Dernière mise à jour le 01/06/2026
SOOLANTRA 10 mg/g, crème
Présentations
> 1 tube(s) polyéthylène aluminium avec fermeture de sécurité enfant de 30 g
Code CIP : 34009 300 241 1 9
Déclaration de commercialisation : 12/02/2018
Cette présentation n'est pas agréée aux collectivités
Service médical rendu (SMR)
Pas de SMR disponible pour ce médicament ( plus d'informations dans l'aide )
Amélioration du service médical rendu (ASMR)
Pas d'ASMR disponible pour ce médicament (plus d'informations dans l'aide )
ANSM - Mis à jour le : 04/08/2020
2. COMPOSITION QUALITATIVE ET QUANTITATIVE
Un gramme de crème contient 10 mg d’ivermectine.
Excipients à effet notoire : 1 gramme de crème contient 35 mg d’alcool cétylique, 25 mg d’alcool stéarylique, 2 mg de parahydroxybenzoate de méthyle (E218), 1 mg de parahydroxybenzoate de propyle (E216) et 20 mg de propylène glycol.
Pour la liste complète des excipients, voir rubrique 6.1.
Crème hydrophile blanche à jaune pâle.
4.1. Indications thérapeutiques
4.2. Posologie et mode d'administration
Une application par jour, jusqu'à 4 mois. Soolantra doit être appliqué quotidiennement tout au long du traitement. Le traitement peut être répété. Il peut être utilisé dans le cadre d’une monothérapie ou être associé à un ou plusieurs traitements (voir rubrique 5.1).
En cas d'absence d'amélioration après 3 mois, le traitement doit être interrompu.
Populations particulières
Insuffisant rénal
Aucun ajustement posologique n’est nécessaire.
Insuffisant hépatique
La prudence s’impose chez les patients présentant une insuffisance hépatique sévère.
Personnes âgées
Aucun ajustement posologique n’est nécessaire chez les personnes âgées (voir également rubrique 4.8).
Population pédiatrique
La sécurité et l’efficacité de Soolantra chez les enfants et adolescents âgés moins de 18 ans n’ont pas été établies. Aucune donnée n’est disponible.
Mode d’administration
Uniquement voie cutanée.
Application cutanée d'une quantité de médicament équivalente à un petit pois sur chacune des cinq zones du visage (à répartir sur le front, le menton, le nez et les deux joues). Le médicament doit être réparti en couche mince sur l'ensemble du visage, en évitant les yeux, les lèvres et les muqueuses.
Soolantra doit être appliqué uniquement sur le visage.
Les mains doivent être lavées après l'application du médicament.
Des cosmétiques peuvent être appliqués après que Soolantra ait séché.
Hypersensibilité à la substance active ou à l’un des excipients mentionnés à la rubrique 6.1.
4.4. Mises en garde spéciales et précautions d'emploi
En cas d'aggravation sévère accompagnée d'une forte réaction cutanée, le traitement doit être interrompu.
Soolantra n'a pas été étudié chez les patients présentant une insuffisance rénale ou hépatique.
Le médicament contient :
de l'alcool cétylique et de l'alcool stéarylique qui peuvent provoquer des réactions cutanées locales (par exemple, dermite de contact),
du parahydroxybenzoate de méthyle (E218) et du parahydroxybenzoate de propyle (E216) qui peuvent provoquer des réactions allergiques (éventuellement retardées),
du propylène glycol qui peut provoquer une irritation cutanée.
4.5. Interactions avec d'autres médicaments et autres formes d'interactions
Aucune étude d’interaction n’a été réalisée (voir rubrique 5.2 Biotransformation).
Des études in vitro ont montré que l'ivermectine est principalement métabolisée par le CYP3A4. Par conséquent, la prudence est recommandée quand l'ivermectine est administrée de manière concomitante avec des puissants inhibiteurs du CYP3A4 car l'exposition plasmatique peut être significativement augmentée.
4.6. Fertilité, grossesse et allaitement
Grossesse
Des études de toxicité par voie orale sur la reproduction ont montré que l'ivermectine est tératogène chez le rat et le lapin (voir rubrique 5.3). Cependant en raison de la faible exposition systémique après administration topique du produit à la posologie proposée, il y a un faible risque pour un fœtus humain. Soolantra n’est pas recommandé pendant la grossesse.
Allaitement
Après administration orale, l'ivermectine est excrétée dans le lait maternel en faibles concentrations. L'excrétion dans le lait maternel après l'administration topique n'a pas été évaluée. Les données pharmacocinétiques / toxicologiques disponibles chez l'animal ont mis en évidence l’excrétion de l'ivermectine dans le lait. Un risque pour les enfants allaités ne peut être exclu. Une décision doit être prise soit d'interrompre l'allaitement soit d'interrompre / de s’abstenir du traitement par Soolantra en tenant compte du bénéfice de l’allaitement pour l'enfant et de celui du traitement pour la mère.
Fertilité
Aucune donnée humaine n’est disponible concernant l'effet de l'ivermectine sur la fertilité. Chez le rat, le traitement par ivermectine n’avait aucun effet sur l'accouplement ou la fertilité.
4.7. Effets sur l'aptitude à conduire des véhicules et à utiliser des machines
Les effets indésirables les plus fréquemment rapportés sont les suivants : sensation de brûlure cutanée, irritation cutanée, prurit et sécheresse cutanée, survenant chacun chez 1 % ou moins des patients traités avec Soolantra dans les études cliniques.
Ils sont généralement d’intensité légère à modérée, et normalement diminuent lorsque le traitement est poursuivi.
Aucune différence significative des profils de sécurité n’a été observée entre les sujets de 18 à 65 ans et les sujets de 65 ans et plus.
Liste des effets indésirables sous forme de tableau
Les effets indésirables sont répertoriés par classe de systèmes d'organes et par fréquence en utilisant la convention suivante : très fréquent (≥ 1/10), fréquent (≥ 1/100, < 1/10), peu fréquent (≥ 1/1 000, < 1/100), rare (≥ 1/10 000, < 1/1 000), très rare (< 1/10 000), fréquence indéterminée (ne peut être estimée sur la base des données disponibles), et ont été rapportés avec l’utilisation de Soolantra dans les études cliniques (voir Tableau 1).
Tableau 1 – Effets indésirables
|
Classe de systèmes d’organes |
Fréquence |
Effets indésirables |
|
Affections de la peau et du tissu sous‑cutané |
Fréquent |
Sensation de brûlure cutanée |
|
Peu fréquent |
irritation cutanée, prurit, sécheresse cutanée, aggravation de la rosacée* |
|
|
fréquence indéterminée |
Erythème, Dermite de contact (allergique ou irritante), Gonflement du visage |
|
|
Investigations |
Fréquence indéterminée |
Augmentation des transaminases* |
* Effet indésirable rapporté après la mise sur le marché
Déclaration des effets indésirables suspectés
La déclaration des effets indésirables suspectés après autorisation du médicament est importante. Elle permet une surveillance continue du rapport bénéfice/risque du médicament. Les professionnels de santé déclarent tout effet indésirable suspecté via le système national de déclaration : Agence nationale de sécurité du médicament et des produits de santé (ANSM) et réseau des Centres Régionaux de Pharmacovigilance - Site internet : www.signalement-sante.gouv.fr.
Il n’y a aucun surdosage rapporté avec Soolantra.
Chez l'Homme, lors d’exposition accidentelle ou significative à des quantités inconnues de préparations vétérinaires d'ivermectine, par ingestion, inhalation, injection, ou exposition des surfaces du corps, les effets indésirables suivants ont été rapportés le plus fréquemment : éruption cutanée, œdème, maux de tête, vertiges, asthénie, nausées, vomissements et diarrhée. D'autres effets indésirables ont été rapportés dont : convulsion, ataxie, dyspnée, douleurs abdominales, paresthésies, urticaire et dermite de contact.
En cas d'ingestion accidentelle, un traitement symptomatique, pourrait inclure : équilibration hydro-électrolytique parentérale, assistance respiratoire (oxygène et ventilation mécanique si nécessaire) et agents hypertensifs en cas d'hypotension cliniquement significative. Des vomissements provoqués et / ou lavage gastrique dès que possible, suivis par des laxatifs et d’autres mesures habituelles anti-poison peuvent être indiqués pour empêcher l'absorption de la substance ingérée, si nécessaire.
5. PROPRIETES PHARMACOLOGIQUES
5.1. Propriétés pharmacodynamiques
Mécanisme d’action
L'ivermectine appartient à la classe des avermectines. Les avermectines ont des effets anti-inflammatoires par inhibition de la production de cytokines inflammatoires induites par le lipopolysaccharide. Les propriétés anti-inflammatoires de l'ivermectine par voie cutanée ont été observées dans des modèles animaux de l'inflammation de la peau. L’ivermectine provoque aussi la mort des parasites, principalement par liaison sélective aux canaux chlorure glutamate-dépendants avec une affinité importante, qui se produit dans les cellules nerveuses et musculaires des invertébrés. Le mécanisme d'action de Soolantra dans le traitement des lésions inflammatoires de la rosacée n’est pas connu mais peut être lié à l’activité anti-inflammatoire de l'ivermectine, ainsi qu’en provoquant la mort des acariens Demodex, discuté comme étant un facteur important dans l'inflammation de la peau au cours de la rosacée.
Efficacité et sécurité clinique
Soolantra appliqué une fois par jour au coucher a été évalué dans le traitement des lésions inflammatoires de la rosacée dans deux études cliniques de même méthodologie, randomisées, en double aveugle, contrôlées versus véhicule. Ces études ont été menées sur 1 371 sujets âgés de 18 ans et plus qui ont été traités une fois par jour pendant 12 semaines avec Soolantra ou le véhicule.
Parmi les sujets, 96 % étaient de type caucasien et 67 % étaient des femmes. D’après l'échelle à 5 niveaux, IGA (Investigator Global Assessment/Evaluation Globale par l’Investigateur), 79 % des sujets ont présenté un score modéré (IGA = 3) et 21 % un score sévère (IGA = 4) à l'inclusion.
Les 2 critères principaux d’efficacité des 2 études cliniques étaient le taux de succès défini par le résultat IGA (% de sujets «guéri» et « presque guéri» à la semaine 12 de l'étude) et la différence absolue du nombre de lésions inflammatoires par rapport à l’inclusion. L'échelle IGA est basée sur les définitions suivantes :
Tableau 2: Echelle Investigator Global Assessment (IGA)
|
Grade |
Score |
Description clinique |
|
Guéri |
0 |
Pas de lésions inflammatoires, pas d’érythème. |
|
Presque guéri |
1 |
Très peu de petites papules/pustules, érythème très léger. |
|
Léger |
2 |
Quelques petites papules/pustules, érythème léger. |
|
Modéré |
3 |
Plusieurs petites ou grandes papules/pustules, érythème modéré. |
|
Sévère |
4 |
Nombreuses petites ou grandes papules/pustules, érythème sévère. |
Les résultats des deux études cliniques ont démontré que Soolantra appliqué une fois par jour pendant 12 semaines était significativement supérieur à son véhicule en termes de taux de succès du score IGA et de différence absolue du nombre de lésions inflammatoires (p < 0,001, voir tableau 3 et figure 1, figure 2, figure 3 et figure 4).
Les tableaux et figures suivants présentent l'efficacité des résultats des deux études.
Tableau 3: Résultats d’efficacité
|
|
Etude 1 |
Etude 2 |
||
|
Soolantra (N=451) |
Véhicule (N=232) |
Soolantra (N=459) |
Véhicule (N=229) |
|
|
Investigator Global Assessment |
|
|
|
|
|
Nombre (%) de sujets guéris ou Presque guéris selon l’IGA à la semaine 12 |
173 (38,4) |
27 (11,6) |
184 (40,1) |
43 (18,8) |
|
Lésions inflammatoires |
|
|
|
|
|
Nombre moyen de lésions inflammatoires à l’inclusion |
31,0 |
30,5 |
33,3 |
32,2 |
|
Nombre moyen de lésions inflammatoires à la semaine 12 |
10,6 |
18,5 |
11,0 |
18,8 |
|
Différence absolue moyenne (% de différence) du nombre de lésions inflammatoires entre l’inclusion et la semaine 12 |
-20,5 (-64,9) |
-12,0 (-41,6) |
-22,2 (-65,7) |
-13,4 (-43,4) |
Figures 1 et 2: Taux de succès selon l’IGA au cours du temps, en semaine
Etude 1 Etude 2
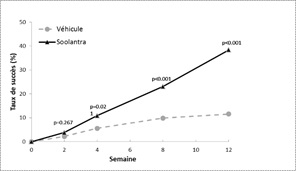
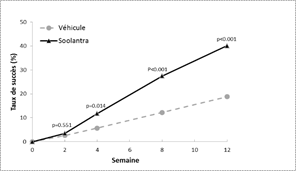
Figures 3 et 4: Différence absolue moyenne du nombre de lésions inflammatoires depuis l’inclusion au cours du temps, en semaine
Etude 1 Etude 2
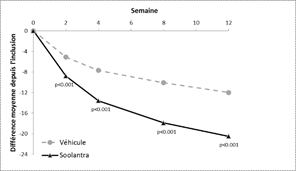
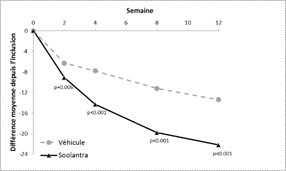
Soolantra était significativement supérieur à son véhicule pour les 2 critères principaux d’efficacité avec un début d'efficacité dès 4 semaines de traitement (p < 0,05).
L’IGA a été évaluée lors de l'extension de 40 semaines des deux études cliniques et les pourcentages de sujets traités par Soolantra atteignant un score IGA de 0 ou 1 a continué à augmenter jusqu'à la semaine 52. Le taux de succès (IGA = 0 ou 1) à la semaine 52 était de 71 % et 76 % dans les études 1 et 2, respectivement.
L'efficacité et la tolérance du médicament dans le traitement des lésions inflammatoires de la rosacée ont également été évaluées dans une étude clinique randomisée, investigateur aveugle, contrôlée versus comparateur actif. L'étude a été menée sur 962 sujets âgés de 18 ans et plus qui ont été traités pendant 16 semaines avec Soolantra une fois par jour ou une crème de métronidazole dosée à 7,5 mg/g, deux fois par jour. Dans cette étude, 99,7 % des sujets étaient de type caucasien et 65,2 % étaient des femmes ; sur l'échelle IGA, à l’inclusion 83,3 % des sujets avait un score modéré (IGA = 3) et 16,7 % un score sévère (IGA = 4) (voir figure 5).
Les résultats de cette étude ont montré que Soolantra était statistiquement supérieur à la crème de métronidazole dosée à 7,5 mg/g sur le critère principal d'efficacité (différence moyenne du pourcentage de lésions inflammatoires) avec une réduction de 83,0 % et de 73,7 % pour les groupes Ivermectine et Métronidazole respectivement (p < 0,001), entre l’inclusion et après 16 semaines de traitement. La supériorité de Soolantra à la semaine 16 a été confirmée par le taux de réussite défini par l’IGA et la différence absolue du nombre de lésions inflammatoires (critères secondaires (p <0,001)).
Figure 5: Différence moyenne en pourcentage au cours du temps, en semaine
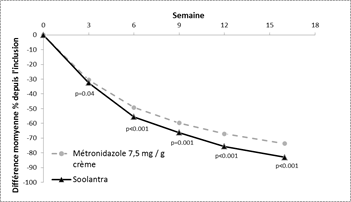
Environ 300 sujets âgés de 65 ans et plus ont été traités avec le médicament au cours des études cliniques. Aucune différence significative du profil d'efficacité et de tolérance n'a été observée entre les sujets âgés et les sujets de 18 à 65 ans.
Le profil de tolérance, comme décrit dans la rubrique 4.8, est resté stable dans des conditions d'utilisation à long terme comme observé dans les traitements jusqu'à un an.
Traitement associant l’ivermectine et 40 mg de doxycycline en gélules à libération modifiée.
L'étude ANSWER a évalué l'efficacité relative de Soolantra (IVM) en association avec des gélules de doxycycline à libération modifiée (DLM) de 40 mg par rapport à l'IVM associée à un placebo de la DLM (PBO) dans le traitement de la rosacée sévère. Il s'agissait d'une étude d’une durée de 12 semaines, randomisée, investigateur aveugle, contrôlée et en groupes parallèles, menée auprès de 273 sujets masculins et féminins âgés de plus de 18 ans, présentant 20 à 70 lésions inflammatoires (papules et pustules) sur le visage et un score IGA de 4.
Le critère principal d’efficacité était la différence en pourcentage du nombre de lésions inflammatoires à la semaine 12 par rapport à l’inclusion. Un pourcentage moyen de diminution du nombre de lésions inflammatoires, significativement plus élevé, a été observé pour IVM + DLM par rapport à IVM + PBO (moyenne ± écart type : -80,29 ± 21,65 % vs -73,56 ± 30,52 % ; p=0,032).
Population pédiatrique
L’Agence européenne des médicaments (EMEA) a accordé une dérogation à l’obligation de soumettre les résultats d’études réalisées avec Soolantra dans tous les sous-groupes de la population pédiatrique (voir rubrique 4.2 pour les informations concernant l’usage pédiatrique.
5.2. Propriétés pharmacocinétiques
L'absorption de l'ivermectine de Soolantra a été évaluée dans une étude clinique chez des sujets adultes atteints de rosacée papulopustuleuse sévère dans des conditions d'utilisation maximales. À l'état d'équilibre (après 2 semaines de traitement), la concentration plasmatique maximale (± écart-type) de l'ivermectine a été atteinte dans les 10 ± 8 heures après l’application (Cmax : 2,1 ± 1,0 ng / mL : de 0,7 à 4,0 ng / mL), et l’aire sous la courbe moyenne (± écart-type) ASC0-24h était de 36 ± 16 ng.hr/mL (intervalle : 14-75ng.hr/mL). Les taux d’exposition systémique de l’ivermectine ont atteint un plateau après deux semaines de traitement (conditions de l'état d'équilibre). Lors de traitement de plus longues durées (études cliniques de phase III), les taux d’exposition systémique de l’ivermectine étaient similaires à ceux observés après 2 semaines de traitement. À l'état d'équilibre, les niveaux d'exposition systémique de l’ivermectine (ASC0-24h: 36 ± 16 ng.hr/mL) étaient inférieurs à ceux obtenus après une dose orale unique de 6 mg d'ivermectine chez des volontaires sains (ASC0-24h: 134 ± 66 ng.hr/mL).
Distribution
Une étude in vitro a montré que l'ivermectine est liée aux protéines plasmatiques à plus de 99 % et est liée essentiellement à l'albumine sérique humaine. Aucune liaison significative de l'ivermectine aux érythrocytes n’a été observée.
Biotransformation
Des études in vitro utilisant des microsomes hépatiques humains et des enzymes du CYP450 recombinantes ont montré que l'ivermectine est principalement métabolisée par le CYP3A4.
Les études in vitro montrent que l'ivermectine n’inhibe pas les isoenzymes du CYP450 : 1A2, 2A6, 2B6, 2C8, 2C9, 2C19, 2D6, 3A4, 4A11 ou 2E1. L'ivermectine n’induit pas l'expression des enzymes du CYP450 (1A2 , 2B6 , 2C9 ou 3A4) dans les hépatocytes humains en culture .
Deux principaux métabolites de l'ivermectine ont été identifiés dans une étude clinique de pharmacocinétique en utilisation maximale et évalués au cours des études cliniques de phase II (3 '' - O -déméthyl ivermectine et 4a -hydroxy ivermectine). Comme pour le composé parent, les métabolites ont atteint l'état d'équilibre à 2 semaines de traitement, avec aucun signe d'accumulation jusqu'à 12 semaines. De plus, les expositions systémiques des métabolites (estimées avec Cmax et ASC) obtenues à l'état d'équilibre étaient beaucoup plus faibles que celles observées après l'administration orale de l’ivermectine.
Élimination
La demi-vie terminale était en moyenne de 6 jours (moyenne: 145 heures, intervalle de 92 à 238 heures) chez les patients recevant une application cutanée quotidienne du médicament pendant 28 jours, dans l’étude clinique de pharmacocinétique en utilisation maximale. L'élimination après traitement topique par Soolantra est dépendante de l’absorption. La pharmacocinétique de l'ivermectine n’a pas été étudiée chez les patients présentant une insuffisance rénale et hépatique.
5.3. Données de sécurité préclinique
L'ivermectine n’est pas génotoxique dans une batterie de tests in vitro et in vivo. Une étude de cancérogénicité sur 2 ans par application cutanée de l'ivermectine 10 mg / g de crème chez la souris n'a montré aucune augmentation de l’incidence des tumeurs.
Les études de toxicité sur la reproduction après administration orale d'ivermectine ont montré des effets tératogènes chez le rat (fentes palatines) et le lapin (flexion carpienne) à des doses élevées (marge d’exposition à la NOAEL d’au moins 70 fois par rapport à l'exposition clinique).
Dans les études par voie orale chez le rat, la toxicité néonatale n’était pas liée à l'exposition in utero, mais à l'exposition postnatale par le lait maternel qui a entraîné des niveaux élevés d’ivermectine dans le cerveau et dans le plasma des nouveau-nés.
Chez le cobaye, Ivermectine 10 mg / g crème a montré une irritation de la peau, une sensibilisation et une photosensibilisation, mais n’était pas phototoxique.
Évaluation du risque environnemental
L’ivermectine est très toxique pour les invertébrés et un risque a été identifié pour le milieu aquatique, les sédiments et le compartiment terrestre. Des précautions doivent être prises pour éviter la contamination de l'environnement, notamment dans le milieu aquatique.
Après première ouverture : à utiliser dans les 6 mois.
6.4. Précautions particulières de conservation
Ce médicament ne nécessite pas de précautions particulières de conservation.
6.5. Nature et contenu de l'emballage extérieur
Tubes blancs en plastique laminé Polyéthylène (PE)/Aluminium (Al)/Polyéthylène (PE) avec :
une ouverture blanche en polyéthylène haute densité (PEHD) et une fermeture de sécurité enfant en polypropylène (PP) pour les tubes de 15g, 30g, 45g et 60g.
un bouchon blanc en polypropylène (PP) pour le tube de 2g (sans fermeture de sécurité enfant)
Présentations : 1 tube de 2g, 15g, 30g, 45g et 60g.
Toutes les présentations peuvent ne pas être commercialisées.
6.6. Précautions particulières d’élimination et de manipulation
Des mesures d'atténuation devraient être prises pour prévenir ou réduire la contamination, en particulier les milieux aquatiques.
Tout médicament non utilisé ou déchet doit être éliminé conformément à la réglementation en vigueur.
7. TITULAIRE DE L’AUTORISATION DE MISE SUR LE MARCHE
Tour Europlaza
20, avenue André Prothin
La Défense 4
92927 PARIS LA DEFENSE Cedex
8. NUMERO(S) D’AUTORISATION DE MISE SUR LE MARCHE
34009 300 241 1 9 : 30 g en tube (PE/Aluminium/PE) muni d'un bouchon sécurité enfant (Polypropylène), boîte de 1.
34009 300 241 2 6 : 45 g en tube (PE/Aluminium/PE) muni d'un bouchon sécurité enfant (Polypropylène), boîte de 1.
34009 300 241 4 0 : 60 g en tube (PE/Aluminium/PE) muni d'un bouchon sécurité enfant (Polypropylène), boîte de 1.
9. DATE DE PREMIERE AUTORISATION/DE RENOUVELLEMENT DE L’AUTORISATION
[à compléter ultérieurement par le titulaire]
10. DATE DE MISE A JOUR DU TEXTE
[à compléter ultérieurement par le titulaire]
Sans objet.
12. INSTRUCTIONS POUR LA PREPARATION DES RADIOPHARMACEUTIQUES
Liste II
ANSM - Mis à jour le : 04/08/2020
Ivermectine
· Gardez cette notice. Vous pourriez avoir besoin de la relire.
· Si vous avez d’autres questions, interrogez votre médecin ou votre pharmacien.
· Ce médicament vous a été personnellement prescrit. Ne le donnez pas à d’autres personnes. Il pourrait leur être nocif, même si les signes de leur maladie sont identiques aux vôtres.
· Si vous ressentez un quelconque effet indésirable, parlez-en à votre médecin ou votre pharmacien. Ceci s’applique aussi à tout effet indésirable qui ne serait pas mentionné dans cette notice. Voir rubrique 4.
1. Qu'est-ce que SOOLANTRA et dans quels cas est-il utilisé ?
2. Quelles sont les informations à connaître avant d'utiliser SOOLANTRA ?
3. Comment utiliser SOOLANTRA ?
4. Quels sont les effets indésirables éventuels ?
5. Comment conserver SOOLANTRA ?
6. Contenu de l’emballage et autres informations.
1. QU’EST-CE QUE SOOLANTRA 10mg/g, crème ET DANS QUELS CAS EST-IL UTILISE ?
La crème est à appliquer sur la peau pour traiter les boutons (papules et pustules) liés à la rosacée.
Soolantra doit être utilisé seulement chez les adultes (18 ans ou plus).
2. QUELLES SONT LES INFORMATIONS A CONNAITRE AVANT D’UTILISER SOOLANTRA 10mg/g, crème ?
si vous êtes allergique à l’ivermectine ou à l’un des autres composants contenus dans ce médicament, mentionnés dans la rubrique 6.
Avertissements et précautions
Adressez-vous à votre médecin ou pharmacien avant d’utiliser Soolantra.
Au début du traitement, certains patients peuvent présenter une aggravation des symptômes de la rosacée. Cependant, ces effets sont rares et se résolvent généralement au bout d’une semaine en poursuivant le traitement. Adressez-vous à votre médecin si cela vous arrive.
Autres médicaments et Soolantra
D’autres médicaments pourraient avoir un effet sur Soolantra, vous devez donc informer votre médecin si vous utilisez, avez récemment utilisé ou pourriez utiliser tout autre médicament.
Soolantra avec des aliments et boissons
Sans objet.
Soolantra n’est pas recommandé pendant la grossesse.
Si vous allaitez, vous ne devez pas utiliser ce médicament, sinon vous devez arrêter l’allaitement avant de commencer le traitement par Soolantra. Vous devez consulter votre médecin pour vous aider à décider entre l’utilisation de Soolantra et l’allaitement, en tenant compte des bénéfices du traitement et de ceux de l’allaitement.
Conduite de véhicules et utilisation de machines
Soolantra a une influence nulle ou négligeable sur l'aptitude à conduire des véhicules et à utiliser des machines.
Soolantra contient :
de l’alcool cétylique et de l’alcool stéarylique qui peuvent provoquer des réactions cutanées locales (ex : dermite de contact),
du parahydroxybenzoate de méthyle (E218) et du parahydroxybenzoate de propyle (E216) qui peuvent provoquer des réactions allergiques (éventuellement retardées),
du propylène glycol qui peut induire des irritations de la peau
3. COMMENT UTILISER SOOLANTRA 10mg/g, crème ?
Important : Soolantra est destiné aux adultes et seulement pour une application sur la peau du visage. Ne pas utiliser ce médicament sur d'autres parties de votre corps, surtout pas sur des surfaces humides du corps, par exemple : vos yeux, votre bouche ou les muqueuses. Ne pas avaler.
La dose recommandée est une application par jour sur la peau du visage. Appliquer une quantité de médicament équivalente à un petit pois sur chacune des cinq zones du visage (front, menton, nez et deux joues). Puis étaler la crème en couche mince sur l'ensemble du visage.
Veillez à éviter les paupières, les lèvres et toute muqueuse telle que l'intérieur du nez, la bouche et les yeux. Si vous mettez accidentellement de la crème dans les yeux ou près des yeux, les paupières, les lèvres, la bouche ou les muqueuses lavez immédiatement et abondamment la zone avec de l'eau.
Vous ne devez pas appliquer de cosmétiques (tels que des crèmes pour le visage ou du maquillage) avant l’application quotidienne de Soolantra. Les produits cosmétiques peuvent être utilisés après que la crème Soolantra ait séché.
Lavez‑vous les mains immédiatement après avoir appliqué la crème Soolantra.
Vous devez utiliser Soolantra quotidiennement tout au long du traitement, le traitement peut être répété. Votre médecin vous dira combien de temps vous aurez besoin d'utiliser Soolantra. La durée de traitement peut varier d’une personne à l’autre et dépend de la sévérité de l’affection de la peau.
Vous pouvez remarquer une amélioration après 4 semaines de traitement. En cas d'absence d'amélioration après 3 mois, vous devez cesser Soolantra et consultez votre médecin.
Insuffisance hépatique
Si vous avez des problèmes hépatiques (au niveau du foie), veuillez consulter votre médecin avant d'utiliser Soolantra.
Utilisation chez les enfants et les adolescents
Soolantra ne doit pas être utilisé chez les enfants et les adolescents.
Comment ouvrir un tube muni d'un bouchon de sécurité enfants :
Pour éviter de répandre accidentellement, ne pas comprimer le tube à l’ouverture ou à la fermeture.
Enfoncez le bouchon et faites‑le tourner dans le sens contraire des aiguilles d'une montre (vers la gauche). Tirez ensuite le bouchon.
Comment fermer un tube muni d'un bouchon sécurité enfants :
Enfoncez le bouchon et faites‑le tourner dans le sens des aiguilles d'une montre (vers la droite).
Si vous avez utilisé plus de Soolantra que vous n’auriez dû
Si vous avez utilisé plus que la dose quotidienne recommandée, veuillez consultez immédiatement votre médecin, qui vous conseillera sur les mesures à prendre.
Si vous oubliez d’utiliser Soolantra
N’utilisez pas de dose double pour compenser la dose oubliée.
Si vous arrêtez d’utiliser Soolantra
Les boutons ne diminueront qu'après plusieurs applications de ce médicament. Il est important que vous continuiez à utiliser Soolantra aussi longtemps que prescrit par votre médecin.
Si vous avez d’autres questions sur l’utilisation de ce médicament, demandez plus d’informations à votre médecin ou à votre pharmacien.
4. QUELS SONT LES EFFETS INDESIRABLES EVENTUELS ?
Comme tous les médicaments, ce médicament peut provoquer des effets indésirables, mais ils ne surviennent pas systématiquement chez tout le monde.
Effets indésirables fréquents (pouvant affecter jusqu'à 1 patient sur 10):
sensation de brûlure au niveau de la peau
Effets indésirables peu fréquents (pouvant affecter jusqu'à 1 patient sur 100) :
irritation de la peau
démangeaison de la peau
peau sèche
Aggravation de la rosacée (veuillez consulter votre médecin)
Fréquence indéterminée (ne peut être estimée sur la base des données disponibles) :
rougeur de la peau
Inflammation de la peau
Gonflement du visage
Augmentation des enzymes hépatiques (ALAT / ASAT)
Déclaration des effets secondaires
Si vous ressentez un quelconque effet indésirable, parlez-en à votre médecin ou votre pharmacien. Ceci s’applique aussi à tout effet indésirable qui ne serait pas mentionné dans cette notice. Vous pouvez également déclarer les effets indésirables directement via le système national de déclaration : Agence nationale de sécurité du médicament et des produits de santé (ANSM) et réseau des Centres Régionaux de Pharmacovigilance - Site internet:www.signalement-sante.gouv.fr.
En signalant les effets indésirables, vous contribuez à fournir davantage d’informations sur la sécurité du médicament.
5. COMMENT CONSERVER SOOLANTRA 10mg/g, crème ?
Tenir ce médicament hors de la vue et de la portée des enfants.
N’utilisez pas ce médicament après la date de péremption indiquée sur l’emballage et le tube après EXP. La date de péremption fait référence au dernier jour de ce mois.
Ce médicament ne nécessite pas de conditions particulières de conservation.
Après première ouverture du tube, utiliser ce produit dans les 6 mois.
Ne jetez aucun médicament au tout-à-l’égout ou avec les ordures ménagères. Demandez à votre pharmacien d’éliminer les médicaments que vous n’utilisez plus. Ces mesures contribueront à protéger l’environnement.
6. CONTENU DE L’EMBALLAGE ET AUTRES INFORMATIONS
Ce que contient Soolantra 10mg/g, crème
· La substance active est : Ivermectine. 1 gramme de crème contient 10mg d’ivermectine.
· Les autres composants sont : glycérol, palmitate d’isopropyle, carbomère, diméticone, édétate disodique, acide citrique monohydraté, alcool cétylique, alcool stéarylique, éther cétostéarylique de macrogol, stéarate de sorbitan, parahydroxybenzoate de méthyle (E218), parahydroxybenzoate de propyle (E216), phénoxyéthanol, propylène glycol, alcool oléique, hydroxyde de sodium, eau purifiée.
Qu’est-ce que Soolantra 10mg/g, crème et contenu de l’emballage extérieur
Soolantra est une crème blanche à jaune pâle, disponible en tubes de 2, 15, 30, 45 ou 60 grammes de crème. Les plus grands tubes ont une fermeture de sécurité enfant tandis que le tube de 2g ne l’a pas.
Boîte de 1 tube.
Toutes les présentations peuvent ne pas être commercialisées.
Titulaire de l’autorisation de mise sur le marché
TOUR EUROPLAZA
20, AVENUE ANDRE PROTHIN
LA DEFENSE 4
92927 PARIS LA DEFENSE CEDEX
Exploitant de l’autorisation de mise sur le marché
GALDERMA INTERNATIONAL
TOUR EUROPLAZA
20, AVENUE ANDRE PROTHIN
LA DEFENSE 4
92927 PARIS LA DEFENSE CEDEX
ZI MONTDESIR
74540 ALBY-SUR-CHERAN
Noms du médicament dans les Etats membres de l'Espace Economique Européen
Ce médicament est autorisé dans les Etats membres de l'Espace Economique Européen sous les noms suivants : Conformément à la réglementation en vigueur.
[À compléter ultérieurement par le titulaire]
La dernière date à laquelle cette notice a été révisée est :
[à compléter ultérieurement par le titulaire]
Sans objet.
Des informations détaillées sur ce médicament sont disponibles sur le site Internet de l’ANSM (France).
[1] A préciser pour les demandes de modification et indiquer le code la modification selon les lignes directrices https://ec.europa.eu/health//sites/health/files/files/eudralex/vol-2/c_2013_2008/c_2013_2008_pdf/c_2013_2804_fr.pdf