Dernière mise à jour le 03/08/2026
ZYMAD 50 000 UI, solution buvable en ampoule
Présentations
> 1 ampoule(s) en verre brun de 2 ml
Code CIP : 34009 300 872 9 9
Déclaration de commercialisation : 29/10/2018
Cette présentation est agréée aux collectivités
- Prix hors honoraire de dispensation : 1,24 €
- Honoraire de dispensation : 1,02 €
- Prix honoraire compris : 2,26 €
- Taux de remboursement :65%
> 2 ampoule(s) en verre brun de 2 ml
Code CIP : 34009 301 991 3 8
Déclaration de commercialisation : 20/11/2020
Cette présentation est agréée aux collectivités
- Prix hors honoraire de dispensation : 2,17 €
- Honoraire de dispensation : 1,02 €
- Prix honoraire compris : 3,19 €
- Taux de remboursement :65 %
> 4 ampoule(s) en verre brun de 2 ml
Code CIP : 34009 302 199 9 7
Déclaration de commercialisation : 16/03/2022
Cette présentation est agréée aux collectivités
- Prix hors honoraire de dispensation : 3,99 €
- Honoraire de dispensation : 1,02 €
- Prix honoraire compris : 5,01 €
- Taux de remboursement :65 %
Service médical rendu (SMR)
Les libellés affichés ci-dessous ne sont que des résumés ou extraits issus des avis rendus par la Commission de la Transparence. Seul l'avis complet de la Commission de la Transparence fait référence.
Cet avis est consultable à partir du lien `Avis du jj/mm/aaaa` ou encore sur demande auprès de la HAS (plus d'informations dans l'aide). Les avis et synthèses d'avis contiennent un paragraphe sur la place du médicament dans la stratégie thérapeutique.
| Valeur du SMR | Avis | Motif de l'évaluation | Résumé de l'avis |
|---|---|---|---|
| Important | Avis du 08/09/2021 | Inscription (CT) | Le service médical rendu par ZYMAD 50 000 UI (cholécalciférol) est important dans l’indication de l’AMM. |
| Important | Avis du 22/04/2020 | Inscription (CT) | Le service médical rendu par ZYMAD 50 000 UI, solution buvable en ampoule est important dans l’indication de l’AMM. |
| Important | Avis du 09/11/2017 | Inscription (CT) | Le service médical rendu par ZYMAD 50 000 UI, solution buvable en ampoule est important dans l’indication de l’AMM. |
Amélioration du service médical rendu (ASMR)
Les libellés affichés ci-dessous ne sont que des résumés ou extraits issus des avis rendus par la Commission de la Transparence. Seul l'avis complet de la Commission de la Transparence fait référence.
Cet avis est consultable à partir du lien `Avis du jj/mm/aaaa` ou encore sur demande auprés de la HAS (plus d'informations dans l'aide). Les avis et synthéses d'avis contiennent un paragraphe sur la place du médicament dans la stratégie thérapeutique.
| Valeur de l'ASMR | Avis | Motif de l'évaluation | Résumé de l'avis |
|---|---|---|---|
| V (Inexistant) | Avis du 08/09/2021 | Inscription (CT) | Cette spécialité est un complément de gamme qui n’apporte pas d’amélioration du service médical rendu (ASMR V) par rapport aux présentations déjà inscrites. |
| V (Inexistant) | Avis du 22/04/2020 | Inscription (CT) | Cette spécialité est un complément de gamme qui n’apporte pas d’amélioration du service médical rendu (ASMR V) par rapport à la présentation déjà inscrite. |
| V (Inexistant) | Avis du 09/11/2017 | Inscription (CT) | Cette spécialité est un complément de gamme qui n’apporte pas d’amélioration du service médical rendu (ASMR V) par rapport aux présentations déjà inscrites. |
ANSM - Mis à jour le : 23/04/2025
ZYMAD 50 000 UI, solution buvable en ampoule
2. COMPOSITION QUALITATIVE ET QUANTITATIVE
Cholécalciférol (vitamine D3).............................................................................................. 50 000 UI
Pour une ampoule.
Pour la liste complète des excipients, voir rubrique 6.1.
Solution buvable en ampoule.
4.1. Indications thérapeutiques
Traitement et/ou prophylaxie de la carence en vitamine D.
4.2. Posologie et mode d'administration
Posologie
Ø Enfant de plus de 2 ans et adolescent
Prophylaxie :
Il est préférable d’administrer des doses quotidiennes en gouttes.
En cas de non-observance à la supplémentation quotidienne :
o 1 ampoule tous les trimestres ou 2 ampoules en une prise deux fois par an (en automne et en hiver) chez les enfants en bonne santé.
o 1 ampoule toutes les 6 semaines ou 2 ampoules en une prise tous les trimestres en cas de disponibilité réduite de la vitamine D (obésité, peau pigmentée, absence d'exposition au soleil) ou d’un apport réduit (régime végétalien).
Ø Adulte et sujet âgé
Prophylaxie : 1 ampoule tous les 1 à 2 mois
Traitement : 1 ampoule toutes les 1 à 2 semaines selon l'intensité de la carence, jusqu'à normalisation de la calcémie et de la phosphorémie, en surveillant la calciurie pour éviter un surdosage, puis traitement prophylactique selon le schéma ci-dessus, sans dépasser 600 000 Ul par an.
Mode d’administration
Voie orale.
Le contenu de l'ampoule peut être pris pur dans une petite cuillère ou mélangé dans un aliment liquide ou semi-liquide.
· Hypersensibilité à la substance active ou à l’un des excipients mentionnés à la rubrique 6.1,
· Hypercalcémie, hypercalciurie, lithiase calcique,
· Hypervitaminose D,
· Pathologie et/ou conditions entraînant une hypercalcémie et/ou une hypercalciurie.
4.4. Mises en garde spéciales et précautions d'emploi
Surveillance
· Pour éviter tout surdosage, tenir compte de l’apport total en vitamine D toutes sources confondues, qu’il s’agisse de sources nutritionnelles, d’autres supplémentations contenant déjà cette vitamine, ou en cas d'utilisation de lait supplémenté en vitamine D, ainsi que des médicaments susceptibles d’interagir avec ZYMAD 50 000 UI, solution buvable en ampoule (voir rubrique 4.5).
· Dans des indications nécessitant des doses fortes et répétées, surveiller la calciurie et la calcémie et arrêter les apports de vitamine D si la calcémie dépasse 106 mg/l (2,65 mmol/l) ou si la calciurie dépasse 300 mg/24 h chez l’adulte ou 4-6 mg/kg/j chez l’enfant.
Lorsque la prescription de ce médicament est envisagée chez des patients atteints de sarcoïdose, une évaluation attentive du bénéfice du traitement au regard des risques pour ces patients est nécessaire en raison du risque d’augmentation du métabolisme de la vitamine D en sa forme active (1,25-dihydroxyvitamine D). Lorsque sa prescription est justifiée chez des patients présentant une sarcoïdose stable (y compris en rémission) en l’absence d’insuffisance rénale et d’hypercalcémie, l’utilisation de ce médicament doit être réalisée avec prudence. Chez ces patients, il convient de surveiller étroitement la calcémie et la calciurie.
Insuffisance rénale
Ce médicament doit être utilisé avec prudence chez les patients présentant une insuffisance rénale et l'effet sur les taux de calcium et de phosphate doit être surveillé.
Le risque de calcification des tissus mous doit être pris en compte.
Patients sous digoxine
L’effet de la vitamine D sur la calcémie devra inciter à une surveillance accrue des patients sous digoxine vis-à-vis du risque d'arythmies cardiaques.
Patients sous diurétiques thiazidiques
Il existe un risque accru d'hypercalcémie lors de l'utilisation concomitante de diurétiques thiazidiques car ils réduisent l'excrétion urinaire du calcium.
Toxicité de la vitamine D en situation d’hypervitaminose D
Le risque de toxicité de la vitamine D est augmenté chez les patients présentant une pathologie et/ou des troubles entraînant une hypercalcémie et/ou une hypercalciurie, et chez les patients ayant une sensibilité accrue à la vitamine D, d’origine génétique, donnant lieu à une hypervitaminose D (voir rubrique 4.9).
4.5. Interactions avec d'autres médicaments et autres formes d'interactions
Précaution d’emploi
+ ANTICONVULSIVANTS INDUCTEURS ENZYMATIQUES (carbamazépine, fosphénytoïne, phénobarbital, phénytoïne, primidone ou barbituriques)
Diminution des concentrations de vitamine D plus marquée qu’en l’absence d’inducteur enzymatique.
Dosage des concentrations de vitamine D et supplémentation si nécessaire.
+ GLUCOCORTICOÏDES
Les glucocorticoïdes réduisent l'effet de la vitamine D en raison d'un métabolisme accru. Dosage des concentrations de vitamine D et supplémentation vitaminocalcique si nécessaire.
+ RESINES CHELATRICES
La prise de résine chélatrice peut diminuer l’absorption intestinale et, potentiellement, l’efficacité d’autres médicaments pris simultanément. D’une façon générale, la prise de la résine doit se faire à distance de celle des autres médicaments, en respectant un intervalle de plus de 2 heures, si possible.
+ RIFAMPICINE
Diminution des concentrations plasmatiques de vitamine D plus marquée qu’en l’absence de traitement par la rifampicine.
Dosage des concentrations plasmatiques de vitamine D et supplémentation si nécessaire.
A prendre en compte
+ ORLISTAT
Diminution de l'absorption de la vitamine D.
+ LAXATIFS
Avec les laxatifs, notamment en vue d’explorations endoscopiques: risque de diminution de l’efficacité du médicament administré avec le laxatif. Eviter la prise d’autres médicaments pendant et après l’ingestion dans un délai d’au moins 2 h après la prise du laxatif, voire jusqu’à la réalisation de l’examen.
4.6. Fertilité, grossesse et allaitement
Grossesse
Les données sont limitées sur l’utilisation du cholécalciférol chez la femme enceinte.
Les études sur l'animal ont mis en évidence des effets tératogènes à des doses très élevées.
Un surdosage en cholécalciférol doit être évité pendant la grossesse, en raison du risque d’hypercalcémie prolongée pouvant entraîner un retard de développement physique et mental, une sténose aortique supravalvulaire et une rétinopathie chez l'enfant.
Le cholécalciférol n’est pas recommandé dans la prophylaxie de la carence chez les femmes enceintes. En cas de carence, ZYMAD 50 000 Ul, solution buvable en ampoule n’est pas recommandée chez les femmes enceintes en raison de son fort dosage.
Allaitement
Le cholécalciférol et ses métabolites sont excrétés dans le lait maternel. Aucun surdosage induit par une mère allaitante n'a été observé chez le nouveau-né ; cependant, lors de prescription d'un supplément de cholécalciférol à un nouveau-né allaité, le médecin doit tenir compte de toute dose supplémentaire de cholécalciférol prise par la mère. ZYMAD 50 000 UI, solution buvable en ampoule n’est pas recommandée chez les femmes allaitantes en raison de son fort dosage.
Fertilité
Il n’y a pas de donnée concernant l’effet de la vitamine D sur la fertilité.
4.7. Effets sur l'aptitude à conduire des véhicules et à utiliser des machines
Les effets indésirables de ZYMAD sont listés selon la classification MedDRA par système classe-organe. Au sein de chaque système classe-organe, les événements indésirables sont présentés par ordre décroissant de fréquence selon la convention suivante : très fréquent (≥1/10), fréquent (>1/100, <1/10), peu fréquent (>1/1 000, <1/100), rare (>1/10 000, <1/1 000), très rare (<1/10 000), fréquence indéterminée (ne peut être estimée sur la base des données disponibles).
|
Fréquence |
Effet indésirable |
|
|
Troubles du système immunitaire |
Indéterminée |
Réactions d’hypersensibilité |
|
Affections de la peau et du tissu sous-cutané |
Indéterminée |
Prurit, rash, érythème, angiœdème |
|
Troubles généraux et anomalies au site d’administration |
Indéterminée |
Œdème, lithiase calcique |
|
Troubles du métabolisme et de la nutrition |
Indéterminée |
Hypercalcémie |
|
Affections du rein et des voies urinaires |
Indéterminée |
Hypercalciurie, néphrolithiase, néphrocalcinose |
Déclaration des effets indésirables suspectés
La déclaration des effets indésirables suspectés après autorisation du médicament est importante. Elle permet une surveillance continue du rapport bénéfice/risque du médicament. Les professionnels de santé déclarent tout effet indésirable suspecté via le système national de déclaration : Agence nationale de sécurité du médicament et des produits de santé (ANSM) et réseau des Centres Régionaux de Pharmacovigilance - Site internet : https://signalement.social-sante.gouv.fr/.
Le risque de surdosage est particulièrement élevé si le patient reçoit plusieurs sources de vitamine D et que la supplémentation globale en vitamine D ne correspond pas aux besoins individuels du patient ; ainsi que chez les patients ayant une sensibilité accrue à la vitamine D, donnant lieu à une hypervitaminose D (voir rubrique 4.4).
Signes cliniques :
Ce sont les signes associés à l’hypercalcémie
· céphalées, asthénie, anorexie, amaigrissement, arrêt de croissance,
· douleurs osseuses,
· faiblesse musculaire,
· nausées, vomissements,
· constipation, douleurs abdominales,
· polyurie, polydipsie, déshydratation, soif,
· hypertension artérielle,
· lithiase calcique, calcifications tissulaires, en particulier rénales (néphrocalcinose) et vasculaires,
· insuffisance rénale,
· arythmie cardiaque dans les cas graves,
· une hypercalcémie extrême peut entraîner le coma et la mort.
Signes biologiques :
· hypercalcémie, hypercalciurie, hyperphosphatémie, concentration basse en hormone parathyroïdienne et élevée en 25-hydroxyvitamine D.
Conduite à tenir :
Cesser l'administration de vitamine D, réduire les apports calciques, augmenter la diurèse, boissons abondantes.
5. PROPRIETES PHARMACOLOGIQUES
5.1. Propriétés pharmacodynamiques
Classe pharmacothérapeutique : VITAMINE D ET ANALOGUES, code ATC : A11CC05
Le rôle essentiel de la vitamine D s'exerce sur l'intestin, dont elle augmente la capacité à absorber le calcium et les phosphates, et sur le squelette, dont elle favorise la minéralisation.
5.2. Propriétés pharmacocinétiques
La vitamine D est absorbée dans l'intestin grêle de façon passive, puis rejoint la circulation générale par voie lymphatique, incorporée aux chylomicrons.
Après absorption, elle se lie à une protéine porteuse spécifique et est transportée jusqu'au foie pour y être convertie en 25-hydroxyvitamine D. Cette dernière se lie à son tour à la même protéine porteuse et est transportée jusqu'aux reins où elle est transformée en sa forme active, la 1,25-dihydroxyvitamine D.
Ses sites de stockage essentiels sont le tissu adipeux, les muscles, mais aussi le sang. La 25-hydroxyvitamine D liée à sa protéine porteuse est le témoin circulant du statut vitaminique et des réserves. Sa demi-vie dans le sang est de 15 à 40 jours.
L'élimination de la vitamine D et de ses métabolites se fait par voie fécale, sous forme non transformée ou sous forme hydrosoluble (acide calcitroïque, dérivés glycuroconjugués).
5.3. Données de sécurité préclinique
Des effets n'ont été observés chez l'animal qu'à des expositions considérées comme très largement supérieures à l'exposition maximale préconisée en usage thérapeutique chez l’Homme.
Une étude de génotoxicité sur Salmonella typhimurium s'est révélée négative.
Deux études de cancérogenèse chez le rat ont montré une possible relation entre une consommation chronique de fortes doses de vitamine D3 et une augmentation de l'incidence de phéochromocytome.
Des études d'embryotoxicité et de teratogénicité ont montré :
· Une dégénérescence des cellules musculaires lisses chez le porc.
· Une diminution des propriétés élastiques et contractiles de l'aorte chez le rat.
· Une diminution du nombre et du poids moyen des portées ainsi que du temps de gestation chez la souris albinos.
Des études de toxicité à doses répétées ont montré une minéralisation étendue des tissus mous, en particulier des reins, des poumons et du myocarde.
Huile essentielle d'orange douce, glycérides polyglycolysés insaturés, huile d'olive raffinée pour préparations injectables.
Sans objet.
3 ans.
6.4. Précautions particulières de conservation
A conserver à une température ne dépassant pas 25°C.
Conserver l'ampoule dans l'emballage extérieur, à l'abri de la lumière.
6.5. Nature et contenu de l'emballage extérieur
2 ml en ampoule (verre brun de type III) de 5 ml avec pointe autocassable ; boîte de 1, 2 ou 4.
6.6. Précautions particulières d’élimination et de manipulation
Tout produit non utilisé ou déchet doit être éliminé conformément à la réglementation en vigueur
7. TITULAIRE DE L’AUTORISATION DE MISE SUR LE MARCHE
VIATRIS MEDICAL
1 BIS PLACE DE LA DEFENSE – TOUR TRINITY
92400 COURBEVOIE
8. NUMERO(S) D’AUTORISATION DE MISE SUR LE MARCHE
· 34009 300 872 9 9 : 2 ml en ampoule (verre brun). Boîte de 1
· 34009 301 991 3 8 : 2 ml en ampoule (verre brun). Boîte de 2
· 34009 302 199 9 7: 2 ml en ampoule (verre brun). Boîte de 4
9. DATE DE PREMIERE AUTORISATION/DE RENOUVELLEMENT DE L’AUTORISATION
[à compléter ultérieurement par le titulaire]
10. DATE DE MISE A JOUR DU TEXTE
[à compléter ultérieurement par le titulaire]
Sans objet.
12. INSTRUCTIONS POUR LA PREPARATION DES RADIOPHARMACEUTIQUES
Sans objet.
Liste II.
ANSM - Mis à jour le : 23/04/2025
ZYMAD 50 000 UI, solution buvable en ampoule
Cholécalciférol
Veuillez lire attentivement cette notice avant de prendre ce médicament car elle contient des informations importantes pour vous.
· Gardez cette notice. Vous pourriez avoir besoin de la relire.
· Si vous avez d’autres questions, interrogez votre médecin ou votre pharmacien.
· Ce médicament vous a été personnellement prescrit. Ne le donnez pas à d’autres personnes. Il pourrait leur être nocif, même si les signes de leur maladie sont identiques aux vôtres.
· Si vous ressentez un quelconque effet indésirable, parlez-en à votre médecin ou votre pharmacien. Ceci s’applique aussi à tout effet indésirable qui ne serait pas mentionné dans cette notice. Voir rubrique 4.
1. Qu'est-ce que ZYMAD 50 000 UI, solution buvable en ampoule et dans quels cas est-il utilisé ?
2. Quelles sont les informations à connaître avant de prendre ZYMAD 50 000 UI, solution buvable en ampoule ?
3. Comment prendre ZYMAD 50 000 UI, solution buvable en ampoule ?
4. Quels sont les effets indésirables éventuels ?
5. Comment conserver ZYMAD 50 000 UI, solution buvable en ampoule ?
6. Contenu de l’emballage et autres informations.
1. QU’EST-CE QUE ZYMAD 50 000 UI, solution buvable en ampoule ET DANS QUELS CAS EST-IL UTILISE ?
Classe pharmacothérapeutique : VITAMINE D ET ANALOGUES, code ATC : A11CC05
Ce médicament est indiqué dans le traitement et/ou la prévention de la carence en vitamine D.
2. QUELLES SONT LES INFORMATIONS A CONNAITRE AVANT DE PRENDRE ZYMAD 50 000 UI, solution buvable en ampoule
Ne prenez jamais ZYMAD 50 000 UI, solution buvable en ampoule dans les cas suivants :
· Si vous êtes allergique (hypersensible) à la substance active ou à l'un des autres composants contenus dans ce médicament, mentionnés dans la rubrique 6 ;
· Si vous présentez un excès de calcium dans le sang (hypercalcémie), un excès de calcium dans les urines (hypercalciurie), des calculs rénaux (lithiase calcique), des dépôts de calcium dans les reins (néphrocalcinose) ;
· Si vous présentez une hypervitaminose D (intoxication par la vitamine D) ;
· Si vous avez une maladie et/ou des troubles pouvant entraîner une hypercalcémie et/ou une hypercalciurie.
Avertissements et précautions :
Adressez-vous à votre médecin, à votre pharmacien ou à votre infirmier/ère avant de prendre ZYMAD 50 000 UI, solution buvable en ampoule.
Pour éviter tout surdosage, tenir compte des doses totales de vitamine D quelle qu’en soit la source, telles que des sources nutritionnelles, des compléments alimentaires de vitamine D, des médicaments contenant déjà cette vitamine ou susceptibles d’interagir avec ZYMAD 50 000 UI, solution buvable en ampoule, ou en cas d'utilisation de lait supplémenté en vitamine D.
Toxicité de la vitamine D en situation d’hypervitaminose D (voir rubrique 3)
Le risque de toxicité de la vitamine D est augmenté si vous présentez une maladie et/ou des troubles entraînant un excès de calcium dans le sang et/ou dans les urines, et si vous présentez une sensibilité accrue à la vitamine D, d’origine génétique, donnant lieu à une hypervitaminose D.
Sarcoïdose
Si vous souffrez de sarcoïdose, votre médecin décidera si vous pouvez prendre ce médicament après une évaluation attentive du bénéfice de ce médicament par rapport aux risques notamment en cas de production anormale de vitamine D par votre organisme entraînant un excès de calcium dans le sang et les urines. Votre taux de calcium dans le sang et dans les urines sera étroitement surveillé.
Insuffisance rénale
Si vous présentez une insuffisance rénale, l’effet sur les taux de calcium et de phosphate doit être surveillé. Le risque de calcification des tissus mous doit être pris en compte.
Autres médicaments et ZYMAD 50 000 UI, solution buvable en ampoule :
Informez votre médecin ou pharmacien si vous prenez, avez récemment pris ou pourriez prendre tout autre médicament.
En particulier, si vous prenez de la digoxine (médicament utilisé dans le traitement de l’insuffisance cardiaque et de certains troubles du rythme cardiaque) ou des diurétiques (médicaments utilisés dans le traitement de l’hypertension artérielle et de l’insuffisance cardiaque), informez votre médecin.
Grossesse et Allaitement
Grossesse
Les données sont limitées sur l’utilisation du cholécalciférol chez la femme enceinte.
Il faut éviter tout surdosage de vitamine D pendant la grossesse, car une hypercalcémie (concentration accrue de calcium dans le sang) prolongée peut provoquer un retard physique et mental, ainsi que des effets néfastes au niveau du cœur et des yeux chez l’enfant à naitre
ZYMAD 50 000 Ul, solution buvable en ampoule n’est pas recommandée chez les femmes enceintes en cas de prévention ou de traitement de la carence en raison de son fort dosage.
Demandez conseil à votre médecin ou à votre pharmacien avant de prendre tout médicament.
Allaitement
La vitamine D passe dans le lait maternel.
ZYMAD 50 000 UI, solution buvable en ampoule n’est pas recommandée chez les femmes allaitantes en cas de prévention ou de traitement de la carence en raison de son fort dosage.
Demandez conseil à votre médecin ou à votre pharmacien avant de prendre tout médicament.
Conduite de véhicules et utilisation de machines
La vitamine D n’a aucun effet ou un effet négligeable sur l’aptitude à conduire des véhicules et à utiliser des machines.
3. COMMENT PRENDRE ZYMAD 50 000 UI, solution buvable en ampoule ?
Posologie
Enfant de plus de 2 ans et l’adolescent
Prévention
Il est préférable d’administrer des doses quotidiennes en gouttes.
Dans le cas où la supplémentation quotidienne n’est pas respectée :
o 1 ampoule tous les trimestres ou 2 ampoules en une prise deux fois par an (en automne et en hiver) chez les enfants en bonne santé.
o 1 ampoule toutes les 6 semaines ou 2 ampoules en une prise tous les trimestres en cas de disponibilité réduite de la vitamine D (obésité, peau pigmentée, absence d'exposition au soleil) ou d’un apport réduit (régime végétalien).
Adulte et sujet âgé
Prévention
1 ampoule par mois ou tous les 2 mois.
Traitement
1 ampoule par semaine ou toutes les 2 semaines selon l’intensité de la carence, sans dépasser 600 000 UI par an, soit 12 ampoules par an.
Mode et voie d'administration
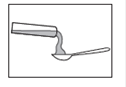
Voie orale. Le contenu de l'ampoule peut être pris pur dans une petite cuillère ou mélangé dans un aliment liquide ou semi-liquide.
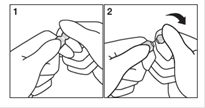
Les ampoules autocassables peuvent générer lors de l’ouverture des débris de verre.
Pour éviter l’ingestion de débris de verre, il est important de ne pas ouvrir l’ampoule au-dessus de la cuillère ou de l’aliment et suivre les instructions suivantes :
1. Placez votre main dominante sur la tête de l’ampoule (partie renflée) et l’autre sur le corps de l’ampoule près du trait circulaire. Vous pouvez placer l’ampoule dans un tissu propre pour vous aider,
2. Exercez une pression franche sur la partie renflée de l’ampoule,
3. Versez lentement le liquide en inclinant légèrement l’ampoule.
![]()
Si vous avez pris plus de ZYMAD 50 000 UI, solution buvable en ampoule que vous n'auriez dû :
Consultez immédiatement votre médecin ou votre pharmacien.
Un surdosage en vitamine D lors d’un traitement à court ou à long terme peut entraîner une hypervitaminose D. Cette intoxication médicamenteuse par la vitamine D peut provoquer des taux anormalement élevés de calcium dans le sang, ce qui peut finir par endommager gravement les tissus mous de l’organisme (qui ne font pas partie des os du squelette) et les reins. Ce risque est particulièrement élevé si vous recevez plusieurs sources de vitamine D et que cela ne correspond pas à vos besoins ou si vous avez une sensibilité accrue à la vitamine D, d’origine génétique, donnant lieu à une hypervitaminose D (voir rubrique 2).
Les signes cliniques d'un surdosage en vitamine D sont :
· maux de tête, fatigue, perte de l'appétit, amaigrissement, arrêt de la croissance ;
· état confus ;
· douleurs osseuses ;
· faiblesse musculaire ;
· nausées, vomissements ;
· constipation, douleurs abdominales ;
· urines abondantes, soif intense, déshydratation, soif ;
· hypertension artérielle ;
· calcul, calcifications des tissus, en particulier du rein et des vaisseaux ;
· la persistance de taux de calcium élevés peut entraîner des lésions irréversibles et une calcification des tissus mous ;
· insuffisance rénale (défaillance des fonctions du rein) ;
· arythmie cardiaque (battements du cœur anormaux ou irréguliers) dans les cas graves ;
· une hypercalcémie extrême peut entraîner le coma et la mort.
Les signes biologiques d'un surdosage en vitamine D sont :
· augmentation du taux de calcium dans le sang et les urines, augmentation du taux de phosphore dans le sang et les urines, concentration sanguine basse en hormone parathyroïdienne et élevée en 25-hydroxyvitamine D.
En cas de surdosage, il faut arrêter le traitement, boire abondamment, limiter les apports en calcium (laitage) et consulter un médecin pour une prise en charge médicale.
Si vous oubliez de prendre ZYMAD 50 000 UI, solution buvable en ampoule
Ne prenez pas de dose double pour compenser la dose que vous avez oublié de prendre.
4. QUELS SONT LES EFFETS INDESIRABLES EVENTUELS ?
Comme tous les médicaments, ce médicament peut provoquer des effets indésirables, mais ils ne surviennent pas systématiquement chez tout le monde.
Fréquence indéterminée (ne peut être estimée sur la base des données disponibles) :
· allergie (réactions d’hypersensibilité)
· prurit, éruption cutanée, rougeur de la peau (érythème), gonflement rapide sous la peau dans des zones telles que le visage, la gorge, les bras et les jambes, qui peut mettre la vie en danger si le gonflement de la gorge bloque les voies respiratoires (angiœdème)
· œdème, formation de calculs de calcium (lithiase calcique)
· taux anormalement élevé de calcium dans le sang (hypercalcémie)
· taux anormalement élevé de calcium dans les urines (hypercalciurie)
· calculs rénaux (néphrolithiase)
· calcification rénale (néphrocalcinose)
Déclaration des effets secondaires
Si vous ressentez un quelconque effet indésirable, parlez-en à votre médecin ou votre pharmacien. Ceci s’applique aussi à tout effet indésirable qui ne serait pas mentionné dans cette notice. Vous pouvez également déclarer les effets indésirables directement via le système national de déclaration : Agence nationale de sécurité du médicament et des produits de santé (ANSM) et réseau des Centres Régionaux de Pharmacovigilance - Site internet : https://signalement.social-sante.gouv.fr/
En signalant les effets indésirables, vous contribuez à fournir davantage d’informations sur la sécurité du médicament.
5. COMMENT CONSERVER ZYMAD 50 000 UI, solution buvable en ampoule ?
Tenir ce médicament hors de la vue et de la portée des enfants.
N’utilisez pas ce médicament après la date de péremption indiquée sur l’emballage et sur l’ampoule après EXP. La date de péremption fait référence au dernier jour de ce mois.
A conserver à une température ne dépassant pas 25°C.
Conserver l'ampoule dans l'emballage extérieur, à l'abri de la lumière.
Ne jetez aucun médicament au tout-à-l’égout ou avec les ordures ménagères. Demandez à votre pharmacien d’éliminer les médicaments que vous n’utilisez plus. Ces mesures contribueront à protéger l’environnement.
6. CONTENU DE L’EMBALLAGE ET AUTRES INFORMATIONS
Ce que contient ZYMAD 50 000 UI, solution buvable en ampoule
· La substance active est :
Cholécalciférol (vitamine D3).............................................................................................. 50 000 UI
· Les autres composants sont :
Huile essentielle d'orange douce, glycérides polyglycolysés insaturés, huile d'olive raffinée
Qu’est-ce que ZYMAD 50 000 UI, solution buvable en ampoule et contenu de l’emballage extérieur
Ce médicament se présente sous forme d'une solution buvable en ampoule. Boîte de 1, 2 ou 4 ampoules de 2 ml.
Titulaire de l’autorisation de mise sur le marché
VIATRIS MEDICAL
1 BIS PLACE DE LA DEFENSE – TOUR TRINITY
92400 COURBEVOIE
Exploitant de l’autorisation de mise sur le marché
VIATRIS SANTE
1 BIS PLACE DE LA DEFENSE – TOUR TRINITY
92400 COURBEVOIE
LABORATOIRE RENAUDIN
ZONE ARTISANALE ERROBI
64250 ITXASSOU
Noms du médicament dans les Etats membres de l'Espace Economique Européen
Sans objet.
La dernière date à laquelle cette notice a été révisée est :
[à compléter ultérieurement par le titulaire]
{mois AAAA}.
Des informations détaillées sur ce médicament sont disponibles sur le site Internet de l’ANSM (France).