Dernière mise à jour le 28/04/2026
NOVOFEMME, comprimé pelliculé
Ce médicament n'est ou ne sera bientôt plus disponible sur le marché.
Si vous prenez actuellement ce médicament, il vous est recommandé d'en parler avec votre médecin
ou avec votre pharmacien qui pourra vous orienter vers un autre traitement.
Indications thérapeutiques
NOVOFEMME est un traitement hormonal substitutif (THS) combiné séquentiel, qui doit être pris chaque jour sans interruption. NOVOFEMME est destiné aux femmes ménopausées dont les dernières règles remontent à au moins 6 mois.
NOVOFEMME contient deux hormones, un estrogène (estradiol) et un progestatif (acétate de noréthistérone). L’estradiol contenu dans NOVOFEMME est identique à celui fabriqué par les ovaires et appartient à la classe des estrogènes naturels. L’acétate de noréthistérone est un progestatif synthétique qui agit de façon similaire à la progestérone, une autre hormone sexuelle féminine importante.
NOVOFEMME est utilisé pour :
Le soulagement des symptômes de la ménopause
Durant la ménopause, la quantité d’estrogènes produits par le corps de la femme diminue. Cette diminution peut provoquer certains symptômes tels que des sensations de chaleur au niveau du visage, du cou et de la poitrine (« bouffées de chaleur »). NOVOFEMME soulage ces symptômes après la ménopause. NOVOFEMME ne vous sera prescrit que si vos symptômes altèrent votre vie quotidienne de manière importante.
La prévention de l’ostéoporose
Après la ménopause, certaines femmes peuvent développer une fragilité des os (ostéoporose). Vous devez discuter de toutes les éventualités de traitement avec votre médecin.
Si vous présentez un risque accru de fractures dues à l’ostéoporose et que les autres médicaments ne vous conviennent pas, vous pouvez utiliser NOVOFEMME pour la prévention de l’ostéoporose après la ménopause.
L’expérience de ce traitement chez les femmes âgées de plus de 65 ans est limitée.
Service médical rendu (SMR)
Les libellés affichés ci-dessous ne sont que des résumés ou extraits issus des avis rendus par la Commission de la Transparence. Seul l'avis complet de la Commission de la Transparence fait référence.
Cet avis est consultable à partir du lien `Avis du jj/mm/aaaa` ou encore sur demande auprès de la HAS (plus d'informations dans l'aide). Les avis et synthèses d'avis contiennent un paragraphe sur la place du médicament dans la stratégie thérapeutique.
| Valeur du SMR | Avis | Motif de l'évaluation | Résumé de l'avis |
|---|---|---|---|
| Important | Avis du 24/09/2025 | Réévaluation suite saisine Ministères (CT) | Le service médical rendu par ACTIVELLE 1 mg/ 0,5 mg (estradiol, acétate de noréthistérone), KLIOGEST 2 mg/ 1 mg (estradiol, acétate de noréthistérone), NOVOFEMME 1 mg/1 mg (estradiol, acétate de noréthistérone) et TRISEQUENS 2 mg/ 2 mg-1 mg/ 1 mg (estradiol, acétate de noréthistérone) reste important dans le traitement hormonal substitutif (THS) des symptômes de déficit en estrogènes chez les femmes ménopausées et dans la prévention de l'ostéoporose post-ménopausique chez les femmes ayant un risque accru de fracture ostéoporotique et présentant une intolérance ou une contre-indication aux autres traitements indiqués dans la prévention de l'ostéoporose. |
| Important | Avis du 13/04/2016 | Renouvellement d'inscription (CT) | Le service médical rendu par les spécialités ACTIVELLE, KLIOGEST, NOVOFEMME et TRISEQUENS dans : • le traitement hormonal substitutif (THS) des symptômes de déficit en estrogènes chez les femmes ménopausées reste important chez les patientes dont les troubles du climatère sont ressentis comme suffisamment gênant pour altérer leur qualité de vie, lorsque ces spécialités sont utilisées selon les préconisations de la Commission. • la prévention de l'ostéoporose post-ménopausique chez les femmes ayant un risque accru de fracture ostéoporotique et présentant une intolérance ou une contre-indication aux autres traitements indiqués dans la prévention de l'ostéoporose en cas de troubles du climatère et de ménopause récente, après une fracture mineure ou s’il existe un T-score bas, lorsque ces spécialités sont utilisées selon les préconisations de la Commission. |
Amélioration du service médical rendu (ASMR)
Les libellés affichés ci-dessous ne sont que des résumés ou extraits issus des avis rendus par la Commission de la Transparence. Seul l'avis complet de la Commission de la Transparence fait référence.
Cet avis est consultable à partir du lien `Avis du jj/mm/aaaa` ou encore sur demande auprés de la HAS (plus d'informations dans l'aide). Les avis et synthéses d'avis contiennent un paragraphe sur la place du médicament dans la stratégie thérapeutique.
| Valeur de l'ASMR | Avis | Motif de l'évaluation | Résumé de l'avis |
|---|---|---|---|
| V (Inexistant) | Avis du 24/09/2025 | Réévaluation suite saisine Ministères (CT) | ACTIVELLE (estradiol, acétate de noréthistérone), KLIOGEST (estradiol, acétate de noréthistérone), NOVOFEMME (estradiol, acétate de noréthistérone) et TRISEQUENS (estradiol, acétate de noréthistérone) n’apportent pas d’amélioration du service médical rendu (ASMR V) dans la stratégie thérapeutique. |
ANSM - Mis à jour le : 03/10/2024
NOVOFEMME, comprimé pelliculé
2. COMPOSITION QUALITATIVE ET QUANTITATIVE
Un comprimé pelliculé rouge contient :
1 mg d’estradiol (sous forme d’estradiol hémihydraté).
Un comprimé pelliculé blanc contient :
1 mg d’estradiol (sous forme d’estradiol hémihydraté) et 1 mg d’acétate de noréthistérone.
Excipient à effet notoire
Chaque comprimé pelliculé rouge contient 37,3 mg de lactose monohydraté.
Chaque comprimé pelliculé blanc contient 36,8 mg de lactose monohydraté.
Pour la liste complète des excipients, voir rubrique 6.1.
Comprimé pelliculé blanc, biconvexe gravé avec NOVO 283 – Diamètre : 6 mm.
Comprimé pelliculé rouge, biconvexe gravé avec NOVO 282 – Diamètre : 6 mm.
4.1. Indications thérapeutiques
Prévention de l'ostéoporose post-ménopausique chez la femme ayant un risque accru de fracture ostéoporotique et présentant une intolérance, ou une contre-indication, aux autres traitements indiqués dans la prévention de l'ostéoporose (voir aussi rubrique 4.4).
L’expérience de ce traitement chez les femmes âgées de plus de 65 ans est limitée.
4.2. Posologie et mode d'administration
Prendre un comprimé par jour dans l’ordre suivant :
· 1 comprimé rouge (estradiol seul) pendant 16 jours ;
· puis 1 comprimé blanc (associant estradiol et acétate de noréthistérone) pendant les 12 jours suivants.
Après la prise du dernier comprimé blanc, poursuivre le traitement avec le premier comprimé rouge de la plaquette suivante, sans interruption de traitement entre les plaquettes. Des saignements s’apparentant aux règles surviennent habituellement au début d’un nouveau cycle de traitement.
S’il s’agit d’une première prescription chez les femmes n’ayant jamais pris de THS ou s’il s’agit d’un relais d’un THS combiné continu, le traitement par NOVOFEMME peut être commencé n’importe quel jour. Par contre, s’il s’agit d’un relais d’un autre THS séquentiel, le traitement doit être commencé le lendemain du dernier jour du traitement précédent.
Pour débuter ou poursuivre un traitement dans l’indication des symptômes post-ménopausiques, la dose minimale efficace doit être utilisée pendant la plus courte durée possible (voir aussi rubrique 4.4).
Un passage à un produit combiné plus fortement dosé peut être indiqué si après 3 mois de traitement le soulagement attendu des symptômes est insuffisant.
Si la patiente a oublié de prendre un comprimé, le comprimé doit être pris le plus tôt possible dans les 12 heures qui suivent l’heure habituelle de la prise. Si plus de 12 heures se sont écoulées, le comprimé doit être jeté. L’oubli d’un comprimé peut augmenter la survenue de métrorragies et de « spottings ».
· Cancer du sein connu ou suspecté ou antécédent de cancer du sein
· Tumeurs malignes estrogéno-dépendantes connues ou suspectées ou antécédent de tumeurs malignes estrogéno-dépendantes (exemple : cancer de l’endomètre)
· Hémorragie génitale non diagnostiquée
· Hyperplasie endométriale non traitée
· Antécédent d’accident thrombo-embolique veineux ou accident thrombo-embolique veineux en évolution (thrombose veineuse profonde, embolie pulmonaire)
· Troubles thrombophiliques connus (par exemple déficit en protéine C, en protéine S ou en antithrombine, voir rubrique 4.4)
· Antécédent d’accident thrombo-embolique artériel ou accident thrombo-embolique artériel en évolution (exemple : angor, infarctus du myocarde)
· Affection hépatique aiguë ou antécédents d’affection hépatique, jusqu’à la normalisation des tests hépatiques
· Hypersensibilité connue aux substances actives ou à l’un des excipients
· Porphyrie.
4.4. Mises en garde spéciales et précautions d'emploi
Les données concernant les risques associés à la prise d’un THS lors du traitement de la ménopause précoce sont limitées. Cependant, en raison du faible niveau de risque absolu chez les femmes plus jeunes, la balance bénéfices-risques chez ces femmes pourrait être plus favorable que chez les femmes plus âgées.
Examen clinique et surveillance
Avant de débuter ou de recommencer un traitement hormonal substitutif (THS), il est indispensable d’effectuer un examen clinique et gynécologique complet (y compris le recueil des antécédents médicaux personnels et familiaux), en tenant compte des contre-indications et des précautions particulières d’emploi. Pendant toute la durée du traitement, des examens réguliers sont recommandés, leur nature et leur fréquence étant adaptées à chaque patiente.
Les femmes doivent être informées du type d’anomalies mammaires pouvant survenir sous traitement ; ces anomalies doivent être signalées au médecin traitant ou à l’infirmière (voir « Cancer du sein » ci-dessous). Les examens, y compris les outils d’imagerie appropriée (mammographie), doivent être pratiqués selon les recommandations en vigueur, et adaptés à chaque patiente.
Conditions nécessitant une surveillance
Si l’une des affections suivantes survient, est survenue précédemment, et/ou s’est aggravée au cours d’une grossesse ou d’un précédent traitement hormonal, la patiente devra être étroitement surveillée. Les affections suivantes peuvent réapparaître ou s’aggraver au cours du traitement par NOVOFEMME, en particulier :
· Léiomyome (fibrome utérin) ou endométriose.
· Facteurs de risque thrombo-emboliques (voir ci-dessous).
· Facteurs de risque de tumeurs estrogéno-dépendantes, par exemple : 1er degré d’hérédité pour le cancer du sein.
· Hypertension artérielle.
· Troubles hépatiques (par exemple : adénome hépatique).
· Diabète avec ou sans atteinte vasculaire.
· Lithiase biliaire.
· Migraines ou céphalées (sévères).
· Lupus érythémateux disséminé.
· Antécédent d’hyperplasie endométriale (voir ci-dessous).
· Epilepsie.
· Asthme.
· Otospongiose.
Arrêt immédiat du traitement
Le traitement doit être arrêté immédiatement en cas de survenue d’une contre-indication ou dans les cas suivants :
· Ictère ou altération de la fonction hépatique.
· Augmentation significative de la pression artérielle.
· Céphalée inhabituelle de type migraine.
· Grossesse.
Hyperplasie endométriale et cancer de l’endomètre
Chez les femmes non hystérectomisées, le risque d’hyperplasie endométriale et de cancer de l’endomètre augmente en cas d’administration prolongée d’estrogènes seuls. Le risque de cancer de l’endomètre chez les utilisatrices d’estrogènes seuls est multiplié par 2 à 12 comparativement aux non-utilisatrices, selon la durée du traitement et la dose d’estrogène (voir rubrique 4.8). Après l’arrêt du traitement, le risque peut rester élevé pendant au moins 10 ans.
L’ajout d’un progestatif de manière cyclique pendant au moins 12 jours par mois/cycle de 28 jours ou un traitement combiné continu estroprogestatif chez les femmes non hystérectomisées prévient le risque supplémentaire associé au THS à base d’estrogènes seuls.
Des métrorragies et des « spottings » peuvent survenir au cours des premiers mois de traitement. Si ces évènements persistent après les premiers mois de traitement, débutent un certain temps après le début du traitement ou s’ils persistent après l’arrêt du traitement, leur cause doit être recherchée. Cette démarche peut nécessiter une biopsie endométriale afin d’éliminer une pathologie maligne.
Cancer du sein
Les données disponibles montrent une augmentation du risque de cancer du sein chez les femmes prenant un traitement estroprogestatif ou chez celles prenant un THS à base d’estrogènes seuls, ce risque étant dépendant de la durée du traitement.
L’essai randomisé contrôlé versus placebo Women’s Health Initiative Study (WHI) et une méta-analyse des études épidémiologiques prospectives montrent une augmentation du risque de survenue de cancer du sein chez les femmes traitées par un THS estroprogestatif combiné qui devient significative après environ 3 (1-4) années d’utilisation (voir rubrique 4.8).
Les résultats d’une importante méta-analyse ont montré qu’après avoir arrêté le traitement, le risque additionnel diminue dans le temps et la durée nécessaire pour qu’il revienne à la normale dépend de la durée de la prise du THS. Lorsqu’un THS a été suivi pendant plus de 5 ans, le risque peut perdurer 10 ans ou plus.
Les THS, particulièrement les traitements estroprogestatifs, augmentent la densité mammaire à la mammographie, ce qui pourrait gêner le diagnostic de cancer du sein.
Cancer ovarien
Le cancer ovarien est beaucoup plus rare que le cancer du sein.
Les données épidémiologiques provenant d'une importante méta-analyse suggèrent une légère augmentation du risque chez les femmes prenant un THS par oestrogènes seuls ou par une combinaison d'oestrogènes et de progestatifs, qui apparaît dans les cinq ans suivant le début de l'utilisation du produit et diminue progressivement après l'arrêt du traitement.
D'autres études, y compris l'essai WHI (Women's Health Initiative), suggèrent qu'un risque similaire ou légèrement inférieur peut être associé avec une utilisation de THS combinés (voir rubrique 4.8).
Thrombo-embolie veineuse
Le THS est associé à un risque multiplié par 1,3 à 3 de développer une thrombo-embolie veineuse (TEV), c’est-à-dire une thrombose veineuse profonde ou une embolie pulmonaire. La probabilité de survenue d’un tel événement est plus élevée au cours de la première année d’utilisation du THS (voir rubrique 4.8).
Les patientes ayant une maladie thrombophilique connue ont un risque majoré de TEV, et les THS peuvent accroître ce risque. Par conséquent, l’utilisation d’un THS est contre-indiquée chez ces patientes (voir rubrique 4.3).
Les facteurs de risque de TEV généralement reconnus incluent l’utilisation d’estrogènes, l’âge avancé, une chirurgie lourde, l’immobilisation prolongée, l’obésité (IMC > 30 kg/m2), la période de grossesse/postpartum, le lupus érythémateux disséminé (LED) et le cancer. Il n’existe pas de consensus concernant le rôle possible des varices dans la TEV.
Comme chez tous les patients en période post-opératoire, des mesures prophylactiques doivent être appliquées pour prévenir une TEV après la chirurgie. Si une immobilisation prolongée doit suivre une chirurgie programmée, l’arrêt provisoire du THS 4 à 6 semaines avant l’intervention est recommandé. Le traitement ne doit pas être réintroduit avant que la patiente ait retrouvé toute sa mobilité.
Chez les femmes sans antécédent personnel de TEV mais ayant un parent du premier degré avec un antécédent de thrombo-embolie veineuse à un âge jeune, un dépistage peut être proposé après une revue approfondie de ses limites (seules certaines anomalies thrombophiliques sont identifiées par dépistage).
Si une maladie thrombophilique est identifiée, associée à une thrombo-embolie veineuse chez des membres de la famille, ou si le déficit est « sévère » (par exemple déficit en antithrombine, en protéine S ou en protéine C, ou combinaison de déficits), le THS est contre-indiqué.
Chez les femmes sous traitement anticoagulant chronique, le rapport bénéfice/risque de l’utilisation du THS doit être soigneusement évalué.
Si une TEV se développe après l’initiation du traitement, ce dernier devra être arrêté. Il devra être recommandé aux patientes de contacter immédiatement leur médecin en cas de symptômes évoquant un événement thromboembolique (notamment gonflement douloureux d’une jambe, douleur soudaine dans la poitrine ou dyspnée).
Maladie coronarienne
Il n’existe pas de données d’études contrôlées randomisées démontrant une protection contre l’infarctus du myocarde chez les femmes avec ou sans maladie coronarienne ayant reçu un THS estroprogestatif ou à base d’estrogènes seuls.
Le risque relatif de maladie coronarienne au cours de l’utilisation d’un THS estroprogestratif est légèrement augmenté. Le risque absolu de base de maladie coronarienne étant fortement dépendant de l’âge, le nombre de cas supplémentaires dus à l’utilisation d’un THS estroprogestatif est très faible chez les femmes en bonne santé proches de la ménopause, mais il augmente avec l’âge.
Accidents vasculaires cérébraux ischémiques
Le traitement estroprogestatif et le traitement estrogénique seul sont associés à une augmentation du risque d’accident vasculaire cérébral ischémique pouvant atteindre un facteur de 1,5. Le risque relatif n’évolue pas avec l’âge ni avec le temps écoulé depuis la ménopause. Toutefois, le risque de base d’AVC étant fortement âge-dépendant, le risque global d’AVC chez les femmes utilisant un THS augmente avec l’âge (voir rubrique 4.8).
Hypothyroïdie
Les patientes nécessitant une hormonothérapie thyroïdienne substitutive doivent faire contrôler régulièrement leur fonction thyroïdienne pendant la prise du THS afin de s’assurer que les taux d'hormones thyroïdiennes restent dans une mesure acceptable.
Autres pathologies
Les estrogènes pouvant provoquer une rétention hydrique, les patientes présentant une insuffisance rénale ou cardiaque doivent être étroitement surveillées.
Les femmes avec une hypertriglycéridémie préexistante doivent être étroitement surveillées pendant le traitement estrogénique substitutif ou pendant le traitement hormonal substitutif. De rares cas d’augmentation importante du taux plasmatique des triglycérides conduisant à une pancréatite ont été observés sous estrogénothérapie.
Les estrogènes exogènes peuvent provoquer ou aggraver les symptômes des angiœdèmes héréditaire et acquis.
Au cours du traitement par les estrogènes, une augmentation des taux plasmatiques de la TBG (thyroid binding globulin) est observée, elle conduit à une élévation des taux plasmatiques des hormones thyroïdiennes totales mesurés par PBI (protein-bound iodine), de la T4 totale (mesurée sur colonne ou par RIA (radioimmunoassay)) et de la T3 totale (mesurée par RIA). La fixation de la T3 sur la résine est diminuée, reflétant l’augmentation de la TBG. Les concentrations des fractions libres de T4 et de T3 restent inchangées. Les taux sériques d’autres protéines de liaison telles que la CBG (corticoid binding globulin) et la SHBG (sex-hormone binding globulin) peuvent être augmentés entraînant, respectivement, une augmentation des taux circulants de corticostéroïdes et de stéroïdes sexuels. Les concentrations des fractions libres ou actives des hormones restent inchangées. D’autres protéines plasmatiques peuvent également être augmentées (angiotensinogène/substrat de la rénine, alpha-1-antitrypsine et céruloplasmine).
Le THS n’améliore pas les fonctions cognitives. Certaines données indiquent une augmentation du risque de probable démence chez les femmes débutant un traitement combiné continu ou à base d’estrogènes seuls après 65 ans.
Augmentations de l’ALAT
Au cours des essais cliniques menés avec des patientes traitées pour des infections par le virus de l’hépatite C (VHC) avec l'association ombitasvir/paritaprévir/ritonavir avec et sans dasabuvir, des élévations de l'ALAT supérieures à 5 fois la limite supérieure de la normale (LSN) ont été significativement plus fréquentes chez les femmes utilisant des médicaments contenant de l'éthinylestradiol, tels que les CHC. Par ailleurs, également chez les patientes traitées par glécaprévir/pibrentasvir, des élévations de l’ALAT ont été observées chez les femmes utilisant des médicaments contenant de l’éthinylestradiol, tels que des CHC. Les femmes utilisant des médicaments contenant des estrogènes autres que l'éthinylestradiol, tels que l'œstradiol, ont présenté un taux d’élévation de l’ALAT similaire à celui des femmes ne recevant aucun estrogène. Cependant, en raison du nombre limité de femmes prenant ces autres estrogènes, la prudence est de mise lors de la co-administration avec l’association médicamenteuse ombitasvir/paritaprévir/ritonavir avec ou sans dasabuvir et également avec l’association médicamenteuse glécaprevir/pibrentasvir. Voir rubrique 4.5.
Les comprimés NOVOFEMME contiennent du lactose. Ce médicament ne doit pas être utilisé chez les patients présentant une intolérance au galactose, un déficit total en lactase ou un syndrome de malabsorption du glucose et du galactose (maladies héréditaires rares).
4.5. Interactions avec d'autres médicaments et autres formes d'interactions
Le métabolisme des estrogènes et des progestatifs peut être augmenté par l’utilisation concomitante de médicaments inducteurs enzymatiques, en particulier des iso-enzymes du cytochrome P450, comme les anticonvulsivants (phénobarbital, phénytoïne, carbamazépine) et les anti-infectieux (rifampicine, rifabutine, névirapine, éfavirenz).
Le ritonavir, le télaprévir et le nelfinavir, bien que connus comme de puissants inhibiteurs enzymatiques, ont paradoxalement des propriétés inductrices quand ils sont utilisés avec des hormones stéroïdiennes. Les préparations à base de plantes contenant du millepertuis (Hypericum perforatum) pourraient modifier le métabolisme des estrogènes et des progestatifs.
L’augmentation du métabolisme des estrogènes et des progestatifs peut conduire à une diminution de l’effet thérapeutique et à une modification du profil des saignements utérins.
Effet du THS à base d’estrogènes sur d’autres médicaments
Il a été démontré que les contraceptifs hormonaux contenant des estrogènes diminuent significativement les concentrations plasmatiques de lamotrigine lorsqu’ils sont administrés de façon concomitante, en raison de l’induction de la glucuronidation de la lamotrigine. Cela peut réduire le contrôle des crises. Bien que l’interaction potentielle entre le traitement hormonal substitutif et la lamotrigine n’ait pas été étudiée, on s’attend à ce qu’une interaction similaire existe, ce qui peut conduire à une réduction du contrôle des crises chez les femmes prenant les deux médicaments en association.
Interactions pharmacodynamiques
Certaines analyses de laboratoire peuvent être modifiées par les traitements à base d’estrogènes, telles que les tests de tolérance au glucose ou de la fonction thyroïdienne.
Les médicaments qui inhibent les enzymes hépatiques microsomales, comme le kétoconazole, peuvent augmenter les taux circulants des principes actifs de NOVOFEMME.
L’administration concomitante de ciclosporine et de NOVOFEMME peut entraîner une augmentation des taux sanguins de ciclosporine, de créatinine et des transaminases, suite à la diminution du métabolisme hépatique de la ciclosporine.
4.6. Fertilité, grossesse et allaitement
Grossesse
NOVOFEMME n’a pas d’indication au cours de la grossesse.
La découverte d’une grossesse au cours du traitement par NOVOFEMME impose l’arrêt immédiat du traitement.
Les données cliniques sur un nombre limité de grossesses exposées révèlent des effets indésirables de la noréthistérone sur le fœtus. A des doses supérieures à celles habituellement contenues dans les contraceptifs oraux et les THS, des cas de masculinisation de fœtus femelles ont été observés.
A ce jour, la plupart des études épidémiologiques n’ont pas mis en évidence d’effet tératogène ou fœtotoxique chez les femmes enceintes exposées par mégarde à des doses thérapeutiques d’estrogènes et de progestatifs.
NOVOFEMME n’a pas d’indication au cours de l’allaitement.
4.7. Effets sur l'aptitude à conduire des véhicules et à utiliser des machines
NOVOFEMME n’a pas d’effet connu sur l’aptitude à conduire des véhicules et à utiliser des machines.
Expérience clinique
Les effets indésirables les plus fréquemment rapportés au cours des essais cliniques réalisés avec un TSH similaire à NOVOFEMME étaient des tensions mammaires et des céphalées (rapportées chez au moins 10% des patientes).
Les effets secondaires ci-dessous peuvent également survenir lors d’un traitement estroprogestatif.
Les fréquences sont issues d’essais cliniques conduits avec une spécialité similaire à NOVOFEMME et d’une étude de suivi après commercialisation de NOVOFEMME.
|
Système classe organe |
Très fréquent ≥ 1/10 |
Fréquent ≥ 1/100, <1/10 |
Peu fréquent ≥ 1/1 000, <1/100 |
Rare ≥ 1/10 000, < 1/1 000 |
|
Infections et infestations |
|
Candidose vaginale |
|
|
|
Affections du système immunitaire |
|
|
|
Réaction allergique |
|
Affections psychiatriques |
|
|
|
Nervosité |
|
Affections du système nerveux |
Céphalée |
Etourdissement insomnie, dépression |
Migraine, troubles de la libido (non spécifiques) |
Vertiges |
|
Affections vasculaires |
|
Augmentation de la pression artérielle, aggravation d’une hypertension artérielle |
Thrombose et embolie périphériques |
|
|
Affections gastro-intestinales |
|
Dyspepsie, douleur abdominale, flatulence, nausée |
Vomissement |
Diarrhées, ballonnement |
|
Affections hépatobiliaires |
|
|
Maladie de la vésicule biliaire, calculs biliaires |
|
|
Affections de la peau et du tissu sous-cutané |
|
Rash, prurit |
Alopécie |
Acné |
|
Affections musculo-squelettiques et systémiques |
|
|
Crampes musculaires |
|
|
Affections des organes de reproduction et du sein |
Tension mammaire |
Hémorragie vaginale, fibromes utérins aggravés |
|
Fibrome utérin |
|
Troubles généraux et anomalies au site d’administration |
|
Œdème |
|
|
|
Investigations |
|
Prise de poids |
|
|
Expérience après commercialisation
En plus des événements ci-dessus, les effets indésirables présentés ci-après ont été rapportés spontanément et il est possible qu’ils soient en relation avec le traitement par NOVOFEMME. La fréquence de ces effets indésirables ne peut être estimée sur la base des données disponibles :
· Tumeurs bénignes et malignes (dont kystes et polypes) : cancer de l’endomètre
· Affections du système immunitaire : réactions d’hypersensibilité généralisées (par exemple réaction/choc anaphylactique)
· Affections psychiatriques : anxiété
· Affections du système nerveux : accident vasculaire cérébral
· Affections visuelles : troubles visuels
· Affections cardiaques : infarctus du myocarde
· Affections vasculaires : aggravation de l’hypertension arterielle
· Affections hépatobiliaires : aggravation de lithiase biliaire, réapparition de lithiase biliaire
· Affections cutanées et sous-cutanées : séborrhée, œdème angioneurotique, hirsutisme
· Affections des organes de reproduction et du sein : hyperplasie endométriale, prurit vulvo-vaginal
· Investigations : perte de poids.
D’autres effets indésirables sont rapportés lors de l’administration d’un traitement estroprogestatif :
· Affections de la peau et du tissu sous-cutané : chloasma, érythème polymorphe, érythème noueux, éruption hémorragique, purpura vasculaire
· Probable démence après 65 ans (voir rubrique 4.4)
· Sécheresse oculaire
· Changements de la composition du film lacrymal.
Risque de cancer du sein
Une augmentation jusqu’à 2 fois du risque de cancer a été rapportée chez les femmes ayant pris une association estroprogestative pendant plus de 5 ans.
L’augmentation du risque est plus faible chez les utilisatrices d’estrogènes seuls comparativement aux utilisatrices d’associations estroprogestatives.
Le niveau de risque dépend de la durée du traitement (voir rubrique 4.4).
Les estimations du risque absolu basées sur les résultats du plus grand essai randomisé contrôlé versus placebo (étude WHI) et de la plus grande méta-analyse des études épidémiologiques prospectives sont présentées ci-après :
Plus importante méta-analyse d’études épidémiologiques prospectives
Estimation du risque additionnel de cancer du sein après 5 ans de traitement chez des femmes ayant un IMC de 27 (kg/m2)
|
Âge au début du THS (ans) |
Incidence pour 1 000 femmes non utilisatrices de THS sur une période de 5 ans (50-54 ans)* |
Risque relatif |
Nombre de cas supplémentaires pour 1 000 utilisatrices de THS après 5 ans |
|
Estrogènes seuls |
|||
|
50 |
13,3 |
1,2 |
2,7 |
|
Association estroprogestative |
|||
|
50 |
13,3 |
1,6 |
8,0 |
* Issu des taux d’incidence de base en Angleterre en 2015 chez des femmes ayant un IMC de 27 (kg/m2).
Remarque : étant donné que l’incidence de base du cancer du sein diffère selon les pays de l’UE, le nombre de cas supplémentaires de cancer du sein variera proportionnellement.
Estimation du risque additionnel de cancer du sein après 10 ans de traitement chez des femmes ayant un IMC de 27 (kg/m2)
|
Âge au début du THS (ans) |
Incidence pour 1 000 femmes non utilisatrices de THS sur une période de 10 ans (50-59 ans)* |
Risque relatif |
Nombre de cas supplémentaires pour 1 000 utilisatrices de THS après 10 ans |
|
THS à base d’estrogènes seuls |
|||
|
50 |
26,6 |
1,3 |
7,1 |
|
Association estroprogestative |
|||
|
50 |
26,6 |
1,8 |
20,8 |
*Issu des taux d’incidence de base en Angleterre en 2015 chez des femmes ayant un IMC de 27 (kg/m2)
Remarque : étant donné que l’incidence de base du cancer du sein diffère selon les pays de l’UE, le nombre de cas supplémentaires de cancer du sein variera proportionnellement.
Etudes WHI aux Etats-Unis – Risque additionnel de cancer du sein après 5 ans de traitement
|
Age (ans) |
Incidence pour 1 000 femmes dans le bras placebo sur 5 ans |
Risque relatif (IC 95 %) |
Nombre de cas supplémentaires pour 1 000 utilisatrices de THS sur 5 ans (IC 95 %) |
|
Estrogènes seuls (estrogènes conjugués équins (CEE)) |
|||
|
50-79 |
21 |
0,8 (0,7-1,0) |
-4 (-6-0)* |
|
Estrogènes et progestatif (estrogènes conjugués équins (CEE) + acétate de médroxyprogestérone (MPA))** |
|||
|
50-79 |
17 |
1,2 (1,0-1,5) |
4 (0-9) |
* Etude WHI chez les femmes hystérectomisées, n’ayant pas montré d’augmentation du risque de cancer du sein.
** Lorsque l’analyse était limitée aux femmes n’ayant pas utilisé de THS avant l’étude, il n’était pas observé d’augmentation du risque au cours des 5 premières années de traitement. Après 5 ans, le risque était plus élevé que chez les non-utilisatrices.
Risque de cancer de l’endomètre
Le risque de cancer de l’endomètre est d’environ 5 pour 1 000 femmes ayant un utérus intact et n’utilisant pas de THS.
Chez les femmes ayant un utérus intact, l’utilisation d’un THS à base d’estrogènes seuls n’est pas recommandée car cela augmente le risque de cancer de l’endomètre (voir rubrique 4.4).
Dans les études épidémiologiques, l’augmentation du risque de cancer de l’endomètre dépendait de la durée de traitement à base d’estrogènes seuls et de la dose d’estrogène et variait entre 5 et 55 cas supplémentaires diagnostiqués pour 1 000 femmes âgées de 50 à 65 ans.
L’ajout d’un progestatif au traitement par estrogènes seuls pendant au moins 12 jours par cycle permet de prévenir cette augmentation du risque. Dans l’étude « Million Women Study », l’utilisation pendant 5 ans d’un THS combiné (séquentiel ou continu) n’a pas augmenté le risque de cancer de l’endomètre (RR de 1,0 (0,8-1,2)).
Risque de cancer ovarien
L'utilisation d'un THS par oestrogènes seuls ou par une combinaison d'oestrogènes et de progestatifs a été associée à une légère augmentation du risque de cancer ovarien diagnostiqué (voir rubrique 4.4).
Une méta-analyse portant sur 52 études épidémiologiques a signalé un risque accru de cancer ovarien chez les femmes prenant actuellement un THS par rapport aux femmes n'en ayant jamais pris (RR 1,43, IC 95 % 1,31-1,56). Chez les femmes âgées de 50 à 54 ans, prendre un THS pendant cinq ans entraîne l'apparition d'un cas supplémentaire pour 2 000 utilisatrices. Chez les femmes âgées entre 50 à 54 ans qui ne prennent pas de THS, un diagnostic de cancer ovarien sera posé chez environ 2 femmes sur 2 000 sur une période de cinq ans.
Risque d’accident thrombo-embolique veineux
Le THS est associé à une augmentation de 1,3 à 3 fois du risque de survenue d’un accident thrombo-embolique veineux (TEV), c’est-à-dire thrombose veineuse profonde ou embolie pulmonaire. La probabilité de survenue d’un tel événement est plus élevée au cours de la première année d’utilisation du THS (voir rubrique 4.4). Les résultats des études WHI sont présentés ci-après :
Etudes WHI - Risque additionnel d’accident thrombo-embolique veineux sur 5 ans de traitement
|
Age (ans) |
Incidence pour 1 000 femmes dans le bras placebo sur 5 ans |
Risque relatif (IC 95 %) |
Nombre de cas supplémentaires pour 1 000 utilisatrices de THS sur 5 ans (IC 95 %) |
|
Estrogènes seuls par voie orale* |
|||
|
50-59 |
7 |
1,2 (0,6-2,4) |
1 (-3-10) |
|
Association estroprogestative orale |
|||
|
50-59 |
4 |
2,3 (1,2-4,3) |
5 (1-13) |
* Etude chez les femmes hystérectomisées.
Risque de maladie artérielle coronarienne
Le risque de maladie artérielle coronarienne est légèrement augmenté chez les utilisatrices de THS estroprogestatif au-delà de 60 ans (voir rubrique 4.4).
Risque d’accident vasculaire cérébral ischémique
L’utilisation d’un THS à base d’estrogènes seuls ou d’une association estroprogestative est associée à une augmentation jusqu’à 1,5 fois du risque relatif d’AVC ischémique. Le risque d’AVC hémorragique n’est pas augmenté lors de l’utilisation d’un THS.
Ce risque relatif ne dépend pas de l’âge ni de la durée du traitement, mais comme le risque de base est fortement dépendant de l’âge, le risque global d’AVC chez les femmes prenant un THS augmente avec l’âge (voir rubrique 4.4).
Etudes WHI combinées – Risque additionnel d’AVC* sur 5 ans de traitement
|
Age (ans) |
Incidence pour 1 000 femmes dans le bras placebo sur 5 ans |
Risque relatif (IC 95 %) |
Nombre de cas supplémentaires pour 1 000 utilisatrices de THS sur 5 ans (IC 95 %) |
|
50-59 |
8 |
1,3 (1,1-1,6) |
3 (1-5) |
* Sans distinction entre les AVC ischémiques et hémorragiques.
Déclaration des effets indésirables suspectés
La déclaration des effets indésirables suspectés après autorisation du médicament est importante. Elle permet une surveillance continue du rapport bénéfice/risque du médicament. Les professionnels de santé déclarent tout effet indésirable suspecté via le système national de déclaration : Agence nationale de sécurité du médicament et des produits de santé (ANSM) et réseau des Centres Régionaux de Pharmacovigilance - Site internet : https://signalement.social-sante.gouv.fr/.
Les symptômes d'un surdosage en estrogènes oraux sont une tension mammaire, des nausées, des vomissements et/ou des métrorragies. Un surdosage en progestatifs peut entraîner une humeur dépressive, de la fatigue, de l'acné et de l'hirsutisme. Le traitement doit être symptomatique.
5. PROPRIETES PHARMACOLOGIQUES
5.1. Propriétés pharmacodynamiques
Estradiol : le principe actif, 17β-estradiol de synthèse, est chimiquement et biologiquement identique à l’estradiol endogène humain. Il remplace l’arrêt de production des estrogènes chez les femmes ménopausées et soulage les symptômes climatériques de la ménopause.
Les estrogènes préviennent la perte osseuse liée à la ménopause ou à une ovariectomie.
L’acétate de noréthistérone : Progestatif de synthèse. Les estrogènes stimulent la croissance de l’endomètre et majorent le risque d’hyperplasie et de cancer de l’endomètre. L’association d’un progestatif chez les femmes non hystérectomisées entraîne une réduction du risque d’hyperplasie de l’endomètre induit par les estrogènes.
Le soulagement des symptômes de la ménopause survient au cours des premières semaines de traitement.
Dans une étude après commercialisation, des saignements de privation réguliers d’une durée moyenne de 3-4 jours sont survenus chez 91 % des femmes traitées par NOVOFEMME pendant 6 mois. Ces saignements surviennent habituellement quelques jours après le dernier comprimé de la phase progestative.
Le déficit en estrogène à la ménopause est associé à un renouvellement osseux accru et à une diminution de la masse osseuse. L’effet des estrogènes sur la densité minérale osseuse est dose-dépendant. L’effet protecteur est efficace aussi longtemps que le traitement est poursuivi. A l’arrêt du THS, la masse osseuse diminue à une vitesse similaire à celle observée chez les femmes non traitées.
Les résultats de l’étude WHI et d’une méta-analyse de plusieurs études montrent que l’utilisation d’un traitement estrogénique substitutif, seul ou en association avec un progestatif – principalement chez des femmes en bonne santé – diminue le risque de fractures de la hanche, des vertèbres et d’autres fractures ostéoporotiques. Des données limitées suggèrent que les THS pourraient également prévenir des fractures chez des femmes ayant une faible densité minérale osseuse et/ou une ostéoporose établie.
Des études randomisées en double aveugle versus placebo ont montré qu’une dose de 1 mg d’estradiol prévient la déminéralisation osseuse post-ménopausique et augmente la densité minérale osseuse. Après deux ans de traitement avec 1 mg de 17-β estradiol, l’augmentation de la densité minérale osseuse est respectivement de 2,8 %, 1,6 % et 2,5 % au niveau vertébral, au niveau de la tête fémorale et au niveau du trochanter.
5.2. Propriétés pharmacocinétiques
Après l’administration orale de 17β-estradiol sous forme micronisée, l’absorption digestive est rapide. Le métabolisme est important en raison de l’effet de premier passage hépatique et du passage au niveau des autres organes entériques. Après administration orale d’une dose unique de 1 mg, un pic de concentration plasmatique d’environ 27 pg/ml (entre 13 et 40 pg/ml), est atteint en 6 heures. L’aire sous la courbe (ASC(0-tz)) est de 629 h x pg/ml. La demi-vie du 17β-estradiol est d’environ 25 heures. Il circule lié à la SHBG (37 %) et à l’albumine (61 %), alors qu’environ 1-2 % sont sous forme non liée. Le métabolisme du 17β-estradiol s’effectue principalement dans le foie et l’intestin, mais également dans les organes cibles et il implique la formation de métabolites moins actifs ou inactifs, en particulier l’estrone, les catécholestrogènes et plusieurs sulfates et glycuronides d’estrogènes. Les estrogènes sont éliminés avec la bile, où ils sont hydrolysés et réabsorbés (circulation entéro-hépatique), et principalement dans les urines sous forme biologiquement inactive.
Après administration orale, l’acétate de noréthistérone est rapidement absorbé et transformé en noréthistérone (NET). Il subit un métabolisme de premier passage hépatique et des autres organes entériques. Après administration orale d’une dose unique de 1 mg, un pic de concentration plasmatique d’environ 9 ng/ml (entre 6 et 11 ng/ml) est atteint en 1 heure. L’aire sous la courbe (ASC(0-tz)) est de 29 h x pg/ml. La demi-vie terminale de la NET est d’environ 10 heures. La NET se lie à la SHBG (36 %) et à l'albumine (61 %). Les principaux métabolites sont les isomères de la 5α-dihydro-NET et de la tétrahydro-NET, principalement éliminés dans les urines sous forme de dérivés sulfo- et glucuro-conjugués.
La pharmacocinétique de l’estradiol n’est pas influencée par l’acétate de noréthistérone.
Les propriétés pharmacocinétiques chez les personnes âgées n’ont pas été étudiées.
5.3. Données de sécurité préclinique
Les comprimés rouges et blancs contiennent :
lactose monohydraté, amidon de maïs, hydroxypropylcellulose, talc, stéarate de magnésium.
Pelliculage :
Comprimé blanc : hypromellose, triacétine, talc
Comprimé rouge : hypromellose, oxyde de fer rouge (E172), dioxyde de titane (E171), propylèneglycol, talc.
3 ans
6.4. Précautions particulières de conservation
6.5. Nature et contenu de l'emballage extérieur
1 x 28 comprimés ou 3 x 28 comprimés dans un distributeur journalier.
Le distributeur journalier de 28 comprimés comporte 3 parties :
· un fond en polypropylène coloré opaque,
· un couvercle circulaire en polystyrène transparent,
· un cadran central en polystyrène coloré opaque.
Toutes les présentations peuvent ne pas être commercialisées.
6.6. Précautions particulières d’élimination et de manipulation
Tout médicament non utilisé ou déchet doit être éliminé conformément à la réglementation en vigueur.
7. TITULAIRE DE L’AUTORISATION DE MISE SUR LE MARCHE
10-12 COURS MICHELET
92800 PUTEAUX
FRANCE
8. NUMERO(S) D’AUTORISATION DE MISE SUR LE MARCHE
· 34009 358 479-2 8 : 28 comprimés en distributeur journalier (Polypropylène/Polystyrène). Boîte de 1.
· 34009 358 480-0 0 : 28 comprimés en distributeur journalier (Polypropylène/Polystyrène). Boîte de 3.
9. DATE DE PREMIERE AUTORISATION/DE RENOUVELLEMENT DE L’AUTORISATION
[à compléter ultérieurement par le titulaire]
10. DATE DE MISE A JOUR DU TEXTE
[à compléter ultérieurement par le titulaire]
Sans objet.
12. INSTRUCTIONS POUR LA PREPARATION DES RADIOPHARMACEUTIQUES
Sans objet.
Liste I
ANSM - Mis à jour le : 03/10/2024
estradiol/acétate de noréthistérone
Veuillez lire attentivement cette notice avant de prendre ce médicament car elle contient des informations importantes pour vous.
· Gardez cette notice. Vous pourriez avoir besoin de la relire.
· Si vous avez d’autres questions, interrogez votre médecin ou votre pharmacien.
· Ce médicament vous a été personnellement prescrit. Ne le donnez pas à d’autres personnes. Il pourrait leur être nocif, même si les signes de leur maladie sont identiques aux vôtres.
· Si vous ressentez un quelconque effet indésirable, parlez-en à votre médecin ou à votre pharmacien. Ceci s’applique aussi à tout effet indésirable qui ne serait pas mentionné dans cette notice. Voir rubrique 4.
1. Qu'est-ce que NOVOFEMME, comprimé pelliculé et dans quels cas est-il utilisé ?
2. Quelles sont les informations à connaître avant de prendre NOVOFEMME, comprimé pelliculé ?
3. Comment prendre NOVOFEMME, comprimé pelliculé ?
4. Quels sont les effets indésirables éventuels ?
5. Comment conserver NOVOFEMME, comprimé pelliculé ?
6. Contenu de l’emballage et autres informations.
1. QU’EST-CE QUE NOVOFEMME, comprimé pelliculé ET DANS QUELS CAS EST-IL UTILISE ?
NOVOFEMME est un traitement hormonal substitutif (THS) combiné séquentiel, qui doit être pris chaque jour sans interruption. NOVOFEMME est destiné aux femmes ménopausées dont les dernières règles remontent à au moins 6 mois.
NOVOFEMME contient deux hormones, un estrogène (estradiol) et un progestatif (acétate de noréthistérone). L’estradiol contenu dans NOVOFEMME est identique à celui fabriqué par les ovaires et appartient à la classe des estrogènes naturels. L’acétate de noréthistérone est un progestatif synthétique qui agit de façon similaire à la progestérone, une autre hormone sexuelle féminine importante.
NOVOFEMME est utilisé pour :
Le soulagement des symptômes de la ménopause
Durant la ménopause, la quantité d’estrogènes produits par le corps de la femme diminue. Cette diminution peut provoquer certains symptômes tels que des sensations de chaleur au niveau du visage, du cou et de la poitrine (« bouffées de chaleur »). NOVOFEMME soulage ces symptômes après la ménopause. NOVOFEMME ne vous sera prescrit que si vos symptômes altèrent votre vie quotidienne de manière importante.
La prévention de l’ostéoporose
Après la ménopause, certaines femmes peuvent développer une fragilité des os (ostéoporose). Vous devez discuter de toutes les éventualités de traitement avec votre médecin.
Si vous présentez un risque accru de fractures dues à l’ostéoporose et que les autres médicaments ne vous conviennent pas, vous pouvez utiliser NOVOFEMME pour la prévention de l’ostéoporose après la ménopause.
L’expérience de ce traitement chez les femmes âgées de plus de 65 ans est limitée.
2. QUELLES SONT LES INFORMATIONS A CONNAITRE AVANT DE PRENDRE NOVOFEMME, comprimé pelliculé ?
Antécédents médicaux et examens réguliers
L’utilisation d’un THS comporte des risques qui doivent être pris en considération lors de l’initiation du traitement ou lors de la poursuite du traitement.
L’expérience chez les femmes traitées ayant une ménopause précoce (due à une insuffisance ovarienne ou une chirurgie) est limitée. Si vous avez une ménopause précoce, les risques d’utilisation d’un THS peuvent être différents. Parlez-en à votre médecin.
Avant de débuter (ou reprendre) un THS, votre médecin vous interrogera sur vos antécédents médicaux personnels et familiaux. Votre médecin peut décider de faire un examen clinique. Celui-ci peut inclure l’examen de vos seins et/ou un examen gynécologique, si nécessaire.
Dès que vous commencez NOVOFEMME, vous devez consulter votre médecin pour des examens réguliers (au moins une fois par an). Lors de ces examens médicaux, parlez à votre médecin des bénéfices et des risques encourus si vous continuez à prendre NOVOFEMME.
Faites régulièrement une mammographie, tel que recommandé par votre médecin.
Ne prenez jamais NOVOFEMME, comprimé pelliculé
Si l’une des situations suivantes s’applique à votre cas, si vous n’êtes pas sûre concernant l’un des points ci-dessous, parlez-en à votre médecin avant de prendre NOVOFEMME.
Ne prenez jamais NOVOFEMME :
· si vous avez, avez eu ou si vous pensez avoir un cancer du sein.
· si vous avez, avez eu ou si vous pensez avoir un cancer de la paroi de l’utérus (cancer de l’endomètre), ou un cancer estrogéno-dépendant.
· si vous avez des saignements vaginaux inexpliqués.
· si vous avez un épaississement excessif de la paroi de l’utérus (hyperplasie endométriale) qui n’est pas traité.
· si vous avez ou avez déjà eu un caillot sanguin dans une veine (accident thrombo-embolique veineux), dans la jambe (thrombose veineuse profonde) ou dans les poumons (embolie pulmonaire).
· si vous avez un trouble de la coagulation sanguine (tel qu’un déficit en protéine C, en protéine S ou en antithrombine).
· si vous avez ou avez précédemment eu une maladie causée par des caillots sanguins dans les artères, telle qu’une crise cardiaque, un accident vasculaire cérébral ou une angine de poitrine.
· si vous avez ou avez eu une maladie du foie et que vos analyses hépatiques ne sont pas revenues aux valeurs normales.
· si vous êtes allergique (hypersensible) à l’estradiol, à l’acétate de noréthistérone ou à l’un des autres composants contenus dans NOVOFEMME, mentionnés dans la rubrique 6.
· si vous avez une maladie rare du sang appelée « porphyrie » qui est transmise dans une même famille (héréditaire).
Si une des conditions citées ci-dessus apparaît pour la première fois pendant le traitement avec NOVOFEMME, il faut arrêter le traitement et consulter votre médecin.
Avertissements et précautions
Avant de commencer le traitement, prévenez votre médecin si vous avez déjà eu un des problèmes suivants, car ils pourraient récidiver ou s’aggraver au cours du traitement par NOVOFEMME. Si tel est le cas, vous devrez consulter votre médecin plus souvent pour des examens :
· fibrome dans votre utérus
· développement de la paroi utérine en dehors de votre utérus (endométriose) ou antécédent de développement excessif de la paroi utérine (hyperplasie endométriale)
· risque plus élevé de développer des caillots sanguins (voir « Caillots sanguins dans une veine (accident thrombo-embolique veineux) »)
· risque plus élevé de développer un cancer sensible aux estrogènes (par exemple, si votre mère, votre sœur ou votre grand-mère a développé un cancer du sein)
· pression artérielle élevée
· troubles hépatiques comme une tumeur bénigne du foie
· diabète
· calculs biliaires
· migraine ou maux de tête sévères
· maladie du système immunitaire qui affecte plusieurs organes du corps (lupus érythémateux disséminé, LED)
· épilepsie
· asthme
· maladie affectant le tympan et l’audition (otospongiose)
· taux très élevé de lipides dans votre sang (triglycérides)
· rétention d’eau due à des problèmes cardiaques ou rénaux
· maladie au cours de laquelle votre thyroïde n’arrive pas à produire assez d’hormones thyroïdiennes (hypothyroïdie) et vous êtes traitée avec une hormonothérapie thyroïdienne substitutive
· maladie héréditaire provoquant des épisodes répétés de gonflement sévère (angiœdème héréditaire) ou si vous avez eu des épisodes de gonflement rapide des mains, du visage, des pieds, des lèvres, des yeux, de la langue, de la gorge (obstruction des voies respiratoires) ou du tube digestif (angiœdème acquis)
· intolérance au lactose.
Arrêtez de prendre NOVOFEMME et consultez immédiatement un médecin
Si vous remarquez l’une des situations suivantes lorsque vous prenez un THS :
· l’une des conditions mentionnées dans la rubrique « Ne prenez jamais NOVOFEMME »
· jaunissement de votre peau ou du blanc de vos yeux (jaunisse). Cela peut être le signe d’une maladie du foie
· gonflement du visage, de la langue et/ou de la gorge et/ou difficultés à avaler ou urticaire, associés à des difficultés à respirer, qui suggèrent un angiœdème
· une forte augmentation de votre pression artérielle (les symptômes peuvent être : mal de tête, fatigue, vertiges)
· maux de tête de type migraines qui apparaissent pour la première fois.
· si vous êtes enceinte
· si vous remarquez des signes de caillots sanguins, tels que :
o gonflement douloureux et rougeur des jambes
o douleur soudaine dans la poitrine
o difficultés pour respirer
Pour plus d’informations, voir « Caillots sanguins dans une veine (accident thrombo-embolique veineux) ».
Note : NOVOFEMME n’est pas un contraceptif. Si vos dernières règles remontent à moins de 12 mois ou si vous avez moins de 50 ans, vous pourriez encore avoir besoin d’une contraception supplémentaire afin de ne pas être enceinte. Demandez conseil à votre médecin.
THS et cancer
Epaississement excessif de la paroi de l’utérus (hyperplasie endométriale) et cancer de la paroi de l’utérus (cancer de l’endomètre)
La prise d’un THS à base d’estrogènes seuls augmente le risque d’épaississement excessif de la paroi de l’utérus (hyperplasie endométriale) et de cancer de la paroi de l’utérus (cancer de l’endomètre).
Le progestatif contenu dans NOVOFEMME vous protège contre ce risque supplémentaire.
Pour comparaison
Chez les femmes ayant un utérus intact et ne prenant pas de THS, on estime qu’en moyenne 5 pour 1 000 auront un cancer de l’endomètre diagnostiqué entre 50 et 65 ans.
Chez les femmes de 50 à 65 ans ayant toujours leur utérus intact et prenant un THS à base d’estrogènes seuls, le nombre de cas sera de 10 à 60 pour 1 000 utilisatrices (soit 5 à 55 cas supplémentaires), en fonction de la dose et de la durée d’utilisation.
Saignements inattendus
En prenant NOVOFEMME, vous aurez des saignements une fois par mois (également appelés saignements de privation). Cependant, si vous avez des saignements inattendus ou des gouttes de sang (spottings) en dehors de vos saignements mensuels et qui :
· continuent après les 6 premiers mois d’utilisation,
· commencent alors que vous prenez NOVOFEMME depuis plus de 6 mois,
· continuent alors que vous avez arrêté de prendre NOVOFEMME,
consultez votre médecin dès que possible.
Cancer du sein
Les données montrent une augmentation du risque de cancer du sein chez les femmes prenant une association estroprogestative ou chez les femmes prenant un traitement hormonal substitutif (THS) à base d’estrogènes seuls. Cette augmentation du risque dépend de la durée de suivi du THS. L’augmentation du risque devient significative au bout de 3 ans d’utilisation. Après avoir arrêté le THS, le risque supplémentaire diminuera avec le temps, mais pourra perdurer 10 ans ou plus si vous avez suivi un THS pendant plus de 5 ans.
Pour comparaison
Chez les femmes âgées de 50 à 54 ans qui ne prennent pas de THS, on estime qu’en moyenne 13 à 17 sur 1 000 auront un cancer du sein diagnostiqué sur une période de 5 ans.
Chez les femmes âgées de 50 ans qui débutent un THS à base d'estrogènes seuls pendant 5 ans, on dénombrera 16 à 17 cas sur 1 000 utilisatrices (soit 0 à 3 cas supplémentaires).
Chez les femmes âgées de 50 ans qui débutent un THS estroprogestatif pendant 5 ans, on dénombrera 21 cas pour 1 000 utilisatrices (soit 4 à 8 cas supplémentaires).
Chez les femmes âgées de 50 à 59 ans qui ne prennent pas de THS, un diagnostic de cancer du sein sera posé, en moyenne, chez environ 27 femmes sur 1 000 après une période de dix ans.
Chez les femmes âgées de 50 ans qui débutent un THS à base d'estrogènes seuls pendant 10 ans, on dénombrera 34 cas sur 1 000 utilisatrices (soit 7 cas supplémentaires).
Chez les femmes âgées de 50 ans qui débutent un THS estroprogestatif pendant 10 ans, on dénombrera 48 cas sur 1 000 utilisatrices (soit 21 cas supplémentaires).
Surveillez régulièrement vos seins. Contactez votre médecin si vous observez des changements tels que :
· creux de la peau
· modification du mamelon
· nodule que vous pouvez voir ou sentir.
Par ailleurs, il vous est conseillé de prendre part aux programmes de mammographie de dépistage qui pourront vous être proposés. Lors des mammographies de dépistage, il est important que vous indiquiez au personnel infirmier/professionnel de santé réalisant la radiographie que vous utilisez un THS car ce traitement peut augmenter la densité de vos seins, ce qui peut affecter les résultats de la mammographie. Lorsque la densité du sein est augmentée, la mammographie peut ne pas détecter toutes les grosseurs.
Cancer ovarien
Le cancer de l'ovaire est rare (beaucoup plus rare que le cancer du sein).
L'utilisation d'un THS par oestrogènes seuls ou par une combinaison d'oestrogènes et de progestatifs a été associée à une légère augmentation du risque de cancer ovarien.
Le risque de cancer ovarien varie en fonction de l'âge. Par exemple, chez les femmes âgées entre 50 et 54 ans qui ne prennent pas de THS, un diagnostic de cancer ovarien sera posé chez 2 femmes sur 2 000 en moyenne sur une période de 5 ans. Chez les femmes ayant pris un THS pendant 5 ans, il y aura environ 3 cas sur 2 000 utilisatrices (soit environ un cas supplémentaire).
Effet du THS sur le cœur et la circulation sanguine
Caillots sanguins dans une veine (accident thrombo-embolique veineux)
Le risque de caillots sanguins dans les veines est multiplié par environ 1,3 à 3 chez les utilisatrices d’un THS par rapport aux non-utilisatrices, en particulier au cours de la première année de traitement.
Les caillots sanguins peuvent être graves et si l’un d’eux atteint les poumons, il peut provoquer une douleur dans la poitrine, un essoufflement, un évanouissement ou même la mort.
Vous avez plus de risques d’avoir un caillot sanguin dans vos veines si vous êtes âgée ou si vous êtes dans l’une des situations suivantes. Informez votre médecin si vous êtes dans l’une des situations suivantes :
· vous ne pouvez pas marcher pendant une longue période à cause d’une opération chirurgicale, d’une blessure ou d’une maladie (voir aussi rubrique 3 « Si vous devez subir une chirurgie »)
· vous êtes en surpoids sévère (IMC > 30 kg/m²)
· vous avez un problème de coagulation nécessitant un traitement à long terme avec un médicament destiné à la prévention des caillots sanguins
· l’un de vos proches a déjà eu un caillot sanguin dans une jambe, un poumon ou un autre organe.
· vous avez un lupus érythémateux disséminé (LED)
· vous avez un cancer.
Pour les signes d’un caillot sanguin, voir « Arrêtez de prendre NOVOFEMME et consultez immédiatement un médecin ».
Pour comparaison
Chez les femmes âgées d’une cinquantaine d’années et ne prenant pas de THS, on estime que sur une période moyenne de 5 ans, en moyenne 4 à 7 sur 1 000 utilisatrices pourraient avoir un caillot sanguin dans une veine.
Chez les femmes âgées d’une cinquantaine d’années et prenant un THS estroprogestatif depuis une période de 5 ans, on estime qu’il y aura 9 à 12 cas pour 1 000 utilisatrices (soit 5 cas supplémentaires).
Maladie du cœur (crise cardiaque)
Rien ne prouve qu’un THS puisse aider à éviter une crise cardiaque. Les femmes âgées de plus de 60 ans prenant un THS estroprogestatif ont légèrement plus de risques de développer une maladie cardiaque que celles ne prenant pas de THS.
Accident vasculaire cérébral (AVC)
Le risque d’AVC est environ 1,5 fois plus élevé chez les utilisatrices de THS par rapport aux non-utilisatrices. Le nombre de cas supplémentaires d’AVC dus à la prise d’un THS augmentera avec l’âge.
Pour comparaison
Chez les femmes âgées d’une cinquantaine d’années et ne prenant pas un THS, on estime qu’environ 8 sur 1 000 devraient faire un AVC sur une période de 5 ans.
Pour les femmes dans la cinquantaine prenant un THS, le nombre de cas sera de 11 pour 1 000 utilisatrices sur une période de 5 ans (soit 3 cas supplémentaires).
Autres maladies
Le THS n’a pas d’effet préventif sur la perte de mémoire. Le risque de probable perte de mémoire pourrait être plus élevé chez les femmes débutant un THS après 65 ans. Demandez conseil à votre médecin.
Autres médicaments et NOVOFEMME, comprimé pelliculé
Certains médicaments peuvent interagir avec l’effet de NOVOFEMME et entraîner des saignements irréguliers. Cela concerne les médicaments suivants :
· Médicaments pour le traitement de l’épilepsie (comme le phénobarbital, la phénytoïne et la carbamazépine)
· Médicaments pour le traitement de la tuberculose (comme la rifampicine et la rifabutine)
· Médicaments pour le traitement de l’infection par le VIH (comme la névirapine, l’éfavirenz, le ritonavir ou le nelfinavir)
· Médicaments pour le traitement de l’hépatite C (comme le télaprévir)
· Les préparations à base de plantes contenant du millepertuis (Hypericum perforatum).
Le THS peut modifier le mode d’action de certains autres médicaments :
· Un médicament contre l’épilepsie (lamotrigine), car il pourrait augmenter la fréquence des crises
· Les médicaments contre le virus de l’hépatite C (VHC) (tels que l’association ombitasvir/ paritaprévir/ritonavir avec ou sans dasabuvir, ainsi que l’association glécaprévir/pibrentasvir) peuvent induire des augmentations au niveau du résultat de certaines analyses sanguines de la fonction hépatique (augmentation de l’enzyme hépatique ALAT) chez les femmes utilisant des contraceptifs hormonaux combinés (CHC) contenant de l’éthinylestradiol. NOVOFEMME contient de l’estradiol à la place de l’éthinylestradiol. Il n’est pas connu qu’une augmentation de l’enzyme hépatique ALAT puisse survenir lors de l’utilisation de NOVOFEMME avec cette association contre le VHC.
D’autres médicaments peuvent augmenter les effets de NOVOFEMME :
· Médicaments contenant du kétoconazole (un fongicide).
NOVOFEMME peut avoir un impact sur un traitement concomitant avec la ciclosporine.
Informez votre médecin ou votre pharmacien si vous prenez ou avez récemment pris tout autre médicament y compris un médicament obtenu sans ordonnance, un médicament à base de plantes ou tout autre produit naturel. Votre médecin vous informera.
Analyses du sang
Si vous devez subir un examen sanguin, dites à votre médecin ou au médecin du laboratoire que vous prenez NOVOFEMME car ce médicament peut modifier les résultats de certaines analyses.
NOVOFEMME, comprimé pelliculé avec des aliments et des boissons
Les comprimés peuvent être pris avec ou sans aliments et boissons.
Grossesse et allaitement
Grossesse
NOVOFEMME est indiqué uniquement chez les femmes ménopausées. Si vous découvrez que vous êtes enceinte, arrêtez de prendre NOVOFEMME et contactez votre médecin.
Allaitement
Vous ne devez pas prendre NOVOFEMME si vous allaitez.
Conduite de véhicules et utilisation de machines
NOVOFEMME n’a pas d’effet connu sur la capacité à conduire des véhicules ou à utiliser des machines.
NOVOFEMME, comprimé pelliculé contient du lactose monohydraté
Si vous avez une intolérance à certains sucres, contactez votre médecin avant de prendre NOVOFEMME.
3. COMMENT PRENDRE NOVOFEMME, comprimé pelliculé ?
Si vous ne preniez pas d’autre traitement hormonal substitutif, vous pouvez commencer votre traitement par NOVOFEMME n’importe quel jour. Cependant, si vous passez d’un autre traitement hormonal substitutif à celui-ci, demandez à votre médecin à quel moment vous devez commencer votre traitement par NOVOFEMME.
Prenez un comprimé par jour, de préférence à la même heure chaque jour.
Chaque distributeur contient 28 comprimés :
· Jours 1 à 16 Prenez un comprimé rouge par jour pendant 16 jours
· Jours 17 à 28 Prenez un comprimé blanc par jour pendant 12 jours
Avalez le comprimé avec un verre d’eau.
Après avoir terminé la plaquette, le traitement doit être poursuivi le lendemain, sans interruption, avec une nouvelle plaquette. Des saignements évoquant des règles apparaissent généralement au commencement d’une nouvelle plaquette.
Pour plus d’informations sur l’utilisation du distributeur journalier, consultez les « INSTRUCTIONS D’UTILISATION » à la fin de cette notice.
Votre médecin doit vous prescrire la plus faible dose avec la durée la plus courte possible qui permettra de soulager vos symptômes. Contactez votre médecin si vous avez l’impression que cette dose est trop forte ou trop faible.
Contactez votre médecin si vos symptômes ne sont pas améliorés après 3 mois de traitement. Continuez le traitement uniquement si les bénéfices sont plus importants que les risques.
Si vous avez pris plus de NOVOFEMME, comprimé pelliculé que vous n’auriez dû :
Si vous avez pris plus de NOVOFEMME que vous n’auriez dû, consultez votre médecin ou votre pharmacien. Les signes de surdosage en estrogènes peuvent se traduire par une tension mammaire, des nausées, des vomissements et/ou des saignements vaginaux irréguliers (métrorragies). Un surdosage en progestatifs peut entraîner une humeur dépressive, de la fatigue, de l'acné et une croissance excessive des poils du corps ou du visage (hirsutisme).
Si vous oubliez de prendre NOVOFEMME, comprimé pelliculé :
Si l’oubli d’un comprimé est constaté dans les 12 heures qui suivent l’heure habituelle de la prise, prenez immédiatement le comprimé oublié. Si plus de 12 heures se sont écoulées, poursuivez le traitement normalement en prenant le comprimé suivant le lendemain au moment habituel. Ne prenez pas de dose double pour compenser le comprimé que vous avez oublié de prendre.
L’oubli d’une dose peut favoriser la survenue de saignements et de gouttes de sang « spottings ».
Si vous arrêtez de prendre NOVOFEMME, comprimé pelliculé :
Si vous souhaitez arrêter de prendre NOVOFEMME, contactez tout d’abord votre médecin. Il vous expliquera les effets liés à l’arrêt du traitement et pourra discuter des alternatives avec vous.
Si vous avez d’autres questions sur l’utilisation de ce médicament, demandez plus d’informations à votre médecin ou à votre pharmacien.
Si vous devez subir une chirurgie
Si vous devez vous faire opérer, indiquez au chirurgien que vous prenez NOVOFEMME. Vous pourriez avoir besoin d’arrêter la prise de NOVOFEMME 4 à 6 semaines environ avant l’opération afin de réduire le risque de caillot sanguin (voir rubrique 2 « Caillots sanguins dans une veine (accident thrombo-embolique veineux) »). Demandez à votre médecin à partir de quand vous pourrez reprendre NOVOFEMME.
4. QUELS SONT LES EFFETS INDESIRABLES EVENTUELS ?
Les maladies suivantes sont rapportées plus fréquemment chez les femmes utilisant un THS comparativement aux femmes qui n’en prennent pas :
· cancer du sein
· développement anormal ou cancer de la paroi de l’utérus (hyperplasie endométriale ou cancer de l’endomètre)
· cancer ovarien
· caillots sanguins dans les veines des jambes ou des poumons (accident thrombo-embolique veineux)
· maladie cardiovasculaire
· accident vasculaire cérébral
· possible perte de mémoire, si le traitement est débuté après 65 ans.
Pour plus d’informations sur ces effets indésirables, voir rubrique 2.
Allergie/hypersensibilité (effet indésirable peu fréquent – survient chez 1 à 10 utilisatrices sur 1 000)
Même si cet effet est peu fréquent, des réactions allergiques/d’hypersensibilité peuvent survenir. Les signes d’allergie/hypersensibilité peuvent inclure un ou plusieurs des symptômes suivants : urticaire, démangeaisons, gonflement, difficultés pour respirer, baisse de la pression artérielle (pâleur et peau froide, battements de cœur rapides), sensations de vertige, sueurs, qui peuvent être les signes d’une réaction ou d’un choc anaphylactique. Si l’un de ces symptômes se manifeste, arrêtez de prendre NOVOFEMME et consultez immédiatement un médecin.
Effets indésirables très fréquents (survient chez plus d’une utilisatrice sur 10)
· Mal de tête
· Tension mammaire.
Effets indésirables fréquents (survient chez 1 à 10 utilisatrices sur 100)
· Augmentation de la pression artérielle ou aggravation d’une hypertension artérielle
· Mycose vaginale (par exemple muguet)
· Etourdissement, insomnie, dépression
· Dyspepsie (indigestion), douleur abdominale, flatulences
· Nausées
· Démangeaisons, éruption cutanée
· Saignements vaginaux (voir rubrique 2, sous-rubrique « Saignements inattendus »)
· Aggravation de fibromes utérins (tumeur bénigne de l’utérus)
· Œdème (gonflement des mains, des chevilles et des pieds)
· Prise de poids.
Effets indésirables peu fréquents (survient chez 1 à 10 utilisatrices sur 1 000)
· Migraine
· Modifications de la libido (changements du désir sexuel)
· Accident thrombo-embolique périphérique et thrombose (caillot sanguin)
· Vomissements
· Lithiase biliaire ou calculs biliaires
· Chute de cheveux (alopécie)
· Crampes musculaires.
Effets indésirables rares (survient chez 1 à 10 utilisatrices sur 10 000)
· Réactions allergiques
· Nervosité
· Vertige (étourdissement)
· Diarrhée
· Ballonnement
· Acné
· Fibromes utérins (tumeur bénigne de l’utérus).
Fréquence non connue (ne peut être déterminée sur la base des données disponibles)
· Hyperplasie de l’endomètre (épaississement excessif de la paroi de l’utérus)
· Augmentation de la pilosité du corps et du visage
· Anxiété
· Troubles de la vision
· Séborrhée
· Démangeaisons vaginales.
Autres effets indésirables d’un THS combiné
Les effets indésirables suivants ont été rapportés avec d’autres THS :
· Troubles cutanés divers :
o décoloration de la peau, en particulier au niveau du visage et du cou, connue sous le terme de « masque de grossesse » (chloasma)
o nodules cutanés rouges et douloureux (érythème noueux)
o éruption cutanée avec rougeurs ou lésions en forme de bulles (érythème polymorphe)
o décolorations ou tâches rouges ou violettes de la peau et/ou des muqueuses (purpura vasculaire)
· Sécheresse des yeux
· Changements de la composition des larmes.
Déclaration des effets secondaires
Si vous ressentez un quelconque effet indésirable, parlez-en à votre médecin ou votre pharmacien. Ceci s’applique aussi à tout effet indésirable qui ne serait pas mentionné dans cette notice. Vous pouvez également déclarer les effets indésirables directement via le système national de déclaration : Agence nationale de sécurité du médicament et des produits de santé (ANSM) et réseau des Centres Régionaux de Pharmacovigilance - Site internet : https://signalement.social-sante.gouv.fr/. En signalant les effets indésirables, vous contribuez à fournir davantage d’informations sur la sécurité du médicament.
5. COMMENT CONSERVER NOVOFEMME, comprimé pelliculé ?
Tenir ce médicament hors de la vue et de la portée des enfants.
N’utilisez pas ce médicament après la date de péremption indiquée sur l’étiquette et l’emballage extérieur après « EXP ». La date de péremption fait référence au dernier jour de ce mois.
À conserver à une température ne dépassant pas 25°C. Ne pas mettre au réfrigérateur.
Conserver le distributeur journalier dans l’emballage extérieur, à l’abri de la lumière.
Ne jetez aucun médicament au tout-à-l’égout ni avec les ordures ménagères. Demandez à votre pharmacien d’éliminer les médicaments que vous n’utilisez plus. Ces mesures contribueront à protéger l’environnement.
6. CONTENU DE L’EMBALLAGE ET AUTRES INFORMATIONS
Ce que contient NOVOFEMME, comprimé pelliculé
· Les substances actives sont :
l’estradiol sous forme d’estradiol hémihydraté et l’acétate de noréthistérone.
Un comprimé pelliculé rouge contient : 1 mg d’estradiol (sous forme d’estradiol hémihydraté).
Un comprimé pelliculé blanc contient : 1 mg d’estradiol (sous forme d’estradiol hémihydraté) et 1 mg d’acétate de noréthistérone.
· Les autres composants sont :
Comprimés blancs : lactose monohydraté, amidon de maïs, hydroxypropylcellulose, talc, stéarate de magnésium. Pelliculage : hypromellose, triacétine, talc.
Comprimés rouges : lactose monohydraté, amidon de maïs, hydroxypropylcellulose, talc, stéarate de magnésium. Pelliculage : hypromellose, oxyde de fer rouge (E172), dioxyde de titane (E171), propylèneglycol, talc.
Qu’est-ce que NOVOFEMME, comprimé pelliculé et contenu de l’emballage extérieur
Chaque distributeur journalier de 28 comprimés contient 16 comprimés rouges et 12 comprimés blancs.
Présentations :
· 1 x 28 comprimés pelliculés
· 3 x 28 comprimés pelliculés
Toutes les présentations peuvent ne pas être commercialisées.
Titulaire de l’autorisation de mise sur le marché
10-12 COURS MICHELET
92800 PUTEAUX
FRANCE
Exploitant de l’autorisation de mise sur le marché
NOVO NORDISK
10-12 COURS MICHELET
92800 PUTEAUX
FRANCE
NOVO ALLÉ
DK-2880 BAGSVÆRD
DANEMARK
Noms du médicament dans les Etats membres de l'Espace Economique Européen
Ce médicament est autorisé dans les Etats membres de l'Espace Economique Européen et au Royaume-Uni (Irlande du Nord) sous les noms suivants : Conformément à la réglementation en vigueur.
[à compléter ultérieurement par le titulaire]
La dernière date à laquelle cette notice a été révisée est :
[à compléter ultérieurement par le titulaire]
Des informations détaillées sur ce médicament sont disponibles sur le site Internet de l’ANSM (France).
INSTRUCTIONS D’UTILISATION
Comment utiliser le distributeur journalier
|
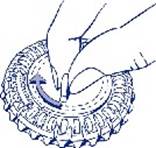
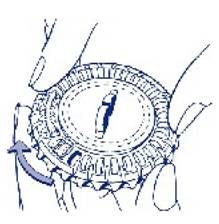
1. Sélection du jour Faites tourner le cadran central du distributeur journalier de façon à placer le jour de la semaine en face de la petite languette en plastique.
|
|
|
|
|
|
2. Prise du premier comprimé Cassez la languette en plastique et faites sortir le premier comprimé.
|
|
|
|
|
|
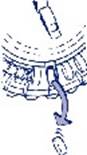
3. Déplacement du cadran tous les jours Le jour suivant, faites simplement avancer d’1 cran le cadran transparent en le tournant dans le sens des aiguilles d’une montre, comme indiqué par la flèche. Faites sortir le comprimé suivant. Rappelez-vous de ne prendre qu’1 seul comprimé par jour. Le cadran transparent ne peut être tourné que lorsque le comprimé se trouvant dans l’ouverture a été retiré. |
|
|
|
|

[1] Indiquer le libellé détaillé qui figure dans l’application form avec le code de la modification selon les lignes directrices https://ec.europa.eu/health//sites/health/files/files/eudralex/vol-2/c_2013_2008/c_2013_2008_pdf/c_2013_2804_fr.pdf