Dernière mise à jour le 01/06/2026
TEMOZOLOMIDE VIATRIS 5 mg, gélule
Ce médicament n'est ou ne sera bientôt plus disponible sur le marché.
Si vous prenez actuellement ce médicament, il vous est recommandé d'en parler avec votre médecin
ou avec votre pharmacien qui pourra vous orienter vers un autre traitement.
Indications thérapeutiques
Classe pharmacothérapeutique : AGENTS ANTINEOPLASIQUES - AUTRES AGENTS ALKYLANTS, code ATC : L01AX03.
TEMOZOLOMIDE VIATRIS est un agent antitumoral.
TEMOZOLOMIDE VIATRIS est utilisé pour le traitement de formes particulières de tumeurs cérébrales :
· chez les adultes atteints de glioblastome multiforme nouvellement diagnostiqué. TEMOZOLOMIDE VIATRIS est tout d'abord utilisé en association avec la radiothérapie (traitement en phase concomitante) puis seul (traitement en phase de monothérapie),
· chez les enfants âgés de 3 ans et plus, et chez les patients adultes atteints de gliome malin, tel que glioblastome multiforme ou astrocytome anaplasique. TEMOZOLOMIDE VIATRIS est utilisé pour ces tumeurs si ces dernières réapparaissent ou s'aggravent après un traitement standard.
Présentations
> sachet(s)-dose(s) de 5 gélule(s)
Code CIP : 269 958-2 ou 34009 269 958 2 9
Déclaration d'arrêt de commercialisation : 11/08/2025
Cette présentation n'est pas agréée aux collectivités
Service médical rendu (SMR)
Les libellés affichés ci-dessous ne sont que des résumés ou extraits issus des avis rendus par la Commission de la Transparence. Seul l'avis complet de la Commission de la Transparence fait référence.
Cet avis est consultable à partir du lien `Avis du jj/mm/aaaa` ou encore sur demande auprès de la HAS (plus d'informations dans l'aide). Les avis et synthèses d'avis contiennent un paragraphe sur la place du médicament dans la stratégie thérapeutique.
| Valeur du SMR | Avis | Motif de l'évaluation | Résumé de l'avis |
|---|---|---|---|
| Important | Avis du 20/11/2013 | Inscription (CT) | Le service médical rendu par TEMOZOLOMIDE MYLAN est important dans les indications de l’AMM. |
| Important | Avis du 22/09/2010 | Inscription (CT) | Le service médical rendu par ces spécialités est important. |
Amélioration du service médical rendu (ASMR)
Les libellés affichés ci-dessous ne sont que des résumés ou extraits issus des avis rendus par la Commission de la Transparence. Seul l'avis complet de la Commission de la Transparence fait référence.
Cet avis est consultable à partir du lien `Avis du jj/mm/aaaa` ou encore sur demande auprés de la HAS (plus d'informations dans l'aide). Les avis et synthéses d'avis contiennent un paragraphe sur la place du médicament dans la stratégie thérapeutique.
| Valeur de l'ASMR | Avis | Motif de l'évaluation | Résumé de l'avis |
|---|---|---|---|
| V (Inexistant) | Avis du 20/11/2013 | Inscription (CT) | Ces spécialités sont des compléments de gamme qui n’apportent pas d’amélioration du service médical rendu (ASMR V, inexistante) par rapport aux autres présentations déjà inscrites. |
| V (Inexistant) | Avis du 22/09/2010 | Inscription (CT) | Absence d’amélioration du service médical rendu (ASMR V) par rapport à TEMODAL 5 mg, 20 mg, 100 mg, 140 mg, 180 mg et 250 mg, gélules. |
Autres informations
- Titulaire de l'autorisation : VIATRIS SANTE
- Conditions de prescription et de délivrance :
- liste I
- médicament nécessitant une surveillance particulière pendant le traitement
- prescription hospitalière
- prescription réservée aux médecins compétents en CANCEROLOGIE
- prescription réservée aux spécialistes et services HEMATOLOGIE
- prescription réservée aux spécialistes et services ONCOLOGIE MEDICALE
- Statut de l'autorisation : Valide
- Type de procédure : Procédure décentralisée
- Code CIS : 6 724 625 8
ANSM - Mis à jour le : 17/07/2024
TEMOZOLOMIDE VIATRIS 5 mg, gélule
2. COMPOSITION QUALITATIVE ET QUANTITATIVE
Chaque gélule contient 5 mg de témozolomide.
Excipient à effet notoire :
Chaque gélule de TEMOZOLOMIDE VIATRIS 5 mg, gélule contient 87 mg de lactose anhydre.
Pour la liste complète des excipients, voir rubrique 6.1.
Gélules de taille 3, avec un corps et une coiffe opaques blancs, poudre blanche à brun clair/rose clair. Les gélules sont marquées de deux rayures vertes sur la coiffe et « T 5 mg » vert sur le corps.
4.1. Indications thérapeutiques
Témozolomide Viatris est indiqué pour le traitement :
· des patients adultes atteints d'un glioblastome multiforme nouvellement diagnostiqué en association avec la radiothérapie (RT) puis en traitement en monothérapie,
· des enfants à partir de 3 ans, des adolescents et des patients adultes atteints d'un gliome malin, tel que glioblastome multiforme ou astrocytome anaplasique, présentant une récidive ou une progression après un traitement standard.
4.2. Posologie et mode d'administration
Témozolomide Viatris doit uniquement être prescrit par des médecins qui ont l'expérience du traitement oncologique des tumeurs cérébrales.
Un traitement antiémétique peut être administré (voir rubrique 4.4).
Posologie
Patients adultes avec un glioblastome multiforme nouvellement diagnostiqué
Témozolomide Viatris est administré en association avec la radiothérapie focale (phase concomitante) suivi de témozolomide (TMZ) en monothérapie jusqu'à 6 cycles (phase de monothérapie).
Phase concomitante
TMZ est administré par voie orale à une dose de 75 mg/m2 quotidiennement pendant 42 jours, en association à la radiothérapie focale (60 Gy administrés en 30 fractions). Aucune réduction de dose n'est recommandée, mais le report ou l'interruption de l'administration de TMZ doit être décidé de manière hebdomadaire selon des critères de toxicité hématologique et non hématologique.
L'administration de TMZ peut être prolongée au-delà de la période concomitante de 42 jours (jusqu'à 49 jours) si toutes les conditions suivantes sont satisfaites :
· taux de neutrophiles (PNN) en valeur absolue ≥ 1,5 x 109/l,
· taux de plaquettes ≥ 100 x 109/l,
· toxicité non hématologique selon la classification « Common Toxicity Criteria » (CTC) ≤ Grade 1 (excepté pour l'alopécie, les nausées et les vomissements).
Pendant le traitement, une numération formule sanguine complète doit être effectuée chaque semaine.
L'administration de TMZ doit être interrompue temporairement ou arrêtée définitivement pendant la phase concomitante selon les critères de toxicité hématologique et non hématologique tels que décrits dans le Tableau 1.
Tableau 1. Interruption temporaire ou arrêt définitif du traitement par TMZ pendant la phase concomitante par radiothérapie et TMZ.
|
Toxicité |
Interruption temporaire du TMZa |
Arrêt définitif du TMZ |
|
Taux de neutrophiles en valeur absolue |
≥ 0,5 et < 1,5 x 109/l |
< 0,5 x 109/l |
|
Taux de plaquettes |
≥ 10 et < 100 x 109/l |
< 10 x 109/l |
|
Toxicité non hématologique CTC |
Grade 2 CTC |
Grade 3 ou 4 CTC |
aLe traitement concomitant avec TMZ peut être repris lorsque toutes les conditions suivantes sont rencontrées: taux de neutrophiles en valeur absolue ≥ 1,5 x 109/l; taux de plaquettes ≥ 100 x 109/l; toxicité non hématologique CTC ≤ Grade 1 (excepté pour l'alopécie, les nausées, les vomissements).
Phase de monothérapie
Quatre semaines après avoir terminé la phase concomitante de traitement par TMZ + RT, TMZ est administré jusqu'à 6 cycles de traitement en monothérapie. La dose pendant le cycle 1 (monothérapie) est de 150 mg/m2 une fois par jour pendant 5 jours suivis de 23 jours sans traitement. Au début du cycle 2, la dose est augmentée à 200 mg/m2 si la toxicité non hématologique CTC pour le cycle 1 est de Grade ≤ 2 (excepté pour l'alopécie, les nausées et les vomissements), le taux de neutrophiles (PNN) en valeur absolue est ≥ 1,5 x 109/l, et le taux de plaquettes est ≥ 100 x 109/l. Si la dose n'est pas augmentée au cycle 2, l'augmentation ne doit pas être effectuée aux cycles suivants. Une fois augmentée, la dose demeure à 200 mg/m2 par jour pour les 5 premiers jours de chaque cycle suivant à moins qu'une toxicité apparaisse. Les diminutions de dose et les arrêts pendant la phase de monothérapie doivent être effectués selon les tableaux 2 et 3.
Pendant le traitement, une numération formule sanguine complète doit être effectuée au Jour 22 (21 jours après la première dose de TMZ). La dose doit être réduite ou l'administration interrompue selon le Tableau 3.
Tableau 2. Différents niveaux de dose de TMZ pour le traitement en monothérapie
|
Niveau de dose |
Dose de TMZ (mg/m2/jour) |
Remarques |
|
-1 |
100 |
Diminution pour toxicité antérieure |
|
0 |
150 |
Dose pendant le cycle 1 |
|
1 |
200 |
Dose pendant les cycles 2 à 6 en l'absence de toxicité |
Tableau 3. Diminution de dose de TMZ ou arrêt pendant le traitement en monothérapie
|
Toxicité |
Diminution de TMZ d'un niveau de dosea |
Arrêt de TMZ |
|
Taux de neutrophiles en valeur absolue |
< 1,0 x 109/l |
Voir annotationb |
|
Taux de plaquettes |
< 50 x 109/l |
Voir annotationb |
|
Toxicité non hématologique CTC |
Grade 3 CTC |
Grade 4b CTC |
ales niveaux de dose de TMZ sont listés dans le Tableau 2.
ble TMZ doit être arrêté si :
· le niveau de posologie -1 (100 mg/m2) provoque toujours une toxicité inacceptable
· la même toxicité non hématologique Grade 3 (excepté pour l'alopécie, les nausées, les vomissements) se reproduit après réduction de dose.
Patients adultes et enfants âgés de 3 ans ou plus atteints de gliome malin en progression ou récidive :
Un cycle de traitement comprend 28 jours. Chez les patients n'ayant pas reçu de chimiothérapie au préalable, TMZ est administré par voie orale à la dose de 200 mg/m2 une fois par jour pendant les 5 premiers jours du cycle puis le traitement devra être arrêté pendant les 23 jours suivants (total de 28 jours). Chez les patients ayant reçu une chimiothérapie préalable, la dose initiale est de 150 mg/m2 une fois par jour, puis est augmentée lors du second cycle à 200 mg/m2 une fois par jour, pendant 5 jours s'il n'y a pas de toxicité hématologique (voir rubrique 4.4).
Populations spéciales
Population pédiatrique
Chez les enfants âgés de 3 ans ou plus, TMZ est uniquement utilisé dans le traitement du gliome malin en progression ou récidive.
L’expérience chez ces enfants est très limitée (voir rubriques 4.4 et 5.1). La sécurité et l’efficacité de TMZ chez les enfants de moins de 3 ans n’a pas été établie. Aucune donnée n’est disponible.
Insuffisance hépatique ou rénale
Les paramètres pharmacocinétiques du TMZ étaient comparables chez les patients ayant une fonction hépatique normale et chez ceux atteints d'insuffisance hépatique faible ou modérée. Aucune donnée n'est disponible concernant l'administration de TMZ chez les patients atteints d'insuffisance hépatique sévère (stade C de la classification de Child) ou d'insuffisance rénale. Sur la base des propriétés pharmacocinétiques du TMZ, il est peu probable qu'une réduction de dose soit nécessaire chez les patients atteints d'insuffisance hépatique sévère ou de n'importe quel degré d'insuffisance rénale. Cependant, des précautions doivent être prises lorsque TMZ est administré chez ces patients.
Personnes âgées
Sur la base d'une analyse pharmacocinétique de population chez des patients âgés de 19 à 78 ans, la clairance du TMZ n'est pas affectée par l'âge. Cependant, les patients âgés (> 70 ans) semblent avoir un risque augmenté de neutropénie et thrombocytopénie (voir rubrique 4.4).
Mode d’administration
Voie orale.
Témozolomide Viatris, gélule doit être administré à jeun.
Les gélules doivent être avalées entières avec un verre d'eau et ne doivent pas être ouvertes ni mâchées.
Si des vomissements surviennent après l'administration de la dose, ne pas administrer une deuxième dose le même jour.
· Hypersensibilité à la substance active ou à l'un des excipients mentionnés à la rubrique 6.1.
· Hypersensibilité à la dacarbazine (DTIC).
· Myélosuppression sévère (voir rubrique 4.4).
4.4. Mises en garde spéciales et précautions d'emploi
Infections opportunistes et réactivation d’infections
Des infections opportunistes (telle que la pneumonie à Pneumocystis jirovecii) et la réactivation d’infections (telles que VHB et CMV) ont été observées pendant le traitement par TMZ (voir rubrique 4.8).
Méningo-encéphalite herpétique
Dans certains cas postérieurs à la mise sur le marché, une méningo-encéphalite herpétique (ayant parfois entraîné une issue fatale) a été observée chez des patients recevant TMZ en association avec une radiothérapie, notamment en cas d'administration concomitante de stéroïdes.
Pneumonie à Pneumocystis jirovecii
Dans un essai pilote, les patients ayant reçu TMZ de manière concomitante à la radiothérapie sur un schéma prolongé de 42 jours ont montré un risque particulier de développer des pneumonies à Pneumocystis jirovecii (PPC). Ainsi, une prophylaxie pour la pneumonie à PPC est requise pour tous les patients recevant TMZ de manière concomitante avec la radiothérapie pour les 42 jours de traitement (avec un maximum de 49 jours) quel que soit le taux de lymphocytes. Si une lymphopénie se produit, ils doivent continuer la prophylaxie jusqu'à retrouver une lymphopénie de Grade ≤ 1.
Il peut y avoir une fréquence plus importante de PPC quand TMZ est administré selon un schéma de traitement plus long. Néanmoins, tous les patients recevant TMZ, particulièrement les patients recevant des stéroïdes, doivent être surveillés attentivement concernant le développement de PPC, quel que soit le schéma de traitement. Des cas d'insuffisance respiratoire fatale ont été rapportés chez des patients utilisant TMZ, en particulier en association avec la dexaméthasone ou d'autres stéroïdes.
VHB
Des cas d’hépatite liée à une réactivation du virus de l’hépatite B (VHB) ont été rapportés, incluant certains cas d’évolution fatale. Les patients présentant une sérologie positive pour l'hépatite B (incluant les patients présentant une hépatite B active) doivent être adressés à un médecin spécialisé en hépatologie avant l’instauration du traitement. Au cours du traitement, les patients devront être surveillés et pris en charge de façon appropriée.
Hépatotoxicité
Des cas d’atteintes hépatiques, dont des cas d’insuffisance hépatique fatale, ont été rapportés chez des patients traités par TMZ (voir rubrique 4.8). Avant l’initiation du traitement, des examens de la fonction hépatique devront être réalisés pour obtenir des valeurs de référence. En cas d’anomalies, les médecins devront évaluer le rapport bénéfice/risque incluant le risque d’insuffisance hépatique fatale, avant d’initier le traitement par le témozolomide. Chez les patients qui ont un cycle de traitement de 42 jours, les examens de la fonction hépatique devront être répétés au milieu du cycle. Les examens de la fonction hépatique devront être réalisés après chaque cycle de traitement chez tous les patients. Chez les patients ayant des perturbations significatives de la fonction hépatique, les médecins devront évaluer le rapport bénéfice/risque de la poursuite du traitement. Une toxicité hépatique peut survenir plusieurs semaines, voire plus, après la dernière prise de témozolomide.
Tumeurs malignes
Des cas de syndromes myélodysplasiques et de tumeurs malignes secondaires, incluant la leucémie myéloïde, ont également été observés, très rarement (voir rubrique 4.8).
Traitement antiémétique
Les nausées et les vomissements sont très fréquemment associés à TMZ.
Un traitement antiémétique peut être administré avant ou après administration de TMZ.
Patients adultes atteints d'un glioblastome multiforme nouvellement diagnostiqué
Une prophylaxie antiémétique est recommandée avant la dose initiale en phase concomitante. Une telle prophylaxie est fortement recommandée pendant la phase de monothérapie.
Patients avec un gliome malin en progression ou récidive
Les patients qui ont eu des vomissements sévères (Grade 3 ou 4) au cours des cycles de traitement antérieurs peuvent nécessiter un traitement antiémétique.
Paramètres biologiques
Chez les patients traités avec TMZ, une myélosuppression peut survenir, y compris une pancytopénie prolongée pouvant entraîner une anémie aplasique qui dans certains cas, a eu une issue fatale. Dans certains cas, l'exposition concomitante à des médicaments pouvant être à l’origine d'une anémie aplasique, y compris la carbamazépine, phénytoïne et sulfamethoxazole/triméthoprime, complique l'évaluation.
Avant administration, les paramètres biologiques doivent être les suivants : PNN ≥ 1,5 x 109/l et taux de plaquettes ≥ 100 x 109/l. Une numération formule sanguine complète doit être effectuée au Jour 22 (21 jours après la première dose) ou dans un délai de 48 heures suivant ce jour, et chaque semaine jusqu'à un taux PNN ≥ 1,5 x 109/l et un taux de plaquettes ≥ 100 x 109/l. Si le taux de PNN tombe à une valeur < 1,0 x 109/l ou le taux de plaquettes est < 50 x 109/l pendant n'importe quel cycle, la dose doit être diminuée d'un niveau au prochain cycle (voir rubrique 4.2). Les niveaux de doses incluent 100 mg/m2, 150 mg/m2, et 200 mg/m2. La plus basse dose recommandée est 100 mg/m2.
Population pédiatrique
Il n'existe aucune expérience clinique de l'utilisation de TMZ chez les enfants de moins de 3 ans. L'expérience chez les enfants plus âgés et les adolescents est très limitée (voir rubriques 4.2 et 5.1).
Patients âgés (> 70 ans)
Les patients âgés présentent un risque plus élevé de neutropénie et de thrombocytopénie comparativement aux patients plus jeunes. Par conséquent, une attention particulière est nécessaire lorsque TMZ est administré chez les patients âgés.
Patients de sexe féminin
Les femmes en âge de procréer doivent utiliser une contraception efficace afin d’éviter toute grossesse lorsqu’elles reçoivent TMZ et pendant au moins 6 mois après la fin du traitement.
Hommes traités
Il est conseillé aux hommes traités par TMZ de ne pas procréer pendant au moins 3 mois après la dernière dose prise et de se renseigner sur la cryoconservation du sperme avant d'initier le traitement (voir rubrique 4.6).
Excipients
Ce médicament contient du lactose. Les patients présentant une intolérance au galactose, un déficit total en lactase ou un syndrome de malabsorption du glucose et du galactose (maladies héréditaires rares) ne doivent pas prendre ce médicament.
Ce médicament contient moins de 1 mmol (23 mg) de sodium par gélule, c.-à-d. qu’il est essentiellement « sans sodium ».
4.5. Interactions avec d'autres médicaments et autres formes d'interactions
Dans une autre étude de phase I, l'administration simultanée de TMZ et de ranitidine ne modifie pas l'absorption du témozolomide ou l'exposition à son métabolite actif le monométhyl triazénoimidazole carboxamide (MTIC).
L'administration de TMZ lors des repas se traduit par une diminution de 33 % de la Cmax et par une diminution de 9 % de l'aire sous la courbe (ASC). Comme on ne peut pas exclure que ce changement de la Cmax ait une signification clinique, Témozolomide Viatris doit être administré en dehors des repas.
Selon une analyse pharmacocinétique de population lors des essais de phase II, l'administration concomitante de dexaméthasone, de prochlorpérazine, de phénytoïne, de carbamazépine, d'ondansétron, d'antagonistes des récepteurs H2, ou de phénobarbital ne modifie pas la clairance du TMZ. L'administration concomitante d'acide valproïque a été associée à une diminution faible mais statistiquement significative de la clairance du TMZ.
Aucune étude n'a été réalisée pour déterminer l'effet du TMZ sur le métabolisme ou l'élimination d'autres médicaments. Cependant, comme le TMZ ne subit pas de métabolisme hépatique et présente une faible liaison aux protéines plasmatiques, il ne devrait pas affecter les paramètres pharmacocinétiques d'autres médicaments (voir rubrique 5.2).
L'utilisation de TMZ en association avec d'autres agents myélosuppresseurs est susceptible d'accroître le risque de myélosuppression.
Population pédiatrique
Les études d’interaction n’ont été réalisées que chez l’adulte.
4.6. Fertilité, grossesse et allaitement
Grossesse
Il n'existe aucune donnée chez la femme enceinte. Lors des études précliniques conduites chez le rat et le lapin ayant reçu une dose de 150 mg/m2 de TMZ, des effets tératogènes et/ou toxiques pour le fœtus ont été démontrés (voir rubrique 5.3). Témozolomide Viatris ne doit pas être administré chez la femme enceinte. Si l'administration est envisagée pendant la grossesse, la patiente doit être prévenue du risque potentiel pour le fœtus.
Allaitement
On ne sait pas si TMZ est excrété dans le lait maternel. Par conséquent, l'allaitement doit être interrompu pendant le traitement par TMZ.
Femmes en âge de procréer
Fertilité masculine
TMZ peut avoir des effets génotoxiques. Par conséquent, les hommes traités par ce dernier doivent utiliser des mesures contraceptives efficaces et il leur est conseillé de ne pas procréer pendant au moins 3 mois après avoir reçu la dernière dose et de se renseigner sur la cryoconservation du sperme avant d'initier le traitement, compte tenu de la possibilité d'infertilité irréversible due à la thérapie avec le TMZ.
4.7. Effets sur l'aptitude à conduire des véhicules et à utiliser des machines
Expérience issue d'essai clinique
Chez les patients présentant un gliome en récidive ou progression, les nausées (43 %) et les vomissements (36 %) ont été généralement de Grade 1 ou 2 (0 à 5 épisodes de vomissements par 24 heures) et ont disparu spontanément, ou ont été rapidement contrôlés par un traitement antiémétique standard. L’incidence des nausées et vomissements sévères a été de 4 %.
Liste tabulée des effets indésirables
Les effets indésirables observés dans les études cliniques et rapportés depuis la commercialisation de TMZ sont listés dans le tableau 4. Ces effets sont listés selon la Classification des Systèmes d'Organe et par fréquence. Les groupes de fréquence sont définis selon la convention suivante : très fréquent (≥ 1/10) ; fréquent (≥ 1/100, < 1/10) ; peu fréquent (≥ 1/1 000, < 1/100) ; rare (≥ 1/10 000, < 1/1 000) ; très rare (< 1/10 000), fréquence indéterminée (ne peut être estimée sur la base des données disponibles). Au sein de chaque groupe de fréquence, les effets indésirables sont présentés suivant un ordre décroissant de gravité.
|
Tableau 4. Effets indésirables chez les patients traités avec témozolomide |
|
|
Infections et infestations |
|
|
Fréquent : |
Infections, zona, pharyngitea, candidose orale |
|
Peu fréquent : |
Infection opportuniste (incluant PPC), sepsis, méningo-encéphalite herpétique, infection à CMV, réactivation du CMV, virus de l’hépatite B, herpès simplex, réactivation d’une infection, infection des plaies, gastro- entériteb |
|
Tumeurs bénignes, malignes et non précisées |
|
|
Peu fréquent : |
Syndrome myélodysplasique (SMD), tumeurs malignes secondaires, incluant la leucémie myéloïde |
|
Affections hématologiques et du système lymphatique |
|
|
Fréquent : |
Neutropénie fébrile, neutropénie, thrombocytopénie, lymphopénie, leucopénie, anémie |
|
Peu fréquent : |
Pancytopénie prolongée, anémie aplasique, pancytopénie, pétéchie. |
|
Affections du système immunitaire |
|
|
Fréquent : |
Réaction allergique. |
|
Peu fréquent : |
Anaphylaxie. |
|
Affections endocriniennes |
|
|
Fréquent : |
Syndrome cushingoïdec |
|
Peu fréquent : |
Diabète insipide |
|
Troubles du métabolisme et de la nutrition |
|
|
Très Fréquent : |
Anorexie |
|
Fréquent : |
Hyperglycémie |
|
Peu fréquent |
Hypokaliémie, augmentation des phosphatases alcalines |
|
Affections psychiatriques |
|
|
Fréquent : |
Agitation, amnésie, dépression, anxiété, confusion, insomnie |
|
Peu fréquent : |
Trouble du comportement, instabilité émotionnelle, hallucination, apathie |
|
Affections du système nerveux |
|
|
Très Fréquent : |
Convulsions, hémiparésie, aphasie/dysphasie, céphalée |
|
Fréquent : |
Ataxie, trouble de l’équilibre, altération de la cognition, concentration altérée, baisse de conscience, étourdissements, hypoesthésie, troubles de la mémoire, troubles neurologiques, neuropathied, paresthésie, somnolence, trouble de la parole, altération du goût, tremblements |
|
Peu fréquent |
Etat de mal épileptique, hémiplégie, trouble extrapyramidal, parosmie, anomalie de la démarche, hyperesthésie, trouble sensoriel, coordination anormale |
|
Affections oculaires |
|
|
Fréquent : |
Hémianopsie, vision floue, trouble de la visione, défaut du champ visuel, diplopie, douleur oculaire |
|
Peu fréquent : |
Acuité visuelle réduite, sécheresse oculaire |
|
Affections de l’oreille et du labyrinthe |
|
|
Fréquent : |
Surditéf, vertige, acouphène, douleur à l’oreilleg |
|
Peu fréquent : |
Baisse de l’audition, hyperacousie, otite moyenne |
|
Affections cardiaques |
|
|
Peu fréquent : |
Palpitation |
|
Affections vasculaires |
|
|
Fréquent : |
Hémorragie, embolie pulmonaire, thrombose veineuse profonde, hypertension |
|
Peu fréquent : |
Hémorragie cérébrale, bouffées vasomotrices, bouffées de chaleur |
|
Affections respiratoires, thoraciques et médiastinales |
|
|
Fréquent : |
Pneumonie, dyspnée, sinusite, bronchite, toux, infection des voies aériennes hautes |
|
Peu fréquent : |
Insuffisance respiratoire, pneumopathies / pneumonie interstitielle, fibrose pulmonaire, congestion nasale |
|
Affections hépatobiliaires |
|
|
Peu fréquent : |
Insuffisance hépatique, atteinte hépatique, hépatite, cholestase, hyperbilirubinémie |
|
Très fréquent : |
Diarrhée, constipation, nausées, vomissements |
|
Fréquent |
Stomatite, douleur abdominaleh, dyspepsie, dysphagie |
|
Peu fréquent : |
Distension abdominale, incontinence fécale, troubles gastro-intestinaux, hémorroïdes, bouche sèche |
|
Affections de la peau et du tissu sous-cutané |
|
|
Très fréquent : |
Rash, alopécie |
|
Fréquent |
Erythème, sécheresse cutanée, prurit |
|
Peu fréquent : |
Nécrolyse épidermique toxique, syndrome de Stevens- Johnson, angiœdème, érythème multiforme, érythrodermie, exfoliation cutanée, réaction de photosensibilité, urticaire, exanthème, dermatite, transpiration accrue, pigmentation anormale |
|
Fréquence indéterminée |
Syndrome d’hypersensibilité médicamenteuse avec éosinophilie et symptômes systémiques (DRESS) |
|
Affections musculo-squelettiques et systémiques |
|
|
Fréquent : |
Myopathie, faiblesse musculaire, arthralgie, douleur dorsale, douleur musculosquelettique, myalgie |
|
Affections du rein et des voies urinaires |
|
|
Fréquent : |
Miction fréquente, incontinence urinaire |
|
Peu fréquent : |
Dysurie |
|
Affections des organes de reproduction et du sein |
|
|
Peu fréquent : |
Hémorragie vaginale, ménorragie, aménorrhée, vaginite, douleur mammaire, impuissance |
|
Troubles généraux et anomalies au site d’administration |
|
|
Très fréquent : |
Fatigue |
|
Fréquent |
Fièvre, symptômes pseudo-grippaux, asthénie, malaise, douleur, œdème, œdème périphériquei |
|
Peu fréquent : |
Aggravation de l’état, raideur, œdème de la face, décoloration de la langue, soif, trouble dentaire |
|
Investigations |
|
|
Fréquent : |
Augmentation des enzymes hépatiquesj, perte de poids, prise de poids |
|
Peu fréquent : |
Augmentation des gamma-glutamyltransférase |
|
Lésions, intoxications et complications liées aux procédures |
|
|
Fréquent : |
Lésions radiquesk |
a Y compris pharyngite, rhinopharyngite, pharyngite à streptocoques
b Y compris gastro-entérite, gastro-entérite virale
c Y compris syndrome cushingoïde, syndrome de Cushing
d Y compris neuropathie, neuropathie périphérique, polyneuropathie, neuropathie sensitive périphérique, neuropathie motrice périphérique
e Y compris déficience visuelle, affections oculaires
f Y compris surdité, surdité bilatérale, surdité neurosensorielle, surdité unilatérale
g Y compris douleur à l’oreille, gêne au niveau de l’oreille
h Y compris douleur abdominale, douleur abdominale basse, douleur abdominale haute, gêne abdominale
i Y compris œdème périphérique, gonflement périphérique
j Y compris augmentation des tests de la fonction hépatique, augmentation de l’alanine aminotransférase, augmentation de l’aspartate aminotransférase, augmentation des enzymes hépatiques
k Y compris lésion radique, lésion radique cutanée
Y compris des cas d’issue fatale
Glioblastome multiforme nouvellement diagnostiqué
Résultats de laboratoire
Une myélosuppression (neutropénie et thrombocytopénie), connue comme étant la toxicité dose-limitante de la plupart des agents cytotoxiques, y compris le TMZ, a été observée. Parmi les anomalies de laboratoire associées à des événements indésirables apparues au cours de la phase concomitante et de monothérapie, les anomalies de neutrophiles de Grade 3 ou 4 incluant les événements neutropéniques étaient observés chez 8 % des patients. Les anomalies de Grades 3 ou 4 des thrombocytes, incluant les évènements de thrombocytopénie ont été observées chez 14 % des patients qui recevaient TMZ.
Gliome malin en récidive ou progression
Résultats de laboratoire
Une thrombocytopénie et une neutropénie de Grade 3 ou 4 sont survenues respectivement chez 19 % et 17 %, des patients traités pour gliome malin. Cela a entraîné une hospitalisation et/ou un arrêt de TMZ respectivement chez 8 % et 4 % des patients.
La myélosuppression était prévisible (habituellement lors des premiers cycles de traitement, avec un nadir entre le Jour 21 et le Jour 28), et rapidement réversible, généralement en 1 - 2 semaines. Il n'a pas été observé de myélosuppression cumulative. La présence de thrombocytopénie peut augmenter le risque de saignements, et la présence de neutropénie ou de leucopénie peut augmenter le risque d'infection.
Genre
Dans une analyse pharmacocinétique de population d'un essai clinique, il y a eu 101 femmes et 169 hommes pour qui les taux de nadir des neutrophiles étaient disponibles et 110 femmes et 174 hommes pour qui les taux de nadir des plaquettes étaient disponibles. Il y a eu des taux supérieurs de neutropénie de Grade 4 (PNN < 0,5 x 109/l), 12 % vs 5 %, et de thrombocytopénie (< 20 x 109/l), 9 % vs 3 %, chez les femmes vs chez les hommes durant le premier cycle du traitement. Dans une série de 400 sujets ayant un gliome en récidive, une neutropénie de Grade 4 est survenue chez 8 % des femmes vs 4 % des hommes et une thrombocytopénie de Grade 4 chez 8 % des femmes vs 3 % des hommes durant le premier cycle du traitement. Dans une étude de 288 sujets avec un glioblastome multiforme nouvellement diagnostiqué, une neutropénie de Grade 4 survenait chez 3 % des femmes vs 0 % des hommes et une thrombocytopénie de Grade 4 chez 1 % des femmes vs 0 % des hommes durant le premier cycle du traitement.
Population pédiatrique
TMZ par voie orale a été étudié chez des enfants (3-18 ans) atteints d’un gliome du tronc cérébral en récidive ou d’un astrocytome de haut grade en récidive, administré quotidiennement pendant 5 jours consécutifs tous les 28 jours. Bien que les données soient limitées, la tolérance attendue chez les enfants devrait être similaire à celle chez l’adulte. La sécurité de TMZ chez les enfants de moins de 3 ans n’a pas été établie.
Déclaration des effets indésirables suspectés
La déclaration des effets indésirables suspectés après autorisation du médicament est importante. Elle permet une surveillance continue du rapport bénéfice/risque du médicament. Les professionnels de santé déclarent tout effet indésirable suspecté via le système national de déclaration : Agence nationale de sécurité du médicament et des produits de santé (ANSM) et réseau des Centres Régionaux de Pharmacovigilance - Site internet : https://signalement.social-sante.gouv.fr/.
5. PROPRIETES PHARMACOLOGIQUES
5.1. Propriétés pharmacodynamiques
Mécanisme d’action
Le TMZ est un dérivé triazène, qui subit une conversion chimique rapide à pH physiologique en monométhyl triazenoimidazole carboxamide (MTIC) actif. La cytotoxicité du MTIC est vraisemblablement due principalement à une alkylation de la guanine en position O6 et à une alkylation supplémentaire en position N7. Les lésions cytotoxiques qui sont développées par la suite sont supposées impliquer une réparation aberrante de l'ADN méthylé.
Efficacité et sécurité clinique
Glioblastome multiforme nouvellement diagnostiqué
Un total de 573 patients ont été randomisés pour recevoir soit TMZ + RT (n=287) soit RT seule (n=286). Les patients dans le bras TMZ + RT ont reçu TMZ concomitant (75 mg/m2) une fois par jour, en commençant le premier jour de la RT jusqu'au dernier jour de la RT, pendant 42 jours (avec un maximum de 49 jours). Ceci a été suivi de TMZ en monothérapie (150 - 200 mg/m2) les Jours 1 - 5 de chaque cycle de 28 jours jusqu'à 6 cycles, commençant 4 semaines après la fin de la RT. Les patients dans le bras contrôle ont reçu seulement de la RT. La prophylaxie de la pneumonie à Pneumocystis jirovecii (PPC) a été exigée pendant le traitement par RT et TMZ associés.
Le TMZ a été administré comme traitement de rattrapage dans la phase de suivi chez 161 des 282 patients (57 %) dans le bras radiothérapie seule, et 62 des 277 patients (22 %) dans le bras TMZ + RT.
Le hazard ratio (HR) pour la survie globale était 1,59 (95 % IC pour HR = 1,33 - 1,91) avec un log-rank p < 0,0001 en faveur du bras TMZ. La probabilité estimée de survie à 2 ans ou plus (26 % vs 10 %) est plus importante pour le bras radiothérapie + TMZ. L'addition de TMZ de manière concomitante à la RT, suivie de TMZ en monothérapie dans le traitement des patients avec un glioblastome multiforme nouvellement diagnostiqué a démontré une amélioration de la survie globale (SG) statistiquement significative comparée à la RT seule (Figure 1).
Figure 1 Courbe de Kaplan-Meier pour la survie globale (population Intent-to-Treat)
|
|
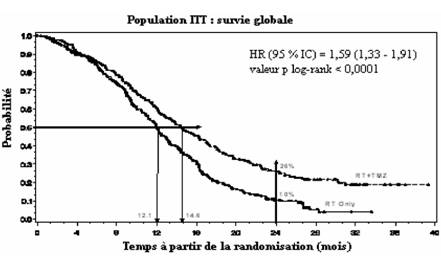
Les résultats de l'étude n'étaient pas cohérents dans le sous-groupe de patients avec un faible « performance status » (OMS, PS=2, n=70), où la survie globale et le temps de progression étaient similaires dans les deux bras. Cependant, aucun risque inacceptable ne semble avoir été décelé dans ce groupe de patients.
Gliome malin en progression ou récidive
Les données d'efficacité clinique chez les patients atteints de glioblastome multiforme (indice de performance Karnofsky [IPK] ≥ 70), en progression ou récidivant après une chirurgie et une radiothérapie, proviennent de deux essais cliniques avec TMZ par voie orale. L'un était un essai non comparatif chez 138 patients (29 % avaient reçu une chimiothérapie préalable), et l'autre était un essai randomisé contrôlé contre référence active comparant TMZ vs procarbazine chez un total de 225 patients (67 % avaient reçu une chimiothérapie préalable à base de nitrosourée). Dans les deux essais, le critère d'évaluation principal était la survie sans progression (SSP) définie par Imagerie de Résonnance Médicale (IRM) ou une aggravation neurologique. Dans l'essai non comparatif, la SSP à 6 mois était de 19 %, la survie médiane sans progression était de 2,1 mois, et la survie médiane globale de 5,4 mois. Le taux de réponse objective (TRO) défini par des IRM était de 8 %.
Dans l'essai randomisé contrôlé contre référence active, la SSP à 6 mois était significativement plus importante pour le TMZ que pour la procarbazine (21 % versus 8 %, respectivement - Chi2: p = 0,008) avec une SSP médiane de 2,89 et 1,88 mois respectivement (log rank p = 0,0063). La survie médiane était de 7,34 et 5,66 mois respectivement pour le TMZ et la procarbazine (log rank p = 0,33). A 6 mois, le pourcentage des patients survivants était significativement plus élevé dans le bras TMZ (60 %) comparé au bras procarbazine (44 %) (Chi2: p = 0,019). Chez les patients préalablement traités par chimiothérapie un bénéfice a été noté pour ceux ayant un IPK ≥ 80.
Les données sur la durée jusqu'à aggravation de l'état neurologique comme celles sur la durée jusqu'à aggravation de l'indice de performance (diminution jusqu'à un IPK < 70 ou diminution d'au moins 30 points) étaient en faveur du TMZ par rapport à la procarbazine. Les temps médians jusqu'à progression pour ces critères d'évaluation sont plus longs pour le TMZ que pour la procarbazine, entre 0,7 et 2,1 mois (log rank p = < 0,01 à 0,03).
Astrocytome anaplasique en récidive
Lors d'un essai de phase II prospectif, multicentrique évaluant la sécurité et l'efficacité du TMZ oral dans le traitement des patients atteints d'astrocytome anaplasique en première rechute, la SSP à 6 mois était de 46 %. La médiane de la SSP était de 5,4 mois. La médiane de la survie globale était de 14,6 mois. Le taux de réponse, basé sur une revue centralisée, était de 35 % (13 RC et 43 RP) sur la population en intention de traiter (ITT) n=162. Chez 43 patients la stabilité de la maladie a été rapportée. La survie sans évènement à 6 mois pour la population en intention de traiter était de 44 % avec une médiane de survie sans évènement de 4,6 mois, ce qui était identique aux résultats de survie sans progression. Pour la population histologiquement éligible, les résultats d'efficacité étaient identiques. L'obtention d'une réponse radiologique objective ou le maintien d'une stabilisation sans progression était fortement associé à une qualité de vie maintenue ou améliorée.
Population pédiatrique
TMZ oral a été étudié chez les enfants (3-18 ans) atteints d'un gliome du tronc cérébral en récidive ou d'un astrocytome de haut grade en récidive, administré quotidiennement pendant 5 jours tous les 28 jours. La tolérance au TMZ était similaire à celle de l'adulte.
5.2. Propriétés pharmacocinétiques
Absorption
Après administration orale chez l'adulte, le TMZ est rapidement absorbé avec des pics de concentration observés parfois dès 20 minutes après administration (temps moyens compris entre 0,5 et 1,5 heure). Après administration orale de TMZ marqué au 14C, l'excrétion fécale moyenne de 14C dans les 7 jours suivant l'administration était de 0,8 % signe d'une absorption complète.
Distribution
Le TMZ est faiblement lié aux protéines plasmatiques (10 à 20 %), et par conséquent, est peu susceptible d'interagir avec des produits très fortement liés aux protéines.
Les études cliniques utilisant la technique de TEP (Tomographie par Emission de Positrons) et les données précliniques suggèrent que le TMZ traverse rapidement la barrière hémato-encéphalique et se retrouve dans le liquide céphalo-rachidien (LCR). La pénétration dans le LCR a été confirmée chez un patient ; sur la base de l'ASC du TMZ l'exposition du LCR était approximativement 30 % de celle du plasma, ce qui est cohérent avec les données chez l'animal.
Élimination
La demi-vie plasmatique (t1/2) est approximativement de 1,8 heure. La voie principale d'élimination du 14C est la voie rénale. Après administration orale, environ 5 à 10 % de la dose sont retrouvés sous forme inchangée dans les urines dans les 24 heures, et le reste est excrété sous forme d'acide témozolomide, de 5-aminoimidazole-4-carboxamide (AIC) ou de métabolites polaires non identifiés.
L'augmentation des concentrations plasmatiques est dose-dépendante. La clairance plasmatique, le volume de distribution et la demi-vie sont indépendants de la dose.
Populations spéciales
Insuffisance rénale et hépatique
L'analyse des paramètres pharmacocinétiques de population du TMZ a montré que la clairance plasmatique du TMZ est indépendante de l'âge, de la fonction rénale ou de la consommation de tabac. Dans une autre étude pharmacocinétique, les profils pharmacocinétiques plasmatiques des patients atteints de dysfonctionnement hépatique faible à modéré étaient identiques à ceux observés chez les patients dont la fonction hépatique était normale.
Population pédiatrique
Les enfants présentent une ASC plus élevée que les adultes ; cependant, la dose maximale tolérée (DMT) est de 1 000 mg/m2 par cycle de traitement à la fois chez l'enfant et chez l'adulte.
5.3. Données de sécurité préclinique
Le TMZ est un agent alkylant embryotoxique, tératogène et génotoxique. Le TMZ est plus toxique chez le rat et le chien que chez l'homme, et la dose thérapeutique est proche de la dose létale minimale chez le rat et le chien. Les diminutions dose-dépendantes du nombre des leucocytes et des plaquettes apparaissent comme des indicateurs sensibles de la toxicité. Différents types de néoplasmes ont été observés lors de l'étude de toxicité après 6 cycles de traitement chez le rat, dont carcinome mammaire, kérato-acanthome cutané et adénome baso-cellulaire alors qu'aucune tumeur, ni aucun changement prénéoplasique n'a été observé au cours des études chez le chien. Le rat semble être particulièrement sensible aux effets oncogènes du TMZ, avec l'apparition des premières tumeurs dans les 3 mois suivant le début du traitement. La période de latence est très courte, même pour un agent alkylant.
Les résultats des tests d'Ames/salmonella et d'aberration chromosomique sur lymphocyte humain démontrent l'existence d'un potentiel mutagène.
Enveloppe de la gélule : gélatine, dioxyde de titane (E171).
Encre d'impression : laque, propylène glycol, dioxyde de titane (E171), oxyde de fer jaune (E172), laque aluminique indigo carmin (E132).
3 ans.
Après ouverture, le médicament doit être utilisé dans les 21 jours.
Sachet-dose
2 ans.
6.4. Précautions particulières de conservation
A conserver à une température ne dépassant pas + 30 °C. A conserver dans l’emballage extérieur d’origine à l’abri de la lumière. Conserver le flacon soigneusement fermé à l’abri de l’humidité.
Sachet-dose
A conserver à une température ne dépassant pas + 25 °C. A conserver dans l’emballage extérieur d’origine à l’abri de la lumière.
6.5. Nature et contenu de l'emballage extérieur
5 ou 20 gélules en flacons de verre teinté munis de bouchon sécurité-enfant en polypropylène blanc avec un scellage d’induction en polyéthylène. L’étui contient un flacon.
Sachet-dose
Chaque sachet-dose contient une gélule et est dispensé dans un étui en carton.
L’étui contient 5 ou 20 gélules, individuellement scellées dans des sachets-dose.
Toutes les présentations peuvent ne pas être commercialisées.
6.6. Précautions particulières d’élimination et de manipulation
Les gélules ne doivent pas être ouvertes. Lorsqu'une gélule est détériorée, tout contact entre la poudre et la peau ou les muqueuses doit être évité. En cas de contact de TEMOZOLOMIDE VIATRIS avec la peau ou les muqueuses, laver immédiatement et abondamment à l'eau et au savon.
Les patients devront être avertis de tenir les gélules hors de la portée et de la vue des enfants, de préférence dans un endroit fermé à clé. L'ingestion accidentelle peut être mortelle pour les enfants.
Tout médicament non utilisé ou déchet doit être éliminé conformément à la réglementation en vigueur.
Le personnel enceinte ne doit pas manipuler ce produit.
7. TITULAIRE DE L’AUTORISATION DE MISE SUR LE MARCHE
1 RUE DE TURIN
69007 LYON
8. NUMERO(S) D’AUTORISATION DE MISE SUR LE MARCHE
· 34009 577 103 8 5 : 5 gélules en flacon (verre teinté).
· 34009 577 104 4 6 : 20 gélules en flacon (verre teinté).
· 34009 269 958 2 9 : 5 gélules en sachet-dose.
· 34009 269 959 9 7 : 20 gélules en sachet-dose.
9. DATE DE PREMIERE AUTORISATION/DE RENOUVELLEMENT DE L’AUTORISATION
[à compléter ultérieurement par le titulaire]
10. DATE DE MISE A JOUR DU TEXTE
[à compléter ultérieurement par le titulaire]
Sans objet.
12. INSTRUCTIONS POUR LA PREPARATION DES RADIOPHARMACEUTIQUES
Liste I.
Médicament soumis à prescription hospitalière. Prescription réservée aux spécialistes en oncologie ou en hématologie ou aux médecins compétents en cancérologie. Médicament nécessitant une surveillance particulière pendant le traitement.
ANSM - Mis à jour le : 17/07/2024
TEMOZOLOMIDE VIATRIS 5 mg, gélule
Témozolomide
· Gardez cette notice. Vous pourriez avoir besoin de la relire.
· Si vous avez d’autres questions, interrogez votre médecin, votre pharmacien ou votre infirmier/ère.
· Ce médicament vous a été personnellement prescrit. Ne le donnez pas à d’autres personnes. Il pourrait leur être nocif, même si les signes de leur maladie sont identiques aux vôtres.
· Si vous ressentez un quelconque effet indésirable, parlez-en à votre médecin, votre pharmacien ou votre infirmier/ère. Ceci s’applique aussi à tout effet indésirable qui ne serait pas mentionné dans cette notice. Voir rubrique 4.
1. Qu'est-ce que TEMOZOLOMIDE VIATRIS 5 mg, gélule et dans quels cas est-il utilisé ?
2. Quelles sont les informations à connaître avant de prendre TEMOZOLOMIDE VIATRIS 5 mg, gélule ?
3. Comment prendre TEMOZOLOMIDE VIATRIS 5 mg, gélule ?
4. Quels sont les effets indésirables éventuels ?
5. Comment conserver TEMOZOLOMIDE VIATRIS 5 mg, gélule ?
6. Contenu de l’emballage et autres informations.
1. QU’EST-CE QUE TEMOZOLOMIDE VIATRIS 5 mg, gélule ET DANS QUELS CAS EST-IL UTILISE ?
Classe pharmacothérapeutique : AGENTS ANTINEOPLASIQUES - AUTRES AGENTS ALKYLANTS, code ATC : L01AX03.
TEMOZOLOMIDE VIATRIS est un agent antitumoral.
TEMOZOLOMIDE VIATRIS est utilisé pour le traitement de formes particulières de tumeurs cérébrales :
· chez les adultes atteints de glioblastome multiforme nouvellement diagnostiqué. TEMOZOLOMIDE VIATRIS est tout d'abord utilisé en association avec la radiothérapie (traitement en phase concomitante) puis seul (traitement en phase de monothérapie),
· chez les enfants âgés de 3 ans et plus, et chez les patients adultes atteints de gliome malin, tel que glioblastome multiforme ou astrocytome anaplasique. TEMOZOLOMIDE VIATRIS est utilisé pour ces tumeurs si ces dernières réapparaissent ou s'aggravent après un traitement standard.
2. QUELLES SONT LES INFORMATIONS A CONNAITRE AVANT DE PRENDRE TEMOZOLOMIDE VIATRIS 5 mg, gélule ?
Ne prenez jamais TEMOZOLOMIDE VIATRIS 5 mg, gélule :
· si vous êtes allergique au témozolomide ou à l'un des autres composants contenus dans ce médicament, mentionnés dans la rubrique 6,
· si vous avez eu une réaction allergique à la dacarbazine (un médicament anticancéreux parfois appelé DTIC). Les signes de réaction allergique incluent une sensation de démangeaison, un essoufflement ou sifflement, un gonflement du visage, des lèvres, de la langue ou de la gorge,
· si certains types de cellules sanguines sont sévèrement diminués (myélosuppression), comme le taux de globules blancs ou le taux de plaquettes. Ces cellules du sang sont importantes pour combattre l'infection et pour une bonne coagulation du sang. Votre médecin contrôlera votre sang afin d'être sûr que vous avez suffisamment de ces cellules avant de débuter le traitement.
Avertissements et précautions
Adressez-vous à votre médecin, pharmacien ou votre infirmier/ère avant de prendre Témozolomide Viatris, gélule :
· car vous devrez être surveillé avec attention pour détecter le développement d'une forme grave d’infection pulmonaire appelée pneumonie à Pneumocystis jirovecii (PPC). Si vous êtes un patient nouvellement diagnostiqué (glioblastome multiforme) vous pouvez recevoir témozolomide pendant 42 jours en association avec de la radiothérapie. Dans ce cas, votre médecin vous prescrira également un médicament pour aider à prévenir ce type de pneumonie (PPC),
· si vous avez eu ou pourriez avoir actuellement une infection par le virus de l’hépatite B. En effet Témozolomide Viatris pourrait rendre cette hépatite B de nouveau active, potentiellement d’évolution fatale dans certains cas. Avant de commencer le traitement, vous serez soigneusement examiné par votre médecin à la recherche de signes de cette infection,
· si vous avez un faible taux de globules rouges (anémie), de globules blancs et de plaquettes, ou des troubles de la coagulation sanguine avant de débuter le traitement, ou si vous les développez lors du traitement. Votre médecin peut décider de diminuer la dose, d'interrompre, d'arrêter ou de changer votre traitement. Vous pouvez également avoir besoin d'autres traitements. Dans certains cas, il sera nécessaire d'arrêter le traitement par témozolomide. Vous serez soumis à de fréquentes prises de sang durant le traitement afin de surveiller les effets indésirables de témozolomide sur les cellules sanguines.
· car vous pouvez avoir un faible risque de modification des cellules sanguines, incluant la leucémie,
· si vous avez des nausées (sensation d'avoir envie de vomir) et/ou vomissez ce qui correspond à des effets indésirables très fréquents de témozolomide (voir rubrique 4), votre médecin peut vous prescrire un médicament (un anti-vomitif) aidant à prévenir les vomissements,
· si vous vomissez fréquemment avant ou pendant votre traitement, demandez conseil à votre médecin sur le meilleur moment pour prendre témozolomide jusqu'à contrôler vos vomissements. Si vous vomissez après avoir pris votre traitement, ne prenez pas une deuxième dose le même jour,
· si vous développez de la fièvre ou les symptômes d'une infection contactez votre médecin immédiatement,
· si vous avez plus de 70 ans, vous pouvez être plus sujet aux infections, aux contusions et aux saignements,
· si vous avez des troubles du foie ou du rein, votre dose de témozolomide peut avoir besoin d'être ajustée.
Enfants et adolescents
Ce médicament ne doit pas être donné aux enfants âgés de moins de 3 ans car cela n’a pas été étudié. On dispose de peu d’informations sur témozolomide chez les patients de plus de 3 ans.
Autres médicaments et TEMOZOLOMIDE VIATRIS 5 mg, gélule
· de l’acide valproïque (un médicament utilisé pour traiter les crises d’épilepsie),
· des médicaments qui suppriment le système immunitaire (tels que la ciclosporine, azathioprine).
TEMOZOLOMIDE VIATRIS 5 mg, gélule avec des aliments et boissons
Sans objet.
Grossesse, allaitement et fertilité
Grossesse
Si vous êtes enceinte ou si vous allaitez, pensez être enceinte ou planifiez une grossesse, demandez conseil à votre médecin ou pharmacien avant de prendre ce médicament. En effet, vous ne devez pas être traitée par témozolomide pendant une grossesse à moins que votre médecin ne vous l’indique clairement.
Allaitement
Vous devez arrêter l’allaitement lorsque vous êtes traitée par témozolomide.
Fertilité
Fertilité masculine :
Témozolomide peut causer une infertilité permanente. Il est conseillé aux hommes d'utiliser des mesures de contraception efficaces et de ne pas procréer pendant au moins 3 mois après l'arrêt du traitement. Il est recommandé de se renseigner sur la cryoconservation du sperme avant d'initier le traitement.
Conduite de véhicules et utilisation de machines
Vous pouvez vous sentir fatigué ou somnolent avec du témozolomide. Dans ce cas, attendez de voir comment ce médicament vous affecte avant de conduire un véhicule ou monter à vélo ou d’utiliser un outil ou une machine (voir rubrique 4).
TEMOZOLOMIDE VIATRIS 5 mg, gélule contient du lactose (un type de sucre) et du sodium.
Si votre médecin vous a informé(e) d’une intolérance à certains sucres, contactez-le avant de prendre ce médicament.
3. COMMENT PRENDRE TEMOZOLOMIDE VIATRIS 5 mg, gélule ?
Posologie et durée du traitement
Votre médecin calculera votre dose de témozolomide. Celle-ci est déterminée en fonction de votre surface corporelle (taille et poids) et de si vous avez une tumeur récidivante et avez eu antérieurement un traitement par chimiothérapie.
Il peut vous prescrire d'autres traitements (anti-vomitifs) à prendre avant et/ou après avoir pris témozolomide pour éviter ou contrôler les nausées et vomissements.
Patients atteints d'un glioblastome nouvellement diagnostiqué :
Si vous êtes un patient nouvellement diagnostiqué, le traitement se déroulera en deux phases :
· tout d'abord le traitement associé avec la radiothérapie (phase concomitante),
· suivi du traitement par témozolomide seul (phase de monothérapie).
Durant la phase concomitante, votre médecin débutera témozolomide à la dose de 75 mg/m2 (dose usuelle).
Vous prendrez cette dose chaque jour pendant 42 jours (au maximum jusqu'à 49 jours) en association avec la radiothérapie. Selon votre numération sanguine et la façon dont vous tolérez votre médicament pendant la phase concomitante, la prise de la dose de témozolomide peut être retardée ou arrêtée définitivement.
Une fois la radiothérapie terminée, vous interromprez votre traitement pendant 4 semaines. Cela permettra à votre corps de récupérer.
Puis, vous commencerez la phase de monothérapie.
Durant la phase de monothérapie, la dose et la manière dont vous prenez témozolomide seront différentes. Votre médecin calculera votre dose exacte. Il peut y avoir jusqu'à 6 périodes (cycles) de traitement. Chacune d'elles dure 28 jours. Vous prendrez votre nouvelle dose de témozolomide seul une fois par jour pendant les cinq premiers jours (« jours de prise ») de chaque cycle. La dose initiale sera de 150 mg/m2. Puis vous aurez 23 jours sans témozolomide. Ceci complète le cycle de traitement de 28 jours.
Après le Jour 28, le cycle suivant commence. Vous prendrez à nouveau témozolomide une fois par jour pendant 5 jours suivis de 23 jours sans témozolomide. La seconde dose sera de 200 mg/m².
Selon votre numération sanguine et la façon dont vous tolérez votre médicament pendant chaque cycle de traitement, la dose de témozolomide peut être ajustée, retardée ou arrêtée définitivement.
Patients atteints de tumeurs ayant réapparues ou s'étant aggravées (gliome malin, tel que le glioblastome multiforme ou l'astrocytome anaplasique), prenant témozolomide seul :
Le cycle de traitement par témozolomide comprend 28 jours.
Vous prendrez témozolomide seul une fois par jour pendant les 5 premiers jours. Cette dose quotidienne dépendra du fait que vous ayez reçu ou non une chimiothérapie antérieure.
Si vous n'avez pas été traité préalablement par chimiothérapie, votre première dose de témozolomide sera de 200 mg/m2 une fois par jour pendant les 5 premiers jours. Si vous avez été préalablement traité par chimiothérapie, votre première dose de témozolomide sera de 150 mg/m2 une fois par jour pendant les 5 premiers jours.
Puis, vous aurez 23 jours sans témozolomide. Ceci complète le cycle de traitement de 28 jours.
Après le Jour 28, le cycle suivant commence. Vous prendrez à nouveau témozolomide une fois par jour pendant 5 jours suivis de 23 jours sans témozolomide.
Avant chaque nouveau cycle de traitement, votre sang sera analysé afin de voir si la dose de Témozolomide Viatris a besoin d'être ajustée. Selon vos résultats d'analyse de sang, votre médecin peut ajuster votre dose pour le cycle suivant.
Mode d'administration
Prenez votre dose prescrite de témozolomide une fois par jour, de préférence à la même heure chaque jour.
Prenez la(les) gélule(s) l'estomac vide ; par exemple, au moins une heure avant de prendre votre petit-déjeuner. Avalez la(les) gélule(s) entière(s) avec un verre d'eau. N'ouvrez pas, n’écrasez pas ou ne mâchez pas les gélules. Si la gélule est détériorée, éviter tout contact avec la peau, les yeux ou le nez. Si vous en mettez accidentellement dans les yeux ou le nez, rincez la zone concernée avec de l'eau.
En fonction de la dose prescrite, vous pouvez avoir à prendre en même temps plus d'une gélule, parfois avec des dosages différents (contenu en substance active, en mg). La couleur de la coiffe et le marquage sur la gélule sont différents pour chaque dosage (voir dans le tableau ci-dessous).
|
Dosage |
Couleur/marquage |
|
TEMOZOLOMIDE VIATRIS 5 mg |
Deux rayures marquées à l'encre verte sur la coiffe et « T 5 mg » marqué à l'encre verte sur le corps |
|
TEMOZOLOMIDE VIATRIS 20 mg |
Deux rayures marquées à l'encre orange sur la coiffe et « T 20 mg » marqué à l'encre orange sur le corps |
|
TEMOZOLOMIDE VIATRIS 100 mg |
Deux rayures marquées à l'encre rose sur la coiffe et « T 100 mg » marqué à l'encre rose sur le corps |
|
TEMOZOLOMIDE VIATRIS 140 mg |
Deux rayures marquées à l'encre bleue sur la coiffe et « T 140 mg » marqué à l'encre bleue sur le corps |
|
TEMOZOLOMIDE VIATRIS 180 mg |
Deux rayures marquées à l'encre rouge sur la coiffe et « T 180 mg » marqué à l'encre rouge sur le corps |
|
TEMOZOLOMIDE VIATRIS 250 mg |
Deux rayures marquées à l'encre noire sur la coiffe et « T 250 mg » marqué à l'encre noire sur le corps |
Vous devez être certain d'avoir compris et de vous rappeler exactement des informations suivantes :
· combien de gélules de chaque dosage vous avez besoin de prendre par jour. Demandez à votre médecin ou à votre pharmacien de l'écrire (avec la couleur),
· quels jours sont vos jours de prise.
Revoyez la dose avec votre médecin à chaque fois que vous commencez un nouveau cycle, puisqu'elle peut être différente de celle du dernier cycle.
Respectez toujours la posologie indiquée par votre médecin. En cas de doute, consultez votre médecin ou votre pharmacien. Les erreurs sur la façon de prendre votre traitement pourraient avoir des conséquences sérieuses pour votre santé.
Si vous avez pris plus de TEMOZOLOMIDE VIATRIS 5 mg, gélule que vous n’auriez dû :
Consultez immédiatement votre médecin ou votre pharmacien.
Si vous prenez accidentellement plus de témozolomide gélules que la quantité prescrite, contactez immédiatement votre médecin, votre pharmacien ou infirmier/ère.
Si vous oubliez de prendre TEMOZOLOMIDE VIATRIS 5 mg, gélule :
Prenez la dose oubliée dès que possible au cours de la même journée. Si une journée entière s'est écoulée, demandez conseil à votre médecin. Ne prenez pas de dose double pour compenser la dose que vous avez oublié de prendre à moins que votre médecin ne vous dise de le faire.
Si vous arrêtez de prendre TEMOZOLOMIDE VIATRIS 5 mg, gélule :
Sans objet.
Si vous avez d’autres questions sur l’utilisation de ce médicament, demandez plus d’informations à votre médecin, votre pharmacien ou infirmier/ère.
4. QUELS SONT LES EFFETS INDESIRABLES EVENTUELS ?
Comme tous les médicaments, ce médicament peut provoquer des effets indésirables, mais ils ne surviennent pas systématiquement chez tout le monde.
Informez immédiatement votre médecin si vous avez un des effets indésirables suivants :
· réactions allergiques sévères soudaines avec difficultés respiratoires, gonflement, sensations vertigineuses, battements de cœur rapides, transpiration excessive et perte de conscience (anaphylaxie),
· saignement non contrôlé (hémorragie),
· épisodes de crampes et niveau de conscience réduit (convulsions),
· crise ou série de crises d’épilepsie, d’une durée supérieure à 5 minutes (état de mal épileptique),
· mal de tête sévère persistant (hémorragie cérébrale),
· réaction potentiellement mortelle avec symptômes grippaux et formation de cloques sur la peau ou éruption cutanée douloureuse, dans la bouche, les yeux et les organes génitaux (nécrolyse épidermique toxique, syndrome de Stevens-Johnson, érythème polymorphe),
· gonflement rapide sous la peau dans des zones telles que le visage, la gorge, les bras et les jambes pouvant être potentiellement mortel si le gonflement de gorge bloque les voies respiratoires (angiœdème),
· symptômes de type grippal accompagnés d’une éruption cutanée, de fièvre, de ganglions enflés, et de résultats anormaux des tests sanguins [dont une augmentation des globules blancs (éosinophilie) et des enzymes hépatiques] [syndrome d’hypersensibilité médicamenteuse avec éosinophilie et symptômes systémiques (DRESS)].
Le traitement par témozolomide peut provoquer une diminution du nombre de certaines cellules sanguines. Cela peut provoquer une augmentation des contusions ou des saignements, une anémie (une diminution du nombre de globules rouges), de la fièvre, et une diminution de la résistance aux infections. La diminution du taux de cellules sanguines est habituellement passagère. Dans certains cas, elle peut être prolongée et peut mener à une forme très sévère d'anémie (anémie aplastique). Votre médecin surveillera régulièrement vos paramètres sanguins pour déterminer tout changement, et décidera si un traitement spécifique est nécessaire. Dans certains cas, votre dose de témozolomide sera diminuée ou le traitement arrêté.
D’autres effets indésirables rapportés sont énumérés ci-dessous :
Les effets indésirables très fréquents (pouvant toucher plus de 1 patient sur 10) sont :
· perte d’appétit (anorexie),
· faiblesse ou incapacité à bouger sur un côté du corps (hémiparésie),
· difficulté à parler (aphasie/dysphasie),
· mal de tête,
· diarrhée,
· constipation,
· nausées,
· vomissements,
· éruption cutanée,
· chute des cheveux et des poils (alopécie),
· fatigue.
Les effets indésirables fréquents (pouvant toucher jusqu’à 1 patient sur 10) sont :
· infections, telles que zona (formation de cloques douloureuses sur une partie du corps) et infections buccales, telles que candidose buccale (muguet, type d’infection fongique causée par des champignons du genre Candida),
· mal de gorge (pharyngite),
· faibles taux de globules blancs avec fièvre en lien avec une infection (neutropénie fébrile),
· nombre réduit de cellules sanguines (neutropénie, thrombopénie, lymphopénie, leucopénie, anémie),
· réaction allergique,
· trouble dont les signes sont une prise de poids, une accumulation de graisse sur le visage et l’apparition de bleus, causé par une quantité excessive d’un type d’hormone stéroïdienne (syndrome de Cushing),
· augmentation du taux de sucre dans le sang (hyperglycémie),
· agitation,
· troubles de la mémoire (amnésie),
· dépression,
· anxiété,
· confusion,
· incapacité à s’endormir ou à rester endormi(e) (insomnie),
· troubles de la coordination (ataxie),
· troubles de l’équilibre,
· difficulté à traiter les informations (trouble de la cognition),
· difficultés de concentration,
· modification de l’état mental ou de la vigilance (diminution du niveau de conscience),
· sensation vertigineuse,
· modification des sensations (hypoesthésie),
· oublis (troubles de la mémoire),
· troubles touchant le cerveau ou les nerfs (trouble neurologique),
· lésion nerveuse (neuropathie),
· sensation de picotements (paresthésie),
· envie de dormir (somnolence),
· troubles de la parole,
· goût anormal (altération du goût),
· tremblements,
· perte partielle de la vue (hémianopsie),
· vision trouble,
· vision anormale (trouble de la vue, défaut du champ visuel),
· vision double (diplopie),
· yeux endoloris (douleur oculaire),
· surdité,
· sensation de tournoiement (vertige),
· bourdonnement d’oreilles (acouphènes),
· douleur à l’oreille,
· caillot sanguin dans les poumons ou les jambes (embolie pulmonaire, thrombose veineuse profonde),
· pression artérielle élevée (hypertension),
· infection des poumons (pneumonie, infection des voies aériennes supérieures),
· essoufflement (dyspnée),
· inflammation des sinus (sinusite),
· inflammation des voies aériennes pulmonaires (bronchite),
· toux,
· inflammation de la muqueuse buccale (stomatite),
· maux d’estomac ou douleurs abdominales,
· brûlure d’estomac (dyspepsie),
· difficulté à avaler (dysphagie),
· rougeur cutanée (érythème),
· peau sèche,
· démangeaisons (prurit),
· lésion musculaire (myopathie),
· faiblesse musculaire,
· douleurs articulaires (arthralgie, douleur musculosquelettique),
· mal de dos,
· douleur musculaire (myalgie, douleur musculosquelettique),
· miction fréquente,
· fuite urinaire (incontinence urinaire),
· fièvre,
· symptômes pseudo-grippaux,
· faiblesse (asthénie),
· sensation générale d’être malade (malaise),
· douleur,
· rétention d’eau (œdème), en particulier au niveau des chevilles et des jambes (œdème périphérique),
· augmentation des enzymes hépatiques,
· perte de poids,
· prise de poids,
· lésion radique.
Les effets indésirables peu fréquents (pouvant toucher jusqu’à 1 patient sur 100) sont :
· infections opportunistes, notamment infections des poumons (pneumonie à Pneumocystis jirovecii),
· circulation de bactéries et de leurs toxines dans le sang entraînant des lésions sur les organes (septicémie),
· infections du cerveau (méningo-encéphalite herpétique) ayant parfois une issue fatale,
· apparition ou réactivation d’infections à cytomégalovirus,
· réactivation d’infections par le virus de l’hépatite B,
· infection virale de la bouche, tels que les boutons de fièvre, ou des organes génitaux (herpès simplex),
· réactivation d’infections,
· infections de plaies,
· infections entraînant des diarrhées et des vomissements (gastro-entérite),
· production insuffisante de cellules sanguines ou plaquettes saines par la moelle osseuse (syndromes myélodysplasiques),
· cancers secondaires, y compris leucémie,
· diminution des taux de cellules sanguines (pancytopénie, anémie aplasique),
· taches rouges sous la peau (pétéchies),
· diabète insipide (dont les symptômes incluent augmentation des mictions et une sensation de soif),
· faible taux de potassium dans le sang (hypokaliémie),
· augmentation des enzymes hépatiques (augmentation des taux de phosphatase alcaline et de gamma‑glutamyltransférase),
· trouble du comportement,
· sautes d’humeur (labilité émotionnelle),
· hallucinations,
· manque d’intérêt ou d’énergie (apathie),
· paralysie partielle (hémiplégie),
· effets sur une partie du cerveau qui régule les mouvement, ce qui peut entraîner des tremblements, des spasmes musculaires ou des troubles des mouvements (trouble extrapyramidal),
· modification de l’odorat (parosmie),
· démarche inhabituelle (anomalie de la démarche),
· augmentation de la perception du toucher, de la douleur et de la température (hyperesthésie),
· trouble sensoriel,
· coordination anormale,
· baisse de l’acuité visuelle,
· sécheresse des yeux,
· trouble de l’audition,
· sensibilité aux sons (hyperacousie),
· infection de l’oreille moyenne (otite moyenne),
· palpitations (perception des battements du cœur),
· rougeur de la peau,
· bouffées de chaleur,
· incapacité des poumons à fonctionner correctement (insuffisance respiratoire),
· inflammation des poumons entraînant un essoufflement et de la toux (pneumopathie interstitielle/pneumopathie),
· lésion des poumons avec épaississement et durcissement du tissu (fibrose pulmonaire),
· nez bouché (congestion nasale),
· ballonnement (distension abdominale),
· incapacité à contrôler l’émission des selles (incontinence fécale),
· troubles touchant l’estomac et les intestins (trouble gastro-intestinal),
· hémorroïdes,
· bouche sèche,
· défaillance fatale du foie (insuffisance hépatique),
· lésion du foie (lésion hépatique),
· inflammation du foie (hépatite),
· réduction de l’écoulement de la bile produite dans le foie en raison d’une obstruction (cholestase),
· taux élevés de bilirubine dans le sang, pouvant causer un jaunissement de la peau et des yeux, indiquant des problèmes hépatiques (hyperbilirubinémie),
· rougeur et peau qui pèle sur une large zone du corps, avec possibles démangeaisons ou douleurs (érythrodermie, exfoliation cutanée),
· augmentation de la sensibilité au soleil (réaction de photosensibilité),
· rash avec démangeaisons (urticaire, exanthème),
· inflammation de la peau (dermatite),
· augmentation de la transpiration,
· changement de la couleur de la peau (pigmentation anormale),
· miction douloureuse (dysurie),
· saignement vaginal (hémorragie vaginale),
· saignement menstruel abondant (ménorragie),
· absence de règles (aménorrhée),
· inflammation et irritation vaginales (vaginite),
· douleur au sein,
· impuissance sexuelle,
· aggravation de l’état,
· frissons (rigidité),
· gonflement du visage (œdème du visage),
· décoloration de la langue,
· soif,
· trouble dentaire.
Déclaration des effets secondaires
Si vous ressentez un quelconque effet indésirable, parlez-en à votre médecin, votre pharmacien ou à votre infirmier/ère. Ceci s’applique aussi à tout effet indésirable qui ne serait pas mentionné dans cette notice. Vous pouvez également déclarer les effets indésirables directement via le système national de déclaration : Agence nationale de sécurité du médicament et des produits de santé (ANSM) et réseau des Centres Régionaux de Pharmacovigilance - Site internet : https://signalement.social-sante.gouv.fr/.
En signalant les effets indésirables, vous contribuez à fournir davantage d’informations sur la sécurité du médicament.
5. COMMENT CONSERVER TEMOZOLOMIDE VIATRIS 5 mg, gélule ?
Tenir ce médicament hors de la vue et de la portée des enfants, de préférence dans un endroit fermé à clé. Une ingestion accidentelle peut être mortelle pour les enfants.
N’utilisez pas ce médicament après la date de péremption indiquée sur l’étiquette et la boîte après EXP. La date de péremption fait référence au dernier jour de ce mois.
Flacon
A conserver à une température ne dépassant pas + 30 °C, dans l’emballage extérieur d’origine à l’abri de la lumière. Conserver le flacon soigneusement fermé à l’abri de l’humidité.
Après ouverture, le médicament doit être utilisé dans les 21 jours.
Sachets-dose :
A conserver à une température ne dépassant pas + 25 °C, dans l’emballage extérieur d’origine à l’abri de la lumière.
Prévenez votre pharmacien si vous constatez un changement dans l’apparence des gélules.
Ne jetez aucun médicament au tout-à-l’égout ou avec les ordures ménagères. Demandez à votre pharmacien d’éliminer les médicaments que vous n’utilisez plus. Ces mesures contribueront à protéger l’environnement.
6. CONTENU DE L’EMBALLAGE ET AUTRES INFORMATIONS
Ce que contient TEMOZOLOMIDE VIATRIS 5 mg, gélule
Le témozolomide. Chaque gélule contient 5 mg de témozolomide.
· Les autres composants sont :
Contenu de la gélule : lactose anhydre, carboxyméthylamidon sodique type A, silice colloïdale anhydre, acide tartrique, acide stéarique.
Enveloppe de la gélule : gélatine, dioxyde de titane (E171), laque, propylène glycol, laque aluminique indigo carmin (E132), oxyde de fer jaune (E172).
Qu’est-ce que TEMOZOLOMIDE VIATRIS 5 mg, gélule et contenu de l’emballage extérieur
Les gélules de TEMOZOLOMIDE VIATRIS 5 mg ont un corps et une coiffe opaques blancs, elles sont marquées de deux rayures vertes sur la coiffe et « T 5 mg » vert sur le corps.
Packs de flacon
Les gélules destinées à la voie orale se présentent en flacon de verre teinté contenant 5 ou 20 gélules. L’étui contient un flacon.
Sachets-dose
Les gélules destinées à la voie orale se présentent en étui en carton contenant 5 ou 20 gélules individuellement scellées dans des sachets-dose.
Toutes les présentations peuvent ne pas être commercialisées.
Titulaire de l’autorisation de mise sur le marché
1 RUE DE TURIN
69007 LYON
Exploitant de l’autorisation de mise sur le marché
1 RUE DE TURIN
69007 LYON
PHARMACEUTICAL SCIENCES VIALE PASTEUR, 10
20014 NERVIANO (Ml)
ITALIE
OU
HAUPT PHARMA AMAREG GmbH
DONAUTRAUFER STRASSE 378
93055 REGENSBURG
ALLEMAGNE
OU
DELORBIS PHARMACEUTICALS LTD
17 ATHINON STREET
ERGATES INDUSTRIAL AREA
2643 ERGATES
P.O.BOX 28629
2081 LEFKOSIA
CHYPRE
Noms du médicament dans les Etats membres de l'Espace Economique Européen
[À compléter ultérieurement par le titulaire]
La dernière date à laquelle cette notice a été révisée est :
[à compléter ultérieurement par le titulaire]
Des informations détaillées sur ce médicament sont disponibles sur le site Internet de l’ANSM (France).