Dernière mise à jour le 01/06/2026
SALVACYL L.P. 11,25 mg, poudre et solvant pour suspension injectable à libération prolongée
Indications thérapeutiques
SALVACYL contient de la triptoréline, qui est semblable à une hormone appelée l’hormone entraînant la libération des gonadotrophines (GnRH). C’est une forme à libération prolongée conçue pour libérer lentement 11,25 mg de triptoréline sur une période de 12 semaines. SALVACYL diminue le taux de l’hormone sexuelle mâle (la testostérone). Il est utilisé pour diminuer les pulsions sexuelles chez l’homme souffrant d’un comportement sexuel déviant.
Le traitement par SALVACYL doit être instauré et contrôlé par un psychiatre. Le traitement doit être associé à une psychothérapie dans le but de diminuer le comportement sexuel déviant.
Présentations
> 1 flacon(s) en verre - 1 ampoule(s) en verre de 2 ml avec 1 seringue(s) polypropylène avec 2 aiguille(s)
Code CIP : 380 814-5 ou 34009 380 814 5 9
Déclaration de commercialisation : 18/11/2009
Cette présentation est agréée aux collectivités
- Prix hors honoraire de dispensation : 300,08 €
- Honoraire de dispensation : 1,02 €
- Prix honoraire compris : 301,10 €
- Taux de remboursement :100%
Service médical rendu (SMR)
Les libellés affichés ci-dessous ne sont que des résumés ou extraits issus des avis rendus par la Commission de la Transparence. Seul l'avis complet de la Commission de la Transparence fait référence.
Cet avis est consultable à partir du lien `Avis du jj/mm/aaaa` ou encore sur demande auprès de la HAS (plus d'informations dans l'aide). Les avis et synthèses d'avis contiennent un paragraphe sur la place du médicament dans la stratégie thérapeutique.
| Valeur du SMR | Avis | Motif de l'évaluation | Résumé de l'avis |
|---|---|---|---|
| Important | Avis du 15/10/2014 | Renouvellement d'inscription (CT) | Le service médical rendu par SALVACYL L.P. reste important dans l’indication de l’AMM. |
Amélioration du service médical rendu (ASMR)
Les libellés affichés ci-dessous ne sont que des résumés ou extraits issus des avis rendus par la Commission de la Transparence. Seul l'avis complet de la Commission de la Transparence fait référence.
Cet avis est consultable à partir du lien `Avis du jj/mm/aaaa` ou encore sur demande auprés de la HAS (plus d'informations dans l'aide). Les avis et synthéses d'avis contiennent un paragraphe sur la place du médicament dans la stratégie thérapeutique.
| Valeur de l'ASMR | Avis | Motif de l'évaluation | Résumé de l'avis |
|---|---|---|---|
| IV (Mineur) | Avis du 18/02/2009 | Inscription (CT) | SALVACYL L.P en injection intramusculaire apporte une amélioration mineure du service médical rendu (ASMR de niveau IV) dans la prise en charge des pulsions sexuelles, en association à une psychothérapie, chez l'homme adulte ayant des déviances sexuelles sévères. |
Autres informations
- Titulaire de l'autorisation : IPSEN PHARMA
- Conditions de prescription et de délivrance :
- liste I
- prescription réservée aux spécialistes et services PSYCHIATRIE
- Statut de l'autorisation : Valide
- Type de procédure : Procédure de reconnaissance mutuelle
- Code CIS : 6 984 799 4
ANSM - Mis à jour le : 18/06/2025
SALVACYL L.P. 11,25 mg, poudre et solvant pour suspension injectable à libération prolongée
2. COMPOSITION QUALITATIVE ET QUANTITATIVE
Triptoréline (sous forme d’embonate de triptoréline)............................................................. 11,25 mg
Pour un flacon de poudre.
Après reconstitution dans 2 ml de solvant, la suspension reconstituée contient 11,25 mg de triptoréline sous forme d’embonate de triptoréline.
Excipient à effet notoire : ce médicament contient du sodium mais moins de 1 mmol (23 mg) par flacon.
Pour la liste complète des excipients, voir rubrique 6.1.
Poudre et solvant pour suspension injectable à libération prolongée.
· Poudre : poudre blanche à jaunâtre.
· Solvant : solution limpide.
4.1. Indications thérapeutiques
Le traitement par SALVACYL doit être instauré et contrôlé par un psychiatre. Le traitement doit être associé à une psychothérapie dans le but de diminuer le comportement sexuel déviant.
4.2. Posologie et mode d'administration
La dose recommandée de SALVACYL est de 11,25 mg de triptoréline (1 flacon) administrée toutes les douze semaines par injection intramusculaire unique.
Population pédiatrique
La sécurité et l’efficacité de SALVACYL chez le nouveau-né, le nourrisson, l’enfant et l’adolescent n’ont pas encore été établies, c’est pourquoi SALVACYL n’est pas indiqué dans ces populations.
Insuffisants rénaux ou hépatiques
Aucune adaptation posologique n’est nécessaire chez les patients atteints d’insuffisance rénale ou hépatique.
Mode d’administration
Pour les instructions concernant la reconstitution du médicament avant administration, voir la rubrique 6.6
Précautions à prendre avant la manipulation ou l’administration du médicament
SALVACYL est une suspension de microgranules, son injection accidentelle par voie intravasculaire doit être strictement évitée.
SALVACYL doit être administré sous contrôle médical (personnel soignant ou médecin).
Le bénéfice thérapeutique sera suivi régulièrement, par exemple avant une nouvelle injection.
Il est nécessaire de changer périodiquement de site d’injection.
· Ostéoporose grave
· Hypersensibilité à la substance active, à la GnRH, à ses analogues ou à l’un des excipients mentionnés à la rubrique 6.1 (voir aussi la rubrique 4.8).
4.4. Mises en garde spéciales et précautions d'emploi
Au début du traitement, la triptoréline peut entraîner une augmentation transitoire du taux de testostérone sérique. Ainsi, lors de la phase initiale du traitement, le patient sera suivi attentivement par le médecin traitant et l’administration additionnelle d’un anti-androgène adapté sera envisagée si nécessaire, ceci afin de neutraliser l’augmentation initiale du taux sérique de testostérone et ainsi la possible augmentation des pulsions sexuelles.
A l’arrêt du traitement, il existe un risque d’augmentation de la sensibilité à la testostérone, ce qui peut conduire à une forte augmentation des pulsions sexuelles. Dès lors, l’administration additionnelle d’un anti-androgène adapté avant l’arrêt de SALVACYL doit être envisagée.
Une fois le taux de castration de la testostérone atteint à la fin du premier mois, ce taux est maintenu aussi longtemps que les patients reçoivent leur injection toutes les douze semaines.
L’évaluation de l’efficacité du traitement est essentiellement clinique. Une évaluation clinique de l’efficacité sera faite régulièrement, par exemple avant chaque injection trimestrielle. En cas de doute sur l’efficacité du traitement, cette dernière pouvant être liée à l’observance du traitement ou à un problème technique lors de l’injection, les taux sériques de testostérone peuvent être mesurés.
La prudence est requise chez les patients traités par des anticoagulants, en raison du risque potentiel d’hématome au site d’injection.
L’administration de triptoréline aux doses thérapeutiques conduit à une suppression du système hypophyso-gonadique. Un retour à la normale est généralement obtenu après l’interruption du traitement. Des tests diagnostiques de la fonction hypophyso-gonadique effectués pendant le traitement et après l’interruption de la thérapie avec un agoniste de la LH-RH peuvent donc être erronés.
L’inhibition prolongée de la sécrétion androgénique qu’elle soit induite par orchidectomie bilatérale ou par administration d’analogue de la GnRH est associée à un risque élevé de perte osseuse et peut conduire à une ostéoporose et à un risque accru de fracture osseuse.
Des données préliminaires suggèrent que l’utilisation d’un bisphosphonate en association avec un analogue de la GnRH pourrait réduire le risque de déminéralisation osseuse. Des précautions particulières sont nécessaires pour la prise en charge des patients présentant des facteurs de risque d’ostéoporose (alcoolisme chronique, tabagisme, traitement au long cours par des médicaments responsables d’une diminution de la densité minérale osseuse, tels que antiépileptiques ou corticoïdes, antécédent familial d’ostéoporose, malnutrition).
La densité minérale osseuse peut être mesurée avant le début du traitement et suivie régulièrement pendant le traitement.
Afin de prévenir la perte osseuse liée au traitement, un changement du mode de vie, comprenant l’arrêt du tabac, une consommation modérée d’alcool et des exercices physiques réguliers entraînant une charge sur le squelette, est recommandé. Un régime alimentaire adéquat en calcium et en vitamine D est également recommandé.
Dans de rares cas, le traitement par des analogues de la LH-RH peut révéler la présence jusque-là inconnue d’un adénome hypophysaire gonadotrope. Les patients concernés peuvent présenter une apoplexie pituitaire qui se caractérise par des céphalées d’apparition brutale, des vomissements, une altération de la vision et une ophtalmoplégie.
Une augmentation des lymphocytes a été rapportée chez des patients traités par des analogues de la LH-RH. Cette lymphocytose secondaire est apparemment liée à la castration induite par la LH-RH et semble indiquer que les hormones gonadiques sont impliquées dans l’involution thymique.
Il y a un risque accru de survenue d’une dépression (potentiellement sévère) chez les patients traités par les agonistes de la GnRH, comme la triptoréline. Les patients doivent être informés en conséquence et traités de manière appropriée si des symptômes apparaissent.
Les patients présentant une dépression connue doivent être surveillés de près pendant le traitement.
Le traitement par suppression androgénique peut provoquer un allongement de l’intervalle QT.
Chez les patients avec des antécédents ou présentant des facteurs de risque d’allongement de l’intervalle QT et chez les patients traités par un médicament susceptible de prolonger l’intervalle QT (voir rubrique 4.5), le rapport bénéfice /risque incluant le risque de torsades de pointe devra être évalué par le médecin avant l’initiation d’un traitement par SALVACYL.
De plus, des données épidémiologiques ont mis en évidence que les patients pouvaient présenter des changements métaboliques (ex : intolérance au glucose, une stéatose hépatique), ou un risque plus élevé de maladies cardiovasculaires pendant le traitement inhibant la sécrétion androgénique. Cependant, les données prospectives ne confirment pas le lien entre le traitement par analogue de la GnRH et l’augmentation de la mortalité cardiovasculaire. Les patients présentant un risque élevé de maladies métaboliques ou cardiovasculaires doivent faire l’objet d’une évaluation attentive avant d’instaurer le traitement et doivent faire l’objet d’un suivi adapté pendant le traitement inhibant la sécrétion androgénique.
Du fait de la suppression androgénique, le traitement par les analogues de la GnRH peut augmenter le risque d’anémie. Ce risque doit être évalué chez les patients traités et pris en charge de façon appropriée.
Ce médicament contient moins de 1 mmol (23 mg) de sodium par dose, c.-à-d. qu’il est essentiellement « sans sodium ».
4.5. Interactions avec d'autres médicaments et autres formes d'interactions
Lorsque la triptoreline est administrée avec des médicaments agissant sur la sécrétion pituitaire des gonadotrophines le patient doit faire l’objet d’une surveillance particulière, notamment de son statut hormonal.
L’inhibition de la sécrétion androgénique pouvant provoquer un allongement de l’intervalle QT, l’utilisation concomitante de triptoréline et de médicaments connus pour allonger l’intervalle QT ou de médicaments susceptibles de provoquer des torsades de pointe tels les médicaments antiarythmiques de classe IA (par exemple : quinidine, disopyramide) ou de classe III (par exemple : amiodarone, sotalol, dofetilide, ibutilide), la méthadone, la moxifloxacine, les antipsychotiques, etc doit être évaluée avec attention (voir rubrique 4.4).
4.6. Fertilité, grossesse et allaitement
SALVACYL n’est pas indiqué chez la femme.
Des études chez l’animal ont mis en évidence des effets sur les paramètres de l’appareil reproducteur (voir rubrique 5.3).
4.7. Effets sur l'aptitude à conduire des véhicules et à utiliser des machines
Cependant, l’aptitude à conduire des véhicules et à utiliser des machines peut être altérée en cas de survenue de sensations vertigineuses, d’une somnolence et de troubles de la vision qui sont des effets indésirables possibles du traitement ou qui peuvent résulter de la pathologie traitée.
Comme il a été observé avec d’autres agonistes de la LH-RH ou après castration chirurgicale, les effets indésirables les plus fréquemment observés lors de traitement avec la triptoréline étaient dus à ses effets pharmacologiques attendus. Ces effets incluaient des bouffées de chaleur, une dysfonction érectile (observée chez plus de 10% des patients). A l’exception de réactions d’hypersensibilité (rare) et de douleur au site d’injection (<5%), tous les effets indésirables sont connus pour être liés aux changements de la testostéronémie. L’utilisation au long terme des analogues de synthèse de la LH-RH peut être associée à une augmentation de la perte osseuse et conduire à une ostéoporose et augmenter le risque de fracture osseuse.
Les effets indésirables suivants ont été rapportés et sont considérés comme étant au moins possiblement reliés au traitement lors des études cliniques réalisées chez des hommes atteints de cancer avancé de la prostate ou chez des hommes volontaires sains. La plupart de ces effets sont connus comme étant liés à une castration biochimique ou chirurgicale. La fréquence de ces effets indésirables peut être classée comme suit : très fréquent (≥ 1/10) ; fréquent (≥ 1/100 à < 1/10) ; peu fréquent (≥ 1/1000 à < 1/100), rare (≥ 1/10000 à < 1/1000), fréquence indéterminée (ne peut être estimée sur la base des données disponibles).
|
Classes des systèmes d’organe |
Fréquent |
Peu fréquent |
Rare |
Après commercialisation Fréquence indéterminée |
|
|
Infections et infestations |
|
|
|
Rhinopharyngite |
|
|
Affections hématologiques et du système lymphatique |
|
|
Thrombocytose |
|
Anémie |
|
Affections du système immunitaire |
|
Hypersensibilité |
|
Réaction anaphylactique |
Choc anaphylactique |
|
Troubles du métabolisme et de la nutrition |
|
|
Anorexie Diabète Goutte Hyperlipidémie Augmentation de l’appétit |
|
|
|
Affections psychiatriques |
Diminution de la libido |
Perte de la libido Dépression * Changements d’humeur * |
Insomnie
Irritabilité |
Etat confusionnel Baisse de l’activité Humeur euphorique |
Anxiété |
|
Système nerveux |
Paresthésie des membres inférieurs |
Sensation vertigineuse Céphalées |
Paresthésies |
Troubles mnésiques |
|
|
Affections oculaires |
|
|
Défauts visuels |
Sensation anormale de l’oeil Perturbation visuelle |
|
|
Affections de l'oreille et du labyrinthe |
|
|
Acouphènes Vertige |
|
|
|
Affections endocriniennes |
|
|
|
|
Apoplexie hypophysaire** |
|
Affections cardiaques |
|
|
Palpitations |
|
Allongement de l’intervalle QT*(voir rubriques 4.4 et 4.5) |
|
Affections vasculaires |
Bouffées de chaleur |
Hypertension |
|
Hypotension |
|
|
Affections respiratoires, thoraciques et médiastinales |
|
|
Dyspnée Epistaxis |
Orthopnée |
|
|
Affections gastro-intestinales |
|
Bouche sèche Nausées |
Douleur abdominale |
Distension abdominale Dysgueusie Flatulence |
|
|
Constipation |
|||||
|
Diarrhée |
|||||
|
Vomissement |
|||||
|
Affections de la peau et du tissu sous-cutané |
Hyperhidrose |
|
Acné Alopécie Erythème Prurit Rash Urticaire |
Eruption bullaire Purpura |
Œdème angioneurotique |
|
Affections musculo-squelettiques et systémiques |
Douleur dorsale |
Douleur musculo-squelettique Extrémités douloureuses |
Arthralgie Douleur osseuse Crampes musculaires Faiblesse musculaire Myalgie |
Raideur articulaire Gonflement articulaire Raideur musculo-squelettique Ostéoarthrite |
|
|
Affections du rein et des voies urinaires |
|
|
Nycturie Rétention urinaire |
|
Incontinence urinaire |
|
Affections des organes de reproduction et du sein |
Dysérection (incluant anéjaculation, trouble de l’éjaculation) |
Douleur pelvienne |
Gynécomastie Atrophie testiculaire Douleur testiculaire Douleur mammaire |
|
|
|
Troubles généraux et anomalies au site d'administration |
Asthénie |
Réaction au site d’injection (incluant érythème, inflammation et douleur) Œdème |
Œdèmes périphériques Douleur Frissons Somnolence Léthargie |
Douleur thoracique Dystasie Syndrome pseudo-grippal Fièvre |
Malaise |
|
Investigations |
|
Poids augmenté |
Alanine aminotransférase augmentée Aspartate aminotransférase augmentée Créatininémie augmentée Urémie augmentée Pression artérielle augmentée Gamma-glutamyl transférase augmentée Poids abaissé |
Phosphatase alcaline augmentée |
|
* La fréquence de ces effets de classe est basée sur la fréquence commune aux agonistes de la GnRH.
** Rapporté après l’administration initiale chez des patients avec un adénome hypophysaire.
Déclaration des effets indésirables suspectés
La déclaration des effets indésirables suspectés après autorisation du médicament est importante. Elle permet une surveillance continue du rapport bénéfice/risque du médicament. Les professionnels de santé déclarent tout effet indésirable suspecté via le système national de déclaration : Agence nationale de sécurité du médicament et des produits de santé (ANSM) et réseau des Centres Régionaux de Pharmacovigilance - Site internet : https://signalement.social-sante.gouv.fr
5. PROPRIETES PHARMACOLOGIQUES
5.1. Propriétés pharmacodynamiques
Mécanisme d’action et effets pharmacodynamiques
La triptoréline, un agoniste de la LH-RH, agit comme inhibiteur puissant de la sécrétion des gonadotrophines lorsqu’elle est donnée de façon continue et à doses thérapeutiques. Chez l’homme, des études ont montré qu’après administration de triptoréline, survient dans un premier temps une augmentation transitoire des taux circulant de l’hormone lutéinique (LH), de l’hormone de stimulation folliculaire (FSH) et de la testostérone.
Cependant, une administration chronique et continue de triptoréline provoque une diminution de la sécrétion de la LH et de la FSH ainsi que la suppression de la synthèse des stéroïdes testiculaires. Une diminution des taux sériques de testostérone à des taux observés chez les hommes castrés chirurgicalement survient approximativement entre 2 et 4 semaines après le début du traitement. Ceci entraîne une atrophie des organes sexuels accessoires. Ces effets sont généralement réversibles après arrêt du médicament.
La testostérone joue un rôle majeur dans la régulation de la sexualité, des agressions, de la cognition, des émotions et de la personnalité. En particulier, c’est un déterminant majeur du désir, des fantasmes et du comportement sexuel et de façon fondamentale, du contrôle de la fréquence, de la durée et de l’ampleur des érections spontanées. Les effets de la testostérone (et de son métabolite, le 5α-dihydrotestostérone [DHT]) agissent par leur action sur les récepteurs androgéniques intracellulaires.
Efficacité et sécurité clinique
L’administration par voie intramusculaire d’un total de 3 doses (9 mois) de SALVACYL chez des patients ayant un cancer avancé de la prostate permet d’atteindre, après quatre semaines, des taux de testostérone à un niveau de castration chez 97,6% d’entre eux, taux maintenus du deuxième au neuvième mois de traitement chez 94,1% des patients.
5.2. Propriétés pharmacocinétiques
Après une injection unique par voie intramusculaire de SALVACYL, le tmax était de 2 (2-6) heures et le Cmax (0-85 jours) de 37,1 (22,4-57,4) ng/ml. La triptoréline ne s’est pas accumulée après 9 mois de traitement.
Distribution
Les résultats d’études pharmacocinétiques conduites chez l’homme sain indiquent qu’après une administration par voie intraveineuse en bolus, la triptoréline est distribuée et éliminée selon un modèle à trois compartiments et les demi-vies correspondantes sont d’environ 6 minutes, 45 minutes et 3 heures.
Le volume de distribution à l’état d’équilibre de la triptoréline après administration intraveineuse de 0,5 mg de triptoréline est approximativement de 30 l chez les volontaires sains masculins.
Biotransformation
Le métabolisme de la triptoréline n’a pas été déterminé chez l’homme.
Elimination
La triptoréline est éliminée par le foie et les reins. Après administration intraveineuse de 0,5 mg de triptoréline chez le volontaire sain, 42% de la dose était éliminée dans les urines sous forme de triptoréline inchangée. Chez ces volontaires sains, la demi-vie terminale de la triptoréline est de 2,8 heures et la clairance totale de 212 ml/min.
Populations particulières
La clairance à la triptoréline diminue chez l’insuffisant rénal ou hépatique. Après administration intraveineuse de 0,5 mg de triptoréline chez les sujets avec une insuffisance rénale modérée (Clcreat 40 ml/min), la triptoréline avait une clairance de 120 ml/min ; 88.6 ml/min chez les sujets avec une insuffisance rénale sévère (Clcreat 8,9 ml/min) et 57.8 ml/min chez les patients avec une insuffisance hépatique faible à modérée (Clcreat 89,9 ml/min).
Au vu de l’importante marge thérapeutique de SALVACYL aucun ajustement de la dose n’est recommandé chez les patients souffrant d’insuffisance rénale ou hépatique.
Les effets de l’âge et de l’ethnie sur la pharmacocinétique de la triptoréline n’ont pas été étudiés.
Relations pharmacocinétiques/pharmacodynamiques
La relation pharmacocinétique/pharmacodynamique de la triptoréline n’est pas simple à déterminer car elle est non linéaire et dépendante de la durée d’administration. Toutefois, après administration aigüe chez des sujets non traités, la triptoréline induit une augmentation dose dépendante de la réponse en termes de LH et de FSH. Lors de l’administration d’une formulation à libération prolongée, la triptoréline stimule la sécrétion de LH et FSH durant les premiers jours et par conséquent la sécrétion de testostérone. Les différentes études de bioéquivalence montrent que l’augmentation maximale de la testostérone est atteinte après environ 4 jours avec un Cmax équivalent, qui est indépendant de la vitesse de libération de la triptoréline. La réponse initiale n’est pas maintenue malgré l’exposition continue à la triptoréline et est suivie par une diminution progressive des taux de testostérone. Dans ce cas également, le degré d’exposition à la triptoréline peut varier largement sans affecter l’effet global sur les taux de testostérone sériques.
5.3. Données de sécurité préclinique
La toxicité de la triptoréline sur les organes extragénitaux est faible.
Les effets observés sont principalement liés à une exacerbation des effets pharmacologiques de la triptoréline.
Lors d’études de toxicologie chronique, la triptoréline induit à doses cliniques des changements macro et microscopiques sur les organes de la reproduction chez les rats, les chiens et les singes mâles. Ceux-ci sont considérés comme reflétant la suppression de la fonction gonadique due à l’activité pharmacologique du principe actif. Ces changements sont partiellement réversibles pendant la période de récupération. Après administration chez le rat par voie sous cutanée de 10 microgrammes/kg du jour 6 à 15 de la gestation, la triptoréline n’a pas démontré d’embryotoxicité, de tératogénicité, ou d’effet sur le développement de la progéniture (génération F1) ou sur leur performance reproductrice. Une diminution de l’augmentation du poids maternel et une augmentation des résorptions ont été observées à 100 microgrammes/kg.
La triptoréline n’est pas mutagène in vitro ou in vivo. Aucun effet oncogène n’a été observé chez la souris à des doses de triptoréline allant jusqu’à 6000 microgrammes/kg, après 18 mois de traitement. Une étude de carcinogénicité conduite chez le rat pendant 23 mois, a montré une incidence de près de 100% des tumeurs bénignes pituitaires à chaque dose, conduisant à une mort prématurée. L’augmentation de l’incidence des tumeurs pituitaires chez le rat est un effet fréquemment associé au traitement par les agonistes de la LH-RH. La pertinence clinique de cette observation n’est pas connue.
Poudre : polymère-d, l-lactide-co-glycolide, mannitol, carmellose sodique, polysorbate 80.
Solvant : eau pour préparations injectables.
Après reconstitution : la stabilité physico-chimique de la suspension reconstituée a été démontrée pendant 24 heures à 25°C. D’un point de vue microbiologique, le produit doit être utilisé immédiatement. En cas d’utilisation non immédiate, les durées et conditions de conservation après reconstitution et avant utilisation relèvent de la seule responsabilité de l’utilisateur et ne devraient pas dépasser 24 heures à une température comprise entre 2-8°C.
6.4. Précautions particulières de conservation
A conserver à une température ne dépassant pas 25°C.
Pour les conditions de conservation du médicament après reconstitution, voir la rubrique 6.3.
6.5. Nature et contenu de l'emballage extérieur
Flacon de poudre : flacon (verre type I) de 6 ml transparent légèrement brun à septum avec un bouchon en bromobutyle et une capsule aluminium recouverte d’un flip-off jaune-vert.
Ampoule de solvant : ampoule (verre type I) transparente et incolore contenant 2 ml de solvant stérile pour la reconstitution de la suspension.
Boîte de 1 flacon de poudre et 1 ampoule de solvant conditionnés avec un blister contenant une une seringue vide en polypropylène et deux aiguilles (une avec un système de sécurité pour l’injection et une sans système de sécurité pour la reconstitution).
6.6. Précautions particulières d’élimination et de manipulation
La suspension pour injection doit être reconstituée en conditions aseptiques et en utilisant exclusivement l’ampoule de solvant pour injection.
Il faut suivre strictement les instructions pour la reconstitution mentionnées ci-après et dans la notice.
La totalité du solvant doit être aspirée dans la seringue fournie en utilisant l’aiguille pour la reconstitution (20G, sans système de sécurité) et transférée dans le flacon contenant la poudre. La suspension doit être reconstituée en agitant le flacon doucement d’un mouvement circulaire assez longtemps pour obtenir une suspension laiteuse et homogène. Ne pas retourner le flacon.
Il est important de vérifier qu’il n’y a pas d’agglomérats dans le flacon. La suspension obtenue doit être aspirée dans la seringue sans retourner le flacon. L’aiguille pour la reconstitution doit être remplacée par l’aiguille pour injection (20G avec système de sécurité) utilisée pour l’administration du produit.
Le produit est sous la forme d’une suspension, il doit être injecté immédiatement après la reconstitution pour éviter sa précipitation.
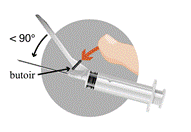
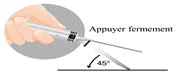
Pour administration unique seulement. Après utilisation, le système de sécurité doit être activé d’une seule main. Soit en poussant le butoir avec le doigt, ou en poussant la gaine de protection sur une surface plane. Dans les deux cas, appuyer avec un mouvement ferme rapide jusqu’à l’audition d’un clic. Il doit être vérifié visuellement que l’aiguille est complètement prise dans le manchon de protection.
Les aiguilles utilisées, toute suspension non utilisée ou déchet doivent être éliminés conformément à la réglementation en vigueur.
7. TITULAIRE DE L’AUTORISATION DE MISE SUR LE MARCHE
70, RUE BALARD
75015 PARIS
8. NUMERO(S) D’AUTORISATION DE MISE SUR LE MARCHE
9. DATE DE PREMIERE AUTORISATION/DE RENOUVELLEMENT DE L’AUTORISATION
[à compléter ultérieurement par le titulaire]
10. DATE DE MISE A JOUR DU TEXTE
[à compléter ultérieurement par le titulaire]
Sans objet.
12. INSTRUCTIONS POUR LA PREPARATION DES RADIOPHARMACEUTIQUES
Liste I
Médicament à prescription réservée aux spécialistes en psychiatrie
ANSM - Mis à jour le : 18/06/2025
SALVACYL L.P. 11,25 mg, poudre et solvant pour suspension injectable à libération prolongée.
Triptoréline
Veuillez lire attentivement cette notice avant d’utiliser ce médicament car elle contient des informations importantes pour vous.
· Gardez cette notice. Vous pourriez avoir besoin de la relire.
· Si vous avez d’autres questions, interrogez votre médecin ou votre pharmacien.
· Ce médicament vous a été personnellement prescrit. Ne le donnez pas à d’autres personnes. Il pourrait leur être nocif, même si les signes de leur maladie sont identiques aux vôtres.
· Si vous ressentez un quelconque effet indésirable, parlez-en à votre médecin ou votre pharmacien. Ceci s’applique aussi à tout effet indésirable qui ne serait pas mentionné dans cette notice. Voir rubrique 4.
1. Qu'est-ce que SALVACYL L.P. 11,25 mg, poudre et solvant pour suspension injectable à libération prolongée et dans quel cas est-il utilisé ?
2. Quelles sont les informations à connaître avant d'utiliser SALVACYL L.P. 11,25 mg, poudre et solvant pour suspension injectable à libération prolongée ?
3. Comment utiliser SALVACYL L.P. 11,25 mg, poudre et solvant pour suspension injectable à libération prolongée ?
4. Quels sont les effets indésirables éventuels ?
5. Comment conserver SALVACYL L.P. 11,25 mg, poudre et solvant pour suspension injectable à libération prolongée ?
6. Contenu de l’emballage et autres informations.
1. QU’EST-CE QUE SALVACYL L.P. 11,25 mg, poudre et solvant pour suspension injectable à libération prolongée ET DANS QUELS CAS EST-IL UTILISE ?
SALVACYL contient de la triptoréline, qui est semblable à une hormone appelée l’hormone entraînant la libération des gonadotrophines (GnRH). C’est une forme à libération prolongée conçue pour libérer lentement 11,25 mg de triptoréline sur une période de 12 semaines. SALVACYL diminue le taux de l’hormone sexuelle mâle (la testostérone). Il est utilisé pour diminuer les pulsions sexuelles chez l’homme souffrant d’un comportement sexuel déviant.
Le traitement par SALVACYL doit être instauré et contrôlé par un psychiatre. Le traitement doit être associé à une psychothérapie dans le but de diminuer le comportement sexuel déviant.
2. QUELLES SONT LES INFORMATIONS A CONNAITRE AVANT D’UTILISER SALVACYL L.P. 11,25 mg, poudre et solvant pour suspension injectable à libération prolongée ?
· Si vous êtes allergique (hypersensible) à la triptoréline, à l’hormone entraînant la libération des gonadotrophines (GnRH), aux autres analogues de la GnRH ou à l’un des autres composants contenus dans ce médicament, mentionnés dans la rubrique 6.
· Si vous souffrez d’ostéoporose sévère (maladie qui affecte la résistance des os).
Avertissements et précautions
Lors de l’initiation du traitement, une augmentation du taux de testostérone va se produire dans votre corps. Ceci peut entrainer une augmentation de vos pulsions sexuelles. Afin de neutraliser cet effet, votre médecin peut vous donner un médicament (anti-androgène).
Adressez-vous à votre médecin avant d’utiliser SALVACYL L.P. 11,25 mg :
· Si votre humeur devient dépressive. Des dépressions parfois sévères ont été rapportées chez des patients traités par SALVACYL L.P. 11,25 mg. Votre médecin peut vouloir surveiller votre dépression pendant le traitement.
· Si vous prenez des anticoagulants (médicaments qui inhibent la coagulation du sang), à cause du risque d’hématome au site d’injection.
· Si vous êtes un grand consommateur d’alcool, si vous êtes fumeur, si vous souffrez d’ostéoporose (maladie qui affecte la résistance des os) ou s’il y a des cas d’ostéoporose dans votre famille, si vous avez une mauvaise alimentation ou si vous prenez des antiépileptiques (médicaments utilisés pour traiter diverses formes d'épilepsies) ou des corticoïdes. L’utilisation à long terme de SALVACYL, engendre une augmentation du risque de diminution de la résistance des os, plus particulièrement si vous vous trouvez dans une des situations décrites ci-dessus. Afin de prévenir la fragilisation des os, une hygiène de vie, comprenant l’arrêt du tabac, une consommation modérée d’alcool et des exercices physiques réguliers qui entraînent une charge sur le squelette et en augmentent la résistance (p. ex. la marche, le jogging, d’autres formes de sports induisant une charge sur le squelette) est recommandée. Un régime alimentaire adéquat en calcium et en vitamine D est également recommandé.
· Si vous devez passer des tests diagnostiques de la fonction gonadotrope hypophysaire ou des organes sexuels durant le traitement ou après interruption du traitement par SALVACYL, les résultats peuvent être faussés.
· Si vous ressentez des maux de tête d’apparition soudaine, des vomissements, des problèmes de vue ou une paralysie de vos yeux. Ces effets peuvent être les symptômes d’une tumeur bénigne de la glande hypophysaire qui se révèle sous l’effet de la mise sous traitement par SALVACYL.
· Si vous êtes diabétique, ou si vous souffrez d’une maladie cardiaque ou vasculaire.
· Si vous avez des troubles cardiovasculaires, y compris des troubles du rythme cardiaque (arythmie), ou si vous êtes traité par des médicaments pour soigner ce type de problème. Le risque de troubles du rythme cardiaque peut être augmenté par l’utilisation de SALVACYL.
· Les médicaments diminuant la testostérone peuvent provoquer des modifications de l'ECG associées à des anomalies du rythme cardiaque (allongement de l'intervalle QT).
· Le traitement avec des analogues de la GnRH, dont SALVACYL L.P. 11,25 mg, pourrait augmenter le risque d'anémie (définie comme une diminution du nombre de globules rouges).
A l’arrêt du traitement, le taux de testostérone redevient normal et vos pulsions sexuelles peuvent être augmentées de nouveau. Pour cette raison, votre médecin peut vous donner un autre médicament afin de neutraliser cet effet.
Enfants et adolescents
SALVACYL L.P. 11,25 mg n’est pas indiqué chez les nouveau-nés, les enfants et les adolescents.
Autres médicaments et SALVACYL L.P. 11,25 mg, poudre et solvant pour suspension injectable à libération prolongée
Informez votre médecin ou pharmacien si vous prenez, avez récemment pris ou pourriez prendre tout autre médicament.
Lorsque la triptoréline est administrée avec des médicaments agissant sur la sécrétion pituitaire d’hormones, votre médecin peut être amené à réaliser des contrôles supplémentaires.
SALVACYL peut interagir avec des médicaments utilisés pour traiter les problèmes de rythme cardiaque (par exemple quinidine, procaïnamide, amiodarone et sotalol) ou peut augmenter le risque de troubles du rythme cardiaque quand il est utilisé avec d’autres médicaments (par exemple la méthadone (utilisée pour soulager la douleur et en traitement de substitution des pharmacodépendances), la moxifloxacine (un antibiotique), des antipsychotiques (utilisés pour des troubles mentaux graves)).
SALVACYL L.P. 11,25 mg, poudre et solvant pour suspension injectable à libération prolongée avec des aliments et boissons
Sans objet.
Grossesse, allaitement et fertilité
SALVACYL n’est pas indiqué chez la femme.
Conduite de véhicules et utilisation de machines
Vous pouvez ressentir des sensations vertigineuses, de la fatigue et des problèmes de vue telle qu’une vision trouble. Il peut s’agir d’effets secondaires liés au traitement. Si vous ressentez un de ces effets secondaires, vous ne devez pas conduire de véhicule ou utiliser des machines.
SALVACYL L.P. 11,25 mg, poudre et solvant pour suspension injectable à libération prolongée contient du sodium mais moins de 1 mmol (23 mg) de sodium par flacon, c.-à-d. qu’il est essentiellement « sans sodium » et peut être administré si vous suivez un régime alimentaire pauvre en sodium.
3. COMMENT UTILISER SALVACYL L.P. 11,25 mg, poudre et solvant pour suspension injectable à libération prolongée ?
Si vous avez l’impression que les effets de SALVACYL sont trop faibles ou trop forts, veuillez en informer votre médecin.
Si vous avez pris plus de SALVACYL L.P. 11,25 mg, poudre et solvant pour suspension injectable à libération prolongée que vous n’auriez dû :
Consultez immédiatement votre médecin ou votre pharmacien.
Si vous oubliez de prendre SALVACYL L.P. 11,25 mg, poudre et solvant pour suspension injectable à libération prolongée :
Sans objet.
Si vous arrêtez de prendre SALVACYL L.P. 11,25 mg, poudre et solvant pour suspension injectable à libération prolongée :
Sans objet.
Si vous avez d’autres questions sur l’utilisation de ce médicament, demandez plus d’informations à votre médecin ou à votre pharmacien.
4. QUELS SONT LES EFFETS INDESIRABLES EVENTUELS ?
Beaucoup de ces effets indésirables sont attendus car liés à la modification des taux de testostérone dans votre corps. Ils incluent les bouffées de chaleur et l’impuissance.
Dans de rares cas (peut affecter jusqu’à 1 patient sur 1000), vous pouvez présenter une réaction allergique sévère et de manière fréquente (peut affecter jusqu’à 1 patient sur 10) vous pouvez développer une réaction allergique. Arrêtez de prendre SALVACYL et contactez votre médecin ou allez aux urgences les plus proches immédiatement, si vous présentez l’un des symptômes suivants : des problèmes pour avaler ou respirer, un gonflement des lèvres, de votre visage, de votre gorge ou de votre langue, une éruption cutanée.
D’autres effets indésirables peuvent survenir :
Très fréquent (peut affecter plus d’1 patient sur 10) :
· Bouffées de chaleur
· Sensation de faiblesse
· Transpiration excessive
· Douleur dans le dos
· Fourmillements dans les jambes
· Diminution de la libido
· Impuissance
Fréquent (peut affecter jusqu’à 1 patient sur 10) :
· Nausées, bouche sèche
· Douleur, contusion, rougeur et chaleur au site d’injection, douleurs osseuses et musculaires, douleurs dans les bras et les jambes, œdèmes (accumulation de liquides dans les tissus), douleur dans la partie inférieure de l’abdomen
· Réaction allergique
· Hypertension
· Augmentation du poids
· Sensation vertigineuse, maux de tête
· Diminution de la libido, dépression, changements d’humeur
Peu fréquent (peut affecter jusqu’à 1 patient sur 100) :
· Augmentation du nombre de plaquettes dans le sang
· Palpitations
· Sifflement d’oreilles, vertige (sensation de tournoiement)
· Vision trouble
· Douleur abdominale, constipation, diarrhée, vomissement
· Somnolence, frissonnements importants associés à de la sueur et de la fièvre, état de sommeil profond (léthargie), douleur
· Tests sanguins de laboratoire perturbés (notamment les tests de la fonction hépatique)
· Augmentation de la pression artérielle
· Perte de poids
· Diminution de l’appétit, augmentation de l’appétit, goutte (douleur sévère et gonflement des articulations généralement du gros orteil), diabète, excès de lipides dans le sang
· Douleur articulaire, crampe musculaire, faiblesse musculaire et douleur musculaire, tuméfaction et sensibilité au toucher, douleur osseuse
· Fourmillements ou engourdissements
· Insomnie, irritabilité
· Gonflement des seins, douleurs des seins, réduction de la taille des testicules, douleurs testiculaires
· Difficulté à respirer
· Acné, perte de cheveux, démangeaisons, éruption cutanée, rougeur de la peau, urticaire
· Réveil nocturne pour uriner, difficulté d’uriner
· Saignements de nez
Rare (peut affecter jusqu’à 1 patient sur 1000)
· Coloration rouge ou violette de la peau
· Sensation anormale dans les yeux ou perturbation visuelle
· Distension de l’abdomen, excès de gaz, anomalie du goût
· Douleur thoracique
· Difficulté à se tenir debout
· Pathologie pseudo-grippale, fièvre
· Inflammation du nez ou de la gorge (rhinopharyngite)
· Augmentation d’une enzyme présente notamment dans les os et dans le foie
· Augmentation de la température corporelle
· Raideur et gonflement des articulations, raideurs musculo-squelettiques, arthrose
· Atteinte de la mémoire
· État confusionnel, diminution de l’activité, humeur euphorique
· Difficulté à respirer en position couchée
· Ampoules cutanées
· Diminution de la pression artérielle
Fréquence indéterminée (la fréquence ne peut être estimée sur la base des données disponibles)
· Modifications de l’électrocardiogramme (allongement de l’intervalle QT)
· Sensation de malaise général
· Anxiété
· Incontinence urinaire
· En cas de présence d’une tumeur hypophysaire, risque accru de saignement au niveau de la tumeur (apoplexie hypophysaire).
· Anémie (diminution du nombre de globules rouges).
Une augmentation des globules blancs sanguins peut être observée, comme avec tout traitement à base d’analogue de la GnRH, lors d’un traitement par SALVACYL L.P. 11,25 mg.
Déclaration des effets secondaires
Si vous ressentez un quelconque effet indésirable, parlez-en à votre médecin ou votre pharmacien. Ceci s’applique aussi à tout effet indésirable qui ne serait pas mentionné dans cette notice. Vous pouvez également déclarer les effets indésirables directement via le système national de déclaration : Agence nationale de sécurité du médicament et des produits de santé (ANSM) et réseau des Centres Régionaux de Pharmacovigilance - Site internet : https://signalement.social-sante.gouv.fr.
En signalant les effets indésirables, vous contribuez à fournir davantage d’informations sur la sécurité du médicament.
5. COMMENT CONSERVER SALVACYL L.P. 11,25 mg, poudre et solvant pour suspension injectable à libération prolongée ?
Tenir ce médicament hors de la vue et de la portée des enfants.
N’utilisez pas ce médicament après la date de péremption indiquée sur l’emballage, l’ampoule et le flacon après EXP. La date d’expiration fait référence au dernier jour de ce mois.
A conserver à une température ne dépassant pas 25 °C.
La stabilité physico-chimique de la suspension reconstituée a été démontrée pendant 24 heures à 25°C. D’un point de vue microbiologique, le produit doit être utilisé immédiatement. En cas d’utilisation non immédiate, les durées et conditions de conservation après reconstitution et avant utilisation relèvent de la seule responsabilité de l’utilisateur et ne devraient pas dépasser 24 heures à une température comprise entre 2-8°C.
Ne jetez aucun médicament au tout-à-l’égout ou avec les ordures ménagères. Demandez à votre pharmacien d’éliminer les médicaments que vous n’utilisez plus. Ces mesures contribueront à protéger l’environnement.
6. CONTENU DE L’EMBALLAGE ET AUTRES INFORMATIONS
· La substance active est :
Triptoréline (sous forme d’embonate de triptoréline)....................................................... 11,25 mg
Pour 1 flacon de poudre.
La suspension reconstituée (2 ml) contient 11,25 mg de triptoréline sous forme d’embonate de triptoréline.
· Les autres composants sont :
Poudre : Polymère-d, l-lactide-co-glycolide, mannitol, carmellose sodique, polysorbate 80.
Solvant : eau pour préparations injectables.
Ce médicament se présente sous forme de poudre et solvant pour suspension injectable à libération prolongée. La poudre est blanche à jaunâtre et le solvant est une solution limpide. Une boîte contient un flacon de poudre, une ampoule contenant 2 ml de solvant, une seringue, et deux aiguilles.
Titulaire de l’autorisation de mise sur le marché
IPSEN PHARMA
70, RUE BALARD
75015 PARIS
Exploitant de l’autorisation de mise sur le marché
IPSEN PHARMA
70, RUE BALARD
75015 PARIS
IPSEN PHARMA BIOTECH
PARC D’ACTIVITES DU PLATEAU DE SIGNES
CHEMIN DEPARTEMENTAL N°402
83870 SIGNES
Noms du médicament dans les Etats membres de l'Espace Economique Européen
Ce médicament est autorisé dans les Etats membres de l’Espace Economique Européen sous les noms suivants : Conformément à la réglementation en vigueur.
[A compléter ultérieurement par le titulaire]
La dernière date à laquelle cette notice a été révisée est :
[à compléter ultérieurement par le titulaire]
Sans objet.
Des informations détaillées sur ce médicament sont disponibles sur le site Internet de l’ANSM (France).
%-------------------------------------------------------------------------------------------------------------------------------
Les informations suivantes sont destinées exclusivement aux professionnels de la santé (voir rubrique 3) :
|
1 – PREPARATION DU PATIENT AVANT LA RECONSTITUTION |
|
|
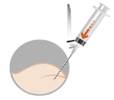
· Préparer le patient en désinfectant le muscle fessier au site d’injection. Cette étape doit être réalisée en premier car la suspension doit être injectée immédiatement après la reconstitution. |
|
|
2 – PREPARATION DE L’INJECTION |
|
|
Deux aiguilles sont fournies dans la boîte : · Aiguille 1 : aiguille de 20G (38 mm) sans système de sécurité à utiliser pour la reconstitution · Aiguille 2 : aiguille de 20G (38 mm) avec un système de sécurité à utiliser pour l’injection
Aiguille 1 – 38 mm Aiguille 2 – 38 mm
|
|
|
|
|
|
La présence de bulles au-dessus du lyophilisat est un aspect normal du produit. Les étapes suivantes doivent être effectuées les unes après les autres sans interruption. |
|
|
2 a · Sortir l'ampoule contenant le solvant. Si une partie de la solution est bloquée dans le haut de l’ampoule, la tapoter afin de la faire descendre dans le corps de l’ampoule. · Visser l’Aiguille 1 (sans système de sécurité) sur la seringue. Ne pas retirer le capuchon de l’aiguille à ce stade. · Casser le haut de l’ampoule en positionnant le point face à soi. · Retirer le capuchon de l’Aiguille 1. Insérer l’aiguille dans l’ampoule et prélever la totalité du solvant dans la seringue. · Mettre de côté la seringue contenant le solvant. |
|
|
2b · Sortir le flacon contenant la poudre. Tapoter pour ramener au fond du flacon la poudre éventuellement accumulée en haut du flacon. · Retirer le capuchon en plastique du flacon. · Reprendre la seringue contenant le solvant et enfoncer verticalement l’aiguille au travers du bouchon en élastomère du flacon. Injecter le solvant lentement, de façon à, si possible, rincer toute la partie supérieure du flacon. |
|
|
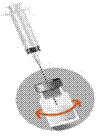
2c · Remonter l'Aiguille 1 au-dessus du niveau du liquide. Ne pas retirer l’aiguille du flacon. · Agiter le temps nécessaire (au moins 30 secondes) à l’obtention d’une suspension homogène et laiteuse. · Important : Vérifier l’absence d’agglomérats (en cas d’agglomérats poursuivre l’agitation jusqu’à complète homogénéisation). |
|
|
2d · Quand la suspension est homogène, descendre l’aiguille et, sans retourner le flacon, aspirer toute la suspension. Une petite quantité de suspension restera dans le flacon et devra être éliminée. Un excès de poudre et de solvant est prévu lors de la fabrication pour autoriser cette perte lors de la préparation de la seringue. · Enlever l’Aiguille 1 utilisée pour la reconstitution en la saisissant par l’embout coloré. Visser sur la seringue l’Aiguille 2 avec le système de sécurité. · Faire basculer le manchon de protection de l’aiguille vers le corps de la seringue. Le manchon de protection reste dans la position que vous lui donnez. · Enlever le capuchon de l’aiguille. · Amorcer l’aiguille en vidant l’air contenu dans la seringue et injecter immédiatement. |
|
|
3 – INJECTION INTRAMUSCULAIRE |
|
|
Pour éviter la sédimentation, injecter immédiatement dans la zone désinfectée le plus rapidement possible (dans la minute suivant la reconstitution). |
|
|
4 – APRES UTILISATION |
|
|
· Activer le système de sécurité d’une seule main o Remarque : garder constamment votre doigt derrière le butoir
Il existe deux méthodes pour activer le système de sécurité : § Méthode A : pousser le butoir avec votre doigt ou § Méthode B : pousser la gaine de protection sur une surface plane. o Dans les deux cas, appuyer avec un mouvement ferme rapide jusqu’à l’audition d’un clic. o Vérifier visuellement que l’aiguille est complètement prise dans le manchon de protection. · Les aiguilles utilisées, toute suspension non utilisée ou déchet doivent être éliminés conformément à la réglementation en vigueur.
|
Méthode A
Ou
Méthode B
|