Dernière mise à jour le 01/06/2026
AXITINIB TEVA 1 mg, comprimé pelliculé
Indications thérapeutiques
Classe pharmacothérapeutique : Antinéoplasiques, inhibiteurs de protéine kinase - code ATC : L01EK01
AXITINIB TEVA est un médicament contenant la substance active axitinib. L’axitinib réduit l’apport de sang à la tumeur et ralentit le développement du cancer.
AXITINIB TEVA est indiqué dans le traitement du cancer du rein avancé (carcinome rénal avancé) chez les adultes, quand un autre médicament (appelé sunitinib ou cytokine) n’empêche plus la progression de la maladie.
Si vous avez des questions sur la façon dont ce médicament agit ou si vous souhaitez savoir pourquoi ce médicament vous a été prescrit, adressez-vous à votre médecin.
Présentations
> 56 plaquettes prédécoupées unitaires OPA : polyamide orienté aluminium PVC-Aluminium de 1 comprimé
Code CIP : 34009 302 982 9 9
Déclaration de commercialisation : 03/07/2025
Cette présentation est agréée aux collectivités
- Prix hors honoraire de dispensation : 241,79 €
- Honoraire de dispensation : 1,02 €
- Prix honoraire compris : 242,81 €
- Taux de remboursement :100 %
Service médical rendu (SMR)
Amélioration du service médical rendu (ASMR)
Ce médicament étant un générique, l'ASMR n'a pas été évalué par la commission de la transparence (CT), il est possible de se référer à la /aux spécialité(s) de référence du groupe générique auquel appartient ce médicament (cliquez ici pour aller à la rubrique des groupes génériques)
Autres informations
- Titulaire de l'autorisation : TEVA BV
- Conditions de prescription et de délivrance :
- liste I
- médicament nécessitant une surveillance particulière pendant le traitement
- prescription hospitalière
- prescription réservée aux médecins compétents en CANCEROLOGIE
- prescription réservée aux spécialistes et services ONCOLOGIE MEDICALE
- Statut de l'autorisation : Valide
- Type de procédure : Procédure décentralisée
- Code CIS : 6 379 182 7
ANSM - Mis à jour le : 02/07/2024
AXITINIB TEVA 1 mg, comprimé pelliculé
2. COMPOSITION QUALITATIVE ET QUANTITATIVE
Axitinib.................................................................................................................................... 1 mg
Pour un comprimé pelliculé.
Excipient(s) à effet notoire : chaque comprimé pelliculé contient 33 mg de lactose monohydraté.
Pour la liste complète des excipients, voir rubrique 6.1.
Comprimé pelliculé.
Comprimé enrobé de couleur rouge, rond, biconvexe, d’environ 6 mm de diamètre, et comportant la mention « A7TI » gravée en creux sur une face et la mention « 1 » gravée en creux sur l’autre face.
4.1. Indications thérapeutiques
AXITINIB TEVA est indiqué dans le traitement des patients adultes atteints de cancer du rein (RCC) avancé après échec d’un traitement antérieur par sunitinib ou cytokine.
4.2. Posologie et mode d'administration
Le traitement par AXITINIB TEVA doit être instauré par un médecin expérimenté dans l’utilisation des traitements anticancéreux.
Posologie
La dose d’axitinib recommandée est de 5 mg deux fois par jour.
Le traitement doit être poursuivi tant qu’un bénéfice clinique est observé ou jusqu’à la survenue d’une toxicité inacceptable ne pouvant être prise en charge par des médicaments concomitants ou des ajustements posologiques.
En cas de vomissement ou d’oubli d’une dose, le patient ne doit pas prendre de dose supplémentaire. La dose prescrite suivante doit être prise à l’heure habituelle.
Ajustements posologiques
Une augmentation ou une diminution de la dose est recommandée en fonction de la sécurité et de la tolérance individuelles.
Chez les patients qui tolèrent la dose initiale de 5 mg d’axitinib deux fois par jour sans effet indésirable de grade > 2 (c'est-à-dire sans effet indésirable sévère selon la Common Terminology Criteria for Adverse Events [CTCAE] version 3.0) pendant deux semaines consécutives, la dose peut être augmentée à 7 mg deux fois par jour, sauf si la pression artérielle du patient est > 150/90 mmHg ou si le patient reçoit un traitement antihypertenseur. Par la suite, selon les mêmes critères, chez les patients qui tolèrent une dose de 7 mg deux fois par jour, la dose d’axitinib peut être augmentée jusqu’à un maximum de 10 mg deux fois par jour.
La prise en charge de certains effets indésirables peut nécessiter l’interruption temporaire ou définitive de l’administration d’axitinib et/ou une réduction de dose (voir rubrique 4.4). Si une réduction de dose est nécessaire, la dose d’axitinib peut être diminuée à 3 mg deux fois par jour, puis si nécessaire à 2 mg deux fois par jour.
Aucun ajustement posologique n’est nécessaire en fonction de l’âge, de l’origine ethnique, du sexe ou du poids corporel du patient.
Administration concomitante d’inhibiteurs puissants du CYP3A4/5
L’administration concomitante d’axitinib et d’inhibiteurs puissants du CYP3A4/5 peut augmenter les concentrations plasmatiques de l’axitinib (voir rubrique 4.5). Il est recommandé d’opter pour un autre médicament concomitant ayant peu ou pas d’effet sur l’inhibition du CYP3A4/5.
Bien que l’ajustement posologique de l’axitinib n’ait pas été étudié chez des patients recevant des inhibiteurs puissants du CYP3A4/5, une diminution d'environ 50 % de la posologie d’axitinib (par exemple, diminution de la dose initiale de 5 mg deux fois par jour à 2 mg deux fois par jour) est recommandée si un inhibiteur puissant du CYP3A4/5 doit être administré de façon concomitante. La prise en charge de certains effets indésirables peut nécessiter l’interruption temporaire ou définitive de l’administration d’axitinib (voir rubrique 4.4). Si l’administration concomitante de l’inhibiteur puissant est interrompue, un retour à la dose d’axitinib utilisée avant l’instauration de l’inhibiteur puissant du CYP3A4/5 devra être envisagé (voir rubrique 4.5).
Administration concomitante d’inducteurs puissants du CYP3A4/5
L’administration concomitante d’axitinib et d’inducteurs puissants du CYP3A4/5 peut diminuer les concentrations plasmatiques d’axitinib (voir rubrique 4.5). Il est recommandé d’opter pour un autre médicament concomitant ayant peu ou pas d’effet sur l’induction du CYP3A4/5.
Bien que l’ajustement posologique de l’axitinib n’ait pas été étudié chez les patients recevant des inducteurs puissants du CYP3A4/5, une augmentation progressive de la dose d’axitinib est recommandée si un inducteur puissant du CYP3A4/5 doit être administré de façon concomitante. Une induction maximale du CYP3A4/5 a été observée au cours de la première semaine de traitement avec les inducteurs du CYP3A4/5 à doses élevées. Si la dose d’axitinib est augmentée, le patient doit faire l’objet d’une étroite surveillance en vue de déceler d’éventuelles toxicités. La prise en charge de certains effets indésirables peut nécessiter l’interruption temporaire ou définitive de l’axitinib et/ou une réduction de dose (voir rubrique 4.4). Si l’administration concomitante de l’inducteur puissant est interrompue, un retour à la dose d’axitinib utilisée avant l’instauration de l’inducteur puissant du CYP3A4/5 doit être envisagé (voir rubrique 4.5).
Populations particulières
Personnes âgées (≥ 65 ans)
Aucun ajustement posologique n’est nécessaire (voir rubriques 4.4 et 5.2).
Insuffisants rénaux
Aucun ajustement posologique n’est nécessaire (voir rubrique 5.2). Pratiquement aucune donnée n'est disponible concernant le traitement par axitinib chez les patients dont la clairance de la créatinine est < 15 mL/min.
Insuffisants hépatiques
Aucun ajustement posologique n’est nécessaire lors de l’administration d'axitinib à des patients présentant une insuffisance hépatique légère (classe A de Child-Pugh). Une diminution de la dose est recommandée lors de l’administration d'axitinib à des patients présentant une insuffisance hépatique modérée (classe B de Child-Pugh) (par exemple, réduction de la dose initiale de 5 mg deux fois par jour à 2 mg deux fois par jour). L’axitinib n’a pas été étudié chez les patients présentant une insuffisance hépatique sévère (classe C de Child-Pugh) et ne doit pas être administré dans cette population (voir rubriques 4.4 et 5.2).
Population pédiatrique
La sécurité et l’efficacité d’AXITINIB TEVA chez les enfants et les adolescents âgés de moins de 18 ans n’ont pas été établies. Aucune donnée n’est disponible.
Mode d’administration
L’axitinib doit être administré par voie orale. Les comprimés doivent être pris oralement deux fois par jour, à intervalles d’environ 12 heures, au cours ou en dehors des repas (voir rubrique 5.2). Ils doivent être avalés entiers avec un verre d’eau.
Hypersensibilité à l’axatinib ou à l’un des excipients mentionnés à la rubrique 6.1.
4.4. Mises en garde spéciales et précautions d'emploi
Des événements spécifiques doivent faire l’objet d’une surveillance avant l’instauration du traitement par axitinib, et régulièrement pendant toute la durée de celui-ci, tel que décrit ci-dessous.
Evénements d’insuffisance cardiaque
Des événements d’insuffisance cardiaque (incluant insuffisance cardiaque, insuffisance cardiaque congestive, insuffisance cardiopulmonaire, dysfonctionnement ventriculaire gauche, diminution de la fraction d’éjection et insuffisance du ventricule droit) ont été rapportés lors d’études cliniques portant sur l’axitinib dans le traitement de patients atteints de RCC (voir rubrique 4.8).
Les signes ou symptômes d’insuffisance cardiaque doivent être surveillés régulièrement pendant le traitement par axitinib. La prise en charge des événements d’insuffisance cardiaque peut nécessiter l’interruption temporaire ou définitive et/ou une diminution de la dose d’axitinib.
Hypertension
Une hypertension a été très fréquemment rapportée au cours des études cliniques portant sur l’axitinib dans le traitement de patients atteints de RCC (voir rubrique 4.8).
Au cours d’une étude clinique contrôlée, une hypertension (pression artérielle systolique > 150 mmHg ou pression artérielle diastolique > 100 mmHg) est survenue en moyenne au cours du premier mois de traitement par axitinib, et des augmentations de la pression artérielle ont été observées dès le quatrième jour suivant le début du traitement.
La pression artérielle doit être correctement contrôlée avant l’instauration du traitement par axitinib. Il conviendra de surveiller la pression artérielle des patients et de les traiter, le cas échéant, par un traitement antihypertenseur standard. La dose d’axitinib doit être réduite si l’hypertension persiste malgré l’instauration d’un traitement antihypertenseur. En cas d’hypertension sévère, le traitement par axitinib doit être interrompu temporairement puis repris à une dose inférieure après normalisation de la pression artérielle. Il conviendra de surveiller les patients recevant un traitement antihypertenseur en raison d’un risque d’hypotension (voir rubrique 4.2).
En cas d’hypertension artérielle sévère ou persistante et de symptômes évocateurs d’un syndrome d’encéphalopathie postérieure réversible (SEPR) (voir ci-dessous), une imagerie par résonance magnétique (IRM) cérébrale à visée diagnostique doit être envisagée.
Dysfonctionnement thyroïdien
Des cas d’hypothyroïdie et, moins fréquemment, d’hyperthyroïdie ont été rapportés au cours des études cliniques portant sur l’axitinib dans le traitement de patients atteints de RCC (voir rubrique 4.8).
La fonction thyroïdienne doit être contrôlée avant l’instauration du traitement par axitinib et régulièrement pendant toute la durée de celui-ci. Les patients atteints d’hypothyroïdie ou d’hyperthyroïdie doivent être traités conformément à la pratique médicale standard afin de maintenir un état euthyroïdien.
Evénements artériels emboliques et thrombotiques
Des événements artériels emboliques et thrombotiques (dont accident ischémique transitoire, infarctus du myocarde, accident vasculaire cérébral et occlusion de l’artère rétinienne) ont été rapportés au cours des études cliniques portant sur l’axitinib (voir rubrique 4.8).
L’axitinib doit être utilisé avec prudence chez les patients à risque ou présentant des antécédents de ce type d’événement. L’axitinib n’a pas été étudié chez les patients ayant présenté un événement artériel embolique ou thrombotique au cours des 12 mois précédant l’instauration du traitement.
Evénements veineux emboliques et thrombotiques
Des événements veineux emboliques et thrombotiques (dont embolie pulmonaire, thrombose veineuse profonde et occlusion/thrombose de la veine rétinienne) ont été rapportés au cours des études cliniques portant sur l’axitinib (voir rubrique 4.8).
L’axitinib doit être utilisé avec prudence chez les patients à risque ou présentant des antécédents de ce type d’événement. L’axitinib n’a pas été étudié chez les patients ayant présenté un événement veineux embolique ou thrombotique au cours des 6 mois précédant l’instauration du traitement.
Elévation de l’hémoglobine ou de l’hématocrite
Des augmentations de l’hémoglobine ou de l’hématocrite, reflétant des augmentations de la masse des globules rouges, peuvent survenir au cours du traitement par axitinib (voir rubrique 4.8, polycythémie). Une augmentation de la masse des globules rouges peut aggraver le risque d’événements emboliques et thrombotiques.
L’hémoglobine ou l’hématocrite doit être contrôlée avant l’instauration du traitement par axitinib et régulièrement pendant toute la durée de celui-ci. En cas d’élévation de l’hémoglobine ou de l’hématocrite au-dessus de la normale, les patients doivent être traités conformément à la pratique médicale standard afin de ramener l’hémoglobine ou l’hématocrite à une valeur acceptable.
Hémorragie
Des événements hémorragiques ont été rapportés au cours des études cliniques portant sur l’axitinib (voir rubrique 4.8).
L’axitinib n’a pas été étudié chez les patients présentant des métastases cérébrales non traitées ou des hémorragies gastro-intestinales actives récentes, et ne doit pas être administré à ces patients. En cas d'hémorragie nécessitant une intervention médicale, l’administration d’axitinib doit être temporairement interrompue.
Anévrismes et dissections artérielles
L’utilisation d’inhibiteurs des voies du VEGF chez les patients souffrant ou non d’hypertension peut favoriser la formation d’anévrismes et/ou de dissections artérielles. Avant l’instauration d’AXITINIB TEVA, ce risque doit être soigneusement pris en compte chez les patients présentant des facteurs de risque tels que l’hypertension ou des antécédents d’anévrisme.
Perforations gastro-intestinales et formation de fistules
Des cas de perforations intestinales et de fistules ont été rapportés au cours des études cliniques portant sur l’axitinib (voir rubrique 4.8).
Des symptômes de perforation gastro-intestinale ou de fistule doivent être régulièrement recherchés pendant toute la durée du traitement par axitinib.
Complications de la cicatrisation des plaies
Aucune étude formelle de l’effet d’axitinib sur la cicatrisation des plaies n’a été menée.
Le traitement par axitinib doit être interrompu au moins 24 heures avant une intervention chirurgicale programmée. La décision de reprendre le traitement par axitinib après une intervention chirurgicale doit reposer sur l’appréciation clinique de la cicatrisation adéquate de la plaie.
Syndrome d’encéphalopathie postérieure réversible (SEPR)
Des cas de SEPR ont été rapportés au cours des études cliniques portant sur l’axitinib (voir rubrique 4.8).
Le SEPR est un trouble neurologique qui peut se manifester par des céphalées, des convulsions, une léthargie, une confusion, une cécité et d’autres troubles visuels et neurologiques. Une hypertension légère à sévère peut être présente. Une imagerie par résonance magnétique est nécessaire pour confirmer le diagnostic de SEPR. Le traitement par axitinib doit être interrompu temporairement ou définitivement chez les patients présentant des signes ou symptômes de SEPR. La sécurité de la reprise du traitement par axitinib chez les patients ayant déjà présenté un SEPR n'est pas connue.
Protéinurie
Des cas de protéinurie, y compris de sévérité de grade 3 et 4, ont été rapportés au cours des études cliniques portant sur l’axitinib (voir rubrique 4.8).
Il est recommandé de rechercher une protéinurie avant l’instauration du traitement par axitinib, puis régulièrement pendant toute la durée de celui-ci. Une réduction de dose ou une interruption temporaire du traitement par axitinib est recommandée chez les patients présentant une protéinurie modérée à sévère (voir rubrique 4.2). Le traitement par axitinib doit être arrêté si le patient développe un syndrome néphrotique.
Effets indésirables hépatiques
Des effets indésirables hépatiques ont été rapportés au cours d’une étude clinique contrôlée évaluant l’axitinib chez des patients atteints de RCC. Les effets indésirables hépatiques les plus fréquemment rapportés ont été des augmentations de l’alanine aminotransférase (ALAT), de l’aspartate aminotransférase (ASAT) et de la bilirubinémie (voir rubrique 4.8). Aucune élévation concomitante de l’ALAT (> 3 fois la limite supérieure de la normale [LSN]) et de la bilirubinémie (> 2 fois la LSN) n’a été observée.
Lors d’une étude clinique de recherche de dose, des élévations concomitantes de l’ALAT (12 fois la LSN) et de la bilirubinémie (2,3 fois la LSN) considérées comme une hépatotoxicité liée au médicament, ont été observées chez un patient qui recevait l’axitinib à une dose initiale de 20 mg deux fois par jour (4 fois la dose initiale recommandée).
Les tests de la fonction hépatique doivent être contrôlés avant l’instauration du traitement par axitinib, puis régulièrement pendant toute la durée de celui-ci.
Insuffisance hépatique
Au cours des études cliniques portant sur l’axitinib, l’exposition systémique à l’axitinib a été environ deux fois plus élevée chez les sujets présentant une insuffisance hépatique modérée (classe B de Child-Pugh) que chez les sujets présentant une fonction hépatique normale. Une diminution de la dose d’axitinib est recommandée chez les patients présentant une insuffisance hépatique modérée (classe B de Child-Pugh) (voir rubrique 4.2).
L’axitinib n’a pas été étudié chez les patients présentant une insuffisance hépatique sévère (classe C de Child-Pugh) et ne doit pas être utilisé dans cette population.
Personnes âgées (> 65 ans) et origine ethnique
Au cours d’une étude clinique contrôlée portant sur l’axitinib dans le traitement de patients atteints de RCC, 34 % des patients traités par axitinib étaient âgés de > 65 ans. Les patients étaient majoritairement caucasiens (77 %) ou asiatiques (21 %). Bien qu’une sensibilité plus élevée à la survenue d’effets indésirables ne puisse être exclue chez les patients âgés et les patients asiatiques, aucune différence majeure de la sécurité et de l’efficacité d’axitinib n’a été globalement observée entre les patients âgés de > 65 ans et les plus jeunes et entre les patients caucasiens et ceux d’autres origines ethniques.
Aucun ajustement posologique n’est recommandé en fonction de l’âge ou de l’origine ethnique du patient (voir rubriques 4.2 et 5.2).
Excipients
Lactose
Ce médicament contient du lactose. Les patients présentant une intolérance au galactose, un déficit total en lactase ou un syndrome de malabsorption du glucose et du galactose (maladies héréditaires rares) ne doivent pas prendre ce médicament.
Sodium
Ce médicament contient moins de 1 mmol (23 mg) de sodium par comprimé pelliculé, c’est-à-dire qu’il est essentiellement « sans sodium ».
4.5. Interactions avec d'autres médicaments et autres formes d'interactions
Inhibiteurs du CYP3A4/5
Le kétoconazole, un inhibiteur puissant du CYP3A4/5, administré à la dose de 400 mg une fois par jour pendant 7 jours, a augmenté de 2 fois l’aire moyenne sous la courbe (ASC) et de 1,5 fois la Cmax de l’axitinib administré à raison d’une dose orale unique de 5 mg à des volontaires sains. Les concentrations plasmatiques de l’axitinib peuvent augmenter en cas d’administration concomitante avec des inhibiteurs puissants du CYP3A4/5 (par exemple, kétoconazole, itraconazole, clarithromycine, érythromycine, atazanavir, indinavir, néfazodone, nelfinavir, ritonavir, saquinavir et télithromycine).
Le pamplemousse peut également augmenter les concentrations plasmatiques de l’axitinib. Il est recommandé d’opter pour des médicaments concomitants ayant peu ou pas d’effet sur l’inhibition du CYP3A4/5. Si un inhibiteur puissant du CYP3A4/5 doit être administré de façon concomitante, un ajustement posologique de l’axitinib est recommandé (voir rubrique 4.2).
Inhibiteurs des CYP1A2 et CYP2C19
Les CYP1A2 et CYP2C19 constituent des voies mineures (< 10 %) du métabolisme de l’axitinib. L’effet des inhibiteurs puissants de ces isoenzymes sur la pharmacocinétique de l’axitinib n’a pas été étudié. La prudence est de mise en raison du risque d’augmentation des concentrations plasmatiques de l’axitinib chez les patients prenant des inhibiteurs puissants de ces isoenzymes.
Inducteurs du CYP3A4/5
La rifampicine, un inducteur puissant du CYP3A4/5, administrée à la dose de 600 mg une fois par jour pendant 9 jours, a diminué de 79 % l'ASC moyenne et de 71 % la Cmax de l’axitinib administré à raison d’une dose orale unique de 5 mg à des volontaires sains.
Les concentrations plasmatiques de l’axitinib peuvent diminuer en cas d’administration concomitante d’inducteurs puissants du CYP3A4/5 (par exemple, rifampicine, dexaméthasone, phénytoïne, carbamazépine, rifabutine, rifapentine, phénobarbital et Hypericum perforatum [millepertuis]). Il est recommandé d’opter pour des médicaments concomitants ayant peu ou pas d’effet sur l’induction du CYP3A4/5. Si un inducteur puissant du CYP3A4/5 doit être administré de façon concomitante, un ajustement posologique de l’axitinib est recommandé (voir rubrique 4.2).
Etudes in vitro de l’inhibition et de l’induction du CYP et de l’UGT
Des études in vitro ont indiqué que l’axitinib n’inhibait pas les CYP2A6, CYP2C9, CYP2C19, CYP2D6, CYP2E1, CYP3A4/5 ou UGT1A1 aux concentrations plasmatiques thérapeutiques.
Des études in vitro ont indiqué que l’axitinib pouvait inhiber le CYP1A2. Par conséquent, l’administration concomitante d’axitinib et de substrats du CYP1A2 (théophylline, par exemple) pourrait donc augmenter les concentrations plasmatiques de ces substrats.
Des études in vitro ont également indiqué que l’axitinib pouvait inhiber le CYP2C8. Cependant, l’administration concomitante d’axitinib et de paclitaxel, substrat connu du CYP2C8, n’a pas entraîné d’augmentation des concentrations plasmatiques du paclitaxel chez des patients atteints de cancer avancé, ce qui témoigne de l’absence d’inhibition clinique du CYP2C8.
Des études in vitro sur des hépatocytes humains ont également indiqué que l’axitinib n’induisait pas les CYP1A1, CYP1A2 ou CYP3A4/5. Par conséquent, l'administration concomitante de l’axitinib ne devrait donc pas réduire la concentration plasmatique de substrats des CYP1A1, CYP1A2 ou CYP3A4/5 in vivo.
Etudes in vitro avec la glycoprotéine P
Des études in vitro ont indiqué que l’axitinib inhibait la glycoprotéine P. Cependant, l’axitinib ne devrait pas inhiber la glycoprotéine P aux concentrations plasmatiques thérapeutiques. L'administration concomitante d’axitinib ne devrait donc pas augmenter la concentration plasmatique de la digoxine ou d’autres substrats de la glycoprotéine P in vivo.
4.6. Fertilité, grossesse et allaitement
Grossesse
Il n’existe aucune donnée sur l'utilisation de l’axitinib chez la femme enceinte. Compte tenu de ses propriétés pharmacologiques, l’axitinib peut être nocif pour le fœtus s’il est administré pendant la grossesse. Les études effectuées chez l’animal ont mis en évidence une toxicité sur la reproduction, y compris des malformations (voir rubrique 5.3). L’axitinib ne doit pas être utilisé pendant la grossesse à moins que la situation clinique de la femme ne justifie le traitement par ce médicament.
Les femmes en capacité de procréer doivent utiliser une contraception efficace pendant le traitement et jusqu'à 1 semaine après l’arrêt du traitement.
On ne sait pas si l’axitinib est excrété dans le lait maternel. Un risque pour l’enfant allaité ne peut être exclu. L’axitinib ne doit pas être utilisé pendant l’allaitement.
Fertilité
Selon des données non cliniques, l’axitinib pourrait altérer la fonction de reproduction et la fertilité chez l’être humain (voir rubrique 5.3).
4.7. Effets sur l'aptitude à conduire des véhicules et à utiliser des machines
L’axitinib a une influence mineure sur l’aptitude à conduire des véhicules et à utiliser des machines. Les patients doivent être avertis du risque de sensations vertigineuses et/ou de fatigue pendant le traitement par axitinib.
Résumé du profil de sécurité
Les risques suivants, ainsi que les conduites à tenir appropriées, sont discutés de manière plus détaillée dans la rubrique 4.4 : événements d’insuffisance cardiaque, hypertension, dysfonctionnement thyroïdien, événements thrombo-emboliques artériels, événements thrombo-emboliques veineux, élévation de l’hémoglobine ou de l’hématocrite, hémorragie, perforation gastro-intestinale et formation de fistules, complications de la cicatrisation des plaies, SEPR, protéinurie et élévation des enzymes hépatiques.
Les effets indésirables les plus fréquents (≥ 20 %) observés sous traitement par axitinib ont été : diarrhée, hypertension, fatigue, appétit diminué, nausées, poids diminué, dysphonie, érythrodysesthésie palmo-plantaire (syndrome main-pied), hémorragie, hypothyroïdie, vomissements, protéinurie, toux et constipation.
Tableau listant les effets indésirables
Le tableau 1 présente les effets indésirables rapportés dans un ensemble de données regroupées de 672 patients ayant reçu l’axitinib au cours des études cliniques pour le traitement des patients atteints de RCC (voir rubrique 5.1). Les effets indésirables identifiés au cours des études cliniques, après la commercialisation, sont également inclus.
Les effets indésirables sont listés par classe de systèmes d'organes, fréquence et grade de sévérité. Les catégories de fréquence sont définies de la façon suivante : très fréquent (≥ 1/10), fréquent (≥ 1/100, < 1/10), peu fréquent (≥ 1/1 000, < 1/100), rare (≥ 1/10 000, < 1/1 000), très rare (< 1/10 000) et fréquence indéterminée (ne peut être estimée sur la base des données disponibles). La base de données de pharmacovigilance ne permet pas actuellement d’établir la liste des effets indésirables rares et très rares pour l’axitinib.
Des catégories ont été assignées sur la base des fréquences absolues des données d’études cliniques combinées. Dans chaque classe de systèmes d’organes, les effets indésirables de fréquence identique sont présentés par ordre décroissant de gravité.
Tableau 1. Effets indésirables rapportés au cours des études chez les patients atteints de RCC ayant reçu l’axitinib (N = 672)
|
Classe de systèmes d’organes |
Catégorie de fréquence |
Effets indésirablesa |
Tous gradesb % |
Grade 3b % |
Grade 4b % |
|
Affections hématologiques et du système lymphatique |
Fréquent |
Anémie |
6,3 |
1,2 |
0,4 |
|
Thrombopénie |
1,6 |
0,1 |
0 |
||
|
Polycythémiec |
1,5 |
0,1 |
0 |
||
|
Peu fréquent |
Neutropénie |
0,3 |
0,1 |
0 |
|
|
Leucopénie |
0,4 |
0 |
0 |
||
|
Affections endocriniennes |
Très fréquent |
Hypothyroïdiec |
24,6 |
0,3 |
0 |
|
Fréquent |
Hyperthyroïdiec |
1,6 |
0,1 |
0,1 |
|
|
Troubles du métabolisme et de la nutrition |
Très fréquent |
Appétit diminué |
39,0 |
3,6 |
0,3 |
|
Fréquent |
Déshydratation |
6,7 |
3,1 |
0,3 |
|
|
Hyperkaliémie |
2,7 |
1,2 |
0,1 |
||
|
Hypercalcémie |
2,2 |
0,1 |
0,3 |
||
|
Affections du système nerveux |
Très fréquent |
Céphalées |
16,2 |
0,7 |
0 |
|
Dysgueusie |
11,5 |
0 |
0 |
||
|
Fréquent |
Sensations vertigineuses |
9,1 |
0,6 |
0 |
|
|
Peu fréquent |
Syndrome d’encéphalopathie postérieure réversiblee |
0,3 |
0,1 |
0 |
|
|
Affections de l’oreille et du labyrinthe |
Fréquent |
Acouphènes |
3,1 |
0 |
0 |
|
Affections cardiaques |
Fréquent |
Evénements d’insuffisance cardiaque c,d,f |
1,8 |
0,3 |
0,7 |
|
Affections vasculaires |
Très fréquent |
Hypertensiong |
51,2 |
22,0 |
1,0 |
|
Hémorrhagiec,d,h |
25,7 |
3,0 |
1,0 |
||
|
Fréquent |
Evénements veineux emboliques et thrombotiquesc,d,i |
2,8 |
0,9 |
1,2 |
|
|
Evénements artériels emboliques et thrombotiquesc,d,j |
2,8 |
1,2 |
1,3 |
||
|
Fréquence indéterminée |
Anévrismes et dissections artériellesd |
- |
- |
- |
|
|
Affections respiratoires, thoraciques et médiastinales |
Très fréquent |
Dyspnéed |
17,1 |
3,6 |
0,6 |
|
Toux |
20,4 |
0,6 |
0 |
||
|
Dysphonie |
32,7 |
0 |
0,1 |
||
|
Fréquent |
Douleur oropharyngée |
7,4 |
0 |
0 |
|
|
Affections gastro-intestinales |
Très fréquent |
Diarrhée |
55,4 |
10,1 |
0,1 |
|
Vomissements |
23,7 |
2,7 |
0,1 |
||
|
Nausées |
33,0 |
2,2 |
0,1 |
||
|
Douleur abdominale |
14,7 |
2,5 |
0,3 |
||
|
Constipation |
20,2 |
1,0 |
0 |
||
|
Stomatite |
15,5 |
1,8 |
0 |
||
|
Dyspepsie |
11,2 |
0,1 |
0 |
||
|
Fréquent |
Douleur abdominale haute |
9,4 |
0,9 |
0 |
|
|
Flatulences |
4,5 |
0 |
0 |
||
|
Hémorroïdes |
3,3 |
0 |
0 |
||
|
Glossodynie |
2,8 |
0 |
0 |
||
|
Perforation gastro-intestinale et fistulec,k |
1,9 |
0,9 |
0,3 |
||
|
Affections hépatobiliaires |
Fréquent |
Hyperbilirubinémie |
1,3 |
0,1 |
0,1 |
|
Cholécystiten |
1,0 |
0,6 |
0,1 |
||
|
Affections de la peau et du tissu sous-cutané |
Très fréquent |
Erythrodysesthésie palmo-plantaire (syndrome main-pied) |
32,1 |
7,6 |
0 |
|
Rash |
14,3 |
0,1 |
0 |
||
|
Sécheresse cutanée |
10,1 |
0,1 |
0 |
||
|
Fréquent |
Prurit |
6,0 |
0 |
0 |
|
|
Erythème |
3,7 |
0 |
0 |
||
|
Alopécie |
5,7 |
0 |
0 |
||
|
Affections musculo-squelettiques et systémiques |
Très fréquent |
Arthralgie |
17,7 |
1,9 |
0,3 |
|
Douleur aux extrémités |
14,1 |
1,0 |
0,3 |
||
|
Fréquent |
Myalgie |
8,2 |
0,6 |
0,1 |
|
|
Affections du rein et des voies urinaires |
Très fréquent |
Protéinuriel |
21,1 |
4,8 |
0,1 |
|
Fréquent |
Insuffisance rénalem |
1,6 |
0,9 |
0,1 |
|
|
Troubles généraux et anomalies au site d’administration |
Très fréquent |
Fatigue |
45,1 |
10,6 |
0,3 |
|
Asthénied |
13,8 |
2,8 |
0,3 |
||
|
Inflammation des muqueuses |
13,7 |
1,0 |
0 |
||
|
Investigations |
Très fréquent |
Poids diminué |
32,7 |
4,9 |
0 |
|
Fréquent |
Lipase augmentée |
3,7 |
0,7 |
0,7 |
|
|
Alanine aminotransférase augmentée |
6,5 |
1,2 |
0 |
||
|
Amylase augmentée |
3,4 |
0,6 |
0,4 |
||
|
Aspartate aminotransférase augmentée |
6,1 |
1,0 |
0 |
||
|
Phosphatase alcaline augmentée |
4,8 |
0,3 |
0 |
||
|
Créatinine augmentée |
5,7 |
0,4 |
0 |
||
|
Thyrotrophine augmentée |
7,9 |
0 |
0 |
a Les effets indésirables sont présentés par fréquence d'apparition sous traitement, toutes causes confondues.
b National Cancer Institute Common Terminology Criteria for Adverse Events, version 3.0.
c Voir la description des effets indésirables sélectionnés.
d Des cas fatals (grade 5) ont été rapportés.
e Incluant leuco-encéphalopathie.
f Incluant insuffisance cardiaque, insuffisance cardiaque congestive, insuffisance cardiopulmonaire, fraction d'éjection diminuée, dysfonctionnement ventriculaire gauche et insuffisance ventriculaire droite.
g Incluant hypertension artérielle accélérée, pression artérielle augmentée, hypertension et crise hypertensive.
h Incluant temps de céphaline activée allongé, hémorragie anale, hémorragie artérielle, présence de sang dans l’urine, hémorragie du système nerveux central, hémorragie cérébrale, durée de coagulation prolongée, hémorragie conjonctivale, contusion, diarrhée hémorragique, métrorragies fonctionnelles, épistaxis, hémorragie gastrique, hémorragie gastro-intestinale, saignement gingival, hématémèse, hématochézie, hématocrite diminué, hématome, hématurie, hémoglobine diminuée, hémoptysie, hémorragie, hémorragie de l’artère coronaire, hémorragie des voies urinaires, hémorragie hémorroïdale, hémostase, tendance ecchymotique augmentée, rapport normalisé international augmenté, hémorragie gastro-intestinale basse, méléna, pétéchies, hémorragie pharyngée, temps de prothrombine allongé, hémorragie pulmonaire, purpura, hémorragie rectale, globules rouges diminués, hémorragie rénale, hémorragie sclérale, hématocèle scrotale, hématome de la rate, hémorragie linéaire sous-unguéale, hémorragie sous-arachnoïdienne, hémorragie de la langue, hémorragie gastro-intestinale haute et hémorragie vaginale.
i Incluant syndrome de Budd-Chiari, thrombose veineuse profonde, thrombose de la veine jugulaire, thrombose veineuse pelvienne, embolie pulmonaire, occlusion de la veine rétinienne, thrombose de la veine rétinienne, thrombose de la veine sous-clavière, thrombose veineuse et thrombose veineuse d’un membre.
j Incluant infarctus du myocarde aigu, embolie, infarctus du myocarde, occlusion de l’artère rétinienne et accident ischémique transitoire.
k Les perforations gastro-intestinales et les fistules incluent les termes préférentiels suivants : abcès abdominal, abcès anal, fistule anale, fistule, fuite d'une anastomose gastro-intestinale, perforation gastro-intestinale, perforation du gros intestin, fistule œsophagobronchique et péritonite.
l La protéinurie inclut les termes préférentiels suivants : protéine urinaire, présence de protéine dans l’urine et protéinurie.
m Incluant insuffisance rénale aiguë.
n Cholécystite incluant cholécystite aiguë, cholécystite, cholécystite infectieuse.
Description des effets indésirables sélectionnés
Evénements d’insuffisance cardiaque (voir rubrique 4.4)
Au cours d’une étude clinique contrôlée portant sur l’axitinib (N = 359) dans le traitement de patients atteints de RCC, des événements d’insuffisance cardiaque ont été rapportés chez 1,7 % des patients sous axitinib, incluant insuffisance cardiaque (0,6 %), insuffisance cardiopulmonaire (0,6 %), dysfonctionnement ventriculaire gauche (0,3 %) et insuffisance ventriculaire droite (0,3 %). Des effets indésirables d’insuffisance cardiaque de grade 4 ont été rapportés chez 0,6 % des patients sous axitinib. Une insuffisance cardiaque fatale a été rapportée chez 0,6 % des patients sous axitinib.
Au cours des études portant sur l’axitinib en monothérapie (N = 672) dans le traitement de patients atteints de RCC, des événements d’insuffisance cardiaque (incluant insuffisance cardiaque, insuffisance cardiaque congestive, insuffisance cardiopulmonaire, dysfonctionnement ventriculaire gauche, fraction d’éjection diminuée et insuffisance ventriculaire droite) ont été rapportés chez 1,8 % des patients sous axitinib. Des événements d’insuffisance cardiaque de grade 3/4 ont été rapportés chez 1,0 % des patients et des événements d’insuffisance cardiaque fatals ont été rapportés chez 0,3 % des patients sous axitinib.
Dysfonctionnement thyroïdien (voir rubrique 4.4)
Au cours d’une étude clinique contrôlée portant sur l’axitinib dans le traitement de patients atteints de RCC, une hypothyroïdie a été rapportée chez 20,9 % des patients et une hyperthyroïdie a été rapportée chez 1,1 % des patients. L’effet indésirable « thyrotrophine (TSH) augmentée » a été rapporté chez 5,3 % des patients sous axitinib. Des analyses de laboratoire de routine ont indiqué que, chez des patients présentant un taux de TSH < 5 μU/mL avant traitement, 32,2 % des patients sous axitinib ont présenté des élévations de la TSH ≥ 10 μU/mL.
Au cours des études cliniques combinées portant sur l’axitinib (N = 672) dans le traitement de patients atteints de RCC, l’hypothyroïdie a été rapportée chez 24,6 % des patients sous axitinib. L’hyperthyroïdie a été rapportée chez 1,6 % des patients sous axitinib.
Evénements veineux emboliques et thrombotiques (voir rubrique 4.4)
Au cours d’une étude clinique contrôlée portant sur l’axitinib dans le traitement de patients atteints de RCC, des événements veineux emboliques et thrombotiques, incluant embolie pulmonaire (2,2 %), occlusion/thrombose de la veine rétinienne (0,6 %) et thrombose veineuse profonde (0,6 %), ont été rapportés chez 3,9 % des patients sous axitinib.
Des événements veineux emboliques et thrombotiques de grade 3/4 ont été rapportés chez 3,1 % des patients sous axitinib. Une embolie pulmonaire fatale a été rapportée chez un patient (0,3 %) sous axitinib.
Au cours des études cliniques combinées portant sur l’axitinib (N = 672) dans le traitement de patients atteints de RCC, des événements veineux emboliques et thrombotiques ont été rapportés chez 2,8 % des patients sous axitinib. Des événements veineux emboliques et thrombotiques de grade 3 ont été rapportés chez 0,9 % des patients. Des événements veineux emboliques et thrombotiques de grade 4 ont été rapportés chez 1,2 % des patients. Des événements veineux emboliques et thrombotiques fatals ont été rapportés chez 0,1 % des patients sous axitinib.
Evénements artériels emboliques et thrombotiques (voir rubrique 4.4)
Des événements artériels emboliques et thrombotiques, incluant infarctus du myocarde (1,4 %), accident ischémique transitoire (0,8 %) et accident vasculaire cérébral (0,6 %) ont été rapportés chez 4,7 % des patients sous axitinib au cours d’une étude clinique contrôlée portant sur l’axitinib dans le traitement de patients atteints de RCC. Des événements artériels emboliques et thrombotiques de grade 3/4 ont été rapportés chez 3,3 % des patients sous axitinib. Un infarctus du myocarde aigu et un accident vasculaire cérébral fatals ont été rapportés chacun chez un patient (0,3 %). Au cours des études portant sur l’axitinib en monothérapie, des effets indésirables artériels emboliques et thrombotiques (incluant accident ischémique transitoire, infarctus du myocarde et accident vasculaire cérébral) ont été rapportés chez 5,3 % des patients sous axitinib.
Au cours des études cliniques combinées portant sur l’axitinib (N = 672) dans le traitement de patients atteints de RCC, des événements artériels emboliques et thrombotiques ont été rapportés chez 2,8 % des patients sous axitinib. Des événements artériels emboliques et thrombotiques de grade 3 ont été rapportés chez 1,2 % des patients. Des événements artériels emboliques et thrombotiques de grade 4 ont été rapportés chez 1,3 % des patients. Des événements artériels emboliques et thrombotiques fatals ont été rapportés chez 0,3 % des patients sous axitinib.
Polycythémie (voir Elévation de l’hémoglobine ou de l’hématocrite à la rubrique 4.4)
Au cours d’une étude clinique contrôlée portant sur l’axitinib dans le traitement de patients atteints de RCC, une polycythémie a été rapportée chez 1,4 % des patients sous axitinib. Les analyses de laboratoire de routine ont montré une élévation de l’hémoglobine au-dessus de la LSN chez 9,7 % des patients sous axitinib. Au cours de quatre études cliniques portant sur l’axitinib dans le traitement de patients atteints de RCC (N = 537), une élévation de l’hémoglobine au-dessus de la LSN a été observée chez 13,6 % des patients sous axitinib.
Au cours des études cliniques combinées portant sur l’axitinib (N = 672) dans le traitement de patients atteints de RCC, une polycythémie a été rapportée chez 1,5 % des patients sous axitinib.
Hémorragie (voir rubrique 4.4)
Au cours d’une étude clinique contrôlée portant sur l’axitinib dans le traitement de patients atteints de RCC, où les patients présentant des métastases cérébrales non traitées étaient exclus, des effets indésirables hémorragiques ont été rapportés chez 21,4 % des patients sous axitinib. Les effets indésirables hémorragiques rapportés chez les patients traités par axitinib ont inclus : épistaxis (7,8 %), hématurie (3,6 %), hémoptysie (2,5 %), hémorragie rectale (2,2 %), hémorragie gingivale (1,1 %), hémorragie gastrique (0,6 %), hémorragie cérébrale (0,3 %) et hémorragie gastro-intestinale basse (0,3 %). Des effets indésirables hémorragiques de grade ≥ 3 ont été rapportés chez 3,1 % des patients sous axitinib (incluant hémorragie cérébrale, hémorragie gastrique, hémorragie gastro-intestinale basse et hémoptysie). Une hémorragie fatale a été rapportée chez un patient (0,3 %) sous axitinib (hémorragie gastrique). Dans les études portant sur l’axitinib en monothérapie (N = 850), une hémoptysie a été rapportée chez 3,9 % des patients ; une hémoptysie de grade ≥ 3 a été rapportée chez 0,5 % des patients.
Au cours des études cliniques combinées portant sur l’axitinib (N = 672) dans le traitement de patients atteints de RCC, des événements hémorragiques ont été rapportés chez 25,7 % des patients sous axitinib. Des effets indésirables hémorragiques de grade 3 ont été rapportés chez 3 % des patients. Des effets indésirables hémorragiques de grade 4 ont été rapportés chez 1 % des patients et des hémorragies fatales ont été rapportées chez 0,4 % des patients sous axitinib.
Perforations gastro-intestinales et formation de fistules (voir rubrique 4.4)
Au cours d’une étude clinique contrôlée portant sur l’axitinib dans le traitement de patients atteints de RCC, des événements de type perforation gastro-intestinale, incluant fistule anale (0,6 %), fistule (0,3 %) et perforation gastro-intestinale (0,3 %), ont été rapportés chez 1,7 % des patients sous axitinib. Au cours des études cliniques portant sur l’axitinib en monothérapie (N = 850), des événements de type perforation gastro-intestinale ont été rapportés chez 1,9 % des patients et une perforation gastro-intestinale fatale a été rapportée chez un patient (0,1 %).
Au cours des études cliniques combinées portant sur l’axitinib (N = 672) dans le traitement de patients atteints de RCC, des perforations gastro-intestinales et des fistules ont été rapportées chez 1,9 % des patients sous axitinib.
Déclaration des effets indésirables suspectés
La déclaration des effets indésirables suspectés après autorisation du médicament est importante. Elle permet une surveillance continue du rapport bénéfice/risque du médicament. Les professionnels de santé déclarent tout effet indésirable suspecté via le système national de déclaration : Agence nationale de sécurité du médicament et des produits de santé (ANSM) et réseau des Centres Régionaux de Pharmacovigilance - Site internet : https://signalement.social-sante.gouv.fr/.
Il n’existe aucun traitement spécifique en cas de surdosage par axitinib.
Au cours d’une étude clinique contrôlée portant sur l’axitinib dans le traitement de patients atteints de RCC, un patient a accidentellement reçu une dose de 20 mg deux fois par jour pendant 4 jours et a présenté des sensations vertigineuses (grade 1).
Au cours d’une étude clinique de recherche de dose portant sur l’axitinib, des patients ayant reçu une dose initiale de 10 mg d’axitinib deux fois par jour ou de 20 mg deux fois par jour ont présenté des effets indésirables incluant hypertension, convulsions associées à une hypertension et hémoptysie fatale.
En cas de suspicion de surdosage, l’administration d’axitinib doit être suspendue et un traitement symptomatique instauré.
5. PROPRIETES PHARMACOLOGIQUES
5.1. Propriétés pharmacodynamiques
Classe pharmacothérapeutique : Antinéoplasiques, inhibiteurs de protéine kinase, code ATC : L01EK01.
Mécanisme d’action
L’axitinib est un inhibiteur de tyrosine kinase puissant et sélectif des récepteurs du facteur de croissance de l’endothélium vasculaire (VEGFR-1, VEGFR-2 et VEGFR-3). Ces récepteurs sont impliqués dans l’angiogénèse pathologique, la croissance tumorale et la progression métastatique de cancers. Il a été démontré que l’axitinib inhibait de façon puissante la prolifération et la survie des cellules endothéliales assurées par la médiation du VEGF. L’axitinib a inhibé la phosphorylation du VEGFR-2 dans le système vasculaire tumoral de xénogreffes ayant exprimé la cible in vivo, et a entraîné un retard et une régression de la croissance tumorale et une inhibition des métastases dans de nombreux modèles expérimentaux de cancers.
Effet sur l’intervalle QTc
Au cours d’une étude randomisée et réalisée en cross over, 35 sujets sains ont reçu une dose orale unique d’axitinib (5 mg) en l’absence et en présence de 400 mg de kétoconazole pendant 7 jours. Les résultats de cette étude ont indiqué que des expositions plasmatiques d’axitinib jusqu’à deux fois supérieures aux niveaux thérapeutiques attendus après une dose de 5 mg n’avaient pas induit d’allongement cliniquement significatif de l’intervalle QT.
Efficacité et sécurité clinique
La sécurité et l’efficacité de l’axitinib ont été évaluées au cours d’une étude de phase III multicentrique, randomisée et réalisée en ouvert. Des patients (N = 723) atteints d’un RCC avancé ayant progressé pendant ou après un traitement systémique antérieur à base de sunitinib, bévacizumab, temsirolimus ou cytokine, ont été randomisés (1/1) pour recevoir l’axitinib (N = 361) ou le sorafénib (N = 362). Le critère d’évaluation principal, la survie sans progression (SSP), a été évalué en aveugle par un comité centralisé indépendant.
Les critères d’évaluation secondaires incluaient le taux de réponse objective (TRO) et la survie globale (SG).
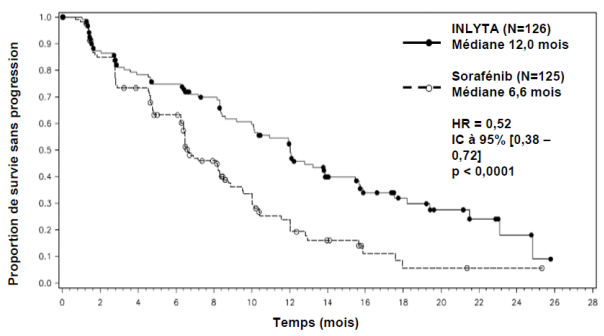
Parmi les patients inclus dans cette étude, 389 patients (53,8 %) avaient reçu antérieurement un traitement à base de sunitinib, 251 patients (34,7 %) un traitement à base de cytokine (interleukine-2 ou interféron-alpha), 59 patients (8,2 %) un traitement à base de bévacizumab et 24 patients (3,3 %) un traitement à base de temsirolimus. Les caractéristiques démographiques et les caractéristiques de la maladie à l’inclusion ont été similaires entre les groupes axitinib et sorafénib en ce qui concerne l’âge, le sexe, l’origine ethnique, l’indice de performance ECOG (Eastern Cooperative Oncology Group), la région géographique et le traitement antérieur.
Dans la population totale de patients et dans les deux sous-groupes principaux (traitement antérieur par sunitinib et traitement antérieur par cytokine), une amélioration statistiquement significative du critère d’évaluation principal de SSP a été observée sous axitinib par rapport au sorafénib (voir tableau 2 et figures 1, 2 et 3). L’amplitude de l’effet sur la SSP médiane a été différente selon les sous-groupes définis en fonction du traitement antérieur. Les effectifs de deux des sous-groupes ont été trop faibles pour générer des résultats suffisamment fiables (traitement antérieur par temsirolimus ou traitement antérieur par bévacizumab). Il n’a pas été observé de différence statistiquement significative entre les deux bras en termes de SG, que ce soit dans la population globale ou dans les sous-groupes définis en fonction du traitement antérieur.
Tableau 2. Résultats d’efficacité
|
Critère d’évaluation/population de l’étude |
axitinib |
sorafénib |
HR (IC à 95 %) |
Valeur de p |
|
Population globale ITT |
N = 361 |
N = 362 |
|
|
|
SSP médianea,b en mois |
6,8 (6,4 ; 8,3) |
4,7 (4,6 ; 6,3) |
0,67 (0,56 ; 0,81) |
< 0,0001c |
|
(IC à 95 %) |
|
|
|
|
|
SG médianed en mois |
20,1 (16,7 ; 23,4) |
19,2 (17,5 ; 22,3) |
0,97 (0,80 ; 1,17) |
NS |
|
(IC à 95 %) |
|
|
|
|
|
TROb,e % (IC à 95 %) |
19,4 (15,4 ; 23,9) |
9,4 (6,6 ; 12,9) |
2,06f (1,41 ; 3,00) |
0,0001g |
|
Traitement antérieur à base de sunitinib |
N = 194 |
N = 195 |
|
|
|
SSP médianea,b en mois |
4,8 (4,5 ; 6,5) |
3,4 (2,8 ; 4,7) |
0,74 (0,58 ; 0,94) |
0,0063h |
|
(IC à 95 %) |
|
|
|
|
|
SG médianed en mois |
15,2 (12,8 ; 18,3) |
16,5 (13,7 ; 19,2) |
1,00 (0,78 ; 1,27) |
NS |
|
(IC à 95 %) |
|
|
|
|
|
TROb,e % (IC à 95 %) |
11,3 (7,2 ; 16,7) |
7,7 (4,4 ; 12,4) |
1,48f (0,79 ; 2,75) |
NS |
|
Traitement antérieur à base de cytokine |
N = 126 |
N = 125 |
|
|
|
SSP médianea,b en mois |
12,0 (10,1 ; 13,9) |
6,6 (6,4 ; 8,3) |
0,52 (0,38 ; 0,72) |
< 0,0001h |
|
(IC à 95 %) |
|
|
|
|
|
SG médianed en mois |
29,4 (24,5 ; NE) |
27,8 (23,1 ; 34,5) |
0,81 (0,56 ; 1,19) |
NS |
|
(IC à 95 %) |
|
|
|
|
|
TROb,e % (IC à 95 %) |
32,5 (24,5 ; 41,5) |
13,6 (8,1 ; 20,9) |
2,39f (1,43 ; 3,99) |
0,0002i |
IC = intervalle de confiance ; HR = Hazard Ratio (axitinib/sorafénib) ; ITT = intention de traiter ; NE = non estimable ; NS = non statistiquement significatif ; TRO = taux de réponse objective ; SG = survie globale ; SSP = survie sans progression.
a Temps écoulé de la randomisation jusqu’à la progression ou le décès toutes causes confondues selon l’événement survenant en premier. Date du gel des données : 3 juin 2011.
b Evaluation radiologique par un comité central indépendant selon les critères RECIST (Response Evaluation Criteria in Solid Tumours).
c Valeur unilatérale de p issue d’un test du log-rank stratifié selon l’indice de performance ECOG et le traitement antérieur.
d Date du gel des données : 1er novembre 2011.
e Date du gel des données : 31 août 2010.
f Le rapport de risque est utilisé pour le TRO. Un rapport de risque > 1 indique une probabilité plus élevée de réponse dans le bras axitinib ; un rapport de risque < 1 indique une probabilité plus élevée de réponse dans le bras sorafénib.
g Valeur unilatérale de p issue d’un test de Cochran-Mantel-Haenszel stratifié selon l’indice de performance ECOG et le traitement antérieur.
h Valeur unilatérale de p issue d’un test du log-rank stratifié selon l’indice de performance ECOG.
i Valeur unilatérale de p issue d’un test de Cochran-Mantel-Haenszel stratifié selon l’indice de performance ECOG.
Figure 1. Courbe de Kaplan-Meier de la survie sans progression dans la population globale évaluée par un comité indépendant
IC à 95 % Axitinib
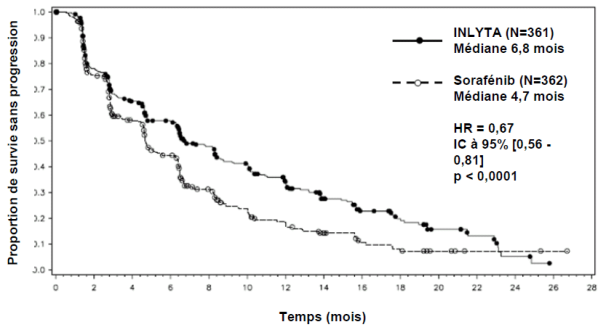
Figure 2. Courbe de Kaplan-Meier de la survie sans progression dans le sous-groupe de traitement antérieur à base de sunitinib évaluée par un comité indépendant
IC à 95 % = Axitinib
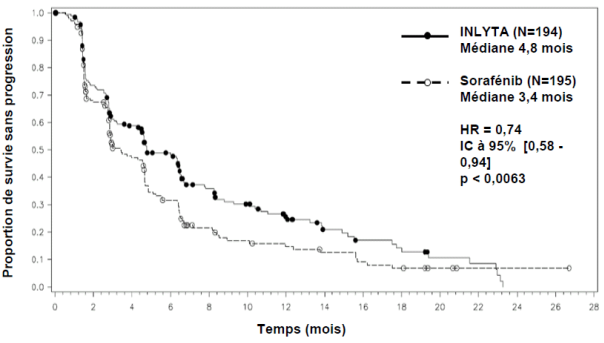
Figure 3. Courbe de Kaplan-Meier de la survie sans progression dans le sous-groupe de traitement antérieur à base de cytokine évaluée par un comité indépendant
IC à 95 % Axitinib
Population pédiatrique
L'Agence européenne des médicaments a accordé une dérogation à l'obligation de soumettre les résultats d’études réalisées avec l’axitinib dans tous les sous-groupes de la population pédiatrique pour le traitement du cancer du rein et du bassinet (à l'exclusion du néphroblastome, de la néphroblastomatose, du sarcome à cellules claires, du néphrome mésoblastique, du carcinome médullaire du rein et de la tumeur rhabdoïde du rein) (voir rubrique 4.2 pour les informations concernant l’usage pédiatrique).
5.2. Propriétés pharmacocinétiques
Après administration orale de comprimés d’axitinib, la biodisponibilité absolue moyenne a été de 58 % par rapport à une administration intraveineuse. La demi-vie plasmatique de l’axitinib est de 2,5 à 6,1 heures. L’administration d’axitinib à la dose de 5 mg deux fois par jour n’a pas entraîné un doublement de l’accumulation par rapport à l’administration d’une dose unique. En raison de la courte demi-vie de l’axitinib, l’état d’équilibre est attendu 2 à 3 jours après la dose initiale.
Absorption et distribution
Les concentrations plasmatiques maximales (Cmax) de l’axitinib sont généralement atteintes dans les 4 heures suivant l’administration orale d’axitinib, avec un Tmax médian allant de 2,5 à 4,1 heures. L’administration d’axitinib au cours d’un repas modérément riche en graisses a entraîné une diminution de l’exposition de 10 % par rapport à l’administration après un jeûne nocturne. Un repas riche en graisses et en calories a entraîné une augmentation de l’exposition de 19 % par rapport à l’administration après un jeûne nocturne. L’axitinib peut être administré au cours ou en dehors des repas (voir rubrique 4.2).
La Cmax et l’ASC moyennes ont augmenté de façon proportionnelle à la dose d’axitinib dans l’intervalle posologique de 5 à 10 mg.
La liaison in vitro de l’axitinib aux protéines plasmatiques humaines est supérieure à 99 %, avec une liaison préférentielle à l’albumine et une liaison modérée à l’α1-glycoprotéine acide. A la dose de 5 mg deux fois par jour administrée en période postprandiale, les moyennes géométriques des concentrations plasmatiques maximales et de l’ASC sur 24 heures ont été respectivement de 27,8 ng/mL et de 265 ng.h/mL chez des patients atteints de RCC avancé. Les moyennes géométriques de la clairance orale et du volume de distribution apparent ont été respectivement de 38 L/h et de 160 L.
Biotransformation et élimination
L’axitinib est principalement métabolisé dans le foie par le CYP3A4/5 et, dans une moindre mesure, par les CYP1A2, CYP2C19 et UGT1A1.
Après l’administration orale d’une dose de 5 mg d’axitinib radiomarqué, 30 à 60 % de la radioactivité ont été retrouvés dans les fèces et 23 % dans les urines. L’axitinib sous forme inchangée, représentant 12 % de la dose, a été le principal composant identifié dans les fèces. L’axitinib sous forme inchangée n’a pas été retrouvé dans les urines ; les métabolites acide carboxylique et sulfoxyde ont représenté la majorité de la radioactivité dans les urines. Dans le plasma, le métabolite N-glucuronide a représenté la fraction radioactive prédominante (50 % de la radioactivité circulante), et l’axitinib sous forme inchangée et le métabolite sulfoxyde ont représenté chacun approximativement 20 % de la radioactivité circulante.
La puissance inhibitrice in vitro des métabolites sulfoxyde et N-glucuronide sur le VEGFR-2 a été respectivement environ 400 et 8 000 fois inférieure à celle de l’axitinib.
Populations particulières
Personnes âgées, sexe et origine ethnique
Des analyses de pharmacocinétique de population menées chez des patients atteints de cancer avancé (notamment de RCC avancé) et chez des volontaires sains ont indiqué l’absence d’effet cliniquement pertinent de l’âge, du sexe, du poids corporel, de l’origine ethnique, de la fonction rénale, du génotype d’UGT1A1 ou du génotype de CYP2C19.
Population pédiatrique
L’axitinib n’a pas été étudié chez des patients âgés de < 18 ans.
Insuffisants hépatiques
Des données in vitro et in vivo indiquent que l’axitinib est principalement métabolisé par le foie.
Par rapport à des sujets présentant une fonction hépatique normale, l'exposition systémique après prise unique d’axitinib a été similaire chez les sujets présentant une insuffisance hépatique légère (classe A de Child-Pugh), et supérieure (environ deux fois supérieure) chez les sujets présentant une insuffisance hépatique modérée (classe B de Child-Pugh). L’axitinib n’a pas été étudié chez les sujets présentant une insuffisance hépatique sévère (classe C de Child-Pugh) et ne doit pas être administré dans cette population (voir rubrique 4.2 pour les recommandations relatives aux ajustements posologiques).
Insuffisants rénaux
L’axitinib sous forme inchangée n’a pas été détecté dans les urines.
L’axitinib n’a pas été étudié chez les sujets présentant une insuffisance rénale. Au cours des études cliniques portant sur l’axitinib dans le traitement de patients atteints de RCC, les patients présentant une créatinémie > 1,5 fois la LSN ou une clairance de la créatinine calculée < 60 mL/min ont été exclus. Les analyses de pharmacocinétique de population ont révélé que la clairance de l’axitinib n’était pas modifiée chez les sujets présentant une insuffisance rénale et par conséquent, aucun ajustement posologique de l’axitinib n’est nécessaire.
5.3. Données de sécurité préclinique
Toxicité de doses répétées
Les principales toxicités observées chez les souris et les chiens suite à l’administration de doses répétées allant jusqu’à 9 mois ont concerné les systèmes gastro-intestinal, hématopoïétique, reproducteur, squelettique et dentaire. La dose sans effet nocif observé (NOAEL, No Observed Adverse Effect Levels) a été approximativement équivalente ou inférieure à l’exposition attendue chez l’être humain à la dose initiale recommandée en clinique (sur la base des valeurs de l’ASC).
Carcinogénicité
Aucune étude de carcinogénicité n’a été menée avec l’axitinib.
Génotoxicité
L’axitinib n’a été ni mutagène ni clastogène lors de tests conventionnels de génotoxicité in vitro. Une augmentation significative de la polyploïdie a été observée in vitro à des concentrations supérieures à 0,22 μg/mL, et une augmentation des érythrocytes polychromatiques micronucléés a été observée in vivo avec une dose sans effet observé (NOEL) correspondant à 69 fois l’exposition attendue chez l’être humain. Les résultats de génotoxicité ne sont pas considérés comme cliniquement pertinents aux niveaux d’exposition observés chez l’être humain.
Toxicité sur la reproduction
Les effets liés à l’axitinib sur les testicules et l’épididyme ont inclus une diminution du poids, une atrophie ou une dégénérescence des organes, une diminution du nombre des cellules germinales, une hypospermie ou des spermatozoïdes de morphologie anormale et une réduction du nombre et de la densité des spermatozoïdes. Ces effets ont été observés chez la souris à des niveaux d’exposition environ 12 fois supérieurs à l'exposition attendue chez l’être humain et chez le chien à des niveaux inférieurs à celle-ci. Aucun effet sur l’accouplement ou la fertilité n’a été constaté chez la souris mâle à des niveaux d’exposition correspondant à environ 57 fois l’exposition attendue chez l’être humain. Les effets observés chez les femelles ont inclus des signes de retard de la maturité sexuelle, une réduction ou une absence des corps jaunes, une diminution du poids de l’utérus et une atrophie utérine à des expositions approximativement équivalentes à l’exposition attendue chez l’être humain. Une réduction de la fertilité et une diminution de la viabilité embryonnaire ont été observées chez les souris femelles à toutes les doses testées, avec des niveaux d’exposition à la plus faible dose correspondant à environ 10 fois l’exposition attendue chez l’être humain.
Des souris gravides exposées à l’axitinib ont révélé une augmentation de l’apparition des malformations à type de fente palatine et de variations de l’ossification du squelette, dont un retard d’ossification, à des niveaux d’exposition inférieurs à l’exposition attendue chez l’être humain. Aucune étude de la toxicité sur le développement périnatal et postnatal n’a été menée.
Observations toxicologiques chez des animaux juvéniles
Une dysplasie physaire réversible a été observée chez la souris et le chien lors de traitements d’au moins 1 mois par axitinib à des niveaux d’exposition environ six fois plus élevés que l’exposition attendue chez l’être humain. Des caries dentaires partiellement réversibles ont été observées chez des souris traitées pendant plus de 1 mois à des niveaux d’exposition similaires à l’exposition attendue chez l’être humain. D’autres toxicités potentiellement préoccupantes pour les patients pédiatriques n’ont pas été évaluées chez des animaux juvéniles.
Cellulose microcristalline, lactose monohydraté, croscarmellose sodique, stéarate de magnésium.
Pelliculage du comprimé
Hypromellose, dioxyde de titane (E 171), lactose monohydraté, triacétine (E1518), oxyde de fer rouge (E 172).
3 ans
6.4. Précautions particulières de conservation
6.5. Nature et contenu de l'emballage extérieur
Toutes les présentations peuvent ne pas être commercialisées.
6.6. Précautions particulières d’élimination et de manipulation
Tout médicament non utilisé ou déchet doit être éliminé conformément à la réglementation en vigueur.
7. TITULAIRE DE L’AUTORISATION DE MISE SUR LE MARCHE
SWENSWEG 5
2031 GA HAARLEM
PAYS-BAS
8. NUMERO(S) D’AUTORISATION DE MISE SUR LE MARCHE
· 34009 302 982 8 2 : 56 comprimés sous plaquettes (OPA/Aluminium/PVC/Aluminium).
· 34009 302 982 9 9 : 56 x 1 comprimés sous plaquettes prédécoupées unitaires (OPA/Aluminium/ PVC/Aluminium).
· 34009 302 983 0 5 : 60 comprimés sous plaquettes (OPA/Aluminium/PVC/Aluminium).
9. DATE DE PREMIERE AUTORISATION/DE RENOUVELLEMENT DE L’AUTORISATION
[à compléter ultérieurement par le titulaire]
10. DATE DE MISE A JOUR DU TEXTE
[à compléter ultérieurement par le titulaire]
Sans objet.
12. INSTRUCTIONS POUR LA PREPARATION DES RADIOPHARMACEUTIQUES
Médicament soumis à prescription hospitalière.
Prescription réservée aux spécialistes en oncologie ou aux médecins compétents en cancérologie.
Médicament nécessitant une surveillance particulière pendant le traitement.
ANSM - Mis à jour le : 02/07/2024
AXITINIB TEVA 1 mg, comprimé pelliculé
Axitinib
· Gardez cette notice. Vous pourriez avoir besoin de la relire.
· Si vous avez d’autres questions, interrogez votre médecin, votre pharmacien ou votre infirmier/ère.
· Ce médicament vous a été personnellement prescrit. Ne le donnez pas à d’autres personnes. Il pourrait leur être nocif, même si les signes de leur maladie sont identiques aux vôtres.
· Si vous ressentez un quelconque effet indésirable, parlez-en à votre médecin, votre pharmacien ou votre infirmier/ère. Ceci s’applique aussi à tout effet indésirable qui ne serait pas mentionné dans cette notice. Voir rubrique 4.
1. Qu'est-ce que AXITINIB TEVA 1 mg, comprimé pelliculé et dans quels cas est-il utilisé ?
2. Quelles sont les informations à connaître avant de prendre AXITINIB TEVA 1 mg, comprimé pelliculé ?
3. Comment prendre AXITINIB TEVA 1 mg, comprimé pelliculé ?
4. Quels sont les effets indésirables éventuels ?
5. Comment conserver AXITINIB TEVA 1 mg, comprimé pelliculé ?
6. Contenu de l’emballage et autres informations.
1. QU’EST-CE QUE AXITINIB TEVA 1 mg, comprimé pelliculé ET DANS QUELS CAS EST-IL UTILISE ?
Classe pharmacothérapeutique : Antinéoplasiques, inhibiteurs de protéine kinase - code ATC : L01EK01
AXITINIB TEVA est un médicament contenant la substance active axitinib. L’axitinib réduit l’apport de sang à la tumeur et ralentit le développement du cancer.
AXITINIB TEVA est indiqué dans le traitement du cancer du rein avancé (carcinome rénal avancé) chez les adultes, quand un autre médicament (appelé sunitinib ou cytokine) n’empêche plus la progression de la maladie.
Si vous avez des questions sur la façon dont ce médicament agit ou si vous souhaitez savoir pourquoi ce médicament vous a été prescrit, adressez-vous à votre médecin.
2. QUELLES SONT LES INFORMATIONS A CONNAITRE AVANT DE PRENDRE AXITINIB TEVA 1 mg, comprimé pelliculé ?
Ne prenez jamais AXITINIB TEVA 1 mg, comprimé pelliculé :
Si vous êtes allergique à l’axitinib ou à l’un des autres composants contenus dans ce médicament, mentionnés dans la rubrique 6. Si vous pensez que vous pourriez être allergique, demandez l’avis de votre médecin.
Avertissements et précautions
Adressez-vous à votre médecin, votre pharmacien ou votre infirmier/ère avant de prendre AXITINIB TEVA
· Si votre pression artérielle est élevée.
AXITINIB TEVA peut augmenter votre pression artérielle. Il est important de contrôler votre pression artérielle avant de prendre ce médicament, et à intervalles réguliers tout au long du traitement. Si votre pression artérielle est élevée (hypertension), vous pouvez être traité(e) par des médicaments destinés à la faire baisser. Votre médecin doit vérifier que votre pression artérielle est sous contrôle avant que vous ne commenciez votre traitement par AXITINIB TEVA et au cours de celui-ci.
· Si vous avez des problèmes de thyroïde.
AXITINIB TEVA peut provoquer des problèmes de thyroïde. Informez votre médecin si vous vous sentez fatigué(e) plus facilement, si vous avez généralement plus froid que d’autres personnes ou si votre voix devient plus grave pendant la prise de ce médicament. Votre fonction thyroïdienne doit être contrôlée avant de commencer votre traitement par AXITINIB TEVA et régulièrement pendant votre traitement. Si votre thyroïde ne produit pas suffisamment d’hormone avant ou pendant votre traitement par ce médicament, vous devez être traité(e) par une hormone thyroïdienne de substitution.
· Si vous avez récemment eu un problème de caillots sanguins dans vos veines et dans vos artères (types de vaisseaux sanguins), tels qu’un accident vasculaire cérébral, une crise cardiaque, une embolie ou une thrombose.
Demandez immédiatement une aide médicale d’urgence et contactez votre médecin si vous présentez des symptômes tels qu’une douleur ou une oppression à la poitrine, des douleurs dans les bras, le dos, le cou ou la mâchoire, un essoufflement, un engourdissement ou une faiblesse d’un côté du corps, des troubles de la parole, des maux de tête, des troubles de la vision ou des sensations vertigineuses au cours de votre traitement par ce médicament.
· Si vous présentez des saignements.
AXITINIB TEVA peut aggraver votre risque de saignement. Informez votre médecin si vous saignez ou si vous avez une toux accompagnée de sang ou de crachat sanglant au cours de votre traitement par ce médicament.
· Si vous souffrez ou avez souffert d’un anévrisme (élargissement et affaiblissement de la paroi d’un vaisseau sanguin) ou d’une déchirure dans la paroi d’un vaisseau sanguin.
· Si, au cours de votre traitement par ce médicament, vous souffrez de maux de ventre ou d’estomac intenses qui ne disparaissent pas.
AXITINIB TEVA peut augmenter le risque de perforation de l’intestin ou de l’estomac ou de formation de fistule (communication anormale [conduit] entre une cavité du corps et une autre cavité du corps ou de la peau). Informez votre médecin si vous souffrez de maux de ventre intenses durant votre traitement par ce médicament.
· Si vous devez subir une intervention chirurgicale ou si vous avez une plaie qui n'a pas cicatrisé.
Votre médecin doit arrêter AXITINIB TEVA au moins 24 heures avant votre intervention chirurgicale car ce médicament peut affecter la cicatrisation d’une plaie. Votre traitement par ce médicament devra être repris quand votre plaie aura cicatrisé correctement.
· Si, au cours de votre traitement par ce médicament, vous souffrez de maux de tête, de confusion, de convulsions (crises d’épilepsie) ou de troubles de la vision avec ou sans pression artérielle élevée.
Demandez immédiatement une aide médicale d’urgence et contactez votre médecin. Ces troubles pourraient être dus à un effet indésirable neurologique rare appelé syndrome d’encéphalopathie postérieure réversible.
· Si vous avez des problèmes de foie.
Votre médecin doit vous prescrire des analyses de sang pour vérifier le fonctionnement de votre foie avant et pendant votre traitement par AXITINIB TEVA.
· Si, au cours de votre traitement par ce médicament, vous souffrez d’une fatigue excessive, d’un gonflement de l’abdomen, des jambes ou des chevilles, d’un essoufflement ou si les veines de votre cou sont saillantes.
AXITINIB TEVA peut augmenter le risque de développer des événements d’insuffisance cardiaque. Votre médecin doit surveiller l’apparition de signes ou de symptômes d’événements d’insuffisance cardiaque de façon régulière et tout au long du traitement par axitinib.
Utilisation chez les enfants et les adolescents
AXITINIB TEVA n’est pas recommandé chez les personnes âgées de moins de 18 ans. Ce médicament n’a pas été étudié chez les enfants et les adolescents.
Autres médicaments et AXITINIB TEVA 1 mg, comprimé pelliculé
Certains médicaments peuvent affecter AXITINIB TEVA ou être affectés par AXITINIB TEVA. Veuillez informer votre médecin, votre pharmacien ou votre infirmier/ère de tous les médicaments que vous avez récemment pris, que vous prenez actuellement ou que vous pourriez prendre, y compris des médicaments obtenus sans ordonnance, des vitamines et des médicaments à base de plantes. Les médicaments mentionnés dans cette notice ne sont pas les seuls qui pourraient interagir avec AXITINIB TEVA.
Les médicaments suivants peuvent augmenter le risque d’effets indésirables avec AXITINIB TEVA :
· kétoconazole ou itraconazole, utilisés pour le traitement de mycoses ;
· clarithromycine, érythromycine ou télithromycine, des antibiotiques utilisés pour le traitement d’infections bactériennes ;
· atazanavir, indinavir, nelfinavir, ritonavir ou saquinavir, utilisés pour le traitement de l'infection par le VIH/sida ;
· néfazodone, utilisée pour le traitement de la dépression.
Les médicaments suivants peuvent réduire l’efficacité d’AXITINIB TEVA :
· rifampicine, rifabutine ou rifapentine, utilisées pour le traitement de la tuberculose (TB) ;
· dexaméthasone, un médicament corticoïde prescrit pour de nombreuses maladies différentes, dont certaines graves ;
· phénytoïne, carbamazépine ou phénobarbital, antiépileptiques utilisés pour la prévention de convulsions ou crises d’épilepsie ;
· millepertuis (Hypericum perforatum), une plante utilisée pour le traitement de la dépression.
Vous ne devez pas prendre ces médicaments pendant votre traitement par AXITINIB TEVA. Informez votre médecin, votre pharmacien ou votre infirmier/ère si vous prenez l’un d'entre eux. Votre médecin pourra modifier la dose de ces médicaments ou celle d’AXITINIB TEVA, ou vous prescrire un médicament différent.
AXITINIB TEVA peut augmenter les effets indésirables de la théophylline, utilisée pour le traitement de l’asthme et d’autres maladies des poumons.
AXITINIB TEVA 1 mg, comprimé pelliculé avec des aliments et boissons
Ne prenez pas ce médicament avec du pamplemousse ou du jus de pamplemousse, car cela peut augmenter le risque d'effets indésirables.
· Si vous êtes enceinte ou que vous allaitez, si vous pensez être enceinte ou planifiez une grossesse, demandez conseil à votre médecin, pharmacien ou infirmier/ère avant de prendre ce médicament.
· AXITINIB TEVA pourrait être nocif pour l’enfant à naître ou le nourrisson allaité au sein.
· Ne prenez pas ce médicament si vous êtes enceinte. Si vous êtes enceinte ou si vous êtes susceptible de l’être, parlez-en à votre médecin avant de prendre ce médicament.
· Utilisez une méthode de contraception fiable pendant votre traitement par AXITINIB TEVA et pendant 1 semaine après la dernière dose de ce médicament afin d’éviter une grossesse.
· N’allaitez pas pendant le traitement par AXITINIB TEVA. Si vous allaitez, votre médecin vous indiquera si vous devez arrêter l’allaitement ou le traitement par AXITINIB TEVA.
Conduite de véhicules et utilisation de machines
Si vous ressentez des sensations vertigineuses et/ou une fatigue pendant votre traitement par AXITINIB TEVA, soyez très prudent(e) en conduisant ou en utilisant des machines.
AXITINIB TEVA 1 mg, comprimé pelliculé contient du lactose
Si votre médecin vous a informé(e) d'une intolérance à certains sucres, contactez-le avant de prendre ce médicament.
AXITINIB TEVA 1 mg, comprimé pelliculé contient du sodium
Ce médicament contient moins de 1 mmol (23 mg) de sodium par comprimé pelliculé, c’est-à-dire qu’il est essentiellement « sans sodium ».
3. COMMENT PRENDRE AXITINIB TEVA 1 mg, comprimé pelliculé ?
La dose recommandée est de 5 mg deux fois par jour. Votre médecin pourra ensuite augmenter ou diminuer votre dose en fonction de votre tolérance au traitement par AXITINIB TEVA.
Avalez les comprimés entiers avec de l’eau, au cours ou en dehors des repas. Prenez les doses d’AXITINIB TEVA environ toutes les 12 heures.
Utilisation chez les enfants et les adolescents
AXITINIB TEVA n'est pas recommandé chez les personnes âgées de moins de 18 ans. Ce médicament n'a pas été étudié chez les enfants et les adolescents.
Si vous avez pris plus d’AXITINIB TEVA 1 mg, comprimé pelliculé que vous n’auriez dû
Si vous avez pris accidentellement trop de comprimés ou une dose plus élevée que vous n’auriez dû, demandez immédiatement conseil à un médecin. Si possible, montrez la boîte ou cette notice au médecin. Vous pourriez avoir besoin de soins médicaux.
Si vous oubliez de prendre AXITINIB TEVA 1 mg, comprimé pelliculé
Si vous vomissez en prenant AXITINIB TEVA 1 mg, comprimé pelliculé
Si vous vomissez, ne prenez pas de dose additionnelle. Prenez la dose prescrite suivante au moment habituel.
Si vous arrêtez de prendre AXITINIB TEVA 1 mg, comprimé pelliculé
Si vous ne pouvez plus prendre ce médicament comme votre médecin vous l’a prescrit, ou si vous pensez que vous n’en avez plus besoin, contactez immédiatement votre médecin.
Si vous avez d’autres questions sur l’utilisation de ce médicament, demandez plus d’informations à votre médecin, à votre pharmacien ou à votre infirmier/ère.
4. QUELS SONT LES EFFETS INDESIRABLES EVENTUELS ?
Certains effets indésirables peuvent être graves. Vous devez immédiatement contacter votre médecin si vous présentez n’importe lequel des effets indésirables graves suivants (voir également rubrique 2 : « Quelles sont les informations à connaître avant de prendre AXITINIB TEVA 1 mg, comprimé pelliculé ? ») :
· Evénements d’insuffisance cardiaque. Avertissez votre médecin si vous ressentez une fatigue excessive, un gonflement de l’abdomen, des jambes ou des chevilles, un essoufflement ou si les veines de votre cou sont saillantes.
· Caillots sanguins dans vos veines et dans vos artères (types de vaisseaux sanguins), tels qu’un accident vasculaire cérébral, une crise cardiaque, une embolie ou une thrombose. Demandez immédiatement une aide médicale d’urgence ou contactez votre médecin si vous présentez des symptômes tels qu’une douleur ou une oppression à la poitrine, des douleurs dans les bras, le dos, le cou ou la mâchoire, un essoufflement, un engourdissement ou une faiblesse d’un côté du corps, des troubles de la parole, des maux de tête, des troubles de la vision ou des sensations vertigineuses.
· Hémorragie. Avertissez immédiatement votre médecin si vous présentez l’un des symptômes suivants ou un problème de saignement grave au cours de votre traitement par AXITINIB TEVA : selles noires, toux accompagnée de sang ou de crachat sanglant ou modification de votre état mental.
· Perforation de l’intestin ou de l’estomac ou formation de fistule (communication anormale [conduit] entre une cavité du corps et une autre cavité du corps ou de la peau). Avertissez votre médecin si vous avez des maux de ventre sévères.
· Elévation sévère de la pression artérielle (crise hypertensive). Avertissez votre médecin si vous avez une pression artérielle très élevée ou si vous ressentez des maux de tête sévères ou une douleur sévère dans la poitrine.
· Œdème réversible du cerveau (syndrome d’encéphalopathie postérieure réversible). Demandez immédiatement une aide médicale d’urgence et contactez votre médecin si vous présentez des symptômes tels que maux de tête, confusion, convulsions (crises épileptiques) ou troubles de la vision avec ou sans pression artérielle élevée.
Les autres effets indésirables d’AXITINIB TEVA peuvent inclure :
Très fréquents : pouvant affecter plus de 1 personne sur 10
· Pression artérielle élevée ou augmentation de la pression artérielle
· Diarrhée, nausées ou vomissements, douleur au niveau de l’estomac, indigestion, douleur au niveau de la bouche, de la langue ou de la gorge, constipation
· Essoufflement, toux, enrouement
· Manque d’énergie, sensation de faiblesse ou de fatigue
· Diminution de la fonction thyroïdienne (pouvant être révélée par des analyses de sang)
· Rougeur et gonflement des paumes des mains et des plantes des pieds (syndrome main-pied), éruption cutanée, sécheresse de la peau
· Douleurs dans les articulations, douleurs dans les mains ou les pieds
· Perte d’appétit
· Protéines dans les urines (pouvant être révélées par des analyses d’urine)
· Perte de poids
· Maux de tête, troubles du goût ou perte du goût
Fréquents : pouvant affecter jusqu’à 1 personne sur 10
· Déshydratation (perte de liquides corporels)
· Insuffisance rénale
· Flatulences (vents), hémorroïdes, saignement des gencives, saignement du rectum, sensation de brûlure ou de picotement dans la bouche
· Hyperactivité de la glande thyroïdienne (pouvant être révélée par des analyses de sang)
· Maux de gorge ou irritation du nez et de la gorge
· Douleurs dans les muscles
· Saignement du nez
· Démangeaison de la peau, rougeur de la peau, perte de cheveux
· Bourdonnement/bruits dans les oreilles (acouphènes)
· Diminution du nombre des globules rouges sanguins (pouvant être révélée par des analyses de sang)
· Diminution du nombre de plaquettes sanguines (cellules qui aident le sang à coaguler) (pouvant être révélée par des analyses de sang)
· Présence de globules rouges dans les urines (pouvant être révélée par des analyses d’urine)
· Modifications des concentrations de différentes substances chimiques et enzymes dans le sang (pouvant être révélées par des analyses de sang)
· Augmentation du nombre des globules rouges sanguins (pouvant être révélée par des analyses de sang)
· Gonflement de l’abdomen, des jambes ou des chevilles, veines du cou saillantes, fatigue excessive, essoufflement (signes d'événements d’insuffisance cardiaque)
· Fistule (communication anormale [conduit] entre une cavité du corps et une autre cavité du corps ou de la peau)
· Sensations vertigineuses
· Inflammation de la vésicule biliaire
Peu fréquents : pouvant affecter jusqu’à 1 personne sur 100
· Diminution du nombre des globules blancs sanguins (pouvant être révélée par des analyses de sang)
Fréquence indéterminée : ne peut être estimée sur la base des données disponibles
· Elargissement et affaiblissement de la paroi d’un vaisseau sanguin ou déchirure dans la paroi d’un vaisseau sanguin (anévrismes et dissections artérielles).
Déclaration des effets secondaires
Si vous ressentez un quelconque effet indésirable, parlez-en à votre médecin, à votre pharmacien ou à votre infirmier/ère. Ceci s’applique aussi à tout effet indésirable qui ne serait pas mentionné dans cette notice. Vous pouvez également déclarer les effets indésirables directement via le système national de déclaration : Agence nationale de sécurité du médicament et des produits de santé (ANSM) et réseau des Centres Régionaux de Pharmacovigilance - Site internet: https://signalement.social-sante.gouv.fr/
En signalant les effets indésirables, vous contribuez à fournir davantage d’informations sur la sécurité du médicament.
5. COMMENT CONSERVER AXITINIB TEVA 1 mg, comprimé pelliculé ?
Tenir ce médicament hors de la vue et de la portée des enfants.
N’utilisez pas ce médicament après la date de péremption indiquée sur la boîte et la feuille en aluminium de la plaquette ou sur le flacon après « EXP ». La date de péremption fait référence au dernier jour de ce mois.
A conserver dans l’emballage d'origine, à l’abri de l’humidité. Ce médicament ne nécessite pas de précautions particulières de conservation concernant la température.
N’utilisez pas un emballage qui est endommagé ou présente des signes de détérioration.
Ne jetez aucun médicament au tout-à-l’égout ou avec les ordures ménagères. Demandez à votre pharmacien d’éliminer les médicaments que vous n’utilisez plus. Ces mesures contribueront à protéger l’environnement.
6. CONTENU DE L’EMBALLAGE ET AUTRES INFORMATIONS
Ce que contient AXITINIB TEVA 1 mg, comprimé pelliculé
· La substance active est :
Axitinib.............................................................................................................................. 1 mg
Pour un comprimé pelliculé.
· Les autres composants sont :
Cellulose microcristalline, lactose monohydraté (voir rubrique 2 « AXITINIB TEVA 1 mg, comprimé pelliculé contient du lactose »), croscarmellose sodique (voir rubrique 2 « AXITINIB TEVA 1 mg, comprimé pelliculé contient du sodium »), stéarate de magnésium, hypromellose, dioxyde de titane (E171), triacétine (E1518), oxyde de fer rouge (E 172).
Qu’est-ce que AXITINIB TEVA 1 mg, comprimé pelliculé et contenu de l’emballage extérieur
Les comprimés pelliculés d’AXITINIB TEVA 1 mg sont de couleur rouge, ronds, biconvexes, mesurant environ 6 mm de diamètre, et comportant la mention « A7TI » gravée en creux sur une face et la mention « 1 » gravée en creux sur l’autre face.
Plaquettes et plaquettes prédécoupées unitaires en OPA/Aluminium/PVC/Aluminium (Aluminium/Aluminium) conditionnées dans des boîtes en carton. Présentations de 56, 60, 56 × 1 ou 120 × 1 comprimés pelliculés.
Toutes les présentations peuvent ne pas être commercialisées.
Titulaire de l’autorisation de mise sur le marché
SWENSWEG 5
2031 GA HAARLEM
PAYS-BAS
Exploitant de l’autorisation de mise sur le marché
TEVA SANTE
100-110 ESPLANADE DU GENERAL DE GAULLE
92931 PARIS LA DEFENSE CEDEX
c/ Castelló, 1
08830 - Sant Boi de Llobregat
Barcelona
Espagne
Synthon B.V.
Microweg 22
Nijmegen
6545 CM
PAYS-BAS
Balkanpharma-Dupnitsa AD
Ul Samokovsko Shose 3
2600 - Dupnitsa
BulgariE
Noms du médicament dans les Etats membres de l'Espace Economique Européen
Ce médicament est autorisé dans les Etats membres de l'Espace Economique Européen sous les noms suivants : Conformément à la réglementation en vigueur.
[À compléter ultérieurement par le titulaire]
La dernière date à laquelle cette notice a été révisée est :
[à compléter ultérieurement par le titulaire]
{mois AAAA}.
Des informations détaillées sur ce médicament sont disponibles sur le site Internet de l’ANSM (France).