Dernière mise à jour le 29/06/2026
TYAVAX, suspension et solution pour suspension injectable en seringue préremplie. Vaccin de l'hépatite A (inactivé, adsorbé) et typhoïdique polyosidique
Ce médicament n'est ou ne sera bientôt plus disponible sur le marché.
Si vous prenez actuellement ce médicament, il vous est recommandé d'en parler avec votre médecin
ou avec votre pharmacien qui pourra vous orienter vers un autre traitement.
Indications thérapeutiques
Classe pharmacothérapeutique : Vaccins combinés bactériens et viraux - code ATC : J07CA10.
La fièvre typhoïde est une maladie infectieuse qui peut être transmise par des aliments ou boissons contenant les bactéries (appelées Salmonella enterica, sérotype typhi) qui causent la maladie. Il s'agit d'une infection grave qui peut être fatale si elle n'est pas rapidement traitée.
L'infection par l'hépatite A est due à un virus qui attaque le foie. Elle peut être transmise par des aliments ou boissons contenant le virus. La jaunisse et une sensation de malaise généralisé font partie des symptômes.
Quand vous recevez une injection de TYAVAX, les défenses naturelles de votre corps élaborent une protection contre la fièvre typhoïde et l'infection causée par le virus de l'hépatite A.
Composition en substances actives
-
Compartiment 1 ( Composition pour une dose de vaccin mélangé )
- > polyoside capsulaire Vi de Salmonella typhi (souche Ty2) 25 microgrammes
-
Compartiment 2 ( Composition pour une dose de vaccin mélangé )
- > virus de l'hépatite A souche GBM inactivé adsorbé 160 unités antigéniques
Service médical rendu (SMR)
Pas de SMR disponible pour ce médicament ( plus d'informations dans l'aide )
Amélioration du service médical rendu (ASMR)
Pas d'ASMR disponible pour ce médicament (plus d'informations dans l'aide )
ANSM - Mis à jour le : 08/02/2021
TYAVAX, suspension et solution pour suspension injectable en seringue préremplie
Vaccin de l’hépatite A (inactivé, adsorbé) et typhoïdique polyosidique
2. COMPOSITION QUALITATIVE ET QUANTITATIVE
Après reconstitution, 1 dose (1 mL) contient :
Initialement contenu dans la suspension :
Virus de l’hépatite A, souche GBM1 (inactivé) 2 ........................................................................ 160 U 3
1 Produite sur cellules diploïdes humaines MRC-5
2 Adsorbé sur de l’hydroxyde d’aluminium hydraté (0,3 milligramme d’Al)
3 En l’absence de référence internationale standardisée, le contenu antigénique est exprimé à l’aide d’une référence interne.
Initialement contenu dans la solution :
Polyosides capsulaires Vi de Salmonella typhi (souche Ty2).................................... 25 microgrammes
Excipient à effet notoire (voir rubrique 4.4) :
Phénylalanine........................................................................................................ 10 microgrammes
Pour la liste complète des excipients, voir rubrique 6.1.
TYAVAX peut contenir des traces de néomycine, utilisée au cours du procédé de fabrication (voir rubrique 4.3).
Suspension et solution pour suspension injectable en seringue préremplie.
Le vaccin hépatite A inactivé est une suspension trouble et blanchâtre; le vaccin typhoïdique polyosidique est une solution limpide et incolore.
4.1. Indications thérapeutiques
TYAVAX doit être administré conformément aux recommandations officielles.
4.2. Posologie et mode d'administration
La dose recommandée pour les sujets à partir de 16 ans est de 1 millilitre de vaccin reconstitué.
La protection initiale est obtenue après une seule injection de TYAVAX. Les taux d’anticorps protecteurs ne sont obtenus que 14 jours après l’administration du vaccin.
Afin d'obtenir une protection à long terme contre l'infection causée par le virus de l'hépatite A, une seconde dose (rappel) d'un vaccin hépatite A inactivé doit être administrée. TYAVAX peut être utilisé pour dispenser une ou deux doses de vaccin hépatite A, comme suit :
· Chez les sujets ayant reçu une dose de TYAVAX :
o Une dose de vaccin hépatite A monovalent doit être administrée dans les 36 mois et de préférence dans les 6 à 12 mois (voir rubrique 5.1).
o Ou, si une protection contre la fièvre typhoïde est toujours requise, une deuxième dose de TYAVAX peut être administrée à condition que 36 mois environ se soient écoulés depuis la première dose.
· Chez les sujets ayant reçu une dose de vaccin hépatite A monovalent :
TYAVAX peut être utilisé pour la deuxième dose (rappel) de vaccin hépatite A, si une protection contre la fièvre typhoïde est aussi souhaitée. Il doit être alors administré dans les 36 mois qui suivent le vaccin hépatite A et de préférence dans les 6 à 12 mois.
Il est estimé que les anticorps anti-VHA persistent plusieurs années (au-delà de 10 ans) après la seconde dose (rappel).
Chez les sujets qui restent exposés au risque de fièvre typhoïde, la vaccination par une dose unique d’un vaccin typhoïdique polyosidique Vi purifié doit être renouvelée tous les 3 ans (voir rubrique 5.1).
Population pédiatrique
La sécurité et l'efficacité de TYAVAX chez l'enfant et l'adolescent de moins de 16 ans n'ont pas encore été établies. Aucune donnée n'est disponible.
Mode d’administration
TYAVAX doit être administré par voie intramusculaire dans le muscle deltoïde en injection lente.
TYAVAX ne doit pas être administré par voie intravasculaire.
TYAVAX ne doit pas être injecté dans le muscle fessier en raison de la quantité variable de tissus graisseux dans cette partie du corps, ni par voie intradermique, ces modes d'administration pouvant induire une réponse immunitaire plus faible.
TYAVAX peut être administré par voie sous-cutanée chez les patients souffrant de thrombocytopénie ou de risque hémorragique.
Pour les instructions concernant la reconstitution du médicament avant administration, voir la rubrique 6.6.
La vaccination devra être différée en cas de maladie fébrile sévère aiguë.
4.4. Mises en garde spéciales et précautions d'emploi
Une syncope (évanouissement), en réaction psychogène à l’injection avec une aiguille, peut survenir après, voire avant, toute vaccination, en particulier chez les adolescents. Cela peut s’accompagner de plusieurs signes neurologiques comme des troubles transitoires de la vision, des paresthésies et des mouvements tonico-cloniques des membres durant la phase de récupération. Il est important que des mesures soient mises en place afin d’éviter toute blessure en cas d’évanouissement.
L’immunogénicité du vaccin peut être réduite en cas de traitement immunosuppresseur ou de déficit immunitaire. Dans ce cas, il est recommandé de différer la vaccination jusqu'à la fin de tout traitement immunosuppresseur. Les sujets ayant une immunodéficience chronique, telle qu’une infection par le VIH, peuvent être vaccinés si l’immunodéficience permet l’induction, même limitée, d’une réponse en anticorps.
Du fait de la durée d’incubation de l'hépatite A, l’infection peut être présente mais cliniquement asymptomatique au moment de la vaccination. Dans ce cas, il se peut que TYAVAX ne prévienne pas l’hépatite A.
TYAVAX ne protège pas contre les infections causées par d’autres agents pathogènes hépatiques comme les virus des hépatites B, C ou E.
TYAVAX ne protège pas contre les infections causées par Salmonella enterica autre que le sérotype typhi.
Comme pour tous les vaccins, la réponse protectrice immune n’est pas acquise par tous les sujets vaccinés.
TYAVAX contient de la phénylalanine, de l’éthanol, du potassium et du sodium
TYAVAX contient 10 microgrammes de phénylalanine par dose de 1 mL équivalent à 0,17 microgrammes/kg chez une personne de 60 kg. La phénylalanine peut être dangereuse pour les personnes atteintes de phénylcétonurie (PCU), une maladie génétique rare caractérisée par l’accumulation de la phénylalanine ne pouvant être éliminée correctement.
TYAVAX contient 2 mg d’alcool (éthanol) par dose de 1 mL. La faible quantité d’alcool contenue dans ce médicament n’est pas susceptible d’entraîner d’effet notable.
TYAVAX contient moins de 1 mmol (39 mg) de potassium et moins de 1 mmol (23 mg) de sodium par dose, c’est-à-dire qu’il est essentiellement « sans potassium » et « sans sodium ».
Traçabilité
Afin d'améliorer la traçabilité des médicaments biologiques, le nom et le numéro de lot du produit administré doivent être clairement enregistrés.
4.5. Interactions avec d'autres médicaments et autres formes d'interactions
L’administration concomitante du vaccin contre la fièvre jaune et de TYAVAX n’a pas été spécifiquement évaluée. Cependant, à partir des données obtenues lors de l’administration concomitante des vaccins monovalents (vaccin typhoïdique polyosidique Vi et vaccin hépatite A inactivé) avec le vaccin contre la fièvre jaune, aucune interférence des réponses immunitaires n’est attendue pour ces 3 antigènes.
Associations faisant l'objet de précautions d’emploi
TYAVAX ne doit pas être mélangé avec d’autres vaccins dans la même seringue.
L’administration concomitante de TYAVAX avec le vaccin combiné, adsorbé, diphtérique (dose réduite), tétanique et poliomyélitique (inactivé) (dT-Polio) en deux sites séparés a démontré une non-infériorité en termes de réponse immunitaire obtenue un mois après la vaccination, comparée à l'administration séparée des deux vaccins à des dates différentes pour toutes les valences, sauf pour la valence Vi. Néanmoins, le taux de séroconversion anti-Vi (augmentation ≥ 4 fois) lors de l'administration concomitante était non-inférieur à l'administration séparée chez les sujets non séroprotégés avant la vaccination (voir rubrique 5.1). Le taux de séroprotection (pourcentage de sujets ayant atteint le seuil de protection ≥ 1 µg/mL vis-à-vis des anticorps anti-Vi) étant conforme aux taux prévisibles des réponses lorsque TYAVAX est administré seul, il est peu probable que l'administration concomitante de TYAVAX avec le dT-Polio en des sites séparés ait des conséquences cliniques. Par conséquent, l'administration concomitante de TYAVAX avec le dT-Polio en deux sites séparés est possible.
Associations déconseillées
L’effet de l’administration concomitante d’immunoglobulines sur l’immunogénicité de TYAVAX n’a pas été évalué. Par conséquent, une interférence avec la réponse immune de TYAVAX ne peut être exclue. Les données obtenues lors de l’administration concomitante d'immunoglobulines avec un vaccin hépatite A inactivé monovalent ont montré que les taux de séroconversion n’étaient pas modifiés même si les titres en anticorps anti-VHA pouvaient être plus bas qu’après une vaccination avec un vaccin monovalent seul.
4.6. Fertilité, grossesse et allaitement
Grossesse
Les données sur un nombre limité de grossesses exposées (plus de 150 cas pour le vaccin typhoïdique polyosidique Vi monovalent, plus de 40 cas pour le vaccin hépatite A inactivé monovalent et plus de 10 cas pour TYAVAX ou les 2 composants administrés simultanément) ne montrent aucun effet délétère de TYAVAX sur la grossesse ou sur le fœtus / nouveau-né.
A ce jour, il n'existe aucune autre donnée épidémiologique pertinente.
Les études effectuées chez l'animal n'ont pas montré d'effet délétère direct ou indirect sur la gestation, le développement embryonnaire ou fœtal, l'accouchement et le développement post-natal (voir rubrique 5.3).
TYAVAX ne sera administré qu'avec prudence chez la femme enceinte.
Quand la patiente est considérée comme un sujet à risque seulement pour l'hépatite A ou la fièvre typhoïde, un vaccin monovalent doit alors être utilisé.
L'excrétion de TYAVAX dans le lait maternel est inconnue. L'excrétion de TYAVAX dans le lait n'a pas été étudiée chez les animaux. La décision de continuer/interrompre l'allaitement ou d'administrer ou ne pas administrer TYAVAX doit être prise en tenant compte du bénéfice de l'allaitement pour l'enfant et du bénéfice de TYAVAX pour la femme.
Fertilité
Aucune donnée concernant la fertilité n'est disponible.
4.7. Effets sur l'aptitude à conduire des véhicules et à utiliser des machines
TYAVAX a une influence mineure sur l'aptitude à conduire des véhicules et à utiliser des machines.
Des cas peu fréquents (≥1/1000, <1/100) de sensations vertigineuses ont été observés suite à l'administration de ce vaccin (voir rubrique 4.8).
a. Résumé du profil de tolérance
Au cours des études cliniques, les réactions le plus fréquemment rapportées étaient celles survenant au site d’injection.
Une douleur au site d’injection de TYAVAX a été rapportée chez 89,9 % des sujets (sévère dans 4,5 % des cas). Pour les sujets ayant reçu les deux vaccins monovalents simultanément en 2 sites d’injection séparés, la douleur a été rapportée chez 83,2 % des sujets (sévère dans 5,0 % des cas) pour les 2 sites d’injection combinés. Une douleur a été rapportée par 79,3 % des sujets au site d'injection du vaccin typhoïdique (sévère dans 5,0 % des cas) et par 50,3 % des sujets au site d'injection du vaccin hépatite A (sévère dans 0,6 % des cas).
Une douleur au site d’injection persistant plus de 3 jours a été rapportée par 17,4 % des sujets après une vaccination par TYAVAX, par 2,8 % des sujets pour un vaccin monovalent Vi et par 0,6 % des sujets pour un vaccin hépatite A monovalent.
Des œdèmes/indurations sévères (>5 cm) au site d’injection ont été rapportés chez 7,9 % des sujets vaccinés par TYAVAX. Pour les sujets qui ont reçu simultanément les deux vaccins monovalents en des sites d’injection séparés, des œdèmes/indurations sévères ont été rapportés chez 1,7 % des sujets pour les deux sites d'injection combinés (chez 1,1 % des sujets, au site d’injection du vaccin typhoïdique et chez 0,6 % au site d’injection du vaccin hépatite A).
L’incidence globale des réactions systémiques était similaire entre les sujets vaccinés avec TYAVAX et les sujets ayant reçu simultanément les 2 vaccins monovalents en 2 sites d’injection séparés.
Toutes les réactions ont guéri sans séquelles.
b. Liste tabulée des réactions indésirables
Les données sur les réactions indésirables sont issues des études cliniques et de l'expérience après la commercialisation dans le monde.
Dans chaque classe de systèmes d'organes les réactions indésirables sont classées en fonction de leur fréquence, les réactions les plus fréquentes en premier, selon la convention suivante :
Très fréquent (≥1/10),
Fréquent (≥1/100, <1/10),
Peu fréquent (≥1/1000, <1/100),
Rare (≥1/10000, <1/1000),
Très rare (<1/10000),
Fréquence indéterminée : ne peut être estimée sur la base des données disponibles. Basés sur des déclarations spontanées, ces effets indésirables ont été rapportés très rarement pendant la commercialisation de TYAVAX. Les effets étant rapportés spontanément à partir d’une population de taille incertaine, il n’est pas toujours possible d’estimer leur fréquence de façon fiable ou d’établir une relation causale avec l’exposition au vaccin.
Au sein de chaque groupe de fréquence, les réactions indésirables sont présentées suivant un ordre décroissant de gravité.
Affections du système immunitaire
Fréquence indéterminée : réactions anaphylactiques, anaphylactoïdes, y compris le choc ; maladie sérique.
Affections du système nerveux
Très fréquent : céphalées.
Peu fréquent : sensations vertigineuses.
Fréquence indéterminée : syncope vasovagale en réponse à l'injection, paresthésie.
Affections gastro-intestinales
Fréquent : nausées, diarrhées.
Fréquence indéterminée : vomissements, douleurs abdominales.
Affections de la peau et du tissu sous-cutané
Peu fréquent : prurit, rash.
Fréquence indéterminée : urticaire.
Affections musculo-squelettiques et systémiques
Très fréquent : myalgies.
Fréquent : arthralgies.
Troubles généraux et anomalies au site d'administration
Très fréquent : malaise, asthénie, troubles au site d'injection (douleur, induration, œdème, érythème).
Fréquent : fièvre.
Investigations
Fréquence indéterminée : élévation des transaminases sériques (légère et transitoire).
Les réactions indésirables suivantes n'ont pas été rapportées pendant la commercialisation de TYAVAX mais ont été rapportées après utilisation du vaccin typhoïdique polyosidique Vi monovalent et du vaccin hépatite A inactivé monovalent, respectivement.
Affections respiratoires, thoraciques et médiastinales
Fréquence indéterminée : aggravation d'asthme.
Troubles généraux et anomalies au site d'administration
Très rare : nodule au site d'injection.
Population pédiatrique
Aucune donnée concernant la tolérance de TYAVAX chez l’enfant et l’adolescent de moins de 16 ans n’est disponible.
Déclaration des effets indésirables suspectés
La déclaration des effets indésirables suspectés après autorisation du médicament est importante. Elle permet une surveillance continue du rapport bénéfice/risque du médicament. Les professionnels de santé déclarent tout effet indésirable suspecté via le système national de déclaration : Agence nationale de sécurité du médicament et des produits de santé (ANSM) et réseau des Centres Régionaux de Pharmacovigilance - Site internet : www.signalement-sante.gouv.fr.
5. PROPRIETES PHARMACOLOGIQUES
5.1. Propriétés pharmacodynamiques
Classe pharmacothérapeutique : Vaccins combinés bactériens et viraux, code ATC : J07CA10.
Quatre études cliniques ont fourni des résultats sur la réponse immunitaire avec TYAVAX. Un total de 1090 sujets a été inclus, chaque étude comprenant 179, 610, 243 et 58 sujets vaccinés.
Après primovaccination, le taux de séroprotection vis à vis du virus de l’hépatite A (VHA)
(% ≥ 20 mUI/mL) se situait entre 95,6 % et 99,4 % après 14 jours et entre 98,7 % et 100 % après 28 jours.
Le taux de séroprotection vis à vis de l’antigène Vi (% ≥ 1 µg/mL) se situe entre 83 % et 89 % après 14 jours et entre 69,8 % et 91 % après 28 jours.
Dans une étude, qui a évalué le taux de séroprotection vis à vis de l’antigène Vi, 1, 2 ou 3 ans après la première dose de TYAVAX et après une revaccination avec TYAVAX à 3 ans, les résultats sont les suivants :
|
|
TYAVAX |
|||
|
Première année |
Deuxième année |
Troisième année |
28 jours après revaccination à 3 ans |
|
|
Nombre de vaccinés |
139 |
124 |
112 |
46 |
|
% de vaccinés séroprotégés |
44,6 |
40,3 |
32,1 |
69,6 |
|
(IC 95 %) |
(36,2 – 53,3) |
(31,6 – 49,5) |
(23,6 – 41,6) |
(54,2 – 82,3) |
Les données sérologiques montrent une protection continue contre l'hépatite A jusqu'à 36 mois chez les sujets ayant répondu à la première dose de TYAVAX. Les taux de séroprotection vis à vis du virus de l’hépatite A, 1, 2 ou 3 ans après la première dose de TYAVAX et après une revaccination avec TYAVAX à 3 ans, sont les suivants :
|
|
TYAVAX |
|||
|
Première année |
Deuxième année |
Troisième année |
28 jours après revaccination à 3 ans |
|
|
Nombre de vaccinés |
140 |
124 |
112 |
46 |
|
% ≥ 20 mU/mL |
99,3 |
98,4 |
99,1 |
100 |
|
(IC 95%) |
(96,1 – 100) |
(94,3 – 99,8) |
(95,1 – 100) |
(92,3 – 100) |
Des résultats similaires ont été observés aux mêmes dates dans le groupe contrôle qui a reçu les vaccins monovalents typhoïdique polyosidique Vi et hépatite A inactivé, administrés de façon concomitante.
Lors d’une étude randomisée ouverte, l’immunogénicité de TYAVAX et du vaccin combiné adsorbé diphtérique (dose réduite), tétanique et poliomyélite (inactivé) (dT-Polio), administrés de façon concomitante en deux sites séparés a été comparée à leur administration séparée à des dates différentes chez des adultes sains. Les taux de séroconversion/séroprotection observés 28 jours après la vaccination chez les sujets Per Protocole étaient les suivants :
|
|
Groupe A (Administration concomitante) |
Groupe B (Administration séparée) |
|
|
N=161 |
N=154 |
|
Taux de séroconversion anti-VHA (≥ 20 mUI/mL) n (%) |
139 (100 %)* |
127 (100 %)** |
|
[IC 95 %] |
[97,3 ; 100,0] |
[97,1 ; 100,0] |
|
Taux de séroconversion anti-Vi (augmentation ≥ 4 fois) n (%) |
134 (83,2 %) |
135 (87,7 %) |
|
[IC 95 %] |
[76,7 ; 88,2] |
[81,5 ; 92,0] |
|
Taux de séroconversion anti-D (≥ 0,1 UI/mL) n (%) |
158 (98,1 %) |
149 (96,8 %) |
|
[IC 95 %] |
[94,7 ; 99,4] |
[92,6 ; 98,6] |
|
Taux de séroconversion anti-T (≥ 0,1 UI/mL) n (%) |
161 (100 %) |
154 (100 %) |
|
[IC 95 %] |
[97,7 ; 100,0] |
[97,6 ; 100,0] |
|
Taux de séroconversion anti -Polio 1 {1/dil} (≥ 5) n (%) |
161 (100 %) |
154 (100 %) |
|
[IC 95 %] |
[97,7 ; 100,0] |
[97,6 ; 100,0] |
|
Taux de séroconversion anti -Polio 2 {1/dil} (≥ 5) n (%) |
161 (100 %) |
154 (100 %) |
|
[IC 95 %] |
[97,7 ; 100,0] |
[97,6 ; 100,0] |
|
Taux de séroconversion anti -Polio 3 {1/dil} (≥ 5) n (%) |
161 (100 %) |
154 (100 %) |
|
[IC 95 %] |
[97,7 ; 100,0] |
[97,6 ; 100,0] |
* N=139 (sujets initialement séronégatifs vis à vis du VHA)
** N=127 (sujets initialement séronégatifs vis à vis du VHA)
La non infériorité de l’administration concomitante des vaccins TYAVAX et dT-Polio comparée à l’administration séparée a été démontrée pour toutes les valences, à l’exception de la valence Vi.
Pour la valence Vi, les taux de séroprotection (titres anti-Vi ≥ 1 μg/mL) ont augmenté de 7,5 % dans le groupe A et de 7,1 % dans le groupe B à J0, à 86,3 % et 94,8 % respectivement 28 jours après la vaccination. Chez les sujets non séroprotégés initialement (titres anti-Vi < 1 μg/mL), les taux de séroconversion observés 28 jours après la vaccination étaient les suivants :
|
|
Groupe A (Administration concomitante) |
Groupe B (Administration séparée) |
|
|
N=149 |
N=143 |
|
Taux de séroconversion anti-Vi (augmentation ≥ 4 fois) n (%) |
132 (88,6 %) |
128 (89,5 %) |
|
[IC 95 %] |
[82,5 ; 92,8] |
[83,4 ; 93,5] |
Chez les sujets non séroprotégés initialement, le taux de séroconversion anti-Vi (augmentation ≥ 4 fois) lors de l’administration concomitante des vaccins était non inférieur à celui de l’administration séparée.
Population pédiatrique
Aucune donnée concernant l’efficacité de TYAVAX chez l’enfant et l’adolescent de moins de 16 ans n’est disponible.
5.2. Propriétés pharmacocinétiques
5.3. Données de sécurité préclinique
Composants du vaccin de l’hépatite A inactivé :
2-phénoxyéthanol
Ethanol
Formaldéhyde
Milieu 199 Hanks (sans rouge de phénol)* supplémenté avec du polysorbate 80.
* Le milieu 199 Hanks (sans rouge de phénol) est un mélange complexe d’acides aminés (incluant de la phénylalanine), des sels minéraux (incluant le potassium), des vitamines et autres composants (comme le glucose), dilués dans de l’eau pour préparations injectables et avec un pH ajusté avec de l’acide chlorhydrique ou de l’hydroxyde de sodium.
Composants du vaccin typhoïdique polyosidique Vi
Solution tampon phosphate :
Chlorure de sodium
Phosphate disodique dihydraté
Phosphate monosodique dihydraté
Eau pour préparations injectables
3 ans
6.4. Précautions particulières de conservation
A conserver au réfrigérateur (entre 2°C et 8°C). Ne pas congeler.
A conserver dans l'emballage extérieur, à l'abri de la lumière.
6.5. Nature et contenu de l'emballage extérieur
Une seringue préremplie à double compartiment (verre de type I) : 0,5 mL de suspension dans le compartiment le plus proche du piston et 0,5 mL de solution dans le compartiment le plus proche de l'aiguille, avec un bouchon-piston en élastomère (mélange de caoutchouc chlorobutyle et bromobutyle), un protège-embout (élastomère) et un bouchon by-pass (élastomère).
Boîte de 1 ou 10 seringues préremplies fournies avec ou sans aiguille.
Toutes les présentations peuvent ne pas être commercialisées.
6.6. Précautions particulières d’élimination et de manipulation
Les deux vaccins monovalents doivent être mélangés immédiatement avant l’injection.
Agiter avant le mélange et de nouveau avant l'injection afin d'obtenir une suspension homogène. Le contenu des 2 compartiments est mélangé en poussant doucement le piston. Le volume final à injecter est de 1 millilitre.
Le vaccin doit être inspecté visuellement avant administration pour vérifier l'absence de particules étrangères.
Le vaccin mélangé est une suspension opalescente blanchâtre. Le vaccin ne doit pas être utilisé en cas de particules étrangères.
Tout médicament non utilisé ou déchet doit être éliminé conformément à la réglementation en vigueur.
7. TITULAIRE DE L’AUTORISATION DE MISE SUR LE MARCHE
14 ESPACE HENRY VALLÉE
69007 LYON
FRANCE
8. NUMERO(S) D’AUTORISATION DE MISE SUR LE MARCHE
· 34009 360 780 8 6: 0,5 mL de suspension + 0,5 mL de solution en seringue préremplie à double compartiment (verre de type I) avec une aiguille, un bouchon-piston en élastomère (mélange de caoutchouc chlorobutyle et bromobutyle), avec un protège-embout (élastomère) et un bouchon by-pass (élastomère) - Boîte de 10.
· 34009 360 781 4 7: 0,5 mL de suspension + 0,5 mL de solution en seringue préremplie à double compartiment (verre de type I) sans aiguille, avec un bouchon-piston en élastomère (mélange de caoutchouc chlorobutyle et bromobutyle), un protège-embout (élastomère) et un bouchon by-pass (élastomère) - Boîte de 1.
· 34009 360 782 0 8: 0,5 mL de suspension + 0,5 mL de solution en seringue préremplie à double compartiment (verre de type I) sans aiguille, avec un bouchon-piston en élastomère (mélange de caoutchouc chlorobutyle et bromobutyle), un protège-embout (élastomère) et un bouchon by-pass (élastomère) - Boîte de 10.
9. DATE DE PREMIERE AUTORISATION/DE RENOUVELLEMENT DE L’AUTORISATION
[à compléter ultérieurement par le titulaire]
10. DATE DE MISE A JOUR DU TEXTE
[à compléter ultérieurement par le titulaire]
Sans objet.
12. INSTRUCTIONS POUR LA PREPARATION DES RADIOPHARMACEUTIQUES
Liste I
ANSM - Mis à jour le : 08/02/2021
TYAVAX, suspension et solution pour suspension injectable en seringue préremplie
Vaccin de l'hépatite A (inactivé, adsorbé) et typhoïdique polyosidique
· Gardez cette notice. Vous pourriez avoir besoin de la relire.
· Si vous avez d’autres questions, interrogez votre médecin, votre pharmacien ou votre infirmier/ère.
· Ce médicament vous a été personnellement prescrit. Ne le donnez pas à d’autres personnes.
· Si vous ressentez un quelconque effet indésirable, parlez-en à votre médecin, ou votre pharmacien ou votre infirmier/ère. Ceci s’applique aussi à tout effet indésirable qui ne serait pas mentionné dans cette notice. Voir rubrique 4.
2. Quelles sont les informations à connaître avant d'utiliser TYAVAX, suspension et solution pour suspension injectable en seringue préremplie ?
3. Comment utiliser TYAVAX, suspension et solution pour suspension injectable en seringue préremplie ?
4. Quels sont les effets indésirables éventuels ?
5. Comment conserver TYAVAX, suspension et solution pour suspension injectable en seringue préremplie ?
6. Contenu de l’emballage et autres informations.
1. QU’EST-CE QUE TYAVAX, suspension et solution pour suspension injectable en seringue préremplie ET DANS QUELS CAS EST-IL UTILISE ?
Classe pharmacothérapeutique : Vaccins combinés bactériens et viraux - code ATC : J07CA10.
La fièvre typhoïde est une maladie infectieuse qui peut être transmise par des aliments ou boissons contenant les bactéries (appelées Salmonella enterica, sérotype typhi) qui causent la maladie. Il s'agit d'une infection grave qui peut être fatale si elle n'est pas rapidement traitée.
L'infection par l'hépatite A est due à un virus qui attaque le foie. Elle peut être transmise par des aliments ou boissons contenant le virus. La jaunisse et une sensation de malaise généralisé font partie des symptômes.
Quand vous recevez une injection de TYAVAX, les défenses naturelles de votre corps élaborent une protection contre la fièvre typhoïde et l'infection causée par le virus de l'hépatite A.
2. QUELLES SONT LES INFORMATIONS A CONNAITRE AVANT D’UTILISER TYAVAX, suspension et solution pour suspension injectable en seringue préremplie ?
N’utilisez jamais TYAVAX, suspension et solution pour suspension injectable en seringue préremplie :
· si vous êtes allergique à la (aux) substance(s) active(s) ou à l’un des autres composants contenus dans ce médicament, mentionnés dans la rubrique 6.
· si vous êtes allergique à la néomycine (antibiotique utilisé lors de la fabrication du vaccin et pouvant être présent dans celui-ci en petites quantités).
· si vous avez une maladie avec une température élevée. La vaccination doit être différée après la guérison.
Avertissements et précautions
Adressez-vous à votre médecin, pharmacien ou votre infirmier/ère avant d’utiliser TYAVAX.
· Si vous avez une faible réponse immunitaire parce que vous avez pris ou vous prenez un traitement susceptible d'affaiblir votre système immunitaire, comme les corticoïdes, les médicaments cytotoxiques ou la radiothérapie, votre médecin souhaitera peut-être attendre la fin du traitement.
· Si votre système immunitaire est déficient à cause d'une infection par le virus de l'immunodéficience humaine (VIH), il sera possible de vous administrer TYAVAX, mais le vaccin peut ne pas vous protéger aussi bien qu'il protège les personnes dont le système immunitaire fonctionne normalement.
· Ce vaccin ne protègera pas contre d'autres virus connus pour infecter le foie (comme les virus de l'hépatite B, l'hépatite C ou l'hépatite E). De même, si vous êtes déjà infecté(e) par le virus de l'hépatite A lors de l'administration de TYAVAX, la vaccination peut ne pas fonctionner correctement.
· Ce vaccin ne protègera pas contre les infections causées par des bactéries Salmonella autres que le type particulier responsable de la fièvre typhoïde.
· Ce vaccin ne peut pas causer les infections contre lesquelles il protège.
· Comme avec tous les vaccins, les personnes recevant TYAVAX ne seront pas toutes protégées de façon certaine contre l'hépatite A et la fièvre typhoïde.
· Un évanouissement peut survenir (surtout chez les adolescents) après, voire avant, toute injection avec une aiguille. Aussi, parlez-en à votre médecin ou infirmier(ère) si vous ou votre enfant vous êtes évanoui lors d’une précédente injection
Enfants et adolescents
Un évanouissement peut survenir (surtout chez les adolescents) après, voire avant, toute injection avec une aiguille. Aussi, parlez-en à votre médecin ou infirmier(ère) si vous ou votre enfant vous êtes évanoui lors d’une précédente injection.
Autres médicaments et TYAVAX, suspension et solution pour suspension injectable en seringue préremplie
Informez votre médecin ou pharmacien si vous utilisez, avez récemment utilisé ou pourriez utiliser tout autre médicament.
Comme TYAVAX ne contient aucune bactérie ou aucun virus vivant, il peut, en général, être administré en même temps que d'autres vaccins, mais en un site d'injection différent (une autre partie du corps, comme l'autre bras ou une jambe). TYAVAX ne doit pas être mélangé à un autre vaccin dans une même seringue.
La protection obtenue en administrant TYAVAX en même temps que des immunoglobulines (anticorps obtenus à partir de dons du sang) n'a pas été évaluée. Si vous avez besoin d'une injection d'immunoglobulines, elle peut être administrée en même temps que TYAVAX ou quelques semaines après. Cependant il se peut que vous ne produisiez pas autant d'anticorps contre le virus de l'hépatite A qu'en temps normal, mais il est probable que vous soyez toutefois protégé(e) contre l'infection.
TYAVAX, suspension et solution pour suspension injectable en seringue préremplie avec des aliments, boissons et de l’alcool
Sans objet.
Grossesse, allaitement et fertilité
Si vous êtes enceinte ou que vous allaitez, si vous pensez être enceinte ou planifiez une grossesse, demandez conseil à votre médecin ou pharmacien avant de prendre ce médicament.
Bien que TYAVAX ne soit pas supposé nuire au futur bébé, votre médecin ou votre infirmier/ère décidera si vous devez être vaccinée tout de suite ou après la naissance du bébé.
Conduite de véhicules et utilisation de machines
Ce vaccin a une influence mineure sur l'aptitude à conduire des véhicules et à utiliser des machines.
Des cas de vertige ayant été rapportés chez certaines personnes (moins d'1 sur 100 mais plus d'1 sur 1000) après administration de TYAVAX, il convient donc d'être prudent si vous conduisez un véhicule ou utilisez une machine.
TYAVAX contient de la phénylalanine, de l’éthanol, du potassium et du sodium
TYAVAX contient 10 microgrammes de phénylalanine par dose de 1 mL équivalent à 0,17 microgrammes/kg chez une personne de 60 kg. La phénylalanine peut être dangereuse pour les personnes atteintes de phénylcétonurie (PCU), une maladie génétique rare caractérisée par l’accumulation de la phénylalanine ne pouvant être éliminée correctement.
TYAVAX contient 2 mg d’alcool (éthanol) par dose de 1 mL. La faible quantité d’alcool contenue dans ce médicament n’est pas susceptible d’entraîner d’effet notable.
TYAVAX contient moins de 1 mmol (39 mg) de potassium et moins de 1 mmol (23 mg) de sodium par dose, c’est-à-dire qu’il est essentiellement « sans potassium » et « sans sodium ».
3. COMMENT UTILISER TYAVAX, suspension et solution pour suspension injectable en seringue préremplie ?
Veillez à toujours utiliser ce médicament en suivant exactement les instructions de cette notice ou les indications de votre médecin, pharmacien ou infirmier/ère. Vérifiez auprès de votre médecin, pharmacien ou infirmier/ère en cas de doute.
Posologie
La dose recommandée est d'un millilitre de vaccin mélangé, aux personnes âgées de 16 ans et plus. La protection initiale est atteinte avec une seule dose de vaccin.
Ce vaccin commencera à vous protéger contre l'hépatite A environ 14 jours après la première dose. Vous aurez besoin d'une deuxième injection (rappel) de vaccin hépatite A inactivé pour obtenir une protection à long terme. Ce rappel vous protégera contre l'hépatite A au-delà de dix ans. La dose de rappel devra être administrée dans les 36 mois et de préférence dans les 6 à 12 mois après la première dose.
Ce vaccin peut vous être administré pour renforcer votre immunité vis-à-vis de l'hépatite A si vous avez déjà reçu une première dose de vaccin hépatite A inactivé 6 à 36 mois plus tôt, à condition que vous ayez également besoin d'une protection contre la fièvre typhoïde. Cependant, si la première dose de vaccin hépatite A a été administrée sous forme d'un vaccin typhoïdique et hépatite A combinés, alors la deuxième dose de vaccin combiné est habituellement administrée environ 36 mois après la première dose.
Ce vaccin commencera à vous protéger contre la fièvre typhoïde environ 14 jours après l'injection, et la protection peut durer environ 3 ans. Si, après 3 ans, il existe toujours un risque que vous contractiez la fièvre typhoïde, vous devez faire en sorte de recevoir une autre injection de vaccin typhoïdique polyosidique Vi.
Les liquides des deux compartiments doivent être mélangés dans la seringue juste avant l'injection. Une fois les liquides mélangés, votre médecin ou votre infirmier/ère agitera la seringue et s'assurera que le liquide est une suspension trouble blanchâtre et qu'aucune particule étrangère ne s'y trouve.
Mode et voie d'administration
Ce vaccin vous sera administré en injection lente dans un muscle (voie intramusculaire (IM)) de la partie supérieure externe de votre bras. Votre médecin ou votre infirmier/ère évitera de vous injecter le vaccin dans la peau ou dans un vaisseau sanguin. Ce vaccin ne doit pas être administré dans la fesse.
Si vous souffrez d'hémophilie (maladie au cours de laquelle vous avez des bleus ou saignez facilement) ou de toute autre maladie qui implique que vous ne devez pas recevoir une injection dans le muscle, celle-ci peut se faire sous la peau.
Si vous avez utilisé plus de TYAVAX, suspension et solution pour suspension injectable en seringue préremplie que vous n’auriez dû
Consultez immédiatement votre médecin ou votre pharmacien.
Dans certains cas, il a été utilisé plus que la dose recommandée. Dans ces cas, lorsque des effets indésirables ont été rapportés, ils étaient de même nature que ceux décrits dans la rubrique 4.
Si vous oubliez d’utiliser TYAVAX, suspension et solution pour suspension injectable en seringue préremplie
Si vous arrêtez d’utiliser TYAVAX, suspension et solution pour suspension injectable en seringue préremplie
Sans objet.
Si vous avez d’autres questions sur l’utilisation de ce médicament, demandez plus d’informations à votre médecin, à votre pharmacien ou à votre infirmier/ère.
4. QUELS SONT LES EFFETS INDESIRABLES EVENTUELS ?
Des réactions allergiques graves ont été rapportées :
- Réaction allergique sévère (anaphylaxie), pouvant inclure un ou plusieurs des symptômes suivants :
· urticaire, éruptions cutanées,
· gonflement du visage et/ou du cou, difficulté à respirer, coloration bleue de la langue ou des lèvres,
· tension artérielle basse, fréquence cardiaque rapide et pouls faible, refroidissement de la peau, sensations vertigineuses et potentiellement collapsus.
Quand ces signes ou symptômes apparaissent, c'est en général très rapidement après l'injection alors que la personne atteinte se trouve encore à la clinique ou au cabinet du médecin.
Si un de ces symptômes apparait après que vous ayez quitté le lieu où l'injection vous a été administrée, vous devez IMMEDIATEMENT consulter un médecin.
- Maladie sérique :
· douleurs articulaires, éruptions cutanées, ganglions lymphatiques augmentés et sensation de malaise général.
Quand ces symptômes apparaissent, c'est en général 2-4 semaines après avoir reçu le vaccin.
Si ces symptômes apparaissent vous devez consulter un médecin aussi vite que possible.
Réactions très fréquentes (pouvant toucher plus d'1 personne sur 10)
· douleur dans la zone où le vaccin a été injecté, persistant quelquefois plus de trois jours. La douleur peut être sévère chez jusqu'à 1 personne sur 10 (fréquent),
· rougeur, gonflement et durcissement de la zone où le vaccin a été injecté. Le gonflement et le durcissement peuvent être sévères chez jusqu'à 1 personne sur 10 (fréquent),
· maux de tête,
· sensation de faiblesse,
· sensation de malaise général,
· douleurs musculaires.
Réactions fréquentes (pouvant toucher jusqu'à 1 personne sur 10)
· nausées,
· diarrhée,
· douleurs articulaires,
· fièvre (température élevée).
Réactions peu fréquentes (pouvant toucher jusqu'à 1 personne sur 100)
· démangeaisons de la peau,
· éruptions,
· sensations vertigineuses.
Réactions très rares (pouvant toucher jusqu'à 1 personne sur 10 000)
· bosse au site d'injection.
Réactions de fréquence indéterminée (la fréquence ne peut être estimée sur la base des données disponibles)
· aggravation de l’asthme chez les personnes qui en souffrent,
· évanouissement en réponse à l'injection,
· sensation d’engourdissement ou de picotements de la peau,
· éruptions qui sont parfois gonflées et qui démangent,
· vomissements, douleurs à l’estomac,
· modifications des analyses sanguines mesurant l’activité du foie.
Déclaration des effets secondaires
Si vous ressentez un quelconque effet indésirable, parlez-en à votre médecin, votre pharmacien ou à votre infirmier/ère. Ceci s’applique aussi à tout effet indésirable qui ne serait pas mentionné dans cette notice. Vous pouvez également déclarer les effets indésirables directement via le système national de déclaration : Agence nationale de sécurité du médicament et des produits de santé (ANSM) et réseau des Centres Régionaux de Pharmacovigilance - Site internet: www.signalement-sante.gouv.fr
En signalant les effets indésirables, vous contribuez à fournir davantage d’informations sur la sécurité du médicament.
5. COMMENT CONSERVER TYAVAX, suspension et solution pour suspension injectable en seringue préremplie ?
Tenir ce médicament hors de la vue et de la portée des enfants.
N'utilisez pas ce vaccin après la date de péremption indiquée sur la boîte et sur l'étiquette de la seringue après EXP. La date de péremption fait référence au dernier jour de ce mois.
A conserver au réfrigérateur (entre 2°C et 8°C). Ne pas congeler. Conserver le vaccin dans l'emballage extérieur à l'abri de la lumière.
N’utilisez pas ce médicament si vous remarquez qu'il contient des particules étrangères.
Ne jetez aucun médicament au tout-à-l’égout ou avec les ordures ménagères. Demandez à votre pharmacien d’éliminer les médicaments que vous n’utilisez plus. Ces mesures contribueront à protéger l’environnement.
6. CONTENU DE L’EMBALLAGE ET AUTRES INFORMATIONS
Ce que contient TYAVAX, suspension et solution pour suspension injectable en seringue préremplie
· Les substances actives sont :
Virus de l'hépatite A, souche GBM1 (inactivé) 2............................................................................................................. 160 unités antigène
1 produit sur cellules diploïdes humaines MRC-5
2 adsorbé sur de l'hydroxyde d'aluminium hydraté (0,3 milligrammes d'Al)
L'hydroxyde d'aluminium est inclus dans le vaccin en tant qu'adsorbant. Les adsorbants sont des substances incluses dans certains vaccins pour accélérer, améliorer et/ou prolonger l'effet protecteur de ce vaccin.
Polyosides capsulaires Vi de Salmonella typhi (souche Ty2).................................... 25 microgrammes
· Les autres composants sont : chlorure de sodium, phosphate disodique dihydraté, phosphate monosodique dihydraté, 2-phénoxyéthanol, éthanol, formaldéhyde, milieu 199 Hanks sans rouge de phénol (mélange d'acides aminés comprenant phénylalanine (voir rubrique 2), sels minéraux (incluant le potassium), vitamines et autres composants) supplémenté avec du polysorbate 80, et eau pour préparations injectables.
Toutes les présentations ne sont pas commercialisées.
Le vaccin hépatite A inactivé est une suspension blanche, trouble, et le vaccin typhoïdique polyosidique est une solution claire, incolore.
Titulaire de l’autorisation de mise sur le marché
14 ESPACE HENRY VALLÉE
69007 LYON
FRANCE
Exploitant de l’autorisation de mise sur le marché
14 ESPACE HENRY VALLÉE
69007 LYON
FRANCE
14 ESPACE HENRY VALLÉE
69007 LYON
FRANCE
Noms du médicament dans les Etats membres de l'Espace Economique Européen
Ce médicament est autorisé dans les Etats membres de l'Espace Economique Européen sous les noms suivants : Conformément à la réglementation en vigueur.
[à compléter ultérieurement par le titulaire]
La dernière date à laquelle cette notice a été révisée est :
[à compléter ultérieurement par le titulaire]
Des informations détaillées sur ce médicament sont disponibles sur le site Internet de l’ANSM (France).
Les informations suivantes sont destinées exclusivement aux professionnels de santé:
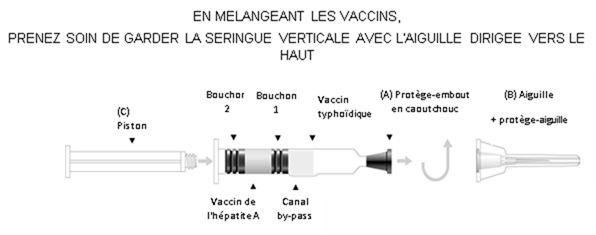
Instructions d'utilisation – seringue à double compartiment (Voir le schéma ci-après)
TYAVAX, suspension et solution pour suspension injectable en seringue préremplie
Vaccin de l'hépatite A (inactivé, adsorbé) et typhoïdique polyosidique
1. Enlever le protège embout (A).
2. Fixer l'aiguille et le protège-aiguille (B) à la seringue.
3. Visser le piston (C) dans le bouchon piston (Bouchon 2).
4. Agiter la seringue ; puis mélanger les composants du vaccin en poussant lentement le piston, en gardant l'aiguille vers le haut. Le vaccin contenu dans la chambre inférieure passe dans la chambre supérieure par le canal by-pass.
5. Agiter vigoureusement jusqu'à obtenir une suspension homogène.
6. En tenant le protège-aiguille par l'extrémité, l'enlever en tirant vers le haut sans tourner.
7. Procéder immédiatement à l'injection. Un test de retour veineux peut être réalisé en tirant légèrement sur le piston. Les bouchons peuvent se séparer mais assurez-vous que le bouchon 2 n'atteigne pas le canal by pass afin d'éviter toute perte de liquide. Si un vaisseau sanguin a été pénétré, du sang sera aspiré dans la seringue.
|
|
Voir rubrique 3. Comment utiliser TYAVAX, suspension et solution pour suspension injectable en seringue préremplie.