMYCINE ZENTIVA.
Si vous prenez TOBRAMYCINE ZENTIVA, les souches de Pseudomonas peuvent devenir résistantes au traitement avec le temps. Ceci peut signifier que, sur le long terme, le médicament pourrait ne pas être aussi efficace qu’il ne le devrait.
Si ceci vous inquiète, parlez-en à votre médecin.
Si vous recevez de la tobramycine en injection, elle peut parfois provoquer une perte d’audition, des sensations vertigineuses et une atteinte rénale et elle peut être nocive pour l’enfant à naître.
Enfants et adolescents
TOBRAMYCINE ZENTIVA peut être utilisé par les enfants et adolescents âgés de 6 ans et plus. TOBRAMYCINE ZENTIVA ne doit pas être administré à des enfants âgés de moins de 6 ans.
Patients âgés
Si vous êtes âgé de 65 ans ou plus, votre médecin pourra réaliser des examens supplémentaires pour décider si TOBRAMYCINE ZENTIVA vous convient.
Autres médicaments et TOBRAMYCINE ZENTIVA 300 mg/5 mL, solution pour inhalation par nébuliseur
Informez votre médecin ou pharmacien si vous prenez, avez récemment pris ou pourriez prendre tout autre médicament.
Vous ne devez pas prendre les médicaments ci-dessous pendant que vous prenez TOBRAMYCINE ZENTIVA :
· Furosémide ou acide éthacrynique, diurétiques (médicaments faisant uriner),
· Urée ou mannitol par voie intraveineuse,
· D’autres médicaments qui peuvent avoir des effets nocifs sur le système nerveux, les reins ou sur l’audition.
Les médicaments suivants peuvent augmenter les risques d’apparition d’effets nocifs s’ils vous sont administrés pendant que vous recevez des injections de tobramycine :
· Amphotéricine B, céfalotine, ciclosporine, tacrolimus, polymyxines : ces médicaments peuvent altérer vos reins.
· Composés à base de platine (tels que carboplatine et cisplatine) : ces médicaments peuvent altérer vos reins ou votre audition.
· Anticholinestérases (tels que néostigmine et pyridostigmine), ou toxine botulique : ces médicaments peuvent entraîner l’apparition ou l’aggravation d’une faiblesse musculaire.
Si vous prenez un ou plusieurs des médicaments cités ci-dessus, discutez-en avec votre médecin avant de prendre TOBRAMYCINE ZENTIVA.
Vous ne devez pas mélanger ou diluer TOBRAMYCINE ZENTIVA avec un autre médicament dans votre nébuliseur.
Si vous prenez plusieurs traitements différents pour la mucoviscidose, vous devez les prendre dans l’ordre suivant :
1. bronchodilatateur, tel que le salbutamol
2. kinésithérapie respiratoire
3. autres médicaments inhalés
4. TOBRAMYCINE ZENTIVA en dernier lieu.
Veuillez vérifier également cet ordre avec votre médecin.
TOBRAMYCINE ZENTIVA 300 mg/5 mL, solution pour inhalation par nébuliseur avec des aliments, boissons et de l’alcool
Sans objet.
Si vous êtes enceinte ou que vous allaitez, si vous pensez être enceinte ou planifiez une grossesse, demandez conseil à votre médecin ou pharmacien avant de prendre ce médicament.
Si vous êtes enceinte, si vous pensez être enceinte ou planifiez une grossesse, vous devez parler avec votre médecin de la possibilité d’effets nocifs que ce médicament peut entraîner pour vous ou l’enfant à naître.
On ne sait pas si l’inhalation de ce médicament peut avoir des effets indésirables pendant la grossesse.
Lorsqu’ils sont administrés par injection, la tobramycine et les autres antibiotiques de la famille des aminosides peuvent être néfastes pour l’enfant à naître, et provoquer par exemple une surdité.
Si vous allaitez votre enfant, demandez conseil à votre médecin avant d’utiliser votre médicament.
Conduite de véhicules et utilisation de machines
TOBRAMYCINE ZENTIVA ne devrait pas altérer votre aptitude à conduire et utiliser des machines.
TOBRAMYCINE ZENTIVA 300 mg/5 mL, solution pour inhalation par nébuliseur contient :
Sans objet.
3. COMMENT UTILISER TOBRAMYCINE ZENTIVA 300 mg/5 mL, solution pour inhalation par nébuliseur ?
Quelle dose de ce médicament devez-vous prendre et à quelle fréquence ?
· La dose recommandée est la même pour tous les patients âgés de 6 ans et plus.
· Vous devez utiliser deux ampoules par jour pendant 28 jours. Inhalez l’intégralité du contenu d’une ampoule le matin et d’une ampoule le soir. Idéalement, il doit y avoir un intervalle de 12 heures entre les doses.
· Vous devez respecter un délai d’au moins 6 heures entre deux inhalations de TOBRAMYCINE ZENTIVA.
· Après avoir pris votre médicament pendant 28 jours, vous faites une pause de 28 jours pendant laquelle vous n’inhalez pas de TOBRAMYCINE ZENTIVA, puis vous recommencez une autre cure.
· Il est important de bien respecter l’utilisation du médicament deux fois par jour pendant les 28 jours de traitement et de respecter le cycle de 28 jours avec traitement suivi de 28 jours sans traitement.
|
|
|
|
PERIODE DE TRAITEMENT PAR TOBRAMYCINE ZENTIVA |
PERIODE SANS TRAITEMENT PAR TOBRAMYCINE ZENTIVA |
|
Prenez TOBRAMYCINE ZENTIVA deux fois par jour chaque jour, pendant 28 jours |
Ne prenez pas TOBRAMYCINE ZENTIVA pendant les 28 jours suivants |
|
|
|
Répétez le cycle
Si vous avez pris plus de TOBRAMYCINE ZENTIVA 300 mg/5 mL, solution pour inhalation par nébuliseur que vous n’auriez dû
Si vous inhalez une trop grande quantité de TOBRAMYCINE ZENTIVA, vous pourrez avoir un fort enrouement de la voix. Prévenez votre médecin le plus rapidement possible. Si vous avalez TOBRAMYCINE ZENTIVA, prévenez votre médecin le plus rapidement possible.
Si vous oubliez de prendre TOBRAMYCINE ZENTIVA 300 mg/5 mL, solution pour inhalation par nébuliseur
Si vous oubliez de prendre TOBRAMYCINE ZENTIVA et que vous devez prendre la dose suivante dans 6 heures ou plus, prenez la dose dès que possible. Sinon, attendez la prochaine dose. Ne prenez pas de dose double pour compenser la dose que vous avez oublié de prendre.
Si vous arrêtez de prendre TOBRAMYCINE ZENTIVA 300 mg/5 mL, solution pour inhalation par nébuliseur
Sans objet.
Instructions d'utilisation de TOBRAMYCINE ZENTIVA
Cette partie de la notice vous explique comment utiliser, entretenir et manipuler TOBRAMYCINE ZENTIVA. Veuillez lire attentivement et suivre ces instructions.
Si vous avez d’autres questions sur l’utilisation de ce médicament, demandez plus d’informations à votre médecin ou à votre pharmacien.
Matériel nécessaire pour l’inhalation de TOBRAMYCINE ZENTIVA
TOBRAMYCINE ZENTIVA doit être utilisé avec un nébuliseur réutilisable propre et sec.
Le nébuliseur LC PLUS (fabriqué par PARI GmbH) est adapté pour faire les inhalations de TOBRAMYCINE ZENTIVA.
Votre médecin ou votre kinésithérapeute pourra vous conseiller sur la manière correcte d’utiliser TOBRAMYCINE ZENTIVA et le matériel nécessaire. Vous pourriez avoir besoin de nébuliseurs différents pour vos autres médicaments inhalés dans la mucoviscidose.
Préparation de l’inhalation de TOBRAMYCINE ZENTIVA
· Lavez-vous soigneusement les mains à l’eau et au savon.
· Chaque poche métallisée de TOBRAMYCINE ZENTIVA contient un plateau de 7 ampoules. Coupez ou déchirez la poche pour l’ouvrir. Sortez une ampoule de TOBRAMYCINE ZENTIVA du plateau en la détachant délicatement des autres ampoules au niveau des languettes du bas.
· Replacez le plateau dans la poche métallisée et conservez-la au réfrigérateur.
· Etalez toutes les pièces de votre nébuliseur sur du papier ou une serviette de toilette propre et sec.
· Assurez-vous d’avoir le compresseur approprié et le tuyau pour raccorder le nébuliseur et le compresseur.
· Respectez soigneusement le mode d’emploi de votre modèle de nébuliseur ; lisez attentivement la notice du fabricant fournie avec le nébuliseur. Vérifiez que le nébuliseur et le compresseur fonctionnent correctement selon les instructions du fabricant avant de commencer à prendre votre médicament.
Utilisation de TOBRAMYCINE ZENTIVA avec le nébuliseur LC PLUS (PARI GmbH)
Pour des instructions plus détaillées sur l’utilisation et l’entretien du nébuliseur PARI LC PLUS, veuillez lire la notice fournie avec l’appareil.
1. Détachez la partie supérieure de la partie inférieure du nébuliseur en tournant dans le sens inverse des aiguilles d’une montre et en la soulevant. Placez la partie supérieure sur la serviette et maintenez la partie inférieure du nébuliseur en position verticale sur la serviette.
2. Branchez une extrémité du tuyau sur la sortie d’air du compresseur. Assurez-vous que le tuyau soit bien ajusté. Branchez le compresseur à la prise électrique.
3. Ouvrez l’ampoule de TOBRAMYCINE ZENTIVA en la tenant d’une main par le bas et en tournant la partie supérieure avec l’autre main. Pressez le contenu entier de l’ampoule dans la partie inférieure du nébuliseur.
|
|
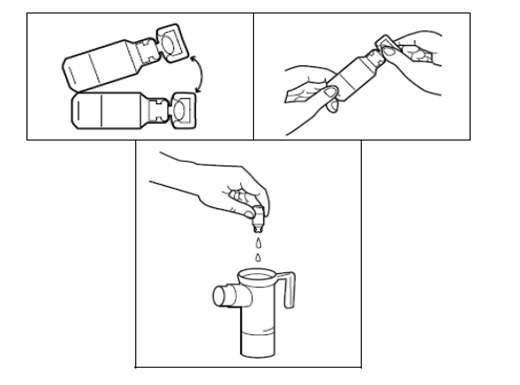
4. Replacez la partie supérieure du nébuliseur, fixez l’embout buccal et insérez le bouchon de la valve d’inspiration sur le nébuliseur et branchez ensuite le compresseur comme il est indiqué dans la notice du nébuliseur PARI LC PLUS.
5. Mettez le compresseur en marche. Vérifiez que la vaporisation sortant de l’embout buccal soit régulière. Si rien ne sort, vérifiez que le tuyau est bien branché et que le compresseur fonctionne correctement.
6. En position assise ou debout, tenez-vous droit de façon à pouvoir respirer normalement.
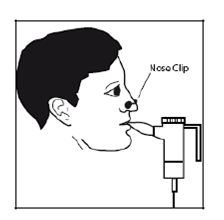
7. Placez l’embout buccal entre vos dents et sur la partie supérieure de votre langue. Respirez normalement mais uniquement par la bouche (vous pouvez utiliser une pince pour le nez, avec l’accord de votre médecin). Essayer de ne pas bloquer l’arrivée d’air avec votre langue.
|
|
8. Continuez jusqu’à ce qu’il ne reste plus de solution de TOBRAMYCINE ZENTIVA et que la vaporisation cesse. La durée complète de traitement est d’environ 15 minutes. Vous pourrez entendre un bruit de crachotements lorsque le réservoir du nébuliseur sera vide.
9. N’oubliez pas de nettoyer et de désinfecter votre nébuliseur après le traitement selon les instructions du fabricant. N’utilisez jamais un nébuliseur sale ou obstrué. Ne prêtez pas votre nébuliseur à une autre personne.
Si vous êtes interrompu, si vous avez besoin de tousser ou de vous reposer pendant votre traitement, arrêtez le compresseur pour conserver le médicament. Remettez le compresseur en marche lorsque vous êtes prêt à recommencer votre traitement. N’utilisez pas le reste de cette dose si la prochaine inhalation doit être effectuée dans moins de 6 heures.
4. QUELS SONT LES EFFETS INDESIRABLES EVENTUELS ?
Certains effets indésirables peuvent être graves.
Si vous présentez l'un des symptômes suivants, arrêtez de prendre TOBRAMYCINE ZENTIVA et parlez-en immédiatement à votre médecin :
· Difficultés respiratoires inhabituelles avec respiration sifflante ou toux et oppression dans la poitrine,
· Réactions allergiques, incluant urticaire et démangeaisons.
Si vous présentez l'un des symptômes suivants, parlez-en immédiatement à votre médecin :
· Perte de l'audition (les bourdonnements d'oreilles sont un signe d'alerte possible d'une perte auditive), bruits (tels que sifflements) dans les oreilles.
Votre maladie pulmonaire sous-jacente peut être aggravée au cours du traitement par TOBRAMYCINE ZENTIVA. Cela peut être dû à un manque d’efficacité. Parlez-en immédiatement à votre médecin si cela vous arrive.
Certains effets indésirables sont très fréquents.
Ces effets indésirables peuvent affecter plus de 1 patient sur 10.
· Nez qui coule ou nez bouché, éternuements,
· Modification de la voix (enrouement),
· Modification de la couleur de la substance que vous crachez (expectorations),
· Aggravation des résultats des tests de la fonction respiratoire.
Si vous ressentez l’un de ces effets comme grave, parlez-en à votre médecin.
Certains effets indésirables sont fréquents.
Ces effets indésirables peuvent affecter jusqu’à 1 patient sur 10.
· Sensation générale de malaise,
· Douleurs musculaires,
· Altération de la voix avec mal de gorge et difficulté à avaler (laryngite).
Si vous ressentez l’un de ces effets comme grave, parlez-en à votre médecin.
Autres effets indésirables :
· Démangeaisons,
· Eruption prurigineuse,
· Eruption cutanée,
· Perte de la voix,
· Troubles du goût,
· Mal de gorge.
Si vous ressentez l’un de ces effets comme grave, parlez-en à votre médecin.
Chez les patients ayant pris TOBRAMYCINE ZENTIVA en même temps, ou après avoir reçu par voie injectable des cures répétées de tobramycine ou d’un autre antibiotique de la famille des aminosides, une perte d’audition a été rapportée comme effet indésirable.
Les injections de tobramycine ou d’autres aminosides peuvent provoquer des réactions allergiques, des troubles de l’audition et des troubles rénaux.
Les patients atteints de mucoviscidose présentent de nombreux symptômes de la maladie. Ils peuvent quand même survenir pendant le traitement par TOBRAMYCINE ZENTIVA, mais ils ne devraient pas être plus fréquents ou plus sévères qu’avant le traitement.
Déclaration des effets secondaires
Si vous ressentez un quelconque effet indésirable, parlez-en à votre médecin, à votre pharmacien ou à votre infirmier/ère. Ceci s’applique aussi à tout effet indésirable qui ne serait pas mentionné dans cette notice. Vous pouvez également déclarer les effets indésirables directement via le système national de déclaration : Agence nationale de sécurité du médicament et des produits de santé (ANSM) et réseau des Centres Régionaux de Pharmacovigilance - Site internet : https://signalement.social-sante.gouv.fr
En signalant les effets indésirables, vous contribuez à fournir davantage d’informations sur la sécurité du médicament.
5. COMMENT CONSERVER TOBRAMYCINE ZENTIVA 300 mg/5 mL, solution pour inhalation par nébuliseur ?
Tenir ce médicament hors de la vue et de la portée des enfants.
N’utilisez pas ce médicament après la date de péremption indiquée sur l’emballage ou la poche ou figurant sur l’ampoule.
N’utilisez pas ce médicament si vous remarquez qu’il est devenu trouble ou s’il contient des particules dans la solution.
À conserver entre 2°C et 8°C (au réfrigérateur). Si vous ne disposez pas d’un réfrigérateur (par exemple lorsque vous transportez votre médicament) vous pouvez conserver les poches métallisées (ouvertes ou non ouvertes) à température ambiante (ne dépassant pas 25°C) pendant 28 jours au maximum.
Ne pas utiliser d’ampoules de TOBRAMYCINE ZENTIVA qui ont été conservées à température ambiante pendant plus de 28 jours.
Conserver vos ampoules dans l’emballage d’origine car ce médicament est sensible à la forte lumière. La solution de ce médicament est normalement jaune clair, mais cela peut varier et elle peut parfois être jaune plus foncé. Ceci ne modifie pas l’efficacité de ce médicament si les instructions de conservation ont été respectées.
Ne conserver jamais une ampoule ouverte. Une fois ouverte, l’ampoule doit être utilisée immédiatement et tout le produit restant doit être éliminé.
6. CONTENU DE L’EMBALLAGE ET AUTRES INFORMATIONS
Ce que contient TOBRAMYCINE ZENTIVA 300 mg/5 mL, solution pour inhalation par nébuliseur
· La substance active est :
Tobramycine.................................................................................................................. 300 mg
Pour une ampoule à dose-unique de 5 mL.
· Les autres composants sont :
Chlorure de sodium, eau pour préparations injectables, acide sulfurique (pour l’ajustement du pH), hydroxyde de sodium (pour l’ajustement du pH).
TOBRAMYCINE ZENTIVA est une solution claire, légèrement jaune qui est présentée en ampoules prêtes à l’emploi.
Les ampoules sont conditionnées dans des poches métallisées ; une poche métallisée contient 7 ampoules, ce qui correspond à 3,5 jours de traitement.
TOBRAMYCINE ZENTIVA se présente en boîte de 56, 112 ou 168 ampoules, ce qui correspond respectivement à un, deux ou trois cycles de traitement de 28 jours.
Toutes les présentations peuvent ne pas être commercialisées.
Titulaire de l’autorisation de mise sur le marché
ZENTIVA FRANCE
35 RUE DU VAL DE MARNE
75013 PARIS
Exploitant de l’autorisation de mise sur le marché
ZENTIVA FRANCE
35 RUE DU VAL DE MARNE
75013 PARIS
ALTAN PHARMACEUTICALS, S.A.
POLÍGONO INDUSTRIAL DE BERNEDO S/N
01118 BERNEDO, ÁLAVA
Ou
ALTAN PHARMACEUTICALS, S.A.
AVDA. DE LA CONSTITUCION, 198-199,
POLIGONO INDUSTRIAL MONTE BOYAL,
CASARRUBIOS DEL MONTE, 45950 TOLEDO,
ESPAGNE
Noms du médicament dans les Etats membres de l'Espace Economique Européen
Ce médicament est autorisé dans les Etats membres de l'Espace Economique Européen sous les noms suivants : Conformément à la réglementation en vigueur.
[à compléter ultérieurement par le titulaire]
La dernière date à laquelle cette notice a été révisée est :
[à compléter ultérieurement par le titulaire]
Des informations détaillées sur ce médicament sont disponibles sur le site Internet de l’ANSM (France).
CONSEILS / EDUCATION SANITAIRE
QUE SAVOIR SUR LES ANTIBIOTIQUES ?
Les antibiotiques sont efficaces pour combattre les infections dues aux bactéries. Ils ne sont pas efficaces contre les infections dues aux virus.
Aussi, votre médecin a choisi de vous prescrire cet antibiotique parce qu’il convient précisément à votre cas et à votre maladie actuelle.
Les bactéries ont la capacité de survivre ou de se reproduire malgré l’action d’un antibiotique. Ce phénomène est appelé résistance : il rend certains traitements antibiotiques inactifs.
La résistance s’accroît par l’usage abusif ou inapproprié des antibiotiques.
Vous risquez de favoriser l’apparition de bactéries résistantes et donc de retarder votre guérison ou même de rendre inactif ce médicament, si vous ne respectez pas :
· la dose à prendre,
· les moments de prise,
· et la durée de traitement.
En conséquence, pour préserver l’efficacité de ce médicament :
1- N’utilisez un antibiotique que lorsque votre médecin vous l’a prescrit.
2- Respectez strictement votre ordonnance.
3- Ne réutilisez pas un antibiotique sans prescription médicale même si vous pensez combattre une maladie apparemment semblable.
4- Ne donnez jamais votre antibiotique à une autre personne, il n’est peut-être pas adapté à sa maladie.
5- Une fois votre traitement terminé, rapportez à votre pharmacien toutes les boîtes entamées pour une destruction correcte et appropriée de ce médicament.