Dernière mise à jour le 01/06/2026
LETYBO 50 unités, poudre pour solution injectable
Indications thérapeutiques
Classe pharmacothérapeutique : autres myorelaxants à action périphérique - code ATC : M03AX01.
Letybo contient la substance active « toxine botulinique de type A », qui agit en bloquant les impulsions nerveuses vers les muscles dans lesquels il a été injecté. Cela empêche la contraction des muscles, entraînant une paralysie temporaire.
Letybo est utilisé chez les adultes de moins de 75 ans pour l’amélioration temporaire de l’apparence des rides verticales intersourcilières modérées à sévères lorsque leur présence entraîne chez eux un retentissement psychologique important.
Présentations
> 1 flacon en verre de 5 mL
Code CIP : 34009 302 469 0 0
Déclaration de commercialisation : 23/06/2022
Cette présentation n'est pas agréée aux collectivités
Service médical rendu (SMR)
Pas de SMR disponible pour ce médicament ( plus d'informations dans l'aide )
Amélioration du service médical rendu (ASMR)
Pas d'ASMR disponible pour ce médicament (plus d'informations dans l'aide )
Autres informations
- Titulaire de l'autorisation : CROMA PHARMA GMBH
- Conditions de prescription et de délivrance :
- liste I
- prescription réservée aux spécialistes et services CHIRURGIE DE LA FACE ET DU COU
- prescription réservée aux spécialistes et services CHIRURGIE MAXILLO-FACIALE
- prescription réservée aux spécialistes et services CHIRURGIE PLASTIQUE, RECONSTRUCTRICE ET ESTHETIQUE
- prescription réservée aux spécialistes et services DERMATOLOGIE
- prescription réservée aux spécialistes et services OPHTALMOLOGIE
- réservé à l'usage professionnel selon l'article R.5121-80 du code de la santé publique
- Statut de l'autorisation : Valide
- Type de procédure : Procédure décentralisée
- Code CIS : 6 987 836 3
ANSM - Mis à jour le : 27/02/2026
LETYBO 50 unités, poudre pour solution injectable
2. COMPOSITION QUALITATIVE ET QUANTITATIVE
Chaque flacon contient 50 unités de toxine botulinique de type A produit par Clostridium botulinum.
Après reconstitution, chaque 0,1 mL de solution contient 4 unités.
Pour la liste complète des excipients, voir rubrique 6.1.
Poudre pour solution injectable.
Poudre blanche.
4.1. Indications thérapeutiques
4.2. Posologie et mode d'administration
Posologie
La dose recommandée est un total de 20 unités réparties en cinq injections de 4 unités (0,1 mL) chacune : 2 injections dans chaque muscle corrugator supercilii et 1 injection dans le muscle procerus.
Les unités de toxine botulinique ne sont pas interchangeables d’un produit à l’autre.
Les doses recommandées diffèrent des autres préparations de toxine botulinique.
L’intervalle entre les traitements doit être d’au moins trois mois.
En l’absence d’effets indésirables consécutifs à une séance de traitement, il est possible d’entamer une nouvelle séance après un délai d’au moins trois mois.
En cas d’échec du traitement un mois après une première séance, c’est-à-dire en l’absence d’amélioration significative par rapport à la situation initiale, les approches suivantes peuvent être envisagées :
· L’analyse des causes de l’échec, par exemple l’injection dans les mauvais muscles, une technique d’injection inappropriée, la formation d’anticorps neutralisant la toxine ou un dosage insuffisant.
· La réévaluation de la pertinence du traitement par toxine botulinique de type A.
L’efficacité et la sécurité des injections répétées de Letybo au-delà d’une période de 12 mois n’ont pas été évaluées.
Populations particulières
Sujets âgées
Il n’existe pas de données cliniques concernant l’utilisation de Letybo chez les patients de plus de 75 ans. Aucun ajustement spécifique de la dose n’est nécessaire pour l’utilisation chez les personnes âgées de plus de 65 ans (voir rubrique 5.1).
Population pédiatrique
Il n’y a pas d’utilisation justifiée de Letybo dans la population pédiatrique (voir rubrique 5.1).
Mode d’administration
Voie intramusculaire.
Après reconstitution, Letybo ne doit être utilisé que pour une seule séance d’injection(s) par patient.
Pour les instructions concernant la dilution, l’utilisation, la manipulation et l’élimination des flacons, voir rubrique 6.6.
Les injections intramusculaires doivent être réalisées à l’aide d’une seringue stérile du type utilisé pour l’insuline ou la tuberculine, avec une capacité de 1 mL, des graduations de 0,01 mL et une aiguille de calibre 30 à 31 G.
Aspirer dans la seringue stérile 0,5 mL de la solution dûment reconstituée et expulser toute bulle d’air présente dans le cylindre. L’aiguille utilisée pour reconstituer le médicament doit être retirée et remplacée pour l’administration.
Veiller à ce que Letybo ne soit pas injecté dans un vaisseau sanguin.
Afin de réduire le risque de blépharoptose, éviter les injections à proximité du muscle levator palpebrae superioris, en particulier chez les patients présentant des puissants complexes abaisseurs des sourcils. Lors de l’injection dans deux sites pour chaque muscle corrugator supercilii, réaliser la première injection juste au-dessus du bord médial des sourcils. La seconde injection sera pratiquée à environ 1 cm au-dessus du bord supra-orbitaire (limites osseuses rigides palpables au-dessus de la marge haute de la paupière supérieure), au point de rencontre des lignes médianes des sourcils. Le site d’injection pour le muscle procerus se situe juste au-dessus de la ligne médiane de l’arête nasale, à l’endroit où se forment les rides horizontales entre les extrémités médiales des sourcils. Lors de l’administration dans les extrémités médiales des muscles corrugator supercilii et sur les lignes médianes des sourcils, les sites d’injection doivent être éloignés d’au moins 1 cm du bord supra-orbitaire (limites osseuses rigides palpables au-dessus de la marge haute de la paupière supérieure).
![]()
![]()
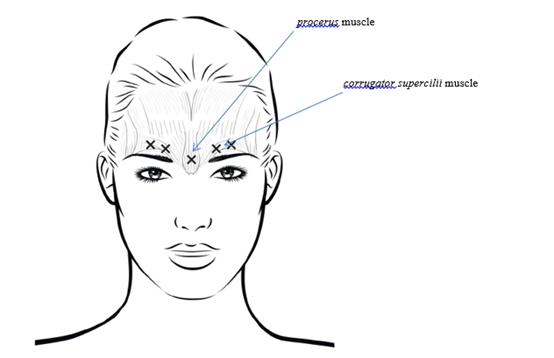
Lors de l’administration, agir avec prudence pour éviter toute injection intravasculaire. Avant l’injection, il est possible de placer fermement le pouce ou l’index sous le bord orbitaire pour éviter l’épanchement du médicament dans cette zone. Orienter l’aiguille vers le haut et médialement.
Hypersensibilité à la substance active ou à l’un des excipients mentionnés à la rubrique 6.1.
Troubles généralisés de l’activité musculaire (par exemple, myasthénie grave, syndrome de Lambert-Eaton, sclérose latérale amyotrophique).
Présence d’une infection ou d’une inflammation aiguë aux sites d’injection proposés.
4.4. Mises en garde spéciales et précautions d'emploi
Avant toute administration de Letybo, il importe de comprendre l’anatomie des muscles et des structures vasculaires et nerveuses environnantes dans la région glabellaire, ainsi que toute altération anatomique liée à des interventions chirurgicales antérieures. Éviter l’injection dans des structures anatomiques vulnérables.
Il existe un risque de ptosis après le traitement. Voir les instructions d’administration à la rubrique 4.2 pour limiter cette éventualité.
Événements liés à la procédure
Après le traitement par d’autres toxines botuliniques, une douleur et/ou une anxiété liée à l’aiguille ont donné lieu à des réactions vasovagales, notamment à une hypotension symptomatique transitoire et à une syncope.
Troubles neuromusculaires préexistants
Les patients atteints de troubles neuromusculaires non reconnus peuvent présenter un risque accru d’effets systémiques cliniquement significatifs à l’administration de doses standard de toxine botulinique de type A, notamment une dysphagie sévère et une atteinte respiratoire.
Réactions d’hypersensibilité
Dans de très rares cas, une réaction anaphylactique peut se produire après l’injection de toxine botulinique. Il convient donc d’avoir à disposition de l’épinéphrine (adrénaline) ou toute autre mesure anti-anaphylactique.
Diffusion locale ou à distance des effets de la toxine
Des effets indésirables potentiellement liés à la diffusion de la toxine à distance du site d’administration ont été signalés dans de très rares cas pour la toxine botulinique (voir rubrique 4.8). Les patients recevant des doses thérapeutiques peuvent présenter une faiblesse musculaire excessive.
Les difficultés à avaler et à respirer sont graves et peuvent entraîner la mort. L’injection de Letybo n’est pas recommandée chez les patients présentant des antécédents de dysphagie et d’inhalation.
Des cas de botulisme iatrogène ont été signalés après l’injection de produits à base de toxine botulinique. Les patients doivent être avertis de la nécessité d’une prise en charge médicale immédiate en cas d’apparition de signes ou de symptômes indiquant une diffusion de l’effet de la toxine botulinique ou en présence de troubles de la déglutition, de troubles de la parole ou de troubles respiratoires (voir rubrique 4.9).
Formation d’anticorps
L’administration de doses trop rapprochées ou trop élevées peut accroître le risque de formation d’anticorps. La formation d’anticorps peut entraîner l’échec du traitement par toxine botulinique de type A, même dans le cadre d’autres indications.
Troubles hémorragiques
Agir avec prudence lorsque Letybo est utilisé chez des patients présentant des troubles hémorragiques, car l’injection peut générer des contusions.
Ce médicament contient moins de 1 mmol (23 mg) de sodium par dose, c.-à-d. qu’il est essentiellement « sans sodium ».
Traçabilité
Afin d’améliorer la traçabilité des médicaments biologiques, le nom et le numéro de lot du produit administré doivent être clairement enregistrés.
4.5. Interactions avec d'autres médicaments et autres formes d'interactions
Théoriquement, l’effet de la toxine botulinique peut être potentialisé par les antibiotiques du groupe des aminosides, la spectinomycine ou d’autres médicaments qui perturbent la transmission neuromusculaire (par exemple, les agents bloquants neuromusculaires).
4.6. Fertilité, grossesse et allaitement
Grossesse
Il n’existe pas de données appropriées sur l’utilisation de la toxine botulinique de type A chez la femme enceinte. Les études effectuées chez l’animal ont mis en évidence une toxicité sur la reproduction à haute dose (voir rubrique 5.3). Le risque potentiel pour l’homme n’est pas connu. Letybo n’est pas recommandé pendant la grossesse et chez les femmes en âge de procréer n’utilisant pas de contraception.
Allaitement
On ne sait pas si Letybo est excrété dans le lait maternel. L’utilisation de Letybo pendant l’allaitement n’est pas recommandée.
Fertilité
Il n’existe pas de données appropriées concernant les effets de la toxine botulinique de type A sur la fertilité chez les femmes en âge de procréer. Les études effectuées chez des rats mâles et femelles ont montré une diminution de la fertilité (voir rubrique 5.3).
4.7. Effets sur l'aptitude à conduire des véhicules et à utiliser des machines
Résumé du profil de sécurité
La sécurité de Letybo a été évaluée dans trois études cliniques déterminantes de phase 3 comprenant toutes une partie contrôlée par placebo (cycle 1) et une partie d’extension à long terme (cycles 2-4) couvrant jusqu’à une année et incluant 1 162 patients traités par Letybo. En outre, il existe des données probantes issues d’une étude de phase 3 sur les rides glabellaires réalisée en Corée, ainsi que des données post-commercialisation.
Les effets indésirables peuvent être liés au médicament à l’étude (Letybo), à la procédure d’injection ou aux deux. En général, les effets indésirables sont transitoires et surviennent dans les premiers jours suivant l’injection. La plupart des événements indésirables signalés étaient d’une intensité légère à modérée. Les réactions indésirables au médicament les plus fréquentes (signalées chez au moins 2 patients traités par Letybo au cours du cycle 1) dans les trois études pivots sur Letybo dans le traitement des rides glabellaires étaient la céphalée (1,7 % des patients), la douleur au site d’injection (0,3 % des patients) et le ptosis, le blépharospasme, la gêne de la tête et la contusion (0,2 % des patients pour chaque).
Des cas de douleur localisée, d’inflammation, de paresthésie, d’hypoesthésie, de sensibilité au toucher, de gonflement/œdème, d’érythème, d’infection localisée, d’hémorragie et/ou de contusion ont été associés à l’injection. Des cas de fièvre et de syndrome de grippe ont également été signalés après des injections de toxine botulinique (voir rubrique 4.4).
Tableau récapitulatif des effets indésirables
Des informations sur la fréquence des effets indésirables, issues de l’expérience clinique, sont présentées ci-après. Les catégories de fréquence sont définies comme suit : très fréquent (≥ 1/10) ; fréquent (≥ 1/100, < 1/10) ; peu fréquent (≥ 1/1 000, < 1/100) ; rare (≥ 1/10 000, < 1/1 000) ; très rare (< 1/10 000).
Tableau 1 Effets indésirables signalés dans les études cliniques et post-commercialisation après l’administration de Letybo
|
Fréquence |
Effet indésirable |
|
|
Infections et infestations |
Peu fréquent |
Rhinopharyngite |
|
Rare |
Herpès buccal, folliculite* |
|
|
Affections du système nerveux |
Fréquent |
Céphalée |
|
Peu fréquent |
Gêne de la tête* |
|
|
Rare |
Migraine, sensation vertigineuse, paresthésie, défaut du champ visuel, dysarthrie |
|
|
Affections oculaires |
Peu fréquent |
Ptosis, blépharospasme, œdème périorbitaire |
|
Rare |
Hémorragie conjonctivale*, sécheresse oculaire, vision trouble, douleur oculaire*, trouble de la sensibilité palpébrale** |
|
|
Affections respiratoires, thoraciques et médiastinales |
Rare |
Hypoesthésie pharyngée |
|
Affections gastro-intestinales |
Rare |
Constipation, nausée |
|
Affections de la peau et du tissu sous-cutané |
Rare |
Ptose sourcilière, sécheresse cutanée, urticaire |
|
Troubles musculo-squelettiques et du tissu conjonctif |
Peu fréquent |
Effet Mephisto (élévation latérale du sourcil) |
|
Troubles généraux et anomalies au site d’administration |
Fréquent |
Réaction au site d’injection |
|
Peu fréquent |
Douleur au site d’injection, contusion au site d’injection, gonflement au site d’administration*, prurit au site d’injection, tuméfaction au site d’injection, pression au site d’injection** |
|
|
Rare |
Douleur faciale*, syndrome grippal, fièvre |
|
|
Investigations |
Rare |
Potassium sanguin augmenté |
|
Lésions, intoxications et complications liées aux procédures |
Peu fréquent |
Contusion, hématome périorbitaire* |
Remarque : sur les 1 162 patients traités par Letybo, 1 seul a connu des événements rares.
Une « approche du pire des cas » a été utilisée pour qualifier la fréquence des événements survenus lors des études cliniques et post-commercialisation.
*Réaction indésirable au médicament liée à la procédure d’injection. À noter que ces informations n’ont pas été recueillies pour l’étude coréenne post-commercialisation.
**Étude post-commercialisation uniquement.
Description de certains effets indésirables
Effets indésirables liés à l’application
Les effets indésirables liés à l’application qui ont été signalés après l’administration de Letybo sont peu fréquents lorsqu’ils sont pris individuellement, mais fréquents lorsqu’ils sont additionnés. Les réactions peu fréquentes au site d’injection comprennent la douleur, la contusion, le gonflement, le prurit, la tuméfaction et la pression. Les événements rares au niveau du site d’injection comprennent la douleur et la gêne.
Risque de diffusion de la toxine à distance du site d’administration
Des effets indésirables potentiellement liés à la diffusion de la toxine à distance du site d’administration ont été signalés dans de très rares cas pour la toxine botulinique (par exemple faiblesse musculaire, dysphagie, constipation ou pneumopathie d’inhalation possiblement fatale) (voir rubrique 4.4).
Déclaration des effets indésirables suspectés
La déclaration des effets indésirables suspectés après autorisation du médicament est importante. Elle permet une surveillance continue du rapport bénéfice/risque du médicament. Les professionnels de santé déclarent tout effet indésirable suspecté via le système national de déclaration : Agence nationale de sécurité du médicament et des produits de santé (ANSM) et réseau des Centres Régionaux de Pharmacovigilance - Site internet: https://signalement.social-sante.gouv.fr/
Symptômes de surdosage
Le surdosage de Letybo dépend de la dose, du site d’injection et des propriétés des tissus sous-jacents.
Aucun cas de toxicité systémique résultant d’une injection accidentelle de toxine botulinique de type A n’a été observé. Des doses trop élevées peuvent engendrer une paralysie neuromusculaire généralisée et profonde, locale ou à distance. Aucun cas d’ingestion de toxine botulinique de type A n’a été signalé.
Les signes de surdosage peuvent ne pas être visibles immédiatement après l’injection.
Gestion du surdosage
En cas d’injection ou d’ingestion accidentelle, ou de suspicion de surdosage ou de diffusion de la toxine, le patient doit être mis sous observation médicale afin de déceler tout signe ou symptôme de faiblesse générale ou de paralysie musculaire. L’hospitalisation doit être envisagée si le patient présente des symptômes d’intoxication par la toxine botulinique de type A (faiblesse généralisée, ptosis, diplopie, troubles de la déglutition et de la parole, ou parésie des muscles respiratoires).
5. PROPRIETES PHARMACOLOGIQUES
5.1. Propriétés pharmacodynamiques
Classe pharmacothérapeutique : Relaxants musculaires, Autres relaxants musculaires, Agents à action périphérique, Code ATC : M03AX01.
Mécanisme d’action
La neurotoxine de type A sécrétée par Clostridium botulinum bloque la libération périphérique du neurotransmetteur acétylcholine au niveau des terminaisons nerveuses cholinergiques présynaptiques des jonctions neuromusculaires en clivant la SNAP-25, une protéine nécessaire à la fixation et à la libération de l’acétylcholine par les vésicules situées dans les terminaisons nerveuses, conduisant ainsi à une dénervation du muscle et à une paralysie flasque.
Après l’injection, il se produit une liaison initiale rapide et extrêmement forte de la toxine à des récepteurs cellulaires de surface spécifiques. Cette étape est suivie d’un passage de la toxine à travers la membrane plasmatique par endocytose médiée par récepteurs. Enfin, la toxine est libérée dans le cytosol, entraînant une inhibition progressive de la libération d’acétylcholine. Les signes cliniques sont visibles au bout de 2 à 3 jours, le pic d’effet étant observé 4 semaines après l’injection. Le rétablissement survient généralement dans les 3 à 4 mois suivant l’injection, lorsque les terminaisons nerveuses recréent leurs connexions avec les plaques basales.
Données cliniques
La sécurité et l’efficacité de Letybo ont été analysées dans 3 études pivots de phase 3 en double aveugle (BLESS I, BLESS II et BLESS III) au cours desquelles un total de 955 patients ont été traités par Letybo et 317 patients ont été traités par placebo dans le cadre de 1 traitement. En outre, des données sont disponibles pour 854 patients traités par Letybo dans une extension en ouvert des études BLESS I et II dans le cadre de 1 à 3 traitements supplémentaires. Le programme de développement clinique en Corée a permis d’obtenir des données complémentaires sur les rides glabellaires. Il comprenait une étude de phase 3 (HG-11-01) portant sur 137 patients et une étude post-commercialisation (HG-13-02) portant sur 815 patients.
Au moment de l’inclusion dans les études BLESS I, BLESS II et BLESS III, tous les patients présentaient des rides glabellaires modérées (27 % des sujets) ou sévères (73 % des sujets) lors du froncement maximal des sourcils. À la dose de 20 unités, Letybo a réduit de manière significative la sévérité des rides glabellaires observées au froncement maximal des sourcils, telle que mesurée à travers une évaluation par l’investigateur et par le patient sur une échelle des rides du visage en 4 points (FWS). Un taux de réponse statistiquement significatif a été observé avec Letybo même en utilisant le critère d’évaluation strict exigeant une amélioration de 2 points du score FWS. Des taux de réponse élevés en faveur de Letybo ont également été observés en appliquant la définition de la réponse cliniquement significative, à savoir l’obtention d’un score FWS de 0 ou 1 (rides absentes ou légères) selon l’évaluation de l’investigateur à la semaine 4 (voir tableau 2).
Tableau 2 Taux de réponse à la semaine 4 par rapport à l’inclusion au froncement maximal des sourcils selon l’échelle des rides du visage (FWS) dans les études BLESS I, BLESS II et BLESS III – Analyse des données complètes
|
|
BLESS I |
BLESS II |
BLESS III |
|||
|
Évaluation par : |
Letybo |
Placebo |
Letybo |
Placebo |
Letybo |
Placebo |
|
Taux de réponse (n [%]) : Réduction du score FWS de modéré ou sévère à nul ou léger (amélioration ≥ 2 points requise)a |
||||||
|
Investigateur ET patient |
246 (46,5 %)* |
0 (0 %) |
78 (48,8 %)* |
1 (1,9 %) |
172 (64,7 %)* |
0 (0,0 %) |
|
Investigateur |
348 (65,8 %)* |
1 (0,6 %) |
120 (75,0 %)* |
1 (1,9 %) |
209 (78,6 %)* |
1 (1,1 %) |
|
Patient |
290 (54,8 %)* |
0 (0 %) |
83 (51,9 %)* |
1 (1,9 %) |
183 (68,8 %)* |
0 (0,0 %) |
|
Taux de réponse (%) : Réduction du score FWS de modéré ou sévère à nul ou légerb |
||||||
|
Investigateur |
393 (74,3 %)* |
3 (1,7 %) |
136 (85,0 %)* |
2 (3,8 %) |
218 (82,0 %)* |
1 (1,1 %) |
*Valeur p < 0,001 au test de Cochran-Mantel-Haenszel pour la différence entre Letybo et le placebo ;
N = nombre de patients randomisés ; n = nombre de répondeurs
a Critère principal d’évaluation de l’efficacité
b Analyse post hoc
Au total, 38,3 % des sujets traités par Letybo présentaient une amélioration de 3 points du score de sévérité des rides, passant de sévère (grade 3 de la FWS) au moment de l’inclusion à nul (grade 0 de la FWS) à la semaine 4, selon l’évaluation de l’investigateur.
L’amélioration des rides glabellaires (basée sur une réduction ≥ 2 points du score FWS au froncement maximal des sourcils selon l’évaluation du patient et de l’investigateur) a débuté pendant la semaine suivant l’injection, avec un effet maximal au cours de la deuxième semaine suivant l’injection. On peut considérer que l’effet dure entre 12 et 16 semaines (voir figure 1).
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
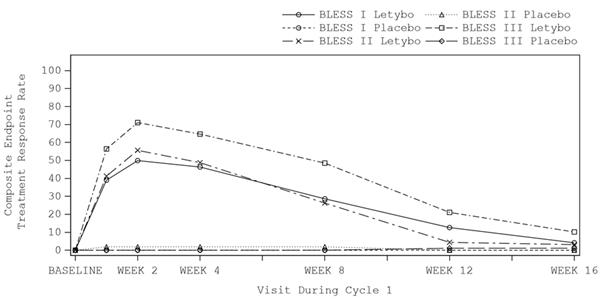
Figure 1 Évolution dans le temps du taux de répondeurs (amélioration ≥ 2 points du score FWS requise selon l’évaluation du patient et de l’investigateur) pendant le cycle 1 pour le traitement actif par rapport au placebo dans les études pivots BLESS
Il a pu être démontré que, sur le plan statistique, le taux de répondeurs avec une réduction ≥ 1 point du score FWS au repos était significativement plus élevé dans le groupe sous Letybo que dans le groupe sous placebo : quatre semaines après l’injection, les investigateurs ont estimé que 63,1 %, 59,4 % et 61,3 % des patients traités par Letybo et 15,4 %, 5,7 % et 9,0 % des patients traités par placebo avaient connu une amélioration ≥ 1 point du score FWS au repos dans les études BLESS I, BLESS II et BLESS III, respectivement (la valeur p des différences entre les traitements était < 0,001 pour toutes les études).
Les données en ouvert sur les doses répétées à long terme ont confirmé que les taux de réponse après les deuxième, troisième et quatrième traitements par Letybo sur la période étudiée d’une année sont restés élevés même si, d’après la méthodologie de l’étude, les cycles de retraitement comportaient un biais de non-réponse.
Selon la nouvelle échelle Skindex-16 modifiée qui évalue la qualité de vie des personnes présentant des rides glabellaires, au moment de l’inclusion dans les études, les rides glabellaires avaient un retentissement psychologique négatif modéré ou sévère chez plus de 85 % des participants, et un retentissement léger chez environ 15 % d’entre eux.
Comme le montrent les scores de l’échelle Skindex-16 modifiée, une nette amélioration du retentissement psychologique a été observée chez les patients traités par Letybo par rapport à ceux traités par placebo.
Des résultats cosmétiques largement favorables, corroborés par des taux élevés de satisfaction, ont été enregistrés par les patients.
Sécurité
Au cours du traitement en double aveugle dans les études BLESS I, BLESS II et BLESS III, 33 patients (3,5 %) ont présenté des EIAT considérés au minimum comme possiblement liés à Letybo, et 8 patients (2,5 %) ont présenté des EIAT considérés au minimum comme possiblement liés au placebo. Au cours du traitement en ouvert, 46 patients (5,4 %) ont présenté des EIAT considérés au minimum comme possiblement liés à Letybo (avec jusqu’à 3 cycles de traitement). Aucun de ces EI connexes n’a été considéré comme grave. Les résultats étaient cohérents avec ceux de l’étude de phase 3 HG-11-01 sur les rides glabellaires.
Dans les études BLESS I, BLESS II et BLESS III, la formation d’anticorps a été évaluée avant chaque traitement, 4 semaines après chaque traitement et lors de la visite finale. Aucun anticorps neutralisant n’a été détecté chez les patients après l’administration de Letybo.
Données post-commercialisation
Les données post-commercialisation, y compris celles d’une étude post-commercialisation sur les rides glabellaires (HG-13-02) impliquant 815 patients, sont conformes à celles observées dans les études cliniques.
Sujets âgées
Sur l’ensemble des études BLESS I, BLESS II et BLESS III, 152/1 272 patients (11,91 %) étaient âgés de 65 ans ou plus au moment de la sélection. Aucun patient n’était âgé de plus de 75 ans. Toujours sur l’ensemble des études BLESS I, BLESS II et BLESS III, le taux de répondeurs composite à la semaine 4 (critère d’évaluation principal) parmi les patients traités par Letybo était plus faible chez les patients de 65 ans ou plus, avec 46/118 (39,0 %), que chez les patients de moins de 65 ans, avec 450/839 (53,6 %). Il demeurait néanmoins à un niveau élevé et cliniquement pertinent. Aucune différence majeure n’a été observée au niveau du taux global de patients présentant des EIAT considérés comme liés au traitement en double aveugle par Letybo au sein des 3 études combinées (3,7 % et 1,7 % chez les patients de moins de 65 ans et de 65 ans ou plus, respectivement, lorsque les EIAT liés au médicament et/ou à la procédure d’injection étaient pris en compte).
Population pédiatrique
L’Agence européenne des médicaments a accordé une dérogation à l’obligation de soumettre les résultats d’études réalisées avec Letybo dans tous les sous-groupes de la population pédiatrique pour le traitement des rides d’origine musculaire conformément à la décision du Plan d’investigation pédiatrique, dans l’indication autorisée EMEA-001788-PIP01-15 (voir rubrique 4.2 pour les informations concernant l’utilisation pédiatrique).
5.2. Propriétés pharmacocinétiques
5.3. Données de sécurité préclinique
Dans une étude sur le développement embryo-fœtal avec injections intramusculaires quotidiennes de BoNT/A-DP jusqu’à la dose de 8 U/kg entre les jours 5 et 16 de la gestation chez des rates gravides, une paralysie musculaire dépendant de la dose a été mise en évidence chez les mères, entraînant une atrophie musculaire, une réduction du poids corporel et une région périnéale souillée. Un retard de l’ossification fœtale et une réduction du poids corporel fœtal (≥ 20 %) sans malformation ont été détectés. En conformité avec l’expérience acquise avec d’autres produits à base de toxine botulinique de type A, ils ont été interprétés comme des conséquences secondaires de la toxicité maternelle. Les effets sur le développement péri-/post-natal n’ont pas été évalués.
Chez les rats mâles et femelles, des atteintes de la fertilité ont été observées avec d’autres produits contenant de la toxine botulinique de type A à doses élevées.
Aucune étude de génotoxicité, d’antigénicité, de carcinogénicité ou de fertilité n’a été réalisée concernant BoNT/A-DP.
Chlorure de sodium
Flacon avant ouverture
3 ans.
Solution reconstituée
La stabilité physico-chimique en cours d’utilisation a été démontrée pendant 24 heures entre 2 °C et 8 °C.
D’un point de vue microbiologique, le produit doit être utilisé immédiatement, sauf si la méthode d'ouverture/de reconstitution/de dilution exclut le risque de contamination microbienne. En cas d’utilisation non immédiate, les durées et conditions de conservation en cours d’utilisation relèvent de la responsabilité de l’utilisateur.
6.4. Précautions particulières de conservation
À conserver et transporter réfrigéré (entre 2 °C et 8 °C).
Pour les conditions de conservation du médicament après reconstitution, voir rubrique 6.3.
6.5. Nature et contenu de l'emballage extérieur
Flacon transparent de 5 mL (en verre de type I) muni d’un bouchon (caoutchouc chlorobutyle) et d’un opercule d’inviolabilité (en aluminium).
Boîtes contenant 1 ou 2 flacons. Conditionnement multiple de 2 (2 boîtes de 1) flacons.
Conditionnement multiple de 6 (6 boîtes de 1) flacons.
Toutes les présentations peuvent ne pas être commercialisées.
6.6. Précautions particulières d’élimination et de manipulation
Les instructions d’utilisation, de manipulation et d’élimination doivent être strictement respectées.
La reconstitution doit être effectuée conformément aux bonnes pratiques cliniques, notamment en ce qui concerne les techniques d’asepsie.
Pour reconstituer Letybo, utiliser 1,25 mL d’une solution injectable de chlorure de sodium à 9 mg/mL (0,9 %) comme diluant.
Il est recommandé de reconstituer le contenu du flacon et de préparer la seringue au-dessus de serviettes en papier à doublure en plastique pour récupérer tout déversement éventuel. Aspirer la solution injectable de chlorure de sodium à 9 mg/mL (0,9 %) dans une seringue et l’injecter délicatement dans le flacon pour éviter la formation de mousse/bulles ou toute agitation vigoureuse qui pourrait dénaturer le produit. Si le vide n’aspire pas le solvant dans le flacon, celui-ci doit être éliminé. Après reconstitution, Letybo se présente comme une solution limpide, incolore et pratiquement dépourvue de particules. Avant utilisation, inspecter visuellement le flacon pour s’assurer que le produit est exempt de particules étrangères.
Letybo ne doit pas être utilisé si la solution reconstituée est trouble ou contient des particules.
Toute solution injectable qui a été conservée plus de 24 heures doit être éliminée.
Tout médicament non utilisé ou déchet doit être éliminé conformément à la réglementation en vigueur.
Procédure à suivre pour une élimination en toute sécurité des flacons, des seringues et des matériels utilisés
Pour une élimination en toute sécurité, la fraction de Letybo non reconstituée doit être reconstituée dans le flacon avec une petite quantité d’eau, puis autoclavée. Les flacons vides ou contenant un résidu de solution, les seringues et les déversements doivent être autoclavés. La fraction restante de Letybo peut sinon être inactivée à l’aide d’une solution diluée d’hydroxyde de sodium (0,1 N NaOH) ou d’hypochlorite de sodium (NaOCl à 0,5 % ou 1 %).
Après inactivation, les flacons, seringues et matériels usagés ne doivent pas être vidés, mais placés dans des récipients appropriés et éliminés conformément à la réglementation en vigueur.
Recommandations en cas d’accident lors de la manipulation de la toxine botulinique
· Tout déversement du produit doit être essuyé : soit à l’aide d’un matériau absorbant imprégné d’une solution d’hypochlorite de sodium dans le cas du produit en poudre, soit à l’aide d’un matériau absorbant sec dans le cas du produit reconstitué.
· Les surfaces contaminées doivent être nettoyées avec un matériau absorbant imprégné d’une solution d’hypochlorite de sodium, puis séchées.
· Si un flacon est cassé, recueillir soigneusement les morceaux de verre et essuyer le produit comme indiqué ci-dessus, en évitant toute coupure de la peau.
· En cas de contact du médicament avec la peau, laver la zone concernée avec une solution d’hypochlorite de sodium, puis rincer abondamment à l’eau.
· En cas de contact du médicament avec les yeux, rincer abondamment à l’eau ou avec une solution de rinçage oculaire.
· En cas de contact du médicament avec une plaie, une coupure ou de la peau éraflée, rincer abondamment à l’eau et prendre les mesures médicales appropriées en fonction de la dose injectée.
7. TITULAIRE DE L’AUTORISATION DE MISE SUR LE MARCHE
Industriezeile 6
2100 Leobendorf
Autriche
8. NUMERO(S) D’AUTORISATION DE MISE SUR LE MARCHE
· 34009 302 469 0 0 : 1 flacon de poudre en verre. Boîte de 1.
· 34009 302 469 2 4 : 2 (2 boîtes de 1) flacons de poudre en verre. (Conditionnement Multiple).
· 34009 302 469 3 1 : 6 (6 boîtes de 1) flacons de poudre en verre. (Conditionnement Multiple).
· 34009 302 747 5 0 : 2 flacons de poudre en verre. Boîte de 2.
9. DATE DE PREMIERE AUTORISATION/DE RENOUVELLEMENT DE L’AUTORISATION
[à compléter ultérieurement par le titulaire]
10. DATE DE MISE A JOUR DU TEXTE
[à compléter ultérieurement par le titulaire]
Sans objet.
12. INSTRUCTIONS POUR LA PREPARATION DES RADIOPHARMACEUTIQUES
Liste I.
Médicament réservé à l'usage professionnel selon l'article R 5121-80 du Code la Santé Publique.
Prescription réservée aux spécialistes en chirurgie plastique, reconstructrice et esthétique, en dermatologie, en chirurgie de la face et du cou, en chirurgie maxillo-faciale et en ophtalmologie.
ANSM - Mis à jour le : 27/02/2026
LETYBO 50 unités, poudre pour solution injectable
Toxine botulinique de type A
· Gardez cette notice. Vous pourriez avoir besoin de la relire.
· Si vous avez d’autres questions, interrogez votre médecin ou votre pharmacien.
· Si vous ressentez un quelconque effet indésirable, parlez-en à votre médecin ou votre pharmacien. Ceci s’applique aussi à tout effet indésirable qui ne serait pas mentionné dans cette notice. Voir rubrique 4.
1. Qu'est-ce que LETYBO 50 unités, poudre pour solution injectable et dans quels cas est-il utilisé ?
2. Quelles sont les informations à connaître avant d'utiliser LETYBO 50 unités, poudre pour solution injectable ?
3. Comment utiliser LETYBO 50 unités, poudre pour solution injectable ?
4. Quels sont les effets indésirables éventuels ?
5. Comment conserver LETYBO 50 unités, poudre pour solution injectable ?
6. Contenu de l’emballage et autres informations.
1. QU’EST-CE QUE LETYBO 50 unités, poudre pour solution injectable ET DANS QUELS CAS EST-IL UTILISE ?
Classe pharmacothérapeutique : autres myorelaxants à action périphérique - code ATC : M03AX01.
Letybo contient la substance active « toxine botulinique de type A », qui agit en bloquant les impulsions nerveuses vers les muscles dans lesquels il a été injecté. Cela empêche la contraction des muscles, entraînant une paralysie temporaire.
Letybo est utilisé chez les adultes de moins de 75 ans pour l’amélioration temporaire de l’apparence des rides verticales intersourcilières modérées à sévères lorsque leur présence entraîne chez eux un retentissement psychologique important.
2. QUELLES SONT LES INFORMATIONS A CONNAITRE AVANT D’UTILISER LETYBO 50 unités, poudre pour solution injectable ?
N’utilisez jamais LETYBO 50 unités, poudre pour solution injectable :
· si vous êtes allergique à la toxine botulinique de type A ou à l’un des autres composants contenus dans ce médicament (mentionnés dans la rubrique 6) ;
· si vous présentez des troubles de l’activité musculaire, par exemple une myasthénie grave, un syndrome de Lambert-Eaton ou une sclérose latérale amyotrophique ;
· si vous présentez une infection ou une inflammation aiguë au niveau des sites d’injection proposés.
Avertissements et précautions
Adressez-vous à votre médecin avant d’utiliser Letybo si vous présentez :
· tout trouble affectant les muscles et/ou leur contrôle direct par le système nerveux ;
· des difficultés à avaler ou à respirer, présentes ou passées ;
· un trouble de la coagulation.
Si vous avez des antécédents de ces pathologies, Letybo n’est pas recommandé dans votre cas.
La douleur liée aux aiguilles et/ou la peur des injections peuvent entraîner une syncope due à une chute soudaine de la pression artérielle.
Des cas d’effets secondaires dus à la diffusion de la toxine botulinique hors du site d’injection et au botulisme ont été rapportés très rarement avec la toxine botulinique (par exemple une vision double, une vision trouble et/ou des paupières tombantes, des difficultés à respirer ou à parler, une faiblesse musculaire excessive, des difficultés à avaler, ou une fausse route d’aliments ou de liquide dans les voies respiratoires). Les difficultés à avaler et à respirer sont graves et peuvent entraîner la mort.
Si vous avez des problèmes pour avaler, parler ou respirer, consultez immédiatement un médecin.
Enfants et adolescents
L’utilisation de Letybo n’est pas recommandée chez les enfants et les adolescents de moins de 18 ans.
Autres médicaments et LETYBO 50 unités, poudre pour solution injectable
Informez votre médecin ou pharmacien si vous utilisez, avez récemment utilisé ou pourriez utiliser tout autre médicament.
Les médicaments suivants peuvent affecter ou être affectés par Letybo :
· les médicaments interférant avec la transmission des impulsions nerveuses aux muscles ;
· certains médicaments utilisés pour traiter les infections bactériennes, comme la spectinomycine ou les antibiotiques du groupe des aminosides ;
· tout autre médicament contenant une toxine botulinique.
Grossesse et allaitement
Si vous êtes enceinte ou que vous allaitez, si vous pensez être enceinte ou planifiez une grossesse, demandez conseil à votre médecin avant d’utiliser ce médicament.
Letybo n’est pas recommandé pendant la grossesse ou l’allaitement ni chez les femmes en âge de procréer n’utilisant pas de contraception.
Conduite de véhicules et utilisation de machines
La toxine botulinique de type A peut provoquer faiblesse, vertiges et troubles de la vision. Ne conduisez pas de véhicule et n’utilisez pas de machine si votre capacité de réaction est réduite.
LETYBO 50 unités, poudre pour solution injectable contient du sodium
Ce médicament contient moins de 1 mmol (23 mg) de sodium par dose, c.-à-d. qu’il est essentiellement « sans sodium ».
3. COMMENT UTILISER LETYBO 50 unités, poudre pour solution injectable ?
Letybo ne vous est administré que par un médecin dûment qualifié qui dispose des équipements nécessaires. La description détaillée de la préparation de la solution et du mode d’emploi figure à la rubrique « Les informations suivantes sont destinées exclusivement aux professionnels de la santé » à la fin de cette notice.
La dose recommandée est de
20 unités réparties en cinq injections de 0,1 mL (4 unités). Chaque injection est pratiquée dans les muscles situés au-dessus ou entre les sourcils.
Letybo est injecté par voie intramusculaire (IM).
Après reconstitution de la solution, le flacon ne doit être utilisé que pour une seule séance par patient. Toute solution non administrée doit être éliminée comme indiqué après la rubrique 6 dans les informations destinées aux professionnels de santé.
Un intervalle minimum de 3 mois est recommandé entre deux traitements par Letybo.
Si vous avez utilisé plus de LETYBO 50 unités, poudre pour solution injectable que vous n’auriez dû
Un surdosage peut entraîner une paralysie des muscles et/ou des nerfs. Les signes de surdosage peuvent ne pas être visibles immédiatement après l’injection.
En cas de surdosage, le médecin surveillera vos symptômes éventuels, comme une faiblesse générale ou une paralysie musculaire. Vous serez hospitalisé(e) si les symptômes d’une intoxication à la toxine botulinique de type A se manifestent, par exemple :
· faiblesse généralisée ;
· paupière supérieure tombante ou vision double ;
· troubles de la déglutition et de la parole ;
· paralysie partielle des muscles contrôlant la respiration.
Si vous avez d’autres questions sur l’utilisation de ce médicament, demandez plus d’informations à votre médecin ou à votre pharmacien.
4. QUELS SONT LES EFFETS INDESIRABLES EVENTUELS ?
La plupart des effets indésirables sont d’intensité légère à modérée. Ils surviennent dans les premiers jours suivant l’injection et sont temporaires.
Certains effets indésirables pourraient être très graves. Si vous présentez l’un des effets indésirables suivants, informez-en immédiatement votre médecin ou demandez à vos proches de l’informer et rendez-vous au service des urgences le plus proche :
Peu fréquent, peut affecter jusqu’à 1 personne sur 100
· Paupière supérieure tombante, spasme de la paupière
Rare, peut affecter jusqu’à 1 personne sur 1 000
· Trouble de la sensibilité des paupières, sourcils tombants
· Saignement conjonctival
· Douleur oculaire, sécheresse oculaire, défaut du champ visuel, vision trouble
· Sensations diminuées dans la gorge
· Constipation
· Altération de phonème.
Très rare, peut affecter jusqu’à 1 personne sur 10 000
· Faiblesse musculaire
· Difficultés à avaler
· Infection causée par l’inhalation d’aliments ou de liquide dans les voies aériennes ou les poumons
· Difficultés à respirer.
Outre ces effets indésirables possibles, une réaction allergique grave peut entraîner les symptômes suivants :
· Difficultés à avaler, à respirer ou à parler en raison d’un gonflement du visage, des lèvres, de la bouche ou de la gorge. Outre ces symptômes, une éruption urticarienne peut survenir (voir rubrique 2).
D’autres effets indésirables connus peuvent survenir selon les catégories de fréquence suivantes. Informez votre médecin ou pharmacien s’ils deviennent graves :
Fréquent, peut affecter jusqu’à 1 personne sur 10
· Céphalée
· Réactions au site d’injection.
Peu fréquent, peut affecter jusqu’à 1 personne sur 100
· Gêne au niveau de la tête
· Gonflement localisé, par exemple de la paupière, du visage ou du contour des yeux
· Site d’injection : douleur, bleus, gonflement, démangeaisons, masse, pression
· Bleus, par exemple autour des yeux
· Infection, par exemple une infection virale des voies aériennes supérieures telle qu’un rhume
· Effet Mephisto (élévation de la partie externe des sourcils).
Rare, peut affecter jusqu’à 1 personne sur 1 000
· Migraine
· Inflammation des follicules pileux
· Vertiges
· Sensations anormales, par exemple picotements, fourmillements et démangeaisons
· Nausée
· Peau sèche, urticaire, démangeaisons
· Douleur du visage
· Fièvre
· Herpès buccal
· Potassium sanguin augmenté
· Syndrome grippal.
Déclaration des effets secondaires
Si vous ressentez un quelconque effet indésirable, parlez-en à votre médecin ou votre pharmacien. Ceci s’applique aussi à tout effet indésirable qui ne serait pas mentionné dans cette notice. Vous pouvez également déclarer les effets indésirables directement via le système national : Agence nationale de sécurité du médicament et des produits de santé (ANSM) et réseau des Centres Régionaux de Pharmacovigilance - Site internet: https://signalement.social-sante.gouv.fr/
En signalant les effets indésirables, vous contribuez à fournir davantage d’informations sur la sécurité du médicament.
5. COMMENT CONSERVER LETYBO 50 unités, poudre pour solution injectable ?
Tenir ce médicament hors de la vue et de la portée des enfants.
N’utilisez pas ce médicament après la date de péremption indiquée sur l’étiquette et l’emballage après EXP. La date de péremption fait référence au dernier jour de ce mois.
À conserver et transporter réfrigéré (entre 2 °C et 8 °C).
Solution reconstituée
La stabilité physico-chimique en cours d’utilisation a été démontrée pendant 24 heures entre 2 °C et 8 °C.
D’un point de vue microbiologique, le produit doit être utilisé immédiatement, sauf si la méthode d'ouverture/de reconstitution/de dilution exclut le risque de contamination microbienne. En cas d’utilisation non immédiate, les durées et conditions de conservation en cours d’utilisation relèvent de la responsabilité de l’utilisateur.
Ne jetez aucun médicament au tout-à-l’égout ou avec les ordures ménagères. Demandez à votre pharmacien d’éliminer les médicaments que vous n’utilisez plus. Ces mesures contribueront à protéger l’environnement.
6. CONTENU DE L’EMBALLAGE ET AUTRES INFORMATIONS
Ce que contient LETYBO 50 unités, poudre pour solution injectable
· La substance active est : Toxine botulinique de type A.
Chaque flacon contient 50 unités de toxine botulinique de type A produit par Clostridium botulinum.
Après reconstitution, chaque 0,1 mL de solution contient 4 unités
· Les autres composants sont : albumine humaine et chlorure de sodium.
Qu’est-ce que LETYBO 50 unités, poudre pour solution injectable et contenu de l’emballage extérieur
Letybo se présente sous forme d’une poudre pour solution injectable blanche dans un flacon en verre transparent muni d’un bouchon en caoutchouc et d’un opercule inviolable en aluminium.
Une boîte contient un ou deux flacons.
Conditionnement multiple contenant 2 boîtes, chaque boîte contient un flacon.
Conditionnement multiple contenant 6 boîtes, chaque boîte contient un flacon.
Toutes les présentations peuvent ne pas être commercialisées.
Titulaire de l’autorisation de mise sur le marché
Industriezeile 6
2100 Leobendorf
Autriche
Exploitant de l’autorisation de mise sur le marché
Industriezeile 6
2100 Leobendorf
Autriche
Industriezeile 6
2100 Leobendorf
Autriche
Noms du médicament dans les Etats membres de l'Espace Economique Européen
Ce médicament est autorisé dans les Etats membres de l'Espace Economique Européen et au Royaume-Uni (Irlande du Nord) sous les noms suivants : Conformément à la réglementation en vigueur.
[À compléter ultérieurement par le titulaire]
La dernière date à laquelle cette notice a été révisée est :
[à compléter ultérieurement par le titulaire]
{mois AAAA}.
Des informations détaillées sur ce médicament sont disponibles sur le site Internet de l’ANSM (France).
‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑
Les informations suivantes sont destinées exclusivement aux professionnels de la santé :
Les unités de toxine botulinique ne sont pas interchangeables d’un produit à l’autre. Les doses recommandées (exprimées en unités) diffèrent des autres préparations de toxine botulinique.
Les instructions d’utilisation, de manipulation et d’élimination doivent être strictement respectées.
Préparation de la solution
La reconstitution doit être effectuée conformément aux bonnes pratiques cliniques, notamment en ce qui concerne les techniques d’asepsie.
Pour reconstituer Letybo, utiliser 1,25 mL d’une solution injectable de chlorure de sodium à 9 mg/mL (0,9 %) comme diluant.
Il est recommandé de reconstituer le contenu du flacon et de préparer la seringue au-dessus de serviettes en papier à doublure en plastique pour récupérer tout déversement éventuel. Aspirer la solution injectable de chlorure de sodium à 9 mg/mL (0,9 %) dans une seringue et l’injecter délicatement dans le flacon pour éviter la formation de mousse/bulles ou toute agitation vigoureuse qui pourrait dénaturer le produit. Si le vide n’aspire pas le solvant dans le flacon, celui-ci doit être éliminé. Après reconstitution, Letybo se présente comme une solution limpide, incolore et pratiquement dépourvue de particules. Avant utilisation, inspecter visuellement le flacon pour s’assurer que le produit est exempt de particules étrangères.
Letybo ne doit pas être utilisé si la solution reconstituée est trouble ou contient des particules.
Solution reconstituée
La stabilité physico-chimique en cours d’utilisation a été démontrée pendant 24 heures entre 2 °C et 8 °C.
D’un point de vue microbiologique, le produit doit être utilisé immédiatement, sauf si la méthode d'ouverture/de reconstitution/de dilution exclut le risque de contamination microbienne. En cas d’utilisation non immédiate, les durées et conditions de conservation en cours d’utilisation relèvent de la responsabilité de l’utilisateur. Toute solution injectable qui a été conservée plus de 24 heures doit être éliminée.
Tout médicament non utilisé ou déchet doit être éliminé conformément à la réglementation en vigueur.
Mode d’emploi
Les injections intramusculaires doivent être réalisées à l’aide d’une seringue stérile du type utilisé pour l’insuline ou la tuberculine, avec une capacité de 1 mL, des graduations de 0,01 mL et une aiguille de calibre 30 à 31 G.
Aspirer dans la seringue stérile 0,5 mL de la solution de Letybo dûment reconstituée et expulser toute bulle d’air présente dans le cylindre. L’aiguille utilisée pour reconstituer le médicament doit être retirée et remplacée pour l’administration.
Veiller à ce que Letybo ne soit pas injecté dans un vaisseau sanguin.
Afin de réduire le risque de blépharoptose, éviter les injections à proximité du muscle levator palpebrae superioris, en particulier chez les patients présentant d’importants complexes abaisseurs des sourcils. Lors de l’injection dans deux sites pour chaque muscle corrugator supercilii, réaliser la première injection juste au-dessus du bord médial des sourcils. La seconde injection sera pratiquée à environ 1 cm au-dessus du bord supra-orbitaire (limites osseuses rigides palpables au-dessus de la marge haute de la paupière supérieure), au point de rencontre des lignes médianes des sourcils. Le site d’injection pour le muscle procerus se situe juste au-dessus de la ligne médiane de l’arête nasale, à l’endroit où se forment les rides horizontales entre les extrémités médiales des sourcils. Lors de l’administration dans les extrémités médiales des muscles corrugator supercilii et sur les lignes médianes des sourcils, les sites d’injection doivent être éloignés d’au moins 1 cm du bord supra-orbitaire (limites osseuses rigides palpables au-dessus de la marge haute de la paupière supérieure).
![]()
![]()
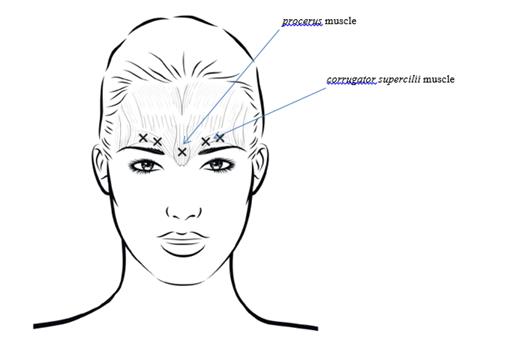
Lors de l’administration, agir avec prudence pour éviter toute injection intravasculaire. Avant l’injection, il est possible de placer fermement le pouce ou l’index sous le bord orbitaire pour éviter l’épanchement du médicament dans cette zone. Orienter l’aiguille vers le haut et médialement.
En cas d’échec du traitement un mois après une première séance, c’est-à-dire en l’absence d’amélioration significative par rapport à la situation initiale, les approches suivantes peuvent être envisagées :
· L’analyse des causes de l’échec, par exemple l’injection dans les mauvais muscles, une technique d’injection inappropriée, la formation d’anticorps neutralisant la toxine ou un dosage insuffisant.
· La réévaluation de la pertinence du traitement par toxine botulinique de type A.
En l’absence d’effets indésirables consécutifs à une séance de traitement, il est possible d’entamer une nouvelle séance de traitement après un délai d’au moins trois mois.
Procédure à suivre pour une élimination en toute sécurité des flacons, des seringues et des matériels utilisés
Pour une élimination en toute sécurité, la fraction de Letybo non reconstituée doit être reconstituée dans le flacon avec une petite quantité d’eau, puis autoclavée. Les flacons vides ou contenant un résidu de solution, les seringues et les déversements doivent être autoclavés. La fraction restante de Letybo peut sinon être inactivée à l’aide d’une solution diluée d’hydroxyde de sodium (0,1 N NaOH) ou d’hypochlorite de sodium (NaOCl à 0,5 % ou 1 %).
Après inactivation, les flacons, seringues et matériels usagés ne doivent pas être vidés, mais placés dans des récipients appropriés et éliminés conformément à la réglementation en vigueur.
Recommandations en cas d’accident lors de la manipulation de la toxine botulinique
· Tout déversement du produit doit être essuyé : soit à l’aide d’un matériau absorbant imprégné d’une solution d’hypochlorite de sodium dans le cas du produit en poudre, soit à l’aide d’un matériau absorbant sec dans le cas du produit reconstitué.
· Les surfaces contaminées doivent être nettoyées avec un matériau absorbant imprégné d’une solution d’hypochlorite de sodium, puis séchées.
· Si un flacon est cassé, recueillir soigneusement les morceaux de verre et essuyer le produit comme indiqué ci-dessus, en évitant toute coupure de la peau.
· En cas de contact du médicament avec la peau, laver la zone concernée avec une solution d’hypochlorite de sodium, puis rincer abondamment à l’eau.
· En cas de contact du médicament avec les yeux, rincer abondamment à l’eau ou avec une solution de rinçage oculaire.
· En cas de contact du médicament avec une plaie, une coupure ou de la peau éraflée, rincer abondamment à l’eau et prendre les mesures médicales appropriées en fonction de la dose concernée.