Dernière mise à jour le 01/06/2026
ACLOTINE 100 UI/mL, poudre et solvant pour solution injectable
Indications thérapeutiques
Classe pharmacothérapeutique : Agents antithrombotiques, groupe héparine - code ATC : B01AB02
ACLOTINE est un médicament antithrombotique (anticoagulant). La substance active est l’antithrombine humaine. L’antithrombine est un composant naturel du plasma humain et joue un rôle important d’inhibiteur de la coagulation du sang.
ACLOTINE est indiqué pour traiter :
· Les déficits congénitaux (de naissance) en antithrombine :
o Prévention des thromboses veineuses et accidents thrombo-emboliques, en cas de situations cliniques à risques élevés (notamment lors d'une chirurgie ou d'une grossesse) lorsque le risque hémorragique ne permet pas d'utiliser des doses suffisantes d'héparine, si indiquée.
o Prévention de la progression d’une thrombose veineuse profonde et d’une thromboembolie en association avec de l’héparine, comme indiqué.
· Le déficit acquis sévère (< 60 %) en antithrombine.
Présentations
> 1 flacon(s) en verre de poudre - 1 flacon(s) en verre de solvant de 5 ml avec dispositif de transfert
Code CIP : 561 937-1 ou 34009 561 937 1 4
Déclaration de commercialisation : 22/02/1999
Cette présentation est agréée aux collectivités
Inscription sur la liste de rétrocession au titre de son AMM, selon les conditions précisées au Journal Officiel. Prix de cession publié au Journal Officiel.
> 1 flacon(s) en verre de poudre - 1 flacon(s) en verre de solvant de 10 ml avec dispositif de transfert
Code CIP : 561 938-8 ou 34009 561 938 8 2
Déclaration de commercialisation : 22/02/1999
Cette présentation est agréée aux collectivités
Inscription sur la liste de rétrocession au titre de son AMM, selon les conditions précisées au Journal Officiel. Prix de cession publié au Journal Officiel.
Service médical rendu (SMR)
Pas de SMR disponible pour ce médicament ( plus d'informations dans l'aide )
Amélioration du service médical rendu (ASMR)
Pas d'ASMR disponible pour ce médicament (plus d'informations dans l'aide )
Autres informations
- Titulaire de l'autorisation : LFB-BIOMEDICAMENTS
- Conditions de prescription et de délivrance :
- liste I
- prescription hospitalière
- Statut de l'autorisation : Valide
- Type de procédure : Procédure nationale
- Code CIS : 6 099 157 3
ANSM - Mis à jour le : 24/02/2026
ACLOTINE 100 UI/mL, poudre et solvant pour solution injectable
2. COMPOSITION QUALITATIVE ET QUANTITATIVE
Pour 1 mL de solution reconstituée
ACLOTINE se présente sous forme de poudre et de solvant pour solution injectable. Un flacon contient 500 UI ou 1000 UI d'antithrombine dérivée du plasma humain.
Le produit contient environ 100 UI/mL d'antithrombine dérivée du plasma humain lorsqu'il est reconstitué avec le solvant (eau pour préparations injectables) correspondant, c’est à dire 5 mL pour 500 UI ou 10 mL pour 1000 UI.
L'activité (UI) est déterminée par le test chromogénique de la Pharmacopée Européenne. L'activité spécifique est ≥ 3 UI/mg de protéines.
Produit à partir de plasma de donneurs humains.
Pour la liste complète des excipients, voir rubrique 6.1.
Poudre et solvant pour solution injectable.
4.1. Indications thérapeutiques
1. Déficits constitutionnels en antithrombine :
· Prévention des thromboses veineuses et accidents thrombo-emboliques, en cas de situations cliniques à risques élevés (notamment lors d'une chirurgie ou d'une grossesse) lorsque le risque hémorragique ne permet pas d'utiliser des doses suffisantes d'héparine, si indiquée.
· Prévention de la progression d’une thrombose veineuse profonde et d’une thromboembolie en association avec de l’héparine, comme indiqué.
2. Déficit acquis sévère (< 60 %) en antithrombine.
4.2. Posologie et mode d'administration
Posologie
En cas de déficit congénital, la posologie doit être individualisée pour chaque patient en tenant compte des antécédents familiaux en matière d'événements thromboemboliques, des facteurs de risque clinique réels et des examens de laboratoire.
La posologie et la durée du traitement de substitution dans les déficits acquis dépendent du taux plasmatique en antithrombine, de la présence de signes d'augmentation du renouvellement, de la maladie sous-jacente et de la sévérité de l'état clinique. La quantité à administrer et la fréquence d'administration doivent toujours être basées sur l'efficacité clinique et l'évaluation de laboratoire pour chaque individu.
Le nombre d'unités d'antithrombine administrées est exprimé en unités internationales (UI), standards actuels de l'OMS pour l'antithrombine. L'activité de l’antithrombine dans le plasma est exprimée soit en pourcentage (par rapport au plasma humain normal), soit en unités internationales (par rapport à la norme internationale pour l'antithrombine dans le plasma).
Une unité internationale (UI) d'antithrombine humaine est équivalente à la quantité d'antithrombine présente dans 1 mL de plasma humain normal.
Le calcul de la dose d'antithrombine nécessaire est basé sur l’observation empirique selon laquelle 1 unité internationale (UI) d'antithrombine par kg de poids corporel augmente l'activité de l’antithrombine plasmatique d'environ 2 %.
La dose initiale est déterminée à l'aide de la formule suivante :
Nombre d’unités à administrer = poids corporel (kg) x (niveau cible - activité réelle de l’antithrombine [%]) x 0,5.
L’activité initiale à atteindre de l’antithrombine dépend de la situation clinique. Lorsque l'indication de substitution de l'antithrombine est établie, la posologie doit être suffisante pour atteindre l’activité souhaitée de l’antithrombine et maintenir un niveau efficace. La posologie doit être déterminée et contrôlée sur la base des mesures de laboratoire de l'activité antithrombine. Celles-ci doivent être effectuées au moins deux fois par jour jusqu'à ce que le patient soit stabilisé, puis une fois par jour, de préférence immédiatement avant la perfusion suivante. La correction de la posologie doit tenir compte à la fois des signes d'augmentation du renouvellement de l'antithrombine selon les contrôles de laboratoire et de l'évolution clinique.
Un taux circulant d'antithrombine de 80 % doit être maintenu pendant toute la durée du traitement, sauf si les signes cliniques indiquent un niveau d'efficacité différent.
A titre indicatif, la posologie est :
· Dans le déficit constitutionnel :
o En traitement prophylactique : 30 à 50 UI/kg, lors d'une situation à risque thrombo‑embolique (grossesse, chirurgie). La posologie et le rythme d'injection sont adaptés à l'évolution clinique et biologique ;
o En traitement curatif : 40 à 50 UI/kg tous les jours ou tous les 2 jours chez l'adulte selon l'évolution clinique et biologique ;
· Dans le déficit acquis sévère :
o En traitement curatif : dose initiale de 40 à 50 UI/kg voire 100 UI/kg. Les doses ultérieures, la fréquence des injections et la durée du traitement seront adaptées à l'état clinique et au suivi biologique.
Mode d’administration
Précautions à prendre avant de manipuler ou d'administrer le médicament :
ACLOTINE se présente sous la forme d'une poudre à reconstituer extemporanément avec de l’eau pour préparations injectables. Pour les instructions concernant la reconstitution du médicament avant administration, voir la rubrique 6.6.
Injecter exclusivement par voie intraveineuse stricte, en une seule fois, immédiatement après reconstitution, sans dépasser un débit de 4 mL/minute.
Ne pas injecter de solution présentant un aspect non homogène ou contenant un dépôt.
Hypersensibilité à la substance active ou à l’un des excipients mentionnés à la rubrique 6.1.
4.4. Mises en garde spéciales et précautions d'emploi
En cas de réaction de type allergique ou anaphylactique, l'administration doit être interrompue immédiatement et les patients doivent contacter leur médecin.
En cas de choc anaphylactique, le traitement médical standard doit être administré.
Les mesures habituelles de prévention du risque de transmission d'agents infectieux par les médicaments préparés à partir de sang ou de plasma humain comprennent la sélection clinique des donneurs, la recherche des marqueurs spécifiques d'infection sur chaque don et sur les mélanges de plasma ainsi que la mise en œuvre dans le procédé de fabrication d'étapes efficaces pour l'inactivation/élimination virale.
Cependant, lorsque des médicaments préparés à partir de sang ou de plasma humain sont administrés, le risque de transmission d'agents infectieux ne peut pas être totalement exclu. Ceci s'applique également aux virus inconnus ou émergents ou autres types d'agents infectieux.
Les mesures prises sont considérées comme efficaces vis-à-vis des virus enveloppés tels que le virus de l’immunodéficience humaine (VIH), le virus de l’hépatite B (VHB) et le virus de l’hépatite C (VHC), et vis-à-vis des virus non-enveloppés de l’hépatite A (VHA) et le parvovirus B19.
Une vaccination appropriée (hépatites A et B) doit être envisagée pour les patients recevant régulièrement ou de manière répétée des produits à base d’antithrombine dérivée du plasma humain.
Traçabilité
A chaque administration d’ACLOTINE, le nom et le numéro de lot du produit mentionnés sur le flacon doivent être enregistrés afin de maintenir un lien entre le patient et le numéro de lot du médicament.
· Pour adapter la dose d’héparine et éviter une hypocoagulabilité excessive, des contrôles du degré d’anticoagulation (TCA et, le cas échéant, de l’activité anti-FXa) doivent être réalisés régulièrement, à intervalles rapprochés et en particulier au cours des premières minutes/heures suivant le début de l'administration de l'antithrombine.
· Mesure quotidienne du taux d'antithrombine afin d’adapter la dose individuelle, en raison du risque de diminution du taux d'antithrombine en cas de traitement prolongé avec de l'héparine non fractionnée.
L'association d'un traitement par l'antithrombine humaine avec un autre anticoagulant utilisé à dose curative peut faire courir un risque hémorragique au patient lorsqu'il existe des circonstances favorisantes telles que : mise en place ou présence de voies d'abord vasculaires profondes, chirurgie récente, ponction lombaire ou anesthésie rachidienne, association avec d'autres troubles de l'hémostase en particulier thrombopénie sévère ou hypofibrinogénémie.
Population pédiatrique
Les données d’essais cliniques et de revues systématiques concernant l’utilisation de l’antithrombine III dans le traitement des nouveau-nés prématurés dans l’indication non approuvée de syndrome de détresse respiratoire du nouveau-né (SDR) suggèrent l’existence d’un risque accru d’hémorragie intracrânienne et de mortalité en l’absence d’un effet bénéfique démontré.
Ce médicament contient du sodium
ACLOTINE contient du sodium. Ce médicament contient environ 0,28 mg de sodium par mL de produit (2,8 mg de sodium par flacon de 10 mL, 1,4 mg de sodium par flacon de 5 mL). Aux doses habituelles d’ACLOTINE, la quantité apportée de sodium est inférieure à 23 mg (1 mmol de sodium), c’est-à-dire « sans sodium ».
4.5. Interactions avec d'autres médicaments et autres formes d'interactions
Héparine : un traitement de substitution de l’antithrombine lors de l’administration d’héparine à des doses thérapeutiques augmente le risque d’hémorragie. L'effet de l'antithrombine est fortement amplifié par l'héparine. La demi-vie de l’antithrombine peut être considérablement réduite par un traitement concomitant par héparine en raison du taux de renouvellement accéléré de l’antithrombine. Dès lors, l'administration simultanée d'héparine et d’antithrombine à un patient qui présente un risque d'hémorragie accru doit faire l'objet d'une surveillance clinique et biologique (voir rubrique 4.4).
4.6. Fertilité, grossesse et allaitement
Grossesse et allaitement
L'expérience concernant la sécurité des produits à base d’antithrombine humaine au cours de la grossesse est limitée.
ACLOTINE ne doit être administré à la femme enceinte et allaitante ayant un déficit en antithrombine que si cela est clairement indiqué, en tenant compte du fait que, chez ces patientes, la grossesse comporte un risque plus élevé d’événements thromboemboliques.
Fertilité
Aucune information n'est disponible sur les effets possibles de l'antithrombine sur la fertilité féminine et masculine.
4.7. Effets sur l'aptitude à conduire des véhicules et à utiliser des machines
Aucun effet sur l'aptitude à conduire et à utiliser des machines n'a été observé.
a. Résumé du profil de sécurité
Des réactions d’hypersensibilité ou allergiques (comprenant les signes et symptômes suivants : œdème de Quincke, urticaire (y compris généralisé), hypotension, léthargie, respiration sifflante, oppression thoracique, agitation, tachycardie, bouffées vasomotrices, frissons, vomissements, nausées, fourmillements, brûlures et picotements au site d’injection) ont rarement été observées. Dans certains cas, ces réactions peuvent évoluer vers une anaphylaxie sévère (y compris un choc).
Des cas de céphalées et de fièvre ont été observés fréquemment.
Pour la sécurité relative aux agents transmissibles, voir rubrique 4.4.
b. Liste tabulée des effets indésirables
Les effets indésirables sont présentés dans le tableau ci-dessous conformément à la classification MedDRA (Classe de Systèmes d’Organes et Termes Préférentiels).
Les effets indésirables associés à ACLOTINE ont été obtenus à partir de 7 études cliniques (avec 148 patients exposés à ACLOTINE) et de la surveillance post-marketing.
La fréquence d’apparition d’effets indésirables a été estimée selon la convention suivante : très fréquent ((³1/10) ; fréquent (³1/100 to <1/10) ; peu fréquent (≥1/1,000 to <1/100); rare (³1/10,000 to <1/1,000); très rare (<1/10,000); fréquence indéterminée (ne peut pas être estimée sur la base des données disponibles).
|
Classes de systèmes d’organes selon MedDRA |
Effets indésirables (Termes préférentiels) |
Fréquence des effets indésirables |
|
Affections gastro-intestinales |
Brûlures œsophagiennes* Nausées* |
Peu fréquent Peu fréquent |
|
Troubles généraux et anomalies au site d'administration |
Fièvre |
Fréquent |
|
Affections du système immunitaire |
Indéterminée |
|
|
Affections du système nerveux |
Céphalées Etourdissements* |
Fréquent Peu fréquent |
|
Affections de la peau et du tissu sous-cutané |
Eruption cutanée |
Peu fréquent |
Déclaration des effets indésirables suspectés
La déclaration des effets indésirables suspectés après autorisation du médicament est importante.
Elle permet une surveillance continue du rapport bénéfice/risque du médicament. Les professionnels de santé déclarent tout effet indésirable suspecté via le système national de déclaration : Agence nationale de sécurité du médicament et des produits de santé (ANSM) et réseau des Centres Régionaux de Pharmacovigilance - Site internet : https://signalement.social-sante.gouv.fr/.
Aucun effet indésirable de surdosage n’a été rapporté avec ACLOTINE.
5. PROPRIETES PHARMACOLOGIQUES
5.1. Propriétés pharmacodynamiques
Classe pharmacothérapeutique : sang et organes hématopoïétiques, Agents antithrombotiques, groupe héparine. Code ATC : B01AB02
L'antithrombine contient deux domaines fonctionnels importants. Le premier contient le centre réactif et offre un site de clivage pour les protéases telles que la thrombine qui est nécessaire pour former un complexe inhibiteur-protéase stable. Le second est un domaine de liaison aux glycosaminoglycanes responsables de l'interaction avec l'héparine et les substances apparentées, qui accélèrent l'inhibition de la thrombine. Les complexes inhibiteurs enzymatiques de la coagulation sont éliminés par le système réticulo-endothélial.
L'activité de l'antithrombine chez l'adulte est de 80 à 120 % et, chez le nouveau-né, les taux sont d’environ 40 à 60 %.
5.2. Propriétés pharmacocinétiques
La demi-vie d’ACLOTINE est de 55 ± 14 heures. Cette demi-vie peut être diminuée lors d'un traitement concomitant par l'héparine ou dans certaines situations pathologiques.
5.3. Données de sécurité préclinique
Les données précliniques ne laissent supposer aucune potentialité mutagène d'ACLOTINE.
Poudre : glycine et chlorure de sodium.
Solvant : eau pour préparations injectables.
Après reconstitution, le produit doit être utilisé immédiatement.
6.4. Précautions particulières de conservation
A conserver à une température ne dépassant pas 25°C et à l'abri de la lumière. Ne pas congeler.
Pour les conditions de conservation du médicament après reconstitution, voir la rubrique 6.3.
6.5. Nature et contenu de l'emballage extérieur
Poudre en flacon (verre de type I) muni d’un bouchon (bromobutyle) et d’une capsule de protection + 5 mL de solvant en flacon (verre de type I) muni d’un bouchon (chlorobutyle) et d’une capsule de protection avec un système de transfert - boîte de 1.
Poudre en flacon (verre de type I) muni d’un bouchon (bromobutyle) et d’une capsule de protection + 10 mL de solvant en flacon (verre de type I) muni d’un bouchon (chlorobutyle) et d’une capsule de protection avec un système de transfert - boîte de 1.
6.6. Précautions particulières d’élimination et de manipulation
Reconstitution :
Respecter les règles d’asepsie habituelles.
Ne jamais utiliser les flacons dès la sortie du réfrigérateur.
· Amener les deux flacons (poudre et solvant) à une température ne dépassant pas 25°C.
· Retirer la capsule protectrice du flacon de solvant (eau pour préparations injectables) et du flacon de poudre.
· Désinfecter la surface de chaque bouchon.
· Retirer l’opercule du dispositif Mix2Vial. Sans extraire le dispositif de son emballage, enclencher l’extrémité bleue du Mix2Vial sur le bouchon du flacon de solvant.
· Retirer puis jeter l’emballage. Prendre soin de ne pas toucher la partie désormais exposée du dispositif.
· Retourner l’ensemble flacon de solvant-dispositif et l’enclencher sur le flacon de poudre par la partie transparente du dispositif. Le solvant est transféré automatiquement dans le flacon de poudre. Maintenir l’ensemble et agiter doucement, d’un mouvement circulaire, pour dissoudre totalement le produit.
· En maintenant la partie produit reconstituée d’une main et la partie solvant de l’autre, séparer les flacons en dévissant le dispositif Mix2Vial.
La mise en solution ainsi opérée est généralement instantanée et doit être totale en moins de 10 minutes. Le produit reconstitué doit être inspecté visuellement pour vérifier l'absence de particules avant l'administration
La solution doit être limpide ou légèrement opalescente. Ne pas utiliser de solution présentant un aspect non homogène ou contenant un dépôt.
Administration :
· Tenir le flacon de produit reconstitué verticalement, en vissant une seringue stérile sur le dispositif Mix2Vial. Aspirer ensuite lentement le produit dans la seringue.
· Une fois le produit transféré dans la seringue, tenir celle-ci fermement (piston dirigé vers le bas) dévisser le dispositif Mix2Vial et le remplacer par une aiguille intraveineuse ou une aiguille épicrânienne.
· Expulser l’air de la seringue et piquer la veine après désinfection.
· Injecter lentement par voie intraveineuse ou en perfusion, en une seule fois, immédiatement après reconstitution, sans dépasser un débit de 4 mL/minute.
Tout médicament non utilisé ou déchet doit être éliminé conformément à la réglementation en vigueur.
7. TITULAIRE DE L’AUTORISATION DE MISE SUR LE MARCHE
3 AVENUE DES TROPIQUES
ZA DE COURTABOEUF
91940 LES ULIS
FRANCE
Tél : +33 (0)1 69 82 70 04
E-mail : infomed@lfb.fr
8. NUMERO(S) D’AUTORISATION DE MISE SUR LE MARCHE
· 34009 561 938 8 2 : Poudre en flacon (verre) + 10 mL de solvant en flacon (verre) avec un système de transfert - boîte de 1.
9. DATE DE PREMIERE AUTORISATION/DE RENOUVELLEMENT DE L’AUTORISATION
[à compléter ultérieurement par le titulaire]
10. DATE DE MISE A JOUR DU TEXTE
[à compléter ultérieurement par le titulaire]
Sans objet.
12. INSTRUCTIONS POUR LA PREPARATION DES RADIOPHARMACEUTIQUES
Des informations détaillées sur ce médicament sont disponibles sur le site Internet de l’ANSM.
Liste I.
Médicament soumis à prescription hospitalière.
ANSM - Mis à jour le : 24/02/2026
ACLOTINE 100 UI/mL, poudre et solvant pour solution injectable
Antithrombine humaine
· Gardez cette notice. Vous pourriez avoir besoin de la relire.
· Si vous avez d’autres questions, interrogez votre médecin, votre pharmacien ou votre infirmier/ère.
· Ce médicament vous a été personnellement prescrit. Ne le donnez pas à d’autres personnes. Il pourrait leur être nocif, même si les signes de leur maladie sont identiques aux vôtres.
· Si vous ressentez un quelconque effet indésirable, parlez-en à votre médecin, votre pharmacien ou votre infirmier/ère. Ceci s’applique aussi à tout effet indésirable qui ne serait pas mentionné dans cette notice. Voir rubrique 4
2. Quelles sont les informations à connaître avant d'utiliser ACLOTINE 100 UI/mL, poudre et solvant pour solution injectable ?
3. Comment utiliser ACLOTINE 100 UI/mL, poudre et solvant pour solution injectable ?
4. Quels sont les effets indésirables éventuels ?
5. Comment conserver ACLOTINE 100 UI/mL, poudre et solvant pour solution injectable ?
6. Contenu de l’emballage et autres informations.
1. QU’EST-CE QUE ACLOTINE 100 UI/mL, poudre et solvant pour solution injectable ET DANS QUELS CAS EST-IL UTILISE ?
Classe pharmacothérapeutique : Agents antithrombotiques, groupe héparine - code ATC : B01AB02
ACLOTINE est un médicament antithrombotique (anticoagulant). La substance active est l’antithrombine humaine. L’antithrombine est un composant naturel du plasma humain et joue un rôle important d’inhibiteur de la coagulation du sang.
ACLOTINE est indiqué pour traiter :
· Les déficits congénitaux (de naissance) en antithrombine :
o Prévention des thromboses veineuses et accidents thrombo-emboliques, en cas de situations cliniques à risques élevés (notamment lors d'une chirurgie ou d'une grossesse) lorsque le risque hémorragique ne permet pas d'utiliser des doses suffisantes d'héparine, si indiquée.
o Prévention de la progression d’une thrombose veineuse profonde et d’une thromboembolie en association avec de l’héparine, comme indiqué.
· Le déficit acquis sévère (< 60 %) en antithrombine.
2. QUELLES SONT LES INFORMATIONS A CONNAITRE AVANT D’UTILISER ACLOTINE 100 UI/mL, poudre et solvant pour solution injectable ?
N’utilisez jamais ACLOTINE 100 UI/mL, poudre et solvant pour solution injectable :
· Si vous êtes allergique (hypersensible) à la substance active (l’antithrombine humaine) ou à l’un des autres composants contenus dans ce médicament, mentionnés dans la rubrique 6.
Avertissements et précautions
Adressez-vous à votre médecin, votre pharmacien ou votre infirmier/ère avant d’utiliser ACLOTINE.
Le traitement substitutif du déficit en antithrombine implique votre prise en charge par un spécialiste de l'hémostase.
Risque de réactions allergiques :
Comme pour tout produit protéique intraveineux, des réactions d'hypersensibilité allergiques sont possibles.
Si un des signes annonciateurs d’une réaction allergique survient, le traitement doit être immédiatement arrêté, et le traitement approprié à la nature et à la gravité de la réaction devra être mis en place par votre médecin.
Veuillez-vous référer à la section 4 de cette notice (Quels sont les effets indésirables éventuels ?) pour retrouver la liste des symptômes/signaux pouvant apparaitre en cas de réaction allergique.
Surveillance clinique et biologique lorsque l’antithrombine est utilisée conjointement à de l’héparine :
La surveillance clinique et biologique comprendra une surveillance étroite des paramètres de la coagulation afin d'éviter un risque de saignement excessif.
Lorsque l'antithrombine humaine est associée à un traitement anticoagulant (héparine), vous serez surveillé étroitement, en particulier, dans les minutes ou les heures qui suivent la perfusion d'antithrombine humaine afin d'adapter la posologie de l'héparine. Il existe en effet un risque de surdosage en héparine non fractionnée ou de bas poids moléculaire au début du traitement par l'antithrombine humaine chez les patients recevant de l'héparine.
Une mesure quotidienne des taux d'antithrombine doit être effectuée afin d'adapter les posologies, les traitements prolongés par l'héparine non fractionnée pouvant entraîner une diminution du taux d'antithrombine circulante.
L'association d'un traitement par l'antithrombine humaine avec un autre anticoagulant utilisé à dose curative peut faire courir un risque hémorragique au patient lorsqu'il existe des circonstances favorisantes telles que : mise en place ou présence de voies d'abord vasculaires profondes, chirurgie récente, ponction lombaire ou analgésie rachidienne, association avec d'autres troubles de l'hémostase en particulier thrombopénie (diminution du taux de cellules sanguines ayant un rôle dans la coagulation) sévère ou hypofibrinogénémie (diminution du taux de fibrinogène dans le sang).
Informations sur les mesures de sécurité liées à l’origine d’ACLOTINE :
Lorsque des médicaments sont préparés à partir de sang ou de plasma humain, des mesures de prévention de la transmission d'agents infectieux sont mises en place. Celles-ci comprennent :
· Une sélection soigneuse par entretien médical des donneurs de sang et de plasma de façon à exclure les donneurs risquant d'être porteurs d'infections.
· Le contrôle de chaque don et des mélanges de plasma pour la présence de virus/d'infection.
· La mise en œuvre des étapes capables d'éliminer ou d'inactiver les virus dans le procédé de fabrication du médicament.
Malgré ces mesures, lorsque des médicaments préparés à partir de sang ou de plasma humain sont administrés, le risque de transmission de maladies infectieuses ne peut être totalement exclu. Ceci s'applique également à tous les virus inconnus ou émergents ou autres types d'agents infectieux.
Les mesures prises pour ACLOTINE sont considérées comme efficaces pour lutter contre le risque d'infection par le virus de l'immunodéficience humaine (VIH), le virus de l'hépatite B, le virus de l'hépatite C, le virus de l'hépatite A et le parvovirus B19.
Il est recommandé que les patients recevant régulièrement ACLOTINE soient correctement vaccinés contre l'hépatite A et l'hépatite B.
A chaque administration d’ACLOTINE, le nom et le numéro de lot du produit mentionnés sur le flacon doivent être enregistrés afin de maintenir un lien entre le patient et le numéro de lot du médicament.
Enfants
Sans objet.
Autres médicaments et ACLOTINE 100 UI/mL, poudre et solvant pour solution injectable
Informez votre médecin ou votre pharmacien si vous utilisez, avez récemment utilisé ou pourriez utiliser tout autre médicament.
Un traitement par ACLOTINE peut augmenter l’effet anticoagulant de l’héparine, et le risque de saignements peut être accru. Si vous présentez un risque élevé de saignements, l’administration conjointe d’héparine doit être très soigneusement évaluée. Si votre médecin décide que vous devez recevoir de l’héparine, vous ferez l’objet d’une surveillance étroite par le biais d’analyses de laboratoire.
ACLOTINE 100 UI/mL, poudre et solvant pour solution injectable avec des aliments, boissons et de l’alcool
Sans objet.
Grossesse, allaitement et fertilité
Si vous êtes enceinte ou que vous allaitez, si vous pensez être enceinte ou planifiez une grossesse, demandez conseil à votre médecin ou pharmacien avant de prendre ce médicament.
L'innocuité d'ACLOTINE au cours de la grossesse et de l'allaitement n'a pas été évaluée par des essais cliniques contrôlés. L'expérimentation animale est insuffisante pour établir la sécurité vis-à-vis de la reproduction, du déroulement de la grossesse, du développement de l'embryon ou du fœtus et du développement péri- et postnatal. Par conséquent, ACLOTINE ne sera prescrit au cours de la grossesse ou de l'allaitement qu'en cas de nécessité bien établie.
Conduite de véhicules et utilisation de machines
Aucun effet sur l’aptitude à conduire des véhicules ou à utiliser des machines n’a été observé
ACLOTINE 100 UI/mL, poudre et solvant pour solution injectable contient du sodium.
Ce médicament contient environ 0,28 mg de sodium par mL de produit (2,8 mg de sodium par flacon de 10 mL, 1,4 mg de sodium par flacon de 5 mL). Aux doses habituelles d’ACLOTINE, la quantité apportée de sodium est inférieure à 23 mg (1 mmol de sodium), c’est-à-dire « sans sodium ».
3. COMMENT UTILISER ACLOTINE 100 UI/mL, poudre et solvant pour solution injectable ?
Posologie
La posologie et la durée du traitement de substitution dépendent de la sévérité des signés cliniques et de l’importance du déficit en antithrombine. La quantité à administrer et la fréquence d'administration doivent toujours être basées sur l'efficacité clinique et l'évaluation de laboratoire pour chaque individu.
Veillez à toujours utiliser ce médicament en suivant exactement les indications de votre médecin, pharmacien ou infirmier/ère. Vérifiez auprès de votre médecin, pharmacien ou infirmier/ère en cas de doute.
La dose de ACLOTINE à utiliser sera déterminée par votre médecin.
A titre indicatif, la dose recommandée est :
· Dans le déficit congénital (de naissance) :
o En traitement prophylactique : 30 à 50 UI/kg, lors d'une situation à risque thrombo-embolique (grossesse, chirurgie). La posologie et le rythme d'injection sont adaptés à l'évolution clinique et biologique.
o En traitement curatif : 40 à 50 UI/kg tous les jours ou tous les 2 jours chez l'adulte selon l'évolution clinique et biologique.
· Dans le déficit acquis sévère :
o En traitement curatif : dose initiale de 40 à 50 UI/kg voire 100 UI/kg. Les doses ultérieures, la fréquence des injections et la durée du traitement seront adaptées à l'état clinique et au suivi biologique.
Si vous avez utilisé plus de ACLOTINE 100 UI/mL, poudre et solvant pour solution injectable que vous n’auriez dû :
Aucun effet indésirable connu lié à un surdosage accidentel d'ACLOTINE n'a été rapporté.
Consultez immédiatement votre médecin ou votre pharmacien.
Si vous oubliez d’utiliser ACLOTINE 100 UI/mL, poudre et solvant pour solution injectable :
Votre médecin a la responsabilité de surveiller l’administration du médicament et de garder vos valeurs biologiques dans la plage spécifiée.
Si vous arrêtez d’utiliser ACLOTINE 100 UI/mL, poudre et solvant pour solution injectable :
Consultez toujours votre médecin, si vous envisagez d’arrêter. Si vous avez d’autres questions sur l’utilisation de ce médicament, demandez plus d’informations à votre médecin, votre pharmacien ou à votre infirmier/ère.
4. QUELS SONT LES EFFETS INDESIRABLES EVENTUELS ?
· Gonflement du visage et/ou de la langue et/ou de la gorge (œdème de Quincke)
· Éruption cutanée localisée (urticaire) ou sur tout le corps (urticaire généralisée),
· Chute de la pression artérielle (hypotension),
· Sensation de fatigue extrême (léthargie),
· Respiration sifflante
· Oppression thoracique,
· Agitation,
· Accélération du rythme du cœur (tachycardie),
· Rougeur du visage (bouffées vasomotrices),
· Frissons,
· Vomissements,
· Nausées,
· Fourmillements (paresthésies),
· Sensation de brûlure et picotement au site d’injection,
En cas de suspicion d’une allergie ou d’une réaction d’hypersensibilité, l’administration doit être interrompue immédiatement. En cas de choc anaphylactique, le traitement symptomatique de l’état de choc devra être instauré.
Effets indésirables observés avec ACLOTINE par ordre de fréquence :
Effets indésirables fréquents (survenant chez 1 personne sur 10) :
· Fièvre
· Maux de tête
Effets indésirables peu fréquents (peuvent toucher jusqu'à 1 personne sur 100) :
· Nausées
· Brûlures œsophagiennes
· Étourdissements
· Éruption cutanée
Autres effets indésirables observés avec ACLOTINE avec une fréquence indéterminée :
· Sensation de fourmillement, d'engourdissement ou de brûlure de la gorge (paresthésie pharyngée)
· Sensation de gorge serrée
Déclaration des effets secondaires
Si vous ressentez un quelconque effet indésirable, parlez-en à votre médecin, votre pharmacien ou à votre infirmier/ère. Ceci s’applique aussi à tout effet indésirable qui ne serait pas mentionné dans cette notice. Vous pouvez également déclarer les effets indésirables directement via le système national de déclaration : Agence nationale de sécurité du médicament et des produits de santé (ANSM) et réseau des Centres Régionaux de Pharmacovigilance - Site internet: https://signalement.social-sante.gouv.fr/.
En signalant les effets indésirables, vous contribuez à fournir davantage d’informations sur la sécurité du médicament.
5. COMMENT CONSERVER ACLOTINE 100 UI/mL, poudre et solvant pour solution injectable ?
Tenir ce médicament hors de la vue et de la portée des enfants.
N’utilisez pas ce médicament après la date de péremption indiquée sur le flacon. La date de péremption fait référence au dernier jour de ce mois.
Après reconstitution, le produit doit être utilisé immédiatement.
Conserver le conditionnement primaire dans l’emballage extérieur à une température ne dépassant pas 25°C et à l’abri de la lumière. Ne pas congeler.
N’utilisez pas ACLOTINE si vous constatez que la solution présente un aspect non homogène ou qu’elle contient un dépôt.
Ne jetez aucun médicament au tout-à-l’égout ou avec les ordures ménagères. Demandez à votre pharmacien ou votre infirmier/ère d’éliminer les médicaments que vous n’utilisez plus. Ces mesures contribueront à protéger l’environnement.
6. CONTENU DE L’EMBALLAGE ET AUTRES INFORMATIONS
Ce que contient ACLOTINE 100 UI/mL, poudre et solvant pour solution injectable
· La substance active est :
Antithrombine humaine (100 UI/mL de solution reconstituée). L’activité spécifique est ³ 3 UI/mg de protéines totales.
Un flacon de 5 mL contient 500 UI d’antithrombine humaine.
Un flacon de 10 mL contient 1000 UI d’antithrombine humaine.
· Les autres composants sont :
Poudre : glycine et chlorure de sodium.
Solvant : eau pour préparations injectables.
Ce médicament se présente sous forme de poudre et de solvant pour solution injectable. Boîte de 1.
Flacon de 5 mL ou de 10 mL.
Titulaire de l’autorisation de mise sur le marché
3 AVENUE DES TROPIQUES
ZA DE COURTABOEUF
91940 LES ULIS
FRANCE
Exploitant de l’autorisation de mise sur le marché
LFB BIOMEDICAMENTS
3 AVENUE DES TROPIQUES
ZA DE COURTABOEUF
91940 LES ULIS
FRANCE
LFB BIOMEDICAMENTS
3 AVENUE DES TROPIQUES
ZA DE COURTABOEUF
91940 LES ULIS
FRANCE
Noms du médicament dans les Etats membres de l'Espace Economique Européen
Sans objet.
La dernière date à laquelle cette notice a été révisée est :
[à compléter ultérieurement par le titulaire]
Des informations détaillées sur ce médicament sont disponibles sur le site Internet de l’ANSM (France).
Les informations suivantes sont destinées exclusivement aux professionnels de santé :
Reconstitution :
Respecter les règles d'asepsie habituelles.
|
|
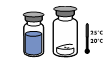
Si nécessaire, amener les deux flacons (poudre et solvant) à une température ne dépassant pas 25°C. |
|
|
Retirer la capsule protectrice du flacon de solvant (eau pour préparations injectables) et du flacon de poudre.
Désinfecter la surface de chaque bouchon. |
|
|
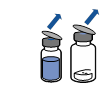
Retirer l’opercule du dispositif Mix2Vial. Sans extraire le dispositif de son emballage, enclencher l’extrémité bleue du Mix2Vial sur le bouchon du flacon de solvant. |
|
|
Retirer puis jeter l’emballage. Prendre soin de ne pas toucher la partie désormais exposée du dispositif.
|
|
|
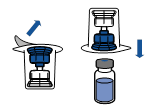
Retourner l’ensemble flacon de solvant-dispositif et l’enclencher sur le flacon de poudre par la partie transparente du dispositif. Le solvant est transféré automatiquement dans le flacon de poudre. Maintenir l’ensemble et agiter doucement, d’un mouvement circulaire, pour dissoudre totalement le produit.
|
|
|
En maintenant la partie produit reconstituée d’une main et la partie solvant de l’autre, séparer les flacons en dévissant le dispositif Mix2Vial. |
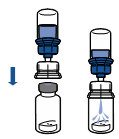
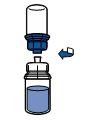
La mise en solution est généralement instantanée et doit être totale en moins de 10 minutes.
La solution est limpide ou légèrement opalescente. Ne pas utiliser de solution présentant un aspect non homogène ou contenant un dépôt.
Administration :
|
|
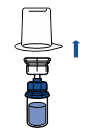
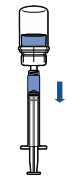
Tenir le flacon du produit reconstitué verticalement en vissant une seringue stérile sur le dispositif Mix2Vial. Aspirer ensuite lentement le produit dans la seringue.
Une fois le produit transféré dans la seringue, tenir celle-ci fermement (piston dirigé vers le bas), dévisser le dispositif Mix2Vial et le remplacer par une aiguille intraveineuse ou une aiguille épicrânienne.
Expulser l’air de la seringue et piquer la veine après désinfection.
Injecter lentement par voie intraveineuse en une seule fois, immédiatement après reconstitution, sans dépasser un débit de 4 mL/minute.
|
Tout médicament non utilisé ou déchet doit être éliminé conformément à la réglementation en vigueur.