Dernière mise à jour le 01/06/2026
DICLOFENAC ARROW CONSEIL 1 %, gel
Ce médicament n'est ou ne sera bientôt plus disponible sur le marché.
Si vous prenez actuellement ce médicament, il vous est recommandé d'en parler avec votre médecin
ou avec votre pharmacien qui pourra vous orienter vers un autre traitement.
Indications thérapeutiques
Ce médicament est un anti-inflammatoire et un antalgique (il calme la douleur), sous forme de gel pour application sur la peau, uniquement au niveau de la région douloureuse.
Ce médicament est indiqué, chez l’adulte (à partir de 15 ans), comme traitement local de courte durée en cas de traumatisme bénin : entorse (foulure), contusion.
Vous devez vous adresser à votre médecin si vous ne ressentez aucune amélioration ou si vous vous sentez moins bien après 4 jours.
Service médical rendu (SMR)
Amélioration du service médical rendu (ASMR)
Ce médicament étant un générique, l'ASMR n'a pas été évalué par la commission de la transparence (CT), il est possible de se référer à la /aux spécialité(s) de référence du groupe générique auquel appartient ce médicament (cliquez ici pour aller à la rubrique des groupes génériques)
ANSM - Mis à jour le : 23/05/2024
DICLOFENAC ARROW CONSEIL 1 %, gel
2. COMPOSITION QUALITATIVE ET QUANTITATIVE
Diclofénac sodique................................................................................................................ 1,00 g
Sous forme de diclofénac de diéthylamine.............................................................................. 1,16 g
Pour 100 g de gel
Excipient à effet notoire : propylène glycol.
Pour la liste complète des excipients, voir rubrique 6.1.
4.1. Indications thérapeutiques
4.2. Posologie et mode d'administration
Réservé à l’adulte (à partir de 15 ans).
La survenue d’effets indésirables peut être minimisée par l’utilisation de la dose la plus faible possible pendant la durée de traitement la plus courte nécessaire au soulagement des symptômes.
1 application locale, 2 à 3 fois par jour, pour une durée maximale de 4 jours. Si la douleur persiste au-delà, un médecin doit être consulté.
Sur avis médical, la durée maximale de traitement est de 7 jours.
La dose administrée à chaque application ne doit pas dépasser 2,5 g de gel (soit 6 cm de gel environ).
Population pédiatrique
DICLOFENAC ARROW CONSEIL 1 %, gel est contre-indiqué chez les enfants âgés de moins de 15 ans (voir rubrique 4.3).
Sujets âgés
Ce médicament doit être utilisé avec précaution chez les personnes âgées qui sont d’avantage sujettes aux effets indésirables.
Voie cutanée.
Faire pénétrer le gel en fine couche par un massage doux et prolongé sur la région douloureuse ou inflammatoire.
Après application, les mains doivent être essuyées avec du papier absorbant, puis lavées, sauf s’il s’agit du site à traiter.
Le papier absorbant doit être jeté à la poubelle après utilisation afin d’éviter que du produit non utilisé n’atteigne l’environnement aquatique.
Les patients doivent attendre, quelques minutes, que DICLOFENAC ARROW CONSEIL 1 %, gel sèche avant d’effectuer un bandage, de prendre une douche ou un bain.
Ce médicament est contre-indiqué dans les cas suivants :
· hypersensibilité à la substance active ou à l'un des excipients mentionnés à la rubrique 6.1 ;
· grossesse, à partir du début du 6ème mois (au-delà de 24 semaines d’aménorrhée) (voir rubrique 4.6) ;
· peau lésée, quelle que soit la lésion : dermatoses suintantes, eczéma, lésion infectée, brûlure ou plaie ;
· antécédents d’asthme, d’angiœdème, d’urticaire ou de rhinite aiguë déclenchée par le diclofénac ou substance d’activité proche telle que l’acide acétylsalicylique ou d’autres médicaments anti-inflammatoires non stéroïdiens (AINS) ;
· chez l’enfant et l’adolescent de moins de 15 ans.
4.4. Mises en garde spéciales et précautions d'emploi
Ne pas appliquer sur les muqueuses, ni sur les yeux.
L'apparition d'une éruption cutanée après application impose l'arrêt immédiat du traitement.
Le port de gants par le masseur kinésithérapeute, en cas d'utilisation intensive, est recommandé.
Excipient à effet notoire
Ce médicament contient 5 g de propylène glycol pour 100 g de gel.
4.5. Interactions avec d'autres médicaments et autres formes d'interactions
En raison du faible passage systémique lors d'un usage normal du gel, les interactions médicamenteuses signalées pour le diclofénac per os sont peu probables.
4.6. Fertilité, grossesse et allaitement
Grossesse
L’inhibition de la synthèse des prostaglandines par les AINS peut affecter le déroulement de la grossesse et/ou le développement de l’embryon ou du fœtus.
Risques associés à l’utilisation au cours du 1er trimestre
Les données des études épidémiologiques suggèrent une augmentation du risque de fausse-couche, de malformations cardiaques et de gastroschisis, après traitement par un inhibiteur de la synthèse des prostaglandines en début de grossesse. Le risque absolu de malformation cardiovasculaire est passé de moins de 1 % dans la population générale, à approximativement 1,5 % chez les personnes exposées aux AINS. Le risque paraît augmenter en fonction de la dose et de la durée du traitement. Chez l’animal, il a été montré que l’administration d’un inhibiteur de la synthèse des prostaglandines provoquait une perte pré et post-implantatoire accrue et une augmentation de la létalité embryo-fœtale. De plus, une incidence supérieure de certaines malformations, y compris cardiovasculaires, a été rapportée chez des animaux ayant reçu un inhibiteur de la synthèse des prostaglandines au cours de la phase d’organogénèse de la gestation.
Risques associés à l’utilisation à partir de la 12ème semaine d’aménorrhée et jusqu’à la naissance
A partir de la 12ème semaine d’aménorrhée et jusqu’à la naissance, tous les AINS, par l’inhibition de la synthèse des prostaglandines, peuvent exposer le fœtus à une atteinte fonctionnelle rénale :
· in utero pouvant s'observer dès 12 semaines d'aménorrhée (mise en route de la diurèse fœtale) : oligoamnios (le plus souvent réversible à l'arrêt du traitement), voire anamnios en particulier lors d'une exposition prolongée ;
· à la naissance, une insuffisance rénale (réversible ou non) peut persister en particulier en cas d'exposition tardive et prolongée (avec un risque d'hyperkaliémie sévère retardée).
Risques associés à l’utilisation au-delà de la 24ème semaine d’aménorrhée et jusqu’à la naissance
Au-delà de la 24ème semaine d’aménorrhée, les AINS peuvent exposer le fœtus à une toxicité cardio-pulmonaire (fermeture prématurée du canal artériel et hypertension artérielle pulmonaire). La constriction du canal artériel peut survenir à partir du début du 6ème mois (au-delà de la 24ème semaine d’aménorrhée) et peut conduire à une insuffisance cardiaque droite fœtale ou néonatale voire à une mort fœtale in utero. Ce risque est d'autant plus important que la prise est proche du terme (moindre réversibilité). Cet effet existe même pour une prise ponctuelle.
En fin de grossesse, la mère et le nouveau-né peuvent présenter
· Un allongement du temps de saignement du fait d’une action anti-agrégante pouvant survenir même après administration de très faibles doses de médicament.
· Une inhibition des contractions utérines entraînant un retard de terme ou un accouchement prolongé.
En conséquence
Sauf nécessité absolue, ce médicament ne doit pas être prescrit chez une femme qui envisage une grossesse ou au cours des 5 premiers mois de grossesse (24 premières semaines d’aménorrhée). Si ce médicament est administré chez une femme souhaitant être enceinte ou enceinte de moins de 6 mois, la dose devra être la plus faible possible et la durée du traitement la plus courte possible. Une prise prolongée est fortement déconseillée.
A partir du début du 6ème mois (au-delà de 24 semaines d'aménorrhée): toute prise de ce médicament, même ponctuelle, est contre-indiquée. Une prise par mégarde à partir de cette date justifie une surveillance cardiaque et rénale, fœtale et/ou néonatale selon le terme d'exposition. La durée de cette surveillance sera adaptée à la demi-vie d'élimination de la molécule.
Allaitement
Les AINS passant dans le lait maternel, ce médicament est déconseillé chez la femme qui allaite.
En cas d'allaitement, ce médicament ne doit en aucun cas être appliqué sur la poitrine.
Fertilité
Comme tous les AINS, l'utilisation de ce médicament peut temporairement altérer la fertilité féminine en agissant sur l’ovulation ; il est donc déconseillé chez les femmes souhaitant concevoir un enfant. Chez les femmes rencontrant des difficultés pour concevoir ou réalisant des tests de fertilité, l'arrêt du traitement doit être envisagé.
4.7. Effets sur l'aptitude à conduire des véhicules et à utiliser des machines
Les effets indésirables sont classés par ordre de fréquence décroissant, selon les conventions suivantes : très fréquent (≥ 1/10) ; fréquent (≥ 1/100 à < 1/10) ; peu fréquent (≥ 1/1 000 à < 1/100) ; rare (≥ 1/10 000 à < 1/1 000) ; très rare (< 1/10 000), fréquence indéterminée : ne peut être estimée à partir des données disponibles. Au sein de chaque groupe de fréquence, les effets indésirables sont présentés par ordre décroissant de gravité.
|
Effet indésirable |
Fréquence |
|
|
Infections et infestations |
Eruption pustuleuse |
Très rare |
|
Affections du système immunitaire |
Angiœdème (œdème de Quincke), réactions d’hypersensibilité (dont urticaire) |
Très rare |
|
Affections respiratoires, thoraciques et médiastinales |
Crise d’asthme* |
Très rare |
|
Affections de la peau et du tissu sous-cutané |
Dermatite (incluant les dermatites de contact), éruptions cutanées, érythèmes, eczéma, prurit |
Fréquent |
|
Dermatose bulleuse |
Rare |
|
|
Réactions de photosensibilité, purpura et ulcérations locales |
Très rare |
* La survenue de crise d'asthme peut être liée chez certains sujets à une allergie à l'aspirine ou à un AINS. Dans ce cas, ce médicament est contre-indiqué.
Autres effets systémiques des AINS
Ils sont fonction du passage transdermique du principe actif et donc de la quantité de gel appliquée, de la surface traitée, du degré d'intégrité cutanée, de la durée du traitement et de l'utilisation ou non d'un pansement occlusif (effets digestifs, rénaux).
Déclaration des effets indésirables suspectés
La déclaration des effets indésirables suspectés après autorisation du médicament est importante. Elle permet une surveillance continue du rapport bénéfice/risque du médicament. Les professionnels de santé déclarent tout effet indésirable suspecté via le système national de déclaration : Agence nationale de sécurité du médicament et des produits de santé (ANSM) et réseau des Centres Régionaux de Pharmacovigilance - Site internet : https://signalement.social-sante.gouv.fr/
En cas d’ingestion accidentelle ou volontaire, des effets similaires à ceux observés en cas de surdosage de diclofénac par voie orale et ayant pour conséquence des effets indésirables peuvent survenir. Les mesures thérapeutiques qui s'imposent sont celles généralement adoptées en cas d'intoxication avec les AINS. Les recommandations à suivre seront celles indiquées par le Centre Antipoison régional, en fonction des quantités ingérées et des caractéristiques du patient.
5. PROPRIETES PHARMACOLOGIQUES
5.1. Propriétés pharmacodynamiques
Le diclofénac est un anti-inflammatoire non stéroïdien (AINS), dérivé de l'acide phénylacétique du groupe des acides arylcarboxyliques doté de propriétés analgésiques, anti-inflammatoires et antipyrétiques. Comme pour tous les AINS, le mécanisme d’action exact du diclofénac n’est pas complètement élucidé mais repose essentiellement sur l'inhibition de la biosynthèse des prostaglandines, substances jouant un rôle majeur dans la genèse de l’inflammation, de la douleur et de la fièvre, par l'inhibition des deux cyclooxygénases (COX-1 et COX-2).
Sous forme de gel, il possède une activité locale anti-inflammatoire et antalgique.
5.2. Propriétés pharmacocinétiques
Appliqué localement sous forme de gel, le diclofénac est absorbé à travers la peau.
Absorption
La quantité de diclofénac absorbée à travers la peau est proportionnelle à la surface traitée et dépend à la fois de la dose totale appliquée et du degré d’hydratation cutanée.
Le passage systémique du gel, par rapport à celui des formes orales de diclofénac chez les volontaires sains, est de l'ordre de 6 %, par estimation d'après son excrétion urinaire et celle de ses métabolites hydroxylés, après administration unique. Une occlusion de 10 heures conduit à une multiplication par trois de la quantité de diclofénac absorbée.
Le passage systémique du gel, par rapport à celui des formes orales de diclofénac chez les volontaires sains, est de l'ordre de 13,9 % après administration réitérée.
Les concentrations mesurées dans le liquide synovial, de même que dans le tissu synovial, sont 40 fois supérieures aux concentrations plasmatiques.
Distribution
Les concentrations de diclofénac ont été mesurées dans le plasma, le tissu synovial et le liquide synovial après administration locale du gel sur les articulations de la main et du genou. Les concentrations plasmatiques maximales sont environ 100 fois inférieures à celles mesurées après l’administration orale de la même quantité de diclofénac. 99,7 % du diclofénac sont liés à des protéines sériques, essentiellement l’albumine (99,4 %).
Le diclofénac s’accumule dans la peau, qui agit comme un réservoir à partir duquel le médicament est libéré de manière durable dans les tissus sous-jacents. De là, le diclofénac est distribué et persiste préférentiellement dans les tissus enflammés profonds, comme l’articulation, où on le trouve à des concentrations pouvant atteindre 20 fois celle mesurée dans le plasma.
Biotransformation
La biotransformation du diclofénac fait intervenir en partie une glucurono-conjugaison de la molécule intacte, mais essentiellement une hydroxylation unique et multiple de plusieurs métabolites phénoliques, dont la majorité est convertie en conjugués glucuronides. Deux des métabolites phénoliques sont biologiquement actifs, mais dans une moindre mesure comparé au diclofénac.
Élimination
La clairance systémique totale du diclofénac depuis le plasma est de 263 ± 56 mL/min. Les demi-vies plasmatiques terminales sont de 1 à 2 heures. Quatre des métabolites, y compris les deux métabolites actifs, présentent également des demi-vies plasmatiques courtes comprises entre 1 et 3 heures. Un métabolite, le 3'-hydroxy-4'-méthoxy-diclofénac, possède une demi-vie supérieure mais il est pratiquement inactif. Le diclofénac et ses métabolites sont excrétés essentiellement dans l’urine.
Caractéristiques chez les patients
Aucune accumulation de diclofénac et de ses métabolites n’est attendue chez les patients présentant une altération rénale. Chez les patients souffrant d’une hépatite chronique ou d’une cirrhose non décompensée, la cinétique et le métabolisme du diclofénac sont les mêmes que chez les patients sans atteinte hépatique.
5.3. Données de sécurité préclinique
Des études ont démontré que le diclofénac de diéthylamine 1,16 g/100 g sous forme de gel est bien toléré. Il n'a pas été observé de potentiel phototoxique chez la souris et le cobaye avec le diclofénac de diéthylamine 1,16 g/100 g sous forme de gel et ce dernier n'occasionne pas de sensibilisation cutanée lors des tests chez le cobaye.
3 ans.
Après première ouverture : 2 ans.
6.4. Précautions particulières de conservation
Ce médicament ne nécessite pas de précautions particulières de conservation.
6.5. Nature et contenu de l'emballage extérieur
50 g en tube (Aluminium verni).
6.6. Précautions particulières d’élimination et de manipulation
Pas d’exigences particulières.
7. TITULAIRE DE L’AUTORISATION DE MISE SUR LE MARCHE
26 AVENUE TONY GARNIER
69007 LYON
8. NUMERO(S) D’AUTORISATION DE MISE SUR LE MARCHE
· 34009 344 083 4 2 : 50 g en tube (Aluminium verni).
9. DATE DE PREMIERE AUTORISATION/DE RENOUVELLEMENT DE L’AUTORISATION
[à compléter ultérieurement par le titulaire]
10. DATE DE MISE A JOUR DU TEXTE
[à compléter ultérieurement par le titulaire]
Sans objet.
12. INSTRUCTIONS POUR LA PREPARATION DES RADIOPHARMACEUTIQUES
Médicament non soumis à prescription médicale.
ANSM - Mis à jour le : 23/05/2024
DICLOFENAC ARROW CONSEIL 1 %, gel
Diclofénac sodique
Vous devez toujours utiliser ce médicament en suivant scrupuleusement les informations fournies dans cette notice ou par votre médecin ou votre pharmacien.
· Gardez cette notice. Vous pourriez avoir besoin de la relire.
· Adressez-vous à votre pharmacien pour tout conseil ou information.
· Si vous ressentez un quelconque effet indésirable, parlez-en à votre médecin ou votre pharmacien. Ceci s’applique aussi à tout effet indésirable qui ne serait pas mentionné dans cette notice. Voir rubrique 4.
· Vous devez vous adresser à votre médecin si vous ne ressentez aucune amélioration ou si vous vous sentez moins bien après 4 jours.
1. Qu'est-ce que DICLOFENAC ARROW CONSEIL 1 %, gel et dans quels cas est-il utilisé ?
2. Quelles sont les informations à connaître avant d’utiliser DICLOFENAC ARROW CONSEIL 1 %, gel ?
3. Comment utiliser DICLOFENAC ARROW CONSEIL 1 %, gel ?
4. Quels sont les effets indésirables éventuels ?
5. Comment conserver DICLOFENAC ARROW CONSEIL 1 %, gel ?
6. Contenu de l’emballage et autres informations.
1. QU’EST-CE QUE DICLOFENAC ARROW CONSEIL 1 %, gel ET DANS QUELS CAS EST-IL UTILISE ?
Ce médicament est un anti-inflammatoire et un antalgique (il calme la douleur), sous forme de gel pour application sur la peau, uniquement au niveau de la région douloureuse.
Ce médicament est indiqué, chez l’adulte (à partir de 15 ans), comme traitement local de courte durée en cas de traumatisme bénin : entorse (foulure), contusion.
Vous devez vous adresser à votre médecin si vous ne ressentez aucune amélioration ou si vous vous sentez moins bien après 4 jours.
2. QUELLES SONT LES INFORMATIONS A CONNAITRE AVANT D’UTILISER DICLOFENAC ARROW CONSEIL 1 %, gel ?
N’utilisez jamais DICLOFENAC ARROW CONSEIL 1 %, gel :
· si vous êtes allergique au diclofénac sodique ou à l’un des autres composants contenus dans ce médicament, mentionnés dans la rubrique 6 ;
· si vous êtes enceinte, à partir du début du 6ème mois de grossesse (au-delà de 24 semaines d’aménorrhée) ;
· sur peau lésée, quelle que soit la lésion : lésions suintantes, eczéma, lésion infectée, brûlure ou plaie ;
· en cas d’antécédent d’allergie ou d’hypersensibilité (survenue d’asthme, d’un angiœdème, d’une urticaire ou d’une rhinite aigue) déclenchée par un médicament contenant du diclofénac, de l’acide acétylsalicylique ou d’autres médicaments anti-inflammatoires non stéroïdiens (AINS) ;
· chez l’enfant de moins de 15 ans.
EN CAS DE DOUTE, IL EST INDISPENSABLE DE DEMANDER L'AVIS DE VOTRE MEDECIN OU DE VOTRE PHARMACIEN.
Avertissements et précautions
Adressez-vous à votre médecin ou pharmacien avant d’utiliser DICLOFENAC ARROW CONSEIL 1 %, gel.
Ne pas avaler. Ne pas appliquer sur les muqueuses, ni sur les yeux. Appliquez uniquement sur la région douloureuse, sur une peau intacte et saine ne présentant aucune plaie, lésions ouvertes ou peau présentant une éruption cutanée ou un eczéma.
Se rincer les yeux intensément à l’eau si le produit entre en contact avec les yeux. Consultez votre médecin ou votre pharmacien si une gêne persiste.
Après application, les mains doivent être essuyées avec du papier absorbant, puis lavées, sauf s’il s’agit du site à traiter.
Si une trop grande quantité de gel est accidentellement appliquée, l’excès de gel doit être essuyé avec du papier absorbant.
Le papier absorbant doit être jeté à la poubelle après utilisation, afin d’éviter que du produit non utilisé n’atteigne l’environnement aquatique.
Attendre quelques minutes que DICLOFENAC ARROW CONSEIL 1 %, gel sèche avant d’effectuer un bandage, de prendre une douche ou un bain.
L’apparition d’une éruption cutanée après application impose l’arrêt immédiat du traitement. Si un tel effet survient, contactez votre médecin ou votre pharmacien.
Ne pas appliquer sur une grande surface cutanée et à un dosage plus important que celui recommandé ou pendant une période prolongée sans avis médical.
La forme gel est réservée à l'adulte (à partir de 15 ans).
Chez les adolescents âgés de 15 ans et plus, si les symptômes s'aggravent, il est recommandé de demander l'avis de votre médecin.
Le port de gants par le masseur kinésithérapeute, en cas d'utilisation intensive, est recommandé.
EN CAS DE DOUTE NE PAS HESITER A DEMANDER L'AVIS DE VOTRE MEDECIN OU DE VOTRE PHARMACIEN.
Si vous êtes enceinte ou si vous allaitez
N'utilisez pas ce médicament de votre propre initiative. Demandez l'avis de votre médecin ou de votre pharmacien.
Enfants et adolescents (de moins de 15 ans)
Ce médicament ne doit pas être utilisé chez les enfants de moins de 15 ans (voir rubrique « N'utilisez jamais DICLOFENAC ARROW CONSEIL 1 %, gel »).
Autres médicaments et DICLOFENAC ARROW CONSEIL 1 %, gel
Informez votre médecin ou pharmacien si vous utilisez, avez récemment utilisé ou pourriez utiliser tout autre médicament, même s’il s’agit d’un médicament obtenu sans ordonnance.
|
Ce médicament contient du diclofénac. D'autres médicaments en contiennent et notamment certains médicaments pris par voie orale. Ne les associez pas, afin de ne pas dépasser les doses maximales conseillées (voir la rubrique « Posologie et durée de traitement »). |
DICLOFENAC ARROW CONSEIL 1 %, gel avec des aliments, boissons et de l’alcool
Sans objet.
Grossesse, allaitement et fertilité
Si vous êtes enceinte ou que vous allaitez, si vous pensez être enceinte ou planifiez une grossesse, demandez conseil à votre médecin ou pharmacien avant de prendre tout médicament.
Avant le début du 6ème mois de grossesse (jusqu’à la 24ème semaine d’aménorrhée), vous ne devez pas prendre ce médicament, sauf en cas d’absolue nécessité déterminée par votre médecin, en raison du risque potentiel de fausses couches ou de malformations. Dans ce cas, la dose devra être la plus faible possible et la durée du traitement la plus courte possible.
A partir du début du 6ème mois jusqu’à la fin de la grossesse (au-delà de la 24ème semaine d’aménorrhée), ce médicament est contre-indiqué, vous ne devez EN AUCUN CAS prendre ce médicament, car ses effets sur votre enfant peuvent avoir des conséquences graves voire fatales, notamment sur le cœur, les poumons et/ou les reins, et cela même avec une seule prise.
Si vous avez pris ce médicament alors que vous étiez enceinte, parlez-en immédiatement à votre gynécologue obstétricien, afin qu’une surveillance adaptée vous soit proposée si nécessaire.
Allaitement
Ce médicament passant dans le lait maternel, il est déconseillé de l'utiliser pendant l'allaitement.
En cas d'allaitement, ce médicament ne doit en aucun cas être appliqué sur les seins.
Fertilité
Ce médicament, comme tous les anti-inflammatoires non stéroïdiens (AINS), peut altérer la fertilité des femmes et entraîner des difficultés pour devenir enceinte, de façon réversible à l’arrêt du traitement. Informez votre médecin si vous planifiez une grossesse ou si vous avez des difficultés à concevoir.
Conduite de véhicules et utilisation de machines
Sans objet.
DICLOFENAC ARROW CONSEIL 1 %, gel contient du propylène glycol
Ce médicament contient 5 g de propylène glycol pour 100 g de gel.
3. COMMENT UTILISER DICLOFENAC ARROW CONSEIL 1 %, gel ?
Posologie et durée de traitement
Réservé à l’adulte (à partir de 15 ans).
La survenue d’effets indésirables peut être minimisée par l’utilisation de la dose la plus faible possible pendant la durée de traitement la plus courte nécessaire au soulagement des symptômes.
1 application locale, 2 à 3 fois par jour, pour une durée maximale de 4 jours. Si la douleur persiste au-delà, un médecin doit être consulté.
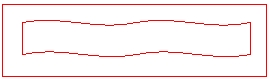
La dose administrée à chaque application ne doit pas dépasser 2,5 g de gel. Cette dose maximale correspond à un ruban de 6 cm de gel (voir schéma à l’échelle).
Populations particulières
Enfants et adolescents de moins de 15 ans
Ce médicament ne doit pas être utilisé chez les enfants et les adolescents de moins de 15 ans (voir rubrique « N’utilisez jamais DICLOFENAC ARROW CONSEIL 1 %, gel »).
Sujets âgés
Ce médicament doit être utilisé avec précaution chez les personnes âgées qui sont d’avantage sujettes aux effets indésirables (voir rubrique 4).
Voie cutanée.
USAGE EXTERNE.
DICLOFENAC ARROW CONSEIL 1 %, gel est uniquement réservé à l’usage cutané.
Faire pénétrer le gel en fine couche par un massage doux et prolongé sur la région douloureuse ou inflammatoire.
Après application, les mains doivent être essuyées avec du papier absorbant, puis lavées, sauf s’il s’agit du site à traiter.
Le papier absorbant doit être jeté à la poubelle après utilisation afin d’éviter que du produit non utilisé n’atteigne l’environnement aquatique.
Attendre quelques minutes que DICLOFENAC ARROW CONSEIL 1 %, gel sèche avant d’effectuer un bandage, de prendre une douche ou un bain.
Si vous avez utilisé plus de DICLOFENAC ARROW CONSEIL 1 %, gel que vous n’auriez dû
En cas de surdosage ou d’intoxication accidentelle, essuyez le surplus de gel avec du papier absorbant puis rincez abondamment à l'eau et consultez immédiatement votre médecin ou votre pharmacien.
Si vous oubliez d’utiliser DICLOFENAC ARROW CONSEIL 1 %, gel
Si vous arrêtez d’utiliser DICLOFENAC ARROW CONSEIL 1 %, gel
Sans objet.
Si vous avez d’autres questions sur l’utilisation de ce médicament, demandez plus d’informations à votre médecin ou à votre pharmacien.
4. QUELS SONT LES EFFETS INDESIRABLES EVENTUELS ?
Exceptionnellement peuvent survenir des réactions pouvant être sévères :
· réactions allergiques cutanées : éruption (avec ou sans boutons), urticaire, bulles,
· problèmes respiratoires de type crise d'asthme (souffle bruyant et court, impression de capacité respiratoire diminuée),
· manifestations générales de type anaphylaxie (gonflement de la face, des lèvres, de la langue, de la gorge).
Il faut immédiatement interrompre le traitement et avertir votre médecin ou votre pharmacien.
Plus fréquemment, peuvent survenir des effets indésirables, généralement légers et passagers :
· des effets locaux cutanés de type rougeur, démangeaisons, irritation cutanée, érosion ou ulcérations locales.
Très exceptionnellement, une augmentation de la sensibilité au soleil.
D'autres effets généraux des anti-inflammatoires non stéroïdiens, fonction de la quantité de gel appliquée, de la surface traitée et de son état, de la durée du traitement et de l'utilisation ou non d'un pansement fermé.
Il faut en avertir votre médecin ou votre pharmacien.
Déclaration des effets secondaires
Si vous ressentez un quelconque effet indésirable, parlez-en à votre médecin ou votre pharmacien. Ceci s’applique aussi à tout effet indésirable qui ne serait pas mentionné dans cette notice. Vous pouvez également déclarer les effets indésirables directement via le système national de déclaration : Agence nationale de sécurité du médicament et des produits de santé (ANSM) et réseau des Centres Régionaux de Pharmacovigilance - Site internet : https://signalement.social-sante.gouv.fr/
En signalant les effets indésirables, vous contribuez à fournir davantage d’informations sur la sécurité du médicament.
5. COMMENT CONSERVER DICLOFENAC ARROW CONSEIL 1 %, gel ?
Tenir ce médicament hors de la vue et de la portée des enfants.
N’utilisez pas ce médicament après la date de péremption indiquée sur l’emballage après EXP. La date de péremption fait référence au dernier jour de ce mois.
Ce médicament ne nécessite pas de précautions particulières de conservation.
A utiliser dans les 2 ans après première ouverture.
Ne jetez aucun médicament au tout-à-l’égout ou avec les ordures ménagères. Demandez à votre pharmacien d’éliminer les médicaments que vous n’utilisez plus. Ces mesures contribueront à protéger l’environnement.
6. CONTENU DE L’EMBALLAGE ET AUTRES INFORMATIONS
Ce que contient DICLOFENAC ARROW CONSEIL 1%, gel
· La substance active est :
Diclofénac sodique.......................................................................................................... 1,00 g
Sous forme de diclofénac de diéthylamine........................................................................ 1,16 g
Pour 100 g de gel
· Les autres composants sont : diéthylamine, carbomère 974P, cétomacrogol 1000, caprylocaprate de cocoyle, alcool isopropylique, paraffine liquide, propylène glycol, eau purifiée.
Qu’est-ce que DICLOFENAC ARROW CONSEIL 1 %, gel et contenu de l’emballage extérieur
Ce médicament se présente sous forme de gel.
Tube de 50 g.
Titulaire de l’autorisation de mise sur le marché
26 AVENUE TONY GARNIER
69007 LYON
Exploitant de l’autorisation de mise sur le marché
26 AVENUE TONY GARNIER
69007 LYON
93 ROUTE DE MONNAIE
37210 VOUVRAY
ou
DELPHARM HUNINGUE SAS
26 rue de la Chapelle
68330 Huningue
Noms du médicament dans les Etats membres de l'Espace Economique Européen
Sans objet.
La dernière date à laquelle cette notice a été révisée est :
[à compléter ultérieurement par le titulaire]
Des informations détaillées sur ce médicament sont disponibles sur le site Internet de l’ANSM (France).
CONSEILS D’EDUCATION SANITAIRE
Attention : s’il existe une impotence fonctionnelle complète, c’est-à-dire si vous ne pouvez pas vous servir du membre et en cas d’hématome (« bleu ») important, vous devez consulter sans tarder votre médecin.
Vous venez d'avoir un traumatisme ou une contusion, vous pouvez utiliser le gel pendant une durée maximale de 4 jours pour soulager votre douleur. Ne reprenez pas une activité sportive ou une pratique physique intense avant la disparition complète de la douleur.
Par ailleurs :
En cas d’entorse de la cheville
Vous devez consulter votre médecin qui jugera de la nécessité d'une radiographie et d'un traitement orthopédique :
· si vous ne pouvez absolument pas vous mettre en appui sur la jambe pour faire quatre pas ;
· ou si un hématome (bleu) apparaît dans les 24 h à 48 h ;
· ou s'il existe une déformation ou un œdème (gonflement) très important.
En cas de lésion du genou
Vous devez consulter votre médecin qui jugera de la nécessité d'une radiographie et d'un traitement orthopédique :
· en cas de gonflement important du genou, avec ou sans hématome ;
· et/ou en cas d'impossibilité d'appui.