Dernière mise à jour le 03/08/2026
BASCELLEX 50 mg/g, crème
Ce médicament n'est ou ne sera bientôt plus disponible sur le marché.
Si vous prenez actuellement ce médicament, il vous est recommandé d'en parler avec votre médecin
ou avec votre pharmacien qui pourra vous orienter vers un autre traitement.
Indications thérapeutiques
Votre médecin peut vous prescrire BASCELLEX pour le traitement de :
· kératose actinique.
Les kératoses actiniques sont des lésions cutanées rugueuses chez des personnes ayant été beaucoup exposées au soleil au cours de leur vie. Certaines lésions ont la couleur de la peau, d’autres sont grisâtres, roses, rouges ou brunes. Elles peuvent être planes et squameuses ou surélevées, rugueuses dures et verruqueuses. BASCELLEX doit être utilisé pour les kératoses planes du visage ou du cuir chevelu chez les patients ayant un système immunitaire sain et lorsque le médecin a décidé que BASCELLEX était le traitement le plus approprié pour vous.
BASCELLEX aide votre système immunitaire à produire des substances naturelles qui interviennent dans la lutte contre votre kératose actinique.
Service médical rendu (SMR)
Pas de SMR disponible pour ce médicament ( plus d'informations dans l'aide )
Amélioration du service médical rendu (ASMR)
Pas d'ASMR disponible pour ce médicament (plus d'informations dans l'aide )
ANSM - Mis à jour le : 01/03/2024
2. COMPOSITION QUALITATIVE ET QUANTITATIVE
Chaque sachet contient 12,5 mg d'imiquimod dans 250 mg de crème (5 %).
1 g de crème contient 50 mg d’imiquimod.
Hydroxybenzoate de méthyle (E 218) 2,0 mg/g de crème
Hydroxybenzoate de propyle (E 216) 0,2 mg/g de crème
Alcool cétylique 22,0 mg/g de crème
Alcool stéarylique 31,0 mg/g de crème
Alcool benzylique 50,0 mg/g de crème
Pour la liste complète des excipients, voir rubrique 6.1.
Crème blanche à légèrement jaune.
4.1. Indications thérapeutiques
Veuillez également consulter la rubrique 5.1.
4.2. Posologie et mode d'administration
Le traitement doit être initié et suivi par un médecin. La crème imiquimod doit être appliquée 3 fois par semaine (par exemple : lundi, mercredi et vendredi) pendant 4 semaines avant l’heure du coucher et rester en contact pendant une huitaine d’heures. Il convient d'appliquer une quantité suffisante de crème pour recouvrir toute la zone à traiter. La disparition de la kératose doit être évaluée 4 semaines après la période de traitement. Si des lésions persistent, le traitement doit être répété pendant quatre autres semaines.
La dose maximale recommandée est de un sachet.
Une interruption du traitement doit être envisagée si les réactions inflammatoires locales intenses se développent (voir rubrique 4.4) ou si une infection est observée au site d’application. Dans ce dernier cas, il est nécessaire de prendre d’autres mesures appropriées. Même en cas d’oubli ou de périodes de repos le traitement ne doit pas être étendu au-delà de 4 semaines.
Si la zone traitée n’apparait pas complètement guérie lors de l’examen de suivi à environ 8 semaines après le dernier traitement de 4 semaines, un traitement additionnel de 4 semaines par l’imiquimod peut être envisagé.
Une autre thérapie est recommandée si la (les) lésion(s) traitée(s) montre(nt) une réponse insuffisante à l’imiquimod.
Les lésions de kératose actinique guéries après une ou deux périodes de traitement et qui réapparaissent ultérieurement peuvent être retraitées avec la crème imiquimod pendant une ou deux autres périodes de traitement après une période de repos d’au moins 12 semaines (voir rubrique 5.1).
En cas d’oubli d’une administration, le patient devra appliquer la crème dès que possible puis continuer le rythme habituel d’applications. Cependant, il convient de ne pas appliquer la crème plus d’une fois par jour.
Population pédiatrique
L'utilisation chez l'enfant n'est pas recommandée. Aucune donnée sur l'utilisation de l'imiquimod chez l'enfant et l'adolescent n'est disponible dans l’indication approuvée. Imiquimod ne doit pas être utilisé chez les enfants présentant un molluscum contagiosum en raison de l'absence d'efficacité dans cette indication (voir rubrique 5.1).
Mode d’administration
Avant d'appliquer la crème imiquimod, les patients doivent nettoyer la zone à traiter avec de l'eau et un savon doux et la sécher soigneusement. Il convient d'appliquer une quantité suffisante de crème pour recouvrir toute la zone à traiter. Masser pour faire pénétrer la crème dans la zone à traiter. La crème doit être appliquée avant l'heure normale du coucher et rester en contact avec la peau pendant une huitaine d'heures. Il convient d'éviter les douches et les bains pendant cette période. Il est indispensable ensuite d'enlever la crème imiquimod avec de l'eau et un savon doux. Les sachets ne doivent pas être réutilisés une fois ouverts. Il est nécessaire de bien se laver les mains avant et après l'application de la crème.
4.4. Mises en garde spéciales et précautions d'emploi
Eviter tout contact avec les yeux, les lèvres et les narines.
La crème imiquimod est susceptible d'exacerber les affections cutanées inflammatoires.
La crème imiquimod doit être utilisée avec précaution chez les patients ayant une maladie auto- immune (cf. rubrique 4.5). Le rapport bénéfice risque du traitement par imiquimod doit être évalué chez ces patients au regard du risque d’aggravation de leur condition auto-immunitaire.
La crème imiquimod doit être utilisée avec précaution chez les patients ayant une transplantation d’organe (cf. rubrique 4.5). Le rapport bénéfice risque du traitement par imiquimod doit être évalué chez ces patients au regard de la possibilité de risque de rejet de greffe ou d’une réaction du greffon contre l’hôte.
L'emploi de la crème imiquimod est déconseillé tant que la peau n'a pas cicatrisé après un traitement médicamenteux ou chirurgical antérieur. L’application sur peau lésée pourrait entraîner une augmentation de l’absorption systémique avec un risque accru d’effets indésirables (voir rubriques 4.8 et 4.9)
L'utilisation d'un pansement occlusif est déconseillée avec le traitement par la crème imiquimod.
Les excipients suivants méthylhydroxybenzoate (E 218) et propylhydroxybenzoate (E216) peuvent entraîner des réactions allergiques (éventuellement retardées). L'alcool cétylique et l'alcool stéarylique peuvent provoquer des réactions cutanées locales (par exemple dermatite de contact).
Rarement, des réactions locales inflammatoires intenses, tel qu’un exsudat ou une érosion, peuvent apparaître après quelques applications seulement de la crème imiquimod. Ces réactions inflammatoires locales peuvent être accompagnées ou précédées de signes pseudo grippaux et de symptômes tels que malaise, fièvre, nausée, myalgie et frissons. L’interruption du traitement doit être envisagée.
L’imiquimod doit être utilisé avec précaution chez les patients ayant une réserve hématologique réduite (voir rubrique 4.8d).
Les lésions cliniquement atypiques de KA ou évoquant un cancer doivent être biopsiées pour établir le traitement le plus approprié.
L'imiquimod n'a pas été évalué pour le traitement des kératoses actiniques des paupières, de l’intérieur des narines ou des oreilles, ou de la zone des lèvres à l’intérieur du vermillon.
Il existe très peu de données disponibles sur l’utilisation de l’imiquimod dans le traitement des kératoses actiniques situées ailleurs que sur le visage et le cuir chevelu. Les résultats obtenus dans le traitement des avant-bras et des mains ne confirmant pas son efficacité dans cette indication, son utilisation dans ce cadre n’est pas recommandée.
L’imiquimod n’est pas recommandé pour le traitement des lésions de KA avec hyperkératose ou hypertrophie marquée comme dans le cas des cornes cutanées.
![]() Pendant le traitement et jusqu'à la cicatrisation, la peau peut avoir un aspect très différent de celui de la peau saine. Les réactions cutanées locales sont fréquentes, mais leur intensité diminue généralement en cours de traitement ou elles cessent après l'arrêt de l'application de la crème imiquimod. Une relation a été établie entre le taux d'élimination complète et l'intensité des réactions cutanées locales (un érythème par exemple). Ces réactions cutanées locales peuvent être liées à la stimulation de la réponse immunitaire locale. Si la gêne occasionnée ou l'intensité de la réaction cutanée locale le requièrent, une période de repos de plusieurs jours peut être prescrite. Le traitement par la crème imiquimod peut être repris après atténuation de la réaction cutanée.
Pendant le traitement et jusqu'à la cicatrisation, la peau peut avoir un aspect très différent de celui de la peau saine. Les réactions cutanées locales sont fréquentes, mais leur intensité diminue généralement en cours de traitement ou elles cessent après l'arrêt de l'application de la crème imiquimod. Une relation a été établie entre le taux d'élimination complète et l'intensité des réactions cutanées locales (un érythème par exemple). Ces réactions cutanées locales peuvent être liées à la stimulation de la réponse immunitaire locale. Si la gêne occasionnée ou l'intensité de la réaction cutanée locale le requièrent, une période de repos de plusieurs jours peut être prescrite. Le traitement par la crème imiquimod peut être repris après atténuation de la réaction cutanée.
Chaque période de traitement ne doit pas être prolongée au-delà de 4 semaines, même en cas d’oublis de doses et de périodes de repos.
Le résultat clinique du traitement peut être déterminé après la régénération de la peau traitée, environ 4 à 8 semaines après la fin du traitement.
Nous n’avons aucune expérience clinique de l'utilisation de la crème imiquimod chez les patients immunodéprimés.
Les informations sur le re-traitement des lésions de kératose actinique qui ont guéri après une ou deux cures de traitement et qui réapparaissent ultérieurement sont présentées dans les rubriques 4.2 et 5.1.
Les données d'un essai clinique réalisé en ouvert suggèrent que la présence de plus de 8 lésions de KA a moins de chance de répondre au traitement par imiquimod.
La zone traitée doit être protégée de l’exposition solaire.
Ce médicament contient 12,5 mg d’alcool benzylique par sachet équivalent à 50 mg/g. L’alcool benzylique peut provoquer des réactions allergiques.
4.5. Interactions avec d'autres médicaments et autres formes d'interactions
Aucune étude d’interaction n’a été réalisée avec les immunosuppresseurs. De telles interactions avec des médicaments administrés par voie générale seraient limitées du fait de l'absorption percutanée minime de la crème imiquimod.
Du fait de ses propriétés immunostimulantes, la crème imiquimod doit être utilisée avec précaution chez les patients recevant un traitement immunosuppresseur (cf. rubrique 4.4).
4.6. Fertilité, grossesse et allaitement
Grossesse
Il n’existe pas de données sur l’utilisation de l’imiquimod chez la femme enceinte. Les études chez l’animal n’ont pas montré d’effets délétères directs ou indirects sur la gestation, le développement embryonnaire ou fœtal, accouchement ou le développement post-natal (voir rubrique 5.3). Imiquimod ne sera prescrit qu’avec prudence chez la femme enceinte.
Allaitement
Dans la mesure où aucune concentration quantifiable d’imiquimod (> 5 ng/ml) ne peut être détectée dans le sérum après administration locale unique ou réitérée, aucun conseil spécifique ne peut être donné pour une utilisation éventuelle chez les femmes allaitantes.
4.7. Effets sur l'aptitude à conduire des véhicules et à utiliser des machines
a) Description générale
Dans les études pivot utilisant une application de crème imiquimod 3 fois par semaine pendant 2 cures de 4 semaines chacune, 56 % des patients ont rapporté au moins un événement indésirable. Les réactions indésirables les plus fréquemment rapportées et jugées probablement ou possiblement imputables au traitement par la crème imiquimod ont été des réactions locales au site d’application (22 % des patients traités avec imiquimod). Quelques réactions indésirables systémiques, à type de myalgies (2 %) ont également été rapportées.
Les réactions indésirables rapportées chez les 252 patients traités par la crème imiquimod pour une kératose actinique dans des études cliniques de phase III contrôlées contre placebo sont présentées ci- après. L'imputabilité de ces événements indésirables au traitement par imiquimod est jugée au moins possible.
b) Tableaux des événements indésirables
Les fréquences sont définies comme suit : très fréquent (≥ 1/10) ; fréquent (≥ 1/100 à < 1/10) ; peu fréquent (≥ 1/1 000 à < 1/100). Les fréquences moindres observées dans les essais cliniques ne sont pas rapportées ici.
|
Infections et infestations: |
|
|
Infection |
Peu fréquente |
|
Pustules |
Peu fréquentes |
|
Rhinite |
Peu fréquente |
|
Grippe |
Peu fréquente |
|
Affections hématologiques et du système lymphatique |
|
|
Lymphadénopathie |
Peu fréquente |
|
Troubles du métabolisme et de la nutrition |
|
|
Anorexie |
Fréquente |
|
Affections psychiatriques : |
|
|
Dépression |
Peu fréquente |
|
Affections du système nerveux : |
|
|
Céphalées |
Fréquentes |
|
Affections oculaires |
|
|
Irritation conjonctivale |
Peu fréquente |
|
Œdème des paupières |
Peu fréquent |
|
Affections respiratoires, thoraciques et médiastinales : |
|
|
Congestion nasale |
Peu fréquente |
|
Douleur pharyngo-laryngée |
Peu fréquente |
|
Affections gastro-intestinales : |
|
|
Nausées |
Fréquentes |
|
Diarrhée |
Peu fréquente |
|
Affections de la peau et du tissu sous-cutané |
|
|
Kératose actinique |
Peu fréquente |
|
Erythème |
Peu fréquent |
|
Œdème du visage |
Peu fréquent |
|
Ulcère cutané |
Peu fréquent |
|
Affections musculo-squelettiques et systémiques |
|
|
Myalgies |
Fréquentes |
|
Arthralgies |
Fréquentes |
|
Douleur des extrémités |
Peu fréquente |
|
Troubles généraux et anomalies au site d’administration : |
|
|
Prurit au site d'application |
Très fréquent |
|
Douleurs au site d'application |
Fréquentes |
|
Brûlure au site d'application |
Fréquente |
|
Irritation au site d'application |
Fréquente |
|
Erythème au site d'application |
Fréquente |
|
Réaction au site d’application |
Fréquente |
|
Saignement au site d'application |
Peu fréquent |
|
Papules au site d'application |
Peu fréquentes |
|
Paresthésies au site d'application |
Peu fréquentes |
|
Fatigue |
Fréquente |
|
Fièvre |
Peu fréquente |
|
Asthénie |
Peu fréquente |
|
Frissons |
Peu fréquents |
|
Dermatite au site d’application |
Peu fréquente |
|
Ecoulement au site d'application |
Peu fréquent |
|
Hyperesthésie au site d’application |
Peu fréquente |
|
Œdème au site d'application |
Peu fréquent |
|
Desquamation au site d'application |
Peu fréquente |
|
Cicatrice au site d’application |
Peu fréquente |
|
Tuméfaction au site d'application |
Peu fréquente |
|
Ulcération au site d'application |
Peu fréquente |
|
Vésicules au site d'application |
Peu fréquentes |
|
Chaleur au site d’application |
Peu fréquente |
|
Gêne |
Peu fréquente |
|
Inflammation |
Peu fréquente |
c) Evénements indésirables fréquents
Dans les essais cliniques avec application de la crème imiquimod 3 fois par semaine pendant 4 ou 8 semaines les réactions indésirables les plus fréquentes au site d’application étaient les démangeaisons (14 %) et les brûlures (5 %). Les érythèmes sévères (24 %) et l'excoriation/la desquamation sévères (20 %) ont été très fréquents. Les réactions cutanées locales, tel que l'érythème, sont probablement une conséquence des effets pharmacologiques de la crème imiquimod. Voir 4.2 et 4.4 pour plus de détails sur les périodes de repos.
Des infections cutanées ont été observées pendant le traitement par imiquimod. Bien qu’aucunes séquelles graves n’aient été observées la possibilité d’infection de la peau endommagée doit toujours être envisagée.
Des cas d'hypopigmentation et d'hyperpigmentation localisées ont été rapportés après utilisation de la crème imiquimod. Les données de suivi suggèrent que ces modifications de la coloration de la peau pourraient être définitives chez certains patients.
Les études cliniques examinant l’utilisation de l’imiquimod dans le traitement de la kératose actinique ont détecté l'apparition d'une alopécie dans 0,4 % des cas (5/1214) au site d’application ou la zone environnante.
Des diminutions de l'hémoglobine, du nombre de leucocytes, du nombre absolu de granulocytes neutrophiles et du nombre de plaquettes ont été observées dans les essais cliniques. Ces diminutions ne sont pas jugées cliniquement significatives chez les patients ayant une réserve hématologique normale. Les patients présentant des réserves hématologiques réduites n'ont pas été étudiés dans les essais cliniques. Les rapports de pharmacovigilance ont fait état de diminutions des paramètres hématologiques nécessitant une intervention clinique. Des cas d’élévation des enzymes hépatiques ont été signalés après la mise sur le marché.
De rare cas de poussées de maladies auto-immunes ont été rapportés.
De rares cas de réactions dermatologiques éloignées du site d’application, incluant l’érythème multiforme ont été reportés dans les essais cliniques. Depuis sa première mise sur le marché, imiquimod a fait l’objet de rapports de réactions cutanées graves, dont d’érythème multiforme, de syndrome de Stevens-Johnson ou de lupus érythémateux cutané.
d) Population pédiatrique
L’imiquimod a été étudié dans des études cliniques contrôlées chez des enfants (voir rubriques 4.2 et 5.1). Il n’a pas été observé de réactions systémiques. Des réactions au site d’application sont apparues plus fréquemment avec l’imiquimod qu’avec l’excipent, néanmoins la fréquence et l’intensité de ces réactions n’ont pas été différentes de celles qui sont observées chez l’adulte dans les indications approuvées. Il n’a pas été constaté d’effets indésirables graves causés par l’imiquimod chez l’enfant.
Déclaration des effets indésirables suspectés
La déclaration des effets indésirables suspectés après autorisation du médicament est importante. Elle permet une surveillance continue du rapport bénéfice/risque du médicament. Les professionnels de santé déclarent tout effet indésirable suspecté via le système national de déclaration : Agence nationale de sécurité du médicament et des produits de santé (ANSM) et réseau des Centres Régionaux de Pharmacovigilance - Site internet : www.signalement-sante.gouv.fr.
Après ingestion accidentelle, des nausées, des vomissements, des céphalées, des myalgies et de la fièvre pourraient survenir après une prise unique de 200 mg d'imiquimod, ce qui correspond au contenu d'environ 16 sachets. Les événements indésirables cliniques les plus graves, rapportés après prise orale réitérée de doses supérieures ou égales à 200 mg, ont été représentés par des hypotensions corrigées par administration orale ou intraveineuse de solutés.
5. PROPRIETES PHARMACOLOGIQUES
5.1. Propriétés pharmacodynamiques
Classe pharmaco-thérapeutique: Chimiothérapie à usage topique, antiviraux, Code ATC : D06BB10.
L'imiquimod est un modificateur de la réponse immunitaire. Les études d’affinité évoquent l'existence d'un récepteur membranaire de l'imiquimod à la surface des cellules immunitaires répondeuses.
L'imiquimod n’a pas d'activité antivirale directe. Dans des modèles animaux, l'imiquimod est efficace contre les infections virales et se comporte comme un agent antitumoral, principalement par induction de l'interféron alpha et d'autres cytokines.
Des augmentations de la concentration systémique de l'interféron alpha et d'autres cytokines après application topique d'imiquimod ont été observées dans une étude pharmacocinétique.
Kératose actinique (données disponibles pour le produit à tester, BASCELLEX 50 mg/g, crème).
Efficacité clinique
Une étude clinique randomisée, en double aveugle, sur plusieurs sites, contrôlée contre placebo et réalisée en parallèle, a été conduite pour évaluer l'équivalence thérapeutique entre le produit à tester, BASCELLEX 50 mg/g, crème et le produit de référence, Imiquimod 5% crème chez des patients atteints de Kératose Actinique. Les patients ont été randomisés et traités pendant 8 semaines (deux sessions de 4 semaines).
Les évaluations d'efficacité étaient basées sur le nombre de lésions, observées par l'investigateur lors de toutes les visites cliniques. L'analyse statistique primaire d’intérêt a porté sur la proportion de patients considérés comme complètement guéris, définie comme étant une disparition à 100% de toutes les lésions AK dans la zone de traitement.
Pour l'évaluation de l'équivalence thérapeutique du critère principal dans les populations per protocole et en intention de traiter (ITT) modifiée, les résultats suivants ont été observés: l'intervalle de confiance à 95% pour la différence entre la proportion de patients considérés comme complètement guéris dans le groupe du produit à tester et le groupe du produit de référence a été compris dans l'intervalle d'équivalence de -20% à + 20% dans les deux populations. Concernant les analyses de supériorité du critère principal, les résultats suivants ont été observés: la proportion de patients considérés comme complètement guéris dans le groupe à tester et le groupe de référence était significativement plus élevée de 5% (p < 0,05) par rapport à la proportion de patients considérés comme complètement guéris dans le groupe placebo.
|
Equivalence: population - par protocole |
||||||
|
|
Différence de traitements |
|||||
|
Groupe de traitement |
No |
Proportion de patients avec une élimination partielle de KA (%) |
Différence |
95 % IC-évaluation |
||
|
Test |
187 |
60,43 % |
-1,11 % |
-11,61; 9,39 |
||
|
Reference |
182 |
61,54 % |
||||
|
Equivalence: population mITT |
||||||
|
Groupe de traitement |
No |
Proportion de patients avec une élimination partielle de KA (%) |
Différence |
95 % IC- évaluation |
||
|
Test |
235 |
54,47 % |
-3,29 % |
-12,72 %, 6,13 |
||
|
Reference |
232 |
57,76 % |
||||
|
Supériorité: population mITT |
||||||
|
Groupe de traitement |
No |
Proportion de patients avec une élimination partielle de KA (%) |
Différence |
95 % IC |
valeur-P |
|
|
Placebo |
79 |
39,24 % |
- |
- |
- |
|
|
Test |
235 |
54,47 % |
15,23 % |
1,87; 28,58 |
0,0270 |
|
|
Reference |
232 |
57,76 % |
18,52 % |
5,17; 31,87 |
0,0066 |
|
L’IC à 95% est basé sur l’Ecart-type de la différence en utilisant les réponses individuelles (non groupées); La valeur P est basée sur l’Ecart-type de la différence en utilisant la réponse groupée.
Par conséquent, l'équivalence et la supériorité ont été démontrées pour le critère principal.
Kératose actinique (données disponibles pour le produit de référence, imiquimod 5% crème
Efficacité clinique
L’efficacité d’imiquimod appliqué 3 fois par semaine pendant une ou 2 cures, séparé par une période de repos de 4 semaines, a été étudiée dans deux études cliniques menées en double aveugle contre excipient.Les patients avaient des kératoses actiniques visibles, cliniquement typiques, non confluentes, non hypertrophiques, non hyperkératosiques, sur la calvitie ou sur la face sur une surface traitée contiguë de 25 cm2. Quatre à huit lésions ont été traitées. La disparition complète (effet imiquimod moins effet excipient) dans les essais cumulés est de 46.1% (IC 39.0%,53.1%).
Les données à un an issues de deux études observationnelles indiquent un taux de récidive de 27 % (35/128) chez les patients ne présentant plus de lésions après une ou deux cures de traitement. Le taux de récidive des lésions individuelles était de 5,6 % (41/737). Le taux correspondant de récidive pour l’excipient était de 47% (8/17 patients) et de 7.5% (6/80 lésions).
Deux études cliniques en ouvert, randomisées, contrôlées, ont comparé les effets à long terme de l'imiquimod avec ceux du diclofénac topique chez des patients ayant une kératose actinique à l'égard du risque de progression vers un carcinome à cellules squameuses in situ ou invasif (CCS).
Les traitements ont été administrés selon les recommandations officielles. Si la zone de kératose actinique traitée n'était pas complètement guérie des lésions, des cures de traitement additionnelles pouvaient être initiées. Tous les patients étaient suivis jusqu'à la sortie de l'essai ou jusqu'à 3 ans après la randomisation. Les résultats sont issus d'une méta-analyse des deux études.
Un total de 482 patients ont été inclus dans les études, parmi lesquels 481 patients ont reçu les traitements de l'étude, et dont 243 patients étaient traités avec de l'imiquimod et 238 patients avec du diclofénac topique. La zone de kératose actinique traitée était localisée sur le cuir chevelu avec une calvitie ou sur le visage avec une surface contiguë d'environ 40 cm2 pour les deux groupes de traitement présentant un nombre médian de 7 lésions de kératose actinique cliniquement typiques à l'inclusion. 90 patients ont eu au moins 3 cures de traitement avec l'imiquimod, 80 patients ont reçu au moins 5 cures de traitement avec l'imiquimod durant les 3 ans d'étude.
En ce qui concerne le critère primaire, la progression histologique, au global 13 des 242 patients (5,4 %) du groupe imiquimod et 26 des 237 patients (11,0 %) du groupe diclofénac ont montré une progression histologique vers un CCS in situ ou invasif dans les 3 ans, une différence de -5,6 % (95 % IC : -10,7 % à -0,7 %). De ceux-ci, 4 des 242 patients (1,7 %) du groupe imiquimod et 7 des 237 patients (3,0 %) du groupe diclofénac ont montré une progression histologique vers un CCS invasif dans les 3 ans.
Un total de 126 des 242 patients traités avec imiquimod (52,1 %) et 84 des 237 patients traités par du diclofénac topique (35,4 %) ont montré une guérison clinique complète de la zone de kératose actinique traitée à la semaine 20 (à savoir environ 8 semaines après la fin de la cure de traitement initiale) ; une différence de 16,6 % (95 % IC : 7,7 % à 25,1 %). La récurrence des lésions de kératose actinique a été évaluée chez les patients dont la zone traitée a été cliniquement complètement guérie. Un patient était considéré comme récurrent dans ces études si au moins une lésion de kératose actinique était observée dans la zone complètement guérie et une lésion récurrente pouvait être une lésion qui apparaissait au même endroit que la lésion précédemment guérie ou une nouvelle lésion identifiée n'importe où dans la zone traitée. Le risque de récurrence des lésions de kératose actinique dans la zone traitée (comme définie ci-dessus) était de 39,7 % (50 des 126 patients) jusqu'à 12 mois pour les patients traités avec de l'imiquimod comparé à 50,0 % (42 des 84 patients) pour les patients traités avec du diclofénac topique, une différence de -10,3 % (95 % IC : -23,6 % à 3,3 %) ; et 66,7 % (84 des 126 patients) pour un traitement avec de l'imiquimod et 73,8 % (62 des 84 patients) pour le diclofénac topique jusqu'à 36 mois, une différence de -7,1 % (95 % IC : -19,0 % à 5,7 %).
Un patient avec des lésions de kératose actinique récurrentes (comme définies ci-dessus) dans la zone qui a été complètement guérie avait une probabilité d'environ 80 % d'obtenir à nouveau une guérison complète après une cure de traitement additionnelle par imiquimod comparé à environ 50 % pour un retraitement avec du diclofénac topique.
Population pédiatrique
L’indication approuvée kératose actinique n’est généralement pas observée chez l'enfant et n'a pas été étudiée.
Imiquimod a été évalué dans quatre essais randomisés, contrôlés, en double aveugle chez des enfants de 2 à 15 ans atteints de molluscum contagiosum (imiquimod n = 576, excipient n = 313). Ces études n'ont pas permis de démontrer l'efficacité de l'imiquimod quelque-soit la posologie utilisée (3 x/semaine pendant ≤ 16 semaines ou 7 x/semaine pendant ≤ 8 semaines).
Le bénéfice-risque de BASCELLEX 50 mg/g, crème dans les indications de verrues génitales et péri-anales externes et de petits carcinomes baso-cellulaires superficiels, approuvés pour le produit de référence imiquimod 5% crème est inconnu en raison de l’absence de données.
5.2. Propriétés pharmacocinétiques
L’exposition systémique (pénétration percutanée) a été calculée à partir de l’élimination dans les urines et les fèces de l’imiquimod marqué au carbone 14.
Une faible absorption systémique de la crème imiquimod à 5% à travers la peau de 58 patients présentant une kératose actinique a été observée avec l'administration trois fois par semaine pendant 16 semaines. Le taux d'absorption percutanée n'a pas varié significativement entre la première et la dernière dose de cette étude. Le pic de concentration sérique à la fin de la 16esemaine a été observé entre 9 à 12 heures et a été de 0,1, 0,2 et 1,6 ng/ml respectivement pour les applications sur le visage (12,5 mg, 1 sachet à usage unique), sur le cuir chevelu (25 mg, 2 sachets) et sur les mains/bras (75 mg, 6 sachets). La surface de la zone d'application n'était pas contrôlée dans les groupes d'application sur le cuir chevelu et les mains/bras. Aucune proportionnalité avec la dose n'a été observée. Une demi-vie apparente a été calculée, qui a été 10 fois supérieure environ à la demi-vie de deux heures observée après administration sous-cutanée dans une étude précédente, ce qui suggère une rétention prolongée du médicament dans la peau. L'élimination urinaire a été inférieure à 0,6% de la dose appliquée à la semaine 16 chez ces patients.
Population pédiatrique
Les propriétés pharmacocinétiques de l'imiquimod ont été étudiées après application locale unique ou répétée chez l'enfant atteint de molluscum contagiosum (MC). Les données d'exposition systémique démontrent que l'absorption de l'imiquimod après application locale sur des lésions de MC chez l'enfant de 6 à 12 ans est faible et comparable à celle qui est observée chez le volontaire sain et l'adulte atteint de kératose actinique. Chez des patients plus jeunes de 2-5ans, l'absorption, évaluée par les valeurs de Cmax, est supérieure par rapport à celle de l'adulte.
5.3. Données de sécurité préclinique
Dans une étude de quatre mois de la toxicité dermique chez le rat, on a observé une diminution significative du poids corporel et une augmentation du poids de la rate avec les doses de 0,5 et 2,5 mg/kg; aucun effet comparable n'a été constaté dans une étude de quatre mois de la toxicité dermique chez la souris. Une irritation dermique locale a été observée dans ces deux espèces, en particulier avec les doses élevées.
![]() Lors d’une étude de carcinogénicité de deux ans réalisée chez la souris, l'application cutanée 3 jours par semaine n'a pas entraîné l'apparition de tumeurs dans la zone d'application. Chez les animaux traités cependant, la fréquence des tumeurs hépatocellulaires a été plus élevée que chez les témoins. Le mécanisme n’est pas connu, mais l'absorption systémique de l'imiquimod à partir de la peau humaine étant minime et l'imiquimod n'étant pas mutagène, le risque pour l'homme résultant d'une exposition systémique apparaît faible. De plus, aucune tumeur n'a été décelée en quelque endroit que ce soit dans une étude de carcinogenicité par voie orale pendant deux ans chez le rat.
Lors d’une étude de carcinogénicité de deux ans réalisée chez la souris, l'application cutanée 3 jours par semaine n'a pas entraîné l'apparition de tumeurs dans la zone d'application. Chez les animaux traités cependant, la fréquence des tumeurs hépatocellulaires a été plus élevée que chez les témoins. Le mécanisme n’est pas connu, mais l'absorption systémique de l'imiquimod à partir de la peau humaine étant minime et l'imiquimod n'étant pas mutagène, le risque pour l'homme résultant d'une exposition systémique apparaît faible. De plus, aucune tumeur n'a été décelée en quelque endroit que ce soit dans une étude de carcinogenicité par voie orale pendant deux ans chez le rat.
La crème imiquimod a été évalué dans un test biologique de la photo-carcinogénicité réalisé sur des souris albinos nues exposées à des rayons ultra-violets artificiels. Les animaux ont reçu la crème imiquimod trois fois par semaine et ont été irradiés cinq jours par semaine pendant 40 semaines. Les souris ont été conservées pendant 12 semaines de plus, soit un total de 52 semaines. Des tumeurs sont apparues plus précocement et en plus grand nombre dans le groupe des souris recevant la crème placebo que dans le groupe témoin exposé à un faible rayonnement UV. La signification de cette observation pour l'homme n’est pas connue. L'administration locale de la crème imiquimod n'a pas entraîné d'augmentation des tumeurs, à quelque dose que ce soit, par rapport au groupe recevant la crème placebo.
6.4. Précautions particulières de conservation
A conserver à une température ne dépassant pas 25°C.
Ne pas réutiliser les sachets déjà ouverts.
6.5. Nature et contenu de l'emballage extérieur
Boîte de 12 ou 24 sachets à usage unique en aluminium, contenant 250 mg de crème.
Toutes les tailles de conditionnement peuvent ne pas être commercialisées.
6.6. Précautions particulières d’élimination et de manipulation
Pas d'exigences particulières.
7. TITULAIRE DE L’AUTORISATION DE MISE SUR LE MARCHE
Sun Pharmaceutical Industries Europe B.V.
Polarisavenue 87
2132 JH, Hoofddorp
Pays-Bas
8. NUMERO(S) D’AUTORISATION DE MISE SUR LE MARCHE
· 34009 301 655 2 2 : Crème en sachet (Aluminium) de 250 mg ; boîte de 12.
· 34009 301 655 3 9 : Crème en sachet (Aluminium) de 250 mg ; boîte de 24.
9. DATE DE PREMIERE AUTORISATION/DE RENOUVELLEMENT DE L’AUTORISATION
[à compléter ultérieurement par le titulaire]
10. DATE DE MISE A JOUR DU TEXTE
[à compléter ultérieurement par le titulaire]
Sans objet.
12. INSTRUCTIONS POUR LA PREPARATION DES RADIOPHARMACEUTIQUES
Liste I
ANSM - Mis à jour le : 01/03/2024
imiquimod
Veuillez lire attentivement cette notice avant d’utiliser ce médicament car elle contient des informations importantes pour vous.
· Gardez cette notice. Vous pourriez avoir besoin de la relire
· Si vous avez d’autres questions, interrogez votre médecin ou votre pharmacien.
· Ce médicament vous a été personnellement prescrit. Ne le donnez pas à d’autres personnes. Il pourrait leur être nocif, même si les signes de leur maladie sont identiques aux vôtres.
· Si vous ressentez un quelconque effet indésirable, parlez-en à votre médecin ou à votre pharmacien. Ceci s’applique aussi à tout effet indésirable qui ne serait pas mentionné dans cette notice. Voir rubrique 4.
1. Qu'est-ce que BASCELLEX 50 mg/g, crème et dans quels cas est-il utilisé ?
2. Quelles sont les informations à connaître avant d'utiliser BASCELLEX 50 mg/g, crème ?
3. Comment utiliser BASCELLEX 50 mg/g, crème ?
4. Quels sont les effets indésirables éventuels ?
5. Comment conserver BASCELLEX 50 mg/g, crème ?
6. Contenu de l’emballage et autres informations.
1. QU’EST-CE QUE BASCELLEX 50 mg/g, crème ET DANS QUELS CAS EST-IL UTILISE ?
Votre médecin peut vous prescrire BASCELLEX pour le traitement de :
· kératose actinique.
Les kératoses actiniques sont des lésions cutanées rugueuses chez des personnes ayant été beaucoup exposées au soleil au cours de leur vie. Certaines lésions ont la couleur de la peau, d’autres sont grisâtres, roses, rouges ou brunes. Elles peuvent être planes et squameuses ou surélevées, rugueuses dures et verruqueuses. BASCELLEX doit être utilisé pour les kératoses planes du visage ou du cuir chevelu chez les patients ayant un système immunitaire sain et lorsque le médecin a décidé que BASCELLEX était le traitement le plus approprié pour vous.
BASCELLEX aide votre système immunitaire à produire des substances naturelles qui interviennent dans la lutte contre votre kératose actinique.
2. QUELLES SONT LES INFORMATIONS A CONNAITRE AVANT D’UTILISER BASCELLEX 50 mg/g, crème ?
N’utilisez jamais BASCELLEX 50 mg/g, crème :
Si vous êtes allergique à l'imiquimod ou à l’un des autres composants contenus de ce produit (mentionnés dans la rubrique 6).
Mises en garde et précautions d’emploi
Adressez-vous à votre médecin avant d’utiliser BASCELLEX :
· si vous avez utilisé auparavant BASCELLEX ou d’autres préparations similaires
· si vous avez des problèmes immunitaires
· si vous avez une numération sanguine anormale.
· N'utilisez pas BASCELLEX avant que la zone à traiter ait cicatrisé après un traitement médical ou chirurgical antérieur
· Evitez tout contact avec les yeux, les lèvres et les narines. En cas de contact accidentel, enlever la crème en rinçant à l’eau
· Ne pas appliquer la crème sur les parties internes
· Ne pas appliquer plus de crème que la quantité prescrite par le médecin
· Ne recouvrez pas la zone traitée de bandages ou d’autres pansements après avoir appliqué BASCELLEX
· Si la gêne ressentie au niveau de la zone traitée devient trop pénible, vous devez éliminer la crème par lavage à l’aide d’eau et d’un savon doux. Dès que le problème aura disparu, vous pourrez recommencer à appliquer la crème
· n'utilisez pas de lampe solaire ni de cabine à bronzer et évitez autant que possible de vous exposer au soleil pendant le traitement par BASCELLEX. Portez des vêtements protecteurs et un chapeau à large bord quand vous sortez.
Du fait du mode d'action de BASCELLEX, il est possible que la crème aggrave une inflammation existante dans la zone traitée.
Pendant que vous prenez BASCELLEX et jusqu'à la cicatrisation, la zone traitée est susceptible d'avoir un aspect nettement différent de celui de la peau saine.
Enfants et adolescents
L’utilisation chez l’enfant et l’adolescent n’est pas recommandée.
Autres médicaments et BASCELLEX 50 mg/g, crème
Informez votre médecin ou pharmacien si vous prenez, avez récemment pris ou pourriez prendre tout autre médicament.
Aucun médicament n’est connu comme incompatible avec BASCELLEX.
Grossesse et allaitement
Demandez conseil à votre médecin ou à votre pharmacien avant de prendre tout médicament.
Si vous êtes enceinte ou si vous avez l’intention de le devenir, vous devez en informer votre médecin. Seul celui-ci pourra juger des risques et du bénéfice de l’utilisation de BASCELLEX pendant une grossesse.
N’allaitez pas votre enfant pendant que vous êtes traitée par BASCELLEX parce qu’on ne sait pas si l'imiquimod passe dans le lait maternel.
Conduite de véhicules et utilisation de machines
Ce médicament n'a pas ou peu d'influence sur l'aptitude à conduire et à utiliser des machines.
BASCELLEX 50 mg/g, crème contient du parahydroxybenzoate de méthyle, du parahydroxybenzoate de propyle, de l'alcool cétylique, de l'alcool stéarylique et de l’alcool benzylique
Le parahydroxybenzoate de méthyle (E218) et le parahydroxybenzoate de propyle (E216) peuvent entraîner des réactions allergiques (pouvant être différées). L’alcool cétylique et l’alcool stéarylique peuvent provoquer des reactions cutanées locales (par exemple dermatite de contact). L’alcool benzylique peut provoquer des réactions allergiques.
3. COMMENT UTILISER BASCELLEX 50 mg/g, crème ?
L’utilisation chez l’enfant et l’adolescent n’est pas recommandée.
Adulte
Respectez toujours la posologie indiquée par votre médecin. En cas d’incertitude, consultez votre médecin ou votre pharmacien.
Lavez-vous soigneusement les mains avant et après l'application de la crème. Ne recouvrez pas la zone traitée de bandages ou d’autres pansements après avoir appliqué BASCELLEX.
Ouvrez un nouveau sachet à chaque fois que vous utilisez la crème. Jetez le sachet après utilisation, même s’il reste de la crème. Ne conservez pas un sachet ouvert pour une utilisation ultérieure.
BASCELLEX Crème Instructions d’application
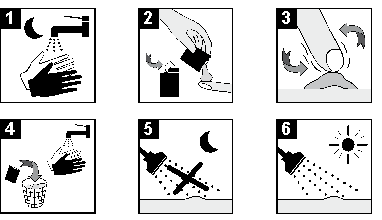
Instructions d’application
1. Avant de vous coucher, lavez vos mains et la zone à traiter avec de l'eau et un savon doux. Séchez- vous soigneusement.
2. Ouvrez un nouveau sachet et prenez de la crème sur le bout des doigts.
3. Appliquez la crème sur la zone à traiter. Massez doucement pour faire pénétrer la crème dans la zone à traiter.
4. Après avoir appliqué la crème, jetez le sachet ouvert. Lavez vos mains avec de l’eau et du savon.
5. Laissez BASCELLEX sur la peau pendant environ 8 heures. Ne pas prendre de bain ni de douche pendant cette période.
6. Après 8 heures, lavez la zone traitée avec de l’eau et un savon doux.
Appliquez BASCELLEX 3 fois par semaine, par exemple lundi, mercredi et vendredi. Un sachet contient assez de crème pour couvrir une zone de 25 cm2. Continuer le traitement pendant 4 semaines. Quatre semaines après avoir terminé ce premier traitement, votre médecin évaluera votre peau. Si les lésions n’ont pas entièrement disparu, 4 semaines supplémentaires de traitement peuvent être nécessaires.
Si vous avez utilisé plus de BASCELLEX 50 mg/g, crème que vous n’auriez dû
Lavez l’excès avec de l’eau et un savon doux. Après la disparition d’une éventuelle réaction cutanée, vous pourrez reprendre votre traitement.
Si vous avalez accidentellement BASCELLEX, contactez votre médecin.
Si vous oubliez d’utiliser BASCELLEX 50 mg/g, crème
Si vous oubliez une application, appliquez la crème dès que vous vous en rendez compte puis reprenez votre schéma d’administration habituel. N’appliquez pas la crème plus d’une fois par jour.
Si vous avez d’autres questions sur l’utilisation de ce médicament, demandez plus d’informations à votre médecin ou à votre pharmacien.
4. QUELS SONT LES EFFETS INDESIRABLES EVENTUELS ?
· Rarement des réactions cutanées graves ont été reportées.
Arrêtez BASCELLEX 50 mg/g, crème et parlez-en à votre médecin rapidement, si vous avez les lésions cutanées ou des tâches sur la peau qui commencent par des petites zones rouges et qui évoluent sous forme de mini cibles, éventuellement avec des symptômes tels que démangeaisons, fièvre, sensation générale de malaise, douleurs articulaires, problèmes visuels, brûlures, yeux douloureux ou qui picotent, bouche irritée, arrêtez BASCELLEX et parlez-en à votre médecin rapidement.
Informez-en votre médecin ou votre pharmacien dès que possible :
· si vous ne vous sentez pas bien pendant la période d’utilisation de BASCELLEX.
· si votre peau réagit mal lors de l’utilisation de BASCELLEX. Vous devez cesser d’appliquer la crème, laver la région concernée avec de l’eau et un savon doux.
· si vous êtes plus sensible aux infections, vous avez facilement de l’ecchymose, ou vous vous sentez fatigué. Ces signes pourraient être les symptômes d’une diminution du nombre de cellules sanguines notée chez certains patients.
Quelques patients ont présenté des modifications de la couleur de la peau dans la région où BASCELLEX a été appliqué. Bien que ces anomalies aient eu tendance à s'améliorer avec le temps, elles peuvent être définitives chez certains patients.
Un petit nombre de patients ont présenté une chute de cheveux sur la zone traitée ou la zone avoisinante.
Autres effets indésirables
Un grand nombre des effets indésirables de BASCELLEX sont dus à son action locale sur la peau. Les réactions cutanées locales peuvent être un signe d'efficacité du médicament.
Effets indésirables très fréquents (peuvent toucher plus d’une personne sur 10)
· la peau traitée peut être légèrement prurigineuse.
Effets indésirables fréquents (peuvent toucher jusqu’à une personne sur 10)
· douleurs
· brûlures
· irritation
· rougeurs
· céphalées
· anorexie
· nausées
· douleurs musculaires
· douleurs articulaires
· fatigue.
Si une réaction cutanée devient trop pénible pendant le traitement, parlez-en à votre médecin. Il se peut qu'il vous conseille d'interrompre l’application de BASCELLEX pendant quelques jours (c'est-à-dire d'avoir une courte période sans traitement). En cas de présence de pus ou d'un autre signe évocateur d'une infection, parlez-en à votre médecin.
Effets indésirables peu fréquents (peuvent toucher jusqu’à une personne sur 100)
· modifications du site d'application (saignements, inflammation, écoulement, sensibilité, oedème, petites tuméfactions cutanées, picotements ou fourmillements, croûte, cicatrice, ulcération ou une sensation de chaleur ou de gêne)
· inflammation de la muqueuse nasale
· obstruction nasale
· symptômes grippaux ou pseudo-grippaux
· dépression
· une irritation oculaire
· un œdème des paupières
· un mal de gorge
· des diarrhées
· une kératose actinique
· un œdème du visage
· des ulcères
· des douleurs des extrémités
· une fièvre
· une faiblesse
· des frissons.
Déclaration des effets secondaires
Si vous ressentez un quelconque effet indésirable, parlez-en à votre médecin, votre pharmacien ou à votre infirmier/ère. Ceci s’applique aussi à tout effet indésirable qui ne serait pas mentionné dans cette notice. Vous pouvez également déclarer les effets indésirables directement via le système national de déclaration : Agence nationale de sécurité du médicament et des produits de santé (ANSM) et réseau des Centres Régionaux de Pharmacovigilance - Site internet : www.signalement-sante.gouv.fr.
En signalant les effets indésirables, vous contribuez à fournir davantage d’informations sur la sécurité du médicament.
5. COMMENT CONSERVER BASCELLEX 50 mg/g, crème ?
Tenir ce médicament hors de la vue et de la portée des enfants.
Ne pas utiliser ce médicament après la date de péremption figurant sur l'emballage. La date de péremption se réfère au dernier jour du mois figurant sur l’emballage.
A conserver à une température ne dépassant pas 25ºC.
Ne pas réutiliser les sachets déjà ouverts.
Ne jetez aucun médicament au tout à l’égout ou avec les ordures ménagères. Demandez à votre pharmacien d’éliminer les médicaments que vous n’utilisez plus. Ces mesures contribueront à protéger l’environnement.
6. CONTENU DE L’EMBALLAGE ET AUTRES INFORMATIONS
Ce que contient BASCELLEX 50 mg/g, crème
· La substance active est l’imiquimod.
Chaque sachet contient 12,5 mg d’imiquimod dans 250 mg de crème (1 g de crème contient 50 mg d’imiquimod).
· Les autres composants sont :
Ll’acide iso-stéarique, l’alcool benzylique, l’alcool cétylique, l’alcool stéarylique, la vaseline blanche, le polysorbate 60, le stéarate de sorbitan, le glycérol, le parahydroxybenzoate de méthyle (E218), le parahydroxybenzoate de propyle (E216), la gomme xanthane, l’eau purifiée.
Qu’est-ce que BASCELLEX 50 mg/g, crème et contenu de l’emballage extérieur
BASCELLEX 50 mg/g, crème est une crème blanche à légèrement jaune.
Chaque boîte renferme 12 ou 24 sachets en aluminium à usage unique (250 mg de crème/sachet).
Toutes les tailles de conditionnement peuvent ne pas être commercialisées.
Titulaire de l’autorisation de mise sur le marché
Sun Pharmaceutical Industries Europe B.V.
Polarisavenue 87
2132 JH, Hoofddorp
Pays-Bas
Exploitant de l’autorisation de mise sur le marché
31 RUE DES POISSONNIERS
92200 NEUILLY-SUR-SEINE
Sun Pharmaceutical Industries Europe B.V.
Polarisavenue 87
2132 JH, Hoofddorp
Pays-Bas
Noms du médicament dans les Etats membres de l'Espace Economique Européen
Ce médicament est autorisé dans les Etats membres de l'Espace Economique Européen sous les noms suivants : Conformément à la réglementation en vigueur.
[À compléter ultérieurement par le titulaire]
La dernière date à laquelle cette notice a été révisée est :
[à compléter ultérieurement par le titulaire]
Des informations détaillées sur ce médicament sont disponibles sur le site Internet de l’ANSM (France).