Dernière mise à jour le 29/06/2026
LIDOCAINE AGUETTANT 20 mg/mL, solution injectable en seringue préremplie
Indications thérapeutiques
LIDOCAÏNE AGUETTANT contient une substance active, le chlorhydrate de lidocaïne
LIDOCAÏNE AGUETTANT est un anesthésique local. Il est utilisé pour anesthésier des parties du corps pendant les interventions chirurgicales. Il empêche temporairement les nerfs de transmettre les messages de douleur au cerveau dans la zone où il est injecté.
LIDOCAÏNE AGUETTANT 20 mg/mL, solution injectable en seringue préremplie peut être utilisé chez les adultes.
Présentations
> 10 seringues préremplies polypropylène de 10 mL, emballées individuellement sous blister
Code CIP : 34009 301 531 6 1
Déclaration de commercialisation : 14/06/2021
Cette présentation est agréée aux collectivités
> 1 seringue(s) préremplie(s) polypropylène de 5 ml
Code CIP : 34009 303 008 6 2
Déclaration de commercialisation : 13/02/2026
Cette présentation n'est pas agréée aux collectivités
> 10 seringue(s) préremplie(s) polypropylène de 5 ml
Code CIP : 34009 303 008 7 9
Déclaration de commercialisation : 29/09/2025
Cette présentation est agréée aux collectivités
Service médical rendu (SMR)
Les libellés affichés ci-dessous ne sont que des résumés ou extraits issus des avis rendus par la Commission de la Transparence. Seul l'avis complet de la Commission de la Transparence fait référence.
Cet avis est consultable à partir du lien `Avis du jj/mm/aaaa` ou encore sur demande auprès de la HAS (plus d'informations dans l'aide). Les avis et synthèses d'avis contiennent un paragraphe sur la place du médicament dans la stratégie thérapeutique.
| Valeur du SMR | Avis | Motif de l'évaluation | Résumé de l'avis |
|---|---|---|---|
| Important | Avis du 12/03/2025 | Extension d'indication | Le service médical rendu par LIDOCAINE AGUETTANT 10 et 20 mg/mL, solution injectable en seringue préremplie, uniquement lors d’une anesthésie locale en cas d’utilisation par voie intraveineuse pour prévenir la douleur liée à l’injection des médicaments chez l’enfant à partir de 12 ans (seulement pour le dosage à 10 mg/ml) et l’adulte. Le service médical rendu par LIDOCAINE AGUETTANT 10 et 20 mg/mL, solution injectable en seringue préremplie de 5 mL, est important lors d’une anesthésie locale en cas d’utilisation par voie intraveineuse pour prévenir la douleur liée à l’injection des médicaments chez l’enfant à partir de 12 ans (seulement pour le dosage à 10 mg/ml) et l’adulte et lors d'anesthésie par blocs nerveux périphériques. |
| Insuffisant | Avis du 12/03/2025 | Extension d'indication | Le service médical rendu par LIDOCAINE AGUETTANT 10 et 20 mg/mL, solution injectable en seringue préremplie, est insuffisant lors d’une anesthésie régionale intraveineuse (ALRIV) des membres supérieurs chez les adultes pour justifier d’une prise en charge par la solidarité nationale au regard de l’alternatives disponibles. Le service médical rendu par LIDOCAINE AGUETTANT 10 et 20 mg/mL, solution injectable en seringue préremplie de 5 mL, est insuffisant lors d’une anesthésie régionale intraveineuse (ALRIV) des membres supérieurs chez les adultes. |
| Important | Avis du 17/10/2018 | Inscription (CT) | le service médical rendu par LIDOCAINE AGUETTANT 10 mg/mL et 20 mg/mL, solution injectable est important dans l’indication de l’AMM. |
Amélioration du service médical rendu (ASMR)
Les libellés affichés ci-dessous ne sont que des résumés ou extraits issus des avis rendus par la Commission de la Transparence. Seul l'avis complet de la Commission de la Transparence fait référence.
Cet avis est consultable à partir du lien `Avis du jj/mm/aaaa` ou encore sur demande auprés de la HAS (plus d'informations dans l'aide). Les avis et synthéses d'avis contiennent un paragraphe sur la place du médicament dans la stratégie thérapeutique.
| Valeur de l'ASMR | Avis | Motif de l'évaluation | Résumé de l'avis |
|---|---|---|---|
| V (Inexistant) | Avis du 12/03/2025 | Extension d'indication | Anesthésie locale en cas d’utilisation par voie intraveineuse pour prévenir la douleur liée à l’injection des médicaments : Compte tenu : • des données limitées issues de méta-analyses ayant suggéré une efficacité modérée de la lidocaïne IV par rapport au placebo sur l’incidence des douleurs liées à l’injection au propofol, prise en compte pour LIDOCAINE AGUETTANT 10 mg/ml SANS CONSERVATEUR, solution injectable . • du besoin médical identifié de disposer de la lidocaïne pour prévenir des douleurs liées à l’injection de médicaments par voie intra-veineuse, notamment le propofol, en respectant les bonnes pratiques cliniques, la Commission considère que LIDOCAINE AGUETTANT 10 et 20 mg/mL, solution injectable en seringue préremplie, n’apporte pas d’amélioration du service médical rendu (ASMR V) par rapport à LIDOCAINE AGUETTANT 10 mg/ml SANS CONSERVATEUR, solution injectable. Complément de gamme : LIDOCAINE AGUETTANT 10 et 20 mg/mL, solution injectable en seringue préremplie de 5 mL sont des compléments qui n’apportent pas d’amélioration du service médical rendu (ASMR V) par rapport/aux présentations déjà inscrites. |
| V (Inexistant) | Avis du 17/10/2018 | Inscription (CT) | Ces spécialités sont des compléments de gamme qui n’apportent pas d’amélioration du service médical rendu (ASMR V) par rapport aux présentations déjà inscrites. |
ANSM - Mis à jour le : 30/07/2024
LIDOCAÏNE AGUETTANT 20 mg/mL, solution injectable en seringue préremplie
2. COMPOSITION QUALITATIVE ET QUANTITATIVE
Chaque millilitre de solution injectable contient 20 mg de chlorhydrate de lidocaïne (sous forme de chlorhydrate de lidocaïne monohydraté).
Chaque seringue préremplie de 5 mL contient 100 mg de chlorhydrate de lidocaïne (sous forme de chlorhydrate de lidocaïne monohydraté).
Chaque seringue préremplie de 10 mL contient 200 mg de chlorhydrate de lidocaïne (sous forme de chlorhydrate de lidocaïne monohydraté).
Excipient à effet notoire :
Chaque mL de solution injectable contient 2,0 mg de sodium, équivalant à 0,09 mmol.
Chaque seringue préremplie de 5 mL contient 10 mg de sodium, équivalent à 0,4 mmol.
Chaque seringue préremplie de 10 mL contient 20 mg de sodium, équivalant à 0,9 mmol.
Pour la liste complète des excipients, voir rubrique 6.1.
Solution injectable (injection).
Solution limpide et incolore.
pH : 5,0 à 6,5
Osmolalité : 270 à 330 mosm/kg
4.1. Indications thérapeutiques
LIDOCAÏNE AGUETTANT 20 mg/mL, solution injectable en seringue préremplie est indiqué pour :
· l’anesthésie locale et le bloc nerveux périphérique chez l’adulte,
· l’anesthésie régionale intraveineuse des membres supérieurs chez l’adulte.
4.2. Posologie et mode d'administration
La dose doit être ajustée en fonction de la réponse du patient, du site d'administration et de la durée prévue de l'intervention chirurgicale.
Il convient d’administrer la concentration et la dose les plus faibles produisant l'effet requis.
Adultes
· Anesthésie locale et anesthésie par bloc nerveux périphérique
Pour l’anesthésie par infiltration et les blocs nerveux périphériques, la dose totale habituelle de lidocaïne à envisager est de 3 à 5 mg/kg.
En général, la dose totale maximale recommandée de lidocaïne ne doit pas dépasser 200 mg, mais selon l'intervention et les facteurs du patient, des doses maximales plus élevées peuvent s'avérer nécessaires.
Le volume de la solution utilisée joue un rôle dans la taille de la zone de diffusion de l'anesthésie.
En cas d’utilisation par voie intraveineuse pour prévenir la douleur liée à l’injection des médicaments, la lidocaïne doit être administrée à une dose de 10 à 40 mg en bolus court avant la procédure douloureuse. Dans ce cas, la dose administrée représente un volume inférieur à une demi-seringue préremplie de 10 mL. Lose dose excédentaire doit être vidée de la seringue de 10 mL avant l'injection. Des seringues plus petites, plus appropriées pour l'administration de la dose recommandée, doivent être prises en considération.
· Anesthésie régionale intraveineuse des membres supérieurs
La dose totale habituelle de lidocaïne est de 3 mg/kg.
En fonction de l'intervention et des facteurs propres au patient, il convient d'utiliser 100 à 200 mg. La dose maximale ne doit pas dépasser 200 mg et 3 mg/kg.
La forme pharmaceutique (seringue préremplie) n'est pas considérée comme adaptée à l'anesthésie régionale intraveineuse des membres inférieurs.
Populations spéciales
Patients âgés
Pour les patients âgés, les doses sont calculées individuellement en fonction de l'âge et du poids corporel du patient. Les doses peuvent nécessiter une adaptation car le débit cardiaque et le débit sanguin hépatique diminuent avec l'âge, ce qui indique une diminution de la clairance de la lidocaïne (voir la rubrique 5.2).
Patients atteints d’une insuffisance rénale
Les patients doivent être surveillés car une insuffisance rénale peut entraîner des effets toxiques en raison de l'accumulation de métabolites actifs (voir les rubriques 4.4 et 5.2). La dose peut nécessiter une adaptation en raison de la réduction de la clairance et de l'augmentation de la demi-vie de la lidocaïne.
Patients atteints d'une insuffisance hépatique
Il peut être nécessaire de réduire la dose de moitié chez les patients atteints d'une insuffisance cardiaque ou hépatique (voir la rubrique 4.4).
Patients atteints d'une insuffisance cardiaque
Il peut être nécessaire de réduire la dose de moitié chez les patients atteints d'une insuffisance cardiaque ou hépatique (voir la rubrique 4.4).
Autre population spéciale
Il peut être nécessaire de réduire les doses chez les patients dont l’état général est altéré, ou chez ceux dont la capacité de liaison aux protéines est réduite (par exemple en cas d’insuffisance rénale, d’insuffisance hépatique, de cancer, ou de grossesse).
Population pédiatrique
LIDOCAÏNE AGUETTANT 20 mg/mL, solution injectable en seringue préremplie ne doit pas être utilisé chez les enfants et les adolescents âgés de moins de 18 ans en raison des risques de réaction toxique (voir rubriques 4.8 et 4.9).
· Anesthésie locale et anesthésie par bloc nerveux périphérique
Enfants et adolescents (2-18 ans)
Pour l'anesthésie par infiltration et les blocs nerveux périphériques, les doses sont calculées individuellement en fonction de l'âge du patient, de son poids corporel et de la nature de l'intervention.
La dose habituelle pour les enfants (2-11 ans) et les adolescents (12-18 ans) est de 3-4 mg/kg de poids corporel. Pour le calcul, le poids-âge moyen doit être pris en compte pour les enfants en surpoids.
La dose administrée peut représenter un volume inférieur à une demi-seringue de 10 mL. La dose requise doit être calculée et la dose excédentaire doit être vidée de la seringue de 10 mL avant l’injection à l’enfant. Pour la dose restant dans la seringue, des injections lentes et progressives sont recommandées.
Le comportement de l’enfant pendant le traitement doit être surveillé attentivement.
Enfants âgés de moins de 2 ans
Lidocaine Aguettant ne doit pas être utilisé chez les enfants âgés de moins de 2 ans car la sécurité et l’efficacité n’ont pas été établies.
En cas d'utilisation par voie intraveineuse pour prévenir la douleur liée à l’injection des médicaments, la posologie pour les adolescents (12-18 ans) est la même que pour les adultes.
Lidocaine Aguettant ne doit pas être utilisé par voie intraveineuse chez les enfants âgés de moins de 12 ans en raison des risques de réaction toxique (voir rubriques 4.8 et 4.9).
· Anesthésie régionale intraveineuse des membres supérieurs
Lidocaine Aguettant ne doit pas être utilisé chez les enfants et les adolescents âgés de moins de 18 ans en raison des risques de réaction toxique (voir rubriques 4.8 et 4.9).
Mode d’administration
La méthode d’administration varie en fonction du type de procédure.
LIDOCAÏNE AGUETTANT 20 mg/mL, solution injectable en seringue préremplie peut être administrée par injection intraveineuse (utilisation intraveineuse) ou par infiltration (intradermique, sous-cutanée ou sous-muqueuse) dans la zone environnante des nerfs périphériques.
LIDOCAÏNE AGUETTANT 20 mg/mL, solution injectable en seringue préremplie est une seringue préremplie prête à être administrée qui n'est pas conçue pour être utilisée avec un pousse-seringue électronique.
Anesthésie régionale intraveineuse
Une technique de garrot spécifique aux membres est essentielle pour la réalisation d’une anesthésie régionale intraveineuse.
4.4. Mises en garde spéciales et précautions d'emploi
La lidocaïne doit être utilisée avec prudence chez les patients souffrant :
· d'épilepsie : les patients atteints de troubles épileptiques cérébraux doivent faire l'objet d'une surveillance très étroite afin de déceler toute manifestation de symptômes nerveux centraux. En outre, de faibles doses de lidocaïne peuvent augmenter l'état de préparation convulsive ;
· d'une insuffisance rénale ou hépatique ;
· de myasthénie grave ;
· d'un blocage du système de la conduction cardiaque dû au fait que les anesthésiques locaux peuvent supprimer la conduction auriculo-ventriculaire ;
· d’une réduction de la fonction cardiovasculaire ;
· de bradycardie ;
· de dépression respiratoire ;
· les personnes âgées et les patients généralement affaiblis ;
· d'une coagulopathie ou traités par des anticoagulants (par ex. l'héparine), des AINS ou des substituts plasmatiques car une lésion accidentelle des vaisseaux sanguins peut entraîner des saignements graves.
Une administration intravasculaire par inadvertance ou un surdosage peuvent provoquer des concentrations sanguines élevées de lidocaïne responsables de symptômes toxiques aigus du système nerveux central et cardiovasculaire.
Des injections intravasculaires accidentelles dans la tête et le cou peuvent provoquer des symptômes cérébraux même à faible dose.
Il faut également faire preuve de prudence si l'anesthésique local doit être injecté dans un tissu enflammé (infecté) en raison d'une absorption systémique accrue due à un débit sanguin plus élevé et d'une diminution de l'effet dû à un pH plus faible du tissu infecté.
Après la commercialisation, quelques cas de chondrolyse ont été rapportés chez des patients qui recevaient en post-opératoire une perfusion intra-articulaire continue d'anesthésiques locaux. La majorité des cas de chondrolyse rapportés concernaient l'articulation de l'épaule. En raison de la multitude de facteurs contributifs et de l'incohérence des données relatives au mode d'action publiées dans les articles scientifiques, il n'a pas pu être établi de lien de causalité.
Un bloc paracervical peut parfois entraîner une bradycardie ou une tachycardie fœtale et il est nécessaire de surveiller attentivement la fréquence cardiaque fœtale (voir rubrique 4.6).
Le retrait du garrot après une anesthésie régionale intraveineuse augmente le risque d’effets indésirables. Le garrot ne doit pas être desserré dans les 20 minutes suivant l’injection.
Ce médicament contient du sodium.
4.5. Interactions avec d'autres médicaments et autres formes d'interactions
Interactions pharmacodynamiques
Antiarythmiques de classe I
Une administration simultanée de lidocaïne et d'autres antiarythmiques de classe I doit être évitée en raison d'un risque d'effets indésirables cardiaques graves.
Autres antiarythmiques
Si la lidocaïne est associée à d'autres antiarythmiques tels que les bêta-bloquants ou les inhibiteurs calciques, l'effet inhibiteur sur la conduction auriculo-ventriculaire et intraventriculaire et sur la contractilité peut être renforcé.
Association avec d'autres anesthésiques locaux
L'association de différents anesthésiques locaux peut entraîner des effets additifs sur le système cardiovasculaire et sur le système nerveux central.
Relaxants musculaires
L'effet des relaxants musculaires (par ex. le suxaméthonium) est prolongé par la lidocaïne.
Sédatifs, hypnotiques
La lidocaïne doit être administrée avec précaution aux patients recevant des sédatifs qui affectent également la fonction du SNC et qui peuvent donc altérer la toxicité de la lidocaïne. Il peut se produire un effet additif entre l'effet anesthésique local et les sédatifs ou hypnotiques.
Anesthésiques volatils
Si de la lidocaïne et des anesthésiques volatils sont administrés simultanément, les effets dépressifs des deux peuvent être intensifiés.
Médicaments pouvant abaisser le seuil convulsif
Comme la lidocaïne elle-même peut réduire le seuil convulsif, son administration concomitante avec d'autres médicaments abaissant le seuil convulsif (par ex. le tramadol ou le bupropion) peut augmenter le risque de convulsions.
Médicaments pouvant élever le seuil convulsif
L'administration simultanée de diazépam élève le seuil convulsif de la lidocaïne. Ceci doit être pris en compte lorsqu'on surveille les patients pour déceler d’éventuels signes de toxicité de la lidocaïne.
Vasoconstricteurs :
L'effet anesthésique local est prolongé par une association avec un vasoconstricteur, par exemple l'adrénaline. Si la lidocaïne est administrée en tant qu’agent antiarythmique, l'administration d'autres médicaments contenant de l'adrénaline ou de la noradrénaline peut entraîner une potentialisation des effets indésirables cardiaques.
Interactions pharmacocinétiques
La lidocaïne est principalement métabolisée par les isoenzymes CYP 3A4 et CYP 1A2 du cytochrome P 450 (voir la rubrique 5.2). Une administration concomitante avec des substances actives qui sont des substrats, des inhibiteurs ou des inducteurs d'enzymes hépatiques, des isoenzymes CYP3A4 et CYP1A2, peut avoir une influence sur la pharmacocinétique de la lidocaïne et donc aussi sur son effet.
Inhibiteurs du CYP 3A4 et/ou du CYP 1A2
Une administration concomitante de lidocaïne et d'inhibiteurs du CYP3A4 et/ou du CYP1A2 peut entraîner une accélération des concentrations plasmatiques de lidocaïne. Il a été signalé une augmentation des concentrations plasmatiques avec, par exemple :
· L'amiodarone (inhibiteur du CYP3A4) : L'amiodarone diminue la puissance du métabolisme hépatique de la lidocaïne, ce qui entraîne un risque d'augmentation du taux de lidocaïne et, par conséquent, une augmentation de la toxicité neurologique et cardiovasculaire. Un suivi clinique, un ECG, et enfin un contrôle de la concentration plasmatique de lidocaïne doivent être réalisés. Au besoin, le taux plasmatique de la lidocaïne doit être surveillée pendant et après le traitement par l'amiodarone.
· La cimétidine (inhibiteur du CYP3A4 et du CYP1A2) : La cimétidine utilisée à des doses égales ou supérieures à 800 mg/jour : augmentation de la concentration plasmatique de lidocaïne avec augmentation subséquente de la toxicité neurologique et cardiovasculaire. Un suivi clinique, un ECG, et enfin un contrôle de la concentration plasmatique de lidocaïne doivent être réalisés. Au besoin, le taux plasmatique de la lidocaïne doit être surveillée pendant et après le traitement par la cimétidine.
· La fluvoxamine (inhibiteur du CYP3A4 et du CYP1A2) : Hausse du taux de lidocaïne, augmentant ainsi le risque de toxicité neurologique et cardiovasculaire. Un suivi clinique, un ECG, et enfin un contrôle de la concentration plasmatique de lidocaïne doivent être réalisés. Au besoin, le taux plasmatique de la lidocaïne doit être surveillée pendant et après l'association.
· Bêtabloquants (excepté l'esmololol) : Lidocaïne intraveineuse : hausse du taux de lidocaïne, avec augmentation subséquente de la toxicité neurologique et cardiovasculaire. Un suivi clinique, un ECG, et enfin un contrôle de la concentration plasmatique de lidocaïne doivent être réalisés. Au besoin, le taux plasmatique de la lidocaïne doit être surveillée pendant et après le traitement par les bêtabloquants.
· Autres inhibiteurs connus du CYP3A4 : inhibiteurs de la protéase (par ex. ritonavir), antibiotiques macrolides (par ex. érythromycine), antifongiques (par ex. kétoconazole, itraconazole).
· Autres inhibiteurs connus du CYP1A2 : ciprofloxacine.
Inducteurs de CYP 3A4 et/ou CYP 1A2
Les substances actives induisant le CYP3A4 et/ou le CYP 1A2 telles que les barbituriques (principalement le phénobarbital), la carbamazépine, la phénytoïne ou la primidone, accélèrent la clairance plasmatique de la lidocaïne et réduisent ainsi l'efficacité de la lidocaïne.
Autres interactions pharmacocinétiques
Les médicaments qui modifient le métabolisme, le débit sanguin hépatique, le débit cardiaque ou la distribution périphérique de la lidocaïne peuvent influencer les concentrations plasmatiques de lidocaïne.
Médicaments provoquant l'hypokaliémie
Les effets électrophysiologiques de la lidocaïne dépendent fortement de la concentration extracellulaire en potassium et peuvent être presque complètement bloqués par l'hypokaliémie. Une utilisation concomitante de médicaments pouvant provoquer une hypokaliémie grave (par ex. acétazolamide, diurétiques de l'anse et thiazides) doit donc être évitée ou utilisée sous une surveillance étroite de la concentration sérique en potassium.
4.6. Fertilité, grossesse et allaitement
Grossesse
Il n'existe pas de données appropriées sur l'utilisation de la lidocaïne chez les femmes enceintes.
La lidocaïne traverse la barrière placentaire (voir la rubrique 5.2). On peut supposer qu'un grand nombre de femmes enceintes et de femmes en âge de procréer ont reçu de la lidocaïne. Aucune perturbation spécifique du processus de reproduction n'a été signalée à ce jour, par exemple, aucune incidence accrue de malformations ou d'effets directs ou indirects sur le fœtus. Toutefois, les risques pour l'homme ne sont pas encore complètement étudiés.
Les études réalisées chez l’animal ont démontré une toxicité pour la reproduction (voir la rubrique 5.3).
Allaitement
La lidocaïne est excrétée en petites quantités dans le lait maternel humain. Il est peu probable qu'elle ait un effet délétère sur le nourrisson lorsqu'elle est utilisée aux doses recommandées. L'allaitement peut donc être poursuivi pendant le traitement par la lidocaïne.
Fertilité
On ne dispose d'aucune donnée humaine sur l'effet de la lidocaïne sur la fertilité.
4.7. Effets sur l'aptitude à conduire des véhicules et à utiliser des machines
Résumé du profil de sécurité d’emploi
La fréquence et la gravité des effets indésirables de la lidocaïne dépendent de la dose, du mode d'administration et de la sensibilité individuelle du patient.
Les effets indésirables liés aux anesthésiques locaux sont rares en l'absence de surdosage, d'absorption systémique rapide anormale ou d'injection intravasculaire accidentelle ; dans de tels cas, ils peuvent être très graves, notamment sur les fonctions cardiaque et neurologique.
Les effets indésirables provoqués par la lidocaïne peuvent être difficiles à distinguer des effets physiologiques du bloc nerveux (par ex. hypotension, bradycardie), des affections provoqués directement (par ex. lésions nerveuses) ou indirectement par l'aiguille.
Des symptômes de toxicité locale peuvent survenir après l'administration de lidocaïne. On peut s'attendre à des effets indésirables systémiques à des concentrations plasmatiques de lidocaïne supérieures à 5- 10 mg/l. Ils se manifestent à la fois sous forme de symptômes du SNC et de symptômes cardiovasculaires.
Les effets indésirables possibles après une administration de lidocaïne comme anesthésique local sont en grande partie identiques à ceux produits par d'autres anesthésiques locaux à liaison amide.
Liste sous forme de tableau des effets indésirables
Les effets indésirables énumérés dans la présente rubrique appartiennent aux catégories de fréquence suivantes : Très fréquents (≥ 1/10) ; fréquents (≥ 1/100 à < 1/10) ; peu fréquents (≥ 1/1 000 à < 1/100) ; rares (≥ 1/10 000 à <1/1 000) ; très rares (<1/10 000) ; indéterminées (ne peuvent être estimées sur la base des données disponibles).
Le tableau suivant énumère les effets indésirables associés à l'utilisation de la lidocaïne comme anesthésique.
|
Classe de systèmes d’organes |
Très fréquents |
Fréquents |
Peu fréquents |
Rares |
Très rares |
Fréquence indéterminée |
|
Affections du sang et du système lymphatique |
|
|
|
|
|
Méthémo-globinémie |
|
Affections du système immunitaire |
|
|
|
réaction allergique*, réactions anaphylactoïdes, broncho-spasme et, dans les cas graves, choc ana-phylactique. |
|
|
|
Affections du système nerveux |
|
Paresthésie, perte de conscience. Symptômes neuro-logiques transitoires. |
|
Neuropathie, convulsions (surdose), anesthésie persistante, parésie, céphalées accompagnées d'acouphènes et photophobie. Lésions du nerf crânien, surdité neuro-sensorielle. Les applications régionales dans la région thoracique ou dans la région de la tête et/ou du cou peuvent induire un blocage sympathique entraînant des symptômes transitoires tels qu'un syndrome de Horner, un syndrome d'Harlequin. |
|
|
|
Affections cardiaques |
|
bradycardie |
|
Arythmie, dépression myocardique ou éven-tuellement arrêt cardiaque (surdose ou injection intravasculaire accidentelle). |
|
|
|
Affections oculaires : |
|
|
|
Vision double |
|
|
|
Affections respiratoires, thoraciques et médiastinales |
|
|
|
Dépression respiratoire |
|
|
|
Affections vasculaires |
|
hypotension, hypertension |
|
|
|
|
|
Affections gastro-intestinales |
nausées |
vomissements |
|
|
|
|
|
Affections de la peau et du tissu sous-cutané |
|
|
|
éruption cutanée, urticaire, œdème |
|
|
* Les tests cutanés pour l'allergie à la lidocaïne ne sont pas considérés comme étant fiables.
Population pédiatrique
On prévoit que la fréquence, le type et la gravité des effets indésirables chez les enfants seront les mêmes que chez les adultes.
Autres populations spéciales
Chez les patients âgés, l'incidence des effets indésirables peut être plus élevée.
Déclaration des effets indésirables suspectés
La déclaration des effets indésirables suspectés après autorisation du médicament est importante. Elle permet une surveillance continue du rapport bénéfice/risque du médicament. Les professionnels de santé déclarent tout effet indésirable suspecté via le système national de déclaration : Agence nationale de sécurité du médicament et des produits de santé (ANSM) et réseau des Centres Régionaux de Pharmacovigilance - Site internet : https://signalement.social-sante.gouv.fr.
En fonction de la sensibilité individuelle, des réactions toxiques se produisent à partir d'une concentration d'environ 5 à 10 mg de lidocaïne par litre de sang veineux.
La concentration plasmatique létale pour les humains est de l'ordre de 6 à 33 mg de lidocaïne par litre.
Une surdose ou une injection intravasculaire accidentelle peut produire des concentrations plasmatiques excessives de lidocaïne, ce qui engendre des signes de toxicité aiguë, pouvant entraîner des effets indésirables très graves. Les effets toxiques de la lidocaïne dépendent du niveau de la concentration plasmatique ; plus la concentration plasmatique est élevée et plus son augmentation est rapide, plus les réactions toxiques seront fréquentes et graves. Ces réactions toxiques concernent le système nerveux central et le système cardiovasculaire.
Symptômes
De faibles surdoses toxiques de lidocaïne entraînent une stimulation du SNC. Un surdosage important, produisant des concentrations plasmatiques toxiques élevées, provoque une dépression des fonctions centrales.
Une toxicité du système nerveux central est une réponse progressive avec des symptômes et des signes de gravité croissante.
Les premiers symptômes observés sont les suivants : étourdissements, vertiges, agitation, hallucination, euphorie, appréhension, bâillements, logorrhée, céphalées, nausées, vomissements, paresthésie labiale, engourdissement de la langue, acouphène et dysarthrie, déficience auditive et visuelle.
D'autres symptômes subjectifs du système nerveux central comprennent : désorientation, sensation occasionnelle de somnolence. Des cas de tachycardie, d'hypertension et de bouffées vasomotrices ont été également rapportés.
Ces signes d'alarme nécessitent une surveillance attentive : contractions musculaires, tremblements, frissons et convulsions généralisées. L'administration simultanée de diazépam élève le seuil épileptogène de la lidocaïne. Ceci doit être pris en compte lorsqu'on surveille les patients pour déceler d’éventuels signes de toxicité de la lidocaïne.
En cas d'administration de doses très élevées : dépression généralisée du système nerveux central, dépression respiratoire, coma et arrêt respiratoire.
Une toxicité cardiovasculaire peut être observée dans les cas graves : troubles du rythme cardiaque tels qu'une extrasystole ventriculaire, fibrillation ventriculaire, pouls impalpable, pâleur, bradycardie majeure, troubles de la conduction auriculo-ventriculaire, diminution de la contractilité cardiaque, hypotension et arrêt cardiaque.
Traitement
Si des signes de toxicité aiguë apparaissent pendant l'administration de l'anesthésique local, l'administration de l'anesthésique doit être immédiatement interrompue. Un intralipide doit être administré afin de prévenir une hypoxie et une acidose qui potentialisent la toxicité systémique de l'anesthésique local et qui exacerbent la progression vers le collapsus cardiovasculaire et les convulsions.
Si des convulsions surviennent, une oxygénation doit être maintenue et la circulation doit être assistée. Au besoin, un anticonvulsif doit être administré. L'utilisation d'une émulsion lipidique intraveineuse doit être envisagée.
Si une dépression cardiovasculaire est évidente (hypotension, bradycardie), il faudra envisager un traitement de substitution intravasculaire par des vasopresseurs, chronotropes et/ou inotropes.
En cas d'arrêt circulatoire, une réanimation cardiopulmonaire immédiate doit être initiée. Pour obtenir un résultat positif, des efforts de réanimation prolongés peuvent s'avérer nécessaires.
Les patients présentant des signes manifestes de toxicité systémique de l'anesthésique local devront être surveillés pendant au moins 12 heures, car la dépression cardiovasculaire peut persister ou réapparaître après le traitement.
Les analeptiques à action centrale sont contre-indiqués.
Il n’existe pas d’antidote spécifique.
La lidocaïne ne peut être éliminée par hémodialyse.
5. PROPRIETES PHARMACOLOGIQUES
5.1. Propriétés pharmacodynamiques
Classe pharmacothérapeutique : anesthésiques locaux, amides, code ATC : N01B B02
La lidocaïne est un anesthésique local de type amide.
La lidocaïne réduit la perméabilité des membranes cellulaires pour les cations, en particulier pour les ions sodium, et également pour les ions potassium à des concentrations plus élevées. Cela conduit, en fonction de la concentration de lidocaïne, à une excitabilité réduite des fibres nerveuses car l'augmentation de la perméabilité du sodium produisant le potentiel d'action est ralentie. La molécule de lidocaïne pénètre, depuis l’intérieur de la cellule, dans le canal sodique ouvert et le bloque en se liant à un récepteur spécifique. Un effet direct de l'incorporation de la lidocaïne dans la membrane cellulaire est beaucoup moins pertinent.
Comme la lidocaïne, avant d'atteindre son site d'action, doit passer dans la cellule, son effet dépend de son pKa et du pH de l'environnement, c'est-à-dire de la proportion de la base libre qui est le fragment migrant principalement à travers les membranes lipophiles des fibres nerveuses.
Dans les tissus enflammés, l'effet anesthésique local est réduit en raison du pH plus bas de ces régions.
5.2. Propriétés pharmacocinétiques
Les concentrations plasmatiques dépendent du site et de la méthode d'administration. Toutefois, il existe un faible rapport entre la quantité d'anesthésique local injecté et les pics plasmatiques.
Les concentrations maximales sont atteintes dans les 30 minutes, et chez la majorité des patients elles sont atteintes en 10 à 20 minutes.
Après une injection intramusculaire de 400 mg de chlorhydrate de lidocaïne monohydraté pour le bloc intercostal, la concentration plasmatique maximale (Cmax) a été déterminée à 6,48 mg/l, atteinte après 5 à 15 min (tmax).
Après une administration sous-cutanée, les valeurs de Cmax ont atteint 4,91 mg/l (injection vaginale) ou 1,95 mg/l (injection abdominale), respectivement. Dans une étude comportant 5 volontaires en bonne santé, après anesthésie par infiltration maxillaire-buccale avec 36 mg de lidocaïne, en utilisant une solution à 2 %, la valeur Cmax a atteint 0,31 mg/l.
Distribution
La lidocaïne suit une cinétique d'élimination biphasique. Après une administration intraveineuse, la substance active est d'abord rapidement distribuée du compartiment central vers les tissus et organes intensément perfusés (phase de distribution alpha). Cette phase est suivie d'une redistribution dans les muscles squelettiques et le tissu adipeux. La demi-vie pendant la phase de distribution alpha est d'environ 4 à 8 minutes. Il est prévu que la distribution dans les tissus périphériques se produise dans les 15 minutes.
Le taux de liaison aux protéines plasmatiques est d'environ 60 à 80 % chez les adultes. Elle dépend de la concentration de substance active et de la concentration d'alpha-1-glycoprotéine acide (AGP). L'AGP est une protéine de phase aiguë qui se lie à la lidocaïne libre et peut être augmentée, par exemple après un traumatisme, une intervention chirurgicale ou des brûlures, selon l'état pathophysiologique du patient. Au contraire, il a été démontré que les concentrations d'AGP sont faibles chez les nouveau-nés et les patients souffrant d'une insuffisance hépatique, ce qui entraîne une réduction marquée de la liaison de la lidocaïne aux protéines plasmatiques.
Le volume de distribution à l'état d’équilibre est de 91 litres. Le volume de distribution peut être modifié chez les patients souffrant d'autres pathologies, comme par exemple une insuffisance cardiaque, une insuffisance hépatique ou une insuffisance rénale.
Biotransformation
La lidocaïne est rapidement métabolisée dans le foie par les mono-oxygénases, principalement par N-désalkylation oxydative, hydroxylation au niveau du cycle aromatique et hydrolyse de la liaison amide. Les dérivés hydroxylés subissent une conjugaison.
Au total, environ 90 % de la lidocaïne est métabolisée en 4-hydroxy-2,6-xylidine, en 4-hydroxy-2,6-xylidine glucuronide et, dans une moindre mesure, en métabolites actifs monoéthylglycine xylidide (MEGX) et glycine xylidide (GX).
Ces derniers peuvent s'accumuler pendant des perfusions de plus longue durée ou en présence d'une insuffisance rénale grave en raison de leur demi-vie plus longue que celle de la lidocaïne elle-même. En présence de pathologies hépatiques, le taux métabolique peut être réduit de 10 à 50 % de la normale.
Les résultats obtenus avec des microsomes hépatiques humains et des isoformes CYP humaines recombinantes ont démontré que les enzymes CYP1A2 et CYP3A4 sont les principales isoformes CYP impliquées dans la N-dééthylation de la lidocaïne.
Élimination
Moins de 10 % de la lidocaïne est excrétée sous forme inchangée dans l'urine, la proportion restante l'étant sous forme de métabolites.
La demi-vie d'élimination est de 1,5 à 2 heures chez les adultes et d'environ 3 heures chez les nouveau-nés. La demi-vie d'élimination peut augmenter en cas d'insuffisance cardiaque grave (jusqu'à 4 à 12 heures) ou de maladie hépatique chronique (jusqu'à 4,5 à 6 heures).
Les temps de demi-vie des métabolites actifs monoéthylglycine xylidide (MEGX) et glycine xylidide (GX) sont respectivement de 2 à 6 heures et de 10 heures. Comme leur demi-vie plasmatique est plus longue que celle de la lidocaïne, une accumulation de métabolites, en particulier de GX, peut se produire pendant une perfusion prolongée.
En outre, la vitesse d'élimination dépend du pH et peut être augmentée par acidification de l'urine. La clairance plasmatique est d'environ 0,95 mL/min.
Le flux sanguin hépatique semble limiter le taux de métabolisme de la lidocaïne.
Populations spéciales
Patients atteints d’une insuffisance rénale
La demi-vie plasmatique de la lidocaïne semble inchangée, à l'exception d'une certaine accumulation de GX pendant une perfusion de 12 heures ou plus. Cette accumulation semble être associée à une administration à long terme du médicament. Toutefois, chez les patients atteints d'une insuffisance rénale grave, la clairance de la lidocaïne est réduite de moitié environ et la demi-vie de la lidocaïne est environ deux fois plus longue que chez les patients en bonne santé.
Patients atteints d'une insuffisance hépatique
La demi-vie plasmatique de la lidocaïne et de ses métabolites peut être prolongée, et il faut s'attendre à des effets importants sur la pharmacocinétique et les exigences posologiques de la lidocaïne chez les patients présentant une altération de la perfusion hépatique, par exemple après un infarctus aigu du myocarde, en présence d'une insuffisance cardiaque, d'une maladie hépatique ou d'une insuffisance cardiaque congestive.
Patients âgés
La demi-vie d'élimination et le volume de distribution peuvent paraître prolongés ou augmentés respectivement chez les patients âgés en raison d'une diminution du débit cardiaque et/ou du débit sanguin hépatique.
Femme enceinte ou allaitante
La lidocaïne traverse le placenta par simple diffusion et atteint le fœtus en quelques minutes après son administration.
Après le bloc paracervical, des concentrations nettement plus élevées de lidocaïne ont été mesurées dans le sang ombilical.
Le fœtus est capable de métaboliser la lidocaïne. Les concentrations dans le sang fœtal représentent environ 60 % des concentrations dans le sang maternel. En raison d'une plus faible liaison aux protéines plasmatiques dans le sang fœtal, la concentration de lidocaïne libre pharmacologiquement active est 1,4 fois plus élevée que la concentration maternelle.
La lidocaïne n'est sécrétée dans le lait maternel qu'en petites quantités.
Population pédiatrique
Chez les nouveau-nés, les niveaux d'α-1-glycoprotéine acide sont faibles et la liaison protéinique peut être réduite. Comme la fraction libre peut être plus élevée, l'utilisation de la lidocaïne chez les nouveau-nés n'est pas recommandée.
5.3. Données de sécurité préclinique
La lidocaïne n'a pas démontré de potentiel génotoxique lors d'essais de génotoxicité in vitro et in vivo. En revanche, la 2,6-xylidine, un métabolite de la lidocaïne, a présenté une activité génotoxique.
Il n'a pas été mené d'études visant à évaluer le potentiel cancérigène de la lidocaïne. La 2,6-xylidine s'est avérée présenter un potentiel cancérigène lors d'études de toxicologie précliniques évaluant l'exposition chronique à ce composé. L'importance clinique de ces résultats n'a pas été déterminée.
Lors d'études de toxicité sur la fonction de reproduction menées chez le lapin, des effets embryotoxiques ou fœtotoxiques de la lidocaïne ont été détectés à des doses de 25 mg/kg administrées en sous-cutané. Chez le rat, la lidocaïne n'a pas eu d'effet sur le développement post-natal de la progéniture à des doses inférieures à la plage toxique pour les mères. Il n'a pas été observé d'altération de la fertilité de rats mâles ou femelles après administration de lidocaïne. La lidocaïne traverse la barrière placentaire par simple diffusion (voir la rubrique 5.2).
Chlorure de sodium,
Hydroxyde de sodium (pour l'ajustement du pH),
Acide chlorhydrique concentré (pour l'ajustement du pH),
Eau pour préparations injectables.
3 ans.
Après ouverture, le médicament doit être utilisé immédiatement.
6.4. Précautions particulières de conservation
Conserver la seringue préremplie dans son blister non ouvert jusqu'à l'utilisation. Ne pas congeler.
6.5. Nature et contenu de l'emballage extérieur
Seringue préremplie (polypropylène) de 5 ou 10 mL, graduée par pas de 0.5 mL de 0 à 5 ou 10 mL, emballée individuellement sous blister.
Boîte en carton de 1 et 10 seringue(s) préremplie(s).
Toutes les présentations peuvent ne pas être commercialisées.
6.6. Précautions particulières d’élimination et de manipulation
Instructions d’utilisation :
La seringue préremplie doit être préparée soigneusement de la façon suivante
La seringue préremplie est à usage unique, c’est-à-dire destinée à un seul patient. Jeter la seringue préremplie après utilisation. NE PAS RÉUTILISER.
Le contenu du blister non ouvert et non endommagé est stérile, et par conséquent il ne doit être ouvert qu’au moment de l’utilisation.
Le produit doit être inspecté visuellement avant l’administration pour déceler la présence de particules et d’une coloration anormale. Seule une solution limpide et incolore dépourvue de particules ou de précipité doit être utilisée.
Le médicament ne doit pas être utilisé si le sceau d'inviolabilité de la seringue est brisé.
La surface externe de la seringue préremplie est stérile jusqu’à l’ouverture du blister.
Lorsqu'il est manipulé en utilisant une technique aseptique, ce médicament peut être placé sur un champ stérile.
1) Retirer la seringue préremplie du blister stérile.
|
|
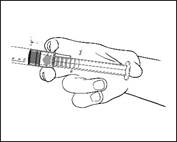
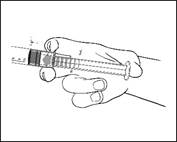
2) Presser la tige du piston pour libérer le joint. Le procédé de stérilisation a pu entraîner une adhérence du joint sur le corps de la seringue préremplie. |
|||
|
|
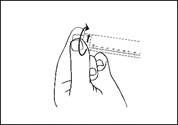
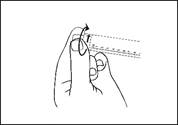
3) Dévisser l'embout protecteur pour rompre l’embout de scellage. Ne pas toucher l’embout de connexion Luer afin d'éviter toute contamination. |
|||
|
|
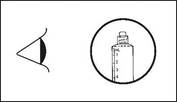
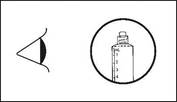
4) Vérifier que l’embout de scellage de la seringue préremplie a bien été totalement retiré. Dans le cas contraire, replacer le protège embout et dévisser à nouveau |
|||
|
|
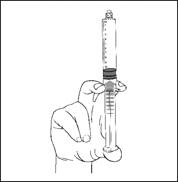
5) Purger l’air de la seringue en poussant légèrement le piston et si nécessaire, éliminer la dose excédentaire avant l’injection. |
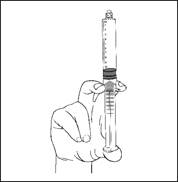
6) Connecter la seringue préremplie à la voie d'accès ou à l'aiguille. Pousser lentement le piston pour injecter le volume requis.
Tout médicament non utilisé ou déchet doit être éliminé conformément à la réglementation en vigueur.
7. TITULAIRE DE L’AUTORISATION DE MISE SUR LE MARCHE
1, rue Alexander Fleming
69007 LYON
France
8. NUMERO(S) D’AUTORISATION DE MISE SUR LE MARCHE
· 34009 303 008 6 2 : Boite de 1 seringue préremplie de 5 ml.
· 34009 303 008 7 9 : Boite de 10 seringues préremplies de 5 ml.
· 34009 301 531 4 7 Seringue préremplie (PP) de 10 mL, emballée individuellement sous blister. Boîte de 1.
· 34009 301 531 6 1 Seringue préremplie (PP) de 10 mL, emballée individuellement sous blister. Boîte de 10.
9. DATE DE PREMIERE AUTORISATION/DE RENOUVELLEMENT DE L’AUTORISATION
[à compléter ultérieurement par le titulaire]
10. DATE DE MISE A JOUR DU TEXTE
[à compléter ultérieurement par le titulaire]
Sans objet.
12. INSTRUCTIONS POUR LA PREPARATION DES RADIOPHARMACEUTIQUES
Liste II
ANSM - Mis à jour le : 30/07/2024
LIDOCAÏNE AGUETTANT 20 mg/mL, solution injectable en seringue préremplie
Chlorhydrate de lidocaïne
· Gardez cette notice. Vous pourriez avoir besoin de la relire.
· Si vous avez d’autres questions, interrogez votre médecin, votre pharmacien ou votre infirmier/ère.
· Si vous ressentez un quelconque effet indésirable, parlez-en à votre médecin, votre pharmacien ou votre infirmier/ère. Ceci s’applique aussi à tout effet indésirable qui ne serait pas mentionné dans cette notice. Voir rubrique 4.
1. Qu'est-ce que LIDOCAÏNE AGUETTANT 20 mg/mL, solution injectable en seringue préremplie et dans quels cas est-il utilisé ?
2. Quelles sont les informations à connaître avant que LIDOCAÏNE AGUETTANT 20 mg/mL, solution injectable en seringue préremplie ne vous soit administré ?
3. Comment LIDOCAÏNE AGUETTANT 20 mg/mL, solution injectable en seringue préremplie est-il administré ?
4. Quels sont les effets indésirables éventuels ?
5. Comment conserver LIDOCAÏNE AGUETTANT 20 mg/mL, solution injectable en seringue préremplie ?
6. Contenu de l’emballage et autres informations.
1. QU’EST-CE QUE LIDOCAÏNE AGUETTANT 20 mg/mL, solution injectable en seringue préremplie ET DANS QUELS CAS EST-IL UTILISE ?
LIDOCAÏNE AGUETTANT contient une substance active, le chlorhydrate de lidocaïne
LIDOCAÏNE AGUETTANT est un anesthésique local. Il est utilisé pour anesthésier des parties du corps pendant les interventions chirurgicales. Il empêche temporairement les nerfs de transmettre les messages de douleur au cerveau dans la zone où il est injecté.
LIDOCAÏNE AGUETTANT 20 mg/mL, solution injectable en seringue préremplie peut être utilisé chez les adultes.
2. QUELLES SONT LES INFORMATIONS A CONNAITRE AVANT D’UTILISER LIDOCAÏNE AGUETTANT 20 mg/mL, solution injectable en seringue préremplie ?
· si vous êtes allergique à la lidocaïne, aux anesthésiques locaux à liaison amide ou à l’un des autres composants contenus dans ce médicament, mentionnés dans la rubrique 6.
Avertissements et précautions
Adressez-vous à votre médecin, pharmacien ou votre infirmier/ère avant que LIDOCAÏNE AGUETTANT 20 mg/mL, solution injectable en seringue préremplie ne vous soit administré :
· si vous souffrez d'épilepsie. Votre médecin vous surveillera de près pour déceler toute manifestation de symptômes ;
· si vous souffrez d'une maladie rénale ou hépatique ;
· si vous souffrez d'une maladie entraînant une faiblesse musculaire (myasthénie grave) ;
· si vous souffrez d'affections cardiaques, y compris de troubles de la conduction, d’un rythme cardiaque lent ;
· si vous souffrez de dépression respiratoire (difficultés respiratoires avec respiration lente et superficielle) ;
· si vous êtes une personne âgée ou si vous avez un mauvais état de santé général ;
· si vous souffrez de troubles de la coagulation ou êtes traité(e) pour de tels troubles.
En outre, votre médecin sait qu'une injection de ce médicament dans un tissu enflammé peut entraîner une augmentation de l'absorption de la substance active dans la circulation et l'effet de la substance active sur votre organisme sera affaibli.
Votre médecin prendra en compte qu'il y a un risque accru d'effets indésirables sur le système nerveux si ce médicament est administré dans la région de la tête et du cou.
Enfants et adolescents
LIDOCAÏNE AGUETTANT 20 mg/mL, solution injectable en seringue préremplie ne doit pas être utilisé chez les enfants et chez les adolescents de moins de 18 ans.
Autres médicaments et LIDOCAÏNE AGUETTANT 20 mg/mL, solution injectable en seringue préremplie
Informez votre médecin ou pharmacien si vous prenez, avez récemment pris ou pourriez prendre tout autre médicament. LIDOCAÏNE AGUETTANT 20 mg/mL, solution injectable en seringue préremplie peut affecter ou être affecté par d'autres médicaments.
En particulier, prévenez votre médecin si vous prenez l’un des médicaments suivants :
· des médicaments destinés au traitement de l'hypertension, tels que des diurétiques (comprimés qui servent à éliminer de l'eau de l'organisme) ;
· des médicaments utilisés pour traiter les affections cardiaques, y compris l'arythmie, comme les bêta-bloquants (par ex. métoprolol, propranolol) ou les inhibiteurs calciques (par ex. amiodarone) ;
· des médicaments qui rétrécissent les vaisseaux sanguins (vasoconstricteurs, par ex. adrénaline, noradrénaline) ;
· des médicaments utilisés pour relaxer les muscles pendant l'anesthésie générale (par ex. le suxaméthonium) ;
· des somnifères et des médicaments qui altèrent votre conscience (sédatifs) ;
· des médicaments qui augmentent le risque de crises d'épilepsie (par ex. tramadol, bupropion) ;
· des médicaments qui réduisent le risque de crises d'épilepsie (par ex. diazépam) ;
· de la cimétidine, un médicament utilisé pour traiter les brûlures d'estomac ;
· des médicaments antiviraux (par ex. ritonavir), des antibiotiques macrolides (par ex. érythromycine) ou des antifongiques (par ex. kétoconazole, itraconazole) ;
· de la ciprofloxacine (antibiotiques) ;
· des médicaments utilisés pour traiter l'épilepsie (phénobarbital, phénytoïne, carbamazépine ou primidone) ;
· de la fluvoxamine, un médicament utilisé dans le traitement des maladies mentales ;
· des médicaments utilisés pour réduire la pression intra-oculaire (par ex. l'acétazolamide) ;
· d'autres anesthésiques, y compris des anesthésiques locaux.
Si vous êtes enceinte ou que vous allaitez, si vous pensez être enceinte ou planifiez une grossesse, demandez conseil à votre médecin ou pharmacien avant que ce médicament ne vous soit administré. Votre médecin déterminera si vous pouvez continuer le traitement.
Grossesse
Votre médecin ne vous administrera ce médicament pendant votre grossesse que s'il le juge nécessaire. La dose doit être aussi faible que possible.
Allaitement
La lidocaïne est excrétée en petites quantités dans le lait maternel humain. Il est peu probable qu'elle ait un effet sur le nourrisson lorsqu'elle est utilisée aux doses recommandées. L'allaitement peut donc être poursuivi pendant le traitement par la lidocaïne.
Conduite de véhicules et utilisation de machines
LIDOCAÏNE AGUETTANT 20 mg/mL, solution injectable en seringue préremplie peut affecter votre capacité à conduire ou à utiliser des machines. Demandez à votre médecin quand la conduite ou l'utilisation de machines peut se faire sans danger.
LIDOCAÏNE AGUETTANT 20 mg/mL, solution injectable en seringue préremplie contient du sodium
Ce médicament contient moins de 1 mmol de sodium (23 mg) par seringue préremplie de 5 ou 10 mL, c’est-à-dire qu’il est essentiellement « sans sodium ».
3. COMMENT UTILISER LIDOCAÏNE AGUETTANT 20 mg/mL, solution injectable en seringue préremplie ?
Votre médecin décidera de la posologie la plus appropriée à votre cas particulier en fonction de votre âge et de votre état de santé ainsi que du site d'injection, de la méthode utilisée et de votre réponse à l'injection.
Utilisation chez les enfants et les adolescents
LIDOCAÏNE AGUETTANT 20 mg/mL, solution injectable en seringue préremplie ne doit pas être utilisé chez les enfants et les adolescents de moins de 18 ans.
Mode d’administration
LIDOCAÏNE AGUETTANT 20 mg/mL, solution injectable en seringue préremplie vous sera administré sous forme d’injection intraveineuse (utilisation intraveineuse) ou d'infiltration (intradermique, sous-cutanée ou sous-muqueuse) dans la zone environnante des nerfs périphériques.
S’il vous est administré plus de LIDOCAÏNE AGUETTANT 20 mg/mL, solution injectable en seringue préremplie que vous n’auriez dû en recevoir
Comme ce médicament vous est administré par un professionnel de santé qualifié, il est peu probable qu'il vous soit administré trop de LIDOCAÏNE AGUETTANT 20 mg/mL, solution injectable en seringue préremplie.
La présence ou non de symptômes de surdosage dépend de la concentration de ce médicament dans votre sang. Plus la concentration de lidocaïne dans votre sang est importante et plus la vitesse d'administration est élevée, et plus les symptômes de surdosage que vous pourriez ressentir seront fréquents et sévères.
Un petit surdosage affecte principalement votre système nerveux central. Les effets indésirables qui se produisent disparaîtront dans la plupart des cas après l'arrêt de l'administration de la lidocaïne.
Toutefois, si vous pensez avoir reçu trop de médicament, ou si vous commencez à présenter des étourdissements ou des vertiges, un engourdissement de la langue, un bourdonnement dans l'oreille, des vomissements ou des frissons, vous devez en informer immédiatement la personne qui vous fait l'injection. Votre médecin saura comment gérer ces symptômes et vous administrer le traitement nécessaire.
Si vous avez d’autres questions sur l’utilisation de ce médicament, demandez plus d’informations à votre médecin, votre pharmacien ou votre infirmier/ère.
4. QUELS SONT LES EFFETS INDESIRABLES EVENTUELS ?
Certains effets indésirables peuvent être graves. Consultez immédiatement un médecin si vous avez une réaction allergique provoquant :
· un gonflement des mains, des pieds, du visage, des lèvres, de la bouche, de la langue ou de la gorge
· des difficultés respiratoires
· une éruption cutanée accompagnée de démangeaisons
· de la fièvre
· une chute de la tension artérielle et un choc.
Ces effets indésirables sont rares (peuvent toucher jusqu’à 1 personne sur 1000).
D'autres effets indésirables peuvent inclure :
Très fréquents (peuvent toucher jusqu’à 1 patient sur 10)
· nausées.
Fréquents (peuvent toucher jusqu’à 1 patient sur 10)
· sensation de fourmillement, de picotement, de brûlure, de démangeaison ou d'engourdissement (paresthésie)
· perte de conscience
· douleurs ou frissons du(e)s aux injections
· rythme cardiaque lent
· hypotension ou hypertension
· vomissements.
Rares (peuvent toucher jusqu’à 1 patient sur 1000) :
· altération des sensations ou faiblesse musculaire (neuropathie)
· convulsions (crises d'épilepsie)
· paralysie partielle
· maux de tête accompagnés d'un bourdonnement ou d'un cliquetis dans les oreilles (acouphènes) et d'une intolérance anormale à la lumière (photophobie).
· perte auditive (surdité)
· dommages aux nerfs cérébraux
· chute des paupières associée à un rétrécissement de vos pupilles et parfois à une réduction de la transpiration (syndrome de Horner). Cela se produit après une administration dans la région tête/cou.
· transpiration et rougissement asymétriques au niveau de la partie supérieure de la poitrine, du cou ou du visage (syndrome d'Harlequin)
· arythmie
· arrêt cardiaque
· vision double
· ralentissement ou arrêt de la respiration
· éruption cutanée ou urticaire.
Fréquence indéterminée (ne peut être estimée sur la base des données disponibles)
· une coloration bleuâtre de la peau, des maux te tête, un essoufflement et une fatigue dus à des quantités anormales de méthémoglobine (une forme d’hémoglobine qui a une capacité réduite à fixer l’oxygène) dans le sang (méthémoglobinémie)
Déclaration des effets secondaires
Si vous ressentez un quelconque effet indésirable, parlez-en à votre médecin, votre pharmacien ou à votre infirmier/ère. Ceci s’applique aussi à tout effet indésirable qui ne serait pas mentionné dans cette notice. Vous pouvez également déclarer les effets indésirables directement via le système national de déclaration : Agence nationale de sécurité du médicament et des produits de santé (ANSM) et réseau des Centres Régionaux de Pharmacovigilance - Site internet : https://signalement.social-sante.gouv.fr
5. COMMENT CONSERVER LIDOCAÏNE AGUETTANT 20 mg/mL, solution injectable en seringue préremplie ?
Tenir ce médicament hors de la vue et de la portée des enfants.
N’utilisez pas ce médicament après la date de péremption indiquée sur l’étiquette de la seringue, le blister et l’étui. La date de péremption fait référence au dernier jour de ce mois.
Conserver la seringue préremplie dans son blister non ouvert jusqu'à l'utilisation. Ne pas congeler.
Après ouverture, le médicament doit être utilisé immédiatement.
N’utilisez pas ce médicament s’il y a des signes visibles de détérioration.
Ne jetez aucun médicament au tout-à-l’égout. Demandez à votre pharmacien d’éliminer les médicaments que vous n’utilisez plus. Ces mesures contribueront à protéger l’environnement.
6. CONTENU DE L’EMBALLAGE ET AUTRES INFORMATIONS
Ce que contient LIDOCAÏNE AGUETTANT 20 mg/mL, solution injectable en seringue préremplie
· La substance active est le chlorhydrate de lidocaïne :
Chaque millilitre de solution injectable contient 20 mg de chlorhydrate de lidocaïne (sous forme de chlorhydrate de lidocaïne monohydraté).
Chaque seringue préremplie de 5 mL contient 100 mg de chlorhydrate de lidocaïne (sous forme de chlorhydrate de lidocaïne monohydraté).
Chaque seringue préremplie de 10 mL contient 200 mg de chlorhydrate de lidocaïne (sous forme de chlorhydrate de lidocaïne monohydraté).
· Les autres composants sont : chlorure de sodium, hydroxyde de sodium, acide chlorhydrique concentré (pour l’ajustement du pH), eau pour préparations injectables.
LIDOCAÏNE AGUETTANT est une solution injectable incolore et claire (injection). LIDOCAÏNE AGUETTANT se présente sous la forme d'une seringue préremplie en polypropylène de 5 ou 10 mL, graduée par pas de 0.5 mL de 0 à 5 ou 10 mL, emballée individuellement dans un blister transparent. Boîte en carton de 1 ou 10 seringue(s) préremplie(s). Toutes les présentations peuvent ne pas être commercialisées.
Titulaire de l’autorisation de mise sur le marché
1, rue Alexander Fleming
69007 LYON
France
Exploitant de l’autorisation de mise sur le marché
1, rue Alexander Fleming
69007 LYON
France
1, rue Alexander Fleming
69007 LYON
France
ou
LABORATOIRE AGUETTANT
LIEU-DIT CHANTECAILLE
07340 CHAMPAGNE
FRANCE
Noms du médicament dans les Etats membres de l'Espace Economique Européen
Ce médicament est autorisé dans les Etats membres de l'Espace Economique Européen sous les noms suivants : Conformément à la réglementation en vigueur.
[À compléter ultérieurement par le titulaire]
La dernière date à laquelle cette notice a été révisée est :
[à compléter ultérieurement par le titulaire]
Des informations détaillées sur ce médicament sont disponibles sur le site Internet de l’ANSM (France).
Les informations suivantes sont destinées exclusivement aux professionnels de santé :
La seringue préremplie doit être préparée soigneusement de la façon suivante
La seringue préremplie est destinée à l'usage unique d'un seul patient. Éliminer la seringue préremplie après utilisation. Ne pas réutiliser.
Le contenu du blister non ouvert et non endommagé est stérile, et par conséquent il ne doit être ouvert qu’au moment de l’utilisation.
Le produit doit être inspecté visuellement avant l’administration pour déceler la présence de particules et d’une coloration anormale. Seule une solution limpide et incolore dépourvue de particules ou de précipité, doit être utilisée.
Le médicament ne doit pas être utilisé si le sceau d'inviolabilité de la seringue est brisé.
La surface externe de la seringue préremplie est stérile jusqu’à l’ouverture du blister.
Lorsqu'il est manipulé en utilisant une technique aseptique, ce médicament peut être placé sur un champ stérile.
1) Retirer la seringue préremplie du blister stérile.
|
|
2) Presser la tige du piston pour libérer le joint. Le procédé de stérilisation a pu entraîner une adhérence du joint sur le corps de la seringue préremplie. |
|||
|
|
3) Dévisser l'embout protecteur pour rompre l’embout de scellage. Ne pas toucher l’embout de connexion Luer afin d'éviter toute contamination. |
|||
|
|
4) Vérifier que l’embout de scellage de la seringue préremplie a bien été totalement retiré. Dans le cas contraire, replacer le protège embout et dévisser à nouveau |
|||
|
|
5) Purger l’air de la seringue en poussant légèrement le piston et si nécessaire, éliminer la dose excédentaire avant l’injection. |
6) Connecter la seringue préremplie à la voie d'accès ou à l'aiguille. Pousser lentement le piston pour injecter le volume requis.