Dernière mise à jour le 29/06/2026
LEVETIRACETAM TILLOMED 100 mg/ml, solution à diluer pour perfusion
Indications thérapeutiques
Classe pharmacothérapeutique : antiépileptiques, autres antiépileptiques, Code ATC : N03AX14.
Le lévétiracétam est un médicament antiépileptique (médicament utilisé pour traiter les crises d’épilepsie).
LEVETIRACETAM TILLOMED 100 mg/ml, solution à diluer pour perfusion est utilisé :
· seul, chez les adultes et les adolescents de 16 ans ou plus présentant une épilepsie nouvellement diagnostiquée, pour traiter une certaine forme d’épilepsie. L’épilepsie est une maladie qui produit des crises à répétition chez les patients (crises d'épilepsie). Le lévétiracétam est utilisé pour traiter la forme d’épilepsie dans laquelle les crises touchent initialement un seul côté du cerveau, mais qui pourraient par la suite s'étendre à des zones plus larges des deux côtés du cerveau (crises partielles avec ou sans généralisation secondaire). Le lévétiracétam vous a été prescrit par votre médecin afin de réduire le nombre de crises
· en complément à d’autres médicaments antiépileptiques pour traiter :
o les crises d’épilepsie partielles, avec ou sans généralisation, chez l’adulte, l’adolescent et l’enfant de 4 ans ou plus ;
o les crises myocloniques (mouvements brefs et saccadés d’un muscle ou d’un groupe de muscles) chez l’adulte et l’adolescent de 12 ans ou plus présentant une épilepsie myoclonique juvénile ;
o les crises tonico-cloniques généralisées primaires (crises généralisées avec perte de connaissance) chez l’adulte et l’adolescent de 12 ans ou plus présentant une épilepsie généralisée idiopathique (un type d’épilepsie que l’on suppose attribué à une cause génétique).
LEVETIRACETAM TILLOMED 100 mg/ml, solution à diluer pour perfusion en solution à diluer pour perfusion représente une forme de traitement alternative chez les patients pour lesquels un traitement par voie orale est temporairement impossible.
Présentations
> 10 flacon(s) en verre de 5 ml
Code CIP : 34009 301 885 8 3
Déclaration de commercialisation : 22/02/2024
Cette présentation est agréée aux collectivités
Service médical rendu (SMR)
Les libellés affichés ci-dessous ne sont que des résumés ou extraits issus des avis rendus par la Commission de la Transparence. Seul l'avis complet de la Commission de la Transparence fait référence.
Cet avis est consultable à partir du lien `Avis du jj/mm/aaaa` ou encore sur demande auprès de la HAS (plus d'informations dans l'aide). Les avis et synthèses d'avis contiennent un paragraphe sur la place du médicament dans la stratégie thérapeutique.
| Valeur du SMR | Avis | Motif de l'évaluation | Résumé de l'avis |
|---|---|---|---|
| Important | Avis du 08/01/2020 | Inscription (CT) | Le service médical rendu par LEVETIRACETAM TILLOMED 100 mg/ml, solution à diluer pour perfusion, est important dans les indications de l’AMM. |
Amélioration du service médical rendu (ASMR)
Les libellés affichés ci-dessous ne sont que des résumés ou extraits issus des avis rendus par la Commission de la Transparence. Seul l'avis complet de la Commission de la Transparence fait référence.
Cet avis est consultable à partir du lien `Avis du jj/mm/aaaa` ou encore sur demande auprés de la HAS (plus d'informations dans l'aide). Les avis et synthéses d'avis contiennent un paragraphe sur la place du médicament dans la stratégie thérapeutique.
| Valeur de l'ASMR | Avis | Motif de l'évaluation | Résumé de l'avis |
|---|---|---|---|
| V (Inexistant) | Avis du 08/01/2020 | Inscription (CT) | Cette spécialité est un générique qui n’apporte pas d’amélioration du service médical rendu (ASMR V) par rapport au princeps KEPPRA 100 mg/ml, solution à diluer pour perfusion (lévétiracétam). |
ANSM - Mis à jour le : 02/06/2026
LEVETIRACETAM TILLOMED 100 mg/ml, solution à diluer pour perfusion
2. COMPOSITION QUALITATIVE ET QUANTITATIVE
Lévétiracétam....................................................................................................................... 100 mg
Pour 1 ml.
Chaque flacon de 5 ml contient 500 mg de lévétiracétam.
Excipient à effet notoire :
Chaque flacon contient 17,13 mg de sodium.
Pour la liste complète des excipients, voir rubrique 6.1.
Solution à diluer pour perfusion.
Liquide limpide, incolore.
pH : 5,00 à 6,00.
Osmolarité : pas plus de 1150 mOsmol/kg.
4.1. Indications thérapeutiques
Le lévétiracétam est indiqué en thérapie adjuvante
· dans le traitement des crises partielles avec ou sans généralisation secondaire chez les adultes, les adolescents et les enfants de 4 ans ou plus présentant une épilepsie,
· dans le traitement des crises myocloniques chez les adultes et les adolescents de 12 ans ou plus présentant une épilepsie myoclonique juvénile,
· dans le traitement des crises tonico-cloniques généralisées primaires chez les adultes et les adolescents de 12 ans ou plus présentant une épilepsie généralisée idiopathique.
Le lévétiracétam en solution à diluer pour perfusion représente une alternative chez les patients chez lesquels un traitement par voie orale est temporairement impossible.
4.2. Posologie et mode d'administration
Le traitement par le lévétiracétam peut être instauré soit par administration intraveineuse, soit par administration orale.
Le passage de l’administration orale à intraveineuse ou inversement peut être fait directement, sans ajustement. La dose totale quotidienne et la fréquence d’administrations doivent être maintenues.
Crises partielles
La dose recommandée en monothérapie (à partir de 16 ans) et en association est la même et est décrite ci-dessous.
Toutes les indications
Adultes (≥18 ans) et adolescents (12 à 17 ans) pesant 50 kg ou plus
La dose thérapeutique initiale est de 500 mg deux fois par jour. Cette dose peut être débutée dès le premier jour de traitement. Toutefois, une dose initiale plus faible de 250 mg deux fois par jour peut être administrée, en fonction de l’évaluation par le médecin de la réduction des crises par rapport aux effets indésirables éventuels. Cette dose peut être augmentée à 500 mg deux fois par jour au bout de deux semaines de traitement.
En fonction de la réponse clinique et de la tolérance, la dose quotidienne peut être augmentée jusqu’à 1 500 mg deux fois par jour. Les augmentations et diminutions posologiques peuvent se faire par paliers de 250 mg ou 500 mg deux fois par jour toutes les deux à quatre semaines.
Adolescents (12 à 17 ans) pesant moins de 50 kg et enfants à partir de 4 ans
Le médecin doit prescrire la forme pharmaceutique, la présentation et le dosage les plus appropriés en fonction du poids, de l’âge et de la dose. Consulter la rubrique Population pédiatrique pour les détails concernant les adaptations posologiques en fonction du poids.
Durée du traitement
Aucune donnée n’est disponible quant à l’administration du lévétiracétam par voie intraveineuse sur une période de plus de 4 jours.
Arrêt du traitement
Si une interruption du traitement par le lévétiracétam s’impose, il est recommandé d’effectuer cette interruption progressivement (p. ex. chez les adultes et les adolescents pesant plus de 50 kg : diminution par paliers de 500 mg deux fois par jour toutes les deux à quatre semaines ; chez les enfants et les adolescents pesant moins de 50 kg : la diminution de la dose doit se faire par paliers pas dépassant 10 mg/kg deux fois par jour toutes les deux semaines)
Populations spéciales
Patients âgés (de 65 ans ou plus)
Une adaptation de la posologie est recommandée chez les patients âgés présentant une insuffisance rénale (voir « Insuffisance rénale » ci-dessous).
Insuffisance rénale
La dose quotidienne doit être individualisée selon la fonction rénale.
Pour les patients adultes, se reporter au tableau qui suit et adapter la posologie comme indiqué. Pour utiliser ce tableau, il est nécessaire de calculer la clairance de la créatinine (CLcr) du patient en ml/min. Chez les adultes et les adolescents pesant plus de 50 kg, la CLcr en ml/min peut être estimée à partir de la valeur de la créatinine sérique (en mg/dl) conformément à la formule suivante :
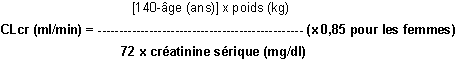
La clairance de la créatinine est ensuite ajustée en fonction de la surface corporelle (SC) comme suit :
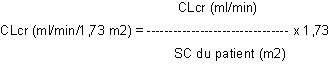
Adaptation de la posologie chez les adultes et les adolescents pesant plus de 50 kg présentant une insuffisance rénale
|
Stade |
Clairance de la créatinine (ml/min/1,73 m2) |
Dose et fréquence des administrations |
|
Fonction rénale normale |
> 80 |
500 à 1 500 mg deux fois par jour |
|
Insuffisance rénale légère |
50-79 |
500 à 1 500 mg deux fois par jour |
|
Insuffisance rénale modérée |
30-49 |
250 à 750 mg deux fois par jour |
|
Insuffisance rénale sévère |
< 30 |
250 à 750 mg deux fois par jour |
|
Insuffisance rénale au stade terminal sous dialyse (1) |
- |
500 à 1 000 mg une fois par jour (2) |
(1) Une dose de charge de 750 mg est recommandée le premier jour du traitement par le lévétiracétam.
(2) Après une séance de dialyse, une dose supplémentaire de 250 à 500 mg est recommandée.
Chez les enfants présentant une insuffisance rénale, la dose de lévétiracétam doit être adaptée selon la fonction rénale, car la clairance du lévétiracétam en dépend. Cette recommandation repose sur une étude menée chez des adultes insuffisants rénaux.
Chez les jeunes adolescents et les enfants, la CLcr (en ml/min/1,73 m²) peut être estimée à partir d’une mesure de la créatinine sérique (en mg/dl) en utilisant la formule suivante (formule de Schwartz):

ks= 0,55 chez les enfants de moins de 13 ans et les adolescentes ; ks= 0,7 chez les adolescents.
Adaptation de la posologie chez les enfants et les adolescents pesant moins de 50 kg présentant une insuffisance rénale
|
Stade |
Clairance de la créatinine (ml/min/1,73 m2) |
Dose et fréquence des administrations |
|
Enfants de 4 ans ou plus et adolescents pesant moins de 50 kg |
||
|
Fonction rénale normale |
>80 |
10 à 30 mg/kg (0,10 à 0,30 ml/kg) deux fois par jour |
|
Insuffisance rénale légère |
50-79 |
10 à 20 mg/kg (0,10 à 0,20 ml/kg) deux fois par jour |
|
Insuffisance rénale modérée |
30-49 |
5 à 15 mg/kg (0,05 à 0,15 ml/kg) deux fois par jour |
|
Insuffisance rénale sévère |
<30 |
5 à 10 mg/kg (0,05 à 0,10 ml/kg) deux fois par jour |
|
Insuffisance rénale au stade terminal sous dialyse |
-- |
10 à 20 mg/kg (0,10 à 0,20 ml/kg) une fois par jour (1) (2)
|
(1) Une dose de charge de 15 mg/kg (0,15 ml/kg) est recommandée le premier jour du traitement par le lévétiracétam.
(2) Après une séance de dialyse, une dose supplémentaire de 5 à 10 mg/kg (0,05 à 0,10 ml/kg) est recommandée.
Insuffisance hépatique
Aucune adaptation de la posologie n’est nécessaire chez les patients présentant une insuffisance hépatique légère à modérée. Chez les patients présentant une insuffisance hépatique sévère, la clairance de la créatinine peut sous-estimer l’insuffisance rénale. Une diminution de 50 % de la dose d’entretien quotidienne est donc recommandée si la clairance de la créatinine est <60 ml/min/1,73 m².
Population pédiatrique
Le médecin doit prescrire la forme pharmaceutique, la présentation et la posologie les plus appropriées pour l’âge, le poids et la dose.
En monothérapie
La sécurité et l’efficacité du lévétiracétam chez l’enfant et l’adolescent de moins de 16 ans n’ont pas été établies en monothérapie.
Pas de donnée disponible.
Adolescents (16 à 17 ans) pesant 50 kg ou plus, ayant des crises partielles avec ou sans généralisation secondaire et présentant une épilepsie nouvellement diagnostiquée
Se référer à la rubrique ci-dessus concernant l’adulte (≥ 18 ans) et l’adolescent (12 à 17 ans) pesant 50 kg ou plus.
Traitement en association chez l’enfants (de 4 à 11 ans) et adolescent (12 à 17 ans) pesant moins de 50 kg
La dose thérapeutique initiale est de 10 mg/kg deux fois par jour.
En fonction de la réponse clinique et de la tolérance, la dose peut être augmentée jusqu’à 30 mg/kg deux fois par jour. Toute Les augmentation et/ou diminutions de doses ne doivent pas dépasser 10 mg/kg deux fois par jour toutes les deux semaines. La dose efficace la plus faible doit être utilisée pour toutes les indications.
La posologie chez l’enfant de 50 kg ou plus est la même que chez l’adulte pour toutes les indications.
Se référer à la rubrique ci-dessus concernant l’adulte (≥ 18 ans) et l’adolescent (12 à 17 ans) pesant 50 kg ou plus pour toutes les indications.
Recommandations posologiques chez les enfants et les adolescents
|
Poids |
Posologie de départ : 10 mg/kg deux fois par jour |
Posologie maximale : 30 mg/kg deux fois par jour |
|
150 mg deux fois par jour |
450 mg deux fois par jour |
|
|
20 kg (1) |
200 mg deux fois par jour |
600 mg deux fois par jour |
|
25 kg |
250 mg deux fois par jour |
750 mg deux fois par jour |
|
À partir de 50 kg (2) |
500 mg deux fois par jour |
1 500 mg deux fois par jour |
(1) Chez les enfants pesant 25 kg ou moins, le traitement doit de préférence être instauré avec lévétiracetam 100 mg/ml, solution buvable.
(2) Chez les enfants et les adolescents pesant 50 kg ou plus, la posologie est la même que chez les adultes.
Thérapie adjuvante chez les nourrissons et les enfants de moins de 4 ans
La sécurité et l’efficacité du lévétiracétam en solution à diluer pour perfusion n’ont pas été établies chez les nourrissons et les enfants de moins de 4 ans.
Les données actuellement disponibles sont décrites aux rubriques 4.8, 5.1 et 5.2 mais aucune recommandation sur la posologie ne peut être donnée.
Mode d’administration
Le lévétiracétam en solution à diluer pour perfusion est uniquement destiné à être utilisé par voie intraveineuse. La dose recommandée doit être préparée par dilution dans au moins 100 ml d’un diluant compatible et administrée par voie intraveineuse en perfusion sur 15 minutes (voir rubrique 6.6).
4.4. Mises en garde spéciales et précautions d'emploi
La posologie du lévétiracétam devra éventuellement être adaptée chez les insuffisants rénaux. Chez les patients présentant une insuffisance hépatique sévère, il est recommandé d’évaluer la fonction rénale avant de sélectionner la posologie à utiliser (voir rubrique 4.2).
Insuffisance rénale aiguë
Une exposition au lévétiracétam a été associée à une insuffisance rénale aiguë dans de très rares cas, le délai de survenue allant de quelques jours à plusieurs mois.
Numération formule sanguine
Des anomalies de la numération formule sanguine (neutropénie, agranulocytose, leucopénie, thrombopénie et pancytopénie) ont été décrites dans de rares cas en association avec l’administration de lévétiracétam, généralement au début du traitement. Il est conseillé d’évaluer la formule sanguine complète chez les patients qui présentent une faiblesse importante, un état fébrile, des infections récurrentes ou des troubles de la coagulation (rubrique 4.8).
Suicide
Des cas de suicide, de tentative de suicide et d’idées et de comportements suicidaires ont été rapportés chez des patients traités par des antiépileptiques (y compris le lévétiracétam). Une méta-analyse d’essais randomisés, contrôlés par placebo portant sur des médicaments antiépileptiques a mis en évidence une légère augmentation du risque de pensées et comportements suicidaires. Le mécanisme sous-jacent n’est pas établi.
Il convient donc de surveiller les patients pour identifier tout signe de dépression et/ou d’idées et de comportements suicidaires et d’envisager d’instaurer un traitement approprié. Il faut également conseiller aux patients (et à leurs aidants) de consulter un médecin à l’émergence de tels signes.
Comportements anormaux et agressifs
Le lévétiracétam peut provoquer des symptômes psychotiques et des troubles du comportement, y compris une irritabilité et une agressivité. Les patients traités par du lévétiracétam doivent être surveillés afin de détecter l’apparition de signes psychiatriques symptomatiques d’importants changements d’humeur et/ou de la personnalité. Si de tels comportements sont observés, l’adaptation au traitement ou l’arrêt progressif du traitement doivent être envisagés. Si une interruption du traitement est envisagée, veuillez-vous référer à la rubrique 4.2.
Aggravation des crises convulsives
Comme avec d’autres types d’antiépileptiques, le lévétiracétam peut, dans de rares cas, accroître la fréquence ou la gravité des crises convulsives. Cet effet paradoxal, principalement signalé au cours du premier mois suivant l’instauration du lévétiracétam ou l’augmentation de la dose, était réversible après l’arrêt du médicament ou la diminution de la dose. Il doit être conseillé aux patients de consulter immédiatement leur médecin en cas d’aggravation des crises convulsives.
Une absence d’efficacité ou une aggravation des crises a par exemple été rapportée chez des patients atteints d’épilepsie associée à des mutations de la sous-unité alpha 8 du canal sodique voltage-dépendant (SCN8A).
Allongement de l'intervalle QT à l'électrocardiogramme
De rares cas d'allongement de l'intervalle QT ECG ont été observés au cours de la surveillance post-commercialisation. Le lévétiracétam doit être utilisé avec prudence chez les patients présentant un allongement de l'intervalle QTc, chez les patients traités de manière concomitante par des médicaments modifiant l'intervalle QTc ou chez les patients présentant une maladie cardiaque préexistante ou des troubles électrolytiques.
Population pédiatrique
Les données disponibles chez les enfants ne suggèrent aucun effet sur la croissance et la puberté. Toutefois, les effets à long terme sur l’apprentissage, l’intelligence, la croissance, les fonctions endocrines, la puberté et l’aptitude à procréer des enfants traités demeurent inconnus.
Excipients
Ce médicament contient 19,1 mg de sodium par flacon (5 ml), ce qui équivaut à 0,796 % de l’apport sodé quotidien maximal recommandé par l’OMS, à savoir 2 g chez les adultes.
À prendre en compte par les patients sous régime contrôlé en sodium
4.5. Interactions avec d'autres médicaments et autres formes d'interactions
Les données obtenues lors d’études cliniques effectuées chez des adultes avant la mise sur le marché indiquent que le lévétiracétam ne modifie pas les teneurs sériques en des médicaments antiépileptiques disponibles (phénytoïne, carbamazépine, acide valproïque, phénobarbital, lamotrigine, gabapentine et primidone) et que ces médicaments n’ont pas d’influence sur la pharmacocinétique du lévétiracétam.
Comme chez les adultes, aucune interaction médicamenteuse cliniquement significative n’a été mise en évidence chez des patients pédiatriques recevant le lévétiracétam à 60 mg/kg/jour au maximum. Une évaluation rétrospective des interactions pharmacocinétiques chez des enfants et adolescents épileptiques (âgés de 4 à 17 ans) a confirmé que le lévétiracétam en thérapie adjuvante orale par n’influence pas les concentrations sériques à l’état d’équilibre de la carbamazépine et du valproate administrés en traitement concomitant. Toutefois, des données suggèrent que la clairance du lévétiracétam est augmentée de 20 % chez les enfants traités par des antiépileptiques inducteurs enzymatiques. Une adaptation de la posologie n’est pas nécessaire.
Probénécide
On a montré que le probénécide (500 mg quatre fois par jour), un inhibiteur de la sécrétion tubulaire rénale, inhibe la clairance rénale du principal métabolite, mais pas celle du lévétiracétam. La concentration de ce métabolite demeure toutefois basse.
Méthotrexate
On a montré que l’administration concomitante de lévétiracétam et de méthotrexate diminue la clairance du méthotrexate, résultant en une augmentation/prolongation des concentrations sanguines du méthotrexate jusqu’à des niveaux potentiellement toxiques. Une surveillance étroite des taux sanguins de méthotrexate et de lévétiracétam s’impose chez les patients qui reçoivent ces deux médicaments en traitement concomitant.
Contraceptifs oraux et autres interactions pharmacocinétiques
À la dose de 1 000 mg par jour, le lévétiracétam n’a pas affecté la pharmacocinétique des contraceptifs oraux (éthynilestradiol et lévonorgestrel) ; les paramètres endocriniens (hormone lutéinisante et progestérone) n’ont pas été modifiés. À la dose de 2 000 mg par jour, le lévétiracétam n’a pas affecté la pharmacocinétique de la digoxine et de la warfarine ; les temps de prothrombine n’ont pas été modifiés. La co-administration de digoxine, de contraceptifs oraux et de warfarine n’a pas affecté la pharmacocinétique du lévétiracétam.
Alcool
Aucune donnée n’est disponible quant aux interactions entre le lévétiracétam et l’alcool.
4.6. Fertilité, grossesse et allaitement
Les femmes en âge de procréer doivent bénéficier de conseils médicaux spécialisés. Une réévaluation du traitement par le lévétiracétam s’impose chez les femmes qui envisagent une grossesse. Comme avec tous les médicaments antiépileptiques, il convient d’éviter tout arrêt brutal du traitement par le lévétiracétam en raison du risque de récidives de crises, qui peuvent avoir de graves conséquences pour la femme et l’enfant à naître. La monothérapie doit être privilégiée dans la mesure du possible. Par comparaison en effet, les traitements d’association par de multiples antiépileptiques sont susceptibles d’être associés à un risque accru de malformations congénitales qui varie selon la combinaison utilisée.
Grossesse
Un grand nombre de données de post-commercialisation concernant les femmes enceintes exposées au lévétiracétam en monothérapie (plus de 1 800, dont plus de 1 500 expositions au cours du 1er trimestre) ne suggère pas d’augmentation du risque de malformations congénitales majeures.
Des données limitées sur le neurodéveloppement des enfants exposés au lévétiracétam en monothérapie in utero sont disponibles. Les données issues de deux études observationnelles basées sur des registres de population, menée principalement sur le même ensemble de données provenant de pays nordiques et incluant plus de 1000 enfants nés de femmes épileptiques ayant été exposées in utero au lévétiracétam en monothérapie, ne suggèrent pas un risque accru de troubles du spectre autistique ou de handicaps intellectuels comparativement aux enfants nés de femmes épileptiques non exposées à un médicament antiépileptique in utero. La durée moyenne de suivi des enfants dans le groupe lévétiracétam est plus courte que pour le groupe des enfants non exposés à un médicament antiépileptique (par ex. 4,4 ans vs 6,8 ans dans l’une des études).
Si après une évaluation attentive le traitement est considéré comme cliniquement nécessaire, le lévétiracétam peut être utilisé pendant la grossesse. Dans ce cas, la dose efficace la plus faible est recommandée..
Allaitement
Le lévétiracétam est excrété dans le lait maternel humain. Par conséquent, l’allaitement n’est pas recommandé. Toutefois, si un traitement par le lévétiracétam s’avère nécessaire, une décision doit être prise soit d’interrompre l’allaitement, soit d’interrompre/de s’abstenir du traitement en prenant en compte le bénéfice de l’allaitement pour l’enfant au regard du bénéfice du traitement pour la femme.
Fertilité
Les études effectuées chez l’animal n’ont mis en évidence aucun impact sur la fertilité (voir rubrique 5.3). Aucune donnée clinique n’est disponible et le risque potentiel pour l’homme inconnu.
4.7. Effets sur l'aptitude à conduire des véhicules et à utiliser des machines
Résumé du profil de sécurité
Les effets indésirables suivants sont ceux qui ont été les plus fréquemment rapportés : rhinopharyngite, somnolence, céphalées, fatigue et étourdissement. Le profil des effets indésirables présenté ci-dessous repose sur l’analyse de l’ensemble des essais cliniques contrôlés par placebo menés avec le lévétiracétam, indépendamment de l’indication, ce qui représente un total de 3 416 patients traités par ce médicament. Ces données sont complétées par celles relatives à l’utilisation du lévétiracétam dans les études d’extension en ouvert correspondantes et celles obtenues à la pharmacovigilance. Le profil de tolérance au lévétiracétam est généralement similaire dans toutes les tranches d’âges (patients adultes et pédiatriques) et pour toutes les indications approuvées dans l’épilepsie. Les données relatives à une exposition au lévétiracétam par voie intraveineuse étant limitées et les formulations orales et intraveineuses étant bioéquivalentes, les informations sur la sécurité du lévétiracétam par voie intraveineuse se fondent sur l’utilisation du lévétiracétam par voie orale.
Liste tabulée des effet indésirables
Les effets indésirables rapportés au cours des études cliniques (adultes, adolescents, enfants et nourrissons de >1 mois) et à la pharmacovigilance sont répertoriés au tableau ci-dessous par classe de systèmes d’organe et par fréquence. Les effets indésirables sont présentés dans l’ordre décroissant de gravité et leur fréquence est définie comme suit : très fréquent (≥ 1/10) ; fréquent (≥ 1/100, < 1/10) ; peu fréquent (≥ 1/1 000, < 1/100) ; rare (≥ 1/10 000, < 1/1 000) et très rare (< 1/10 000).
|
Classes de système d’organes (MedDRA) |
Catégorie de fréquence |
||||
|
Très fréquent |
Fréquent |
Peu fréquent |
Rare |
Très rare |
|
|
Infections et infestations |
Rhinopharyngite |
|
|
Infections |
|
|
Affections hématologiques et du système lymphatique |
|
|
Thrombopénie, leucopénie |
Pancytopénie, neutropénie, agranulocytose |
|
|
Affections du système immunitaire |
|
|
|
Syndrome d’hypersensibilité médicamenteuse avec éosinophilie et symptômes systémiques (DRESS)(1), hypersensibilité (y compris œdème de Quincke et anaphylaxie) |
|
|
Troubles cardiaque |
|
|
|
Electrocardiogramme à QT prolongé |
|
|
Troubles du métabolisme et de la nutrition |
|
Anorexie |
Perte de poids, gain de poids |
Hyponatrémie |
|
|
Affections psychiatriques |
|
Dépression, agressivité/hostilité, anxiété, insomnie, nervosité/irritabilité |
Tentative de suicide, idées suicidaires, troubles psychotiques, troubles du comportement, hallucination, colère, état confusionnel, attaque de panique, labilité émotionnelle/sautes d’humeur, agitation |
Suicide réalisé, troubles de la personnalité, troubles de la pensée, délire |
Trouble obsessionnel compulsif(2) |
|
Affections du système nerveux |
Somnolence, céphalées |
Convulsion, troubles de l’équilibre, étourdissement, léthargie, tremblement |
Amnésie, troubles de mémoire, troubles de la coordination/ataxie, paresthésie, trouble de l’attention |
Choréoathétose, dyskinésie, hyperkinésie, troubles de la démarche, encéphaopathie, aggravation des crises convulsives syndrome malin des neuroleptiques(3) |
|
|
Affections oculaires |
|
|
Diplopie, vision trouble |
|
|
|
Affections de l'oreille et du labyrinthe |
|
Vertige |
|
|
|
|
Affections respiratoires, thoraciques et médiastinales |
|
Toux |
|
|
|
|
Troubles gastro-intestinaux |
|
Douleur abdominale, diarrhée, dyspepsie, vomissements, nausées |
|
Pancréatite |
|
|
Affections hépatobiliaires |
|
|
Anomalies aux tests de la fonction hépatique |
Insuffisance hépatique aiguë, hépatite |
|
|
Affections rénales et urinaires |
|
|
|
Insuffisance rénale aiguë |
|
|
Affections de la peau et du tissu sous-cutané |
|
Éruption cutanée |
Alopécie, eczéma, prurit |
Syndrome de Lyell, syndrome de Stevens-Johnson, érythème polymorphe |
|
|
Affections musculo-squelettiques et systémiques |
|
|
Faiblesse musculaire, myalgie |
Rhabdomyolyse et augmentation du taux sanguin de créatine phosphokinase (CPK)(1) |
|
|
Troubles généraux et anomalies au site d'administration |
|
Asthénie/fatigue |
|
|
|
|
Lésions, intoxications et complications liées aux procédures |
|
|
Lésions |
|
|
(1) Voir la rubrique Description d’effets indésirables sélectionnés.
(2) De très rares cas de développement de troubles obsessionnels compulsifs (TOC) ont été observés chez des patients présentant des antécédents sous-jacents de TOC ou d’affections psychiatriques dans le cadre de la surveillance post-commercialisation.
(3) La prévalence est significativement plus élevée chez les patients japonais par rapport aux patients non japonais.
Description d’effets indésirables sélectionnés
Réactions d’hypersensibilité multiviscérale
Des réactions d’hypersensibilité multiviscérale (également connues sous le nom de syndrome d’hypersensibilité médicamenteuse avec éosinophilie et symptômes systémiques [Drug Reaction with Eosinophilia and Systemic Symptoms, DRESS]) ont été rarement signalées chez des patients traités par lévétiracétam. Les manifestations cliniques peuvent se développer 2 à 8 semaines après le début du traitement. Ces réactions se présentent de différentes manières, mais se manifestent typiquement par de la fièvre, une éruption cutanée, un oedème facial, des adénopathies, des anomalies hématologiques et peuvent être associées à une atteinte de différents systèmes d’organes, dont, principalement, le foie. En cas de suspicion d’une réaction d’hypersensibilité multiviscérale, il convient d’interrompre le traitement par lévétiracétam.
Le risque d’anorexie est plus important lorsque le lévétiracétam est co-administré avec du topiramate. Dans plusieurs cas d’alopécie, une régression a été observée à l’arrêt du lévétiracétam Une aplasie médullaire a été identifiée dans quelques cas de pancytopénie.
Les cas d’encéphalopathie sont généralement survenus en début de traitement (quelques jours à quelques mois) et ont disparu après l’arrêt du traitement.
Population pédiatrique
Au total, 190 patients âgés de 1 mois à moins de 4 ans ont été traités par le lévétiracétam au cours d’études contrôlées par placebo et d’études d’extension en ouvert. Parmi ces patients, 60 ont reçu le lévétiracétam lors d’études contrôlées par placebo. Au total, 645 patients âgés de 4 à 16 ans ont été traités par le lévétiracétam au cours d’études contrôlées par placebo et d’études d’extension en ouvert. Parmi ces patients, 233 ont reçu le lévétiracétam lors d’études contrôlées par placebo. Les données obtenues dans ces deux tranches d’âges pédiatriques sont complétées par l’expérience acquise à la pharmacovigilance avec le lévétiracétam.
Par ailleurs, 101 nourrissons âgés de moins de 12 mois ont été exposés lors d’une étude de sécurité post autorisation. Aucune inquiétude nouvelle n’a été identifiée quant à la sécurité chez les nourrissons épileptiques âgés de moins de 12 mois traités par le lévétiracétam.
Le profil des événements indésirables du lévétiracétam est généralement similaire dans toutes les tranches d’âges et pour toutes les indications approuvées dans l’épilepsie. Les résultats des études cliniques contrôlées par placebo indiquent que les profils de sécurité du lévétiracétam sont concordants entre les populations pédiatrique et adulte, à l’exception des effets indésirables comportementaux et psychiatriques qui sont plus fréquents chez les enfants que chez les adultes. Les effets indésirables suivants ont été signalés plus fréquemment chez les enfants et les adolescents âgés de 4 à 16 ans que dans d’autres tranches d’âges ou dans le profil de sécurité général : vomissements (très fréquent, 11,2 %), agitation (fréquent, 3,4 %), sautes d’humeur (fréquent, 2,1 %), labilité émotionnelle (fréquent, 1,7 %), hostilité (fréquent, 8,2 %), troubles du comportement (fréquent, 5,6 %) et léthargie (fréquent, 3,9 %). Une irritabilité (très fréquent, 11,7 %) et des troubles de la coordination (fréquent, 3,3 %) ont été rapportés plus fréquemment chez les nourrissons et les enfants âgés de 1 mois à moins de 4 ans que dans d’autres tranches d’âges ou dans le profil de sécurité général.
Une étude de non infériorité à double insu et contrôlée par placebo concernant la sécurité dans la population pédiatrique a évalué les effets cognitifs et neuropsychologiques du lévétiracétam chez des enfants âgés de 4 à 16 ans présentant des crises partielles. L’équipe de l’étude a conclu que le lévétiracétam n’est pas différent du placebo (non inférieur) en ce qui concerne le changement, par rapport au départ, du score à un test composite (combinant une épreuve révisée de Leiter sur l’attention et la mémoire et une évaluation de la mémoire) dans la population per protocole. Les résultats relatifs aux fonctions comportementales et émotionnelles ont mis en évidence une aggravation du comportement agressif, évalué d’une manière standardisée et systématique au moyen d’un instrument validé (Inventaire des comportements de l’enfant d’Achenbach ; CBCL, pour Achenbach Child Behavior Checklist) chez les patients traités par le lévétiracétam.
En moyenne, une dégradation des fonctions comportementales et émotionnelles n’a toutefois pas été observée chez les sujets traités par le lévétiracétam dans l’étude de suivi à long terme en ouvert ; en particulier, les mesures du comportement agressif n’ont pas empiré par rapport aux valeurs de départ.
Déclaration des effets indésirables suspectés
La déclaration des effets indésirables suspectés après autorisation du médicament est importante. Elle permet une surveillance continue du rapport bénéfice/risque du médicament. Les professionnels de santé déclarent tout effet indésirable suspecté via le système national de déclaration : Agence nationale de sécurité du médicament et des produits de santé (ANSM) et réseau des Centres Régionaux de Pharmacovigilance - Site internet : https://signalement.social-sante.gouv.fr
Symptômes
Les symptômes suivants ont été signalés en association avec un surdosage par le lévétiracétam : somnolence, agitation, agressivité, diminution du niveau de conscience, dépression respiratoire et coma.
Prise en charge d’un surdosage :
Il n’existe aucun antidote spécifique du lévétiracétam. Le traitement du surdosage sera symptomatique et comprendra éventuellement une hémodialyse. Le taux d’élimination par dialyse est de 60 % pour le lévétiracétam et de 74 % pour le principal métabolite.
5. PROPRIETES PHARMACOLOGIQUES
5.1. Propriétés pharmacodynamiques
Classe pharmacothérapeutique : antiépileptiques, autres antiépileptiques, Code ATC : N03AX14.
La substance active, le lévétiracétam, est un dérivé de la pyrrolidone (énantiomère S de l’acétamide de α-éthyl-2-oxo-1-pyrrolidine) qui n’est pas apparenté sur le plan chimique aux substances actives antiépileptiques disponibles à l’heure actuelle.
Mécanisme d’action
Le mécanisme d’action du lévétiracétam n’est pas encore entièrement élucidé. Des expériences in vitro et in vivo suggèrent que le lévétiracétam ne modifie ni les caractéristiques de base des cellules, ni la neurotransmission normale.
Des études in vitro montrent que le lévétiracétam affecte les concentrations intra-neuronales de Ca2+ par une inhibition partielle des canaux calciques de type N et une réduction de la libération de Ca2+ à partir des réserves intra-neuronales. En outre, le lévétiracétam inverse en partie l’effet inhibiteur du zinc et des β-carbolines sur les canaux GABAergiques et glycinergiques. Par ailleurs, des études in vitro ont montré que le lévétiracétam se lie à un site spécifique du tissu cérébral chez les rongeurs. Ce site de liaison est la protéine 2A de la vésicule synaptique, qui semble être impliquée dans la fusion vésiculaire et l’excrétion cellulaire de neurotransmetteurs. Le degré d’affinité pour la protéine 2A de la vésicule synaptique du lévétiracétam et de ses analogues est en corrélation avec la capacité de protection contre les crises dans le modèle d’épilepsie réflexe audiogénique chez la souris. À la lumière de ces observations, l’interaction entre le lévétiracétam et la protéine 2A de la vésicule synaptique semble contribuer au mécanisme d’action antiépileptique du médicament.
Effets pharmacodynamiques
Le lévétiracétam induit une protection contre les crises dans un grand nombre de modèles animaux d’épilepsie partielle et d’épilepsie généralisée primaire sans exercer un effet proconvulsivant. Le principal métabolite est inactif. Une activité à la fois dans les épilepsies partielles et les épilepsies généralisées chez l’homme (décharges épileptiformes/réponses photoparoxystiques) a confirmé que le lévétiracétam possède un profil pharmacologique à large spectre.
Efficacité et sécurité clinique
Thérapie adjuvante dans le traitement des crises partielles avec ou sans généralisation secondaire chez les adultes, les adolescents et les enfants de 4 ans ou plus présentant une épilepsie :
Chez les adultes, l’efficacité du lévétiracétam a été démontrée dans 3 études en double insu contrôlées par placebo utilisant des doses de 1 000 mg, 2 000 mg ou 3 000 mg par jour en deux prises fractionnées sur une période allant jusqu’à 18 semaines. À l’analyse des données regroupées, une réduction de 50 % ou plus par rapport au départ de la fréquence hebdomadaire des crises partielles sous dose stable (12/14 semaines) a été rapportée chez 27,7 %, 31,6 % et 41,3 %, respectivement, des patients traités par le lévétiracétam à 1 000, 2 000 ou 3 000 mg par jour et chez 12,6 % des témoins sous placebo.
Population pédiatrique
Chez les enfants (âgés de 4 à 16 ans), l’efficacité du lévétiracétam a été établie lors d’une étude en double insu contrôlée par placebo portant sur 198 patients traités pendant 14 semaines. Dans cette étude, les patients ont reçu le lévétiracétam à une dose fixe de 60 mg/kg/jour (en 2 prises fractionnées).
Une réduction de 50 % ou plus par rapport au départ de la fréquence hebdomadaire des crises partielles a été rapportée chez 44,6 % des patients sous lévétiracétam et 19,6 % des témoins sous placebo. Avec une poursuite du traitement à long terme, 11,4 % et 7,2 % des patients ont été exempts de crises pendant au moins 6 mois et au moins un an, respectivement.
Parmi les 35 nourrissons de moins de 1 an présentant des crises partielles exposés lors des études cliniques contrôlées par placebo, 13 seulement étaient âgés de <6 mois.
Monothérapie dans le traitement des crises partielles avec ou sans généralisation secondaire chez les adultes et les adolescents de 16 ans ou plus présentant une épilepsie nouvellement diagnostiquée :
L’efficacité du lévétiracétam en monothérapie a été établie lors d’un essai de non infériorité comparatif contre la carbamazépine à libération prolongée (LP) mené en double insu sur des groupes parallèles chez 576 patients de 16 ans ou plus ayant une épilepsie nouvellement ou récemment diagnostiquée. Seuls des patients présentant des crises partielles non provoquées ou des crises tonico-cloniques généralisées ont été admis. Les patients ont été randomisés pour recevoir la carbamazépine LP à 400-1 200 mg/jour ou le lévétiracétam à 1 000-3 000 mg/jour, selon la réponse thérapeutique, pendant jusqu’à 121 semaines.
Une période sans crises d’une durée de six mois a été rapportée chez 73,0 % des patients sous lévétiracétam et 72,8 % de ceux sous carbamazépine LP ; la différence absolue ajustée entre les traitements a été de 0,2 % (IC à 95 % : 7,8 à 8,2). Plus de la moitié des sujets sont demeurés exempts de crises pendant 12 mois (56,6 % et 58,5 % des patients sous lévétiracétam et sous carbamazépine LP, respectivement).
Dans une étude reflétant la pratique clinique, un arrêt du traitement antiépileptique concomitant a été possible chez un nombre limité de patients qui avaient répondu au lévétiracétam en thérapie adjuvante (36/69 patients adultes).
Thérapie adjuvante dans le traitement des crises myocloniques chez les adultes et les adolescents de 12 ans ou plus présentant une épilepsie myoclonique juvénile :
L’efficacité du lévétiracétam a été établie lors d’une étude en double insu contrôlée par placebo menée sur 16 semaines chez des patients de 12 ans et plus présentant une épilepsie généralisée idiopathique avec crises myocloniques dans différents syndromes. La majorité des patients souffraient d’épilepsie myoclonique juvénile.
Dans cette étude, le lévétiracétam a été administré à la posologie de 3 000 mg/jour en deux prises fractionnées.
Une réduction de 50 % ou plus par rapport au départ de la fréquence hebdomadaire des crises myocloniques a été rapportée chez 58,3 % des patients sous lévétiracétam et 23,3 % des témoins sous placebo. Avec une poursuite du traitement à long terme, 28,6 % et 21,0 % des patients ont été exempts de crises myocloniques pendant au moins 6 mois et au moins 1 an, respectivement.
Thérapie adjuvante dans le traitement des crises tonico-cloniques généralisées primaires chez les adultes et les adolescents de 12 ans ou plus présentant une épilepsie généralisée idiopathique.
L’efficacité du lévétiracétam a été établie lors d’une étude en double insu contrôlée par placebo menée sur 24 semaines chez des adultes, des adolescents et un nombre limité d’enfants présentant une épilepsie généralisée idiopathique avec crises tonico-cloniques généralisées primaires (TCGP) dans différents syndromes épileptiques (épilepsie myoclonique juvénile, épilepsie-absences de l’adolescence, épilepsie-absences de l’enfance ou épilepsie à crises grand mal du réveil). Dans cette étude, le lévétiracétam a été administré en deux prises fractionnées à la dose de 3 000 mg/jour chez les adultes et les adolescents et de 60 mg/kg/jour chez les enfants.
Une réduction de 50 % ou plus par rapport au départ de la fréquence hebdomadaire des crises TCGP t a été rapportée chez 72,2 % des patients sous lévétiracétam et 45,2 % des témoins sous placebo. Avec une poursuite du traitement à long terme, 47,4 % et 31,5 % des patients ont été exempts de crises tonico-cloniques pendant au moins 6 mois et au moins 1 an, respectivement.
5.2. Propriétés pharmacocinétiques
Une étude a évalué des doses allant jusqu’à 4 000 mg diluées dans 100 ml de chlorure de sodium à 0,9 % administrées en perfusion intraveineuse sur 15 minutes et des doses allant jusqu’à 2 500 mg diluées dans 100 ml de chlorure de sodium à 0,9 % administrées en perfusion intraveineuse sur 5 minutes. Les profils de pharmacocinétique et de tolérance n’ont mis en évidence aucun problème de sécurité.
Le lévétiracétam est un composé hautement soluble et perméable. Le profil pharmacocinétique est linéaire et la variabilité intra- et inter-individuelle est faible. La clairance n’est pas modifiée après des administrations répétées. Le lévétiracétam a un profil pharmacocinétique indépendant du temps, une observation qui a également été confirmée après une administration à la posologie de 1 500 mg deux fois par jour en perfusion intraveineuse pendant 4 jours.
Rien n’indique qu’une variabilité pertinente liée au sexe, à la race ou au cycle circadien existe. Le profil pharmacocinétique s’est révélé comparable chez les volontaires sains et les patients épileptiques.
ADULTES ET ADOLESCENTS
Distribution
Le pic des concentrations plasmatiques (Cmax) mesuré chez 17 sujets qui avaient reçu une dose unique de 1 500 mg en perfusion intraveineuse sur 15 minutes a atteint 51 ± 19 μg/ml (moyenne arithmétique ± écart type).
Aucune donnée sur la distribution tissulaire n’est disponible chez l’homme.
Les taux de liaison aux protéines plasmatiques du lévétiracétam et de son principal métabolite ne sont pas significatifs (<10 %). Le volume de distribution du lévétiracétam est de l’ordre de 0,5 à 0,7 l/kg, une valeur qui est proche du volume total d’eau corporelle
Le lévétiracétam n’est pas métabolisé de façon importante chez l’homme. La principale voie métabolique (24 % de la dose) passe par une hydrolyse enzymatique du groupement acétamide. La production du principal métabolite, l’ucb L057, ne fait pas intervenir les isoformes du cytochrome P450 hépatique. Une hydrolyse mesurable du groupement acétamide a été détectée dans un grand nombre de tissus, y compris les cellules sanguines. L’ucb L057 est inactif sur le plan pharmacologique.
Deux métabolites mineurs ont également été identifiés, dont un obtenu par hydroxylation du cycle pyrrolidone (1,6 % de la dose) et l’autre par ouverture du cycle pyrrolidone (0,9 % de la dose). Les autres dérivés, qui n’ont pas été identifiés, représentent 0,6 % seulement de la dose.
Aucune interconversion des énantiomères du lévétiracétam ou de son principal métabolite n’a été mise en évidence in vivo.
Des études in vitro ont montré que le lévétiracétam et son principal métabolite n’inhibent pas l’activité des isoformes majeures du cytochrome P450 hépatique (CYP3A4, 2A6, 2C9, 2C19, 2D6, 2E1 et 1A2), de la glucuronyl-transférase (UGT1A1 et UGT1A6) et de l’époxyde hydroxylase. Par ailleurs, le lévétiracétam n’affecte pas la glucuronidation de l’acide valproïque in vitro.
Dans des hépatocytes humains en culture, les effets du lévétiracétam sur le CYP1A2, la SULTIE1 ou l’UGTIA1 ont été limités ou nuls. Le lévétiracétam a entraîné une légère induction du CYP2B6 et du CYP3A4. À la lumière des données in vitro et des données d’interaction in vivo avec les contraceptifs oraux, la digoxine et la warfarine, aucune induction enzymatique significative ne devrait être observée in vivo. Une interaction du lévétiracétam sur d’autres substances, ou vice versa, est donc peu probable.
Élimination
La demi-vie plasmatique est de 7 ± 1 heures chez l’adulte et elle ne varie pas avec la dose, la voie d’administration ou la répétition des administrations. La clairance corporelle totale moyenne est de 0,96 ml/min/kg.
La principale voie d’élimination est urinaire, représentant en moyenne 95 % de la dose (93 % environ de la dose étant excrétée dans les 48 heures qui suivent l’administration). L’élimination par voie fécale représente 0,3 % seulement de la dose. L’élimination urinaire cumulative du lévétiracétam et de son principal métabolite représente 66 % et 24 % de la dose, respectivement, pendant les 48 premières heures.
La clairance rénale du lévétiracétam et de l’ucb L057 est de 0,6 et 4,2 ml/min/kg, respectivement, ce qui indique que le lévétiracétam est éliminé par filtration glomérulaire puis réabsorption tubulaire ; le principal métabolite est également éliminé par sécrétion tubulaire active et filtration glomérulaire. Une corrélation existe entre l’élimination du lévétiracétam et la clairance de la créatinine.
Patients âgés
La demi-vie est prolongée de 40 % environ (10 à 11 heures) chez les patients âgés en raison de la diminution de la fonction rénale dans cette population (voir rubrique section 4.2).
Insuffisance rénale
Une corrélation existe entre la clairance corporelle apparente du lévétiracétam et de son principal métabolite et la clairance de la créatinine. Il est donc recommandé d’adapter la dose d’entretien quotidienne du lévétiracétam en fonction de la clairance de la créatinine chez les patients présentant une insuffisance rénale modérée à sévère (voir rubrique 4.2).
Chez des adultes présentant une insuffisance rénale anurique au stade terminal, la demi-vie a été d’environ 25 et 3,1 heures entre et pendant les séances de dialyse, respectivement. L’excrétion fractionnelle de lévétiracétam a atteint 51 % au cours d’une séance de dialyse classique de 4 heures.
Insuffisance hépatique
Aucune modification pertinente de la clairance du lévétiracétam n’a été signalée chez des sujets présentant une insuffisance hépatique légère à modérée. Chez la plupart de ceux souffrant d’une insuffisance hépatique sévère, la clairance du lévétiracétam a été réduite de plus de 50 % en raison d’une insuffisance rénale concomitante (voir rubrique 4.2)
Population pédiatrique
Enfants (4 à 12 ans)
La pharmacocinétique après administration intraveineuse n’a pas été étudiée dans la population pédiatrique. Toutefois, sur la base des caractéristiques pharmacocinétiques du lévétiracétam, de la pharmacocinétique après administration intraveineuse chez l’adulte et de la pharmacocinétique après administration orale chez l’enfant, l’aire sous la courbe (AUC) devrait être similaire après une administration intraveineuse ou orale chez les enfants de 4 à 12 ans.
La demi-vie du lévétiracétam a été de 6,0 heures après l’administration d’une dose orale unique (20 mg/kg) à des enfants épileptiques (âgés de 6 à 12 ans). La clairance corporelle apparente ajustée en fonction du poids a été environ 30 % plus élevée que chez des adultes épileptiques.
Le lévétiracétam a été rapidement absorbé après l’administration répétée d’une dose orale (20 à 60 mg/kg/jour) à des enfants épileptiques (âgés de 4 à 12 ans). Le pic des concentrations plasmatiques a été atteint 0,5 à 1 heure après l’administration. Des augmentations linéaires et proportionnelles à la dose ont été mises en évidence pour le pic des concentrations plasmatiques et l’aire sous la courbe. La demi-vie d'élimination a été d’environ 5 heures et la clairance corporelle apparente de 1,1 ml/min/kg.
5.3. Données de sécurité préclinique
Les effets indésirables suivants n’ont pas été observés dans les études cliniques mais ont été signalés chez le rat et, à un degré moindre, la souris à des niveaux d’exposition semblables à ceux utilisés chez l’homme, et ils pourraient avoir une pertinence : modifications hépatiques indicatrices d’une réponse adaptative telles qu’un gain de poids, une hypertrophie centrolobulaire, une infiltration adipeuse et une élévation de l’activité plasmatique des enzymes hépatiques.
Aucun effet indésirable n’a été rapporté sur la fertilité chez le rat mâle ou femelle ni sur les performances de reproduction chez le rat après l’administration de doses allant jusqu’à 1 800 mg/kg/jour [6 x la dose maximale recommandée en clinique (MRHD, pour maximum recommended human dose) rapportée à la surface corporelle en mg/m2 ou à l’exposition] aux parents et à la génération F1.
Deux études sur le développement embryonnaire et fœtal (DEF) ont été effectuées chez le rat traité à des doses de 400, 1 200 et 3 600 mg/kg/jour. Dans une seulement des 2 études sur le DEF, une légère diminution du poids fœtal associée à une augmentation marginale des variations/anomalies mineures du squelette a été rapportée à la dose de 3 600 mg/kg/jour. Aucun effet sur la mortalité embryonnaire et ni augmentation de l’incidence de malformations n’ont été signalés. La dose sans effet nocif observé (NOAEL) a été de 3 600 mg/kg/jour chez la rate gravide (12 x la MRHD rapportée à la surface corporelle en mg/m2) et de 1 200 mg/kg/jour chez les fœtus.
Quatre études sur le développement embryonnaire et fœtal ont été effectuées chez le lapin couvrant les doses de 200, 600, 800, 1 200 et 1 800 mg/kg/jour. Le niveau de dose de 1 800 mg/kg/jour a induit une toxicité maternelle marquée et une diminution du poids fœtal associées à une incidence accrue d’anomalies cardiovasculaires/squelettiques chez les fœtus. La NOAEL a été <200 mg/kg/jour chez les mères et de 200 mg/kg/jour chez les fœtus (égale à la MRHD en mg/m2).
Une étude du développement périnatal et postnatal a été effectuée chez le rat à des doses de lévétiracétam de 70, 350 et 1 800 mg/kg/jour. La NOAEL a été ³ 1 800 mg/kg/jour chez les femelles de la génération F0 et également chez les descendants F1 en ce qui concerne la survie, la croissance et le développement jusqu’au sevrage (6 x la MRHD en mg/m2).
Des études menées chez des rats et des chiens nouveau-nés et juvéniles n’ont mis en évidence aucun effet indésirable sur les critères conventionnels d’évaluation du développement ou de la maturation à des doses allant jusqu’à 1 800 mg/kg/jour (6 17 x la MRHD en mg/m2).
Avant la première ouverture du flacon : 3 ans.
La stabilité physico-chimique en cours d’utilisation a été démontrée pendant 24 heures à 25 °C ± 2 °C (à température ambiante) dans des flacons en polypropylène et dans des poches en PVC.
Du point de vue microbiologique, le produit doit être utilisé immédiatement après dilution. S’il n’est pas utilisé immédiatement, les durées et conditions de conservation après dilution et avant utilisation relèvent de la seule responsabilité de l’utilisateur et ne devraient pas dépasser 24 heures à une température comprise entre 2 °C et 8 °C, sauf si la dilution a été effectuée dans des conditions d’asepsie dûment contrôlées et validées.
6.4. Précautions particulières de conservation
Ce médicament ne nécessite pas de précautions particulières de conservation.
Pour les conditions de conservation du médicament après dilution, voir la rubrique 6.3.
6.5. Nature et contenu de l'emballage extérieur
Flacon de 5 ml en verre de type I fermé par un bouchon en caoutchouc bromobutyle serti par une capsule en aluminium de type « flip-off ».
Les flacons contiennent 5 ml de solution à diluer pour perfusion et sont conditionnés dans une boîte cartonnée.
Chaque boîte contient 1 ou 10 flacons.
Toutes les présentations peuvent ne pas être commercialisées.
6.6. Précautions particulières d’élimination et de manipulation
Le Tableau 1 fournit des recommandations pour la préparation du lévétiracétam en solution à diluer pour perfusion à une dose quotidienne totale de 500 mg, 1 000 mg, 2 000 mg ou 3 000 mg et son administration en deux prises fractionnées.
Tableau 1. Préparation et administration du lévétiracétam en solution à diluer pour perfusion
|
Volume à prélever |
Volume de diluant |
Durée de la perfusion |
Fréquence des administrations |
Dose quotidienne totale |
|
|
250 mg |
2,5 ml (la moitié d’un flacon de 5 ml) |
100 ml |
15 minutes |
Deux fois par jour |
500 mg/jour |
|
500 mg |
5 ml (un flacon de 5 ml) |
100 ml |
15 minutes |
Deux fois par jour |
1 000 mg/jour |
|
1 000 mg |
10 ml (deux flacons de 5 ml) |
100 ml |
15 minutes |
Deux fois par jour |
2 000 mg/jour |
|
1 500 mg |
15 ml (trois flacons de 5 ml) |
100 ml |
15 minutes |
Deux fois par jour |
3 000 mg/jour |
Ce médicament est à usage unique ; toute solution non utilisée doit être jetée.
La solution à diluer pour perfusion de lévétiracetam est compatible physiquement et chimiquement stable pendant 24 heures, stockée dans des sacs en PVC à une température contrôlée de 15°Cà 25°C quand elle est mélangée avec les solvants suivants :
· Sérum physiologique 9 mg/ml (0,9 %) solution pour injection
· Lactate de Ringer solution pour injection
· Dextrose 50 mg/ml (5 %) solution pour injection.
Ne pas utiliser le médicament si la solution présente des particules en suspension ou une coloration anormale.
Tout médicament non utilisé ou déchet doit être éliminé conformément à la réglementation en vigueur.
7. TITULAIRE DE L’AUTORISATION DE MISE SUR LE MARCHE
MITTELSTRAßE 5 / 5A
12529 SCHÖNEFELD
ALLEMAGNE
8. NUMERO(S) D’AUTORISATION DE MISE SUR LE MARCHE
· 34009 301 885 7 6 : 5 mL en Flacon (verre). Boîte de 1.
· 34009 301 885 8 3 : 5 mL en Flacon (verre). Boîte de 10.
9. DATE DE PREMIERE AUTORISATION/DE RENOUVELLEMENT DE L’AUTORISATION
[à compléter ultérieurement par le titulaire]
10. DATE DE MISE A JOUR DU TEXTE
[à compléter ultérieurement par le titulaire]
Sans objet
12. INSTRUCTIONS POUR LA PREPARATION DES RADIOPHARMACEUTIQUES
Liste I
ANSM - Mis à jour le : 02/06/2026
LEVETIRACETAM TILLOMED 100 mg/ml, solution à diluer pour perfusion
Lévétiracétam
· Gardez cette notice. Vous pourriez avoir besoin de la relire.
· Si vous avez d’autres questions, interrogez votre médecin, votre pharmacien ou votre infirmier/ère.
· Si vous ressentez un quelconque effet indésirable, parlez-en à votre médecin ou votre pharmacien. Ceci s’applique aussi à tout effet indésirable qui ne serait pas mentionné dans cette notice. Voir rubrique 4.
1. Qu'est-ce que LEVETIRACETAM TILLOMED 100 mg/ml, solution à diluer pour perfusion et dans quels cas est-il utilisé ?
2. Quelles sont les informations à connaître avant de recevoir LEVETIRACETAM TILLOMED 100 mg/ml, solution à diluer pour perfusion ?
3. Comment LEVETIRACETAM TILLOMED 100 mg/ml, solution à diluer pour perfusion est-il administré ?
4. Quels sont les effets indésirables éventuels ?
5. Comment conserver LEVETIRACETAM TILLOMED 100 mg/ml, solution à diluer pour perfusion ?
6. Contenu de l’emballage et autres informations.
1. QU’EST-CE QUE LEVETIRACETAM TILLOMED 100 mg/ml, solution à diluer pour perfusion ET DANS QUELS CAS EST-IL UTILISE ?
Classe pharmacothérapeutique : antiépileptiques, autres antiépileptiques, Code ATC : N03AX14.
Le lévétiracétam est un médicament antiépileptique (médicament utilisé pour traiter les crises d’épilepsie).
LEVETIRACETAM TILLOMED 100 mg/ml, solution à diluer pour perfusion est utilisé :
· seul, chez les adultes et les adolescents de 16 ans ou plus présentant une épilepsie nouvellement diagnostiquée, pour traiter une certaine forme d’épilepsie. L’épilepsie est une maladie qui produit des crises à répétition chez les patients (crises d'épilepsie). Le lévétiracétam est utilisé pour traiter la forme d’épilepsie dans laquelle les crises touchent initialement un seul côté du cerveau, mais qui pourraient par la suite s'étendre à des zones plus larges des deux côtés du cerveau (crises partielles avec ou sans généralisation secondaire). Le lévétiracétam vous a été prescrit par votre médecin afin de réduire le nombre de crises
· en complément à d’autres médicaments antiépileptiques pour traiter :
o les crises d’épilepsie partielles, avec ou sans généralisation, chez l’adulte, l’adolescent et l’enfant de 4 ans ou plus ;
o les crises myocloniques (mouvements brefs et saccadés d’un muscle ou d’un groupe de muscles) chez l’adulte et l’adolescent de 12 ans ou plus présentant une épilepsie myoclonique juvénile ;
o les crises tonico-cloniques généralisées primaires (crises généralisées avec perte de connaissance) chez l’adulte et l’adolescent de 12 ans ou plus présentant une épilepsie généralisée idiopathique (un type d’épilepsie que l’on suppose attribué à une cause génétique).
LEVETIRACETAM TILLOMED 100 mg/ml, solution à diluer pour perfusion en solution à diluer pour perfusion représente une forme de traitement alternative chez les patients pour lesquels un traitement par voie orale est temporairement impossible.
2. QUELLES SONT LES INFORMATIONS A CONNAITRE AVANT D’UTILISER LEVETIRACETAM TILLOMED 100 mg/ml, solution à diluer pour perfusion ?
N’utilisez jamais LEVETIRACETAM TILLOMED 100 mg/ml, solution à diluer pour perfusion :
· si vous êtes allergique au lévétiracétam, aux dérivés de la pyrrolidone ou à l’un des autres composants contenus dans ce médicament (mentionnés dans la rubrique 6).
Avertissements et précautions
Adressez-vous à votre médecin avant de recevoir LEVETIRACETAM TILLOMED 100 mg/ml, solution à diluer pour perfusion
· Si vous souffrez de problèmes de reins. Suivez les instructions de votre médecin, qui décidera d’adapter ou non votre dose.
· Si vous remarquez tout ralentissement de la croissance ou tout développement pubertaire inattendu chez votre enfant, veuillez consulter votre médecin.
· Un faible nombre de personnes traitées par des antiépileptiques, tels que LEVETIRACETAM TILLOMED 100 mg/ml, solution à diluer pour perfusion, ont développé des idées autodestructrices ou suicidaires. Si vous présentez des symptômes de dépression et/ou des idées suicidaires, veuillez consulter votre médecin.
Si vous avez des antécédents familiaux ou médicaux de rythme cardiaque irrégulier (visible sur un électrocardiogramme), ou si vous avez une maladie et / ou suivez un traitement qui vous rend sujettes à des irrégularités du rythme cardiaque ou à des déséquilibres salins.
Informez votre médecin ou votre pharmacien si l’un des effets secondaires suivants devient grave ou persiste après quelques jours :
· Pensées anormales, irritabilité ou agressivité exacerbée, ou si votre famille, vos amis ou vous remarquez des troubles importants de l’humeur ou du comportement.
· Aggravation de l’épilepsie
Dans de rares cas, vos crises convulsives peuvent s’aggraver ou se produire plus souvent, principalement pendant le premier mois suivant l’instauration du traitement ou l’augmentation de la dose Dans une forme très rare d’épilepsie à début précoce (épilepsie associée à des mutations du SCN8A) qui provoque plusieurs types de crises et une perte d’aptitudes, vous pourriez remarquer que les crises perdurent ou s’aggravent pendant votre traitement.
Enfants et adolescents
LEVETIRACETAM TILLOMED 100 mg/ml, solution à diluer pour perfusion n’est pas indiqué chez les enfants et les adolescents de moins de 16 ans lorsqu’il est administré seul (en monothérapie).
Autres médicaments et LEVETIRACETAM TILLOMED 100 mg/ml, solution à diluer pour perfusion
Informez votre médecin ou pharmacien si vous prenez, avez récemment pris ou pourriez prendre tout autre médicament, y compris des médicaments obtenus sans ordonnance.
Ne prenez pas de macrogol (un médicament utilisé comme laxatif) pendant une période d’une heure avant et d’une heure après la prise de lévétiracétam, car cela pourrait réduire son effet.
Si vous êtes enceinte ou que vous allaitez, si vous pensez être enceinte ou si vous envisagez d’avoir un enfant, demandez conseil à votre médecin avant de prendre ce médicament.
Grossesse
Le lévétiracétam ne peut être utilisé pendant la grossesse, uniquement si votre médecin le juge nécessaire après une évaluation minutieuse.
Vous ne devez pas arrêter votre traitement sans en avoir discuté avec votre médecin.
Un risque de malformations congénitales pour votre enfant à naître ne peut être totalement exclu. Deux études ne suggèrent pas un risque accru d’autisme ou d’handicap intellectuel chez les enfants nés de mères traitées par le lévétiracétam pendant la grossesse. Cependant, les données disponibles sur l’effet du lévétiracétam sur le neurodéveloppement des enfants sont limitées.
Allaitement
L’allaitement n'est pas recommandé durant le traitement.
Conduite de véhicules et utilisation de machines
LEVETIRACETAM TILLOMED 100 mg/ml, solution à diluer pour perfusion peut altérer votre capacité à conduire ou à utiliser des outils ou des machines, car ce médicament peut provoquer une somnolence. Cet effet est plus susceptible de se produire au début du traitement ou après une augmentation de la dose. Vous ne devez ni conduire, ni utiliser des machines jusqu’à ce qu’il soit établi que vos capacités à effectuer ces activités ne sont pas affectées.
LEVETIRACETAM TILLOMED 100 mg/ml, solution à diluer pour perfusion contient du sodium
Ce médicament contient 19,1 mg de sodium (composant principal du sel de cuisine/sel de table) dans chaque flacon (5 ml). Cette quantité équivaut à 0,796 % de l’apport nutritionnel en sodium maximal quotidien recommandé pour un adulte.
3. COMMENT UTILISER LEVETIRACETAM TILLOMED 100 mg/ml, solution à diluer pour perfusion ?
LEVETIRACETAM TILLOMED 100 mg/ml, solution à diluer pour perfusion doit être administré deux fois par jour, une fois le matin et une fois le soir, environ aux mêmes heures chaque jour.
La formulation intraveineuse est une alternative à la formulation orale. Il est possible de passer d’une formulation orale à la formulation intraveineuse, ou vice-versa, sans qu’une adaptation de la dose ne soit nécessaire. La dose quotidienne totale et la fréquence d’administration restent identiques.
Traitement en association et monothérapie (à partir de l’âge de 16 ans)
Adulte (≥ 18 ans) et adolescent (12 à 17 ans) pesant 50 kg ou plus
Posologie recommandée : comprise entre 1 000 mg et 3 000 mg par jour.
Quand vous allez prendre LEVETIRACETAM TILLOMED 100 mg/ml, solution à diluer pour perfusion pour la première fois, votre médecin vous prescrira une dose plus faible que la dose recommandée pendant 2 semaines, ensuite vous prendrez la dose quotidienne efficace la plus petite.
Posologie chez l’enfant (4 à 11 ans) et l’adolescent (12 à 17 ans) pesant moins de 50 kg
Posologie recommandée : entre 20 mg par kg de poids corporel et 60 mg par kg de poids corporel par jour.
Mode et voie d’administration
LEVETIRACETAM TILLOMED 100 mg/ml, solution à diluer pour perfusion est destiné à un usage intraveineux.
La dose recommandée doit être diluée dans au moins 100 ml d’un diluant compatible et perfusée sur une période de 15 minutes. Pour les médecins et le personnel infirmier, des instructions plus détaillées concernant l’utilisation correcte de LEVETIRACETAM TILLOMED 100 mg/ml, solution à diluer pour perfusion sont fournies à la rubrique 6.
Durée du traitement
On ne dispose d’aucune expérience sur l’administration du lévétiracétam par voie intraveineuse pendant une période de plus de 4 jours.
Si vous arrêtez d’utiliser LEVETIRACETAM TILLOMED 100 mg/ml, solution à diluer pour perfusion
En cas d’arrêt de traitement, comme avec d’autres médicaments antiépileptiques, LEVETIRACETAM TILLOMED 100 mg/ml, solution à diluer pour perfusion doit être arrêté progressivement afin d’éviter toute augmentation des crises convulsives. Si votre médecin décide d’arrêter votre traitement par LEVETIRACETAM TILLOMED 100 mg/ml, solution à diluer pour perfusion, il vous fournira des explications sur le processus d’arrêt progressif du médicament.
Si vous avez d’autres questions sur l’utilisation de ce médicament, demandez plus d’informations à votre médecin ou à votre pharmacien.
4. QUELS SONT LES EFFETS INDESIRABLES EVENTUELS ?
Prévenez immédiatement votre médecin ou rendez-vous au service d’urgences de l’hôpital le plus proche si vous présentez les symptômes suivants :
· faiblesse, sensation de tête légère ou d’étourdissement ou difficultés à respirer, car il pourrait s’agir de signes d’une réaction allergique grave (réaction anaphylactique)
· gonflement du visage, des lèvres, de la langue et de la gorge (œdème de Quincke)
· symptômes pseudo-grippaux et éruption cutanée sur le visage suivie d’une éruption cutanée étendue avec une température élevée, une augmentation des taux d’enzymes hépatiques observée dans les test sanguins et une augmentation d’un type de globules blancs (éosinophilie), un gonflement des ganglions lymphatiques et l’atteinte d’autres systèmes d’organes (syndrome d’hypersensibilité médicamenteuse avec éosinophilie et symptômes systémiques [DRESS])
· symptômes tels que faible volume d’urine, fatigue, nausées, vomissements, confusion et gonflement des jambes, des chevilles ou des pieds, car il pourrait s’agir de signes d’une brusque diminution de la fonction rénale ;
· éruption cutanée pouvant former des vésicules et prendre l’aspect de petites cibles (des taches centrales foncées entourées d’une zone plus pâle, avec un cercle sombre autour du bord) (érythème polymorphe) ;
· éruption cutanée généralisée s’accompagnant de vésicules et d’une desquamation de la peau, particulièrement autour de la bouche, du nez, des yeux et des organes génitaux (syndrome de Stevens-Johnson) ;
· forme plus grave d'éruption cutanée entraînant une desquamation de la peau sur plus de 30 % de la surface corporelle (syndrome de Lyell) ;
· signes de modifications mentales graves ou si quelqu’un de votre entourage remarque des signes de confusion, de somnolence (envie de dormir), d'amnésie (perte de mémoire), de troubles de la mémoire (oublis), de comportement anormal ou d’'autres signes neurologiques, notamment des mouvements involontaires ou incontrôlés. Il pourrait s’agir de symptômes d’une encéphalopathie.
Les effets indésirables suivants sont ceux qui ont été les plus fréquemment rapportés : rhinopharyngite, somnolence, maux de tête, fatigue et étourdissement. Au début du traitement ou lors d’une augmentation de la dose, les effets indésirables tels que somnolence, fatigue et étourdissement peuvent être plus fréquents. Toutefois, ces effets devraient s’atténuer avec le temps.
Très fréquents : susceptibles d’affecter plus d’1 personne sur 10
· rhinopharyngite
· somnolence, maux de tête.
Fréquents : susceptibles d’affecter jusqu’à 1 personne sur 10
· Anorexie (perte de l’appétit)
· dépression, hostilité ou agressivité, anxiété, insomnie, nervosité ou irritabilité
· convulsions, trouble de l’équilibre, étourdissement (sensation d’instabilité), léthargie (manque d’énergie et d’enthousiasme), tremblements (tremblements involontaires)
· vertige (sensation de tête qui tourne)
· toux
· douleur abdominale, diarrhée, dyspepsie (indigestion), vomissements, nausées
· éruption cutanée
· asthénie/fatigue.
Peu fréquents : susceptibles d’affecter jusqu’à 1 personne sur 100
· diminution du nombre de plaquettes sanguines, diminution du nombre de globules blancs
· perte de poids, gain de poids
· tentative de suicide et idées suicidaires, trouble mental, comportement anormal, hallucinations, colère, confusion, crise de panique, instabilité émotionnelle/sautes d’humeur, agitation
· amnésie (perte de mémoire), troubles de la mémoire (oublis), troubles de la coordination/ataxie (altération de la coordination des mouvements), paresthésie (picotements), troubles de l’attention (diminution de la concentration)
· diplopie (vision double), vision trouble
· valeurs élevées ou anormales à un test de la fonction hépatique
· chute des cheveux, eczéma, prurit (démangeaisons)
· faiblesse musculaire, myalgies (douleurs musculaires)
· lésions.
Rares : susceptibles d’affecter jusqu’à 1 personne sur 1 000
· infection ;
· diminution du nombre de tous les types de cellules sanguines
· réactions allergiques sévères (syndrome DRESS, réaction anaphylactique [réaction allergique sévère et importante], œdème de Quincke [gonflement du visage, des lèvres, de la langue et de la gorge])
· diminution de la concentration de sodium dans le sang
· suicide, troubles de la personnalité (troubles du comportement), pensées anormales (lenteur de la pensée, incapacité à se concentrer), délire.
· encéphalopathie (voir sous-rubrique « Prévenez immédiatement votre médecin » pour une description détaillée des symptômes)
· aggravation de l’épilepsie ou augmentation de la fréquence des crises convulsives
· spasmes musculaires incontrôlables affectant la tête, le torse et les membres, difficultés à contrôler les mouvements, hyperkinésie (hyperactivité)
·Modification du rythme cardiaque (électrocardiogramme)
· pancréatite
· insuffisance hépatique, hépatite
· brusque diminution de la fonction rénale
· éruption cutanée pouvant former des vésicules et prendre l’aspect de petites cibles (des taches centrales foncées entourées d’une zone plus pâle, avec un cercle sombre autour du bord) (érythème polymorphe), éruption cutanée généralisée s’accompagnant de vésicules et d’une desquamation, particulièrement autour de la bouche, du nez, des yeux et des organes génitaux (syndrome de Stevens Johnson), ainsi qu’une forme plus grave d’éruption corporelle entraînant une desquamation de la peau sur plus de 30 % de la surface corporelle (syndrome de Lyell)
· rhabdomyolyse (destruction des cellules musculaires) et augmentation associée des taux sanguins de créatine phosphokinase. La prévalence est significativement plus élevée chez les patients japonais que chez les patients non japonais
· claudication (boiterie) ou difficultés à marcher.
· association des symptômes de fièvre, raideur musculaire, tension artérielle et fréquence cardiaque instables, confusion, faible niveau de conscience (signes possibles d’un trouble appelé syndrome malin des neuroleptiques). La prévalence est significativement plus élevée chez les patients japonais par rapport aux patients non japonais.
Très rares : pouvant survenir au maximum chez 1 patient sur 10 000
· pensées ou sensations répétées et involontaires ou besoin pressant de faire quelque chose encore et encore (trouble obsessionnel compulsif)
Déclaration des effets secondaires
Si vous ressentez un quelconque effet indésirable, parlez-en à votre médecin ou à votre pharmacien ou à votre infirmier/ère. Ceci s’applique aussi à tout effet indésirable qui ne serait pas mentionné dans cette notice. Vous pouvez également déclarer les effets indésirables directement via le système national de déclaration : Agence nationale de sécurité du médicament et des produits de santé (ANSM) et réseau des Centres Régionaux de Pharmacovigilance - Site internet: https://signalement.social-sante.gouv.fr
En signalant les effets indésirables, vous contribuez à fournir davantage d’informations sur la sécurité du médicament.
5. COMMENT CONSERVER LEVETIRACETAM TILLOMED 100 mg/ml, solution à diluer pour perfusion ?
Tenir ce médicament hors de la vue et de la portée des enfants.
N’utilisez pas ce médicament après la date de péremption indiquée sur l’étiquette et la boîte après « EXP ». La date de péremption fait référence au dernier jour de ce mois.
Ce médicament ne nécessite pas de précautions particulières de conservation.
6. CONTENU DE L’EMBALLAGE ET AUTRES INFORMATIONS
Ce que contient LEVETIRACETAM TILLOMED 100 mg/ml, solution à diluer pour perfusion
· La substance active est :
Lévétiracétam................................................................................................................. 100 mg
Pour 1 ml.
Chaque flacon de 5 ml contient 500 mg de lévétiracétam.
· Les autres composants sont :
Chlorure de sodium, acétate de sodium trihydraté, acide acétique glacial, eau pour préparations injectables.
LEVETIRACETAM TILLOMED solution à diluer pour perfusion est un liquide limpide et incolore.
LEVETIRACETAM TILLOMED solution à diluer pour perfusion est conditionné en boîte de 1 ou 10 flacons de 5 ml.
Toutes les présentations peuvent ne pas être commercialisées.
Titulaire de l’autorisation de mise sur le marché
MITTELSTRAßE 5 / 5A
12529 SCHÖNEFELD
ALLEMAGNE
Exploitant de l’autorisation de mise sur le marché
MEDIPHA SANTE
LES FJORDS - IMMEUBLE OSLO
19 AVENUE DE NORVEGE
VILLEBON-SUR-YVETTE, 91140
MALTA LIFE SCIENCES PARK
LS2.01.06 INDUSTRIAL ESTATE
SAN GWANN, SGN 3000
MALTE
OU
ZENTIVA S.A.
BULEVARDUL PALLADY THEODOR NR. 50,
BUCHAREST, 032266
ROMANIA
Noms du médicament dans les Etats membres de l'Espace Economique Européen
Ce médicament est autorisé dans les Etats membres de l'Espace Economique Européen sous les noms suivants : Conformément à la réglementation en vigueur.
[à compléter ultérieurement par le titulaire]
La dernière date à laquelle cette notice a été révisée est :
[à compléter ultérieurement par le titulaire]
Des informations détaillées sur ce médicament sont disponibles sur le site Internet de l’ANSM (France).
Les informations suivantes sont destinées exclusivement aux professionnels de santé :
Des instructions concernant l’utilisation correcte de LEVETIRACETAM TILLOMED 100 mg/ml, solution à diluer pour perfusion sont fournies à la rubrique 3.
Chaque flacon de 5 ml contient 500 mg de lévétiracétam. Voir Tableau 1 pour les recommandations en matière de préparation et d’administration de LEVETIRACETAM TILLOMED 100 mg/ml, solution à diluer pour perfusion en vue d’atteindre une dose quotidienne totale de 500 mg, 1 000 mg, 2 000 mg ou 3 000 mg en deux doses fractionnées.
Tableau 1. Préparation et administration du lévétiracétam en solution à diluer pour perfusion
|
Dose |
Volume à prélever |
Volume de diluant |
Durée de la perfusion |
Fréquence des administrations |
Dose quotidienne totale |
|
250 mg |
2,5 ml (la moitié d’un flacon de 5 ml) |
100 ml |
15 minutes |
Deux fois par jour |
500 mg/jour |
|
500 mg |
5 ml (un flacon de 5 ml) |
100 ml |
15 minutes |
Deux fois par jour |
1 000 mg/jour |
|
1 000 mg |
10 ml (deux flacons de 5 ml) |
100 ml |
15 minutes |
Deux fois par jour |
2 000 mg/jour |
|
1 500 mg |
15 ml (trois flacons de 5 ml) |
100 ml |
15 minutes |
Deux fois par jour |
3 000 mg/jour |
Ce médicament est à un usage unique ; toute solution inutilisée doit être jetée.
Durée de conservation en cours d’utilisation :
Du point de vue microbiologique, le produit doit être utilisé immédiatement après dilution. S’il n’est pas utilisé immédiatement, les durées de conservation après dilution et avant utilisation relèvent de la seule responsabilité de l’utilisateur et ne devraient pas dépasser 24 heures à une température comprise entre 2 °C et 8 °C, sauf si la dilution a été effectuée dans des conditions d’asepsie dûment contrôlées et validées.
LEVETIRACETAM TILLOMED 100 mg/ml, solution à diluer pour perfusion est physiquement compatible avec les solvants suivants et chimiquement stables après mélange avec ces solvants pour au moins 24 heures dans des poches PVC à température ambiantes 15-25°C :
· Solution de chlorure de sodium à 9 mg/ml (0,9 %) pour préparations injectables ;
· Solution de Ringer lactate pour préparations injectables ;
· Dextrose 50 mg/ml (5 %) pour préparations injectables.