Dernière mise à jour le 01/06/2026
AMIKACINE STRAGEN 250 mg/mL, solution injectable
Indications thérapeutiques
Classe pharmacothérapeutique : autres aminosides - code ATC : J01GB06.
Qu'est-ce que AMIKACINE STRAGEN 250 mg/mL, solution injectable
AMIKACINE STRAGEN contient la substance active amikacine.
C’est un antibiotique qui appartient à la famille des "aminosides".
AMIKACINE STRAGEN est indiqué chez l’adulte, l'adolescent et l'enfant dans le traitement d’infections sévères dues à des bactéries sensibles à l'amikacine. Dans ces conditions, ce médicament peut être utilisé dans les infections suivantes :
· infections au niveau des poumons
· infections au niveau des os et articulations
· infections du système nerveux central
· infections compliquées des voies urinaires
· infections compliquées abdominales
· infections de la peau et des tissus mous, y compris brûlures sévères
· inflammation bactérienne de la paroi interne du cœur (uniquement en association avec d’autres antibiotiques)
Il peut également être utilisé dans le traitement des patients présentant une infection au niveau du sang, associée ou suspectée d’être associée, à l’une des infections précitées.
Présentations
> 10 flacons en verre de 2 mL
Code CIP : 34009 303 272 8 9
Déclaration de commercialisation : 23/04/2026
Cette présentation est agréée aux collectivités
> 10 flacons en verre de 4 mL
Code CIP : 34009 303 273 1 9
Déclaration de commercialisation : 23/04/2026
Cette présentation est agréée aux collectivités
Service médical rendu (SMR)
Les libellés affichés ci-dessous ne sont que des résumés ou extraits issus des avis rendus par la Commission de la Transparence. Seul l'avis complet de la Commission de la Transparence fait référence.
Cet avis est consultable à partir du lien `Avis du jj/mm/aaaa` ou encore sur demande auprès de la HAS (plus d'informations dans l'aide). Les avis et synthèses d'avis contiennent un paragraphe sur la place du médicament dans la stratégie thérapeutique.
| Valeur du SMR | Avis | Motif de l'évaluation | Résumé de l'avis |
|---|---|---|---|
| Important | Avis du 17/12/2025 | Inscription (CT) | Le service médical rendu par AMIKACINE STRAGEN 250 mg/mL (amikacine), solution injectable, est important dans les indications de l’AMM. |
Amélioration du service médical rendu (ASMR)
Les libellés affichés ci-dessous ne sont que des résumés ou extraits issus des avis rendus par la Commission de la Transparence. Seul l'avis complet de la Commission de la Transparence fait référence.
Cet avis est consultable à partir du lien `Avis du jj/mm/aaaa` ou encore sur demande auprés de la HAS (plus d'informations dans l'aide). Les avis et synthéses d'avis contiennent un paragraphe sur la place du médicament dans la stratégie thérapeutique.
| Valeur de l'ASMR | Avis | Motif de l'évaluation | Résumé de l'avis |
|---|---|---|---|
| V (Inexistant) | Avis du 17/12/2025 | Inscription (CT) | Cette spécialité est un eurogénérique qui n’apporte pas d’amélioration du service médical rendu (ASMR V) par rapport aux spécialités injectables à base d’amikacine. |
Autres informations
- Titulaire de l'autorisation : STRAGEN-France
- Conditions de prescription et de délivrance :
- liste I
- prescription hospitalière
- Statut de l'autorisation : Valide
- Type de procédure : Procédure nationale
- Code CIS : 6 389 939 3
ANSM - Mis à jour le : 24/09/2025
AMIKACINE STRAGEN 250 mg/mL, solution injectable
2. COMPOSITION QUALITATIVE ET QUANTITATIVE
Amikacine (sous forme de sulfate d’amikacine)..................................................................... 250 mg
Pour 1 mL de solution injectable.
Chaque flacon de 2 mL contient l’équivalent de 500 mg d’amikacine sous forme de sulfate d’amikacine.
Chaque flacon de 4 mL contient l’équivalent de 1000 mg d’amikacine sous forme de sulfate d’amikacine.
Excipient(s) à effets notoires :
1 mL de solution contient 7,5 mg de sodium et 6,60 mg de métabisulfite de sodium (E223).
Pour la liste complète des excipients, voir rubrique 6.1.
Solution limpide, incolore à jaune pâle.
Le pH est compris entre 3,5 et 5,5.
4.1. Indications thérapeutiques
· Infections respiratoires basses nosocomiales, y compris les pneumonies nosocomiales (PN) dont les pneumonies acquises sous ventilation mécanique (PAVM)
· Infections ostéo-articulaires
· Infections du système nerveux central
· Infections intra-abdominales compliquées
· Infections urinaires compliquées, y compris les pyélonéphrites
· Infections bactériennes aiguës de la peau et des tissus mous, y compris les infections des brûlures
· Endocardite bactérienne (uniquement en association avec d’autres antibiotiques)
AMIKACINE STRAGEN peut également être utilisée dans le traitement des patients présentant une bactériémie associée, ou suspectée d'être associée, à l'une des infections précitées.
AMIKACINE STRAGEN est généralement utilisée en association avec d'autres antibiotiques appropriés, en particulier avec des bêta-lactamines.
Il convient de tenir compte des recommandations officielles concernant l'utilisation appropriée des antibactériens.
4.2. Posologie et mode d'administration
La dose dépend de la gravité du tableau clinique, du terrain, de la fonction rénale du patient et de la bactérie identifiée. La dose est exprimée selon le poids corporel du patient.
Il convient de se référer aux recommandations thérapeutiques.
Il existe plusieurs présentations d’amikacine dont certaines sont plus adaptées aux fortes doses à administrer par voie intraveineuse.
Surveillance des concentrations plasmatiques d’amikacine
Les recommandations thérapeutiques sur la surveillance des concentrations plasmatiques doivent être prises en considération.
L’état de la fonction rénale doit être estimé en mesurant la concentration sérique de la créatinine ou par calcul de la clairance de la créatinine endogène. L’utilisation de l’azote uréique sanguin est beaucoup moins fiable. La fonction rénale doit être régulièrement contrôlée au cours du traitement.
Le suivi du traitement peut nécessiter la surveillance des concentrations plasmatiques d’amikacine pour s’assurer de taux corrects et non excessifs. Il convient de tenir compte des recommandations sur les indications des dosages plasmatiques (dosage au pic plasmatique et dosage de la concentration résiduelle).
Les recommandations préconisent une mesure des concentrations maximales 30 minutes après la fin de la perfusion.
La dose doit être adaptée conformément aux recommandations. Les objectifs de concentrations sont pour l’amikacine de 60 à 80 mg/L au pic (Cmax) et < 2.5 mg/L pour la concentration résiduelle (Cmin) selon les recommandations.
Chez les patients présentant une insuffisance rénale, le suivi des concentrations plasmatiques est fortement recommandé.
Durée du traitement
La durée totale du traitement est limitée à 5 jours. Une prolongation de la durée de traitement doit faire l’objet d’une réévaluation.
Patients dont la fonction rénale est normale (clairance de la créatinine ≥ 50 mL/min)
Il convient de tenir compte des recommandations thérapeutiques, le schéma posologique préférentiel étant la dose unique journalière, soit la totalité de la dose journalière administrée en une seule injection quotidienne. Une dose journalière répartie en 2 à 3 injections quotidiennes est possible, notamment dans certaines situations (en particulier les endocardites).
La dose varie de 15 à 30 mg/kg/jour selon les recommandations officielles, la dose maximale de 30 mg/kg/jour étant surtout recommandée en début de traitement, dans des infections graves et/ou en cas de risque d’infection due à une souche bactérienne de sensibilité diminuée ayant une concentration minimale inhibitrice (CMI) augmentée à l’amikacine.
Adultes, adolescents de 12 ans et plus
Nourrissons et enfants (de 4 semaines à 11 ans)
Une dose journalière administrée par voie intraveineuse (perfusion intraveineuse lente) chez des enfants ayant une fonction rénale normale de 15 à 20 mg/kg de poids corporel peut être administrée en une fois par jour ou répartie en doses de 7,5 mg/kg de poids corporel toutes les 12 heures.
Nouveau-nés (de 0 à 27 jours) et prématurés (voir rubriques 4.4 et 5.2).
Pour les nouveau-nés, il convient d’adapter les posologies selon l’âge post-conceptionnel en tenant compte des recommandations en vigueur.
Patients insuffisants rénaux (clairance de la créatinine < 50 mL/min)
Selon les recommandations, il convient de privilégier chez les patients présentant une altération de la fonction rénale le schéma en dose unique journalière, de pratiquer des dosages plasmatiques (pic et résiduel) pour ajuster les posologies et les intervalles entre chaque injection, de privilégier les durées courtes de traitement (en règle générale : 1 ou 2 injections), de tenir compte des autres facteurs de risque favorisant la néphrotoxicité des aminosides, de surveiller les fonctions rénale et auditive (voir rubriques 4.4 et 4.8). Les patients présentant une altération de la fonction rénale (clairance de la créatinine < 50 mL/min) peuvent être exposés à des concentrations minimales élevées.
Chez les patients présentant une ’insuffisance rénale, les concentrations sériques d’amikacine doivent être contrôlées par des techniques appropriées. Les doses doivent être adaptées en tenant compte des recommandations en vigueur.
Les approches ci-après proposées se basent sur les valeurs de la clairance de la créatinine ou de la créatinine sérique du patient car il a été montré que ces valeurs sont corrélées à la demi-vie des aminosides chez les patients dont la fonction rénale est réduite. Ces schémas posologiques doivent être utilisés conjointement à des examens cliniques et biologiques du patient et doivent être modifiés si nécessaire, notamment lorsque le patient est sous dialyse.
Allongement de l’intervalle entre deux doses normales
Si la clairance de la créatinine n’est pas disponible et que l’état du patient est stable, l’intervalle en heures entre deux doses qui seraient administrées à un patient dont la fonction rénale est normale à raison de 7,5 mg/kg de poids corporel deux fois par jour, se calcule en multipliant par 9 la concentration de créatinine sérique. Par exemple, si la concentration de créatinine est de 2 mg/100 mL, la dose (7,5 mg/kg de poids corporel) doit être administrée toutes les 2 x 9 = 18 heures.
Réduction de la dose administrée à intervalles fixes
Si la détermination des concentrations sériques d’amikacine n’est pas possible, et que l’état du patient est stable, les valeurs de la créatinine sérique et de la clairance de la créatinine sont les indicateurs les plus facilement disponibles permettant de définir le degré d’insuffisance rénale pour déterminer la dose.
Chez des patients présentant une insuffisance rénale chronique et dont la clairance de la créatinine est connue, la dose de la première administration d’amikacine est de 7,5 mg/kg de poids corporel. La dose d’entretien administrée toutes les 12 heures doit être réduite en fonction de la diminution de la clairance de la créatinine du patient et est calculée à l’aide de la formule suivante :
Dose d’entretien d’amikacine =
![]()
(Cl.Cr = Clairance de la créatinine [mL/min])
Cf. Guide pratique sur des doses d’amikacine dans le tableau ci-dessous :
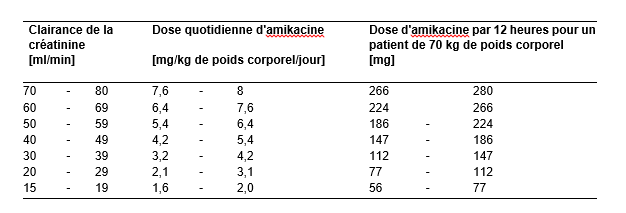
Une autre méthode permettant de déterminer la réduction de la dose avec des intervalles de 12 heures (pour les patients dont les valeurs de la créatinine sérique stables sont connues) est de diviser la dose généralement recommandée par la créatinine sérique du patient.
Patients sous épuration extra-rénale
Les recommandations thérapeutiques sont à prendre en considération.
Patients âgés
Les patients âgés peuvent présenter une altération de la fonction rénale.
L'amikacine étant excrétée par voie rénale, la dose est à adapter en fonction de la fonction rénale.
Patients obèses
L'amikacine diffuse mal dans les tissus adipeux. La dose en mg/kg peut être calculée en se basant sur le poids corporel idéal estimé du patient et en ajoutant 40 % de l’excédent. La dose doit être adaptée en fonction des résultats de la surveillance de la concentration plasmatique.
Formule pour le calcul du poids à prendre en compte pour déterminer la posologie chez les patients obèses (P1) :
P1 = PI + (PA – PI) x 0,4
PI = poids idéal
PA = poids actuel
La dose maximale de 1,5 g par jour ne doit pas être dépassée.
Patients avec ascite
Des doses élevées sont à envisager afin d'obtenir une concentration sérique adéquate compte tenu de la distribution relativement plus importante dans le compartiment liquidien extracellulaire.
Mode d’administration
Voie intraveineuse, et à titre exceptionnel par voie intramusculaire.
Selon les recommandations, la durée de perfusion est de 30 minutes.
La voie intramusculaire est à éviter dans la mesure du possible, mais peut être pratiquée à titre exceptionnel en tenant compte que cette voie n’offre pas les mêmes garanties que la voie intraveineuse.
Si la voie intramusculaire doit être utilisée, les schémas d’administration sont identiques à ceux préconisés pour la voie intraveineuse.
Pour les instructions concernant la dilution du médicament avant administration, voir la rubrique 6.6.
· Hypersensibilité à la substance active ou à l’un des excipients mentionnés à la rubrique 6.1.
· Hypersensibilité à d’autres aminosides.
4.4. Mises en garde spéciales et précautions d'emploi
Il convient de prendre en compte les recommandations thérapeutiques.
La prudence s'impose lors de l'administration à des patients présentant :
· une insuffisance rénale
· des troubles auditifs ou vestibulaires
· des troubles neuromusculaires (par ex : myasthénie, syndromes parkinsoniens, car la faiblesse musculaire peut être aggravée en raison du potentiel effet curarisant des aminosides sur la jonction neuromusculaire)
· chez les patients traités par un autre aminoside immédiatement avant l'amikacine
Les patients traités par aminosides par voie parentérale doivent faire l’objet d’un suivi clinique étroit en raison de l’ototoxicité et de la néphrotoxicité potentielles associées à leur utilisation.
Les effets toxiques des aminosides, y compris l'amikacine, sont plus fréquents chez les patients insuffisants rénaux, si des doses élevées sont administrées et si la durée de traitement est prolongée.
La sécurité du traitement pendant une période de plus de 14 jours n'a pas été établie. Un âge avancé et une déshydratation accroissent également le risque de toxicité des aminosides.
Neuro/Ototoxicité
La neurotoxicité, qui se présente sous la forme d’une ototoxicité vestibulaire et/ou bilatérale, peut survenir chez les patients traités par aminosides. Le risque d’ototoxicité induite par les aminosides est plus élevé chez les patients insuffisants rénaux ou chez ceux dont le traitement est prolongé au-delà de 5 à 7 jours, même chez les patients sains.
Une perte significative de la détection des hautes fréquences est généralement le premier signe d’ototoxicité et ne peut être détectée que par un audiogramme. Les patients peuvent présenter des vertiges, signe d’une atteinte vestibulaire.
Engourdissements, picotements, contractions musculaires et convulsions sont d’autres manifestations de la neurotoxicité.
Les patients développant des troubles cochléaires ou vestibulaires peuvent ne présenter aucun symptôme au cours du traitement les prévenant du développement d’une toxicité du huitième nerf et d’une surdité bilatérale partielle ou totale irréversible, ou de vertiges handicapants susceptibles de survenir après l’arrêt du traitement. Voir également rubrique 4.8.
L’ototoxicité induite par les aminosides est généralement irréversible.
L’utilisation de l’amikacine chez les patients avec un antécédent d’allergie aux aminosides ou chez les patients avec une atteinte infraclinique rénale ou du huitième nerf produite par l’administration antérieure de substances néphrotoxiques et/ou ototoxiques doit être envisagée avec prudence, car la toxicité pourrait être additive.
L’amikacine doit être utilisée chez ces patients uniquement si, selon l’avis du médecin, les avantages thérapeutiques l’emportent sur les risques potentiels.
Il existe un risque accru d’ototoxicité chez les patients présentant des mutations de l’ADN mitochondrial (en particulier la substitution de A par G dans le nucléotide 1555 du gène ARNr 12S), même si les taux sériques d’aminosides se situent dans la fourchette recommandée pendant le traitement. Des alternatives thérapeutiques doivent être envisagées chez ces patients. En cas d’antécédents familiaux de mutations significatives ou de surdité induite par les aminosides, des traitements alternatifs ou des tests génétiques préalables à l’administration doivent être envisagés.
Toxicité neuromusculaire
Etant donné que les aminosides ont des propriétés de blocage neuromusculaire, une attention particulière doit être portée chez les patients ayant une maladie neuromusculaire préexistante (par exemple maladie de Parkinson). Il est impératif d’instaurer un suivi étroit chez de tels patients (voir rubrique 4.8).
Un blocage neuromusculaire et une paralysie respiratoire ont été rapportés à la suite d’une administration parentérale, d’une instillation topique (dans le cas d’une irrigation orthopédique et abdominale ou dans le traitement local de l’empyème) et à la suite de l’utilisation d’aminosides par voie orale.
La possibilité de paralysie respiratoire doit être prise en compte en cas d’administration d’aminosides, quel que soit la voie d’administration, surtout chez les patients recevant simultanément un traitement médicamenteux exerçant des blocages neuromusculaires. Voir également rubrique 4.5.
En cas de blocage neuromusculaire, les sels calciques peuvent neutraliser la paralysie respiratoire mais une assistance respiratoire artificielle peut s’avérer nécessaire. Un blocage neuromusculaire et une paralysie musculaire ont été observés chez les animaux ayant reçu des doses élevées d’amikacine.
L’administration d’aminosides à des patients présentant une maladie neuromusculaire (myasthénie, maladie de Parkinson, par exemple) requiert la plus grande prudence car les aminosides exercent sur la jonction neuromusculaire une action similaire à celle du curare et peuvent, par conséquent, aggraver la faiblesse musculaire.
Toxicité rénale
Les aminosides sont potentiellement néphrotoxiques. La toxicité rénale est indépendante de la concentration plasmatique obtenue au pic (Cmax). Le risque de néphrotoxicité est plus élevé chez les patients insuffisants rénaux et chez ceux recevant des doses élevées ou dont le traitement est prolongé.
Les patients doivent être bien hydratés pendant le traitement. La fonction rénale doit être évaluée avant le début du traitement et surveillée régulièrement pendant le traitement (voir rubrique 4.2).
Les doses quotidiennes doivent être réduites et/ou l’intervalle entre les doses doit être allongé en cas de troubles rénaux tels que : cylindrurie, présence de leucocytes ou d’érythrocytes, albuminurie, diminution de la clairance de la créatinine, diminution de la densité de l’urine, hyperazotémie, élévation de la créatinine sérique et oligurie.
Le traitement doit être interrompu si l’azotémie augmente ou si le volume des urines diminue progressivement.
Les patients âgés présenter une insuffisance rénale susceptible de ne pas être identifiée par des examens de routine comme l’azotémie ou la créatininémie. Une détermination de la clairance de la créatinine peut s’avérer plus utile. Le contrôle de la fonction rénale chez les patients âgés au cours du traitement par aminosides est particulièrement important.
Suivi du patient
La fonction rénale et celle du huitième nerf crânien doivent être attentivement contrôlées, notamment chez les patients présentant une insuffisance rénale suspectée ou connue au début du traitement mais également chez ceux dont la fonction rénale est normale initialement mais qui présentent des signes d’insuffisance rénale au cours du traitement. Les concentrations sériques d’amikacine doivent être contrôlées quand cela est possible pour assurer des taux adéquats et éviter d’atteindre des taux potentiellement toxiques. Les urines doivent être examinées en vue de déceler une diminution de la densité de l’urine, une augmentation de l’excrétion protéique, et la présence de cellules ou de cylindres urinaires. L’azote uréique sanguin, la créatinine sérique ou la clairance de la créatinine doivent être régulièrement mesurés. Si possible, des audiogrammes répétés doivent être effectués chez les patients suffisamment âgés pour être testés, particulièrement chez les patients à haut risque. L’identification d’une ototoxicité (étourdissements, vertiges, acouphènes, bourdonnements dans les oreilles et perte d’audition) ou d’une néphrotoxicité nécessite l’arrêt du médicament ou une adaptation de la dose. Voir rubrique 4.8.
Le traitement par amikacine doit être arrêté si des acouphènes ou une perte d’audition subjective apparaissent ou si les audiogrammes de suivi montrent une perte significative de la détection des hautes fréquences.
Comme avec d’autres antibiotiques, le recours à l’amikacine peut entraîner la prolifération d’organismes résistants. Dans ce cas, un traitement approprié devra être initié.
Autre
Appliqués localement dans le cadre d’une procédure chirurgicale, les aminosides sont absorbés rapidement et presque complètement (à l’exception de la vessie). Des cas de surdité irréversible, d’insuffisance rénale et de décès ont été rapportés à la suite d’un blocage neuromusculaire lié à l’irrigation du champ opératoire avec des préparations à base d’aminosides (quelle que soit la surface).
Un infarctus maculaire entraînant parfois une perte irréversible de la vision a été rapporté à la suite de l’administration intravitréenne (injection dans l’œil) d’amikacine.
Population pédiatrique
Les aminosides doivent être utilisés avec prudence chez les prématurés et les nouveau-nés, en raison de l'immaturité rénale de ces patients qui allonge la demi-vie sérique de ces médicaments.
Interférences avec les tests de laboratoire
Les dosages de la créatinine sérique peuvent donner des valeurs faussement élevées quand les céphalosporines sont administrées de façon concomitante.
L’inactivation mutuelle de l’amikacine et des bêta-lactamines peut persister dans les échantillons (par ex : sérum, liquide céphalo-rachidien, etc.) prélevés pour doser les aminosides, aboutissant ainsi à des résultats incorrects. Les échantillons doivent donc être soit immédiatement analysés après prélèvement, soit réfrigérés ou encore la bêta-lactamine doit être inactivée par l’ajout de bêta-lactamase. L’inactivation de l’aminoside n’est cliniquement pertinente que chez les patients présentant une insuffisance rénale sévère.
Excipients à effet notoire
AMIKACINE STRAGEN contient du métabisulfite de sodium (E 223) et peut, dans de rares cas, provoquer des réactions d'hypersensibilité sévères et des bronchospasmes.
Ce médicament contient 7,5 mg de sodium par mL de solution injectable.
Ce médicament contient 15 mg de sodium par flacon de 2 mL, ce qui équivaut à 0,75 % de l’apport alimentaire quotidien maximal recommandé par l'OMS de 2 g de sodium par adulte.
Ce médicament contient 30 mg de sodium par flacon de 4 mL, ce qui équivaut à 1,5 % de l’apport alimentaire quotidien maximal recommandé par l'OMS de 2 g de sodium par adulte.
4.5. Interactions avec d'autres médicaments et autres formes d'interactions
Autres substances neurotoxiques, ototoxiques ou néphrotoxiques
L'administration concomitante ou subséquente, à la fois systémique ou locale, d'autres substances neurotoxiques, ototoxiques ou néphrotoxiques doivent être évitées en raison des effets additionnels possibles.
La toxicité de l'amikacine peut être augmentée par les substances neurotoxiques et/ou ototoxiques et/ou néphrotoxiques suivantes :
· Autres aminosides par voie parentérale (par ex : kanamycine, paromomycine)
· Autres produits anti-infectieux, comme bacitracine, amphotéricine B, céphalosporines (céphaloridine), vancomycine, polymyxines (polymyxine B, colistine), viomycine
· Cytostatiques à base de platine : carboplatine (à haute dose), cisplatine, oxaliplatine (particulièrement en cas d'insuffisance rénale préexistante)
· Immunosuppresseurs : ciclosporine, tacrolimus
· Diurétiques à action rapide, comme furosémide ou acide éthacrynique (ototoxicité intrinsèque potentielle, toxicité additionnelle des aminosides pouvant être accrue à cause de l’effet de déshydratation des diurétiques et une élévation de la concentration des aminosides dans le sérum et les tissus)
· Anesthésie au méthoxyflurane : les aminosides peuvent accroître les effets néfastes du méthoxyflurane sur les reins. En cas d'usage simultané, des neuropathies extrêmement sévères sont possibles
Lorsque l’amikacine est associée à un agent potentiellement néphrotoxique ou ototoxique, les fonctions auditive et rénale doivent être surveillées attentivement. En cas d’utilisation concomitante avec un diurétique à action rapide, l’hydratation du patient doit être contrôlée.
Une réduction de l’activité sérique peut survenir lorsque des médicaments de la famille des aminosides ou de la pénicilline sont administrés simultanément à l’amikacine in vivo par des voies d’administration séparées.
Bisphosphonates
Le risque d’hypocalcémie est accru quand les aminosides sont administrés avec des bisphosphonates.
Indométacine
L'indométacine peut accroître la concentration plasmatique d'amikacine chez les nouveau-nés.
Amikacine/myorelaxants et autres substances ayant un effet neuromusculaire
En cas de traitement concomitant par amikacine et un myorelaxant (par ex : succinylcholine, décaméthonium, atracurium, rocuronium, vencuronium), des transfusions massives de sang citraté ou un anesthésique, le blocage musculaire induit par ces produits peut être augmenté et conduire à une paralysie respiratoire. En cas de chirurgie, l’anesthésiste doit être informé du traitement du patient par amikacine. Le blocage neuromusculaire doit faire l’objet d’un traitement approprié (voir rubrique 4.9).
Thiamine
La thiamine (vitamine B1) administrée simultanément peut être dégradée par le métabisulfite de sodium réactif contenu dans la formulation du sulfate d'amikacine.
4.6. Fertilité, grossesse et allaitement
Grossesse
Les données disponibles concernant l'utilisation d'aminosides chez les femmes enceintes sont limitées.
Les aminosides peuvent être nocifs pour le fœtus. Ils traversent la barrière placentaire et des cas de surdité congénitale bilatérale totale irréversible ont été rapportés chez des enfants dont les mères ont reçu de la streptomycine durant leur grossesse.
Bien qu'aucun effet indésirable sur le fœtus ou le nouveau-né n'ait été signalé chez des femmes enceintes traitées par d'autres aminosides, il existe une nocivité potentielle. Si l'amikacine est utilisée durant la grossesse ou si la patiente débute une grossesse alors qu'elle prend ce médicament, la patiente doit être informée des risques potentiels pour le fœtus.
AMIKACINE STRAGEN ne doit pas être administrée pendant la grossesse, à moins que l'état clinique de la patiente ne requière un traitement par amikacine. Si le traitement est jugé nécessaire, il doit uniquement avoir lieu sous surveillance médicale (voir rubrique 4.4).
Aucune donnée n'est disponible sur l'éventuelle excrétion de l'amikacine ou de ses métabolites dans le lait maternel. La décision d'interrompre l'allaitement ou d'interrompre l’administration ou de ne pas débuter l’administration d’amikacine doit être prise en tenant compte des avantages de l'allaitement pour l'enfant et des bénéfices du traitement pour la mère.
Fertilité
Des études de toxicité sur la reproduction menées chez la souris et le rat n'ont montré aucun effet sur la fertilité.
4.7. Effets sur l'aptitude à conduire des véhicules et à utiliser des machines
En cas d’administration à des patients non hospitalisés, la prudence s’impose lors de la conduite et de l’utilisation de machines, compte tenu des effets indésirables possibles tels que des troubles de l'équilibre (voir rubrique 4.8), pouvant altérer l’aptitude à conduire des véhicules et utiliser des machines.
Tous les aminosides peuvent potentiellement induire une ototoxicité, une néphrotoxicité et un blocage neuromusculaire. Ces toxicités surviennent plus souvent chez les patients insuffisants rénaux, chez les patients traités par d’autres médicaments ototoxiques ou néphrotoxiques, et chez les patients traités sur de longues périodes et/ou avec des doses supérieures à celles recommandées (voir rubrique 4.4).
Les effets indésirables considérés comme pouvant au moins être liés au traitement sont listés ci-dessous par classe de système d'organes et par fréquence. La terminologie suivante a été utilisée afin de classifier la survenue des effets indésirables :
Très fréquent (≥ 1/10) ;
Fréquent (≥ 1/100 à < 1/10) ;
Peu fréquent (≥ 1/ 1 000 à < 1/100) ;
Rare (≥ 1/ 10 000 à < 1/ 1 000) ;
Très rare (< 1/10 000) ;
Fréquence indéterminée (ne peut être estimée sur la base des données disponibles).
|
Classe de systèmes d’organes |
Fréquence |
Effets indésirables |
|
Infections et infestations |
Peu Fréquent |
Surinfection ou colonisation (par des champignons de type levure ou des germes résistants) |
|
Affections hématologiques et du système lymphatique |
Rare |
Anémie, éosinophilie, leucopénie, granulocytopénie, thrombopénie |
|
Affections du système immunitaire |
Rare |
Réactions d’hypersensibilité3 |
|
Très rare |
Choc anaphylactique |
|
|
Indéterminée |
Allergie croisée entre aminosides |
|
|
Troubles du métabolisme et de la nutrition |
Rare |
Hypomagnésémie |
|
Affections du système nerveux |
Peu fréquent |
Etourdissements1, vertiges1 |
|
Rare |
Tremblements, paresthésies, céphalées, migraines |
|
|
Affections oculaires |
Peu fréquent |
Nystagmus1 |
|
Rare |
Cécité5, infarctus rétinien5 |
|
|
Affections de l'oreille et du labyrinthe |
Peu fréquent |
Acouphènes1, sensation de pression dans l’oreille1, perte auditive1 |
|
Très rare |
Surdité1 |
|
|
Affections vasculaires |
Rare |
Hypotension |
|
Affections respiratoires, thoraciques et médiastinales |
Rare |
Dépression de la fonction respiratoire4 |
|
Très rare |
Paralysie respiratoire4 |
|
|
Indéterminée |
Apnée, bronchospasme |
|
|
Affections gastro-intestinales |
Peu Fréquent |
Nausées1 |
|
Rare |
Vomissement |
|
|
Affections de la peau et du tissu sous-cutané |
Rare |
Rash, exanthème, prurit, urticaire (réactions d’hypersensibilité)3 |
|
Affections musculo-squelettiques et du tissu conjonctif |
Rare |
Arthralgies |
|
Très rare |
Blocage neuromusculaire |
|
|
Affections du rein et des voies urinaires |
Peu Fréquent |
Atteinte des tubules rénaux2, insuffisance rénale2 |
|
Très rare |
Néphropathie toxique, insuffisance rénale aiguë |
|
|
Troubles généraux et anomalies au site d'administration |
Rare |
Fièvre d’origine médicamenteuse3
|
|
Investigations |
Rare |
Aspartate aminotransférase augmentée, alanine aminotransférase augmentée, augmentation (légère et transitoire) des phosphatases alcalines |
Informations complémentaires sur certains effets indésirables :
(1) Ces effets ont été constatés particulièrement en cas de dépassement de la posologie recommandée, lors de traitements de plus de 10 jours ou lorsque la dose n'a pas été réduite de manière appropriée chez les patients insuffisants rénaux. Les premiers symptômes de troubles vestibulaires sont des étourdissements, des nausées et des vomissements. L'examen clinique révèle souvent un nystagmus. Les troubles vestibulaires sont presque toujours réversibles. Les premiers symptômes d'un dysfonctionnement cochléaire incluent souvent une baisse de la perception des hautes fréquences (≥ 4.000 Hertz), qui précède la perte de l'audition et que seule une audiométrie permet de détecter.
(2) L'atteinte des tubules rénaux avec insuffisance rénale est un effet indésirable peu fréquent. Le mécanisme de l'atteinte rénale implique une accumulation dans les lysosomes, une inhibition des phospholipases et une nécrose des cellules tubulaires après une administration répétée d'amikacine. L'atteinte rénale est réversible à des degrés divers mais exacerbe le risque d'un processus d'accumulation qui peut provoquer ou renforcer des effets ototoxiques. Une élévation de la concentration de créatinine dans le sérum, la présence d'albumine, de globules rouges et blancs ou de cylindres dans l'urine, une urémie et une oligurie sont possibles.
(3) Réactions d'hypersensibilité telles qu'un exanthème, des démangeaisons, une urticaire et une fièvre médicamenteuse.
(4) Dans de rares cas, si la perfusion intraveineuse du médicament est trop rapide, la fonction respiratoire peut subir une dépression sévère. Dans des cas isolés, il peut en résulter une paralysie respiratoire. Le risque existe également lorsque l'amikacine est administrée en association avec un anesthésique (voir rubrique 4.5).
(5) L’amikacine n’est pas formulée pour un usage intra-vitréen. Une cécité et un infarctus rétinien ont été rapportés à la suite d’administrations intra-vitréennes (injection dans l’œil) d’amikacine.
Déclaration des effets indésirables suspectés
La déclaration des effets indésirables suspectés après autorisation du médicament est importante. Elle permet une surveillance continue du rapport bénéfice/risque du médicament. Les professionnels de santé déclarent tout effet indésirable suspecté via le système national de déclaration : Agence nationale de sécurité du médicament et des produits de santé (ANSM) et réseau des Centres Régionaux de Pharmacovigilance - Site internet : https://signalement.social-sante.gouv.fr/.
Un surdosage peut entraîner une néphrotoxicité, une ototoxicité ou un effet curarisant (blocage neuromusculaire).
Traitement
En cas de surdosage ou de réaction toxique, la perfusion d'amikacine doit être interrompue et une dialyse péritonéale ou une hémodialyse peuvent être pratiquées afin d'accélérer l'élimination de l'amikacine du sang.
L’hémofiltration artérioveineuse continue peut également favoriser l’élimination de l’amikacine qui s’est accumulée dans le sang.
Chez le nouveau-né, une exsanguino-transfusion peut être envisagée, cependant l’avis d'un expert doit être demandé avant qu'une telle mesure ne soit mise en œuvre.
Les sels de calcium sont indiqués afin de neutraliser l’effet curarisant. En cas de paralysie respiratoire, une ventilation artificielle peut être nécessaire.
5. PROPRIETES PHARMACOLOGIQUES
5.1. Propriétés pharmacodynamiques
Classe pharmacothérapeutique : autres aminosides, code ATC : J01GB06.
L’amikacine est un antibiotique aminoside semi-synthétique dérivé de la kanamycine. Elle est obtenue par acylation avec un acide amino-hydroxybutyrique au niveau du groupe aminé C-1 de la fraction 2déoxystreptamine.
Mécanisme d’action
L’amikacine agit en inhibant la synthèse des protéines au niveau du ribosome bactérien grâce à une interaction avec l’ARN ribosomique, laquelle entraîne une inhibition de la traduction chez les microorganismes sensibles. Il en résulte une action bactéricide.
Rapport pharmacocinétique/pharmacodynamie
Le principal paramètre pharmacocinétique/pharmacodynamique permettant de prédire l’effet bactéricide de l’amikacine est le rapport de la concentration sérique maximale (Cmax) et de la concentration minimale inhibitrice (CMI) de l’agent pathogène concerné. Un ratio Cmax/CMI de 8:1 ou 10:1 est considéré comme permettant l’élimination efficace des bactéries et la prévention d’une nouvelle prolifération bactérienne.
L’amikacine a démontré un effet post-antibiotique in vitro et in vivo. L’effet post-antibiotique permet d’allonger l’intervalle de dose sans perte d’efficacité contre la plupart des bacilles à Gram négatif.
Mécanismes de résistance
Une résistance à l’amikacine peut résulter des mécanismes suivants :
· Inactivation enzymatique : la modification enzymatique des molécules de l’aminoside est le mécanisme de résistance le plus courant. Elle est médiée par des acétyltransférases, des phosphotransférases ou des nucléotidyltransférases, qui sont généralement codées par des plasmides. L’amikacine a démontré une efficacité contre de nombreuses souches résistantes aux aminosides, en raison de sa capacité à résister à une dégradation par des enzymes qui inactivent les aminosides.
· Pénétration réduite et efflux actif : ces mécanismes de résistance sont observés chez Pseudomonas aeruginosa. Des données récentes indiquent l’émergence de mécanismes de résistance similaires chez Acinetobacter spp.
· Altération de la structure cible : occasionnellement, des modifications à l’intérieur des ribosomes sont identifiées comme la cause de la résistance.
L’apparition d’une résistance durant le traitement est peu fréquente. Une résistance croisée partielle entre l’amikacine et d’autres antibiotiques aminosides est possible.
Concentrations critiques
Les concentrations critiques ou seuils de concentrations minimales inhibitrices (CMI) à partir desquels est interprétée la sensibilité de la souche bactérienne, sont établies par le European Committee on Antimicrobial Susceptibility Testing (EUCAST) pour l’amikacine et sont les suivantes :
https://www.ema.europa.eu/documents/other/minimum-inhibitory-concentration-mic-breakpoints_en.xlsx
Spectre d’activité antibactérienne
La prévalence de la résistance acquise peut varier en fonction de la géographie et du temps pour certaines espèces. Il est donc utile de disposer d’informations sur la prévalence de la résistance locale, surtout pour le traitement d’infections sévères. Si nécessaire, il est souhaitable d’obtenir un avis spécialisé principalement lorsque l’intérêt du médicament dans certaines infections peut être mis en cause du fait du niveau de prévalence de la résistance locale.
|
ESPECES HABITUELLEMENT SENSIBLES |
|
Aérobies à Gram positif |
|
Staphylococcus aureus sensible à la méticilline |
|
Aérobies à Gram négatif |
|
Aeromonas spp. |
|
Campylobacter spp. |
|
Citrobacter freundii |
|
Citrobacter koseri |
|
Enterobacter cloacae |
|
Haemophilus influenzae ($) |
|
Francisella tularensis |
|
Klebsiella oxytoca |
|
Morganella morganii |
|
Proteus mirabilis |
|
Proteus vulgaris |
|
Providencia rettgeri |
|
Providencia stuartii |
|
Salmonella enterica |
|
Serratia liquefaciens |
|
Shigella spp. |
|
Yersinia enterocolitica |
|
Yersinia pseudotuberculosis |
|
Bactéries atypiques |
|
Mycobacterium spp. |
|
ESPECES INCONSTAMMENT SENSIBLES RESISTANCE ACQUISE ≥ 10 % |
|
Aérobies à Gram positif |
|
Staphylococcus aureus résistant à la méticilline (+) |
|
Staphylococcus à coagulase négative (+) |
|
Aérobies à Gram négatif |
|
Acinetobacter spp. |
|
A. baumannii |
|
Enterobacter aerogenes |
|
Escherichia coli |
|
Klebsiella pneumoniae |
|
Pseudomonas aeruginosa |
|
Serratia marcescens |
|
Espèces naturellement résistantes |
|
Aérobies à Gram positif |
|
Enterococcus |
|
Streptococcus |
|
- S. pyogenes du groupe A |
|
- S. des groupes B, C, G |
|
- S. pneumoniae |
|
Aérobies à Gram négatif |
|
Burkholderia cepacia |
|
Stenotrophomonas maltophilia |
|
Anaérobies |
|
Bacteroides spp. |
|
Clostridium perfringens |
|
Clostridium difficile |
|
Prevotella spp. |
|
Autres |
|
Chlamydia spp. |
|
Chlamydophila spp. |
|
Mycoplasma spp. |
|
Ureaplasma urealyticum |
Les aminosides peuvent être associés à d’autres antibiotiques dans le traitement d’infections dues à cocci à Gram positif.
5.2. Propriétés pharmacocinétiques
En cas d'administration orale, l'amikacine n'est pratiquement pas absorbée. Elle est utilisée dans le cadre d'une administration parentérale. Le pic sérique est atteint de 1 à 2 heures après la perfusion. La demi-vie sérique varie de 2,2 à 2,4 heures. Une demi-vie plus longue est prévisible chez les patients présentant une insuffisance rénale ainsi que chez les prématurés ou les nouveau-nés.
L'administration d'une dose de 7,5 mg/kg par perfusion intraveineuse continue de 30 minutes entraîne une concentration sérique de 38 µg/mL à la fin de la perfusion. Chez des volontaires sains, l'administration d'une dose de 15 mg/kg par perfusion intraveineuse continue de 30 minutes entraîne une concentration sérique d'environ 77 µg/mL à la fin de la perfusion, de 47 µg/mL 1 heure après la fin de la perfusion et de 1 µg/mL 12 heures après la fin de la perfusion.
Chez les personnes âgées ayant une clairance de la créatinine moyenne de 64 mL/min, l'administration d'une dose de 15 mg/kg par perfusion intraveineuse de 30 minutes entraîne une concentration sérique de 55 µg/mL à la fin de la perfusion, de 5,4 µg/mL 12 heures après la fin de la perfusion et de 1,3 µg/mL 24 heures après la fin de la perfusion.
Dans des études de doses répétées, aucun effet d'accumulation n'a été démontré chez les personnes ayant une fonction rénale normale et recevant une dose quotidienne unique de 15 à 20 mg/kg.
Distribution
Le volume de distribution apparent de l'amikacine est d'environ 24 litres (28 % du poids corporel). Le taux de liaison aux protéines plasmatiques est de 4 à 10 %.
Après administration de la dose recommandée, une concentration thérapeutique d'amikacine est présente dans les os, le cœur, la vésicule biliaire, les tissus pulmonaires, l'urine, la bile, les sécrétions bronchiques, les expectorations, le liquide interstitiel, le liquide pleural et la synovie.
Environ 10 à 20 % de la concentration sérique traverse les méninges saines, pourcentage pouvant atteindre 50 % si les méninges sont enflammées.
L’amikacine s'accumule dans le cortex rénal et le liquide de l'oreille interne et ne s'élimine que lentement de ces compartiments profonds.
L'amikacine traverse la barrière placentaire et est excrétée dans le lait maternel. Une concentration atteignant 20 % de celle mesurée chez la mère a été retrouvée dans le sang fœtal et dans le liquide amniotique.
Biotransformation
L'amikacine n'est pas métabolisée.
Elimination
L'amikacine est éliminée essentiellement par filtration glomérulaire.
Chez les patients dont la fonction rénale est normale, la clairance sérique moyenne de l'amikacine est de 100 mL/min tandis que la clairance rénale est de 94 mL/min.
La plus grande partie du volume (60 % - 82 %) est excrétée sous forme inchangée dans l'urine au cours des 6 premières heures. Seules de très petites quantités sont excrétées dans la bile. Chez les patients dont la fonction rénale est normale, 91 % de la dose d'amikacine (I.M.) sont excrétés dans l'urine sous une forme inchangée après 8 heures et 95 % après 24 heures.
L’amikacine peut être éliminée par hémodialyse et, dans une moindre mesure, par dialyse péritonéale. Selon la méthode de dialyse utilisée, 50 % (intervalle allant de 29 % à 81 %) ou 40 % – 80 % de la dose administrée sont éliminés respectivement en 4 ou 8 heures.
Patients pédiatriques
Des données issues d'essais de pharmacocinétique portant sur différentes doses quotidiennes montrent que chez des nourrissons bien portants, la concentration d’amikacine dans le liquide cérébro-spinal équivaut à 10 à 20 % environ de la concentration sérique, et peut atteindre 50 % en cas de méningite.
Chez les nouveau-nés et plus particulièrement les prématurés, l'élimination rénale de l'amikacine est réduite.
Lors d'une étude menée chez des nouveau-nés (1 à 6 jours d'âge post-natal), répartis en fonction de leur poids de naissance (< 2 000, 2 000 - 3 000 et > 3 000 g), l'amikacine a été administrée par voie intramusculaire et/ou intraveineuse, à la dose de 7,5 mg/kg. Chez les nouveau-nés de plus de 3 000 g, la clairance était de 0,84 mL/min/kg et la demi-vie terminale d’environ 7 heures. Dans ce groupe, le volume de distribution initial et le volume de distribution à l'état d'équilibre étaient respectivement de 0,3 mL/kg et de 0,5 mL/kg. Dans les groupes de plus faibles poids de naissance, la clairance par kilogramme était plus faible et la demi-vie plus longue. L’administration de doses répétées toutes les 12 heures dans tous les groupes précités n'a pas mis en évidence d'accumulation après 5 jours.
5.3. Données de sécurité préclinique
Potentiel mutagène et carcinogène et toxicité sur la reproduction
Aucune étude n'a été menée en ce qui concerne le potentiel mutagène ou carcinogène de l'amikacine.
L'amikacine, après avoir été administrée à des rats à des doses allant jusqu'à 10 fois la dose journalière chez l'homme, n'a pas altéré la fertilité des mâles ou des femelles.
Une diminution de l'activité sérique peut également être observée lorsqu'un aminoside ou un antibiotique de type pénicilline est administré in vivo par une voie distincte. L'inactivation des aminosides n'a d'importance clinique que chez les patients souffrant d'insuffisance rénale grave. L'inactivation peut se poursuivre dans les échantillons de liquide corporel prélevés pour les analyses, ce qui entraîne des mesures inexactes de l'aminoside. Les échantillons doivent être manipulés de manière appropriée (examen direct, congélation ou effet β-lactamase).
2 ans.
Après dilution dans une solution injectable de chlorure de sodium à 0,9 % (9 mg/mL) ou une solution injectable de glucose à 5 % (50 mg/mL), la stabilité chimique et physique pendant l'utilisation a été démontrée pendant 24 heures à 2 – 8 °C et à 25 °C.
D’un point de vue microbiologique, le produit doit être utilisé immédiatement. En cas d'utilisation non immédiate, les durées et conditions de conservation avant utilisation relèvent de la seule responsabilité de l'utilisateur et ne devraient pas dépasser 24 heures à une température comprise entre 2 et 8 °C.
6.4. Précautions particulières de conservation
Ce médicament ne nécessite pas de précautions particulières de conservation.
Pour les conditions de conservation du médicament après dilution, voir la rubrique 6.3.
6.5. Nature et contenu de l'emballage extérieur
2 mL ou 4 mL de solution en flacon en verre de type I incolore muni d’un bouchon en caoutchouc bromobutyle et d’une capsule en aluminium de type « flip-off ».
Boîte de 1, 5 ou 10 flacon(s).
Toutes les présentations peuvent ne pas être commercialisées.
6.6. Précautions particulières d’élimination et de manipulation
AMIKACINE STRAGEN se dissout dans les solutions suivantes :
· Solution injectable de glucose à 5 %
· Solution injectable de chlorure de sodium à 0,9 %
Flacon de 2 mL : En perfusion intraveineuse, les solutions ci-dessus peuvent être utilisées à raison de 500 mg d’amikacine pour 50 mL à 200 mL de solution (solution diluée d’amikacine de 2,5 à 10 mg/mL).
Flacon de 4 mL : En perfusion intraveineuse, les solutions ci-dessus peuvent être utilisées à raison de 1000 mg d’amikacine pour 100 mL à 400 mL de solution (solution diluée d’amikacine de 2,5 à 10 mg/mL).
Toute solution non utilisée doit être éliminée.
Le médicament doit faire l'objet d’une inspection visuelle avant administration, pour détecter la présence éventuelle de particules ou d’une modification de la couleur, lorsque la solution et le récipient le permettent.
Tout produit non utilisé ou déchet doit être éliminé conformément à la réglementation locale en vigueur, immédiatement après usage.
7. TITULAIRE DE L’AUTORISATION DE MISE SUR LE MARCHE
30 rue Edouard Nieuport
69008 LYON
8. NUMERO(S) D’AUTORISATION DE MISE SUR LE MARCHE
· 34009 303 272 5 8 : 2 mL de solution en flacon (verre). Boîte de 1.
· 34009 303 272 6 5 : 2 mL de solution en flacon (verre). Boîte de 5.
· 34009 303 272 8 9 : 2 mL de solution en flacon (verre). Boîte de 10.
· 34009 303 272 9 6 : 4 mL de solution en flacon (verre). Boîte de 1.
· 34009 303 273 0 2 : 4 mL de solution en flacon (verre). Boîte de 5.
· 34009 303 273 1 9 : 4 mL de solution en flacon (verre). Boîte de 10.
9. DATE DE PREMIERE AUTORISATION/DE RENOUVELLEMENT DE L’AUTORISATION
[à compléter ultérieurement par le titulaire]
10. DATE DE MISE A JOUR DU TEXTE
[à compléter ultérieurement par le titulaire]
Sans objet.
12. INSTRUCTIONS POUR LA PREPARATION DES RADIOPHARMACEUTIQUES
Liste I.
Médicament soumis à prescription hospitalière.
ANSM - Mis à jour le : 24/09/2025
AMIKACINE STRAGEN 250 mg/mL, solution injectable
Amikacine
· Gardez cette notice. Vous pourriez avoir besoin de la relire.
· Si vous avez d’autres questions, interrogez votre médecin ou votre pharmacien.
· Ce médicament vous a été personnellement prescrit. Ne le donnez pas à d’autres personnes. Il pourrait leur être nocif, même si les signes de leur maladie sont identiques aux vôtres.
· Si vous ressentez un quelconque effet indésirable, parlez-en à votre médecin ou votre pharmacien. Ceci s’applique aussi à tout effet indésirable qui ne serait pas mentionné dans cette notice. Voir rubrique 4.
1. Qu'est-ce que AMIKACINE STRAGEN 250 mg/mL, solution injectable et dans quels cas est-il utilisé ?
2. Quelles sont les informations à connaître avant d’utiliser AMIKACINE STRAGEN 250 mg/mL, solution injectable ?
3. Comment utiliser AMIKACINE STRAGEN 250 mg/mL, solution injectable ?
4. Quels sont les effets indésirables éventuels ?
5. Comment conserver AMIKACINE STRAGEN 250 mg/mL, solution injectable ?
6. Contenu de l’emballage et autres informations.
1. QU’EST-CE QUE AMIKACINE STRAGEN 250 mg/mL, solution injectable ET DANS QUELS CAS EST-IL UTILISE ?
Classe pharmacothérapeutique : autres aminosides - code ATC : J01GB06.
Qu'est-ce que AMIKACINE STRAGEN 250 mg/mL, solution injectable
AMIKACINE STRAGEN contient la substance active amikacine.
C’est un antibiotique qui appartient à la famille des "aminosides".
AMIKACINE STRAGEN est indiqué chez l’adulte, l'adolescent et l'enfant dans le traitement d’infections sévères dues à des bactéries sensibles à l'amikacine. Dans ces conditions, ce médicament peut être utilisé dans les infections suivantes :
· infections au niveau des poumons
· infections au niveau des os et articulations
· infections du système nerveux central
· infections compliquées des voies urinaires
· infections compliquées abdominales
· infections de la peau et des tissus mous, y compris brûlures sévères
· inflammation bactérienne de la paroi interne du cœur (uniquement en association avec d’autres antibiotiques)
Il peut également être utilisé dans le traitement des patients présentant une infection au niveau du sang, associée ou suspectée d’être associée, à l’une des infections précitées.
2. QUELLES SONT LES INFORMATIONS A CONNAITRE AVANT D’UTILISER AMIKACINE STRAGEN 250 mg/mL, solution injectable ?
N’utilisez jamais AMIKACINE STRAGEN 250 mg/mL, solution injectable :
· si vous êtes allergique à la substance active ou à l’un des autres composants contenus dans ce médicament, mentionnés dans la rubrique 6
· si vous êtes allergique à d’autres substances similaires (autres aminosides).
Avertissements et précautions
Adressez-vous à votre médecin, pharmacien ou votre infirmier/ère avant d’utiliser AMIKACINE STRAGEN.
Des précautions particulières doivent être prises avec AMIKACINE STRAGEN :
· si vous présentez des problèmes rénaux
· si vous présentez des problèmes auditifs
· si vous avez des maladies nerveuses et musculaires, comme un type particulier de faiblesse musculaire (appelée myasthénie)
· si vous avez la maladie de Parkinson
· si vous avez déjà suivi un traitement avec un autre antibiotique similaire à l’amikacine
· si vous ou des membres de votre famille présentez une mutation mitochondriale (une maladie génétique) ou une perte d’audition due à des médicaments antibiotiques, il vous est conseillé d’en informer votre médecin ou votre pharmacien avant de prendre un aminoside; certaines mutations mitochondriales peuvent augmenter votre risque de perte auditive avec ce produit. Votre médecin peut recommander des tests génétiques avant l’administration d’AMIKACINE STRAGEN.
Dans de tels cas, votre médecin fera preuve d'une prudence particulière.
Votre médecin sera en outre particulièrement prudent si vous avez 60 ans et plus ou si vous êtes déshydraté (manque d’eau dans le corps).
Au cours du traitement avec ce médicament, votre médecin surveillera attentivement :
· votre fonction rénale, surtout si vous avez 60 ans et plus ou si vous avez des problèmes rénaux
· votre audition
· le taux d’amikacine dans le sang, si nécessaire
Votre médecin réduira les doses quotidiennes et/ou l’intervalle entre les doses sera allongé si des problèmes rénaux apparaissent ou si les problèmes rénaux s'aggravent. Si le problème rénal devient sévère, le traitement par amikacine sera interrompu.
Le traitement par amikacine sera également interrompu si des acouphènes (bourdonnement au niveau des oreilles) ou une perte d’audition apparaissent.
Afin d’éviter le risque d’altérer le fonctionnement de vos reins, de léser votre nerf auditif ou provoquer une paralysie musculaire, le traitement par amikacine sera limité à 5 jours, à moins que votre médecin estime nécessaire de le prolonger.
Durant le traitement, il faudra s’assurer que vous soyez suffisamment hydraté.
Si vous subissez un lavage de plaie avec des solutions contenant de l’amikacine ou un antibiotique similaire lors d’une intervention chirurgicale, cette application locale sera prise en compte lors de la détermination de la dose d’amikacine.
Enfants
La prudence est de rigueur lorsque le médicament est administré à des nouveau-nés, qu’ils soient nés à terme ou prématurés, en raison de leur fonction rénale immature.
Autres médicaments et AMIKACINE STRAGEN 250 mg/mL, solution injectable
Informez votre médecin ou votre pharmacien si vous prenez, avez récemment pris ou pourriez prendre tout autre médicament.
L'effet nocif de l'amikacine sur les reins et le nerf auditif peut être renforcé par la prise :
· d’autres antibiotiques similaires à l'amikacine (par ex : kanamycine, paromycine)
· d’autres substances utilisées pour traiter les infections, par exemple : bacitracine, amphotéricine B, céphalosporines, vancomycine, polymyxines (polymyxine B, colistine), viomycine
· de médicaments anticancéreux : carboplatine (à haute dose), cisplatine, oxaliplatine (particulièrement en cas d'insuffisance rénale préexistante)
· de substances utilisées pour contrôler des réactions immunitaires indésirables : ciclosporine, tacrolimus
· de médicaments à action rapide qui augmentent le débit urinaire : furosémide ou acide éthacrynique. Une surdité irréversible peut en résulter,
· d’une anesthésie par méthoxyflurane : il convient d’informer l’anesthésiste si vous prenez ou avez pris de l’amikacine ou un antibiotique similaire avant une anesthésie par méthoxyflurane (un gaz anesthésiant), afin d’éviter tant que possible d’utiliser cet agent anesthésiant en raison d’un risque accru de troubles rénaux et nerveux sévères.
Lorsque l'amikacine doit être associée à de telles substances, les fonctions auditive et rénale doivent être surveillées très fréquemment et attentivement. Lorsque l'amikacine est utilisée simultanément à des médicaments à action rapide qui augmentent le débit urinaire, votre équilibre hydrique sera surveillé.
Traitement simultané par amikacine et myorelaxants, d’autres substances agissant sur les muscles et les nerfs
Votre médecin fera preuve d’une attention particulière si vous recevez de l’amikacine avec des myorelaxants (comme succinylcholine, décaméthonium, atracurium, rocuronium, vencurinium), avec des transfusions massives de sang ou avec un anesthésique car votre respiration pourrait se bloquer (paralysie respiratoire).
En cas d’intervention chirurgicale, l’anesthésiste doit être informé du fait que vous être traité par l’amikacine car il existe un risque que le blocage des fonctions nerveuse et musculaire soit fortement accru. Un blocage nerveux ou musculaire dû à un aminoside peut être neutralisé avec un traitement approprié.
Indométacine
Chez les nouveau-nés traités simultanément par amikacine et indométacine (un médicament contre l’inflammation et la douleur), le dosage dans le sang d'amikacine sera contrôlé car l’indométacine peut entraîner une augmentation de la quantité d’antibiotique dans le sang.
Bisphosphonates
Un traitement associé avec des bisphosphonates (utilisés dans le traitement de l’ostéoporose et maladies similaires) peut entrainer une diminution du calcium dans le sang (hypocalcémie).
Thiamine (vitamine B1)
La thiamine (vitamine B1), administrée simultanément à l’amikacine, pourrait perdre son efficacité.
AMIKACINE STRAGEN 250 mg/mL, solution injectable avec des aliments et des boissons
Sans objet.
Si vous êtes enceinte ou que vous allaitez, si vous pensez être enceinte ou planifiez une grossesse, demandez conseil à votre médecin ou pharmacien avant de prendre ce médicament.
Grossesse
Si vous êtes enceinte, ce médicament ne vous sera administré que si votre médecin l'estime absolument nécessaire.
Allaitement
Même s’il est peu probable que l’amikacine soit absorbée par l’intestin des enfants allaités, votre médecin étudiera avec soin si l’allaitement ou le traitement par amikacine doit être interrompu.
Conduite de véhicules et utilisation de machines
Aucune étude dédiée aux effets sur la conduite de véhicules et l'utilisation de machines n'a été réalisée. En cas d'administration à des patients non hospitalisés, la prudence s'impose lors de la conduite et de l'utilisation de machines, compte tenu des effets indésirables possibles tels que des étourdissements et des vertiges.
AMIKACINE STRAGEN 250 mg/mL, solution injectable contient du sodium et du métabisulfite de sodium
Ce médicament contient 7,5 mg de sodium par mL de solution injectable.
Flacon de 2 mL : Ce médicament contient 15 mg de sodium (composant principal du sel de cuisine/de table). Cela équivaut à 0,75 % de l'apport alimentaire quotidien maximal recommandé de sodium pour un adulte.
Flacon de 4 mL : Ce médicament contient 30 mg de sodium (composant principal du sel de cuisine/de table). Cela équivaut à 1,5 % de l'apport alimentaire quotidien maximal recommandé de sodium pour un adulte.
Ce médicament contient du métabisulfite de sodium, qui peut rarement provoquer des réactions d'hypersensibilité (allergie grave) et des bronchospasmes (difficultés respiratoires).
3. COMMENT UTILISER AMIKACINE STRAGEN 250 mg/mL, solution injectable ?
Veillez à toujours utiliser ce médicament en suivant exactement les instructions de cette notice ou les indications de votre médecin, pharmacien ou infirmier/ère. Vérifiez auprès de votre médecin, pharmacien ou infirmier/ère en cas de doute.
AMIKACINE STRAGEN est administrée par perfusion directement dans une veine (perfusion intraveineuse).
Selon les recommandations, la durée de perfusion est de 30 minutes.
La voie intramusculaire est à éviter dans la mesure du possible, mais peut être pratiquée à titre exceptionnel en tenant compte que cette voie n’offre pas les mêmes garanties que la voie intraveineuse.
Pour les instructions concernant la préparation du médicament, voir la rubrique « Informations destinées exclusivement aux professionnels de santé ».
Posologie
La dose dépend de la gravité de l’infection, de votre état de santé, du fonctionnement de vos reins et de la bactérie identifiée. La dose est exprimée selon le poids corporel du patient.
Les recommandations thérapeutiques doivent être prises en considération.
Le schéma posologique préférentiel est la dose unique journalière (totalité de la dose journalière administrée en une seule fois par jour). Dans certaines situations, une dose journalière répartie en 2 à 3 injections quotidiennes est possible.
Les doses peuvent être de 15 à 30 mg/kg/jour ; la dose maximale de 30 mg/kg/jour est surtout recommandée en début de traitement dans des infections graves et/ou en cas de risque d’infection due à une bactérie moins sensible à l’antibiotique.
Votre médecin déterminera la dose d'amikacine qui vous convient et la fréquence à laquelle elle doit être administrée.
Patients avec une fonction rénale normale
Adultes et adolescents de 12 ans et plus
La dose est de 15 mg d’amikacine par kg de poids corporel par 24 heures, administrée en une seule dose quotidienne ou répartie en 2 doses égales : 7,5 mg/kg de poids corporel toutes les 12 heures. La dose journalière peut être portée jusqu’à 30 mg/kg/jour selon les recommandations.
Nourrissons et enfants (de 4 semaines à 11 ans)
Selon les recommandations en vigueur, les doses exprimées en mg/kg chez le nourrisson et l’enfant sont les mêmes que chez l’adulte, et la dose unique journalière est préconisée.
Une dose quotidienne d’amikacine de 15 à 20 mg/kg de poids corporel peut être administrée en une dose une fois par jour ou répartie en doses de 7,5 mg/kg de poids corporel toutes les 12 heures.
Nouveau-nés (de 0 à 27 jours) et prématurés
Pour les nouveau-nés, il convient d’adapter les posologies selon l’âge post-conceptionnel en tenant compte des recommandations en vigueur.
Le suivi du traitement peut nécessiter la surveillance du taux d’amikacine dans votre sang pour adapter votre dose.
Patients présentant des problèmes rénaux
Si vous présentez une insuffisance rénale, le taux d’amikacine dans votre sang ainsi que votre fonction rénale seront surveillés afin d’adapter au mieux votre dose d’amikacine. Votre médecin établira les doses que vous allez recevoir.
Patients âgés
Chez les patients âgés, la fonction rénale peut être diminuée. Votre fonction rénale sera évaluée et la dose sera adaptée si nécessaire.
Patients obèses
Chez ces patients, la dose est calculée sur la base du poids corporel idéal augmenté de 40 % de l’excédent de poids. Ultérieurement, votre dose pourra être adaptée en fonction du taux d’amikacine dans votre sang. La dose maximale est de 1,5 g par jour.
Patients ayant de l’eau dans l’abdomen (ascite)
Une dose plus élevée sera envisagée afin d’obtenir une concentration adéquate dans le sang.
Durée du traitement
La durée totale du traitement est limitée à 5 jours. Une prolongation de la durée de traitement doit faire l’objet d’une réévaluation.
Si vous avez utilisé plus d’AMIKACINE STRAGEN 250 mg/mL, solution injectable que vous n’auriez dû
Si vous pensez avoir utilisé trop d’AMIKACINE STRAGEN, prenez immédiatement contact avec votre médecin.
Un surdosage peut altérer le fonctionnement des reins et altérer les nerfs auditifs ou provoquer un blocage de la fonction musculaire (paralysie). Dans un tel cas, le traitement par ce médicament doit être arrêté et des procédures visant à éliminer le médicament (dialyses) seront envisagées afin d’éliminer l’amikacine de votre sang.
Si vous oubliez d’utiliser AMIKACINE STRAGEN 250 mg/mL, solution injectable
Si vous arrêtez d’utiliser AMIKACINE STRAGEN 250 mg/mL, solution injectable
Sans objet.
Si vous avez d’autres questions sur l’utilisation de ce médicament, demandez plus d’informations à votre médecin, à votre pharmacien ou à votre infirmier/ère.
4. QUELS SONT LES EFFETS INDESIRABLES EVENTUELS ?
Des effets toxiques sur le nerf auditif et les reins ont été observés chez des patients traités par AMIKACINE STRAGEN. Ces effets indésirables peuvent être évités en grande partie en respectant les précautions et les posologies recommandées. Votre médecin vous surveillera afin de repérer les signes éventuels de ces effets indésirables.
Si vous remarquez l’un des effets indésirables sévères suivants, contactez immédiatement votre médecin :
Très rare (pouvant affecter jusqu’à 1 personne sur 10 000) :
· Gonflement du visage, des lèvres ou de la langue, éruption cutanée, difficultés à respirer, car il peut s’agir de signes d’une réaction allergique
· Bourdonnements dans les oreilles ou perte d’audition (surdité)
· Paralysie respiratoire
· Problèmes rénaux, y compris réduction du débit urinaire (insuffisance rénale aiguë)
Autres effets indésirables :
Peu fréquents (pouvant affecter jusqu'à 1 personne sur 100) :
· Infection supplémentaire ou colonisation (par des germes résistants ou champignons de type levure appelés Candida)
· Etourdissements, vertiges
· Mouvements involontaires des yeux (nystagmus)
· Bruit dans les oreilles, pression dans les oreilles, baisse de l’audition
· Nausées
· Altération de certaines parties du rein, altération de la fonction rénale
Rares (pouvant affecter jusqu'à 1 personne sur 1 000) :
· Nombre anormalement bas de globules blancs (leucopénie), de globules rouges (anémie) ou de plaquettes (thrombocytopénie), augmentation du nombre d’un certain type de globules blancs (éosinophiles)
· Réactions allergiques, éruption cutanée, démangeaisons, urticaire
· Faibles taux de magnésium dans le sang
· Maux de tête, migraine, engourdissements, tremblements
· Cécité, ou autres problèmes de vue
· Tension artérielle basse
· Dépression respiratoire
· Vomissements
· Douleurs articulaires
· Fièvre médicamenteuse
· Augmentation de la concentration dans le sang de certaines enzymes présentes dans le foie (ALAT, ASAT, phosphatases alcalines)
Très rares (pouvant affecter jusqu’à 1 personne sur 10 000) :
· Blocage de la fonction musculaire
· Atteinte rénale grave
Fréquence indéterminée (la fréquence ne peut être estimée à partir des données disponibles) :
· Réactions allergiques à toute substance similaire à l’amikacine (aminosides)
· Apnée
· Bronchospasme (contraction brusque et involontaire des muscles des bronches se traduisant par une difficulté à respirer).
Déclaration des effets secondaires
Si vous ressentez un quelconque effet indésirable, parlez-en à votre médecin ou votre pharmacien. Ceci s’applique aussi à tout effet indésirable qui ne serait pas mentionné dans cette notice. Vous pouvez également déclarer les effets indésirables directement via le système national de déclaration : Agence nationale de sécurité du médicament et des produits de santé (ANSM) et réseau des Centres Régionaux de Pharmacovigilance - Site internet : https://signalement.social-sante.gouv.fr.
En signalant les effets indésirables, vous contribuez à fournir davantage d’informations sur la sécurité du médicament.
5. COMMENT CONSERVER AMIKACINE STRAGEN 250 mg/mL, solution injectable ?
Tenir ce médicament hors de la vue et de la portée des enfants.
Ce médicament ne nécessite pas de précautions particulières de conservation.
Après dilution dans une solution injectable de chlorure de sodium à 0,9 % (9 mg/mL) ou une solution injectable de glucose à 5 % (50 mg/mL), la stabilité chimique et physique pendant l'utilisation a été démontrée pendant 24 heures à 2 – 8 °C et à 25 °C.
Du point de vue microbiologique, le produit doit être utilisé immédiatement. En cas d'utilisation non immédiate, les durées et conditions de conservation avant utilisation relèvent de la seule responsabilité de l'utilisateur et ne devraient pas dépasser 24 heures à une température comprise entre 2 et 8 °C.
A usage unique exclusivement.
Jeter toute solution inutilisée.
N’utilisez pas ce médicament après la date de péremption indiquée sur la boîte et le flacon après EXP. La date de péremption fait référence au dernier jour de ce mois.
N’utilisez pas ce médicament si vous remarquez des particules dans la solution et/ou si la solution n’est pas limpide ou n’est pas incolore ou jaune pâle.
Ne jetez aucun médicament au tout-à-l’égout ou avec les ordures ménagères. Demandez à votre pharmacien d’éliminer les médicaments que vous n’utilisez plus. Ces mesures contribueront à protéger l’environnement.
6. CONTENU DE L’EMBALLAGE ET AUTRES INFORMATIONS
Ce que contient AMIKACINE STRAGEN 250 mg/mL, solution injectable
· La substance active est :
Amikacine (sous forme de sulfate d’amikacine)...................................................................... 250 mg
Pour 1 mL de solution injectable.
Chaque flacon de 2 mL contient l’équivalent de 500 mg d’amikacine sous forme de sulfate d’amikacine.
Chaque flacon de 4 mL contient l’équivalent de 1000 mg d’amikacine sous forme de sulfate d’amikacine.
Les autres composants sont : citrate de sodium, métabisulfite de sodium (E223), acide sulfurique, eau pour préparations injectables.
Qu’est-ce que AMIKACINE STRAGEN 250 mg/mL, solution injectable et contenu de l’emballage extérieur
AMIKACINE STRAGEN est une solution injectable limpide, incolore à jaune pâle.
Boîte de 1, 5 ou 10 flacon(s).
Toutes les présentations peuvent ne pas être commercialisées.
Titulaire de l’autorisation de mise sur le marché
30 rue Edouard Nieuport
69008 LYON
Exploitant de l’autorisation de mise sur le marché
STRAGEN FRANCE
30 rue Edouard Nieuport
69008 LYON
ANFARM HELLAS S.A
61st km NAT.RD. ATHENS-LAMIA,
Schimatari Viotias 32009,
Grece
Noms du médicament dans les Etats membres de l'Espace Economique Européen
Sans objet.
La dernière date à laquelle cette notice a été révisée est :
[à compléter ultérieurement par le titulaire]
Des informations détaillées sur ce médicament sont disponibles sur le site Internet de l’ANSM (France).
Conseil d’éducation sanitaire
QUE SAVOIR SUR LES ANTIBIOTIQUES ?
Les antibiotiques sont efficaces pour combattre les infections dues aux bactéries. Ils ne sont pas efficaces contre les infections dues aux virus.
Aussi, votre médecin a choisi de vous prescrire cet antibiotique parce qu'il convient précisément à votre cas et à votre maladie actuelle.
Les bactéries ont la capacité de survivre ou de se reproduire malgré l'action d'un antibiotique. Ce phénomène est appelé résistance : il rend certains traitements antibiotiques inactifs.
La résistance s'accroît par l'usage abusif ou inapproprié des antibiotiques.
Vous risquez de favoriser l'apparition de bactéries résistantes et donc de retarder votre guérison ou même de rendre inactif ce médicament, si vous ne respectez pas :
· la dose à prendre
· les moments de prise
· et la durée de traitement
En conséquence, pour préserver l'efficacité de ce médicament :
1- N’utilisez un antibiotique que lorsque votre médecin vous l’a prescrit.
2- Respectez strictement votre ordonnance.
3- Ne réutilisez pas un antibiotique sans prescription médicale même si vous pensez combattre une maladie apparemment semblable.
4- Ne donnez jamais votre antibiotique à une autre personne, il n’est peut-être pas adapté à sa maladie.
5- Une fois votre traitement terminé, rapportez à votre pharmacien toutes les boîtes entamées pour une destruction correcte et appropriée de ce médicament.
----------------------------------------------------------------------------------------------------------------------------------------
AMIKACINE STRAGEN 250 mg/mL, solution injectable
Les informations suivantes sont destinées exclusivement aux professionnels de santé :
Posologie
La dose dépend de la gravité du tableau clinique, du terrain, de la fonction rénale du patient et de la bactérie identifiée. Il convient de se référer aux recommandations thérapeutiques.
Le schéma posologique préférentiel est la dose unique journalière (totalité de la dose journalière administrée en une seule fois par jour). Dans certaines situations, une dose journalière répartie en 2 à 3 injections quotidiennes est possible.
Les doses peuvent être de 15 à 30 mg/kg/jour ; la dose maximale de 30 mg/kg/jour est surtout recommandée en début de traitement dans des infections graves et/ou en cas de risque d’infection due à une bactérie moins sensible à l’antibiotique.
Mode d’administration
Administration par voie intraveineuse, et à titre exceptionnel par voie intramusculaire.
La durée de perfusion recommandée est de 30 minutes.
La voie intramusculaire est à éviter dans la mesure du possible, mais peut être pratiquée à titre exceptionnel en tenant compte que cette voie n’offre pas les mêmes garanties que la voie intraveineuse. Si la voie intramusculaire doit être utilisée, les schémas d’administration sont identiques à ceux préconisés pour la voie intraveineuse.
Voir rubrique 3 « Comment AMIKACINE STRAGEN 250 mg/mL, solution injectable est-il administré ? »
Incompatibilités
Le mélange in vitro d'aminosides et de bêta-lactamines (pénicillines ou céphalosporines) peut entraîner une inactivation mutuelle significative. Une diminution de l'activité sérique peut également être observée lorsqu'un aminoside ou un antibiotique de type pénicilline est administré in vivo par une voie distincte.
L'inactivation des aminosides n'a d'importance clinique que chez les patients souffrant d'insuffisance rénale grave. L'inactivation peut se poursuivre dans les échantillons de liquide corporel prélevés pour les analyses, ce qui entraîne des mesures inexactes de l'aminoside. Les échantillons doivent être manipulés de manière appropriée (examen direct, congélation ou effet β-lactamase).
Conservation
AMIKACINE STRAGEN dissout dans les solutions suivantes est stable pendant 24 heures à température ambiante et au réfrigérateur (2 °C – 8 °C) :
· Solution injectable de glucose à 5 %
· Solution injectable de chlorure de sodium à 0,9 %
Voir rubrique 5 « Comment conserver AMIKACINE STRAGEN 250 mg/mL, solution injectable ? ».