Dernière mise à jour le 03/08/2026
SPORANOX 10 mg/mL, solution buvable
Ce médicament n'est ou ne sera bientôt plus disponible sur le marché.
Si vous prenez actuellement ce médicament, il vous est recommandé d'en parler avec votre médecin
ou avec votre pharmacien qui pourra vous orienter vers un autre traitement.
Indications thérapeutiques
Sporanox est indiqué dans le traitement de certaines mycoses (infections dues à des champignons microscopiques) localisées au niveau de la bouche, de la gorge et de l'œsophage, chez certaines personnes ayant un système de défense affaibli. Il peut être utilisé pour traiter ces infections ou vous empêcher de les contracter.
Présentations
> 1 flacon(s) de 150 ml en verre brun avec fermeture de sécurité enfant avec gobelet doseur gradué polypropylène
Code CIP : 345 020-6 ou 34009 345 020 6 4
Déclaration d'arrêt de commercialisation : 30/11/2024
Cette présentation n'est pas agréée aux collectivités
- Prix hors honoraire de dispensation : 81,82 €
- Honoraire de dispensation : 1,02 €
- Prix honoraire compris : 82,84 €
- Taux de remboursement :65%
Service médical rendu (SMR)
Les libellés affichés ci-dessous ne sont que des résumés ou extraits issus des avis rendus par la Commission de la Transparence. Seul l'avis complet de la Commission de la Transparence fait référence.
Cet avis est consultable à partir du lien `Avis du jj/mm/aaaa` ou encore sur demande auprès de la HAS (plus d'informations dans l'aide). Les avis et synthèses d'avis contiennent un paragraphe sur la place du médicament dans la stratégie thérapeutique.
| Valeur du SMR | Avis | Motif de l'évaluation | Résumé de l'avis |
|---|---|---|---|
| Important | Avis du 07/06/2017 | Renouvellement d'inscription (CT) | Le service médical rendu par SPORANOX reste important dans les indications de l’AMM. |
Amélioration du service médical rendu (ASMR)
Les libellés affichés ci-dessous ne sont que des résumés ou extraits issus des avis rendus par la Commission de la Transparence. Seul l'avis complet de la Commission de la Transparence fait référence.
Cet avis est consultable à partir du lien `Avis du jj/mm/aaaa` ou encore sur demande auprés de la HAS (plus d'informations dans l'aide). Les avis et synthéses d'avis contiennent un paragraphe sur la place du médicament dans la stratégie thérapeutique.
| Valeur de l'ASMR | Avis | Motif de l'évaluation | Résumé de l'avis |
|---|---|---|---|
| V (Inexistant) | Avis du 20/09/2006 | Inscription (CT) | SPORANOX conserve un intérêt thérapeutique important dans la prise en charge des infections fongiques. Il n'a pas démontré d'amélioration du Service Médical Rendu par rapport aux traitements disponibles (ASMR V). |
Autres informations
- Titulaire de l'autorisation : JANSSEN CILAG
- Conditions de prescription et de délivrance :
- liste I
- prescription initiale hospitalière annuelle
- Statut de l'autorisation : Valide
- Type de procédure : Procédure nationale
- Code CIS : 6 999 815 6
ANSM - Mis à jour le : 02/07/2025
Sporanox 10 mg/mL, solution buvable
2. COMPOSITION QUALITATIVE ET QUANTITATIVE
Pour 100 mL de solution buvable.
1 dose de 10 mL de solution buvable correspond à 100 mg d'itraconazole.
Excipients à effet notoire :
1 dose de 10 mL de solution buvable contient :
· 4000 mg de cyclodextrine,
· 1980 mg de sorbitol,
· 1040 mg de propylène glycol,
· 0,05 mg d’éthanol.
Pour la liste complète des excipients, voir rubrique 6.1.
4.1. Indications thérapeutiques
4.2. Posologie et mode d'administration
Traitement des candidoses orales et/ou œsophagiennes : 200 mg (20 mL) par jour de préférence en 2 prises, ou éventuellement en 1 prise pendant 1 semaine. En l'absence de réponse après 1 semaine, le traitement doit être poursuivi pendant une semaine supplémentaire.
Traitement des candidoses orales et/ou œsophagiennes résistantes au fluconazole : 100 à 200 mg (10 à 20 mL) 2 fois par jour pendant 2 semaines. En l'absence de réponse après deux semaines de traitement, le traitement doit être poursuivi pendant 2 semaines supplémentaires. En l'absence de signes d'amélioration clinique, la dose de 400 mg par jour ne doit pas être utilisée pendant une durée supérieure à 14 jours.
Mode d’administration
Afin d'assurer une absorption optimale, la prise de ce médicament doit s'effectuer en dehors des repas.
La solution doit être laissée en contact avec la bouche pendant quelques instants (environ 20 secondes), puis avalée. Ne pas rincer après avoir avalé (il est recommandé aux patients de s'abstenir de toute prise alimentaire pendant au moins 1 heure après l'administration).
Populations particulières
Utilisation dans la population pédiatrique
Les données cliniques disponibles chez les enfants âgés de moins de 18 ans étant limitées, ce médicament ne sera utilisé chez ces patients que si le bénéfice attendu l’emporte sur les risques potentiels (voir rubrique 5.2).
Utilisation chez le sujet âgé
Les données cliniques sur l’utilisation de Sporanox solution buvable chez les patients âgés étant limitées, ce médicament ne sera utilisé chez ces patients que si le bénéfice attendu l’emporte sur les risques potentiels.
Utilisation chez l'insuffisant hépatique
Les données disponibles sur l’utilisation de l’itraconazole par voie orale chez les patients insuffisants hépatiques sont limitées. La prudence est recommandée lors de l’administration de ce médicament à cette population de patients (voir rubrique 4.4 et 5.2).
Utilisation chez l'insuffisant rénal
L’hémodialyse ou la dialyse rétropéritonéale résulte en une extraction minimale de l’itraconazole.
Les données disponibles sur l’utilisation de l’itraconazole par voie orale chez les patients insuffisants rénaux sont limitées. L’exposition à l’itraconazole peut être plus faible chez certains patients atteints d’une insuffisance rénale. La prudence est recommandée lors de l’administration de ce médicament à cette population de patients et un ajustement de la dose peut être envisagé (voir rubrique 4.4 et 5.2).
· Sporanox solution buvable ne doit pas être administré aux patients ayant une dysfonction ventriculaire démontrée tels qu’une insuffisance cardiaque congestive (ICC) ou des antécédents d’insuffisance cardiaque congestive sauf en cas d’infections sévères ou mettant en jeu le pronostic vital (voir rubrique 4.4).
· Sporanox solution buvable ne doit pas être utilisé pendant la grossesse pour des pathologies ne mettant pas en jeu le pronostic vital (voir rubrique 4.6). Les femmes en âge de procréer, et traitées par Sporanox doivent utiliser un moyen de contraception efficace. Une contraception efficace doit être poursuivie jusqu’au début des règles suivant l’arrêt du traitement par Sporanox.
· La co-administration de Sporanox solution buvable est contre-indiquée avec un certain nombre de substrats du CYP3A4, tels que les exemples énumérés ci-dessous (voir rubrique 4.4 et 4.5). Une augmentation de la concentration plasmatique de ces médicaments, provoquée par la co-administration avec Sporanox solution buvable, peut augmenter ou prolonger les effets thérapeutiques ou indésirables à un tel niveau que des situations potentiellement graves peuvent survenir. Par exemple l’augmentation de la concentration plasmatique de certains de ces médicaments peut entrainer un allongement de l’intervalle QT et des tachycardies ventriculaires incluant la survenue de torsades de pointes, une arythmie potentiellement létale.
|
Analgésiques ; anesthésiques |
||||
|
Alcaloïdes de l’ergot de seigle (par exemple, dihydroergotamine, ergométrine, ergotamine, méthylergométrine) |
|
|||
|
Antibactériens à usage systémique ; antimycobactériens ; antimycotiques à usage systémique |
||||
|
Anthelminthiques ; antiprotozoaires |
||||
|
Halofantrine |
||||
|
Antihistaminiques à usage systémique |
||||
|
Astémizole |
Mizolastine |
Terfénadine |
||
|
Agents antinéoplasiques |
||||
|
Irinotécan |
Vénétoclax (chez les patients atteints de leucémie lymphoïde chronique pendant la phase d’initiation et de titration de dose de vénétoclax) |
|||
|
Agents antithrombotiques |
||||
|
Dabigatran |
Ticagrélor |
|
||
|
Antiviraux à usage systémique |
||||
|
Ombitasvir/Paritaprévir/Ritonavir (avec ou sans Dasabuvir) |
||||
|
Système cardiovasculaire (agents agissant sur le système rénine-angiotensine ; antihypertenseurs ; bêtabloquants ; inhibiteurs des canaux calciques ; traitements cardiaques ; diurétiques) |
||||
|
Aliskiren |
Éplérénone |
Quinidine |
||
|
Bépridil |
Finérénone |
Ranolazine |
||
|
Disopyramide |
Ivabradine |
Sildénafil (hypertension pulmonaire) |
||
|
Dofétilide |
Lercanidipine |
|
||
|
Dronédarone |
Nisoldipine |
|
||
|
Médicaments gastro-intestinaux, y compris antidiarrhéiques, anti-inflammatoires intestinaux/agents anti-infectieux ; antiémétiques et anti-nauséeux ; médicaments pour le traitement de la constipation ; médicaments pour le traitement des troubles gastro-intestinaux fonctionnels |
||||
|
Cisapride |
Dompéridone |
Naloxégol |
||
|
Immunosuppresseurs |
||||
|
Voclosporine |
|
|
||
|
Agents modifiant les lipides |
||||
|
Lovastatine |
Lomitapide |
Simvastatine |
||
|
Psychoanaleptiques ; psycholeptiques (par exemple, antipsychotiques, anxiolytiques et hypnotiques) |
||||
|
Lurasidone |
Pimozide |
Sertindole |
||
|
Midazolam (oral) |
Quétiapine |
Triazolam |
||
|
Urologiques |
||||
|
Avanafil |
Darifénacine |
Solifénacine (chez les patients atteints d’insuffisance rénale sévère ou d’insuffisance hépatique modérée à sévère) |
||
|
Dapoxétine |
Fésotérodine (chez les patients atteints d’insuffisance rénale ou d’insuffisance hépatique modérée à sévère). |
Vardénafil (chez les patients de plus de 75 ans). |
||
|
Médicaments divers et autres substances |
||||
|
Colchicine (chez les patients insuffisants rénaux ou hépatiques) |
Éliglustat (chez les patients métaboliseurs lents (ML) du CYP2D6, métaboliseurs intermédiaires (MI) du CYP2D6 ou métaboliseurs rapides (MR) du CYP2D6 qui prennent un inhibiteur puissant ou modéré du CYP2D6). |
|
||
4.4. Mises en garde spéciales et précautions d'emploi
De très rares cas de toxicité hépatique grave, incluant quelques cas d’insuffisance hépatique aiguë d’évolution fatale ont été rapportés chez des patients traités par Sporanox. La plupart de ces cas sont survenus chez des patients ayant une maladie hépatique pré-existante, traités pour des mycoses systémiques, ayant des pathologies concomitantes significatives et/ou traités par d’autres médicaments hépatotoxiques. Quelques patients ne présentaient pas de facteurs de risque patents de maladie hépatique. Quelques cas ont été observés au cours du 1er mois de traitement, et pour certains au cours de la 1ère semaine de traitement. Il est recommandé de surveiller la fonction hépatique chez les patients traités par Sporanox. Les patients doivent être informés qu’ils doivent avertir très rapidement leur médecin en cas d’apparition de signes et de symptômes suggérant une atteinte hépatique en particulier anorexie, nausées, vomissements, asthénie, douleurs abdominales ou urines foncées. En cas de survenue de l’un de ces symptômes, le traitement doit être immédiatement interrompu et des examens de la fonction hépatique doivent être réalisés.
Chez les patients ayant une élévation des enzymes hépatiques ou présentant une maladie hépatique active, ou chez qui d'autres médicaments ont déjà entraîné une toxicité hépatique, le traitement ne doit pas être débuté à moins que le patient se trouve dans une situation grave ou mettant en jeu le pronostic vital pour laquelle le bénéfice attendu de Sporanox l’emporte sur le risque d’atteinte hépatique. Si le traitement est débuté, la fonction hépatique doit être surveillée.
Effets cardiaques
Une étude réalisée chez le volontaire sain avec Sporanox IV a montré une diminution transitoire asymptomatique de la fraction d'éjection ventriculaire gauche ; se résolvant avant la perfusion suivante. La signification clinique de cette observation, pour les formes orales, n'est pas connue.
L'itraconazole a montré un effet inotrope négatif et Sporanox a été associé à des cas d'insuffisance cardiaque congestive après administration par voie orale.
Dans les notifications spontanées, les insuffisances cardiaques ont été plus fréquemment rapportées lorsque la dose journalière totale d’itraconazole était de 400 mg par rapport à une dose journalière totale plus faible, ce qui suggère que le risque d’insuffisance cardiaque peut être dose-dépendant.
Sporanox ne doit être utilisé chez des patients présentant une insuffisance cardiaque congestive ou des antécédents d'insuffisance cardiaque congestive que si le bénéfice est nettement supérieur aux risques. L'évaluation individuelle du rapport bénéfice/risque doit prendre en compte des facteurs tels que la sévérité de l'indication, la posologie (i.e. la dose totale journalière) et les facteurs de risques individuels d'insuffisance cardiaque congestive. Ces facteurs de risque comprennent des maladies cardiaques telles que les maladies ischémiques et valvulaires, des maladies pulmonaires significatives telles que la bronchopneumopathie chronique obstructive, l'insuffisance rénale et d'autres troubles œdémateux. Ces patients doivent être informés des signes et symptômes de l'insuffisance cardiaque congestive, ils doivent être traités avec précautions et doivent faire l'objet d'un suivi des signes et symptômes de l'insuffisance cardiaque congestive au cours du traitement. Si de tels signes ou symptômes apparaissent au cours du traitement, Sporanox doit être arrêté.
Les inhibiteurs calciques peuvent avoir des effets inotropes négatifs qui peuvent s'ajouter à ceux de l'itraconazole ; l'itraconazole peut inhiber le métabolisme des inhibiteurs calciques. En conséquence, la prudence s'impose en cas de co-administration d'itraconazole et d'inhibiteurs calciques en raison d’un risque augmenté d’insuffisance cardiaque congestive (voir rubrique 4.5).
Hypersensibilité croisée
Les informations relatives à une hypersensibilité croisée entre l’itraconazole et d’autres antifongiques azolés sont limitées. La prudence est de rigueur lorsque ce médicament est prescrit à des patients ayant présenté une hypersensibilité à d’autres azolés.
Excipients à effet notoire
Ce médicament contient 1980 mg de sorbitol pour 10 mL. L’effet additif des produits administrés concomitamment contenant du sorbitol (ou du fructose) et l’apport alimentaire de sorbitol (ou de fructose) doit être pris en compte. La teneur en sorbitol dans les médicaments à usage oral peut affecter la biodisponibilité d’autres médicaments à usage oral administrés de façon concomitante. Les patients présentant une intolérance héréditaire au fructose (IHF) ne doivent pas prendre/recevoir ce médicament. Le sorbitol peut causer une gêne gastro-intestinale et un effet laxatif léger.
Ce médicament contient moins de 1 mmol (23 mg) de sodium pour 10 mL, c.-à-d. qu’il est essentiellement « sans sodium ».
Ce médicament contient 0,05 mg d’alcool (éthanol) pour une dose de 10 mL équivalent à 0,005 mg/mL (0,0005% p/v). La quantité pour 10 mL de ce médicament équivaut à moins de 1 mL de bière ou 1 mL de vin. La faible quantité d’alcool contenue dans ce médicament n’est pas susceptible d’entraîner d’effet notable. L’administration concomitante avec n’importe quel substrat pour l’alcool déshydrogénase comme l’éthanol peut induire des effets indésirables graves chez les enfants âgés de moins de 5 ans (voir rubrique 4.2).
Ce médicament contient 4000 mg de cyclodextrines pour 10 mL. Les cyclodextrines peuvent causer des problèmes digestifs tels que la diarrhée. Il n’existe pas suffisamment d’informations sur les effets des cyclodextrines chez les enfants de moins de 2 ans. Par conséquent, le rapport bénéfice/risque pour le patient doit être évalué au cas par cas (voir rubrique 4.2).
Ce médicament contient 1040 mg de propylène glycol pour 10 mL et ne doit pas être utilisé pendant la grossesse, sauf dans les pathologies mettant en jeu le pronostic vital, où le bénéfice potentiel pour la mère l'emporte sur le préjudice potentiel pour le fœtus (voir rubrique 4.3). L’allaitement est déconseillé en cas de traitement par ce médicament (voir rubrique 4.6).
Une surveillance médicale est requise chez les patients souffrant d’insuffisance rénale ou de troubles de la fonction hépatique, car divers effets indésirables attribués au propylène glycol ont été rapportés tels qu’un dysfonctionnement rénal (nécrose tubulaire aiguë), une insuffisance rénale aiguë et une dysfonction hépatique.
Allaitement
L’allaitement est déconseillé en cas de traitement par ce médicament.
Interactions potentielle
La co-administration de médicaments spécifiques avec l'itraconazole peut entraîner des changements dans l'efficacité ou la sécurité de l'itraconazole et/ou du médicament co-administré. Par exemple, l'utilisation de l'itraconazole avec des inducteurs du CYP3A4 peut conduire à des concentrations plasmatiques sous-thérapeutiques d'itraconazole et donc à l'échec du traitement. En outre, l'utilisation de l'itraconazole avec certains substrats du CYP3A4 peut entraîner une augmentation des concentrations plasmatiques de ces médicaments et des effets indésirables graves et/ou potentiellement mortels, tels que l'allongement de l'intervalle QT et des tachyarythmies ventriculaires, y compris des cas de torsades de pointes, une arythmie potentiellement mortelle. Le prescripteur doit se référer aux informations produits des médicaments co-administrés pour obtenir de plus amples informations sur les effets indésirables graves ou mettant la vie en danger qui pourraient survenir en cas d'augmentation des concentrations plasmatiques de ce médicament. Pour les recommandations concernant la co-administration de médicaments qui sont contre-indiqués, déconseillés ou à utiliser avec précaution en association avec l'itraconazole, veuillez-vous référer aux rubriques 4.3 et 4.5.
Insuffisance rénale
L’hémodialyse ou la dialyse rétropéritonéale résulte en une extraction minimale de l’itraconazole.
Les données disponibles sur l’utilisation de l’itraconazole par voie orale chez les insuffisants rénaux sont limitées. L’exposition à l’itraconazole peut être plus faible chez certains patients atteints d’une insuffisance rénale. La prudence est recommandée lors de l’administration de ce médicament à cette population de patients et un ajustement de la dose peut être envisagé (voir rubriques 4.2 et 5.2).
Insuffisance hépatique
Les données relatives à l’utilisation de l’itraconazole par voie orale chez les patients insuffisants hépatiques sont limitées. La prudence est recommandée lors de l’administration de ce médicament à cette population de patients. Il est préconisé de surveiller étroitement les patients insuffisants hépatiques en cas de prise d’itraconazole (voir rubrique 4.2).
Lors de la décision d’initier un traitement avec d’autres médicaments métabolisés par le CYP3A4, il est recommandé de prendre en compte la prolongation de la demi-vie d’élimination de l’itraconazole observée dans un essai clinique conduit avec l’itraconazole administré à dose unique sous forme de gélules chez des patients cirrhotiques (voir rubrique 5.2).
Sujets immunodéprimés (par exemple : patients neutropéniques, patients infectés par le VIH+)
Les concentrations à l’état stationnaire sont généralement plus faibles chez les immunodéprimés et justifient l’utilisation de posologies élevées (400 mg/jour).
La surveillance des concentrations plasmatiques peut être utile en début de traitement surtout s’il existe des éléments susceptibles de modifier l’absorption (prise à jeun, réaction du greffon contre l’hôte, diarrhée, mucite), et en cas de suspicion d’échec.
Traitement des patients atteints de neutropénie sévère
Ce médicament, dans le traitement des candidoses orales et/ou œsophagiennes, n’a pas été étudié chez les patients atteints de neutropénie sévère. Du fait de ses propriétés pharmacocinétiques (voir rubrique 5.2), ce médicament n’est pas recommandé en initiation de traitement chez des patients présentant un risque immédiat de candidose systémique.
Mucoviscidose
Chez les patients atteints de mucoviscidose, une variabilité des concentrations plasmatiques d’itraconazole a été observée à l’état d’équilibre avec la solution buvable d’itraconazole administrée à la posologie de 2,5 mg/kg 2 fois par jour. A l’état d’équilibre, des concentrations supérieures à 250 ng/mL ont été atteintes chez environ 50 % des patients de plus de 16 ans mais chez aucun patient de moins de 16 ans. En l’absence de réponse au traitement par Sporanox, solution buvable une alternative thérapeutique doit être envisagée.
Perte auditive
Des cas de perte auditive transitoire ou permanente ont été rapportés chez des patients traités par de l’itraconazole. Plusieurs de ces cas comprenaient l’administration concomitante de quinidine, qui est une association contre-indiquée (voir rubriques 4.3 et 4.5). La perte auditive se dissipe généralement à l’arrêt du traitement, mais elle peut persister chez certains patients.
Neuropathie
En cas d’apparition d’une neuropathie qui pourrait être imputée à ce médicament, il convient d’interrompre le traitement.
Résistance croisée
En cas de candidoses systémiques, si des souches de Candida sont suspectées résistantes au fluconazole, on ne peut prédire leur sensibilité à l'itraconazole ; donc celle-ci devra être testée avant d’initier le traitement par l’itraconazole.
Interchangeabilité
Il n’est pas recommandé d’interchanger Sporanox gélule et Sporanox solution buvable car pour une même dose administrée l’exposition à ce médicament est supérieure avec la solution buvable qu’avec les gélules.
Par ailleurs, les effets topiques de l’exposition de la muqueuse digestive à l’itraconazole peuvent être différents entre les 2 formulations. Seule la solution buvable est indiquée dans le traitement de la candidose orale/et /ou œsophagienne chez les patients infectés par le VIH.
4.5. Interactions avec d'autres médicaments et autres formes d'interactions
L’itraconazole est principalement métabolisé par le CYP3A4. D’autres substances qui partagent cette voie métabolique ou modifient l’activité du CYP3A4 peuvent influencer la pharmacocinétique de l’itraconazole. L’itraconazole est un puissant inhibiteur du CYP3A4, un inhibiteur de la P-glycoprotéine et un inhibiteur de la protéine de résistance au cancer du sein (Breast Cancer Resistance Protein (BCRP)).
L’itraconazole peut modifier la pharmacocinétique d’autres substances qui partagent cette voie métabolique ou ces voies de transport de protéines.
Des exemples de médicaments pouvant avoir un impact sur la concentration plasmatique de l’itraconazole sont présentés par classe de médicaments dans le Tableau 1 ci-dessous. Des exemples de médicaments dont les concentrations plasmatiques peuvent être modifiées par l’itraconazole sont présentés dans le Tableau 2 ci-dessous. En raison du nombre d’interactions, les changements potentiels relatifs à la sécurité ou à l’efficacité des médicaments associés ne sont pas inclus. La liste d'exemples d'interactions médicamenteuses figurant dans les tableaux ci-dessous n'est pas exhaustive.
Il convient donc de consulter les informations relatives au produit de chaque médicament administré de manière concomitante avec l'itraconazole pour obtenir des informations sur la voie de métabolisation, les voies d'interaction, les risques potentiels et les mesures spécifiques à prendre en cas de co-administration.
Les interactions décrites dans ces tableaux sont classées « contre-indiqué », « non recommandé » ou « à utiliser avec précaution » avec l’itraconazole en tenant compte de l’ampleur de l’augmentation de la concentration et du profil de sécurité du médicament associé (voir rubriques 4.3 et 4.4 pour plus d’informations). L’interaction potentielle des médicaments listés a été évaluée sur la base d’études pharmacocinétiques chez l’homme avec l’itraconazole et/ou d’études pharmacocinétiques chez l’homme avec d’autres inhibiteurs puissants du CYP3A4 (par exemple le kétoconazole) et/ou des données in vitro :
· « Contre-indiqué » : le médicament ne doit en aucun cas être co-administré avec l’itraconazole, et jusqu’à deux semaines après l’arrêt du traitement par itraconazole.
· « Non recommandé » : l’utilisation du médicament doit être évitée pendant le traitement par itraconazole et jusqu’à deux semaines après l’arrêt du traitement par itraconazole, sauf si les bénéfices ne soient supérieurs aux risques potentiellement accrus d’effets secondaires. Si la co-administration ne peut être évitée, une surveillance clinique des signes ou symptômes d’effets accrus ou prolongés ou d’effets indésirables du médicament administré en concomitance est recommandée, et sa posologie doit être réduite ou interrompue si nécessaire. Le cas échéant, il est recommandé de mesurer les concentrations plasmatiques du médicament co-administré.
· « Utiliser avec précaution » : une surveillance attentive est recommandée lorsque le médicament est co-administré avec l’itraconazole. Lors de la co-administration, il est recommandé de surveiller étroitement les patients pour détecter tout signe ou symptôme d’effets accrus ou prolongés ou d’effets indésirables du médicament associé, et de réduire sa posologie si nécessaire. Le cas échéant, il est recommandé de mesurer les concentrations plasmatiques du médicament co-administré.
Les interactions listées dans ces tableaux ont été caractérisées dans des études réalisées avec les doses recommandées d’itraconazole. Cependant, l’importance de l’interaction peut dépendre de la dose d’itraconazole administrée. Une interaction plus importante peut se produire à une dose supérieure ou avec un intervalle d’administration plus court. L’extrapolation des résultats à d’autres scénarios posologiques ou à d’autres médicaments doit être effectuée avec prudence.
Une fois le traitement arrêté, les concentrations plasmatiques d’itraconazole diminuent jusqu’à une concentration presque indétectable dans un délai de 7 à 14 jours, en fonction de la dose et de la durée du traitement. Chez les patients atteints de cirrhose hépatique ou chez les sujets recevant des inhibiteurs du CYP3A4, la diminution des concentrations plasmatiques peut être encore plus progressive. Ceci est particulièrement important lors de l’instauration d’un traitement avec des médicaments dont le métabolisme est affecté par l’itraconazole (voir rubrique 5.2).
Tableau 1 : Exemples de médicaments pouvant avoir un impact sur la concentration plasmatique d’itraconazole, présentés par classe de médicaments
|
Exemples de médicaments (par voie orale (VO) dose unique, sauf indication contraire) dans la classe |
Effet attendu/potentiel sur les taux d’itraconazole |
Commentaire clinique (voir ci-dessus pour des informations supplémentaires et également les rubriques 4.3 et 4.4) |
|
Antibactériens à usage systémique ; antimycobactériens |
||
|
Isoniazide |
Bien que n’ayant pas fait l’objet d’études directes, l’isoniazide est susceptible de diminuer les concentrations d’itraconazole |
Non recommandé |
|
Rifampicine VO 600 mg 1x/j |
Itraconazole ASC ↓ |
Non recommandé |
|
Rifabutine VO 300 mg 1x/j |
Itraconazole Cmax ↓ 71 %, ASC ↓ 74 % |
Non recommandé |
|
Ciprofloxacine VO 500 mg 2x/j |
Itraconazole Cmax ↑ 53 %, ASC ↑ 82 % |
Utiliser avec précaution |
|
Érythromycine 1 g |
Itraconazole Cmax ↑ 44 %, ASC ↑ 36 % |
Utiliser avec précaution |
|
Clarithromycine VO 500 mg 2x/j |
Itraconazole Cmax ↑ 90 %, ASC ↑ 92 % |
Utiliser avec précaution |
|
Antiépileptiques |
||
|
Carbamazépine, Phénobarbital |
Bien que n’ayant pas fait l’objet d’études directes, ces médicaments sont susceptibles de diminuer les concentrations d’itraconazole. |
Non recommandé |
|
Phénytoïne VO 300 mg 1x/j |
Itraconazole Cmax ↓ 83 %, ASC ↓ 93 % |
Non recommandé |
|
Agents antinéoplasiques |
||
|
Idélalisib |
Bien que n’ayant pas fait l’objet d’études directes, l’idélalisib est susceptible d’augmenter les concentrations d’itraconazole. |
Utiliser avec précaution |
|
Antiviraux à usage systémique |
||
|
Ombitasvir/Paritaprévir/Ritonavir (avec ou sans dasabuvir) |
Bien que n’ayant pas fait l’objet d’études directes, ces médicaments devraient augmenter les concentrations d’itraconazole. |
Contre-indiqué |
|
Éfavirenz 600 mg |
Itraconazole Cmax ↓ 37 %, ASC ↓ 39 % ; Hydroxy-itraconazole Cmax ↓ 35 %, ASC ↓ 37 % |
Non recommandé |
|
Névirapine VO 200 mg 1x/j |
Itraconazole Cmax ↓ 38 %, ASC ↓ 62 % |
Non recommandé |
|
Cobicistat, |
Bien que n’ayant pas fait l’objet d’études directes, ces médicaments sont susceptibles d’augmenter les concentrations d’itraconazole. |
Utiliser avec précaution |
|
Indinavir VO 800 mg 3x/j |
Concentration d’itraconazole ↑ |
Utiliser avec précaution |
|
Inhibiteurs des canaux calciques |
||
|
Diltiazem |
Bien que n’ayant pas fait l’objet d’études directes, le diltiazem est susceptible d’augmenter la concentration d’itraconazole. |
Utiliser avec précaution |
|
Médicaments pour les troubles d’acidité |
||
|
Antiacides (aluminium, calcium, magnésium ou bicarbonate de sodium), |
Itraconazole Cmax ↓, ASC ↓ |
Utiliser avec précaution |
|
Système respiratoire : autres médicaments du système respiratoire |
||
|
Lumacaftor/Ivacaftor |
Concentration d’itraconazole ↓ |
Non recommandé |
|
Divers |
||
|
Millepertuis (Hypericum perforatum) |
Bien que n’ayant pas fait l’objet d’études directes, le millepertuis est susceptible de diminuer la concentration d’itraconazole. |
Non recommandé |
Tableau 2 : Exemples de médicaments dont la concentration plasmatique peut être impactée par l’itraconazole, présentés par classe de médicaments
|
Exemples de médicaments (VO dose unique, sauf indication contraire) dans la classe |
Effet attendu/potentiel sur les taux de médicament |
Commentaire clinique (voir ci-dessus pour des informations supplémentaires et également les rubriques 4.3 et 4.4) |
|
Analgésiques ; anesthésiques |
||
|
Alcaloïdes de l’ergot de seigle (par exemple, dihydroergotamine, ergométrine, ergotamine, méthylergométrine) |
Bien que n’ayant pas fait l’objet d’études directes, l’itraconazole est susceptible d’augmenter les concentrations de ces médicaments. |
Contre-indiqué |
|
Élétriptan, Fentanyl |
Bien que n’ayant pas fait l’objet d’études directes, l’itraconazole est susceptible d’augmenter les concentrations de ces médicaments. |
Non recommandé |
|
Alfentanil, |
Bien que n’ayant pas fait l’objet d’études directes, l’itraconazole est susceptible d’augmenter les concentrations de ces médicaments. |
Utiliser avec précaution |
|
Oxycodone VO 10 mg, |
Oxycodone VO : Cmax ↑ 45 %, ASC ↑ 2,4 fois |
Utiliser avec précaution |
|
Oxycodone IV 0,1 mg/kg |
Oxycodone IV : ASC ↑ 51 % |
Utiliser avec précaution |
|
Antibactériens à usage systémique ; antimycobactériens ; antimycotiques à usage systémique |
||
|
Isavuconazole |
Bien que n’ayant pas fait l’objet d’études directes, l’itraconazole est susceptible d’augmenter les concentrations d’isavuconazole. |
Contre-indiqué |
|
Bédaquiline |
Bien que n’ayant pas fait l’objet d’études directes, l’itraconazole est susceptible d’augmenter les concentrations de bédaquiline. |
Non recommandé |
|
Rifabutine VO 300 mg 1x/j |
Concentration de rifabutine ↑ (ampleur inconnue) |
Non recommandé |
|
Clarithromycine VO 500 mg 2x/j |
Concentration de clarithromycine ↑ |
Utiliser avec précaution |
|
Délamanide |
Bien que n’ayant pas fait l’objet d’études directes, l’itraconazole est susceptible d’augmenter les concentrations de délamanide. |
Utiliser avec précaution |
|
Antiépileptiques |
||
|
Carbamazépine |
Bien que n’ayant pas fait l’objet d’études directes, l’itraconazole est susceptible d’augmenter les concentrations de carbamazépine. |
Non recommandé |
|
Agents anti-inflammatoires et antirhumatismaux |
||
|
Méloxicam 15 mg |
Méloxicam Cmax ↓ 64 %, ASC ↓ 37 % |
Utiliser avec précaution |
|
Anthelminthiques ; Antiprotozoaires |
||
|
Halofantrine |
Bien que n’ayant pas fait l’objet d’études directes, l’itraconazole est susceptible d’augmenter les concentrations de l’halofantrine. |
Contre-indiqué |
|
Artéméther/luméfantrine, Praziquantel |
Bien que n’ayant pas fait l’objet d’études directes, l’itraconazole est susceptible d’augmenter les concentrations de ces médicaments. |
Utiliser avec précaution |
|
Quinine 300 mg |
Quinine Cmax ↔, ASC ↑ 96 % |
Utiliser avec précaution |
|
Antihistaminiques à usage systémique |
||
|
Astémizole, Mizolastine, |
Bien que n’ayant pas fait l’objet d’études directes, l’itraconazole est susceptible d’augmenter les concentrations de ces médicaments. |
Contre-indiqué |
|
Ébastine 20 mg |
Ébastine Cmax ↑ 2,5 fois, ASC ↑ 6,2 fois |
Non recommandé |
|
Bilastine, Rupatadine |
Bien que n’ayant pas fait l’objet d’études directes, l’itraconazole est susceptible d’augmenter les concentrations de ces médicaments. |
Utiliser avec précaution |
|
Agents antinéoplasiques |
||
|
Entrectinib |
Entrectinib Cmax ↑ 73 % ASC ↑ 6,0 fois |
Non recommandé |
|
Irinotécan |
Bien que n’ayant pas fait l’objet d’études directes, l’itraconazole est susceptible d’augmenter les concentrations de l’irinotécan et de son métabolite actif. |
Contre-indiqué |
|
Pemigatinib |
Pemigatinib Cmax ↑ 17 % ASC ↑ 91% |
Utiliser avec précaution |
|
Vénétoclax |
Bien que n’ayant pas fait l’objet d’études directes, l’itraconazole est susceptible d’augmenter les concentrations de vénétoclax |
Contre-indiqué pendant la phase d’initiation et de titration de dose de vénétoclax chez les patients ayant une leucémie lymphoïde chronique. Autrement, non recommandé sauf si les bénéfices l’emportent sur les risques. Consulter les informations de prescription du vénétoclax. |
|
Axitinib, Bosutinib, |
Bien que n’ayant pas fait l’objet d’études directes, l’itraconazole est susceptible d’augmenter les concentrations de ces médicaments, à l’exception du cabazitaxel et du régorafénib. Aucune variation statistiquement significative de l’exposition au cabazitaxel n’a été observée, mais une importante variabilité dans les résultats a été observée. Une diminution de l’ASC du régorafénib est attendue (par estimation de la fraction active) |
Non recommandé |
|
Cobimétinib 10 mg, |
Cobimétinib Cmax ↑ 3,2 fois, ASC ↑ 6,7 fois |
Non recommandé |
|
Olaparib 100 mg |
Olaparib Cmax ↑ 40 %, ASC ↑ 2,7 fois |
Non recommandé |
|
Talazoparib |
Talazoparib Cmax ↑ 40 %, ASC ↑ 56 % |
Non recommandé |
|
Alitrétinoïne (oral), |
Bien que n’ayant pas fait l’objet d’études directes, l’itraconazole est susceptible d’augmenter les concentrations de ces médicaments. |
Utiliser avec précaution |
|
Busulfan 1 mg/kg toutes les 6 h |
Busulfan Cmax ↑, ASC ↑ |
Utiliser avec précaution |
|
Géfitinib 250 mg |
Géfitinib 250 mg Cmax ↑, ASC ↑ 78 % |
Utiliser avec précaution |
|
Agents antithrombotiques |
||
|
Dabigatran, Ticagrélor |
Bien que n’ayant pas fait l’objet d’études directes, l’itraconazole est susceptible d’augmenter les concentrations de ces médicaments. |
Contre-indiqué |
|
Apixaban, Edoxaban, Rivaroxaban, |
Bien que n’ayant pas fait l’objet d’études directes, l’itraconazole est susceptible d’augmenter les concentrations de ces médicaments. |
Non recommandé |
|
Cilostazol, |
Bien que n’ayant pas fait l’objet d’études directes, l’itraconazole est susceptible d’augmenter les concentrations de ces médicaments. |
Utiliser avec précaution |
|
Antiviraux à usage systémique |
||
|
Ombitasvir/Paritaprévir/Ritonavir (avec ou sans dasabuvir) |
L’itraconazole peut augmenter les concentrations de paritaprévir. |
Contre-indiqué |
|
Elbasvir/Grazoprévir, |
Bien que n’ayant pas fait l’objet d’études directes, l’itraconazole est susceptible d’augmenter les concentrations de ces médicaments. |
Non recommandé |
|
Cobicistat, |
Bien que n’ayant pas fait l’objet d’études directes, l’itraconazole est susceptible d’augmenter les concentrations de ces médicaments. |
Utiliser avec précaution |
|
Indinavir PO 800 mg 3x/j |
Indinavir Cmax ↔, ASC ↑ |
Utiliser avec précaution |
|
Système cardiovasculaire (Agents agissant sur le système rénine-angiotensine ; antihypertenseurs ; agents bêta-bloquants ; inhibiteurs des canaux calciques ; traitement cardiaque ; diurétiques) |
||
|
Bépridil, Disopyramide, |
Bien que n’ayant pas fait l’objet d’études directes, l’itraconazole est susceptible d’augmenter les concentrations de ces médicaments. |
Contre-indiqué |
|
Aliskiren 150 mg, |
Aliskiren Cmax ↑ 5,8 fois, ASC ↑ 6,5 fois |
Contre-indiqué |
|
Quinidine 100 mg |
Quinidine Cmax ↑ 59 %, ASC ↑ 2,4 fois |
Contre-indiqué |
|
Félodipine 5 mg |
Félodipine Cmax ↑ 7,8 fois, ASC ↑ 6,3 fois |
Non recommandé |
|
Riociguat, |
Bien que n’ayant pas fait l’objet d’études directes, l’itraconazole est susceptible d’augmenter les concentrations de ces médicaments. |
Non recommandé |
|
Bosentan, Diltiazem, |
Bien que n’ayant pas fait l’objet d’études directes, l’itraconazole est susceptible d’augmenter les concentrations de ces médicaments. |
Utiliser avec précaution |
|
Digoxine 0,5 mg |
Digoxine Cmax ↑ 34 %, ASC ↑ 68 % |
Utiliser avec précaution |
|
Nadolol 30 mg |
Nadolol Cmax ↑ 4,7 fois, ASC ↑ 2,2 fois |
Utiliser avec précaution |
|
Corticostéroïdes à usage systémique ; médicaments pour les maladies obstructives des voies respiratoires |
||
|
Ciclésonide, Salmétérol |
Bien que n’ayant pas fait l’objet d’études directes, l’itraconazole est susceptible d’augmenter les concentrations du salmétérol et du métabolite actif du ciclésonide. |
Non recommandé |
|
Budésonide INH 1 mg SD, |
Budésonide INH Cmax ↑ 65 %, ASC ↑ 4,2 fois ; Budésonide (autres formulations) concentration ↑ |
Utiliser avec précaution |
|
Dexaméthasone IV 5 mg |
Dexaméthasone IV : Cmax ↔, ASC ↑ 3,3 fois |
Utiliser avec précaution |
|
Fluticasone INH 1 mg 2x/j, |
Fluticasone INH concentration ↑ |
Utiliser avec précaution |
|
Méthylprednisolone 16 mg |
Méthylprednisolone VO Cmax ↑ 92 %, ASC ↑ 3,9 fois |
Utiliser avec précaution |
|
Fluticasone nasal |
Bien que n’ayant pas fait l’objet d’études directes, l’itraconazole est susceptible d’augmenter les concentrations du fluticasone administré par voie nasale. |
Utiliser avec précaution |
|
Médicaments utilisés dans le traitement du diabète |
||
|
Répaglinide 0,25 mg |
Répaglinide Cmax ↑ 47 %, ASC ↑ 41 % |
Utiliser avec précaution |
|
Saxagliptine |
Bien que n’ayant pas fait l’objet d’études directes, l’itraconazole est susceptible d’augmenter les concentrations de la saxagliptine. |
Utiliser avec précaution |
|
Médicaments gastrointestinaux, y compris antidiarrhéiques, anti-inflammatoires intestinaux/agents anti-infectieux ; antiémétiques et anti-nauséeux ; médicaments pour le traitement de la constipation ; médicaments pour le traitement des troubles gastro-intestinaux fonctionnels |
||
|
Cisapride, Naloxégol |
Bien que n’ayant pas fait l’objet d’études directes, l’itraconazole est susceptible d’augmenter les concentrations de ces médicaments. |
Contre-indiqué |
|
Dompéridone 20 mg |
Dompéridone Cmax ↑ 2,7 fois, ASC ↑ 3,2 fois |
Contre-indiqué |
|
Aprépitant, Lopéramide, |
Bien que n’ayant pas fait l’objet d’études directes, l’itraconazole est susceptible d’augmenter les concentrations de ces médicaments. |
Utiliser avec précaution |
|
Immunosuppresseurs |
||
|
Voclosporine |
Bien que n’ayant pas fait l’objet d’études directes, l’itraconazole est susceptible d’augmenter les concentrations de voclosporine |
Contre-indiqué |
|
Sirolimus (rapamycine) |
Bien que n’ayant pas fait l’objet d’études directes, l’itraconazole est susceptible d’augmenter les concentrations de sirolimus. |
Non recommandé |
|
Ciclosporine, Tacrolimus |
Bien que n’ayant pas fait l’objet d’études directes, l’itraconazole est susceptible d’augmenter les concentrations de ces médicaments. |
Utiliser avec précaution |
|
Tacrolimus IV 0,03 mg/kg 1x/j |
Tacrolimus IV concentration ↑ |
Utiliser avec précaution |
|
Agents modifiant les lipides |
||
|
Lomitapide |
Bien que n’ayant pas fait l’objet d’études directes, l’itraconazole est susceptible d’augmenter les concentrations de lomitapide. |
Contre-indiqué |
|
Lovastatine 40 mg |
Lovastatine Cmax ↑ 14,5->20 fois, ASC ↑ > 14,8 - > 20 fois |
Contre-indiqué |
|
Simvastatine 40 mg |
Simvastatine acide Cmax ↑ 17 fois, ASC ↑ 19 fois |
Contre-indiqué |
|
Atorvastatine |
Atorvastatine acide : Cmax ↔ à ↑2,5 fois, ASC ↑ 40 % à 3 fois |
Non recommandé |
|
Psychoanaleptiques ; Psycholeptiques (par exemple, antipsychotiques, anxiolytiques et hypnotiques) |
||
|
Lurasidone, Pimozide, Quétiapine, Sertindole |
Bien que n’ayant pas fait l’objet d’études directes, l’itraconazole est susceptible d’augmenter les concentrations de ces médicaments. |
Contre-indiqué |
|
Midazolam (oral) 7,5 mg |
Midazolam (oral) Cmax ↑ 2,5 à 3,4 fois, ASC ↑ 6,6 à 10,8 fois |
Contre-indiqué |
|
Triazolam 0,25 mg |
Triazolam Cmax ↑, ASC ↑ |
Contre-indiqué |
|
Alprazolam 0,8 mg |
Alprazolam Cmax ↔, ASC ↑ 2,8 fois |
Utiliser avec précaution |
|
Aripiprazole 3 mg |
Aripiprazole Cmax ↑ 19 %, ASC ↑ 48% |
Utiliser avec précaution |
|
Brotizolam 0,5 mg |
Brotizolam Cmax ↔, ASC ↑ 2,6 fois |
Utiliser avec précaution |
|
Buspirone 10 mg |
Buspirone Cmax ↑ 13,4 fois, ASC ↑ 19,2 fois |
Utiliser avec précaution |
|
Midazolam (IV) 7,5 mg |
Midazolam (IV) 7,5 mg : concentration ↑ ; |
Utiliser avec précaution |
|
Rispéridone 2-8 mg/jour |
Rispéridone et métabolite actif concentration ↑ |
Utiliser avec précaution |
|
Zopiclone 7,5 mg |
Zopiclone Cmax ↑ 30 %, ASC ↑ 70 % |
Utiliser avec précaution |
|
Eszopiclone |
Bien que n’ayant pas fait l’objet d’études directes, l’itraconazole est susceptible d’augmenter les concentrations de ce médicament |
Augmentation de l'effet sédatif de l’eszopiclone. Contre-indiqué chez les patients âgés. En cas d’association chez les sujets non âgés, une réduction de la dose d’eszopiclone peut être nécessaire. |
|
Cariprazine, Galantamine, |
Bien que n’ayant pas fait l’objet d’études directes, l’itraconazole est susceptible d’augmenter les concentrations de ces médicaments. |
Utiliser avec précaution |
|
Système respiratoire : autres médicaments du système respiratoire |
||
|
Lumacaftor/Ivacaftor VO 200/250 mg 2x/j |
Ivacaftor Cmax ↑ 3,6 fois, ASC ↑ 4,3 fois |
Non recommandé |
|
Ivacaftor |
Bien que n’ayant pas fait l’objet d’études directes, l’itraconazole est susceptible d’augmenter les concentrations d’ivacaftor. |
Utiliser avec précaution |
|
Hormones sexuelles et modulateurs du système génital ; autres médicaments gynécologiques |
||
|
Cabergoline, Diénogest, |
Bien que n’ayant pas fait l’objet d’études directes, l’itraconazole est susceptible d’augmenter les concentrations de ces médicaments. |
Utiliser avec précaution |
|
Urologiques |
||
|
Avanafil, Dapoxétine, |
Bien que n’ayant pas fait l’objet d’études directes, l’itraconazole est susceptible d’augmenter les concentrations de ces médicaments. |
Contre-indiqué |
|
Fésotérodine |
Bien que n’ayant pas fait l’objet d’études directes, l’itraconazole est susceptible d’augmenter les concentrations des métabolites actifs, 5-hydroxyméthyl toltérodine. |
Insuffisance rénale ou hépatique modérée à sévère : contre-indiqué. Insuffisance rénale ou hépatique légère : l’utilisation concomitante doit être évitée. Fonction rénale ou hépatique normale : utiliser avec précaution, avec une dose maximale de fésotérodine de 4 mg. |
|
Solifénacine |
Bien que n’ayant pas fait l’objet d’études directes, l’itraconazole est susceptible d’augmenter les concentrations de solifénacine. |
Insuffisance rénale sévère : contre-indiqué. Insuffisance hépatique modérée à sévère : contre-indiqué. Utiliser avec précaution chez tous les autres patients avec une dose maximale de solifénacine de 5 mg. |
|
Vardénafil |
Bien que n’ayant pas fait l’objet d’études directes, l’itraconazole est susceptible d’augmenter les concentrations de vardénafil. |
Contre-indiqué chez les patients de plus de 75 ans ; autrement, non recommandé. |
|
Alfuzosine, Silodosine, |
Bien que n’ayant pas fait l’objet d’études directes, l’itraconazole est susceptible d’augmenter les concentrations de ces médicaments. |
Non recommandé |
|
Dutastéride, Imidafénacine, |
Bien que n’ayant pas fait l’objet d’études directes, l’itraconazole est susceptible d’augmenter les concentrations de ces médicaments. |
Utiliser avec précaution |
|
Oxybutynine 5 mg |
Oxybutynine Cmax ↑ 2 fois, ASC ↑ 2 fois Après une administration transdermique : Bien que cela n’ayant pas fait l’objet d’études directes, l’itraconazole est susceptible d’augmenter les concentrations d’oxybutynine après une administration transdermique. |
Utiliser avec précaution |
|
Médicaments divers et autres substances |
||
|
Colchicine |
Bien que n’ayant pas fait l’objet d’études directes, l’itraconazole est susceptible d’augmenter les concentrations de colchicine |
Contre-indiqué chez les patients atteints d’insuffisance rénale ou hépatique. Non recommandé chez les autres patients. |
|
Éliglustat |
Bien que n’ayant pas fait l’objet d’études directes, l’itraconazole est susceptible d’augmenter les concentrations d’éliglustat. |
Contre-indiqué chez les métaboliseurs lents (ML) du CYP2D6. Contre-indiqué chez les métaboliseurs intermédiaires (MI) du CYP2D6 ou les métaboliseurs rapides (MR) prenant un inhibiteur puissant ou modéré du CYP2D6. Utiliser avec précaution chez les MI et MR du CYP2D6. |
|
Cinacalcet |
Bien que n’ayant pas fait l’objet d’études directes, l’itraconazole est susceptible d’augmenter les concentrations de cinacalcet. |
Utiliser avec précaution |
4.6. Fertilité, grossesse et allaitement
Grossesse
Les études effectuées chez l’animal ont mis en évidence un effet tératogène (voir rubrique 5.3).
Il a été montré que l’itraconazole traverse le placenta dans un modèle chez le rat.
Des cas d'anomalies congénitales ont été rapportés au cours de la commercialisation de Sporanox, sans qu’aucun lien de cause à effet avec la prise de Sporanox n'ait été établi. Ces cas incluaient notamment des malformations du squelette et des altérations chromosomiques.
Les données cliniques sur l'utilisation de Sporanox pendant la grossesse sont globalement limitées. Par conséquent, Sporanox solution buvable ne doit pas être utilisé pendant la grossesse, sauf dans les pathologies mettant en jeu le pronostic vital, où le bénéfice potentiel pour la mère l'emporte sur le préjudice potentiel pour le fœtus (voir rubrique 4.3).
Femmes en âge de procréer
Les femmes en âge de procréer, et traitées par Sporanox, solution buvable doivent utiliser un moyen de contraception efficace. Une contraception efficace doit être poursuivie jusqu’au début des règles suivant l’arrêt du traitement par Sporanox.
Allaitement
En raison de l’excrétion de l’itraconazole dans le lait maternel et compte-tenu de son profil d’effets secondaires, l’allaitement est déconseillé en cas de traitement par ce médicament.
Fertilité
Les données animales chez le rat n’ont pas mis en évidence un effet de l’itraconazole sur la fertilité mâle ou femelle.
4.7. Effets sur l'aptitude à conduire des véhicules et à utiliser des machines
Résumé du profil de tolérance
Les effets indésirables les plus fréquemment rapportés au cours du traitement par Sporanox solution buvable dans les essais cliniques et/ou à partir des notifications spontanées ont été sensation de vertige, céphalées, dysgueusie, dyspnée, toux, troubles gastro-intestinaux (diarrhée, nausée, vomissement, douleur abdominale, dyspepsie), rash et pyrexie. Les effets indésirables les plus sévères ont été les réactions allergiques sévères, l’insuffisance cardiaque/l’insuffisance cardiaque congestive/œdème pulmonaire, les pancréatites, l’hépatotoxicité sévère (incluant quelques cas d’insuffisance hépatique aiguë d’issue fatale) et réactions cutanées sévères. Se référer à la sous-rubrique « Liste tabulée des effets indésirables » pour les fréquences et pour les autres effets indésirables observés. Se référer à la rubrique 4.4 pour des informations complémentaires sur les autres effets sévères.
Liste tabulée des effets indésirables
Les effets indésirables décrits dans le tableau suivant ont été observés lors des études cliniques réalisées avec Sporanox solution buvable chez 889 patients dans le traitement des candidoses œsophagiennes et oropharyngées, et/ou sont issus des notifications spontanées rapportées après la commercialisation de l’itraconazole, toute formulation confondue.
Le tableau ci-dessous présente les effets indésirables classés par Système organe et fréquence selon la convention suivante : très fréquent (≥ 1/10), fréquent (≥ 1/100 à < 1/10), peu fréquent (≥ 1/1 000 à < 1/100), rare (≥ 1/10 000 à < 1/1 000), très rare (< 1/10 000) et fréquence indéterminée (ne peut être estimée sur la base des données disponibles).
|
Classe de systèmes organes |
Effets indésirables |
||
|
Fréquent (≥ 1/100 à < 1/10) |
Peu fréquent (≥ 1/1 000 à < 1/100) |
Fréquence indéterminée |
|
|
Affections hématologiques et du système lymphatique |
|
Leucopénie, Neutropénie, Thrombopénie |
|
|
Affections du système immunitaire |
|
Hypersensibilité* |
Maladie sérique, Œdème de Quincke, Réaction anaphylactiques et anaphylactoïdes |
|
Affections endocriniennes |
|
|
Pseudohyperaldostéronisme |
|
Troubles du métabolisme et de la nutrition |
|
Hypokaliémie |
Hypertriglycéridémie |
|
Affections du système nerveux |
Sensation vertigineuse, Céphalées, Dysgueusie |
Neuropathie périphérique*, Paresthésie, Hypoesthésie |
Tremblements, Vertiges** |
|
Affections oculaires |
|
Troubles visuels (incluant diplopie et vision floue) |
|
|
Affections de l'oreille et du labyrinthe |
|
Acouphène |
Perte auditive passagère ou permanente* |
|
Affections cardiaques |
|
Insuffisance cardiaque |
Insuffisance cardiaque congestive*, Bradycardie |
|
Affections respiratoires, thoraciques et médiastinales |
Dyspnée, Toux |
|
Œdème pulmonaire |
|
Affections gastro-intestinales |
Douleurs abdominales, Diarrhées, Vomissements, Nausées, Dyspepsie |
Constipation |
Pancréatite, Flatulence** |
|
Affections hépatobiliaires |
|
Augmentation des enzymes hépatiques (notamment ALAT, ASAT), Insuffisance hépatique*, Hépatite*, Hyperbilirubinémie |
Hépatotoxicité sévère (incluant quelques cas d’insuffisance hépatique aiguë d’issue fatale)* |
|
Affections de la peau et du tissu sous-cutané |
Rash |
Urticaire, Prurit |
Nécrolyse épidermique toxique, Syndrome de Stevens-Johnson, Pustulose exanthématique aiguë généralisée, Erythème polymorphe, Dermatite exfoliative, Vascularite leucocytoclasique, Alopécie, Photosensibilité, |
|
Affections musculo-squelettiques et systémiques |
|
Myalgie, Arthralgie |
|
|
Affections du rein et des voies urinaires |
|
|
Pollakiurie, Incontinence urinaire |
|
Affections des organes de reproduction et du sein |
|
Troubles menstruels |
Dysfonction érectile |
|
Troubles généraux et anomalies au site d'administration |
Pyrexie |
Œdème |
|
|
Investigations |
|
|
Augmentation de la créatine phosphokinase sanguine |
* Voir rubrique 4.4
** Effet(s) indésirables associés à l’itraconazole dans les essais cliniques avec Sporanox gélule
Population pédiatrique
Le profil de sécurité d’emploi de Sporanox solution buvable a été évalué chez 250 patients pédiatriques âgés de 6 mois à 14 ans ayant participé à cinq essais cliniques en ouvert.
Sur la base des données de sécurité poolées issues de ces essais cliniques, les effets indésirables très fréquemment rapportés chez les patients pédiatriques ayant reçu au moins une dose de Sporanox solution buvable en prophylaxie des infections fongiques ou pour le traitement d’un muguet buccal ou d’infections fongiques systémiques ont été vomissement (36,0%), pyrexie (30,8%), diarrhée (28,4%), inflammation des muqueuses (23,2%), rash (22,8%), douleur abdominale (17,2%), nausée (15,6%), hypertension (14,0%) et toux (11,2%). La nature des effets indésirables chez les patients pédiatriques est similaire à celle observée chez les patients adultes, mais la fréquence est plus élevée chez les patients pédiatriques.
Déclaration des effets indésirables suspectés
La déclaration des effets indésirables suspectés après autorisation du médicament est importante. Elle permet une surveillance continue du rapport bénéfice/risque du médicament. Les professionnels de santé déclarent tout effet indésirable suspecté via le système national de déclaration : Agence nationale de sécurité du médicament et des produits de santé (ANSM) et réseau des Centres Régionaux de Pharmacovigilance - Site internet : https://signalement.social-sante.gouv.fr.
De manière générale, les effets indésirables rapportés en cas de surdosage ont été cohérents avec ceux rapportés lors de l’utilisation de l’itraconazole (voir rubrique 4.8).
Traitement
En cas de surdosage, un traitement symptomatique sera mis en place.
L'itraconazole n'est pas éliminé par hémodialyse.
Il n'existe aucun antidote spécifique.
5. PROPRIETES PHARMACOLOGIQUES
5.1. Propriétés pharmacodynamiques
Classe pharmacothérapeutique : Antimycosiques à usage systémique, dérivés triazolés et tétrazolés.
Code ATC : J02A C02
Mécanisme d’action
L’itraconazole inhibe l’enzyme fongique lanosterol 14 alpha-demethylase (CYP51), qui catalyse une étape indispensable de la biosynthèse de l’ergostérol, un constituant essentiel de la membrane fongique.
Relation pharmacocinétique (PK) / pharmacodynamique (PD)
Pour les agents triazolés antifongiques, incluant l’itraconazole, le ratio de l’aire sous la courbe des concentrations en fonction du temps/concentration minimale inhibitrice (ASC/CMI) est le paramètre le plus étroitement associé à l’efficacité.
Microbiologie
L’itraconazole a démontré être actif in vitro contre les microorganismes suivants :
Candida spp. (dont Candida albicans, C. tropicalis, C. parapsilosis et Candida dubliniensis).
Candida krusei, Candida glabrata et Candida guillermondii sont généralement les espèces de Candida les moins sensibles, avec quelques isolats ayant montré in vitro une résistance sans équivoque à l’itraconazole
Résistance
Les principaux mécanismes de résistance incluent l’acquisition de mutations entrainant la surexpression du gène codant pour l’enzyme cible lanosterol 14 alpha-demethylase, des mutations ponctuelles résultant en une substitution d’acides aminés de l’enzyme cible conduisant à une diminution de l’affinité pour la cible et/ou une surexpression du transporteur, responsables d’une augmentation de l’efflux.
Concentrations critiques
Les concentrations critiques de l’itraconazole ont été établies dans le document « European Committee on Antimicrobial Susceptibility Testing (EUCAST) breakpoints », version 10.0, valide à compter du 04/02/2020.
|
Espèces de Candida et d’Aspergillus |
Concentration critique de CMI (mg/L) |
|
|
≤ S (sensible) |
> R (résistant) |
|
|
Candida albicans |
0,06 |
0,06 |
|
Candida dubliniensis |
0,06 |
0,06 |
|
Candida parapsilosis |
0,125 |
0,125 |
|
Candida tropicalis |
0,125 |
0,125 |
|
Aspergillus flavus1,2 |
1 |
1 |
|
Aspergillus fumigatus1,2 |
1 |
1 |
|
Aspergillus nidulans1,2 |
1 |
1 |
|
Aspergillus terreus1,2 |
1 |
1 |
Les preuves actuelles sont insuffisantes pour établir des concentrations critiques cliniques pour Candida glabrata3, C. krusei3, C. guilliermondi3, Cryptococcus neoformans et des concentrations critiques non liées à l’espèce pour Candida.
Les preuves actuelles sont insuffisantes pour établir des concentrations critiques cliniques pour Aspergillus niger4,5 et des concentrations critiques non liées à l’espèce pour Aspergillus spp5.
1 Il est recommandé de surveiller les concentrations minimales d’azole chez les patients traités pour une infection fongique.
2 La zone d’incertitude technique (ATU) est : 2. Reporter comme résultat Résistant avec le commentaire suivant : « Dans certaines situations cliniques (formes d’infections non invasives), l’itraconazole peut être utilisé dans la mesure où une exposition suffisante est assurée ».
3 Les valeurs de seuil épidémiologique (ECOFF) pour ces espèces sont généralement supérieures à celles de C. albicans.
4 Les valeurs de seuil épidémiologique (ECOFF) pour ces espèces se situent généralement à une dilution au demi au-dessus de celle d’A. fumigatus.
5 Les valeurs de CMI pour les isolats d’A. niger et d’A. versicolor sont généralement supérieures à celles d’A. fumigatus. Il reste inconnu si cela se traduit par une réponse clinique plus limitée.
Les concentrations critiques d’interprétation de l’itraconazole pour les espèces de Candida et pour les champignons filamenteux n’ont pas été établies par les méthodes du Clinical and Laboratory Standards (CLSI), M60, Performance Standards for Antifungal Susceptibility Testing of Yeasts. 2e édition, 2020.
5.2. Propriétés pharmacocinétiques
Caractéristiques pharmacocinétiques générales
Le pic plasmatique des concentrations d’itraconazole est atteint 2,5 heures après administration de la solution buvable.
En raison de sa pharmacocinétique non linéaire, l’itraconazole s’accumule dans le plasma après administrations répétées. Les concentrations plasmatiques à l’état d’équilibre sont généralement atteintes en approximativement 15 jours avec des valeurs Cmax et ASC 4 à 7 fois plus élevées que celles observées après administration unique.
A l’état d’équilibre, une valeur Cmax d’environ 2 µg/mL est atteinte après administration orale de 200 mg par jour.
La demi-vie d’élimination terminale de l’itraconazole varie généralement de 16 à 28 heures après une administration unique et augmente pour atteindre 34 à 42 heures après administrations répétées. Lorsque le traitement est arrêté, les concentrations plasmatiques en itraconazole diminuent pour atteindre des concentrations presque indétectables en 7 jours à 14 jours, suivant la dose et de la durée du traitement.
La clairance plasmatique totale moyenne de l’itraconazole après administration intraveineuse est de 278 mL/min. La clairance de l’itraconazole diminue aux doses élevées, du fait d’une saturation de son métabolisme hépatique.
Absorption
L’itraconazole est rapidement résorbé après administration de la solution buvable. Le pic plasmatique de l’itraconazole est atteint environ 2,5 heures après administration à jeun de la solution buvable.
La biodisponibilité absolue observée de l’itraconazole administré en présence de nourriture est d’environ 55% et augmente de 30% quand la solution buvable est administrée à jeun.
L’exposition à l’itraconazole est plus élevée avec la solution buvable qu’avec les gélules lorsque la même dose est administrée (voir rubrique 4.4)
Dans certains groupes de patients, la biodisponibilité de l’itraconazole est diminuée d’un facteur 1,5 à 2,5 (sujet HIV, neutropénique, mycose systémique à Aspergillus) pouvant atteindre 3 à 5 (dialyse péritonéale, mycose systémique à Cryptococcus). L’ensemble de ces données se traduit par une très forte variabilité interindividuelle du paramètre biodisponibilité.
Distribution
La majorité de l’itraconazole est liée aux protéines plasmatiques (99,8%) et plus particulièrement à l’albumine (99,6% pour le métabolite hydroxylé). Il présente également une affinité pour les lipides. Seulement 0,2% de l’itraconazole plasmatique est libre.
Le volume de distribution est élevé (700 l) et indique une large pénétration tissulaire associée à un fort tropisme cellulaire.
Les concentrations dans les poumons, les reins, le foie, les os, l’estomac, la rate et les muscles trouvées ont été deux à trois plus élevées que celles dans le plasma. La recapture par les tissus kératineux, et plus particulièrement par la peau est jusqu’à 4 fois plus importante que dans le plasma.
Les concentrations dans le liquide cérébrospinal sont bien plus faibles que dans le plasma.
Biotransformation
L’itraconazole est métabolisé de façon extensive dans le foie en de nombreux métabolites. Les études in vitro ont montré que le CYP 3A4 est la principale enzyme impliquée dans le métabolisme de l’itraconazole.
Le métabolite principal est l’hydroxy-itraconazole qui possède in vitro une activité antifongique comparable à celle de l’itraconazole. Les taux plasmatiques de ce métabolite sont environ 2 fois plus élevés que ceux de l’itraconazole.
Excrétion
L’itraconazole est principalement éliminé sous forme de métabolites inactifs dans les urines (35%) et dans les fécès (54%) dans la semaine après l’administration d’une dose sous forme de solution buvable. L’excrétion rénale de l’itraconazole et de son métabolite actif, l’hydroxy-itraconazole, représente moins de 1% de la dose intraveineuse administrée. Sur la base d’une dose orale d’itraconazole radio marquée, l’excrétion fécale du produit inchangé varie entre 3 et 18% de la dose administrée.
Populations particulières
Insuffisants hépatiques
L’itraconazole est principalement métabolisé par le foie. Une étude pharmacocinétique a été conduite chez 6 sujets sains et 12 sujets cirrhotiques ayant reçu une seule dose d’itraconazole (gélule de 100 mg). Une diminution statistiquement significative de la Cmax moyenne de l’itraconazole (47%) et un doublement de sa demi-vie d’élimination (37± 17 heures versus 16 ± 5 heures) ont été observés chez les sujets cirrhotiques comparativement aux sujets sains.
Toutefois, l’exposition globale à l’itraconazole, calculée sur la base de l’ASC était similaire chez les patients cirrhotiques et chez les sujets sains. Les données sur l’utilisation à long terme de l’itraconazole chez des patients cirrhotiques ne sont pas disponibles (voir rubriques 4.2 et 4.4).
Insuffisants rénaux
Les données disponibles sur l’utilisation de l’itraconazole par voie orale chez des patients insuffisants rénaux sont limitées. Une étude pharmacocinétique dans laquelle l’itraconazole a été administré à la dose unique de 200 mg (quatre gélules de 50 mg) a été conduite dans trois groupes de patients ayant une insuffisance rénale (urémie : n=7 ; hémodialyse : n=7 ; et dialyse péritonéale continue ambulatoire : n=5). Chez les patients urémiques avec une clairance de la créatinine moyenne de 13 mL/min x 1,73 m2, l’exposition, calculée sur la base de l’ASC, était légèrement réduite comparée à celle de la population normale.
Cette étude n’a pas montré d’effet significatif de l’hémodialyse ou de la dialyse péritonéale continue ambulatoire sur la pharmacocinétique de l’itraconazole (Tmax, Cmax et ASC0-8h). Les profils des concentrations plasmatiques en fonction du temps ont montré une large variation interindividuelle entre les trois groupes.
Après administration intraveineuse d’une seule dose, les demi-vies d’élimination terminale moyennes de l’itraconazole chez les patients atteints d’une insuffisance rénale légère (définie dans l’étude par une ClCr de 50-79 mL/min), modérée (définie dans l’étude par une ClCr de 20-49 mL/min), et sévère (définie dans l’étude par une ClCr < 20 mL/min) étaient similaires à celles des sujets sains (intervalle de 42-49 heures chez les patients insuffisants rénaux versus 48 heures chez les patients sains). L’exposition globale à l’itraconazole, calculée sur la base de l’ASC, diminuait d’approximativement 30% et 40 % chez les patients insuffisants rénaux modérés et sévères respectivement, comparée aux sujets avec une fonction rénale normale.
Les données sur l’utilisation à long terme de l’itraconazole chez des patients insuffisants rénaux ne sont pas disponibles La dialyse n’a pas d’effet sur la demi-vie ou la clairance de l’itraconazole ou de l’hydroxy-itraconazole (voir rubriques 4.2 et 4.4).
Population pédiatrique
Deux études de pharmacocinétiques ont été conduites chez des enfants neutropéniques âgés de 6 mois à 14 ans. Lors de ces études, l’itraconazole en solution buvable a été administré à la dose de 5 mg/kg une ou deux fois par jour. L’exposition à l’itraconazole a été quelques peu plus élevée chez les enfants plus âgés (6 à 14 ans) comparés aux enfants plus jeunes. Chez tous les enfants, les concentrations plasmatiques efficaces d’itraconazole ont été atteintes entre 3 et 5 jours après l’instauration du traitement et ont été maintenues pendant toute la durée du traitement.
Hydroxypropyl- β -Cyclodextrine
La biodisponibilité orale de l’hydroxypropyl-β-cyclodextrine utilisée en tant qu’agent solubilisant est en moyenne plus faible que 0,5% et est similaire à celle de l’hydroxypropyl-β-cyclodextrine seule. Cette faible biodisponibilité orale de l’hydroxypropyl-β-cyclodextrine n’est pas modifiée par la présence de nourriture et est similaire après administration unique et administrations répétées.
5.3. Données de sécurité préclinique
· Hydroxypropyl-β-cyclodextrine (HP-β-CD)
Chez la souris, le rat et le chien, les études de toxicité aiguë et de toxicité par administration réitérée montrent une importante marge de sécurité de l'HP-β-CD administrée par voies orale et intraveineuse. La majorité des effets observés sont de caractère adaptatif (modifications histologiques du tractus urinaire, ramollissement des fécès dû à une rétention osmotique aqueuse au niveau du gros intestin, activation du système phagocytaire mononucléaire) et montrent une réversibilité satisfaisante.
De légères modifications hépatiques ont été observées à des doses correspondant à 30 fois la dose d'HP-β-CD proposée en clinique. L'HP-β-CD n'a pas d'effets sur la fertilité, ni d'effets embryotoxiques ou tératogènes directs et est dépourvue d'effets mutagènes.
Dans l'étude de carcinogénèse chez le rat, une augmentation de l'incidence des néoplasmes du gros intestin (à 5000 mg/kg/jour, 3 fois la dose maximale recommandée chez l’homme de 16 g sur la base de mg/m2/jour) et du pancréas exocrine (à toutes les doses testées de 500, 2 000 et 5 000 mg/kg/jour (0,3, 1,2 et 3 fois la dose maximale recommandée chez l’homme de 16 g sur la base de mg/m2/jour, respectivement)) a été observée.
Chez le rat, le développement de tumeurs du pancréas est dû à l'effet mitogène de la cholécystokine.
Ce phénomène n'a pas été observé dans l'étude de cancérogénèse réalisée chez la souris, ni dans l'étude de toxicité à 12 mois chez le chien ou dans l'étude de 2 ans chez le singe cynomolgus femelle. La cholécystokine ne semble pas avoir d'effet mitogène chez l'homme.
Lorsque l'on tient compte des surfaces corporelles, l'exposition à l'HP-β-CD chez l'homme à la posologie initiale recommandée en clinique pour ce médicament correspond à environ 1,7 fois l'exposition observée à la dose la plus basse dans l'étude réalisée chez le rat.
Bien qu'hypothétique, la survenue d'une telle pathologie chez l'homme ne peut être totalement éliminée.
En conséquence, une surveillance de l'amylasémie chez les sujets traités à long terme (pendant plusieurs mois) paraît nécessaire.
· Itraconazole
L'itraconazole a été étudié dans une batterie standard de tests de sécurité non clinique.
L’itraconazole n’est pas un cancérogène primaire chez le rat jusqu’à 13 mg/kg/jour (mâles) et 52 mg/kg/jour (femelles), ou chez la souris jusqu’à 80 mg/kg/jour, (1 fois la dose maximale recommandée chez l’homme en mg/m²/jour).
Les études de toxicité sub-chronique, conduites par voie orale chez le rat et le chien, à des doses d’itraconazole allant de 2,5 à 160 mg/kg/jour, ont révélé plusieurs organes /tissus cibles dont notamment le cortex surrénal, les ovaires, le foie et le système phagocytaire mononucléé. Des anomalies du métabolisme lipidique ont également été notées, se traduisant par des cellules xanthomateuses dans divers organes.
Aux doses de 40 et 80 mg/kg/jour chez le rat (1 et 2 fois la dose maximale recommandée chez l’homme en mg/m2/jour), les analyses histologiques du cortex surrénal ont montré un gonflement réversible avec une hypertrophie cellulaire de la zone réticulée et fasciculée, parfois associée à un amincissement de la zone glomérulée. Au niveau hépatique, des altérations réversibles de type vacuolisation des hépatocytes, ont été observées aux doses de 40 et 160 mg/kg/jour. Les modifications histologiques observées au niveau du système phagocytaire mononucléé consistent en la présence de macrophages présentant un contenu protéique accru dans divers tissus parenchymateux (poumons, foie, tissus lymphoïdes).
Dans une étude de toxicité par administration répétée conduite chez le chien juvénile, une baisse de la densité minérale osseuse globale a été observée après administration chronique de doses de 30 mg/kg/jour (6 fois la dose maximale recommandée chez l’homme en mg/m2/jour) d'itraconazole.
De même, dans plusieurs études de toxicité chronique conduites chez le rat, à des doses d’itraconazole allant de 10 à 80 mg/kg/jour, des anomalies osseuses ont été observées, se caractérisant par une diminution de l’activité des plaques osseuses, un amincissement du tissu osseux compact des os longs et une augmentation de la fragilité osseuse.
Cancérogénicité et mutagénicité
L’itraconazole ne s’est pas révélé génotoxique dans différents essais in vitro et in vivo.
Une étude de 23 mois conduite chez la souris n’a pas révélé de potentiel cancérogène de l’itraconazole jusqu’à la dose de 80 mg/kg/jour. Une autre étude de 24 mois conduite chez le rat n’a pas révélé de potentiel cancérogène de l’itraconazole jusqu’à la dose de 20 mg/kg/jour ; cependant à la dose la plus forte testée (80 mg/kg/jour), une augmentation de l’incidence de sarcome des tissus mous a été observée chez les rats mâles, pouvant possiblement être attribuée à des réactions inflammatoires, secondaires à une accumulation de cholestérol dans le tissu conjonctif.
Toxicologie de la reproduction
Chez le rat, l'itraconazole entraîne à des doses à partir de 40 mg/kg/jour (1 fois la dose maximale recommandée chez l’homme en mg/m²/jour), une augmentation dose-dépendante de la toxicité maternelle, de l'embryotoxicité et de la tératogénicité. Chez la souris, les mêmes observations sont faites pour des doses à partir de 80 mg/kg/jour (1 fois la dose maximale recommandée chez l’homme en mg/m²/jour).
Chez le rat, des anomalies majeures du squelette ont été observées dans la descendance ; et chez la souris, des encéphalocèles et des macroglossies.
Aucun effet tératogène n’a été mis en évidence chez le lapin jusqu’à une dose de 80 mg/kg/jour (4 fois la dose maximale recommandée chez l’homme en mg/m2/jour).
Fertilité
Les données animales disponibles chez le rat n’ont pas mis en évidence d’effet délétère de l'itraconazole sur la fertilité mâle ou femelle (voir rubrique 4.6).
2 ans.
Après première ouverture du flacon, ce médicament doit être conservé maximum un mois.
6.4. Précautions particulières de conservation
A conserver à une température ne dépassant pas 25°C.
6.5. Nature et contenu de l'emballage extérieur
150 mL en flacon (verre brun) avec fermeture de sécurité enfant (polypropylène) et joint (PE) avec gobelet doseur gradué (polypropylène).
6.6. Précautions particulières d’élimination et de manipulation
Sporanox solution buvable est fourni en flacon pourvu d'un bouchon avec fermeture de sécurité enfant. Pour l'ouvrir : appuyer sur le bouchon de plastique tout en le dévissant dans le sens inverse des aiguilles d'une montre.
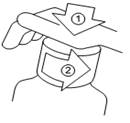
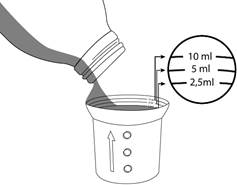
Un gobelet-doseur gradué est fourni avec Sporanox solution buvable. Utiliser ce gobelet-doseur dans le sens où il est posé sur le flacon. S’assurer que le côté avec les graduations (le côté avec le plus petit contenant) soit en haut. Si la flèche sur le côté pointe vers le haut, le gobelet-doseur est dans le bon sens.
Verser la solution buvable jusqu’à la graduation correspondant à la quantité à administrer prescrite.
Après chaque usage, laver le gobelet-doseur soigneusement à l'eau tiède.
7. TITULAIRE DE L’AUTORISATION DE MISE SUR LE MARCHE
167 QUAI DE LA BATAILLE DE STALINGRAD
92130 ISSY-LES-MOULINEAUX
8. NUMERO(S) D’AUTORISATION DE MISE SUR LE MARCHE
· 34009 345 020 6 4 : 150 mL en flacon (verre brun) avec gobelet-doseur gradué (polypropylène).
9. DATE DE PREMIERE AUTORISATION/DE RENOUVELLEMENT DE L’AUTORISATION
[à compléter ultérieurement par le titulaire]
10. DATE DE MISE A JOUR DU TEXTE
[à compléter ultérieurement par le titulaire]
12. INSTRUCTIONS POUR LA PREPARATION DES RADIOPHARMACEUTIQUES
Prescription initiale hospitalière annuelle.
ANSM - Mis à jour le : 02/07/2025
Sporanox 10 mg/mL solution buvable
Itraconazole
Veuillez lire attentivement cette notice avant de prendre ce médicament car elle contient des informations importantes pour vous.
· Gardez cette notice. Vous pourriez avoir besoin de la relire.
· Si vous avez d’autres questions, interrogez votre médecin, ou votre pharmacien.
· Ce médicament vous a été personnellement prescrit. Ne le donnez pas à d’autres personnes. Il pourrait leur être nocif, même si les signes de leur maladie sont identiques aux vôtres.
· Si vous ressentez un quelconque effet indésirable, parlez-en à votre médecin ou votre pharmacien. Ceci s’applique aussi à tout effet indésirable qui ne serait pas mentionné dans cette notice. Voir rubrique 4.
1. Qu'est-ce que Sporanox 10 mg/mL, solution buvable et dans quels cas est-il utilisé ?
2. Quelles sont les informations à connaître avant de prendre Sporanox 10 mg/mL, solution buvable ?
3. Comment prendre Sporanox 10 mg/mL, solution buvable ?
4. Quels sont les effets indésirables éventuels ?
5. Comment conserver Sporanox 10 mg/mL, solution buvable ?
6. Contenu de l’emballage et autres informations.
1. QU’EST-CE QUE Sporanox 10 mg/mL, solution buvable ET DANS QUELS CAS EST-IL UTILISE ?
Sporanox est indiqué dans le traitement de certaines mycoses (infections dues à des champignons microscopiques) localisées au niveau de la bouche, de la gorge et de l'œsophage, chez certaines personnes ayant un système de défense affaibli. Il peut être utilisé pour traiter ces infections ou vous empêcher de les contracter.
2. QUELLES SONT LES INFORMATIONS A CONNAITRE AVANT DE PRENDRE Sporanox 10 mg/mL, solution buvable ?
Ne prenez jamais Sporanox 10 mg/mL, solution buvable dans les cas suivants :
· Si vous êtes allergique (hypersensible) à l’itraconazole ou à l’un des composants contenus dans ce médicament, (mentionnés dans la rubrique 6).
· Si vous êtes enceinte ou si vous pensez être enceinte ou planifiez une grossesse (voir la rubrique sur la grossesse ci-dessous) (sauf si votre médecin sait que vous êtes enceinte et décide quand même de vous traiter par Sporanox car vous en avez besoin).
· Si vous êtes en âge de procréer, pour éviter la survenue d’une grossesse, vous devez utiliser un moyen de contraception efficace pendant votre traitement par Sporanox et le poursuivre jusqu’au début des règles suivant l’arrêt du traitement par Sporanox.
· Si vous présentez une insuffisance cardiaque (également appelée insuffisance cardiaque congestive), Sporanox peut l'aggraver. Cependant, votre médecin peut être amené à vous prescrire Sporanox en cas d'infections sévères ou mettant en jeu le pronostic vital.
Ne prenez jamais Sporanox si l’une des situations ci-dessus s’applique à vous. En cas de doute, consultez votre médecin ou pharmacien avant de prendre ce médicament.
Médicaments que vous ne devez pas prendre en même temps que Sporanox
Ne prenez jamais Sporanox si vous prenez certains médicaments (de même, ne pas prendre dans les 2 semaines suivant l’arrêt de Sporanox).
Exemples de ces médicaments :
Médicaments utilisés pour des problèmes cardiaques, sanguins ou circulatoires
· aliskiren, éplérénone, lercanidipine ou nisoldipine (pour l’hypertension artérielle)
· bépridil, ivabradine ou ranolazine – (pour l’angine de poitrine – douleur thoracique compressive)
· dabigatran ou ticagrélor (pour les caillots sanguins)
· disopyramide, dofétilide, dronédarone ou quinidine (pour les problèmes de rythme cardiaque irrégulier)
· finérénone (pour les problèmes rénaux chez les patients atteints de diabète de type 2)
· lomitapide, lovastatine ou simvastatine (pour diminuer le cholestérol)
· sildénafil en cas d’utilisation pour le traitement de l’hypertension pulmonaire (augmentation de la pression sanguine dans les vaisseaux sanguins des poumons)
Médicaments utilisés pour des problèmes d’estomac ou de constipation
· cisapride (pour les troubles de l’estomac)
· dompéridone (pour les nausées et les vomissements)
· naloxégol (pour les constipations causées par la prise d’analgésiques opiacés)
Médicaments utilisés pour des maux de tête, des troubles du sommeil ou mentaux
· dihydroergotamine ou ergotamine (alcaloïdes de l’ergot de seigle utilisés pour les migraines)
· eszopiclone chez les patients âgés, midazolam (pris par voie orale) ou triazolam (pour l’anxiétéou pour vous aider à dormir)
· lurasidone, pimozide, quétiapine ou sertindole (pour la schizophrénie, le trouble bipolaire ou d’autres troubles mentaux)
Médicaments utilisés pour des troubles de miction ou de vessie
· darifénacine (en cas de perte du contrôle de la vessie)
· fésotérodine ou solifénacine (pour l’irritation de la vessie) en cas d’utilisation chez des patients atteints de certains troubles rénaux ou hépatiques
Médicaments utilisés dans le traitement des allergies
· astémizole, mizolastine ou terfénadine (anti-histaminiques pour les allergies)
Médicaments utilisés pour des troubles de l’érection et de l’éjaculation
· avanafil (pour le trouble de l’érection)
· dapoxétine (pour l’éjaculation précoce)
· vardénafil (pour le trouble de l’érection) en cas d’utilisation chez les hommes âgés de plus de 75 ans
Autres médicaments contenant :
· colchicine (pour la goutte) en cas d’utilisation chez des patients atteints de troubles rénaux ou hépatiques
· alcaloïdes de l’ergot de seigle ergométrine (ergonovine) ou méthylergométrine (méthylergonovine) (utilisés après l’accouchement)
· éliglustat (pour la maladie de Gaucher) en cas d’utilisation chez des patients dont l’organisme ne peut pas dégrader certains médicaments dans le corps
· halofantrine (pour le paludisme)
· irinotécan (pour le cancer)
· isavuconazole (pour les infections fongiques)
· ombitasvir, paritaprévir, ritonavir avec ou sans dasabuvir – (pour le traitement de l’hépatite C).
· vénétoclax (pour la leucémie lymphoïde chronique) lorsque vous commencez un nouveau traitement par vénétoclax ou prenez des doses croissantes au début du traitement
· voclosporine (pour traiter les problèmes de reins liés au lupus)
Ne prenez jamais Sporanox si l’une des situations ci-dessus s’applique à vous. En cas de doute, consultez votre médecin avant de prendre Sporanox solution buvable. De même, lorsque vous avez terminé votre traitement par Sporanox, ne prenez aucun des médicaments mentionnés ci-dessus pendant 2 semaines.
Il ne s’agit pas d’une liste exhaustive, informez donc votre médecin si vous prenez ou prévoyez de prendre l’un de ces médicaments ou tout autre médicament.
EN CAS DE DOUTE, IL EST INDISPENSABLE DE DEMANDER L'AVIS DE VOTRE MEDECIN OU DE VOTRE PHARMACIEN.
Avertissements et précautions
Ce médicament NE DOIT GENERALEMENT PAS être pris :
· Si vous allaitez votre enfant (voir rubrique « Grossesse et Allaitement »).
· Si vous avez une intolérance au fructose (maladie héréditaire rare) car ce médicament contient du sorbitol.
Faites attention aux effets indésirables graves
Arrêtez de prendre Sporanox et contactez immédiatement votre médecin si, lors de la prise de ce médicament :
· vous avez des symptômes tels que perte d'appétit sévère, fatigue inhabituelle, douleur d’estomac, urines très foncées, ou selles décolorées, selles décolorées ou si vous vous sentez ou êtes malade – cela peut être le signe d'un grave problème de foie.
· vous avez des sensations inhabituelles de fourmillements, d'engourdissements ou de faiblesse des mains ou des pieds ou si vous devenez sensible à la lumière.
· vous avez une perte auditive. Dans de très rares cas, des patients prenant Sporanox solution buvable ont rapportés une perte de l’audition temporaire ou permanente
Arrêtez de prendre Sporanox et contactez immédiatement votre médecin si l’une des situations ci-dessus s’applique à vous ou en cas de doute.
Consultez votre médecin avant de prendre Sporanox.
Vous devez consulter votre médecin avant de prendre Sporanox si vous avez déjà eu :
· une réaction allergique à un autre médicament antifongique (médicament agissant contre les champignons microscopiques).
· une maladie cardiaque. Celle-ci peut s’aggraver lors du traitement par Sporanox. Votre médecin peut vous parler des signes d’insuffisance cardiaque qu’il faut surveiller. Si votre médecin décide quand même de vous prescrire Sporanox, et si vous présentez des symptômes tels qu'essoufflement, œdème des membres inférieurs, fatigue inexpliquée, prise de poids inattendue ou difficulté à respirer en position couchée, notamment la nuit, arrêtez de prendre Sporanox et consultez immédiatement votre médecin.
· un problème quelconque au niveau du foie ou une jaunisse (jaunissement de la peau) car il se peut que la posologie de Sporanox doive être modifiée. Votre médecin peut vous informer des symptômes à surveiller et vous prescrire des examens sanguins. Il se peut également que vous ne puissiez pas prendre certains médicaments.
· une insuffisance rénale car il se peut que la posologie de Sporanox solution buvable doive être modifiée. Il se peut également que vous ne puissiez pas prendre certains médicaments.
· mucoviscidose – une maladie héréditaire qui affecte les poumons, le pancréas, le foie, le rein et les intestins.
Il n’est pas recommandé d’interchanger la forme solution buvable de Sporanox avec la forme gélule.
Enfants et adolescents
Chez les enfants et adolescents âgés de moins de 18 ans, ce médicament ne doit pas être utilisé sauf si votre médecin l’a expressément indiqué.
Ce médicament contient des cyclodextrines. Ne pas utiliser chez l’enfant de moins de 2 ans sauf recommandation contraire de votre médecin.
Ce médicament contient du propylène glycol. Si votre enfant a moins de 5 ans, demandez à votre médecin ou à votre pharmacien avant de lui donner ce médicament, en particulier s’il reçoit d’autres médicaments contenant du propylène glycol ou de l’alcool.
Autres médicaments et Sporanox 10 mg/mL, solution buvable
Informez votre médecin ou votre pharmacien si vous prenez, avez récemment pris ou pourriez prendre tout autre médicament.
Médicaments que vous ne devez pas prendre
Il existe certains médicaments que vous ne devez pas prendre en association avec Sporanox solution buvable – ils sont répertoriés ci-dessus sous le titre « Médicaments que vous ne devez pas prendre en même temps que Sporanox solution buvable ».
Médicaments qui ne sont pas recommandés
Votre médecin peut décider que vous ne devez pas prendre certains médicaments en même temps ou dans les 2 semaines qui suivent l’arrêt de traitement par Sporanox solution buvable.
Exemples de ces médicaments :
Médicaments utilisés pour des troubles cardiaques, sanguins ou circulatoires
· apixaban, édoxaban, rivaroxaban ou vorapaxar (pour les caillots sanguins)
· atorvastatine (pour diminuer le cholestérol)
· félodipine (pour l’hypertension artérielle)
· riociguat ou tadalafil en cas d’utilisation pour le traitement de l’hypertension pulmonaire (augmentation de la pression sanguine dans les vaisseaux sanguins des poumons)
Médicaments utilisés pour l’épilepsie, les maux de tête ou les troubles mentaux
· phénytoïne, carbamazépine ou phénobarbital (pour l’épilepsie)
· élétriptan (pour les migraines)
· millepertuis (Hypericum perforatum) (médicament à base de plantes utilisé dans le traitement des troubles mentaux)
Médicaments pour des troubles de la miction ou de la vessie
· tamsulosine (pour l’incontinence urinaire chez l’homme)
· toltérodine (pour l’irritation de la vessie)
Médicaments utilisés pour le cancer
· axitinib, bosutinib, cabazitaxel, cabozantinib, céritinib, cobimétinib, crizotinib, dabrafénib, dasatinib, docétaxel, entrectinib, glasdégib, ibrutinib, lapatinib, nilotinib, olaparib, pazopanib, régorafénib, sunitinib, talazoparib, trabectédine, trastuzumab emtansine, vénétoclax (lorsque vous prenez une dose stable de vénétoclax pour une leucémie lymphoïde chronique ou à tout moment du traitement pour une leucémie aiguë myéloïde) ou vinca-alcaloïdes (comme la vinflunine, la vinorelbine)
Médicaments utilisés pour la tuberculose
· bédaquiline, isoniazide, rifabutine ou rifampicine (pour la tuberculose)
Médicaments utilisés pour le VIH ou l’hépatite
· éfavirenz ou névirapine (pour le VIH/SIDA)
· elbasvir/grazoprévir, ténofovir alafénamide fumarate (TAF), fumarate de ténofovir disoproxil (FTD) (pour le VIH ou l’hépatite)
Médicaments utilisés pour l’inflammation, les troubles pulmonaires ou les allergies
· ciclésonide (pour l’inflammation, l’asthme et les allergies)
· ébastine (pour les allergies)
· salmétérol (pour l’asthme ou la bronchopneumopathie chronique obstructive (BPCO))
Médicaments utilisés pour des troubles de l’érection et de l’éjaculation
· tadalafil ou vardénafil (pour le trouble de l’érection) en cas d’utilisation chez les hommes âgés de 75 ans et moins
Autres médicaments contenant :
· colchicine (pour la goutte)
· fentanyl (pour la douleur)
· évérolimus, rapamycine (également appelée sirolimus) ou temsirolimus (administrés après une greffe d’organe)
· alfuzosine ou silodosine (pour l’hypertrophie bénigne de la prostate)
· lumacaftor/ivacaftor (pour la mucoviscidose)
Informez votre médecin avant de prendre Sporanox si vous prenez l’un des médicaments ci-dessus. De même, lorsque vous avez terminé votre traitement par Sporanox, vous devez attendre 2 semaines avant de prendre ces médicaments.
Il ne s’agit pas d’une liste exhaustive, informez votre médecin si vous prenez ou prévoyez de prendre l’un de ces médicaments, ou tout autre médicament.
Médicaments pour lesquels il peut être nécessaire de modifier la dose
Exemples de ces médicaments :
Médicaments utilisés pour des problèmes cardiaques, sanguins ou circulatoires
· bosentan (pour l’hypertension pulmonaire)
inhibiteurs des canaux calciques tels que les dihydropyridines comme l’amlodipine, l’isradipine, la nifédipine, la nimodipine, le diltiazem ou vérapamil (pour l’hypertension)
· cilostazol (pour les troubles circulatoires)
· « coumarines » telles que la warfarine (pour les caillots sanguins)
· digoxine (pour la fibrillation auriculaire)
· nadolol (pour l’hypertension pulmonaire ou l’angine de poitrine)
Médicaments utilisés pour des troubles de l’estomac ou de la diarrhée
· aprépitant ou nétupitant (pour les nausées et les vomissements)
· lopéramide (pour la diarrhée)
· antiacides comme l’aluminium, le calcium, le magnésium ou le bicarbonate de soude ; antagonistes des récepteurs H2 tels que la cimétidine, la ranitidine et les inhibiteurs de la pompe à protons comme le lansoprazole, l’oméprazole, le rabéprazole (pour le traitement des troubles de l’acidité de l’estomac)
Médicaments utilisés pour des troubles du sommeil ou des troubles mentaux
· alprazolam, brotizolam, buspirone ou midazolam administrés par injection dans une veine (pour l’anxiété ou pour vous aider à dormir)
· eszopiclone chez les patients non âgés ou zopiclone (pour vous aider à dormir)
· réboxétine ou venlafaxine (pour la dépression et l’anxiété)
· aripiprazole, cariprazine, halopéridol ou rispéridone (pour la schizophrénie, le trouble bipolaire ou d’autres troubles mentaux)
· galantamine (pour la maladie d’Alzheimer)
· guanfacine (pour le trouble du déficit de l’attention avec hyperactivité)
Médicaments utilisés pour les problèmes de miction et de vessie
· imidafénacine, fésotérodine, oxybutynine, solifénacine (pour l’irritation de la vessie)
Médicaments utilisés pour le cancer
· bortézomib, brentuximab védotine, busulfan, erlotinib, géfitinib, idélalisib, imatinib, nintédanib, panobinostat, pemigatinib, ponatinib, ruxolitinib, sonidégib ou trétinoïne (administrés par voie orale)
Médicaments utilisés pourles infections
· ciprofloxacine, clarithromycine ou érythromycine (pour les infections bactériennes)
· délamanide (pour la tuberculose)
· artéméther/luméfantrine ou quinine (pour le traitement du paludisme)
· praziquantel (pour la douve et les ténias)
Médicaments utilisés pour le VIH ou l’hépatite
· cobicistat, elvitégravir boosté, maraviroc, ritonavir, darunavir boosté par ritonavir, fosamprénavir boosté par ritonavir, indinavir ou saquinavir (pour le VIH)
· glécaprevir/pibrentasvir (pour l’hépatite)
Médicaments utilisés pour l’inflammation, les troubles pulmonaires, ou les allergies
· bilastine ou rupatadine (pour les allergies)
· méthylprednisolone ou dexaméthasone (médicaments administrés par voie orale ou par injection pour l’inflammation, l’asthme et les allergies
· budésonide ou fluticasone (pour l’asthme et les allergies)
Médicaments utilisés pour des troubles de l’érection et de l’éjaculation
· sildénafil (pour les troubles de l’érection)
Médicaments utilisés pour la douleur
· alfentanil, buprénorphine, oxycodone ou sufentanil (pour la douleur)
· méloxicam (pour l’inflammation articulaire et la douleur)
Autres médicaments contenant :
· ciclosporine ou tacrolimus (administrés après une greffe d’organe)
· dutastéride (pour l’hypertrophie bénigne de la prostate)
· alitrétinoïne (par voie orale) (pour l’eczéma)
· cabergoline (pour la maladie de Parkinson)
· produits à base de cannabis, y compris les médicaments (tels que pour la nausée et les vomissements ou les spasmes musculaires chez les patients atteints de sclérose en plaques)
· cinacalcet (pour l’hyperparathyroïdie)
· diénogest ou ulipristal (contraceptifs)
· éliglustat (pour la maladie de Gaucher) en cas d’utilisation chez des patients dont l’organisme ne peut pas dégrader certains médicaments dans le corps
· ivacaftor (pour la mucoviscidose)
· méthadone (pour le traitement de l’addiction à la drogue)
· répaglinide ou saxagliptine (pour le diabète)
Informez votre médecin avant de prendre certains médicaments, car il peut être nécessaire de modifier la dose de Sporanox ou des autres traitements
Il ne s’agit pas d’une liste exhaustive, informez votre médecin si vous prenez ou prévoyez de prendre l’un de ces médicaments, ou tout autre médicament.
Sporanox 10 mg/mL solution buvable avec des aliments, boissons et de l’alcool
Ne prenez pas Sporanox avec des aliments ou des boissons, car l’absorption du médicament par votre organisme sera moins facile.
Prenez toujours Sporanox solution buvable une heure avant de manger ou de boire car cela aidera l’organisme à absorber le médicament.
Grossesse
Sporanox ne doit pas être pris en cas de grossesse sauf si votre médecin le juge absolument nécessaire.
Si vous êtes en capacité d’avoir des enfants et que vous pourriez débuter une grossesse, parlez-en à votre médecin.
· Utiliser un moyen de contraception efficace pendant votre traitement par Sporanox afin d’éviter de débuter une grossesse alors que vous prenez le médicament.
· Continuer à utiliser d’utiliser un moyen de contraception jusqu’à l’apparition de vos prochaines règles après l’arrêt du traitement par Sporanox, car Sporanox reste un certain temps dans l’organisme après l’arrêt du traitement.
Si vous découvrez que vous êtes enceinte pendant le traitement, consultez rapidement votre médecin.
Allaitement
Il est déconseillé d’allaiter votre enfant lorsque vous êtes traitée par Sporanox, car de petites quantités de médicament peuvent être présentes dans le lait maternel.
Sporanox contient du propylène glycol. Si vous êtes enceinte ou si vous allaitez, ne prenez pas ce médicament sauf avis contraire de votre médecin. Votre médecin pourra procéder à des contrôles supplémentaires pendant que vous prenez ce médicament.
Conduite de véhicules et utilisation de machines
Des vertiges, des troubles visuels (vision double ou vision floue) ou une perte auditive peuvent survenir lors du traitement par Sporanox. Si c’est le cas, ne conduisez pas ou n’utilisez pas de machines ou d’outils.
Sporanox 10 mg/mL solution buvable contient du sorbitol, du propylène glycol, de la cyclodextrine et de l’éthanol.
Ce médicament contient 1980 mg de sorbitol par dose de 10 mL équivalent à 198 mg/mL. Le sorbitol est une source de fructose. Si votre médecin vous a informé(e) que vous (ou votre enfant) présentiez une intolérance à certains sucres ou si vous avez été diagnostiqué(e) avec une intolérance héréditaire au fructose (IHF), un trouble génétique rare caractérisé par l'incapacité à décomposer le fructose, parlez-en à votre médecin avant que vous (ou votre enfant) ne preniez ou ne receviez ce médicament. Le sorbitol peut causer une gêne gastro-intestinale et un effet laxatif léger.
Ce médicament contient 1040 mg de propylène glycol par dose de 10 mL, équivalent à 104 mg/mL. Si votre enfant a moins de 5 ans, demandez à votre médecin ou à votre pharmacien avant de lui donner ce médicament, en particulier s’il reçoit d’autres médicaments contenant du propylène glycol ou de l’alcool (voir rubrique « Enfants et adolescents »). Si vous êtes enceinte ou si vous allaitez, ne prenez pas ce médicament sauf avis contraire de votre médecin. Votre médecin pourra procéder à des contrôles supplémentaires pendant que vous prenez ce médicament (voir rubrique « Grossesse et allaitement »). Si vous souffrez d’une maladie du foie ou du rein, ne prenez ce médicament que sur avis de votre médecin. Votre médecin pourra procéder à des examens complémentaires pendant que vous prenez ce médicament.
Ce médicament contient 4000 mg de cyclodextrine par dose de 10 mL, équivalent à 400 mg/mL. Ne pas utiliser chez l’enfant de moins de 2 ans sauf recommandation contraire de votre médecin (voir rubrique « Enfants et adolescents »). Les cyclodextrines peuvent causer des problèmes digestifs tels que la diarrhée.
Ce médicament contient 0,05 mg d’alcool par dose de 10 mL équivalent à 0,005 mg/mL (0,0005% p/v). La quantité pour 10 mL de ce médicament équivaut à moins de 1 mL de bière ou 1 mL de vin. La faible quantité d’alcool contenue dans ce médicament n’est pas susceptible d’entraîner d’effet notable.
Ce médicament contient moins de 1 mmol (23 mg) de sodium pour une dose de 10 mL, c.-à-d. qu’il est essentiellement « sans sodium ».
3. COMMENT PRENDRE Sporanox 10 mg/mL, solution buvable ?
Quand prendre Sporanox
Prenez toujours Sporanox une heure avant de manger ou de boire, car cela aidera votre organisme à absorber le médicament.
Comment prendre Sporanox
Voie orale.
Effectuez un bain de bouche avec la solution buvable pendant environ 20 secondes environ avant de l’avaler. Ne vous rincez pas la bouche après avoir avalé la solution buvable.
Quantité à prendre
Un gobelet-doseur avec une graduation à 10 mL est fourni. Assurez-vous de remplir le gobelet jusqu’à la graduation de 10 mL.
La posologie habituelle est de deux doses de 10 mL de solution buvable par jour en une ou deux prises pendant une semaine.
Votre médecin pourra décider de doubler la posologie ou de prolonger le traitement dans certains cas.
Respectez toujours la posologie indiquée par votre médecin. En cas de doute, consultez votre médecin ou votre pharmacien.
Votre médecin peut être amené à vous prescrire des examens sanguins réguliers pour surveiller votre fonction hépatique (fonction du foie), s’il le juge nécessaire.
Comment ouvrir le flacon :
Le flacon est pourvu d'un bouchon avec fermeture de sécurité enfant que vous pouvez ouvrir de cette manière :
1. appuyer sur le bouchon de plastique.
2. En même temps, dévisser le bouchon dans le sens inverse des aiguilles d'une montre.
|
|
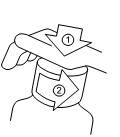
Comment utiliser le gobelet-doseur :
Un gobelet-doseur gradué est fourni avec Sporanox solution buvable. Utiliser ce gobelet-doseur dans le sens où il est posé sur le flacon.
· S’assurer que le côté avec les graduations (le côté avec le plus petit contenant) soit en haut. C’est le côté que vous devez remplir.
· Si la flèche sur le côté pointe vers le haut, le gobelet-doseur est dans le bon sens.
· Verser la solution buvable jusqu’à la graduation correspondant à la quantité prescrite par votre médecin.
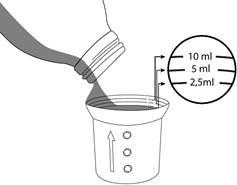
Après chaque usage, laver le gobelet-doseur soigneusement à l'eau tiède.
Par ailleurs, si vous prenez un médicament pouvant limiter l’absorption de Sporanox solution buvable (topiques gastro-intestinaux et adsorbants), vous devrez le prendre au moins 2 heures avant ou 2 heures après la prise de Sporanox solution buvable.
Dans tous les cas, demander conseil à votre pharmacien ou à votre médecin.
Si vous avez pris plus de Sporanox que vous n’auriez dû
Consultez immédiatement votre médecin ou votre pharmacien.
Si vous oubliez de prendre Sporanox
Ne prenez pas de dose double pour compenser la dose que vous avez oublié de prendre.
Si vous arrêtez de prendre Sporanox
Sans objet.
4. QUELS SONT LES EFFETS INDESIRABLES EVENTUELS ?
Effets indésirables graves
Arrêtez de prendre Sporanox et consultez immédiatement votre médecin si, pendant que vous prenez ce médicament :
· vous avez des symptômes qui peuvent traduire une atteinte sévère de la fonction de votre foie tels que perte d'appétit sévère, fatigue inhabituelle, douleurs d’estomac, urines très foncées, selles décolorées ou si vous vous sentez ou êtes malade.
· vous ressentez des sensations inhabituelles de fourmillements, d'engourdissements ou de faiblesse des mains ou des pieds, ou si vous devenez sensible à la lumière.
· vous percevez une perte auditive. Dans de très rares cas, des patients prenant Sporanox solution buvable ont rapportés une perte de l’audition temporaire ou permanente.
· vous avez soudainement des difficultés à respirer, un œdème au niveau du visage, une éruption cutanée, des démangeaisons (en particulier sur tout le corps) ou des problèmes cutanés sévères (éruptions cutanées généralisées avec desquamation de la peau et apparition de cloques au niveau de la bouche, des yeux ou des organes génitaux, ou éruptions cutanées avec présence de petites pustules ou de cloques) : ce sont les signes d’une réaction allergique sévère.
Arrêtez de prendre Sporanox et contactez immédiatement votre médecin si vous présentez l’un des symptômes mentionnés ci-dessus ou en cas de doute.
De même, prévenez votre médecin si vous présentez l’un des effets indésirables suivants :
· Signes d'insuffisance cardiaque - tels que l'essoufflement, une prise de poids inattendue, un gonflement des jambes, une fatigue inhabituelle, des réveils nocturnes répétés.
· Vision floue ou double, bourdonnement dans vos oreilles, perte de contrôle de votre vessie ou besoin accru d’uriner.
· Douleurs intenses dans la partie supérieure de l'estomac, avec le fait souvent de se sentir ou d’être malade en raison d'une inflammation du pancréas.
Contactez immédiatement votre médecin si vous présentez l’un des effets ci-dessus ou en cas de doute.
Autres effets indésirables
Fréquents (peuvant affecter jusqu’à 1 personne sur 10)
· Maux de tête, fièvre ou température élevée,
· Douleurs abdominales, diarrhées, vomissements, nausées, digestion difficile, trouble du goût,
· Eruption cutanée,
· Difficulté à respirer ou de la toux,
· Sensation de vertige.
Peu fréquents (peuvant affecter jusqu’à 1 personne sur 100) :
· Certains problèmes sanguins qui peuvent augmenter le risque de saignements, d'ecchymoses ou d'infections,
· Constipation,
· Urticaire, démangeaisons, gonflement général,
· Crampes musculaires ou battements cardiaques irréguliers,
· Douleur musculaire, douleur articulaire,
· Troubles menstruels (chez la femme),
· Atteinte des nerfs des membres, sensations de picotement, de fourmillement, diminution de la sensibilité cutanée,
· Troubles visuels (incluant vision double et floue),
· Sifflement, bourdonnement dans les oreilles (acouphène),
· Insuffisance cardiaque,
· Augmentation des enzymes du foie (ALAT, ASAT), insuffisance de la fonction du foie, inflammation du foie (hépatite), augmentation du taux de bilirubine dans le sang (se traduisant en général par une jaunisse, des urines foncées ou des selles décolorées).
Fréquence indéterminée (ne pouvant être estimée sur la base des données disponibles)
· Maladie due à une réaction allergique (maladie sérique), brusque gonflement du visage et du cou d’origine allergique (Œdème de Quincke), réactions allergiques,
· Trop de triglycérides dans le sang,
· Tremblements,
· Perte auditive passagère ou permanente,
· Insuffisance cardiaque congestive,
· Œdème pulmonaire,
· Inflammation du pancréas,
· Flatulence,
· Grave toxicité envers les cellules du foie, d’issue fatale dans quelques cas,
· Réactions cutanées graves, de type éruption cutanée diffuse avec desquamation de la peau et apparition de bulles au niveau de la bouche, des yeux et des organes génitaux, ou de type éruption cutanée accompagnée de petites pustules ou bulles,
· Inflammation des artères et des veines,
· Chute de cheveux,
· Réaction cutanée liée à l’exposition au soleil,
· Augmentation de la fréquence des émissions d’urines, incontinence urinaire,
· Troubles de l’érection (chez l’homme),
· Augmentation d’une enzyme appelée créatine phosphokinase dans un bilan sanguin,
· Symptômes d'augmentation du taux d'hormone « aldostérone » (tels que l'hypertension artérielle ou un faible taux de potassium dans le sang), même si le taux d'aldostérone dans le sang est normal ou faible,
· Diminution de la fréquence cardiaque.
Déclaration des effets secondaires
Si vous ressentez un quelconque effet indésirable, parlez-en à votre médecin, votre pharmacien ou à votre infirmier/ère. Ceci s’applique aussi à tout effet indésirable qui ne serait pas mentionné dans cette notice. Vous pouvez également déclarer les effets indésirables directement via le système national de déclaration : Agence nationale de sécurité du médicament et des produits de santé (ANSM) et réseau des Centres Régionaux de Pharmacovigilance - Site internet : https://signalement.social-sante.gouv.fr.
En signalant les effets indésirables, vous contribuez à fournir davantage d’informations sur la sécurité du médicament.
5. COMMENT CONSERVER Sporanox 10 mg/mL, solution buvable ?
Tenir hors de la vue et de la portée des enfants.
N’utilisez pas ce médicament après la date de péremption indiquée sur la boîte. La date de péremption fait référence au dernier jour de ce mois.
A conserver à une température ne dépassant pas 25°C.
Ce médicament ne peut être conservé qu'un mois après ouverture du flacon.
Ne jetez aucun médicament au tout-à-l’égout ou avec les ordures ménagères. Demandez à votre pharmacien d’éliminer les médicaments que vous n’utilisez plus. Ces mesures contribueront à protéger l’environnement.
6. CONTENU DE L’EMBALLAGE ET AUTRES INFORMATIONS
Ce que contient Sporanox 10 mg/mL, solution buvable
· La substance active est :
Itraconazole........................................................................................................................... 1,00 g
Pour 100 mL.
1 dose de 10 mL de solution buvable correspond à 100 mg d'itraconazole
· Les autres composants sont :
Hydroxypropyl-bêta-cyclodextrine, sorbitol à 70 pour cent non cristallisable E 420, propylène glycol E 1520, acide chlorhydrique, arôme cerise 1 (contenant de l’éthanol), arôme cerise 2, le caramel, saccharine sodique, hydroxyde de sodium et eau purifiée. Voir la section 2 pour plus d’informations.
Qu’est-ce que Sporanox 10 mg/mL, solution buvable et contenu de l’emballage extérieur
Titulaire de l’autorisation de mise sur le marché
167 QUAI DE LA BATAILLE DE STALINGRAD
92130 ISSY-LES-MOULINEAUX
Exploitant de l’autorisation de mise sur le marché
167 QUAI DE LA BATAILLE DE STALINGRAD
92130 ISSY-LES-MOULINEAUX
TURNHOUTSEWEG 30
2340 BEERSE
BELGIQUE
Noms du médicament dans les Etats membres de l'Espace Economique Européen
Sans objet.
La dernière date à laquelle cette notice a été révisée est :
[à compléter ultérieurement par le titulaire]
Des informations détaillées sur ce médicament sont disponibles sur le site Internet de l’ANSM (France).