Dernière mise à jour le 01/06/2026
DALBAVANCINE REIG JOFRE 500 mg, poudre pour solution à diluer pour perfusion
Indications thérapeutiques
DALBAVANCINE REIG JOFRE contient la substance active dalbavancine, qui est un antibiotique de la classe des glycopeptides.
DALBAVANCINE REIG JOFRE est utilisé chez les adultes et les enfants âgés de 3 mois et plus dans le traitement d’infections de la peau ou des tissus mous sous la peau.
DALBAVANCINE REIG JOFRE agit en tuant certaines bactéries pouvant être responsables d’infections graves. Il les tue en interférant avec la formation de la paroi des bactéries.
Si d’autres bactéries sont à l’origine de votre infection, votre médecin peut choisir de vous traiter avec d’autres antibiotiques, utilisés en association avec DALBAVANCINE REIG JOFRE.
Présentations
> 1 flacon en verre de 50 mL
Code CIP : 34009 551 013 7 6
Déclaration de commercialisation : 10/03/2026
Cette présentation est agréée aux collectivités
Service médical rendu (SMR)
Amélioration du service médical rendu (ASMR)
Ce médicament étant un générique, l'ASMR n'a pas été évalué par la commission de la transparence (CT), il est possible de se référer à la /aux spécialité(s) de référence du groupe générique auquel appartient ce médicament (cliquez ici pour aller à la rubrique des groupes génériques)
Autres informations
- Titulaire de l'autorisation : LABORATORIO REIG JOFRE SA
- Conditions de prescription et de délivrance :
- liste I
- prescription hospitalière
- Statut de l'autorisation : Valide
- Type de procédure : Procédure décentralisée
- Code CIS : 6 164 415 9
ANSM - Mis à jour le : 08/04/2024
DALBAVANCINE REIG JOFRE 500 mg, poudre pour solution à diluer pour perfusion
2. COMPOSITION QUALITATIVE ET QUANTITATIVE
Pour un flacon de poudre.
Après reconstitution, chaque mL de concentré contient 20 mg de dalbavancine.
La solution diluée pour perfusion doit avoir une concentration finale de 1 à 5 mg/mL de dalbavancine (voir rubrique 6.6).
Pour la liste complète des excipients, voir rubrique 6.1.
Poudre pour solution à diluer pour perfusion (poudre à diluer).
Poudre blanche ou blanc cassé à jaune pâle.
4.1. Indications thérapeutiques
Il convient de tenir compte des recommandations officielles concernant l’utilisation appropriée des antibactériens.
4.2. Posologie et mode d'administration
Adultes
La dose de dalbavancine recommandée est de 1 500 mg administrés, soit en une seule perfusion de 1 500 mg, soit en une perfusion de 1 000 mg suivie d’une perfusion de 500 mg une semaine plus tard (voir rubriques 5.1 et 5.2).
Enfants et adolescents âgés de 6 ans à moins de 18 ans
La dose de dalbavancine recommandée est une dose unique de 18 mg/kg (1 500 mg au maximum).
Nourrissons et enfants âgés de 3 mois à moins de 6 ans
La dose de dalbavancine recommandée est une dose unique de 22,5 mg/kg (1 500 mg au maximum).
Populations particulières
Personnes âgées
Aucun ajustement de la dose n’est nécessaire (voir rubrique 5.2).
Insuffisance rénale
Aucun ajustement de dose n'est nécessaire chez les patients adultes et pédiatriques en insuffisance rénale légère à modérée (clairance de la créatinine ≥ 30 à 79 mL/min). Aucune adaptation de dose n’est nécessaire chez les patients adultes sous hémodialyse régulière (3 fois par semaine) et la dalbavancine peut être administrée sans tenir compte du moment où se déroule l’hémodialyse.
Chez les patients adultes en insuffisance rénale chronique, dont la clairance de la créatinine est < 30 mL/min et qui ne sont pas traités par hémodialyse régulière planifiée, la dose recommandée est réduite à, soit une seule perfusion de 1 000 mg, soit une perfusion de 750 mg, suivie d’une perfusion de 375 mg une semaine plus tard (voir rubrique 5.2).
On ne dispose pas de suffisamment d’informations pour recommander une adaptation de dosage chez les patients âgés de moins de 18 ans dont la clairance de la créatinine est inférieure à 30 mL/min/1,73 m2. Les informations disponibles actuellement sont présentées dans la rubrique 5.2, mais aucune recommandation posologique ne peut être formulée.
Insuffisance hépatique
Aucun ajustement de dose de la dalbavancine n’est recommandé chez les patients en insuffisance hépatique légère (Child-Pugh A). La dalbavancine doit être utilisée avec précaution chez les patients en insuffisance hépatique modérée ou sévère (Child-Pugh B et C), car aucune donnée disponible ne permet de déterminer un dosage approprié (voir rubrique 5.2).
Population pédiatrique
La sécurité et l’efficacité de la dalbavancine chez les enfants âgés de moins de 3 mois n’ont pas encore été établies. Les données actuellement disponibles sont décrites à la rubrique 5.2 mais aucune recommandation posologique ne peut être formulée.
Mode d’administration
Voie intraveineuse.
DALBAVANCINE REIG JOFRE doit être reconstitué, puis dilué avant d’être administré par perfusion intraveineuse pendant 30 minutes. Pour des instructions sur la reconstitution et la dilution du médicament avant administration, voir rubrique 6.6.
Hypersensibilité à la substance active ou à l’un des excipients mentionnés à la rubrique 6.1.
4.4. Mises en garde spéciales et précautions d'emploi
La dalbavancine doit être administrée avec précaution chez les patients présentant une hypersensibilité connue à d’autres glycopeptides, car une hypersensibilité croisée est possible. En cas de survenue d’une réaction allergique à la dalbavancine, il convient d’arrêter l’administration du médicament et d’instaurer un traitement adapté à la réaction allergique.
Diarrhée associée à Clostridioides (anciennement Clostridium) difficile
Des colites associées aux antibactériens et pseudomembraneuses ont été rapportées avec la quasi-totalité des antibactériens et leur sévérité peut varier d’une forme légère à la mise en jeu du pronostic vital. Par conséquent, il est important d’envisager ce diagnostic chez les patients présentant une diarrhée pendant ou après un traitement par dalbavancine (voir rubrique 4.8). Dans cette situation, l’arrêt du traitement par dalbavancine et l’utilisation de traitements symptomatiques, parallèlement à l’administration d’un traitement spécifique contre Clostridioides (anciennement Clostridium) difficile, doivent être envisagés. Les médicaments inhibant le péristaltisme sont contre-indiqués chez ces patients.
Réactions liées à la perfusion
DALBAVANCINE REIG JOFRE doit être administré par perfusion intraveineuse, pendant 30 minutes au total, afin de minimiser le risque de réactions liées à la perfusion. Les perfusions intraveineuses rapides d’antibiotiques glycopeptidiques peuvent provoquer des réactions, notamment des rougeurs de la partie supérieure du corps, une urticaire, un prurit ou une éruption cutanée. Arrêter ou ralentir la perfusion peut entraîner la disparition de ces réactions.
Insuffisance rénale
Les informations relatives à l’efficacité et la sécurité de la dalbavancine chez les patients présentant une clairance de la créatinine < 30 mL/min sont limitées. D’après des simulations, une adaptation de dose est nécessaire chez les patients adultes en insuffisance rénale chronique, dont la clairance de la créatinine est < 30 mL/min et qui ne sont pas traités par hémodialyse régulièrement (voir rubriques 4.2 et 5.2). On ne dispose pas de suffisamment d’informations pour recommander une adaptation de dosage chez les patients âgés de moins de 18 ans dont la clairance de la créatinine est inférieure à 30 mL/min/1,73 m2.
Infections mixtes
Pour les infections mixtes dont on suspecte qu’elles sont provoquées par des bactéries à Gram négatif, les patients doivent également être traités par un ou plusieurs antibactériens adaptés dirigés contre les bactéries à Gram négatif (voir rubrique 5.1).
Microorganismes non sensibles
L’utilisation d’antibiotiques peut favoriser la prolifération excessive de microorganismes non sensibles. Des mesures adaptées doivent être prises en cas de surinfection au cours du traitement.
Limites des données cliniques
Les données concernant la sécurité et l’efficacité de la dalbavancine administrée à plus de deux reprises (à une semaine d’intervalle) sont limitées. Dans le cadre des principaux essais portant sur les infections bactériennes aiguës de la peau et des tissus mous, les types d’infections traités étaient limités aux cellulites ou érysipèles, abcès et infections des plaies. Il n’y a pas d’expérience avec la dalbavancine dans le traitement de patients présentant une immunodépression sévère.
Excipients
Ce médicament contient moins de 1 mmol (23 mg) de sodium par dose, il est donc considéré comme « sans sodium ».
4.5. Interactions avec d'autres médicaments et autres formes d'interactions
Aucune étude d’interaction médicamenteuse clinique n’a été réalisée avec la dalbavancine.
Potentialité d’autres médicaments affectent la pharmacocinétique de la dalbavancine
La dalbavancine n’est pas métabolisée par les enzymes CYP in vitro ; par conséquent, il est peu probable que les inducteurs ou inhibiteurs des CYP aient une influence sur la pharmacocinétique de la dalbavancine.
On ignore si la dalbavancine est un substrat pour la captation hépatique et les transporteurs d’efflux. La coadministration avec des inhibiteurs de ces transporteurs peut augmenter l’exposition à la dalbavancine. Les inhibiteurs des transporteurs incluent notamment les inhibiteurs de protéase boostés, le vérapamil, la quinidine, l’itraconazole, la clarithromycine et la cyclosporine.
Potentialité de la dalbavancine affecte la pharmacocinétique d’autres médicaments
Il est attendu que le potentiel d’interaction de la dalbavancine avec les médicaments métabolisés par les isoenzymes CYP soit faible, car il ne s’agit ni d’un inhibiteur ni d’un inducteur des isoenzymes CYP in vitro. Aucune donnée n’indique que la dalbavancine serait un inhibiteur des isoenzymes CYP2C8.
On ignore si la dalbavancine est un inhibiteur des transporteurs. En cas d’association à la dalbavancine, une augmentation de l’exposition aux substrats de transporteurs sensibles à l’activité des transporteurs inhibés, tels que les statines et la digoxine, ne peut être exclue.
4.6. Fertilité, grossesse et allaitement
Grossesse
L’utilisation de DALBAVANCINE REIG JOFRE est déconseillée pendant la grossesse, sauf si le bénéfice attendu ne prévaut sur le risque potentiel pour le fœtus.
Allaitement
On ignore si la dalbavancine est excrétée dans le lait maternel humain. Cependant, la dalbavancine est excrétée dans le lait des rates qui allaitent et peut l’être dans le lait maternel humain. La dalbavancine n’est pas bien absorbée après administration par voie orale ; toutefois, la possibilité d’un impact sur la flore gastro-intestinale ou buccale d’un nourrisson allaité ne peut être exclue. Une décision doit être prise concernant la poursuite/ l’arrêt de l’allaitement ou la poursuite/ l’arrêt du traitement par DALBAVANCINE REIG JOFRE, en tenant compte des bénéfices de l’allaitement pour l’enfant et du traitement pour la femme.
Fertilité
Des études réalisées chez l’animal ont mis en évidence une réduction de la fertilité (voir rubrique 5.3). Le risque potentiel pour l’espèce humaine est inconnu.
4.7. Effets sur l'aptitude à conduire des véhicules et à utiliser des machines
DALBAVANCINE REIG JOFRE peut avoir une influence mineure sur l’aptitude à conduire des véhicules et à utiliser des machines, car des cas de vertiges ont été rapportés chez un nombre limité de patients (voir rubrique 4.8).
Résumé du profil de sécurité
Dans les études cliniques de phases 2/3, 2 473 patients adultes ont été traités par dalbavancine administrée, soit en une seule perfusion de 1 500 mg, soit en une perfusion de 1 000 mg suivie d’une perfusion de 500 mg une semaine plus tard. Les effets indésirables les plus fréquents survenant chez ≥ 1 % des patients traités par dalbavancine étaient les nausées (2,4 %), la diarrhée (1,9 %) et les céphalées (1,3 %), et ils étaient généralement de sévérité légère à modérée.
Tableau répertoriant les effets indésirables (Tableau 1)
Les effets indésirables suivants ont été identifiés dans le cadre d’essais cliniques de phase 2/3 menés avec la dalbavancine. Ils sont classés par système d’organe et par fréquence. Les catégories de fréquence sont définies selon les conventions suivantes : très fréquent (≥ 1/10), fréquent (≥ 1/100 à < 1/10), peu fréquent (≥ 1/1 000 à < 1/100) et rare (≥ 1/10 000 à < 1/1 000).
Tableau 1.
|
Classe de système d’organe |
Fréquent) |
Peu fréquent |
Rare |
|
Infections et infestations |
|
infection mycosique vulvovaginale, infection urinaire, infection fongique, colite associée à Clostridioides (anciennement Clostridium) difficile, candidose buccale |
|
|
Affections hématologiques et du système lymphatique |
|
anémie, thrombocytose, éosinophilie, leucopénie, neutropénie |
|
|
Affections du système immunitaire |
|
|
réaction anaphylactoïde |
|
Troubles du métabolisme et de la nutrition |
|
diminution de l’appétit |
|
|
Affections psychiatriques |
|
insomnie |
|
|
Affections du système nerveux |
céphalées |
dysgueusie, vertiges |
|
|
Affections vasculaires |
|
rougeurs, phlébite |
|
|
Affections respiratoires, thoraciques et médiastinales |
|
toux |
bronchospasmes |
|
Affections gastro-intestinales |
nausées, diarrhée |
constipation, douleurs abdominales, dyspepsie, gêne abdominale, vomissements |
|
|
Affections de la peau et du tissu sous-cutané |
|
prurit, urticaire, rash cutané |
|
|
Affections des organes de reproduction et du sein |
|
prurit vulvovaginal |
|
|
Troubles généraux et anomalies liées au site d’administration |
|
réactions liées à la perfusion |
|
|
Bilan biologique |
|
augmentation de lacticodéshydrogénase, augmentation de l’alanine aminotransférase, augmentation de l’aspartate aminotransférase, augmentation de l’acide urique, anomalie des tests hépatiques, augmentation des transaminases, augmentation des phosphatases alcalines, augmentation de la numération plaquettaire, augmentation de la température corporelle, augmentation des enzymes hépatiques, augmentation de la gamma glutamyl transférase |
|
Description de certains effets indésirables
Effets indésirables de classe
L’ototoxicité a été associée à l’utilisation de glycopeptides (vancomycine et teicoplanine) Les patients qui reçoivent un traitement concomitant avec un médicament ototoxique, comme un aminoside, peuvent être exposés à un risque accru.
Population pédiatrique
La sécurité de la dalbavancine a été évaluée dans une étude clinique de phase 3 qui incluait 168 patients pédiatriques depuis la naissance jusqu’à moins de 18 ans, présentant des infections bactériennes aiguës de la peau et des tissus mous traités par la dalbavancine (90 patients traités par une dose unique de dalbavancine et 78 autres, tous âgés de 3 mois et plus, traités par un schéma à deux doses de dalbavancine). Globalement, les résultats concernant la sécurité de la dalbavancine chez ces patients pédiatriques étaient similaires à ceux observés chez les adultes
Déclaration des effets indésirables suspectés
La déclaration des effets indésirables suspectés après autorisation du médicament est importante. Elle permet une surveillance continue du rapport bénéfice/risque du médicament. Les professionnels de santé déclarent tout effet indésirable suspecté via le système national de déclaration : Agence nationale de sécurité du médicament et des produits de santé (ANSM) et réseau des Centres Régionaux de Pharmacovigilance - Site internet : https://signalement.social-sante.gouv.fr.
Aucune information particulière n’est disponible à propos du traitement du surdosage par dalbavancine, car aucune toxicité dose-limitante n’a été observée dans les études cliniques réalisées. Dans des études de phase 1, des doses uniques allant jusqu’à 1 500 mg ou des doses cumulées allant jusqu’à 4 500 mg ont été administrées à des volontaires sains pendant un maximum de 8 semaines, sans signe de toxicité ni résultats biologiques représentant une préoccupation clinique. Dans des études de phase 3, des doses uniques allant jusqu’à 1 500 mg ont été administrées aux patients.
Le traitement du surdosage par dalbavancine devrait être symptomatique. Bien qu’aucune information ne soit disponible concernant en particulier l’utilisation de l’hémodialyse pour traiter un surdosage, il convient de souligner que dans l’étude de phase I menée chez des patients en insuffisance rénale, moins de 6 % de la dose de dalbavancine recommandée ont été éliminés après 3 heures d’hémodialyse.
5. PROPRIETES PHARMACOLOGIQUES
5.1. Propriétés pharmacodynamiques
Mécanisme d’action
La dalbavancine est un lipoglycopeptide bactéricide.
Son mécanisme d’action vis-à-vis des bactéries à Gram positif sensibles réside dans l’inhibition de la synthèse de la paroi bactérienne grâce une liaison au résidu terminal D-alanyl-D-alanine du peptide précurseur du peptidoglycane de la paroi cellulaire naissante, empêchant les liaisons covalentes (transpeptidation et transglycosylation) des sous-unités disaccharides, ce qui provoque la mort cellulaire bactérienne.
Mécanisme de résistance
Toutes les bactéries à Gram négatif sont naturellement résistantes à la dalbavancine.
La résistance à la dalbavancine du Staphylococcus spp.et de l’Enterococcus spp. est médiée par VanA, un génotype qui entraîne la modification du peptide cible dans la paroi cellulaire naissante. D’après des études in vitro, l’activité de la dalbavancine n’est pas affectée par d’autres gènes de résistance à la vancomycine.
Les concentrations minimales inhibitrices (CMI) de la dalbavancine sont plus élevées pour les staphylocoques de résistance intermédiaire à la vancomycine (SARIV) que pour les souches pleinement sensibles à la vancomycine. Si les isolats présentant des CMI de la dalbavancine plus élevées représentent des phénotypes stables et sont corrélés à la résistance à d’autres glycopeptides, il est probable que le mécanisme repose sur une augmentation du nombre de cibles glycopeptidiques dans le peptidoglycane naissant.
Aucune résistance croisée entre la dalbavancine et d’autres classes d’antibiotiques n’a été observée dans des études in vitro.
La résistance à la méticilline est sans effet sur l’activité de la dalbavancine.
Interactions avec d’autres antibactériens
Les études in vitro réalisées n’ont mis en évidence aucun antagonisme entre la dalbavancine et d’autres antibiotiques fréquemment utilisés (tels que céfépime, ceftazidime, ceftriaxone, imipénème, méropénème, amikacine, aztréonam, ciprofloxacine, pipéracilline / tazobactam et triméthoprime / sulfaméthoxazole), pour 12 espèces pathogènes à Gram négatif testées (voir rubrique 4.5).
Concentrations critiques
Les concentrations minimales inhibitrices (CMI) critiques déterminées par l’European Committee on Antimicrobial Susceptibility Testing (EUCAST) sont les suivantes :
· Staphylococcus spp : sensible ≤ 0,125 mg/L ; résistant > 0,125 mg/L ;
· Streptocoques bêta-hémolytiques des groupes A, B, C et G : sensible ≤ 0,125 mg/L ; résistant > 0,125 mg/L ;
· Streptocoques viridans (groupe Streptococcus anginosus uniquement) : sensible ≤ 0,125 mg/L ; résistant > 0,125 mg/L.
Rapport pharmacocinétique/pharmacodynamique (PK/PD)
L’activité bactéricide contre les staphylocoques in vitro est temps-dépendante à des concentrations sériques de dalbavancine comparables à celles obtenues avec la dose recommandée chez l’humain. Le rapport PK/PD in vivo de la dalbavancine vis-à-vis de S. aureus a été étudié avec un modèle d’infection chez des animaux neutropéniques. Cette étude a montré que l’activité antibactérienne de la dalbavancine semble particulièrement corrélée avec le rapport de l’aire sous la courbe des concentrations plasmatiques du médicament sous forme libre en fonction du temps à la concentration minimale inhibitrice (ASCl/CMI).
Efficacité clinique contre des bactéries pathogènes spécifiques
L’efficacité du médicament a été démontrée dans des études cliniques portant sur les bactéries pathogènes suivantes en cas d’infections bactériennes aiguës de la peau et des tissus mous sensibles in vitro à la dalbavancine :
· Staphylococcus aureus,
· Streptococcus pyogenes,
· Streptococcus agalactiae,
· Streptococcus dysgalactiae,
· Groupe Streptococcus anginosus (y compris S. anginosus, S. intermedius et S. constellatus),
Activité antibactérienne sur d’autres bactéries pathogènes d’intérêt
L’efficacité clinique contre les bactéries pathogènes suivantes n’a pas été établie, même si des études in vitro semblent indiquer qu’elles sont sensibles à la dalbavancine en l’absence de mécanismes de résistance acquis :
· Streptocoques du groupe G ;
· Clostridium perfringens ;
· Peptostreptococcus spp.
Population pédiatrique
La dalbavancine a été évaluée dans une étude clinique de phase 3, en ouvert, randomisée, contrôlée par un comparateur, conduite chez des patients pédiatriques depuis la naissance jusqu’à moins de 18 ans, présentant des infections bactériennes aiguës de la peau et des tissus mous. L’étude incluait 168 patients traités par la dalbavancine (90 patients traités par une dose unique de dalbavancine et 78 autres, tous âgés de 3 mois et plus, traités par un schéma à deux doses de dalbavancine) et 30 patients traités par un comparateur. L’objectif principal était d’évaluer la sécurité et la tolérance de dalbavancine et les objectifs secondaires incluaient l’évaluation de l’efficacité et de la pharmacocinétique. L’efficacité était un critère d’évaluation descriptif. Le taux de guérisons cliniques lors du test de guérison (population ITTm) était de 95,1 % (78/82) dans le groupe recevant une dose unique de dalbavancine, de 97,3 % (72/74) dans le groupe recevant deux doses de dalbavancine et de 100 % (30/30) dans le groupe recevant le comparateur.
L'Agence Européenne des Médicaments a différé l’obligation de soumettre les résultats d’études réalisées avec la dalbavancine dans le traitement des infections bactériennes aiguës de la peau et des tissus mous dans un ou plusieurs sous-groupes de la population pédiatrique (voir rubriques 4.2 et 5.2 pour les informations concernant l’usage pédiatrique).
5.2. Propriétés pharmacocinétiques
La pharmacocinétique de la dalbavancine a été caractérisée chez des sujets sains, des patients et des populations particulières. Après administration de doses uniques comprises entre 140 et 1 120 mg, l’exposition systémique à la dalbavancine est proportionnelle à la dose, ce qui indique la linéarité de la pharmacocinétique de la dalbavancine. Aucune accumulation de dalbavancine n’a été observée après plusieurs perfusions intraveineuses administrées une fois par semaine pendant un maximum de 8 semaines (1 000 mg au Jour 1, suivis d’un maximum de 7 doses hebdomadaires de 500 mg) à des adultes sains.
La demi-vie moyenne d’élimination terminale (t1/2) était de 372 heures (de 333 à 405). La pharmacocinétique de la dalbavancine peut être mieux définie avec un modèle à trois compartiments (phases de distribution α et β, suivies d’une phase d’élimination terminale). Ainsi, la demi-vie de distribution (t1/2β), qui correspond au profil concentrations-temps le plus cliniquement pertinent, s’étend de 5 à 7 jours, ce qui est cohérent avec une posologie hebdomadaire
Le tableau 2 ci-dessous répertorie les paramètres pharmacocinétiques estimés après administration intraveineuse des schémas à deux doses et à une dose de dalbavancine, respectivement.
Tableau 2. Paramètres pharmacocinétiques moyens (SD) de la dalbavancine pour les adultes, basés sur l’analyse PK de population1
|
Paramètre |
Schéma à deux doses2 |
Schéma à deux doses1 |
|
Cmax (mg/L) |
Jour 1: 281 (52) Jour 8: 141 (26) |
Jour 1: 411 (86)
|
|
AUC0-Jour14 (mgh/L) |
18100 (4600) |
20300 (5300) |
|
CL (L/h) |
0,048 (0,0086) |
0,049 (0,0096) |
|
1 Source: DAL-MS-01. 2 1 000 mg au Jour 1 + 500 mg au Jour 8; sujets de l’étude DUR001-303 avec échantillon PK évaluable. 3 1 500 mg; sujets de l’étude DUR001-303 avec échantillon PK évaluable. |
||
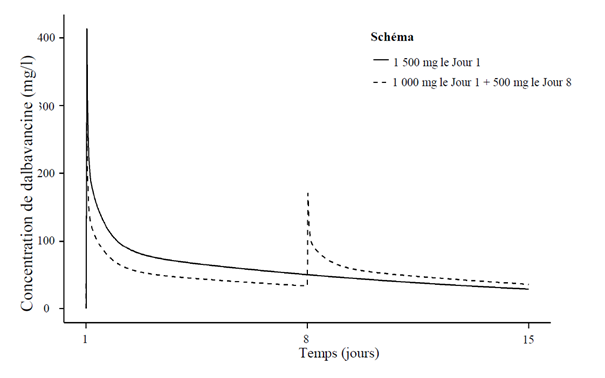
Le rapport temps-concentration plasmatique de dalbavancine après administration des schémas à deux doses et une dose, respectivement, est présenté en Figure 1.
Figure 1. Concentrations plasmatiques de dalbavancine en fonction du temps chez un patient adulte présentant une infection bactérienne aiguë de la peau et des tissus mous typique (simulation utilisant un modèle de pharmacocinétique de population) pour les schémas à une et deux doses.
Distribution
La clairance et le volume de distribution à l’état d’équilibre sont comparables chez les sujets sains et chez les patients présentant des infections. Le volume de distribution à l’état d’équilibre était similaire au volume de liquide extracellulaire. La dalbavancine se lie aux protéines plasmatiques humaines, principalement à l’albumine, de manière réversible. Le taux de liaison de la dalbavancine aux protéines plasmatiques est de 93 % et n’est pas modifié en fonction de la concentration médicamenteuse, de l’insuffisance rénale ou de l’insuffisance hépatique. Après administration intraveineuse d’une dose unique de 1 000 mg à des volontaires sains, l’ASC dans le liquide phlycténulaire (dalbavancine liée et non liée) était d’environ 60 % de l’ASC du plasma 7 jours après administration de la dose.
Biotransformation
Les métabolites n’ont pas été observés en quantités significatives dans le plasma humain. Les métabolites hydroxy-dalbavancine et mannosyl aglycone ont été détectés dans les urines (< 25 % de la dose administrée). Les voies métaboliques responsables de la formation de ces métabolites n’ont pas été identifiées ; cependant, en raison de la contribution relativement faible du métabolisme à l’élimination totale de la dalbavancine, aucune interaction médicamenteuse intervenant par inhibition ou induction du métabolisme de la dalbavancine n’est attendue. L’hydroxy-dalbavancine et le mannosyl aglycone montrent une activité antibactérienne considérablement moindre par rapport à celle de la dalbavancine.
Élimination
Après administration d’une dose unique de 1 000 mg à des sujets sains, de 19 à 33 % de la dose de dalbavancine administrée ont, en moyenne, été excrétés dans les urines sous forme de dalbavancine et de 8 à 12 % l’ont été sous forme de métabolite hydroxy-dalbavancine. Environ 20 % de la dose administrée ont été excrétés dans les selles.
Populations particulières
Insuffisance rénale :
La pharmacocinétique de la dalbavancine a été évaluée chez 28 sujets adultes présentant divers degrés d’insuffisance rénale et chez 15 sujets témoins présentant une fonction rénale normale. Après administration d’une dose unique de 500 mg ou de 1 000 mg de dalbavancine, la clairance plasmatique moyenne (CLT) était réduite de 11 %, 35 % et 47 % chez les sujets en insuffisance rénale légère (CLCR 50- 79 mL/min), modérée (CLCR 30-49 mL/min) et sévère (CLCR < 30 mL/min), respectivement, par rapport aux sujets présentant une fonction rénale normale.
L’ASC moyenne chez les sujets dont la clairance de la créatinine était < 30 mL/min était multipliée par deux environ.
La signification clinique de la réduction de la CLT plasmatique moyenne, et l’augmentation associée de l’ASC0-∞ dans les études pharmacocinétiques menées avec la dalbavancine chez des sujets en insuffisance rénale sévère n’ont pas été établies. La pharmacocinétique de la dalbavancine chez des sujets présentant une néphropathie en phase terminale et traités par dialyse régulière planifiée (3 fois/semaine) était comparable à celle observée chez les sujets en insuffisance rénale légère à modérée, et moins de 6 % de la dose administrée sont éliminés après 3 heures d’hémodialyse. Pour consulter les recommandations posologiques relatives aux sujets adultes en insuffisance rénale, voir rubrique 4.2.
Aucune donnée pharmacocinétique observée n’est disponible chez les patients pédiatriques en insuffisance rénale sévère. L’ASC moyenne estimée de la dalbavancine pour les sujets pédiatriques en insuffisance rénale sévère (CLCR ≤ 30 mL/min/1,73 m2) était environ 13 à 30 % supérieure à celle des patients pédiatriques présentant une fonction rénale normale et traités à la même dose, sur la base d’un modèle de pharmacocinétique de population.
Insuffisance hépatique :
La pharmacocinétique de la dalbavancine a été évaluée chez 17 sujets en insuffisance hépatique légère, modérée et sévère par rapport à 9 sujets témoins sains présentant une fonction hépatique normale. L’ASC moyenne est restée inchangée chez les sujets en insuffisance hépatique modérée par rapport aux sujets présentant une fonction hépatique normale. Cependant, l’ASC moyenne a diminué de 28 % et 31 %, respectivement, chez les sujets en insuffisance hépatique modérée et sévère. La cause et la signification clinique de la diminution de l’exposition chez les sujets en insuffisance hépatique modérée et sévère sont inconnues. Pour consulter les recommandations posologiques relatives aux sujets en insuffisance hépatique, voir rubrique 4.2.
Sexe
Aucune différence cliniquement significative n’a été observée entre les sexes en ce qui concerne la pharmacocinétique de la dalbavancine chez des sujets sains ou chez des patients infectés. Aucune adaptation de dose en fonction du sexe n’est recommandée.
Patients âgés
L’âge n’a pas eu d’influence majeure sur la pharmacocinétique de la dalbavancine. Par conséquent, aucune adaptation posologique en fonction de l’âge n’est nécessaire (voir rubrique 4.2). L’expérience portant sur la dalbavancine chez les personnes âgées est limitée : 276 patients ≥ 75 ans ont été inclus dans des études cliniques de phase 2/3. Parmi eux, 173 ont reçu de la dalbavancine. Des patients de jusqu’à 93 ans ont été inclus dans des études cliniques.
Population pédiatrique
La pharmacocinétique de la dalbavancine a été évaluée chez 218 patients pédiatriques (âgés de 4 jours à 17 ans, dont un nouveau-né prématuré [âge gestationnel de 36 semaines ; n = 1] et des nouveau-nés à terme [âge gestationnel de 37 à 40 semaines ; n = 6]) dont la clairance de la créatinine était supérieure ou égale à 30 mL/min/1,73 m2. Les informations disponibles sont insuffisantes pour évaluer l’exposition à la dalbavancine chez les patients pédiatriques dont la clairance de la créatinine est inférieure à 30 mL/min/1,73 m2. Le modèle prévoyait que l’ASC0-120h plasmatique de la dalbavancine à la naissance chez les nouveau-nés prématurés (âge gestationnel de 26 semaines à < 37 semaines) correspondait à environ 60 % de celle des patients adultes.
|
Tableau 3. Paramètres pharmacocinétiques moyens (SD) simulés de la dalbavancine pour les enfants et les adultes, basés sur l’analyse PK de population1 |
||||||||
|
Paramètre |
Nouveau-né prématuré |
Nouveau-né à terme |
Jeune nourrisson |
Nourrisson |
Jeune enfant |
Enfant |
Adolescent |
Adulte |
|
Tranche d’âge |
AG 26 à 37 semaines |
Naissance à 1 mois |
1 mois à < 3 mois |
3 mois à < 2 ans |
2 ans à < 6 ans |
6 ans à < 12 ans |
12 ans à < 18 ans |
≥ 18 ans |
|
Dose |
22.5 mg/kg |
22.5 mg/kg |
22.5 mg/kg |
22.5 mg/kg |
22.5 mg/kg |
18 mg/kg |
18 mg/kg |
1500 mg |
|
Cmax (mg/L) |
231 (89) |
306 (130) |
306 (130) |
307 (130) |
304 (130) |
259 (110) |
251 (110) |
425 (100) |
|
AUC0-120h (mg·h/L) |
6620 (2000) |
9000 (2900) |
9080 (3000) |
9490 (3100) |
10200 (3200) |
8870 (2900) |
9060 (3100) |
10800 (3200) |
|
1 Source: DAL-MS-02. |
||||||||
|
|
||||||||
Dans toutes les tranches d’âge pédiatriques, 90 % ou plus des patients ont atteint les valeurs PK/PD cibles liées à l’activité in vivo du médicament, à des CMI allant jusqu’à 0,125 mg/L.
5.3. Données de sécurité préclinique
Les études de la reproduction réalisées chez les rats et les lapins n’ont mis en évidence aucun effet tératogène. Chez le rat, à des expositions environ 3 fois supérieures à l’exposition clinique, une réduction de la fertilité et une augmentation de la létalité embryofoetale, une diminution du poids fœtal et de l’ossification du squelette, ainsi qu’une augmentation de la mortalité néonatale ont été observées. Chez les lapins, un avortement spontané associé à une intoxication maternelle s’est produit à une exposition inférieure à celle observée chez l’humain.
Aucune étude de carcinogénicité à long terme n’a été réalisée. La dalbavancine ne s’est révélée ni mutagène ni clastogène lors d’une batterie de tests de génotoxicité réalisés in vivo et in vitro.
Lactose monohydraté
Acide chlorhydrique, concentré (pour ajustement du pH)
Hydroxyde de sodium (pour ajustement du pH).
Ce médicament ne doit pas être mélangé avec d’autres médicaments ou solutions intraveineuses à l’exception de ceux mentionnés dans la rubrique 6.6.
Poudre sèche : 30 mois.
La stabilité chimique et physique de DALBAVANCINE REIG JOFRE a été démontrée pour la solution à diluer reconstituée et pour la solution diluée pendant 48 heures à une température ne dépassant pas 25 °C. La stabilité totale entre la reconstitution et l’administration ne doit pas excéder 48 heures.
Toutefois, du point de vue microbiologique, le produit doit être utilisé immédiatement. En cas d’utilisation non immédiate, les durées et conditions de conservation avant utilisation relèvent de la seule responsabilité de l’utilisateur et ne devraient pas dépasser 24 heures à une température comprise entre 2 °C et 8 °C, sauf si la reconstitution/dilution a été réalisée dans des conditions d’asepsie contrôlées et validées. Ne pas congeler.
6.4. Précautions particulières de conservation
Ce médicament ne nécessite pas de précautions particulières de conservation.
Pour les conditions de conservation du médicament après reconstitution et dilution, voir rubrique 6.3.
6.5. Nature et contenu de l'emballage extérieur
Flacon en verre de type I de 50 mL, muni d’un bouchon en caoutchouc chlorobutyle et d’un joint en aluminium, capuchon vert en proplypropylène.
Chaque boîte contient un flacon.
6.6. Précautions particulières d’élimination et de manipulation
DALBAVANCINE REIG JOFRE doit être reconstitué avec de l’eau pour préparations injectables stérile, puis dilué dans une solution de glucose à 50 mg/mL (5 %) pour perfusion.
Les flacons de DALBAVANCINE REIG JOFRE sont à usage unique.
Instructions pour la reconstitution et la dilution
Des conditions d’asepsie doivent être utilisées pour reconstituer et diluer DALBAVANCINE REIG JOFRE.
1. Le contenu de chaque flacon doit être reconstitué en ajoutant lentement 25 mL d’eau pour préparations injectables.
2. Ne pas agiter. Pour éviter la formation de mousse, alterner entre des mouvements d’agitation douce par rotation et d’inversion du flacon, jusqu’à dissolution complète de son contenu. Le temps de reconstitution peut aller jusqu’à 5 minutes.
3. La solution concentrée reconstituée à diluer dans le flacon contient 20 mg/mL de dalbavancine.
4. La solution concentrée reconstituée à diluer doit être une solution limpide, incolore à jaune pâle, exempt de particules visibles.
5. De plus, la solution concentrée reconstituée doit être diluée dans une solution de glucose à 50 mg/mL (5 %) pour perfusion.
6. Pour diluer la solution concentrée reconstituée, le volume adéquat de 20 mg/mL de cette solution concentrée doit être transféré du flacon dans une poche intraveineuse ou une bouteille contenant une solution de glucose à 50 mg/mL (5 %) pour perfusion. Par exemple : 25 mL de la solution concentrée reconstituée à diluer contient 500 mg de dalbavancine.
7. Après dilution, la solution pour perfusion doit avoir une concentration finale de 1 à 5 mg/mL de dalbavancine.
8. La solution pour perfusion doit être limpide, incolore à jaune pâle, exempte de particules visibles.
9. Si des particules ou un changement de couleur sont observés, la solution doit être éliminée.
DALBAVANCINE REIG JOFRE ne doit pas être mélangé à d’autres médicaments ou solutions intraveineuses. Les solutions contenant du chlorure de sodium peuvent provoquer une précipitation et NE doivent PAS être utilisées pour la reconstitution ou la dilution. La compatibilité de la solution concentrée de DALBAVANCINE REIG JOFRE reconstituée n’a été établie qu’avec une solution de glucose à 50 mg/mL (5 %) pour perfusion.
Si un cathéter intraveineux commun est utilisé pour administrer d’autres médicaments en plus de DALBAVANCINE REIG JOFRE, le cathéter doit être rincé avec une solution de glucose à 5 % pour perfusion avant et après chaque perfusion de DALBAVANCINE REIG JOFRE.
Utilisation dans la population pédiatrique
Pour les patients pédiatriques, la dose of DALBAVANCINE REIG JOFRE variera en fonction de l’âge et du poids de l’enfant, avec une dose maximale de 1 500 mg. Transférer la dose requise de solution de dalbavancine reconstituée en fonction du poids de l’enfant, conformément aux instructions ci-dessus, du flacon dans une poche intraveineuse ou une bouteille contenant une solution de glucose à 50 mg/mL (5 %) pour perfusion. La solution diluée doit avoir une concentration finale de 1 à 5 mg/mL de dalbavancine.
Le tableau 4 ci-dessous fournit des informations pour la préparation d’une solution pour perfusion dont la concentration finale est de 2 mg/mL ou 5 mg/mL (concentration suffisante dans la plupart des cas), à administrer par un pousse-seringue, afin de parvenir à une dose de 22,5 mg/kg chez les patients pédiatriques âgés 3 à 12 mois et pesant entre 3 et 12 kg. D’autres concentrations peuvent être préparées, mais la concentration finale doit être comprise entre 1 et 5 mg/mL de dalbavancine. Se reporter au tableau 4 pour vérifier les calculs. Les valeurs présentées sont approximatives. Il convient de noter que le tableau n’inclut PAS toutes les doses calculées possibles pour chaque tranche d’âge, mais qu’il peut être utilisé pour estimer le volume approximatif afin de vérifier le calcul.
Tableau 4. Préparation de DALBAVANCINE REIG JOFRE (concentration finale de la perfusion de 2 mg/mL ou 5 mg/mL devant être administrée par un pousse-seringue) pour des patients pédiatriques âgés de 3 à 12 mois (dose de 22,5 mg/kg)
|
Patient Weight (kg) |
Dose (mg) to achieve 22.5 mg/kg |
Volume of reconstituted dalbavancin solution (20 mg/mL) to be withdrawn from vial (mL) |
Volume of diluent 50 mg/mL (5 %) glucose solution to add for mixing (mL) |
Final dalbavancin infusion solution concentration |
Total Volume Dosed by syringe pump (mL) |
|
3 |
67.5 |
10 mL |
90 mL |
2 mg/mL |
33.8 |
|
4 |
90.0 |
45.0 |
|||
|
5 |
112.5 |
56.3 |
|||
|
6 |
135.0 |
67.5 |
|||
|
7 |
157.5 |
78.8 |
|||
|
8 |
180.0 |
90.0 |
|||
|
9 |
202.5 |
20 mL |
60 mL |
5 mg/mL |
40.5 |
|
10 |
225.0 |
45.0 |
|||
|
11 |
247.5 |
49.5 |
|||
|
12 |
270.0 |
54.0 |
Élimination
Éliminez tout excédent de solution reconstituée qui demeure inutilisé.
Tout médicament non utilisé ou tout déchet doit être éliminé conformément à la réglementation en vigueur.
7. TITULAIRE DE L’AUTORISATION DE MISE SUR LE MARCHE
Gran Capitan, 10
08970 Sant Joan Despí - Barcelone
ESPAGNE
8. NUMERO(S) D’AUTORISATION DE MISE SUR LE MARCHE
· 34009 551 013 7 6 : Flacon (verre). Boîte de 1 flacon
9. DATE DE PREMIERE AUTORISATION/DE RENOUVELLEMENT DE L’AUTORISATION
[à compléter ultérieurement par le titulaire]
Date de première autorisation:{JJ mois AAAA}
10. DATE DE MISE A JOUR DU TEXTE
[à compléter ultérieurement par le titulaire]
{JJ mois AAAA}
Sans objet.
12. INSTRUCTIONS POUR LA PREPARATION DES RADIOPHARMACEUTIQUES
Liste I
Médicament soumis à prescription hospitalière.
ANSM - Mis à jour le : 08/04/2024
DALBAVANCINE REIG JOFRE 500 mg, poudre pour solution à diluer pour perfusion
Dalbavancine
Veuillez lire attentivement cette notice avant d’utiliser ce médicament car elle contient des informations importantes pour vous.
- Gardez cette notice. Vous pourriez avoir besoin de la relire.
- Si vous avez d’autres questions, interrogez votre médecin, votre pharmacien ou votre infirmier/ère.
- Si vous ressentez un quelconque effet indésirable, parlez-en à votre médecin, votre pharmacien ou votre infirmier/ère. Ceci s’applique aussi à tout effet indésirable qui ne serait pas mentionné dans cette notice. Voir rubrique 4.
1. Qu'est-ce que DALBAVANCINE REIG JOFRE 500 mg, poudre pour solution à diluer pour perfusion et dans quels cas est-il utilisé ?
2. Quelles sont les informations à connaître avant d'utiliser DALBAVANCINE REIG JOFRE 500 mg, poudre pour solution à diluer pour perfusion ?
3. Comment utiliser DALBAVANCINE REIG JOFRE 500 mg, poudre pour solution à diluer pour perfusion ?
4. Quels sont les effets indésirables éventuels ?
5. Comment conserver DALBAVANCINE REIG JOFRE 500 mg, poudre pour solution à diluer pour perfusion ?
6. Contenu de l’emballage et autres informations.
1. QU’EST-CE QUE DALBAVANCINE REIG JOFRE 500 mg, poudre pour solution à diluer pour perfusion ET DANS QUELS CAS EST-IL UTILISE ?
DALBAVANCINE REIG JOFRE contient la substance active dalbavancine, qui est un antibiotique de la classe des glycopeptides.
DALBAVANCINE REIG JOFRE est utilisé chez les adultes et les enfants âgés de 3 mois et plus dans le traitement d’infections de la peau ou des tissus mous sous la peau.
DALBAVANCINE REIG JOFRE agit en tuant certaines bactéries pouvant être responsables d’infections graves. Il les tue en interférant avec la formation de la paroi des bactéries.
Si d’autres bactéries sont à l’origine de votre infection, votre médecin peut choisir de vous traiter avec d’autres antibiotiques, utilisés en association avec DALBAVANCINE REIG JOFRE.
2. QUELLES SONT LES INFORMATIONS A CONNAITRE AVANT D’UTILISER DALBAVANCINE REIG JOFRE 500 mg, poudre pour solution à diluer pour perfusion ?
N’utilisez jamais DALBAVANCINE REIG JOFRE si vous êtes allergique à la dalbavancine ou à l’un des autres composants contenus dans ce médicament (mentionnés dans la rubrique 6).
Avertissements et précautions
Adressez-vous à votre médecin, votre pharmacien ou votre infirmier/ère avant d’utiliser DALBAVANCINE REIG JOFRE :
· si vous avez ou avez présenté des problèmes rénaux. Selon votre âge et l’état de votre rein, votre médecin peut réduire la dose ;
· si vous avez une diarrhée, ou si vous avez déjà eu une diarrhée pendant un traitement antibiotique ;
· si vous êtes allergique à d’autres antibiotiques comme la vancomycine ou la teicoplanine.
Diarrhée pendant ou après le traitement
Si une diarrhée survient pendant ou après le traitement, informez-en immédiatement votre médecin. Ne prenez aucun médicament pour traiter votre diarrhée avant d’en avoir parlé avec votre médecin.
Réactions liées à la perfusion
Les perfusions intraveineuses avec ces types d’antibiotiques peuvent provoquer des rougeurs sur la partie supérieure du corps, une urticaire, des démangeaisons ou une éruption cutanée. Si vous présentez l’un de ces types de réactions, votre médecin peut décider d’arrêter ou de ralentir la perfusion.
Autres infections
Utiliser des antibiotiques peut parfois permettre à des infections nouvelles et différentes de se développer. Dans ce cas, parlez-en à votre médecin qui décidera de ce qu’il convient de faire.
Enfants
Ne donnez pas ce médicament à un enfant âgé de moins de 3 mois. L’utilisation de DALBAVANCINE REIG JOFRE chez l’enfant âgé de moins de 3 mois n’a pas été suffisamment étudiée.
Autres médicaments et DALBAVANCINE REIG JOFRE
Informez votre médecin ou pharmacien si vous prenez, avez pris récemment ou pourriez prendre tout autre médicament.
L’utilisation de DALBAVANCINE REIG JOFRE est déconseillée pendant la grossesse à moins d’une nécessité absolue. Ceci s’explique par le fait que l’on ignore l’effet qu’il pourrait avoir sur un futur nouveau-né. Si vous êtes enceinte ou que vous allaitez, si vous pensez être enceinte ou planifiez une grossesse, demandez conseil à votre médecin avant de prendre ce médicament. Votre médecin et vous-même déterminerez si vous pouvez prendre DALBAVANCINE REIG JOFRE.
On ignore si DALBAVANCINE REIG JOFRE passe dans le lait maternel humain. Demandez conseil à votre médecin avant d’allaiter votre nourrisson. Votre médecin et vous-même déterminerez si vous pouvez prendre DALBAVANCINE REIG JOFRE. Vous ne devriez pas allaiter si vous prenez DALBAVANCINE REIG JOFRE.
Conduite de véhicules et utilisation de machines
DALBAVANCINE REIG JOFRE peut provoquer des vertiges. Soyez prudent si vous conduisez des véhicules ou utilisez des machines après avoir pris ce médicament.
DALBAVANCINE REIG JOFRE contient du sodium
Ce médicament contient moins de 1 mmol (23 mg) de sodium par dose, il est donc considéré comme « sans sodium ».
3. COMMENT UTILISER DALBAVANCINE REIG JOFRE 500 mg, poudre pour solution à diluer pour perfusion ?
DALBAVANCINE REIG JOFRE est administré par un médecin ou un(e) infirmier/ère.
· Adultes : DALBAVANCINE REIG JOFRE est administré, soit en une seule dose de 1 500 mg, soit en deux doses à une semaine d’intervalle : 1 000 mg au Jour 1 et 500 mg au Jour 8.
· Enfants et adolescents âgés de 6 ans à moins de 18 ans : DALBAVANCINE REIG JOFRE est administré en une seule dose de 18 mg/kg (1 500 mg au maximum).
· Nourrissons et enfants âgés de 3 mois à moins de 6 ans : DALBAVANCINE REIG JOFRE est administré en une seule dose de 22,5 mg/kg (1 500 mg au maximum).
La dose pour les enfants âgés de 3 mois à moins de 18 ans sera calculée par le médecin, en fonction de l’âge et du poids de l’enfant.
DALBAVANCINE REIG JOFRE sera administré directement dans votre circulation sanguine à l’aide d’un système de goutte-à-goutte inséré dans une veine (voie intraveineuse), pendant 30 minutes.
Patients présentant des problèmes rénaux chroniques
Si vous avez des problèmes rénaux chroniques, votre médecin peut décider de réduire la dose. Les informations disponibles ne sont pas suffisantes pour recommander l’utilisation de DALBAVANCINE REIG JOFRE chez des enfants présentant des problèmes rénaux chroniques
Si vous avez pris plus de DALBAVANCINE REIG JOFRE que vous n’auriez dû
Informez immédiatement votre médecin ou infirmier/ère si vous pensez avoir pris trop de DALBAVANCINE REIG JOFRE.
Si vous oubliez une dose de DALBAVANCINE REIG JOFRE
Informez immédiatement votre médecin ou infirmier/ère si vous pensez avoir oublié la 2e dose.
Si vous avez d’autres questions sur l’utilisation de ce médicament, demandez plus d’informations à votre médecin, à votre pharmacien ou à votre infirmier/ère.
4. QUELS SONT LES EFFETS INDESIRABLES EVENTUELS ?
Les effets indésirables ci-dessous peuvent survenir avec ce médicament :
Effets indésirables graves
Informez immédiatement votre médecin si vous présentez l’un de ces symptômes – vous pourriez avoir besoin de soins médicaux urgents :
· Soudain gonflement des lèvres, du visage, de la gorge ou de la langue ; éruption cutanée sévère ; démangeaisons ; resserrement de la gorge ; chute de la pression artérielle ; difficulté à avaler et / ou difficulté à respirer. Ces effets peuvent être révélateurs d’une réaction d’hypersensibilité et mettre en jeu votre vie. Cette réaction sévère a été signalée comme un effet secondaire rare. Elle peut concerner jusqu’à 1 personne sur 1 000 10 000.
· Douleurs abdominales (maux de ventre) et/ou diarrhée aqueuse. Ces symptômes peuvent devenir sévères ou persister et les selles peuvent contenir du sang ou du mucus. Ils peuvent être le signe d’une infection intestinale. Dans ce cas, vous ne devez pas prendre de médicaments qui arrêtent ou ralentissent le transit intestinal. L’infection intestinale a été signalée comme un effet indésirable rare. Elle peut concerner jusqu’à 1 personne sur 100.
· Modifications de l’audition. Cette réaction a été signalée comme un effet indésirable avec un médicament comparable. Sa fréquence est inconnue. Elle ne peut pas être estimée sur la base des données disponibles.
Les autres effets indésirables rapportés avec DALBAVANCINE REIG JOFRE sont répertoriés ci-dessous.
Informez immédiatement votre médecin, pharmacien ou infirmier/ère si vous présentez l’un des effets indésirables suivants.
Fréquent - peut concerner jusqu'à 1 personne sur 10 :
· Céphalées
· Nausées (mal au cœur)
· Diarrhée.
Peu fréquent - peut affecter jusqu'à 1 personne sur 100 :
· Infections vaginales, infections fongiques, muguet dans la bouche
· Infections urinaires
· Anémie (faible quantité de globules rouges), taux élevé de plaquettes (thrombocytose), augmentation de certains globules blancs dans le sang, appelés « éosinophiles » (éosinophilie), faible quantité d’autres types de globules blancs dans le sang (leucopénie, neutropénie)
· Modifications des résultats d’autres examens du sang
· Baisse de l’appétit
· Troubles du sommeil
· Vertiges
· Modification du goût
· Inflammation et gonflement des veines superficielles, bouffées vasomotrices
· Toux
· Douleurs et gêne abdominales, indigestion, constipation
· Anomalie des tests hépatiques
· Augmentation des phosphatases alcalines (une enzyme que l'on trouve dans le corps)
· Démangeaisons, urticaire
· Démangeaisons génitales (femmes)
· Douleur, rougeurs ou gonflement à l'endroit où la perfusion a été effectuée
· Sensation de chaleur
· Augmentation de la gamma glutamyl transférase (enzyme produite par le foie et d’autres tissus de l’organisme)
· Éruption cutanée
· Vomissements.
Rare – peut concerner jusqu’à 1 personne sur 1000 :
· Troubles de la respiration (bronchospasmes).
Déclaration des effets secondaires
Si vous ressentez un quelconque effet indésirable, parlez-en à votre médecin ou à votre infirmier/ère. Ceci s’applique aussi à tout effet indésirable qui ne serait pas mentionné dans cette notice. Vous pouvez également déclarer les effets indésirables directement via le système national de déclaration : Agence nationale de sécurité du médicament et des produits de santé (ANSM) et réseau des Centres Régionaux de Pharmacovigilance - Site internet: https://signalement.social-sante.gouv.fr
En signalant les effets indésirables, vous contribuez à fournir davantage d’informations sur la sécurité du médicament.
5. COMMENT CONSERVER DALBAVANCINE REIG JOFRE 500 mg, poudre pour solution à diluer pour perfusion ?
Tenir ce médicament hors de la vue et de la portée des enfants.
N’utilisez pas ce médicament après la date de péremption indiquée sur le flacon après EXP. La date de péremption fait référence au dernier jour de ce mois.
Ce médicament ne nécessite pas de précautions particulières de conservation est conservé scellé dans son conditionnement d’origine.
La solution préparée pour perfusion de DALBAVANCINE REIG JOFRE ne doit pas être utilisée si vous remarquez des signes visibles de particules ou si la solution est trouble.
DALBAVANCINE REIG JOFRE est exclusivement réservé à un usage unique.
Ne jetez aucun médicament au tout-à-l’égout ou avec les ordures ménagères. Demandez à votre pharmacien d’éliminer les médicaments que vous n’utilisez plus. Ces mesures contribueront à protéger l’environnement.
6. CONTENU DE L’EMBALLAGE ET AUTRES INFORMATIONS
Ce que contient DALBAVANCINE REIG JOFRE 500 mg, poudre pour solution à diluer pour perfusion
· La substance active est :
Dalbavancine (sous forme de chlorhydrate)..................................................................... 500 mg
Pour 1 flacon de poudre.
Après reconstitution, chaque mL de concentré contient 20 mg de dalbavancine.
La solution diluée pour perfusion doit avoir une concentration finale de 1 à 5 mg/mL de dalbavancine.
· Les autres composants sont :
Mannitol (E421), le lactose monohydraté, l’acide chlorhydrique concentré ou l’hydroxyde de sodium (pour ajustement du pH uniquement).
DALBAVANCINE REIG JOFRE est disponible dans des boîtes contenant un flacon.
Titulaire de l’autorisation de mise sur le marché
Gran Capitan, 10
08970 Sant Joan Despí - Barcelone
ESPAGNE
Exploitant de l’autorisation de mise sur le marché
LABORATOIRE FORTE PHARMA S.A.M.
41 AVENUE HECTOR OTTO
MC 98000
MONACO
Gran Capitan, 10
08970 Sant Joan Despí - Barcelone
ESPAGNE
Noms du médicament dans les Etats membres de l'Espace Economique Européen
Ce médicament est autorisé dans les Etats membres de l'Espace Economique Européen sous les noms suivants : Conformément à la réglementation en vigueur.
[À compléter ultérieurement par le titulaire]
La dernière date à laquelle cette notice a été révisée est :
[à compléter ultérieurement par le titulaire]
Des informations détaillées sur ce médicament sont disponibles sur le site Internet de l’ANSM (France).
Les informations suivantes sont destinées exclusivement aux professionnels de santé :
Important : veuillez-vous référer au résumé des caractéristiques du produit (RCP) avant prescription.
DALBAVANCINE REIG JOFRE doit être reconstitué avec de l’eau pour préparations injectables stérile, puis dilué dans une solution à base de glucose à 50 mg/mL (5 %) pour perfusion.
Les flacons de DALBAVANCINE REIG JOFRE sont réservés à l’usage unique.
Instructions pour la reconstitution et la dilution
Des conditions d’asepsie doivent être utilisées pour reconstituer et diluer DALBAVANCINE REIG JOFRE.
1. Le contenu de chaque flacon doit être reconstitué en ajoutant lentement 25 mL d’eau pour préparations injectables.
2. Ne pas agiter. Pour éviter la formation de mousse, alterner entre des mouvements d’agitation douce par rotation et d’inversion du flacon, jusqu’à dissolution complète de son contenu. Le temps de reconstitution peut aller jusqu’à 5 minutes.
3. La solution concentrée reconstituée à diluer dans le flacon contient 20 mg/mL de dalbavancine.
4. La solution concentrée reconstituée à diluer doit être une solution limpide, incolore à jaune pâle, exempt de particules visibles.
5. De plus, la solution concentrée reconstituée doit être diluée dans une solution de glucose à 50 mg/mL (5 %) pour perfusion.
6. Pour diluer la solution concentrée reconstituée, le volume adéquat de 20 mg/mL de cette solution concentrée doit être transféré du flacon dans une poche intraveineuse ou une bouteille contenant une solution de glucose à 50 mg/mL (5 %) pour perfusion. Par exemple : 25 mL de la solution concentrée reconstituée à diluer contient 500 mg de dalbavancine.
7. Après dilution, la solution pour perfusion doit avoir une concentration finale de 1 à 5 mg/mL de dalbavancine.
8. La solution pour perfusion doit être limpide, incolore à jaune pâle, exempte de particules visibles.
9. Si des particules ou un changement de couleur sont observés, la solution doit être éliminée.
DALBAVANCINE REIG JOFRE ne doit pas être mélangé à d’autres médicaments ou solutions intraveineuses. Les solutions contenant du chlorure de sodium peuvent provoquer une précipitation et NE doivent PAS être utilisées pour la reconstitution ou la dilution. La compatibilité de la solution à diluer de DALBAVANCINE REIG JOFRE reconstituée n’a été établie qu’avec une solution de glucose à 50 mg/mL (5 %) pour perfusion.
Si un cathéter intraveineux commun est utilisé pour administrer d’autres médicaments en plus de DALBAVANCINE REIG JOFRE, le cathéter doit être rincé avec une solution de glucose à 5 % pour perfusion avant et après chaque perfusion de DALBAVANCINE REIG JOFRE.
Utilisation dans la population pédiatrique
Pour les patients pédiatriques, la dose of DALBAVANCINE REIG JOFRE variera en fonction de l’âge et du poids de l’enfant, avec une dose maximale de 1 500 mg. Transférer la dose requise de solution de dalbavancine reconstituée en fonction du poids de l’enfant, conformément aux instructions ci-dessus, du flacon dans une poche intraveineuse ou une bouteille contenant une solution de glucose à 50 mg/mL (5 %) pour perfusion. La solution diluée doit avoir une concentration finale de 1 à 5 mg/mL de dalbavancine.
Le tableau 1 ci-dessous fournit des informations pour la préparation d’une solution pour perfusion dont la concentration finale est de 2 mg/mL ou 5 mg/mL (concentration suffisante dans la plupart des cas), à administrer par un pousse-seringue, afin de parvenir à une dose de 22,5 mg/kg chez les patients pédiatriques âgés 3 à 12 mois et pesant entre 3 et 12 kg. D’autres concentrations peuvent être préparées, mais la concentration finale doit être comprise entre 1 et 5 mg/mL de dalbavancine. Se reporter au tableau 1 pour vérifier les calculs. Les valeurs présentées sont approximatives. Il convient de noter que le tableau n’inclut PAS toutes les doses calculées possibles pour chaque tranche d’âge, mais qu’il peut être utilisé pour estimer le volume approximatif afin de vérifier le calcul.
Tableau 1. Préparation de DALBAVANCINE REIG JOFRE (concentration finale de la perfusion de 2 mg/mL ou 5 mg/mL devant être administrée par un pousse-seringue) pour des patients pédiatriques âgés de 3 à 12 mois (dose de 22,5 mg/kg)
|
Poids du patient (kg) |
Dose (mg) à atteindre 22,5 mg/kg |
Volume de solution de dalbavancine reconstituée (20 mg/mL) à prélever du flacon (mL) |
Volume de solution de glucose à 50 mg/mL (5 %) à ajouter au mélange comme diluant (mL) |
Concentration finale de la solution pour perfusion de dalbavancine |
Volume total administré par le pousse-seringue (mL) |
|
3 |
67.5 |
10 mL |
90 mL |
2 mg/mL |
33,8 |
|
4 |
90.0 |
45,0 |
|||
|
5 |
112.5 |
56,3 |
|||
|
6 |
135.0 |
67,5 |
|||
|
7 |
157.5 |
78,8 |
|||
|
8 |
180.0 |
90,0 |
|||
|
9 |
202.5 |
20 mL |
60 mL |
5 mg/mL |
40,5 |
|
10 |
225.0 |
45,0 |
|||
|
11 |
247.5 |
49,5 |
|||
|
12 |
270.0 |
54,0 |
Élimination
Éliminez tout excédent de solution qui demeure inutilisé.
Tout médicament non utilisé ou tout déchet doit être éliminé conformément à la réglementation en vigueur.