Dernière mise à jour le 03/08/2026
XEMBIFY 200 mg/mL, solution injectable sous-cutanée

: Ce médicament fait l'objet d'une surveillance renforcée. Pour plus d'informations, cliquez ici
Indications thérapeutiques
Qu’est‑ce que XEMBIFY 200 mg/mL, solution injectable sous-cutanée ?
XEMBIFY est une solution d’immunoglobulines humaines (anticorps, essentiellement des immunoglobulines G) qui aident votre organisme à combattre les infections.
XEMBIFY contient des immunoglobulines qui proviennent du plasma de donneurs sains. Les immunoglobulines aident à combattre les infections causées par des bactéries et des virus. Le médicament agit exactement de la même manière que les immunoglobulines produites par le système immunitaire et présentes naturellement dans le sang humain.
Dans quels cas XEMBIFY 200 mg/mL, solution injectable sous-cutanée est‑il utilisé ?
XEMBIFY vous a été prescrit car votre taux d’immunoglobulines est anormalement bas en raison d’une affection appelée un déficit immunitaire. Les perfusions de XEMBIFY permettent d’augmenter les taux d’immunoglobulines (anticorps), en particulier les taux d’immunoglobulines G (IgG) de votre sang et de les ramener à des taux normaux.
Ce médicament est indiqué chez les adultes, les enfants et les adolescents (de 0 à 18 ans) qui ne possèdent pas suffisamment d’anticorps (traitement substitutif) :
1. patients présentant un déficit immunitaire primitif (DIP), avec un manque d’anticorps existant dès la naissance ;
2. hypogammaglobulinémie (affection au cours de laquelle le taux d’immunoglobulines dans le sang est faible) et infections bactériennes à répétition chez des patients atteints de leucémie lymphoïde chronique (cancer du sang avec production excessive de globules blancs), pour lesquels les antibiotiques administrés à titre préventif n’ont pas été efficaces ;
3. hypogammaglobulinémie et infections bactériennes à répétition dans le myélome multiple (tumeur constituée de cellules dérivées de la moelle osseuse) ;
4. hypogammaglobulinémie chez les patients après une greffe de cellules souches provenant d’un donneur (allogreffe de cellules souches hématopoïétiques, GCSH allogénique).
Présentations
> 1 flacon(s) en verre avec fermeture à témoin d'effraction de 5 ml
Code CIP : 34009 302 440 7 4
Déclaration de commercialisation : 25/03/2024
Cette présentation est agréée aux collectivités
Inscription sur la liste de rétrocession au titre de son AMM, selon les conditions précisées au Journal Officiel. Prix de cession publié au Journal Officiel.
> 1 flacon(s) en verre avec fermeture à témoin d'effraction de 10 ml
Code CIP : 34009 302 440 8 1
Déclaration de commercialisation : 25/03/2024
Cette présentation est agréée aux collectivités
Inscription sur la liste de rétrocession au titre de son AMM, selon les conditions précisées au Journal Officiel. Prix de cession publié au Journal Officiel.
> 1 flacon(s) en verre avec fermeture à témoin d'effraction de 20 ml
Code CIP : 34009 302 440 9 8
Déclaration de commercialisation : 25/03/2024
Cette présentation est agréée aux collectivités
Inscription sur la liste de rétrocession au titre de son AMM, selon les conditions précisées au Journal Officiel. Prix de cession publié au Journal Officiel.
> 1 flacon(s) en verre avec fermeture à témoin d'effraction de 50 ml
Code CIP : 34009 302 441 0 4
Déclaration de commercialisation : 25/03/2024
Cette présentation est agréée aux collectivités
Inscription sur la liste de rétrocession au titre de son AMM, selon les conditions précisées au Journal Officiel. Prix de cession publié au Journal Officiel.
Service médical rendu (SMR)
Les libellés affichés ci-dessous ne sont que des résumés ou extraits issus des avis rendus par la Commission de la Transparence. Seul l'avis complet de la Commission de la Transparence fait référence.
Cet avis est consultable à partir du lien `Avis du jj/mm/aaaa` ou encore sur demande auprès de la HAS (plus d'informations dans l'aide). Les avis et synthèses d'avis contiennent un paragraphe sur la place du médicament dans la stratégie thérapeutique.
| Valeur du SMR | Avis | Motif de l'évaluation | Résumé de l'avis |
|---|---|---|---|
| Important | Avis du 01/06/2022 | Inscription (CT) | Le service médical rendu par XEMBIFY 200 mg/ml (immunoglobuline humaine normale) est important dans les indications de l’AMM. |
Amélioration du service médical rendu (ASMR)
Les libellés affichés ci-dessous ne sont que des résumés ou extraits issus des avis rendus par la Commission de la Transparence. Seul l'avis complet de la Commission de la Transparence fait référence.
Cet avis est consultable à partir du lien `Avis du jj/mm/aaaa` ou encore sur demande auprés de la HAS (plus d'informations dans l'aide). Les avis et synthéses d'avis contiennent un paragraphe sur la place du médicament dans la stratégie thérapeutique.
| Valeur de l'ASMR | Avis | Motif de l'évaluation | Résumé de l'avis |
|---|---|---|---|
| V (Inexistant) | Avis du 01/06/2022 | Inscription (CT) | XEMBIFY 200 mg/ml (immunoglobuline humaine normale) par voie sous-cutanée n’apporte pas d’amélioration du service médical rendu (ASMR V) par rapport aux autres immunoglobulines humaines normales administrées par voie sous-cutanée ou intraveineuse. |
Autres informations
- Titulaire de l'autorisation : INSTITUTO GRIFOLS SA
- Conditions de prescription et de délivrance :
- liste I
- prescription hospitalière
- prescription par un médecin exerçant dans un établissement de transfusion sanguine
- Statut de l'autorisation : Valide
- Type de procédure : Procédure décentralisée
- Code CIS : 6 789 493 1
ANSM - Mis à jour le : 19/03/2026
![]() Ce médicament fait l’objet d’une surveillance supplémentaire qui permettra l’identification rapide de nouvelles informations relatives à la sécurité. Les professionnels de la santé déclarent tout effet indésirable suspecté. Voir rubrique 4.8 pour les modalités de déclaration des effets indésirables.
Ce médicament fait l’objet d’une surveillance supplémentaire qui permettra l’identification rapide de nouvelles informations relatives à la sécurité. Les professionnels de la santé déclarent tout effet indésirable suspecté. Voir rubrique 4.8 pour les modalités de déclaration des effets indésirables.
XEMBIFY 200 mg/mL, solution injectable sous-cutanée
2. COMPOSITION QUALITATIVE ET QUANTITATIVE
Immunoglobuline humaine normale (IgSC)
Immunoglobuline humaine normale ....................................................................................... 200 mg
Pour un mL de solution
(pureté : au moins 98 % d’immunoglobulines de type G [IgG])
Chaque flacon de 5 mL contient : 1 g d’immunoglobuline humaine normale
Chaque flacon de 10 mL contient : 2 g d’immunoglobuline humaine normale
Chaque flacon de 20 mL contient : 4 g d’immunoglobuline humaine normale
Chaque flacon de 50 mL contient : 10 g d’immunoglobuline humaine normale
Distribution des sous‑classes d’IgG (valeurs approximatives) :
IgG1 ............. 62 %
IgG2 ............. 30 %
IgG3 ............. 4,3 %
IgG4 ............. 3,2 %
La teneur maximale en IgA est de 160 microgrammes/mL.
Produit à partir du plasma de donneurs humains.
Pour la liste complète des excipients, voir rubrique 6.1.
Solution injectable sous-cutanée.
La solution est limpide à légèrement opalescente et incolore ou jaune pâle ou marron clair.
XEMBIFY a une osmolalité approximative comprise entre 280 et 404 mOsmol/kg et un pH compris entre 4,1 et 4,8.
4.1. Indications thérapeutiques
· d’un déficit immunitaire primitif (DIP) avec altération de la production d’anticorps (voir rubrique 4.4) ;
· d’une hypogammaglobulinémie avec infections bactériennes récurrentes chez des patients atteints de leucémie lymphoïde chronique (LLC), chez lesquels l’antibioprophylaxie a échoué ou est contre-indiquée ;
· d’une hypogammaglobulinémie avec infections bactériennes récurrentes chez des patients atteints de myélome multiple (MM) ;
· d’une hypogammaglobulinémie chez des patients en pré- et post‑transplantation de cellules souches hématopoïétiques allogéniques (GCSH allogénique).
4.2. Posologie et mode d'administration
Posologie
La dose et la posologie dépendent de l’indication.
Traitement substitutif
Le médicament doit être administré par voie sous‑cutanée.
En traitement substitutif, la dose peut être adaptée à chaque patient en fonction de la réponse pharmacocinétique et clinique. Les posologies suivantes sont données à titre indicatif.
La posologie doit permettre l’obtention d’un taux résiduel d’IgG (mesuré avant la perfusion suivante) d’au moins 5 à 6 g/L et être dans l’intervalle de référence des taux d’IgG sériques pour la tranche d’âge. Une dose de charge d’au moins 0,2 à 0,5 g/kg (1 à 2,5 mL/kg) de poids corporel peut être nécessaire. Elle peut être fractionnée sur plusieurs jours, avec une dose maximale quotidienne de 0,1 à 0,15 g/kg de poids corporel.
Après avoir atteint des taux d’IgG stables, des doses d’entretien sont administrées à intervalles réguliers (environ une fois par semaine) de façon à atteindre une dose mensuelle cumulée de l’ordre de 0,4 à 0,8 g/kg de poids corporel. Chaque dose unique peut nécessiter d’être injectée sur différents sites anatomiques.
Les taux résiduels doivent être mesurés et évalués en fonction de l’incidence des infections. Pour réduire le taux d’infections, il peut être nécessaire d’augmenter la dose et de cibler des taux résiduels plus élevés.
Personnes âgées
La dose étant déterminée en fonction du poids corporel et ajustée à la réponse clinique dans les pathologies susmentionnées, la dose utilisée chez les patients âgés n’est pas différente de celle utilisée chez les patients âgés de 18 à 65 ans.
Dans les études cliniques, XEMBIFY a été évalué chez 5 patients atteints de DIP âgés de plus de 65 ans et aucun ajustement posologique spécifique n’a été nécessaire pour atteindre les taux d’IgG sériques cible.
Population pédiatrique
La posologie chez les enfants et adolescents (de 0 à 18 ans) n’est pas différente de celle des adultes, puisque pour chaque indication, la posologie est déterminée en fonction du poids corporel et ajustée à la réponse clinique dans les indications de traitement substitutif.
XEMBIFY a été évalué chez 43 sujets pédiatriques atteints de DIP âgés de 2 à 16 ans (inclus), parmi lesquels 28 patients étaient âgés de 12 ans ou moins. Dans la population pédiatrique, aucun ajustement posologique spécifique n’a été nécessaire pour atteindre les taux d’IgG sériques cible.
Mode d’administration
Voie sous‑cutanée uniquement.
La perfusion sous‑cutanée pour le traitement à domicile doit être initiée et surveillée par un professionnel de santé expérimenté dans l’accompagnement des patients traités à domicile. Des pompes à perfusion adaptées à l’administration sous‑cutanée des immunoglobulines peuvent être utilisées. Le patient ou un aidant doivent être formés à l’utilisation d’une pompe à perfusion, aux techniques de perfusion, à la tenue d’un carnet de suivi du traitement, à la reconnaissance et à la conduite à tenir en cas de survenue d’effets indésirables sévères.
XEMBIFY peut être injecté dans des sites tels que l’abdomen, la cuisse, le bras et la face latérale de la hanche.
Le débit de perfusion initial recommandé dépend des besoins individuels de chaque patient. Le débit et le volume de perfusion par site sont ajustés selon la tolérance du produit par le patient.
Il est recommandé d'utiliser un débit de perfusion initial de 10 mL/h/site. Si la tolérance est bonne (voir rubrique 4.4), le débit peut être augmenté à des intervalles d'au moins 10 minutes jusqu'à un maximum de 20 mL/h/site chez les patients pédiatriques et jusqu'à un maximum de 25 mL/h/site de perfusion chez les adultes pour les deux premières perfusions.
Si la tolérance est bonne (voir rubrique 4.4) pendant deux perfusions, le débit de perfusion peut être augmenté progressivement à 35 mL/h/site.
Plusieurs pompes peuvent être utilisées simultanément. La quantité de produit perfusé dans un site donné est variable. Chez les nourrissons et les enfants, les sites de perfusion peuvent être changés tous les 5 à 15 mL. Chez les adultes, les volumes de plus de 30 mL peuvent être fractionnés en fonction des préférences du patient. Le nombre de sites de perfusion n’est pas limité. Les sites de perfusion doivent être espacés d’au moins 5 cm. Une rotation des sites de perfusion est nécessaire et les proéminences osseuses doivent être évitées.
Patients ayant eu une réaction anaphylactique ou réaction systémique sévère à l’administration d’une immunoglobuline humaine.
Patients présentant un déficit en IgA avec présence d’anticorps anti‑IgA et antécédent d’hypersensibilité à un traitement par immunoglobulines humaines.
4.4. Mises en garde spéciales et précautions d'emploi
Afin d’améliorer la traçabilité des médicaments biologiques, le nom et le numéro de lot du produit administré doivent être clairement enregistrés.
L’administration accidentelle de XEMBIFY dans un vaisseau sanguin peut provoquer un choc.
Le débit de perfusion recommandé indiqué dans la rubrique 4.2 doit être strictement respecté. Les patients doivent être étroitement suivis et surveillés pendant toute la durée de la perfusion afin que d’éventuels signes d’intolérance puissent être détectés.
Certains effets indésirables peuvent survenir plus fréquemment chez les patients recevant pour la première fois une immunoglobuline humaine normale ou, dans de rares cas, lors d’un changement d’immunoglobuline humaine normale ou si un long délai s’est écoulé depuis la précédente perfusion.
Les complications potentielles peuvent souvent être évitées :
· en commençant par injecter le produit lentement (ne pas dépasser 25 mL/h/site) ;
· en s’assurant que les patients soient étroitement surveillés à la recherche de tout symptômes pendant toute la durée de perfusion. En particulier, chez les patients naïfs de traitement par immunoglobuline humaine normale, lors d’un changement d’immunoglobulines humaines ou si un long délai s’est écoulé depuis la dernière perfusion, ces patients doivent être surveillés pendant toute la durée de la première perfusion et pendant l’heure qui suit la fin de perfusion afin que d’éventuels effets indésirables puissent être détectés.
Tous les autres patients doivent être surveillés pendant au moins 20 minutes après l’administration.
En cas d’effet indésirable, le débit de perfusion doit être diminué ou la perfusion arrêtée. Le traitement nécessaire dépend de la nature et de la sévérité de l’effet indésirable. Les réactions de type allergique ou anaphylactique imposent l’arrêt immédiat de la perfusion.
En cas de choc, un traitement médical standard du choc doit être instauré.
Hypersensibilité
Les vraies réactions allergiques sont rares. Elles peuvent apparaître notamment chez les patients présentant des anticorps anti‑IgA qui doivent être traités avec précaution. Les patients présentant des anticorps anti‑IgA, pour lesquels l’administration par voie sous‑cutanée d’un traitement par IgG demeure la seule option, doivent être traités par XEMBIFY uniquement sous étroite surveillance médicale.
Dans de rares cas, l’immunoglobuline humaine normale peut entraîner une chute de la pression artérielle associée à une réaction anaphylactique, même chez les patients qui avaient toléré un traitement antérieur par l’immunoglobuline humaine normale.
Évènements thromboemboliques
Des évènements thromboemboliques artériels et veineux, incluant infarctus du myocarde, accident vasculaire cérébral, thrombose veineuse profonde et embolie pulmonaire, ont été associés à l’utilisation des immunoglobulines. Les patients doivent être suffisamment hydratés avant de recevoir des immunoglobulines. La prudence s’impose chez les patients présentant des facteurs de risque préexistants d’évènements thrombotiques (tels que : utilisation d’œstrogènes, âge avancé, hypertension, diabète sucré et antécédents de maladie vasculaire ou d’épisodes thrombotiques, patients présentant une thrombophilie acquise ou héréditaire, patients immobilisés durant des périodes prolongées, patients sévèrement hypovolémiques, patients atteints de maladies entraînant une augmentation de la viscosité sanguine).
Il convient d’informer les patients sur les premiers symptômes des évènements thromboemboliques, notamment la difficulté respiratoire, douleur et gonflement d’un membre, déficits neurologiques focaux et douleur thoracique, et de leur conseiller de contacter leur médecin dès l’apparition de ces symptômes.
Syndrome de méningite aseptique (SMA)
Des cas de syndrome de méningite aseptique ont été rapportés en association avec un traitement par immunoglobulines par voie sous‑cutanée; le syndrome apparaît généralement plusieurs heures à 2 jours après le traitement. Le SMA pourrait toucher davantage les femmes que les hommes. Le SMA se caractérise par les signes et symptômes suivants : céphalée sévère, raideur de la nuque, endormissement, fièvre, photophobie, nausée et vomissement. Un examen neurologique approfondi, incluant des analyses du liquide cérébrospinal (LCS) et visant à exclure les autres causes de méningite, doit être effectué chez les patients présentant des signes et symptômes de SMA. Chez les patients présentant des signes et symptômes du SMA, un examen neurologique approfondi, incluant une analyse du LCR doit être réalisé afin d’exclure d’autres causes de méningites. L’interruption du traitement par immunoglobulines peut entraîner la rémission du SMA en quelques jours, sans séquelles.
Les patients doivent être informés des premiers symptômes du SMA. Le SMA peut survenir plus fréquemment lors de traitements à doses élevées et/ou lorsque le débit de perfusion est élevé.
Dysfonction/insuffisance rénale
Des cas d’effets indésirables rénaux sévères ont été rapportés chez des patients recevant un traitement par immunoglobulines, notamment avec les médicaments contenant du saccharose (XEMBIFY ne contient pas de saccharose). Ces effets incluent : insuffisance rénale aiguë, nécrose tubulaire aiguë, néphropathie tubulaire proximale et néphrose osmotique. Les facteurs favorisant la survenue de complications rénales incluent, sans toutefois s’y limiter : insuffisance rénale préexistante, diabète sucré, hypovolémie, administration concomitante de médicaments néphrotoxiques, âgé de plus de 65 ans, sepsis, hyperviscosité et paraprotéinémie.
Une surveillance est également indispensable chez les patients présentant une insuffisance rénale, notamment en cas d’insuffisance rénale préexistante ou de risque d’insuffisance rénale aiguë.
Interférence avec les tests sérologiques
Après une perfusion d’immunoglobulines, l’augmentation transitoire du taux de divers anticorps transférés passivement dans le sang des patients peut entraîner des résultats faussement positifs lors des dosages sérologiques.
La transmission passive d’anticorps anti‑érythrocytaires, tels que les anticorps anti‑A, anti‑B ou anti‑D, peut interférer avec certains tests sérologiques de recherche d’allo-anticorps anti‑érythrocytaires, par example le test direct à l’antiglobuline (TAD, test de Coombs direct). Une hémolyse peut se produire à doses élevées ou chez les patients de groupe sanguin non O. Une surveillance est donc recommandée.
Agents transmissibles
Les mesures habituelles de prévention du risque de transmission d’agents infectieux par les médicaments préparés à partir de sang ou de plasma humain comprennent la sélection des donneurs, la recherche des marqueurs spécifiques d’infection sur chaque don et sur les pools de plasma ainsi que la mise en œuvre d’étapes efficaces pour l’inactivation/élimination virale dans le procédé de fabrication. Cependant, lorsque des médicaments préparés à partir de sang ou de plasma humain sont administrés, le risque de transmission d’agents infectieux ne peut être totalement exclu. Cela s’applique également aux virus inconnus ou émergents et aux autres agents pathogènes.
Les mesures prises sont considérées comme efficaces contre les virus enveloppés tels que le virus de l’immunodéficience humaine (VIH), le virus de l’hépatite B (VHB) et le virus de l’hépatite C (VHC), ainsi que le virus de l’hépatite A (VHA) non enveloppé. Les mesures prises peuvent présenter un intérêt limité contre les virus non enveloppés tels que le parvovirus B19.
L’expérience clinique est rassurante, aucun cas de transmission du virus de l’hépatite A ou du parvovirus B19 par les immunoglobulines n’ayant été rapporté. Il est probable que les anticorps présents contribuent fortement à la sécurité virale.
Il est fortement recommandé que chaque fois que XEMBIFY est administré à un patient, le nom et le numéro de lot du produit soient enregistrés afin de conserver un lien entre le patient et le lot du produit.
Population pédiatrique
Les mises en garde et précautions mentionnées sont valables pour la population adulte et la population pédiatrique.
4.5. Interactions avec d'autres médicaments et autres formes d'interactions
Vaccins à virus vivant atténué
L’administration d’immunoglobulines peut entraver, pendant une période d’au moins 6 semaines et jusqu’à 3 mois, l’efficacité des vaccins à virus vivant atténué, comme les vaccins contre la rougeole, la rubéole, les oreillons et la varicelle. Après administration de ce médicament, un intervalle de 3 mois doit être respecté avant une vaccination constituté de vaccins à virus vivants atténués. Dans le cas de la rougeole, cette diminution de l’efficacité peut persister jusqu’à 1 an.
Par conséquent, le taux d’anticorps doit être contrôlé chez les patients vaccinés contre la rougeole.
Population pédiatrique
Les interactions mentionnées sont valables pour la population adulte et la population pédiatrique.
Personnes âgées
Les interactions mentionnées sont valables pour les personnes âgées.
4.6. Fertilité, grossesse et allaitement
Grossesse
La sécurité de ce médicament chez la femme enceinte n’a pas été établie par des études cliniques contrôlées. Par conséquent, ce médicament doit être prescrit avec prudence chez les femmes enceintes ou qui allaitent.
Les préparations d’immunoglobulines traversent le placenta, particulièrement pendant le troisième trimestre. L’expérience clinique avec les immunoglobulines suggère qu’aucun effet délétère n’est attendu sur le déroulement de la grossesse ni sur le fœtus et le nouveau-né.
Les immunoglobulines sont excrétées dans le lait maternel et peuvent contribuer à la protection des nouveau‑nés contre les agents pathogènes qui pénètrent dans l’organisme par les muqueuses.
Fertilité
L’expérience clinique avec les immunoglobulines suggèere qu’aucun effet délétère n’est attendu sur la fertilité.
4.7. Effets sur l'aptitude à conduire des véhicules et à utiliser des machines
Résumé du profil de sécurité
Des effets indésirables de type frissons, céphalée, sensation vertigineuse, fièvre, vomissement, réactions allergiques, nausée, arthralgie, présion artérielle basse et douleur dans la partie inférieure du dos peuvent survenir occasionnellement.
Dans de rares cas, les immunoglobulines humaines normales peuvent provoquer une chute subite de la pression artérielle et dans des cas isolés, un choc anaphylactique, même si le patient n’avait pas présenté de réaction d’hypersensibilité lors d’une administration antérieure.
Les réactions locales observées aux sites de perfusion telles quelles gonflement, douleur, rougeur, induration, chaleur locale, démangeaison, contusion et rash, peuvent fréquement survenir.
Pour les informations de sécurité relative aux agents transmissibles, voir rubrique 4.4.
Liste tabulée des effets indésirables
La sécurité de XEMBIFY administré par voie sous‑cutanée a été évaluée dans le cadre de deux études prospectives de phase 3, multicentriques, en ouvert, non contrôlées chez 110 patients des deux sexes, âgés de 2 à 72 ans, atteints d’un déficit immunitaire primitif (DIP) et ayant déjà été traités par des IgIV et/ou IgSC. Quarante‑neuf (49) patients ont été inclus dans l’étude nord‑américaine et 61 dans l’étude européenne.
Sur l’ensemble des deux études, huit patients ont arrêté le traitement par XEMBIFY en raison d’effets indésirables, qui étaient tous d’intensité légère ou modérée, à l’exception d’un cas d’insuffisance aortique due à une anomalie congénitale.
Le tableau ci-dessous utilise la classification des systèmes d’organes MedDRA (SOC et terme préférentiel).
Les fréquences ont été évaluées selon la convention suivante : très fréquent (≥ 1/10), fréquent (≥ 1/100 à < 1/10), peu fréquent (≥ 1/1 000 à < 1/100), rare (≥ 1/10 000 à < 1/1 000), très rare (< 1/10 000). Au sein de chaque groupe de fréquence, les effets indésirables sont présentés suivant un ordre décroissant de gravité (tous non graves).
Fréquence des effets indésirables (EI) observés avec XEMBIFY chez au moins 1 % des patients et par perfusion dans les études cliniques
|
Classe de systèmes d’organes (SOC) MedDRA |
Effet indésirable |
Fréquence par patienta (N = 110 patients) |
Fréquence par perfusionb (N = 4 098 perfusions) |
|
Infections et infestations |
Rhinite |
3 (2,7 %) fréquent |
4 (0,0010) peu fréquent |
|
Affections du système nerveux |
Céphalée |
4 (3,6 %) fréquent |
4 (0,0010) peu fréquent |
|
Affections gastro‑intestinales |
Diarrhée |
3 (2,7 %) fréquent |
3 (0,0007) rare |
|
Nausée |
2 (1,8 %) fréquent |
2 (0,0005) rare |
|
|
Affections de la peau et du tissu sous-cutané |
Papule |
2 (1,8 %) fréquent |
2 (0,0005) rare |
|
Prurit |
2 (1,8 %) fréquent |
2 (0,0005) rare |
|
|
Affections musculo-squelettiques et du tissu conjonctif |
Arthralgie |
3 (2,7 %) fréquent |
3 (0,0007) rare |
|
Dorsalgie |
3 (2,7 %) fréquent |
3 (0,0007) rare |
|
|
Troubles généraux et anomalies au site d’administration |
Réaction locale au site de perfusion |
35 (31,8 %) très fréquent |
125 (0,0305) fréquent |
|
Fièvre |
2 (1,8 %) fréquent |
4 (0,0010) peu fréquent |
|
|
Investigations |
Immunoglobulines G sanguines diminuées |
2 (1,8 %) fréquent |
2 (0,0005) rare |
a La fréquence par patient est calculé en divisant le nombre de patients présentant des effets indésirables autres que des infections pour lesquels un lien de causalité avec XEMBIFY était au minimum possible par le nombre total de patients.
b La fréquence par perfusion est calculé en divisant le nombre de perfusions associées à des effets indésirables autres que des infections pour lesquels un lien de causalité avec XEMBIFY était au minimum possible par le nombre total de perfusions.
Expérience après commercialisation
Les effets indésirables suivants ont été identifiés et notifiés depuis la mise sur le marché de XEMBIFY : réaction locale au site de perfusion telle qu’érythème et gonflement, dyspnée, fatigue, douleur, nausée et céphalée. Il n’est pas toujours possible d’estimer la fréquence de ces effets de manière fiable.
Population pédiatrique
La fréquence, le type et la sévérité des effets indésirables dans la population pédiatrique devraient être identiques à celles observées chez les adultes.
Déclaration des effets indésirables suspectés
La déclaration des effets indésirables suspectés après autorisation du médicament est importante. Elle permet une surveillance continue du rapport bénéfice/risque du médicament. Les professionnels de santé déclarent tout effet indésirable suspecté via le système national de déclaration : Agence nationale de sécurité du médicament et des produits de santé (ANSM) et réseau des Centres Régionaux de Pharmacovigilance - Site internet : https://signalement.social-sante.gouv.fr/.
Les conséquences d’un surdosage ne sont pas connues.
5. PROPRIETES PHARMACOLOGIQUES
5.1. Propriétés pharmacodynamiques
Classe pharmacothérapeutique : immunsérums et immunoglobulines : immunoglobulines humaines normales pour administration extravasculaire, code ATC : J06BA01
Mécanisme d’action
XEMBIFY apporte des anticorps de type immunoglobulines G (IgG) capables d’opsoniser et de neutraliser un large spectre d’agents bactériens, viraux, parasitaires et mycoplasmiques ainsi que leurs toxines. Le rôle de ces anticorps et le mécanisme d’action de XEMBIFY n’ont pas été entièrement élucidés.
Effets pharmacodynamiques
L’immunoglobuline humaine normale contient principalement de l’immunoglobuline G (IgG) avec un large spectre d’anticorps dirigés contre les agents infectieux.
L’immunoglobuline humaine normale contient les anticorps IgG présents dans la population normale. Elle est généralement préparée à partir de pools de plasmas provenant d’un minimum de 1 000 donneurs. La répartition des sous‑classes d’immunoglobuline G est étroitement proportionnelle à celle du plasma humain natif. Des doses adéquates de ce médicament permettent de restaurer des taux d’IgG anormalement bas à des taux situés dans la plage normale.
Efficacité clinique dans l’indication de DIP
Dans l’étude européenne, un total de 61 patients atteints de DIP âgés de 2 à 69 ans ont été traités par XEMBIFY pendant une période allant jusqu’à 52 semaines. La dose moyenne administrée chaque semaine était de 125,5 mg/kg de poids corporel. Le maintien de taux résiduels d’IgG à une concentration moyenne de 947,64 mg/dL a ainsi été atteint pendant la période de traitement. Au total, les patients ont reçu 3 045 perfusions hebdomadaires de XEMBIFY . Le taux annuel d’infections bactériennes graves était de 0,017 par patient-année (borne supérieure de l’intervalle de confiance unilatéral à 99 % : 0,036), reflétant le cas d’un patient présentant une pneumonie traité en ambulatoire par des antibiotiques oraux, avec résolution en 4 jours.
Dans l’étude nord‑américaine, un total de 49 patients atteints de DIP âgés de 2 à 72 ans ont été traités par XEMBIFY pendant une période allant jusqu’à 24 semaines. La dose moyenne administrée chaque semaine était de 178,9 mg/kg de poids corporel. Le maintien de taux résiduels d’IgG à une concentration moyenne de 1 244,84 mg/dL a ainsi été atteint pendant la période de traitement. Au total, les patients ont reçu 1 053 perfusions hebdomadaires de XEMBIFY. Le taux annuel d’infections bactériennes graves sous traitement par XEMBIFY était de 0,049 par patient-année (borne supérieure de l’intervalle de confiance unilatéral à 99 % : 0,110), reflétant le cas d’un patient avec sepsis consécutif à une morsure de chat.
Population pédiatrique
La sécurité et l’efficacité de XEMBIFY ont été établies dans la population pédiatrique. XEMBIFY a été évalué chez 28 enfants atteints de DIP âgés de 2 à 12 ans (inclus) et 15 adolescents âgés de plus de 12 ans à moins de 17 ans. Aucune différence dans la pharmacocinétique et dans les profils d’efficacité et de sécurité n’a été observée par rapport aux patients adultes. Dans la population pédiatrique, aucun ajustement posologique spécifique n’a été nécessaire pour atteindre les taux d’IgG sériques cibles. Aucune différence n’a été observée en terme de propriétés pharmacodynamqiues entre les populations adultes et pédiatriques de patients atteints de DIP.
L’Agence européenne des médicaments a accordé une obligation de soumettre les résultats d’études réalisées avec XEMBIFY dans tous les sous-groupes de la population pédiatrique, dans l’indication de déficit immunitaire primitif pour les nouveau‑nés prématurés et/ou à terme (0 à 27 jours) et pour les nourrissons et les tout‑petits (28 jours à 23 mois) (voir rubrique 4.2 pour les informations concernant l’usage pédiatrique).
Personnes âgées
Aucune différence globale de sécurité ou d’efficacité n’a été observée entre les patients atteints de DIP âgés de plus de 65 ans et les patients atteints de DIP âgés de 18 à 65 ans. Dans les études cliniques, XEMBIFY a été évalué chez 5 patients atteints de DIP âgés de plus de 65 ans.
5.2. Propriétés pharmacocinétiques
Après administration de XEMBIFY par voie sous‑cutanée, les pics plasmatiques sont atteints en trois jours environ.
Distribution
Administration hebdomadaire
Au cours d’une étude clinique de XEMBIFY (n = 61) menée en Europe, les patients ont obtenu des taux résiduels d’IgG (médiane : 909,10 mg/dL) ayant persisté pendant une période de 52 semaines après avoir reçu des doses hebdomadaires médianes de 113,0 mg/kg de poids corporel. Les données issues de l’étude clinique de XEMBIFY montrent que les taux sériques résiduels d’IgG peuvent être maintenus à des posologies de 400 mg à 848 mg/kg de poids corporel/4 semaines.
Synthèse des taux résiduels des IgG totales à l’état d’équilibre lors des phases de traitement antérieur et de traitement SC (population IgG)
|
|
Taux résiduel lors de la phase de traitement antérieur (mg/dL) |
Taux résiduel lors de la phase de traitement SC (mg/dL) |
|
|
Statistique |
Taux résiduel moyena |
Taux résiduel moyenb |
Rapport du taux résiduel moyen |
|
n |
59 |
59 |
59 |
|
Moyenne ± ET |
891,37 ± 165,943 |
947,64 ± 150,262 |
1,078 ± 0,1425 |
|
CV% |
18,6 |
15,9 |
13,22 |
|
Médiane |
874,00 |
909,10 |
1,050 |
|
Min, max |
516,5 ; 1 255,0 |
629,2 ; 1 340,8 |
0,83 ; 1,54 |
|
Moyenne géométrique |
875,96 |
936,48 |
1,069 |
a Le taux résiduel moyen de la phase de traitement antérieur est calculé comme la moyenne des taux résiduels pendant le traitement substitutif antérieur par une IgG commercialisée.
b Le taux résiduel moyen de la phase de traitement sous‑cutané (SC) (sous XEMBIFY) est calculé comme la moyenne des taux résiduels lors des visites SC17, SC18, SC20, SC24, SC28, SC32, SC36, SC40, SC44, SC48, SC52 et SC53.
La pharmacocinétique de XEMBIFY a été évaluée lors d’une étude de phase III d’efficacité et de sécurité chez 27 patients adultes atteints d’un DIP. Les résultats pharmacocinétiques sont présentées dans le tableau ci‑dessous.
Paramètres pharmacocinétiques des IgG totales sériques pour XEMBIFY (population PK)
|
Statistique |
Paramètres pharmacocinétiques |
||
|
ASC0-7 jours (h*mg/dL) |
Cmax (mg/dL) |
Tmax (h) |
|
|
n |
27 |
27 |
27 |
|
Moyenne (ET) |
177 445,7 (31 081,89) |
1 126,6 (190,11) |
50,78 (44,596) |
|
CV% |
18 |
17 |
87,8 |
|
Médiane |
172 369,0 |
1 080,0 |
68,80 |
|
Min, max |
132 728 ; 250 410 |
828 ; 1 610 |
0,0 ; 166,8 |
|
Moyenne géométrique |
175 002,1 |
1 112,2 |
|
|
IC à 90 % de la moyenne géométrique |
165 652,5 ; 184 879,5 |
1 055,1 ; 1 172,4 |
|
CV = coefficient de variation ; ET = écart‑type ; IC = intervalle de confiance
Administration hebdomadaire, bimensuelle ou plus fréquente (2 à 7 fois par semaine)
La pharmacocinétique (PK) de l’administration de XEMBIFY à une fréquence au moins bimensuelle a été caractérisée à l’aide de techniques de simulation et de modélisation pharmacocinétiques par approche de population. Les données des taux sériques des IgG consistaient en 1 841 échantillons provenant de 95 patients adultes et pédiatriques atteints de DIP. La modélisation et la simulation PK ont permis de prédire que comparativement à l’administration hebdomadaire, l’administration bimensuelle de XEMBIFY à une dose double de la dose hebdomadaire entraîne un chevauchement des expositions aux IgG pendant toute la durée d’un intervalle de deux semaines. En outre, une modélisation et une simulation PK ont permis de prédire que pour la même dose totale hebdomadaire, XEMBIFY perfusé 2 à 7 fois par semaine (administration fréquente) entraîne également un chevauchement des expositions aux IgG pendant toute la durée de l’intervalle thérapeutique.
Élimination
Les IgG et les complexes d’IgG sont dégradés dans les cellules du système réticulo‑endothélial.
Population pédiatrique
Il n’existe aucune différence théorique ou observée de l’action des immunoglobulines chez les enfants par rapport aux adultes.
5.3. Données de sécurité préclinique
Glycine (E640), polysorbate 80 (E433), eau pour préparations injectables
3 ans.
Une fois le flacon ouvert, il est recommandé d’utiliser la solution immédiatement.
6.4. Précautions particulières de conservation
· A conserver au réfrigérateur (entre 2 °C et 8 °C).
o XEMBIFY peut être conservé à des températures ne dépassant pas 25 °C pendant une période maximum de 6 mois à tout moment avant la date de péremption.
o Le jour de sortie du médicament du réfrigérateur, inscrire dans l’espace prévu à cet effet sur la boîte dans « Date d’élimination » soit la date correspondant à 6 mois à compter de ce jour, soit la date de péremption imprimée sur le rabat de la boîte, selon la date la plus proche.
o Si le médicament est conservé à température ambiante, ne pas remettre le médicament au réfrigérateur. Utiliser le médicament avant la « Date d’élimination » ou l’éliminer.
· Ne pas congeler.
· Conserver le flacon dans l’emballage extérieur à l’abri de la lumière.
· Administrer dès que possible après le transfert de XEMBIFY du flacon dans une seringue.
Pour les conditions de conservation du médicament après la première ouverture, voir la rubrique 6.3.
6.5. Nature et contenu de l'emballage extérieur
Flacon de verre transparent contenant 5, 10, 20 ou 50 mL de solution et muni d’un bouchon en chlorobutyle, d’un opercule en aluminium, d’une capsule en plastique et d’un manchon d’inviolabilité rétractable garantissant l’intégrité du produit.
Présentations
1 ou 10 flacons contenant 1 g d’immunoglobuline humaine normale dans 5 mL solution pour injection sous-cutanée
1,10 ou 20 flacons contenant 2 g d’immunoglobuline humaine normale dans 10 mL solution pour injection sous-cutanée.
1 ou 20 flacons contenant 4 g d’immunoglobuline humaine normale dans 20 mL solution pour injection sous-cutanée.
1 ou 10 flacons contenant 10 g d’immunoglobuline humaine normale dans 50 mL solution pour injection sous-cutanée.
Chaque boîte contient 1,10 ou 20 flacons de XEMBIFY et une notice.
Toutes les présentations peuvent ne pas être commercialisées.
6.6. Précautions particulières d’élimination et de manipulation
Le médicament doit être amené à température ambiante ou corporelle (20 °C à 37 °C) avant utilisation.
Ne pas agiter.
Inspecter visuellement le médicament avant administration. Les solutions présentant une couleur anormale, ayant un aspect trouble ou contenant des dépôts ne doivent pas être utilisées.
Tout médicament non utilisé ou déchet doit être éliminé conformément à la réglementation en vigueur.
Instructions d’utilisation
Uniquement en perfusion sous‑cutanée.
Avant utilisation, laisser la solution atteindre la température ambiante ou corporelle (20 °C à 37 °C).
Ne pas agiter.
Suivre les étapes ci‑dessous et respecter les mesures d’asepsie pour administrer XEMBIFY.
|
1. Inspecter les flacons : vérifier la limpidité, la couleur et la date de péremption. |
|
|
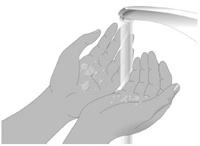
2. Préparer la perfusion Rassembler le matériel : flacon(s) de XEMBIFY, accessoires, collecteur d’aiguilles, carnet de suivi du traitement et pompe à perfusion. Préparer un plan de travail propre. Se laver les mains. |
|
|
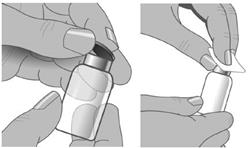
3. Retirer la capsule de protection du flacon pour mettre à nu la partie centrale du bouchon. |
|
|
4. Nettoyer le bouchon à l’alcool et laisser sécher. |
|
|
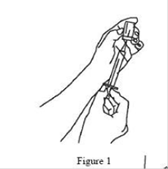
5. À l’aide d’une seringue et d’une aiguille stériles, préparer le prélèvement de XEMBIFY en injectant de l’air dans le flacon d’un volume équivalent au volume de XEMBIFY à prélever. Puis prélever le volume de XEMBIFY souhaité. Si plusieurs flacons sont nécessaires pour que la dose cible soit atteinte, répéter cette étape. (Figure 1) |
|
|
6. Utiliser XEMBIFY dès que possible afin d’éviter la formation de particules éventuelles provenant des seringues en silicone. |
|
|
7. Suivre les instructions du fabricant pour la préparation de la pompe et de la tubulure pour perfusion. Veiller à purger la tubulure pour perfusion afin d’éliminer tout l’air contenu dans la tubulure ou l’aiguille ; pour cela, remplir la tubulure/l’aiguille avec la solution de XEMBIFY. |
|
|
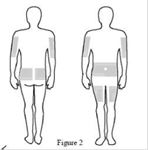
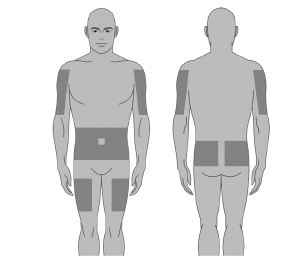
8. Sélectionner le nombre et l’emplacement des sites de perfusion. Effectuer une rotation des sites à chaque administration. (Figure 2) Perfuser XEMBIFY dans l’abdomen, la cuisse, le bras, les côtés, arrières et/ou face latérale de la hanche. Eviter les zones osseuses, les cicatrices, les zones inflammatoires ou infectées ou les vaisseaux sanguins. |
|
|
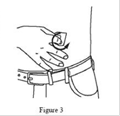
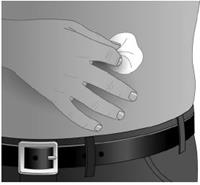
9. Nettoyer le ou les site(s) d’injection à l’aide d’une solution antiseptique en effectuant un mouvement circulaire à partir du centre vers l’extérieur. Les sites doivent être propres, secs, et distants d’au moins 5 cm. (Figure 3) |
|
|
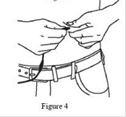
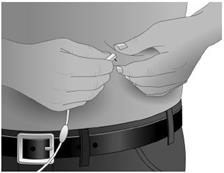
10. Saisir la peau entre deux doigts (pincer au moins 2,5 cm de peau) et introduire l’aiguille dans le tissu sous‑cutané avec un angle de 90 degrés. (Figure 4) |
|
|
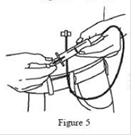
11. Après avoir introduit chaque aiguille, s’assurer qu’aucune aiguille n’a pénétré accidentellement dans un vaisseau sanguin. Fixer une seringue stérile à l’extrémité de la tubulure pour perfusion purgée et tirer sur le piston. En cas de reflux de sang visible, retirer et éliminer l’aiguille et la tubulure pour perfusion. (Figure 5) |
|
|
12. Recommencer les étapes de purge et d’introduction d’une aiguille en utilisant une aiguille et une tubulure pour perfusion neuves et en changeant le site d’administration. Maintenir l’aiguille en place par la pose d’une compresse stérile ou d’un pansement transparent sur le site. |
|
|
13. Pour les deux premières perfusions, le débit commencera à 10 mL par heure par site. Si les perfusions sont bien tolérées et que vous ne présentez pas d'effets secondaires (voir rubrique 4.4), le débit peut être augmenté toutes les 10 minutes jusqu'à un maximum de 20 mL par heure par site de perfusion chez les enfants et les adolescents et de 25 mL par heure par site de perfusion chez les adultes. Si les deux perfusions sont bien tolérées, le débit peut être augmenté progressivement à 35 mL par heure par site de perfusion. Pour tous les patients, quel que soit l’âge, vérifier que les sites de perfusion sont distants d’au moins 5 cm. Le nombre de sites de perfusion est laissé à l’appréciation du professionnel de santé. Chez les adultes, les volumes de plus de 30 mL peuvent être fractionnés en fonction des préférences du patient. Le nombre de sites de perfusion n’est pas limité. Le volume total nécessaire pour une dose spécifique de XEMBIFY sera moins important chez les enfants que chez les adultes (posologie en fonction du poids du patient [mg/kg]). Chez les enfants, le professionnel de santé peut choisir entre un volume plus faible par site ou un nombre moins important de sites de perfusion pour atteindre la dose totale cible, en fonction des besoins de l’enfant. Pour déterminer le nombre de sites de perfusion à utiliser, diviser le volume total de la dose de XEMBIFY par le volume souhaité (mL/site). |
|
|
14. Noter les informations concernant la perfusion (telles que le numéro de lot, la date de péremption, la dose, la date, l’heure, le ou les sites de perfusion, les effets indésirables) dans un carnet de suivi du traitement ou un journal de perfusion du patient. |
|
|
15. Eliminer les aiguilles et tubulures pour perfusion dans un collecteur approprié. Suivre les instructions du fabricant pour le stockage de la pompe à perfusion. |
|
|
16. Eliminer le ou les flacons entamés. |
|
7. TITULAIRE DE L’AUTORISATION DE MISE SUR LE MARCHE
C/CAN GUASCH, 2
POLIGONO INDUSTRIAL LEVANTE
08150 PARETS DEL VALLÈS
BARCELONE
ESPAGNE
8. NUMERO(S) D’AUTORISATION DE MISE SUR LE MARCHE
· 34009 302 440 7 4 : 5 mL en flacon (verre), boite de 1.
· 34009 302 440 8 1 : 10 mL en flacon (verre), boite de 1.
· 34009 302 440 9 8 : 20 mL en flacon (verre), boite de 1.
· 34009 302 441 0 4 : 50 mL en flacon (verre), boite de 1.
· 34009 303 217 9 9 : 5 mL en flacon (verre), boite de 10 flacons
· 34009 303 218 0 5 : 10 mL en flacon (verre), boite de 10 flacons
· 34009 303 218 1 2 : 10 mL en flacon (verre), boite de 20 flacons
· 34009 303 218 3 6 : 20 mL en flacon (verre), boite de 20 flacons
· 34009 303 218 4 3 : 50 mL en flacon (verre), boite de 10 flacons
9. DATE DE PREMIERE AUTORISATION/DE RENOUVELLEMENT DE L’AUTORISATION
[à compléter ultérieurement par le titulaire]
10. DATE DE MISE A JOUR DU TEXTE
[à compléter ultérieurement par le titulaire]
Sans objet.
12. INSTRUCTIONS POUR LA PREPARATION DES RADIOPHARMACEUTIQUES
Des informations détaillées sur ce médicament sont disponibles sur le site Internet de l’ANSM.
Liste I
Médicament soumis à prescription hospitalière.
La prescription par un médecin exerçant dans un établissement de transfusion sanguine autorisé à dispenser des médicaments aux malades qui y sont traités est également autorisée.
: Ce médicament fait l'objet d'une surveillance renforcée. Pour plus d'informations, cliquez ici
ANSM - Mis à jour le : 19/03/2026
XEMBIFY 200 mg/mL, solution injectable sous-cutanée
Immunoglobuline humaine normale (IgSC)
![]() Ce médicament fait l’objet d’une surveillance supplémentaire qui permettra l’identification rapide de nouvelles informations relatives à la sécurité. Vous pouvez y contribuer en signalant tout effet indésirable que vous observez. Voir en fin de rubrique 4 comment déclarer les effets indésirables.
Ce médicament fait l’objet d’une surveillance supplémentaire qui permettra l’identification rapide de nouvelles informations relatives à la sécurité. Vous pouvez y contribuer en signalant tout effet indésirable que vous observez. Voir en fin de rubrique 4 comment déclarer les effets indésirables.
Veuillez lire attentivement cette notice avant d’utiliser ce médicament car elle contient des informations importantes pour vous.
· Gardez cette notice. Vous pourriez avoir besoin de la relire.
· Si vous avez d’autres questions, interrogez votre médecin, votre pharmacien ou votre infirmier/ère.
· Ce médicament vous a été personnellement prescrit. Ne le donnez pas à d’autres personnes. Il pourrait leur être nocif, même si les signes de leur maladie sont identiques aux vôtres.
· Si vous ressentez un quelconque effet indésirable, parlez-en à votre médecin, votre pharmacien ou votre infirmier/ère. Ceci s’applique aussi à tout effet indésirable qui ne serait pas mentionné dans cette notice. Voir rubrique 4.
1. Qu'est-ce que XEMBIFY 200 mg/mL, solution injectable sous-cutanée et dans quels cas est-il utilisé ?
2. Quelles sont les informations à connaître avant d'utiliser XEMBIFY 200 mg/mL, solution injectable sous-cutanée ?
3. Comment utiliser XEMBIFY 200 mg/mL, solution injectable sous-cutanée ?
4. Quels sont les effets indésirables éventuels ?
5. Comment conserver XEMBIFY 200 mg/mL, solution injectable sous-cutanée ?
6. Contenu de l’emballage et autres informations.
1. QU’EST-CE QUE XEMBIFY 200 mg/mL, solution injectable sous-cutanée ET DANS QUELS CAS EST-IL UTILISE ?
Qu’est‑ce que XEMBIFY 200 mg/mL, solution injectable sous-cutanée ?
XEMBIFY est une solution d’immunoglobulines humaines (anticorps, essentiellement des immunoglobulines G) qui aident votre organisme à combattre les infections.
XEMBIFY contient des immunoglobulines qui proviennent du plasma de donneurs sains. Les immunoglobulines aident à combattre les infections causées par des bactéries et des virus. Le médicament agit exactement de la même manière que les immunoglobulines produites par le système immunitaire et présentes naturellement dans le sang humain.
Dans quels cas XEMBIFY 200 mg/mL, solution injectable sous-cutanée est‑il utilisé ?
XEMBIFY vous a été prescrit car votre taux d’immunoglobulines est anormalement bas en raison d’une affection appelée un déficit immunitaire. Les perfusions de XEMBIFY permettent d’augmenter les taux d’immunoglobulines (anticorps), en particulier les taux d’immunoglobulines G (IgG) de votre sang et de les ramener à des taux normaux.
Ce médicament est indiqué chez les adultes, les enfants et les adolescents (de 0 à 18 ans) qui ne possèdent pas suffisamment d’anticorps (traitement substitutif) :
1. patients présentant un déficit immunitaire primitif (DIP), avec un manque d’anticorps existant dès la naissance ;
2. hypogammaglobulinémie (affection au cours de laquelle le taux d’immunoglobulines dans le sang est faible) et infections bactériennes à répétition chez des patients atteints de leucémie lymphoïde chronique (cancer du sang avec production excessive de globules blancs), pour lesquels les antibiotiques administrés à titre préventif n’ont pas été efficaces ;
3. hypogammaglobulinémie et infections bactériennes à répétition dans le myélome multiple (tumeur constituée de cellules dérivées de la moelle osseuse) ;
4. hypogammaglobulinémie chez les patients après une greffe de cellules souches provenant d’un donneur (allogreffe de cellules souches hématopoïétiques, GCSH allogénique).
2. QUELLES SONT LES INFORMATIONS A CONNAITRE AVANT D’UTILISER XEMBIFY 200 mg/mL, solution injectable sous-cutanée ?
N’utilisez jamais XEMBIFY 200 mg/mL, solution injectable sous-cutanée :
· si vous êtes allergique à l’immunoglobuline humaine normale ou à l’un des autres composants contenus dans ce médicament (mentionnés dans la rubrique 6) ;
· si vous avez présenté une réaction allergique sévère (anaphylaxie, par exemple) à une immunoglobuline humaine ;
· si vous avez des anticorps dirigés contre les immunoglobulines A (IgA) dans votre sang. Cela peut se produire si vous avez un déficit en IgA. Dans la mesure où XEMBIFY contient des IgA, vous pourriez présenter une réaction allergique ;
· en l’injectant dans un vaisseau sanguin (voie intraveineuse) ou dans un muscle (voie intramusculaire).
Si vous avez déjà présenté des effets indésirables avec une immunoglobuline ou l’un des autres composants, parlez-en à votre médecin, pharmacien ou infirmier/ère avant la perfusion de XEMBIFY.
Avertissements et précautions
Adressez-vous à votre médecin, pharmacien ou infirmier/ère avant d’utiliser XEMBIFY.
· Indiquez à votre médecin tout antécédent de maladie cardiaque, maladie vasculaire, caillot dans un vaisseau sanguin (tel qu’un accident vasculaire cérébral, crise cardiaque ou embolie pulmonaire), sang épais, diabète sucré, pression artérielle élevée, hémorragie ou trouble de la coagulation, ou si vous avez été immobilisé(e) pendant une période prolongée. Indiquez à votre médecin si vous prenez des oestrogènes (par exemple, pilules contraceptives). Vous pourriez être exposée à un risque plus élevé de formation d’un caillot sanguin après la perfusion de XEMBIFY. Contactez immédiatement votre médecin si vous présentez les symptômes suivants : difficultés à respirer, douleur dans la poitrine, douleur et gonflement d’un bras ou d’une jambe ou faiblesse ou engourdissement d’un côté du corps. Cela pourrait indiquer la présence d’un caillot dans un vaisseau sanguin.
· Contactez votre médecin en cas de : maux de tête sévères, raideur de la nuque, endormissement, fièvre, sensibilité excessive à la lumière, nausées ou vomissements. Ces effets indésirables peuvent survenir quelques heures à quelques jours après la perfusion de XEMBIFY. Vous pourriez présenter un syndrome de méningite aseptique.
· XEMBIFY peut provoquer des problèmes rénaux, tels qu’une insuffisance rénale. Signalez à votre médecin si vous avez une fonction rénale diminuée.
· XEMBIFY peut interférer avec certaines analyses de sang (tests sérologiques). Veuillez informer systématiquement votre médecin de votre traitement par XEMBIFY avant toute analyse de sang.
Réactions allergiques
Les réactions allergiques sont rares. Cependant, vous pourriez être allergique aux immunoglobulines sans le savoir. Les réactions allergiques se manifestant par une diminution soudaine de la pression artérielle ou un choc anaphylactique (chute subite de la pression artérielle associée à d’autres symptômes tels que gonflement de la gorge, difficultés à respirer et éruption cutanée étendue) sont rares, mais peuvent survenir occasionnellement, même si vous n’avez pas présenté d’effets indésirables lors d’une administration antérieure d’immunoglobulines. Votre risque de réactions allergiques est plus élevé si vous avez un déficit en IgA et des anticorps anti‑IgA. Si vous avez un déficit en IgA, assurez vous de le signaler à votre médecin. XEMBIFY contient de petites quantités d’IgA, ce qui pourrait augmenter le risque de réaction allergique. Voir rubrique 4 de la présente notice (« Quels sont les effets indésirables éventuels ») pour les signes et symptômes d’une réaction allergique.
Risque de transmission de maladies
XEMBIFY est purifié à partir de plasma humain provenant de donneurs sains. Lorsque des médicaments biologiques sont administrés, le risque de maladies infectieuses dues à la transmission d’agents pathogènes ne peut être totalement exclu.
Cependant, dans le cas des médicaments préparés à partir de plasma humain, le risque de transmission d’agents pathogènes est réduit par : (1) la surveillance épidémiologique des donneurs de sang et la sélection de chaque donneur par un entretien médical ; (2) le dépistage des marqueurs d’infection virale sur chaque don et sur les mélanges de plasma ; et (3) l’inclusion dans le procédé de fabrication d’étapes ayant fait leurs preuves pour inactiver/éliminer les agents pathogènes.
En dépit de ces mesures, lorsque des médicaments préparés à partir de sang ou de plasma humain sont administrés, le risque de transmission de maladies infectieuses ne peut être totalement exclu. Cela s’applique également aux virus inconnus ou émergents ou aux autres types d’agents infectieux.
Les mesures prises sont jugées efficaces contre les virus enveloppés tels que le virus de l’immunodéficience humaine (VIH), le virus de l’hépatite B et le virus de l’hépatite C, ainsi que le virus de l’hépatite A non enveloppé. Les mesures prises peuvent présenter un intérêt limité contre les virus non enveloppés tels que le parvovirus B19.
Les immunoglobulines n’ont pas été associées à des infections par le virus de l’hépatite A ou le parvovirus B19, probablement en raison de l’effet protecteur conféré par les anticorps contenus dans le médicament.
Chaque fois que vous recevez une dose de XEMBIFY, il est fortement recommandé de noter le nom et le numéro de lot du médicament (figurant sur l’étiquette et sur la boîte après « lot ») afin de garder une trace des lots utilisés.
Enfants et adolescents
Les avertissements et précautions sont valables pour les adultes, les enfants et les adolescents.
Autres médicaments et XEMBIFY 200 mg/mL, solution injectable sous-cutanée
Informez votre médecin ou pharmacien si vous prenez, avez récemment pris ou pourriez prendre tout autre médicament.
XEMBIFY doit toujours être perfusé seul. Ne le mélangez pas à d’autres médicaments.
Avant toute vaccination, signalez au médecin que vous recevez un traitement par XEMBIFY. XEMBIFY peut interférer avec certains vaccins (les vaccins à virus vivant), tels que les vaccins contre la rougeole, les oreillons, la rubéole et la varicelle. Il pourrait être nécessaire d’attendre jusqu’à 3 mois après la perfusion de XEMBIFY pour procéder à la vaccination. En ce qui concerne le vaccin contre la rougeole, ce délai pourrait être étendu à 1 an.
Ces interactions concernent les enfants, les adultes et les patients âgés.
Grossesse, allaitement et fertilité
Si vous êtes enceinte ou que vous allaitez, si vous pensez être enceinte ou planifiez une grossesse, demandez conseil à votre médecin ou pharmacien avant de prendre ce médicament.
XEMBIFY n’a pas été étudié chez la femme enceinte ni chez la femme qui allaite ; votre médecin ou votre pharmacien vous guidera. L'expérience clinique avec les immunoglobulines suggère qu'aucun effet nocif sur le déroulement de la grossesse ou sur le fœtus et le bébé n'est attendu. Si vous allaitez, les immunoglobulines contenues dans XEMBIFY peuvent également être retrouvées dans votre lait. Elles peuvent protéger votre bébé de certaines infections. L'expérience clinique avec les immunoglobulines suggère qu'aucun effet nocif sur la fertilité n'est attendu.
Conduite de véhicules et utilisation de machines
Votre aptitude à conduire des véhicules et à utiliser des machines peut être altérée par certains effets indésirables, tels que les sensations vertigineuses, associés à XEMBIFY. Si vous ressentez des effets indésirables pendant le traitement, attendez qu’ils disparaissent avant de conduire des véhicules ou d’utiliser des machines.
3. COMMENT UTILISER XEMBIFY 200 mg/mL, solution injectable sous-cutanée ?
XEMBIFY doit être perfusé sous la peau (administration sous‑cutanée ou SC).
Le traitement par XEMBIFY sera mis en route par votre médecin ou votre infirmier/ère. Ne commencez pas le traitement par XEMBIFY à domicile tant que vous n’avez pas reçu toutes les instructions.
Dose
La dose recommandée et le calendrier des perfusions sont établis par votre médecin. Votre médecin calculera la dose adaptée pour vous en fonction de votre poids, de tout traitement antérieur éventuel et de votre réponse au traitement. Veillez à toujours utiliser ce médicament en suivant exactement les instructions de cette notice ou les indications de votre médecin, pharmacien ou infirmier/ère. Vérifiez auprès d’eux en cas de doute.
Votre première dose pourrait être ce que nous appelons une « dose de charge » qui vise à augmenter rapidement les taux d’immunoglobulines dans votre sang. Votre médecin déterminera si vous avez besoin d’une « dose de charge » (pour les adultes ou les enfants) d’au moins 1 à 2,5 mL/kg de poids corporel. Vous pouvez recevoir cette dose de charge fractionnée sur plusieurs jours.
Vous recevrez régulièrement XEMBIFY, à une fréquence comprise entre une fois par jour à une fois toutes les deux semaines ; les doses cumulées mensuelles seront d’environ 1,5 à 5 mL/kg de poids corporel. Votre médecin peut ajuster la dose en fonction de votre réponse au traitement.
Ne modifiez pas cette dose ni l’intervalle entre deux doses sans en avoir parlé au préalable à votre médecin.
Si vous pensez que vous devriez recevoir une dose différente ou si vous voulez modifier le calendrier des perfusions, parlez‑en d’abord à votre médecin. Contactez votre médecin si vous avez oublié une dose.
Des tests sanguins seront effectués périodiquement pour contrôler le taux d’immunoglobulines dans votre sang. Discutez du calendrier avec votre médecin.
Il n’y a pas de différence entre la dose utilisée chez les adultes, y compris les patients âgés (65 ans et plus), et celle utilisée chez les enfants et les nourrissons, étant donné que la quantité de XEMBIFY perfusé est déterminé en fonction du poids des patients.
Mode d’administration
Vous recevrez XEMBIFY en perfusion lente sous la peau (perfusion sous‑cutanée) dans le tissu adipeux (« graisseux »). XEMBIFY sera administré à l’aide d’une pompe ou d’un injecteur. La perfusion sous‑cutanée pour le traitement à domicile doit être instaurée et supervisée par un professionnel de santé expérimenté dans l’accompagnement des patients pour le traitement à domicile.
Vous (ou votre aidant) devez recevoir une formation sur :
· l’utilisation d’un dispositif de perfusion, tel qu’un pousse‑seringue, si nécessaire ;
· les techniques de perfusion de manière aseptique (sans germe) ;
· la tenue d’un carnet de suivi du traitement ; et
· l’identification et la conduite à tenir en cas de survenue d’effets indésirables sévères.
Vous devez respecter scrupuleusement les instructions de votre médecin concernant la dose, le débit de perfusion et le calendrier des perfusions de XEMBIFY afin que le traitement soit efficace pour vous.
Sites de perfusion
XEMBIFY doit être administré uniquement par perfusion sous‑cutanée. Perfusez XEMBIFY dans le tissu sous‑cutané des sites tels que :
· abdomen,
· cuisses,
· bras et
· la face latérale de la hanche.
Lors de la sélection des sites de perfusion, évitez : les zones osseuses, les vaisseaux sanguins visibles, les cicatrices et toute zone présentant une inflammation (irritation) ou une infection. Effectuez une rotation des sites à chaque administration.
Pour vos deux premières perfusions, le débit commencera à 10 mL par heure par site de perfusion. Si vous ne ressentez pas d'effet indésirable (voir rubrique 4), le débit peut être augmenté toutes les 10 minutes jusqu'à un maximum de 20 mL par heure par site de perfusion chez les enfants et les adolescents, et de 25 mL par heure par site de perfusion chez les adultes. Après deux séances de perfusion, le débit peut être augmenté progressivement jusqu’à 35 mL par heure et par site de perfusion. Adressez-vous à votre médecin avant d’augmenter le débit de perfusion.
Vous pouvez utiliser plusieurs sites simultanément pour la perfusion sous réserve qu’ils soient distants d’au moins 5 cm. Les adultes peuvent répartir la dose entre plusieurs sites, en particulier si le volume est supérieur à 30 mL. Le nombre de sites de perfusion que vous pouvez utiliser n’est pas limité. Vous pouvez utiliser plusieurs pompes à cette fin.
Instructions d’utilisation
La perfusion sous‑cutanée pour le traitement à domicile doit être instaurée et supervisée par un professionnel de santé expérimenté dans l’accompagnement des patients pour le traitement à domicile. Des pompes à perfusion adaptées à l’administration sous‑cutanée des immunoglobulines peuvent être utilisées. Le patient ou un aidant doit être formé à l’utilisation d’une pompe à perfusion, aux techniques de perfusion, à la tenue d’un carnet de suivi du traitement, à l’identification et la conduite à tenir en cas de survenue d’effets indésirables sévères.
Suivez les étapes ci‑dessous et respectez les règles d’asepsie pour administrer XEMBIFY.
Avant utilisation, laissez la solution atteindre la température ambiante ou corporelle (20 °C à 37 °C). Cela peut prendre 60 minutes ou plus.
Ne pas utiliser d’appareil chauffant ni placer au four à micro‑ondes.
Etape 1 : Rassembler le matériel
Rassembler le ou les flacon(s) de XEMBIFY, les accessoires, le collecteur d’aiguilles, le carnet de suivi du traitement/journal de perfusion et la ou les pompe(s) à perfusion.
Etape 2 : Nettoyer le plan de travail
Installer votre espace de perfusion sur une surface plane, propre, non poreuse, comme un plan de travail de cuisine.
Eviter les surfaces poreuses telles que le bois. Nettoyer la surface avec une lingette imprégnée d’alcool en effectuant un mouvement circulaire du centre vers l’extérieur.
Etape 3 : Lavez-vous les mains.
Lavez-vous les mains et séchez-les complètement avant d’utiliser XEMBIFY.
Votre professionnel de santé peut vous recommander d’utiliser un savon antibactérien ou de porter des gants.
Etape 4 : Vérifier les flacons
Le liquide contenu dans le flacon doit être limpide à légèrement opalescent et incolore ou jaune pâle ou marron clair.
N’utilisez pas le flacon si :
· la solution présente un trouble ou une coloration anormale. La solution doit être limpide à légèrement opalescente et incolore ou jaune pâle ou marron clair ;
· la capsule de protection est absente ou comporte des signes évidents d’effraction. Informez immédiatement votre professionnel de santé ;
· la date de péremption est dépassée.
Etape 5 : Retirer la capsule de protection
Retirer la capsule de protection du flacon pour mettre à nu la partie centrale du bouchon.
Nettoyer le bouchon à l’alcool et laisser sécher.
Etape 6 : Transférer XEMBIFY du ou des flacon(s) vers la seringue
Ne toucher pas avec les doigts et ne laisser pas d’autres objets toucher la tige interne du piston de la seringue, l’embout de la seringue ou d’autres surfaces qui peuvent être en contact avec la solution de XEMBIFY. Veiller à ce que le capuchon des aiguilles reste en place jusqu’à utilisation des aiguilles et que les aiguilles et les seringues restent sur la surface propre préparée à l’étape 2. Cela s’appelle une « technique aseptique » et permet d’empêcher les germes de pénétrer dans XEMBIFY.
Fixer chaque aiguille à l’embout de la seringue en utilisant une technique aseptique.
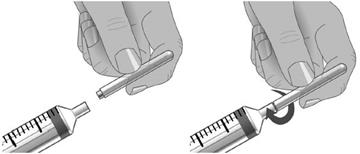
Etape 7 : Préparer la seringue et transférer la solution de XEMBIFY dans la seringue
Retirer le capuchon de l’aiguille.
Tirer le piston de la seringue jusqu’au niveau correspondant au volume de XEMBIFY à prélever dans le flacon.
Placer le flacon de XEMBIFY sur une surface plane et propre et introduire l’aiguille dans le centre du bouchon du flacon.
Injecter l’air dans le flacon. Le volume d’air doit correspondre au volume de XEMBIFY à prélever.
Retourner le flacon de bas en haut et prélevez le volume approprié de XEMBIFY. Si plusieurs flacons sont nécessaires à l’obtention de la dose appropriée, répétez les étapes 4 à 7.
Administrer immédiatement après le transfert de XEMBIFY du flacon vers une seringue.
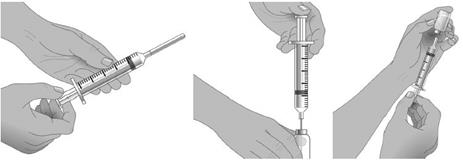
Etape 8 : Préparer la pompe à perfusion
Suivre les instructions du fabricant de la pompe pour la préparation de la pompe à perfusion, de la tubulure pour perfusion et du raccord en Y, si nécessaire.
Purger la tubulure pour perfusion avec la solution de XEMBIFY afin d’éliminer tout l’air contenu dans la tubulure ou l’aiguille. Pour purger, tenez la seringue d’une main et l’aiguille de la tubulure pour perfusion de l’autre main, l’aiguille étant protégée par son capuchon. Pousser doucement sur le piston jusqu’à ce que vous voyiez une goutte de la solution de XEMBIFY sortir de l’aiguille.
Etape 9 : Sélectionner le nombre et l’emplacement des sites de perfusion
Sélectionner un ou plusieurs sites de perfusion selon les instructions de votre professionnel de santé.
Le nombre et l’emplacement des sites de perfusion dépendent du volume de la dose totale.
Les sites de perfusion adaptés sont : abdomen, cuisses, bras, et face latérale des hanches.
Eviter : les zones osseuses, les vaisseaux sanguins visibles, les cicatrices et toute zone présentant une inflammation (irritation) ou une infection.
Effectuer une rotation des sites lors des perfusions suivantes.
Etape 10 : Préparer le site de perfusion
Nettoyer le ou les site(s) de perfusion à l’aide d’une lingette stérile imprégnée d’alcool en effectuant un mouvement circulaire partant du centre et s’étendant vers l’extérieur. Laissez sécher (au moins 30 secondes) le ou les site(s) de perfusion.
Avant la perfusion, les sites doivent être propres, secs, et distants d’au moins 5 cm.
Etape 11 : Introduire l’aiguille.
Saisir la peau entre deux doigts (pincer au moins 2,5 cm de peau) et introduire l’aiguille dans le tissu situé sous la peau (tissu sous cutané) avec un angle de 90 degrés.
Etape 12 : Vérifier que l’aiguille n’est pas dans un vaisseau sanguin
Après avoir introduit chaque aiguille dans le tissu (et avant la perfusion), assurez-vous qu’aucune aiguille n’a pénétré accidentellement dans un vaisseau sanguin. Pour cela, fixer une seringue stérile à l’extrémité de la tubulure pour perfusion purgée. Tirer sur le piston et vérifiez la présence éventuelle d’un reflux de sang dans la tubulure pour perfusion.
Si vous voyez du sang, retirer et éliminer l’aiguille et la tubulure pour perfusion.
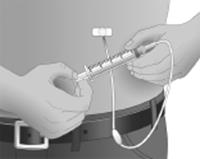
Recommencer les étapes de purge et d’introduction d’une aiguille en utilisant une aiguille et une tubulure pour perfusion neuves et en changeant le site d’administration.
Maintenir l’aiguille en place par la pose d’une compresse stérile ou d’un pansement transparent sur le site.
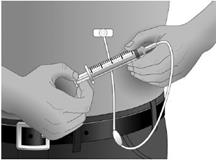
Etape 13 : Recommencer la procédure pour les autres sites, le cas échéant
Etape 14 : Perfuser XEMBIFY
Perfuser XEMBIFY le plus tôt possible après la préparation.
Suivre les instructions du fabricant pour le remplissage de la tubulure et l’utilisation de la pompe à perfusion.
Etape 15 : Après la perfusion
Suivre les instructions du fabricant pour éteindre la pompe.
Retirer et jeter tout pansement ou adhésif.
Retirer délicatement toute aiguille ou tout cathéter.
Eliminer toute solution restante dans un collecteur de déchets approprié conformément aux instructions.
Eliminer tout matériel d’administration usagé dans un collecteur de déchets approprié.
Conserver les accessoires dans un lieu sûr.
Suivre les instructions du fabricant pour l’entretien du pousse seringue.
Etape 16 : Consigner chaque perfusion
Retirer l’étiquette détachable comportant le numéro de lot du produit située sur le flacon de XEMBIFY et utilisez-la pour compléter le dossier patient. Ajouter les informations concernant chaque perfusion, notamment :
· la date et l’heure,
· la dose,
· le ou les numéro(s) de lot,
· les sites de perfusion et
· toute réaction éventuelle.
Penser à apporter votre carnet/journal lorsque vous consultez votre médecin. Votre médecin pourra demander à voir votre carnet de suivi de traitement/journal de perfusion.
Signaler à votre médecin tout problème rencontré lors des perfusions. Appeler votre professionnel de santé pour demander un avis médical sur les effets indésirables.
Utilisation chez les personnes âgées
La dose destinée aux patients âgés n'est pas considérée comme différente de celle administrée aux patients âgés de 18 à 65 ans.
Utilisation chez les enfants et les adolescents
La dose chez les enfants et les adolescents (0 à 18 ans) n'est pas différente de celle des adultes. Chez les nourrissons et les enfants, le site de perfusion peut être changé tous les 5 à 15 mL.
Si vous avez utilisé plus de XEMBIFY 200 mg/mL, solution injectable sous-cutanée que vous n’auriez dû
Contactez votre médecin pour obtenir des instructions.
Si vous oubliez d’utiliser XEMBIFY 200 mg/mL, solution injectable sous-cutanée
Ne prenez pas de dose double pour compenser la dose que vous avez oublié de prendre. Contactez votre médecin pour obtenir des instructions.
Si vous avez d’autres questions sur l’utilisation de ce médicament, demandez à votre médecin.
4. QUELS SONT LES EFFETS INDESIRABLES EVENTUELS ?
Dans de rares cas, les immunoglobulines humaines normales peuvent provoquer une chute brutale de la pression artérielle et, dans des cas isolés, un choc anaphylactique, même si le patient n’a pas présenté de réaction d’hypersensibilité lors d’une administration antérieure.
Les signes ou symptômes de ces réactions allergiques incluent :
· étourdissements et sensations vertigineuses ou syncope;
· éruptions cutanées étendues et démangeaisons, gonflement de la bouche ou de la gorge, difficultés à respirer, sibilances ;
· fréquence cardiaque anormale, douleur dans la poitrine, lèvres ou doigts et orteils bleus.
Si vous remarquez des signes de réaction allergique ou de type anaphylactique pendant la perfusion de XEMBIFY, arrêtez la perfusion et contactez votre médecin ou allez immédiatement à l’hôpital le plus proche. Veuillez consulter la rubrique 2 de la présente notice (Avertissements et précautions). Si vous remarquez l’un de ces signes pendant la perfusion de XEMBIFY, lorsque le médicament est administré par un professionnel de santé, informez immédiatement votre médecin ou votre infirmier/ère. Il/elle décidera s’il convient de diminuer le débit de perfusion ou d’arrêter complètement la perfusion.
Des réactions locales aux sites de perfusion telles que gonflement, douleur, rougeur, induration (zone durcie de la peau), chaleur locale, démangeaisons, contusion et éruption cutanée peuvent survenir.
XEMBIFY peut occasionnellement provoquer : frissons, maux de tête, sensations vertigineuses, fièvre, vomissement, réactions allergiques, sensation de malaise (nausée), douleurs articulaires, pression artérielle basse et douleurs modérées dans la partie inférieure du dos.
L’effet indésirable suivant est très fréquent avec XEMBIFY (peut survenir chez au moins 1 patient sur 10) :
· Réaction locale au site de perfusion
Les effets indésirables suivants sont fréquents avec XEMBIFY (peuvent survenir chez au moins 1 patient sur 100) :
· Maux de tête
· Arthralgies (douleurs articulaires)
· Douleur au dos
· Rhinite (nez qui coule, éternuements et congestion nasale)
· Diarrhée
· Nausée
· Pyrexie (fièvre)
· Diminution des immunoglobulines G sanguines
· Prurit (démangeaisons)
· Papules (peau surélevée sur une petite surface)
Effets secondaires depuis la mise sur le marché
Les effets indésirables suivants ont été identifiés et rapportés depuis la mise sur le marché de XEMBIFY (tous non graves) : dyspnée (difficulté respiratoire), fatigue, douleur, nausées, maux de tête et réaction locale au site de perfusion de types érythème (rougeur de la peau) et gonflement. Il n’est pas toujours possible d’estimer la fréquence de ces effets de manière fiable.
Déclaration des effets secondaires
Si vous ressentez un quelconque effet indésirable, parlez-en à votre médecin ou, votre pharmacien ou à votre infirmier/ère. Ceci s’applique aussi à tout effet indésirable qui ne serait pas mentionné dans cette notice. Vous pouvez également déclarer les effets indésirables directement via le système national de déclaration : Agence nationale de sécurité du médicament et des produits de santé (ANSM) et réseau des Centres Régionaux de Pharmacovigilance - Site internet: https://signalement.social-sante.gouv.fr/.
En signalant les effets indésirables, vous contribuez à fournir davantage d’informations sur la sécurité du médicament.
5. COMMENT CONSERVER XEMBIFY 200 mg/mL, solution injectable sous-cutanée ?
Tenir ce médicament hors de la vue et de la portée des enfants.
· A conserver au réfrigérateur (entre 2 °C et 8 °C).
o XEMBIFY peut être conservé à des températures ne dépassant pas 25 °C pendant une période maximum de 6 mois à tout moment avant la date de péremption.
o Le jour de sortie du médicament du réfrigérateur, inscrire dans l’espace prévu sur la boîte dans « Date d’élimination » soit la date correspondant à 6 mois à compter de ce jour, soit la date de péremption imprimée sur le rabat de la boîte, selon la date la plus proche.
o Si le médicament est conservé à température ambiante, ne pas le remettre au réfrigérateur. Utiliser le médicament avant la « Date d’élimination » ou l’éliminer.
· Ne pas congeler.
· Conserver le flacon dans l’emballage extérieur à l’abri de la lumière.
· Administrer dès que possible une fois que XEMBIFY est transféré du flacon dans une seringue.
N’utilisez pas ce médicament après la date de péremption indiquée sur l’étiquette et sur la boîte.
N’utilisez pas ce médicament si vous remarquez une coloration anormale, si la solution est trouble ou présente des dépôts ou si le médicament a été congelé.
Ne jetez aucun médicament au tout‑à‑l’égout ou avec les ordures ménagères. Demandez à votre pharmacien d’éliminer les médicaments que vous n’utilisez plus. Ces mesures contribueront à protéger l’environnement.
6. CONTENU DE L’EMBALLAGE ET AUTRES INFORMATIONS
Ce que contient XEMBIFY 200 mg/mL, solution injectable sous-cutanée
· La substance active est l’immunoglobuline humaine normale (IgSC).
Un mL contient 200 mg d’immunoglobuline humaine normale, dont au moins 98 % d’IgG.
Les pourcentages des sous‑classes d’IgG sont d’environ 62 % d’IgG1, 30 % d’IgG2, 4,3 % d’IgG3 et 3,2 % d’IgG4. Le médicament contient des IgA en petite quantité (pas plus de 160 microgrammes/mL).
· Les autres composants sont :
Glycine (E640), polysorbate 80 (E433) et eau pour préparations injectables.
XEMBIFY est une solution injectable sous‑cutanée. La solution est limpide à légèrement opalescente et incolore ou jaune pâle ou marron clair.
XEMBIFY est présenté dans une boîte et est contenu dans un flacon de verre transparent muni d’un bouchon, d’un opercule en aluminium, d’une capsule en plastique et d’un manchon d’inviolabilité rétractable garantissant que l’emballage n’a pas été ouvert.
Les présentations de XEMBIFY :
1 ou 10 flacons contenant 1 g d’immunoglobuline humaine normale dans 5 mL solution pour injection sous-cutanée.
1, 10 ou 20 flacons contenant 2 g d’immunoglobuline humaine normale dans 10 mL solution pour injection sous-cutanée.
1 ou 20 flacons contenant 4 g d’immunoglobuline humaine normale dans 20 mL solution pour injection sous-cutanée.
1 ou 10 flacons contenant 10 g d’immunoglobuline humaine normale dans 50 mL solution pour injection sous-cutanée.
Chaque boîte contient 1, 10 ou 20 flacons de XEMBIFY et une notice.
Toutes les présentations peuvent ne pas être commercialisées.
Titulaire de l’autorisation de mise sur le marché et fabricant
C/CAN GUASCH, 2
POLIGONO INDUSTRIAL LEVANTE
08150 PARETS DEL VALLÈS,
BARCELONE
ESPAGNE
Exploitant de l’autorisation de mise sur le marché
24 RUE DE PRONY
75017 PARIS
C/CAN GUASCH, 2
POLIGONO INDUSTRIAL LEVANTE
08150 PARETS DEL VALLÈS,
BARCELONE
ESPAGNE
Noms du médicament dans les Etats membres de l'Espace Economique Européen
Ce médicament est autorisé dans les Etats membres de l'Espace Economique Européen sous les noms suivants : Conformément à la réglementation en vigueur.
[À compléter ultérieurement par le titulaire]
La dernière date à laquelle cette notice a été révisée est :
[à compléter ultérieurement par le titulaire]
Des informations détaillées sur ce médicament sont disponibles sur le site Internet de l’ANSM (France).