Dernière mise à jour le 03/08/2026
MATRIFEN 25 microgrammes/heure, dispositif transdermique
Indications thérapeutiques
Le nom de votre médicament est MATRIFEN.
Les patchs aident à soulager les douleurs très fortes et de longue durée :
· chez les adultes qui ont besoin d’un traitement continu de la douleur
· chez les enfants à partir de l’âge de 2 ans qui ont déjà reçu des médicaments opioïdes et qui ont besoin d’un traitement continu de la douleur.
MATRIFEN contient un médicament appelé fentanyl. Il appartient à la classe des opioïdes, des anti-douleurs puissants.
Présentations
> 5 sachet(s) papier aluminium polytéréphtalate (PET) avec fermeture de sécurité enfant de 1 dispositif(s)
Code CIP : 386 510-8 ou 34009 386 510 8 9
Déclaration de commercialisation : 02/04/2009
Cette présentation est agréée aux collectivités
- Prix hors honoraire de dispensation : 14,83 €
- Honoraire de dispensation : 1,02 €
- Prix honoraire compris : 15,85 €
- Taux de remboursement :65%
- Douleurs chroniques sévères d'origine cancéreuse. ; JOURNAL OFFICIEL ; 01/09/16
Documents de bon usage du médicament
- Prévenir le risque de surdose d’opioïdes
Auteur : Haute autorité de santé
Type : Fiche Bon Usage du Médicament
Date de mise à jour :Avril 2023
- Les médicaments des accès douloureux paroxystiques du cancer
Auteur : Haute autorité de santé
Type : Fiche Bon Usage du Médicament
Date de mise à jour :Juin 2014
Service médical rendu (SMR)
Les libellés affichés ci-dessous ne sont que des résumés ou extraits issus des avis rendus par la Commission de la Transparence. Seul l'avis complet de la Commission de la Transparence fait référence.
Cet avis est consultable à partir du lien `Avis du jj/mm/aaaa` ou encore sur demande auprès de la HAS (plus d'informations dans l'aide). Les avis et synthèses d'avis contiennent un paragraphe sur la place du médicament dans la stratégie thérapeutique.
| Valeur du SMR | Avis | Motif de l'évaluation | Résumé de l'avis |
|---|---|---|---|
| Important | Avis du 10/12/2008 | Inscription (CT) | Le service médical rendu par ces spécialités est important dans les douleurs chroniques d'origine cancéreuse, intenses ou rebelles aux autres antalgiques, en cas de douleurs stables. |
| Insuffisant | Avis du 10/12/2008 | Inscription (CT) | Le service médical rendu par ces spécialités est insuffisant dans les douleurs non cancéreuses compte-tenu : des résultats observés dans les études déposées dans la lombalgie et l'arthrose en termes d'efficacité et de tolérance, de l'absence de donnée dans les autres modèles de douleurs non cancéreuses. |
Amélioration du service médical rendu (ASMR)
Les libellés affichés ci-dessous ne sont que des résumés ou extraits issus des avis rendus par la Commission de la Transparence. Seul l'avis complet de la Commission de la Transparence fait référence.
Cet avis est consultable à partir du lien `Avis du jj/mm/aaaa` ou encore sur demande auprés de la HAS (plus d'informations dans l'aide). Les avis et synthéses d'avis contiennent un paragraphe sur la place du médicament dans la stratégie thérapeutique.
| Valeur de l'ASMR | Avis | Motif de l'évaluation | Résumé de l'avis |
|---|---|---|---|
| V (Inexistant) | Avis du 10/12/2008 | Inscription (CT) | Absence d'amélioration du service médical rendu par rapport aux spécialités DUROGESIC, dispositif transdermique. |
Autres informations
- Titulaire de l'autorisation : ISTITUTO GENTILI S.R.L.
- Conditions de prescription et de délivrance :
- délivrance fractionnée de 14 jours
- prescription en toutes lettres sur ordonnance sécurisée
- prescription limitée à 4 semaines
- stupéfiants
- Statut de l'autorisation : Valide
- Type de procédure : Procédure de reconnaissance mutuelle
- Code CIS : 6 941 929 2
ANSM - Mis à jour le : 19/05/2025
MATRIFEN 25 microgrammes/heure, dispositif transdermique
2. COMPOSITION QUALITATIVE ET QUANTITATIVE
Pour un dispositif transdermique de 8,4 cm2
Un dispositif transdermique diffuse 25 microgrammes/heure de fentanyl.
Pour la liste complète des excipients, voir rubrique 6.1.
Dispositif transdermique rectangulaire, transparent, pourvu d’un film protecteur amovible de taille supérieure à celle du dispositif, portant le nom de marque, la substance active et le dosage imprimés à l’aide d’une encre colorée rouge.
4.1. Indications thérapeutiques
MATRIFEN est indiqué dans le traitement des douleurs chroniques sévères qui nécessitent une administration continue au long cours d’opioïdes.
Chez l’enfant
Traitement au long cours des douleurs chroniques sévères chez les enfants à partir de 2 ans recevant un traitement par opioïdes.
4.2. Posologie et mode d'administration
La dose de MATRIFEN est à ajuster individuellement selon l’état du patient et doit être évaluée à intervalles réguliers après l’application. La dose efficace minimale doit être utilisée. Les patchs sont destinés à délivrer approximativement 12, 25, 50, 75 et 100 microgrammes de fentanyl par heure dans la circulation systémique, ce qui représente respectivement environ 0,3 ; 0,6 ; 1,2 ; 1,8 et 2,4 mg de fentanyl par jour.
Détermination de la posologie initiale
Le choix de la dose initiale optimale de MATRIFEN doit être basé sur le traitement opioïde actuel du patient. Il est recommandé que MATRIFEN soit utilisé chez les patients ayant démontré une tolérance aux opioïdes. D’autres facteurs sont à prendre en compte, tels que l’état général actuel et l’état de santé du patient, incluant la corpulence, l’âge, le degré de sévérité de la maladie ainsi que le degré de tolérance aux opioïdes.
Chez l’adulte
Patients tolérants aux opioïdes
Pour remplacer un traitement par opioïdes oraux ou parentéraux par un traitement par MATRIFEN chez les patients tolérants aux opioïdes, se référer à la conversion des doses équi-analgésiques ci-dessous. Si nécessaire, la posologie peut par la suite être augmentée ou diminuée par paliers de 12 ou 25 microgrammes/heure afin d’atteindre la posologie minimale optimale de MATRIFEN en tenant compte de la réponse au traitement et des besoins en analgésiques supplémentaires.
Patients naïfs d’opioïdes
En général, la voie transdermique n’est pas recommandée chez les patients naïfs d’opioïdes. D’autres voies d’administration (orale, parentérale) doivent être envisagées. Afin d’éviter un surdosage, il est recommandé que les patients naïfs d’opioïdes reçoivent de faibles doses d’opioïdes à libération immédiate (tels que la morphine, l’hydromorphone, l’oxycodone, le tramadol et la codéine) à ajuster jusqu’à atteindre une dose analgésique équivalente à une dose de MATRIFEN de 12 ou 25 microgrammes/heure. Après quoi les patients peuvent changer de traitement pour MATRIFEN.
Dans le cas où il n’est pas possible de débuter par des opioïdes par voie orale et où MATRIFEN représente la seule option de traitement appropriée chez les patients naïfs d’opioïdes, seule l’utilisation de la dose initiale la plus faible (12 microgrammes/heure) est envisageable. Dans ce cas, le patient doit être étroitement surveillé. Il existe un risque d’hypoventilation grave ou potentiellement fatal même si la plus faible dose de MATRIFEN est utilisée pour l’initiation du traitement chez les patients naïfs d’opioïdes (voir rubriques 4.4 et 4.9).
Conversion des doses équi-analgésiques
Chez les patients actuellement traités par des analgésiques opioïdes, la dose initiale de MATRIFEN doit être fonction de la dose journalière du précédent opioïde. Pour calculer la dose initiale optimale de MATRIFEN, il convient de suivre les étapes ci-dessous.
1. Calculer la dose par 24 heures (mg/jour) de l’opioïde actuellement administré.
2. Convertir la quantité ainsi obtenue en dose équi-analgésique de morphine orale par 24 heures à l’aide des facteurs de multiplication du tableau 1 pour la voie d’administration appropriée.
3. Afin d’obtenir le dosage de MATRIFEN correspondant à la dose équi-analgésique calculée de morphine par 24 heures, utiliser les tableaux 2 ou 3 de conversion des doses comme suit :
a. le tableau 2 est destiné aux patients adultes nécessitant une rotation des opioïdes ou cliniquement moins stables (le rapport de conversion de la morphine orale au fentanyl transdermique est d’environ 150:1)
b. le tableau 3 est destiné aux patients adultes dont le traitement par opioïde est stable et bien toléré (le rapport de conversion de la morphine orale au fentanyl transdermique est d’environ 100:1)
Tableau 1 : Tableau de conversion – Facteurs de multiplication pour la conversion de la dose journalière des opioïdes précédents en dose équi-analgésique de morphine orale par 24 heures
(mg/jour de l’opioïde précédent x Facteur = dose équi-analgésique de morphine orale par 24 heures)
|
Opioïde précédent
|
Voie d’administration
|
Facteur de multiplication
|
|
morphine
|
orale |
1a |
|
parentérale |
3 |
|
|
buprénorphine
|
sublinguale |
75 |
|
parentérale |
100 |
|
|
codéine |
orale |
0,15 |
|
parentérale |
0,23b |
|
|
diamorphine |
orale |
0,5 |
|
parentérale |
6b |
|
|
fentanyl
|
orale |
- |
|
parentérale |
300 |
|
|
hydromorphone
|
orale |
4 |
|
parentérale |
20b |
|
|
kétobémidone
|
orale |
1 |
|
parentérale |
3 |
|
|
lévorphanol
|
orale |
7,5 |
|
parentérale |
15b |
|
|
méthadone
|
orale |
1,5 |
|
parentérale |
3b |
|
|
oxycodone
|
orale |
1,5 |
|
parentérale |
3 |
|
|
oxymorphone
|
rectale |
3 |
|
parentérale |
30b |
|
|
péthidine
|
orale |
- |
|
parentérale |
0,4b |
|
|
tapentadol
|
orale |
0,4 |
|
parentérale |
- |
|
|
tramadol
|
orale |
0,25 |
|
parentérale |
0,3 |
a La puissance de la morphine par voie orale/IM est basée sur l’expérience clinique chez les patients présentant une douleur chronique.
b Basé sur des études à dose unique dans lesquelles une dose en IM de chaque substance active listée a été comparée à la morphine pour établir la puissance relative. Les doses orales sont celles recommandées lors du passage d’une voie parentérale à une voie orale.
Tableau 2 : Dose initiale recommandée de MATRIFEN en fonction de la dose orale journalière de morphine (pour des patients nécessitant une rotation des opioïdes ou cliniquement moins stables : rapport de conversion de la morphine orale au fentanyl transdermique d’environ 150:1)1
|
Dose orale de morphine par 24 heures (mg/jour) |
Dosage de MATRIFEN (µg/h) |
|
|
|
|
<90 90-134 |
12 25 |
|
135-224 |
50 |
|
225-314 |
75 |
|
315-404 |
100 |
|
405-494 |
125 |
|
495-584 |
150 |
|
585-674 |
175 |
|
675-764 |
200 |
|
765-854 |
225 |
|
855-944 |
250 |
|
945-1034 |
275 |
|
1035-1124 |
300 |
|
|
|
1 Dans les études cliniques, ces intervalles de doses orales de morphine étaient utilisés comme base pour la conversion en MATRIFEN.
Tableau 3 : Dose initiale recommandée de MATRIFEN en fonction de la dose orale journalière de morphine (pour des patients dont le traitement par opioïde est stable et bien toléré : rapport de conversion de la morphine orale au fentanyl transdermique d’environ 100:1)
|
Dose orale de morphine par 24 heures (mg/jour) |
Dosage de MATRIFEN (µg/h) |
|
|
|
|
≤ 44 |
12 |
|
45-89 |
25 |
|
90-149 |
50 |
|
150-209 |
75 |
|
210-269 |
100 |
|
270-329 |
125 |
|
330-389 |
150 |
|
390-449 |
175 |
|
450-509 |
200 |
|
510-569 |
225 |
|
570-629 |
250 |
|
630-689 |
275 |
|
690-749 |
300 |
L’évaluation initiale de l’effet analgésique maximal de MATRIFEN ne peut être réalisée avant 24 heures de pose du patch. En effet, les concentrations sériques de fentanyl augmentent progressivement pendant les 24 premières heures suivant la pose du premier patch.
Par conséquent, le traitement analgésique précédent doit être progressivement arrêté après l’administration de la première dose, jusqu’à ce que l’efficacité analgésique de MATRIFEN soit atteinte.
Ajustement de la dose et traitement d’entretien
Le patch de MATRIFEN doit être remplacé toutes les 72 heures.
La dose doit être ajustée au cas par cas en fonction de l’utilisation moyenne quotidienne d’analgésiques supplémentaires jusqu’à ce qu’un équilibre entre efficacité analgésique et tolérance soit atteint. Normalement, les adaptations posologiques doivent être effectuées par paliers de 12 ou de 25 microgrammes/heure, bien qu’il faille tenir compte des besoins en analgésiques supplémentaires (morphine orale 45 ou 90 mg/jour équivalent à MATRIFEN 12 ou 25 microgrammes/heure) et de l’intensité de la douleur du patient. Après une augmentation de la dose, il peut s’écouler jusqu’à 6 jours pour que le patient atteigne l’équilibre à la nouvelle dose. Par conséquent, après une augmentation de la dose, les patients doivent porter le patch à la dose augmentée pendant deux périodes de 72 heures avant d’effectuer une autre augmentation de dose.
Il est possible d’utiliser plus d’un patch de MATRIFEN pour des doses supérieures à 100 microgrammes/heure. Les patients peuvent avoir périodiquement besoin de doses supplémentaires d’un analgésique à courte durée d’action, en cas de douleur paroxystique. Certains patients peuvent avoir besoin de méthodes d’administration d’opioïdes supplémentaires ou différentes quand la dose de MATRIFEN dépasse 300 microgrammes/heure.
En l’absence d’un contrôle adéquat de la douleur, l’éventualité d’une hyperalgésie, la tolérance et la progression des pathologies sous-jacentes doivent être prises en considération (voir rubrique 4.4).
Uniquement lors de la première application, si l’effet analgésique est insuffisant, le patch de MATRIFEN peut être remplacé au bout de 48 heures par un patch de la même dose, ou la dose peut être augmentée après 72 heures.
Si le patch doit être remplacé (notamment si le patch se décolle) avant les 72 heures, un patch du même dosage doit être appliqué sur une zone différente de la peau. Une telle situation peut entraîner une augmentation des concentrations sériques (voir rubrique 5.2) et le patient doit être étroitement surveillé.
Durée et objectifs du traitement
Avant l’instauration du traitement par MATRIFEN, une stratégie thérapeutique comprenant la durée et les objectifs du traitement, ainsi qu’un plan pour la fin du traitement, doivent être convenus avec le patient, conformément aux lignes directrices relatives à la prise en charge de la douleur. Pendant le traitement, des contacts fréquents doivent avoir lieu entre le médecin et le patient afin d’évaluer la nécessité de poursuivre le traitement, d’envisager l’arrêt du traitement et d’ajuster les doses si nécessaire. En l’absence d’un contrôle adéquat de la douleur, la possibilité d’hyperalgésie, de tolérance et de progression de la maladie sous-jacente doit être envisagée (voir rubrique 4.4).
Arrêt de MATRIFEN
S’il s’avère nécessaire d’arrêter le traitement par MATRIFEN, le remplacement par d’autres opioïdes doit être progressif, en commençant par une dose faible puis en augmentant progressivement les doses. En effet, les concentrations de fentanyl diminuent progressivement après le retrait du patch de MATRIFEN. Il faut au moins 20 heures pour que les concentrations sériques de fentanyl diminuent de 50 %. En règle générale, il faut arrêter progressivement le traitement analgésique opioïde afin d’éviter les symptômes de sevrage (voir rubriques 4.4 et 4.8). Il a été rapporté qu’un arrêt brutal des analgésiques opioïdes chez des patients physiquement dépendants aux opioïdes a conduit à des symptômes de sevrage significatifs et à une douleur non contrôlée. La diminution de la dose doit se baser sur la dose individuelle, sur la durée du traitement et sur la réponse du patient en matière de douleur et de symptômes de sevrage. Les patients sous traitement à long terme auraient besoin d’une diminution plus progressive. Pour les patients dont le traitement a été de courte durée, une diminution plus rapide peut être envisagée.
Les symptômes de sevrage des opioïdes sont possibles chez certains patients après une conversion ou une adaptation de dose.
Les tableaux 1, 2 et 3 doivent seulement être utilisés pour le passage d’autres opioïdes à MATRIFEN et non pour le passage de MATRIFEN à d’autres traitements afin d’éviter de surestimer la nouvelle dose analgésique et d’entrainer un risque de surdosage.
Populations particulières
Patients âgés
Les patients âgés doivent être étroitement surveillés et la dose doit être ajustée individuellement en fonction de l’état du patient (voir rubriques 4.4 et 5.2).
Chez les patients âgés naïfs d’opioïdes, le traitement doit seulement être envisagé lorsque les bénéfices sont supérieurs aux risques. Dans ces cas, seul MATRIFEN 12 microgrammes/heure doit être utilisé pour initier le traitement.
Insuffisance rénale et hépatique
En cas d’insuffisance hépatique ou rénale, une surveillance étroite est nécessaire et la dose doit être ajustée individuellement en fonction de l’état du patient (voir rubriques 4.4 et 5.2).
Chez les patients naïfs d’opioïdes atteints d’une insuffisance rénale ou hépatique, le traitement doit seulement être envisagé lorsque les bénéfices sont supérieurs aux risques. Dans ces cas, seul MATRIFEN 12 microgrammes/heure doit être utilisé pour initier le traitement.
Population pédiatrique
Enfants âgés de 16 ans et plus
Se reporter à la posologie chez l’adulte.
Enfants âgés de 2 à 16 ans
MATRIFEN doit être administré uniquement aux enfants (âgés de 2 à 16 ans) tolérants aux opioïdes recevant déjà une dose équivalente à au moins 30 mg de morphine orale par jour.
Pour calculer la dose de MATRIFEN à administrer chez les enfants à partir d’opioïdes par voie orale ou parentérale, se référer à la conversion des doses équi-analgésiques (Tableau 1) et à la dose de MATRIFEN recommandée en fonction de la dose journalière de morphine orale (Tableau 4).
Tableau 4 : Dose de MATRIFEN recommandée chez les enfants1 en fonction de la dose journalière de morphine orale2
|
Dose de morphine orale par 24 heures (mg/jour) |
Dosage de MATRIFEN (µg/h) |
|
30 - 44 |
12 |
|
45 - 134 |
25 |
1 La conversion à des dosages de MATRIFEN supérieurs à 25 microgrammes/heure est la même pour les enfants et les adultes (voir Tableau 2).
2 Dans les études cliniques, ces intervalles de dose journalière de morphine orale ont été utilisés comme base de conversion à MATRIFEN.
Dans deux études pédiatriques, la dose nécessaire de fentanyl par voie transdermique a été calculée de façon prudente : 30 mg à 44 mg de morphine orale par jour ou une dose équivalente d’opioïde ont été remplacés par un dispositif transdermique de fentanyl 12 microgrammes/heure. Cette table de conversion chez l’enfant ne s’applique qu’au passage de la morphine orale (ou son équivalent) à un dispositif transdermique de fentanyl. La table de conversion ne doit pas être utilisée pour le passage d’un traitement par du fentanyl transdermique à d’autres opioïdes en raison du risque de surdosage.
L'effet analgésique de la première dose de MATRIFEN ne sera pas optimal pendant les 24 premières heures. C'est pourquoi il est recommandé de poursuivre à doses régulières les analgésiques antérieurement utilisés pendant les 12 heures suivant le remplacement par MATRIFEN. Pendant les 12 heures suivantes, ces analgésiques pourront être utilisés en fonction du besoin clinique.
Une surveillance des effets indésirables du patient, pouvant inclure une hypoventilation, est recommandée pendant au moins 48 heures après l’initiation du traitement par MATRIFEN ou après une augmentation de la dose (voir rubrique 4.4).
MATRIFEN ne doit pas être utilisé chez les enfants âgés de moins de 2 ans car la sécurité et l’efficacité n’ont pas été établies.
Ajustement de la dose et traitement d'entretien chez les enfants
Le patch de MATRIFEN doit être remplacé toutes les 72 heures. La dose doit être ajustée au cas par cas jusqu’à ce qu’un équilibre entre efficacité analgésique et tolérance soit atteint. La posologie ne doit pas être augmentée par paliers de moins de 72 heures. En cas d’effet analgésique insuffisant de MATRIFEN, il peut être nécessaire d’administrer des doses supplémentaires de morphine ou d’un autre opioïde à courte durée d’action. En fonction des besoins analgésiques supplémentaires et de l’état douloureux de l’enfant, il peut être nécessaire d’augmenter la dose. Les adaptations posologiques doivent être réalisées par paliers de 12 microgrammes/heure.
Mode d’administration
MATRIFEN est à usage transdermique.
MATRIFEN doit être appliqué sur une peau non irritée et non irradiée, sur une partie plane du haut du corps ou sur la partie supérieure du bras.
Chez les jeunes enfants, la partie supérieure du dos est l’emplacement privilégié afin d’éviter que l'enfant retire le patch.
Les poils sur le site d’application doivent être coupés (et non rasés) avant l’application (une région glabre de la peau est préférable). Si le site d’application de MATRIFEN nécessite un nettoyage préalable à l’application du patch, il convient de le faire avec de l’eau propre. Les savons, huiles, lotions ou tout autre agent susceptible d’irriter la peau ou d’en altérer ses caractéristiques ne doivent pas être utilisés. La peau doit être parfaitement sèche avant d’appliquer le patch. Les patchs doivent être examinés avec attention avant utilisation. Tout patch découpé ou endommagé de quelque façon que ce soit ne doit pas être utilisé.
MATRIFEN doit être appliqué immédiatement après avoir extrait le patch de l’emballage scellé. Pour enlever le patch de son sachet protecteur, découpez le sachet le long de la ligne pointillée à l’aide de ciseaux. Coupez délicatement et complètement le bord scellé du sachet pour éviter d’endommager le patch. Plier le sachet au niveau de l’encoche puis déchirer soigneusement le sachet. Puis ouvrir le sachet sur deux côtés en le dépliant comme un livre. La pellicule protectrice du patch est découpée. Plier le patch au centre et retirer séparément chaque moitié de la pellicule. Eviter de toucher le côté adhésif du patch. Appliquer le patch sur la peau en appuyant légèrement avec la paume de la main pendant environ 30 secondes. S’assurer que les bordures du patch adhèrent correctement. Se laver ensuite les mains à l’eau propre.
MATRIFEN peut être porté pendant 72 heures consécutives. Après le retrait du précédent patch transdermique le nouveau patch doit être appliqué à un endroit différent. Attendre plusieurs jours avant d’appliquer un nouveau patch sur la même zone de la peau.
Hypersensibilité à la substance active ou à l’un des excipients mentionnés à la rubrique 6.1.
Douleur aiguë ou post-opératoire puisqu’il n’y a aucune possibilité de titration de la dose lors d’une utilisation de courte durée et qu’une hypoventilation grave ou potentiellement fatale pourrait en résulter.
Dépression respiratoire sévère.
4.4. Mises en garde spéciales et précautions d'emploi
Les patients et leur personnel soignant doivent être informés que MATRIFEN contient une quantité de substance active pouvant être fatale, notamment chez l’enfant. En conséquence, ils doivent conserver tous les patchs hors de la vue et de la portée des enfants, avant et après utilisation.
En raison des risques, y compris d’issue fatale, associés à l’ingestion accidentelle, au mésusage et à l’abus, il doit être conseillé aux patients et à leurs soignants de conserver MATRIFEN dans un endroit sûr et sécurisé, non accessible à d’autres personnes.
Patients naïfs d’opioïdes et intolérants aux opioïdes
L’utilisation de MATRIFEN en initiation d’un traitement opioïde chez le patient naïf d’opioïdes a été associée à de très rares cas de dépression respiratoire importante et/ou fatale, particulièrement chez les patients présentant une douleur d’origine non cancéreuse. Le risque d’une hypoventilation grave ou fatale existe même si la plus faible dose de MATRIFEN est utilisée en initiation du traitement chez les patients naïfs d’opioïdes, particulièrement chez les personnes âgées et les patients présentant une insuffisance hépatique ou rénale. La tendance à développer une tolérance varie de façon importante selon les individus. Il est recommandé d’utiliser MATRIFEN chez des patients ayant démontré une tolérance aux opioïdes (voir rubrique 4.2).
Dépression respiratoire
Certains patients peuvent présenter une dépression respiratoire significative avec MATRIFEN ; les patients doivent être surveillés à la recherche de cet effet. La dépression respiratoire peut persister après le retrait du patch de MATRIFEN. L’incidence de la dépression respiratoire augmente avec l’augmentation de la dose de MATRIFEN (voir rubrique 4.9). Les dépresseurs du système nerveux central peuvent accroître la dépression respiratoire (voir rubrique 4.5).
Les opioïdes peuvent entraîner des troubles respiratoires du sommeil, notamment une apnée centrale du sommeil (ACS) et une hypoxie du sommeil. L'utilisation d’opioïdes augmente le risque d’ACS de manière dose-dépendante. Chez les patients présentant une ACS, une réduction de la posologie totale des opioïdes doit être envisagée.
Risque lié à l’utilisation concomitante de dépresseurs du système nerveux central (SNC), incluant les médicaments sédatifs tels que les benzodiazépines ou médicaments apparentés, l’alcool et les médicaments narcotiques dépresseurs du SNC.
L’utilisation concomitante de MATRIFEN et des médicaments sédatifs tels que les benzodiazépines ou des médicaments apparentés, d’alcool ou de médicaments narcotiques dépresseurs du SNC peut entrainer une sédation, une dépression respiratoire, le coma et un décès. En raison de ces risques, la prescription concomitante avec des médicaments sédatifs doit être réservée aux patients pour lesquels aucune autre alternative thérapeutique n’est possible. Si MATRIFEN est prescrit avec des médicaments sédatifs, la plus petite dose efficace devra être administrée et la durée de traitement devra être la plus courte possible.
Les patients doivent être étroitement suivis concernant les signes et symptômes de dépression respiratoire et de sédation. Dans ce cadre, il est fortement recommandé de sensibiliser les patients ainsi que leurs soignants sur ces symptômes (voir rubrique 4.5).
Maladie pulmonaire chronique
MATRIFEN peut entraîner des effets indésirables plus sévères chez les patients atteints d’une maladie pulmonaire chronique obstructive ou autre. Chez ces patients, les opioïdes peuvent diminuer l’activité respiratoire et augmenter la résistance des voies aériennes.
Effets du traitement à long terme et tolérance
Chez tous les patients, une tolérance aux effets des analgésiques, une hyperalgésie ainsi qu’une dépendance physique et psychique peuvent apparaître après l’administration répétée d’opioïdes, alors qu'une tolérance incomplète est développée pour certains effets indésirables comme la constipation induite par les opioïde. Particulièrement chez les patients présentant une douleur chronique d’origine non cancéreuse, il a été rapporté qu’ils pouvaient ne pas ressentir une amélioration significative de l’intensité de la douleur lors d’un traitement opioïde continu au long cours.
Pendant le traitement, des contacts fréquents doivent avoir lieu entre le médecin et le patient afin d’évaluer la nécessité de poursuivre le traitement (voir rubrique 4.2).Lorsqu'il est décidé qu'il n'y a pas d'avantage à poursuivre le traitement, il convient d'appliquer une réduction progressive pour remédier aux symptômes de sevrage.
Ne pas interrompre MATRIFEN de manière brutale chez un patient dépendant aux opioïdes. Un syndrome de sevrage pourrait survenir en cas d’arrêt brutal du traitement ou de réduction de la dose.
Il a été rapporté qu’une diminution rapide de la dose de MATRIFEN chez un patient physiquement dépendant aux opioïdes pouvait conduire à des symptômes de sevrage significatifs et à une douleur non contrôlée (voir rubrique 4.2 et rubrique 4.8). Quand un patient n’a plus besoin de traitement, il est recommandé de diminuer la dose progressivement, afin de minimiser les risques de symptômes de sevrage. Une diminution à partir d’une dose élevée peut prendre des semaines, voire des mois.
Le syndrome de sevrage des opioïdes est caractérisé par une partie ou l’ensemble des symptômes suivants : impatiences, larmoiement, rhinorrhée, bâillements, transpiration, frissons, myalgie, mydriase et palpitations. D’autres symptômes pourraient également se développer, comme de l’irritabilité, de l’agitation, de l’anxiété, de l’hyperkinésie, des tremblements, une faiblesse, de l’insomnie, de l’anorexie, des crampes abdominales, des nausées, des vomissements, de la diarrhée ainsi qu’une tension artérielle, un rythme respiratoire ou un pouls élevé.
Trouble lié à l'utilisation d'opioïdes (abus et dépendance)
L'utilisation répétée de MATRIFEN peut conduire à un trouble lié à l'utilisation des opioïdes (TUO). Une dose plus élevée et une durée plus longue du traitement par opioïdes peuvent augmenter le risque de développer un TUO. Un abus ou un mésusage intentionnel de MATRIFEN peut entraîner un surdosage et/ou un décès. Le risque de développer un TUO est accru chez les patients ayant des antécédents personnels ou familiaux (parents ou frères et sœurs) de troubles liés à la consommation de substances (y compris la consommation d'alcool), chez les consommateurs actuels de tabac ou chez les patients ayant des antécédents personnels d'autres troubles de santé mentale (par exemple dépression majeure, anxiété et troubles de la personnalité).
Avant l’instauration du traitement par MATRIFEN et pendant le traitement, les objectifs du traitement et un plan d’arrêt doivent être convenus avec le patient (voir rubrique 4.2). Avant et pendant le traitement, le patient doit également être informé des risques et des signes de TUO. En cas d’apparition de ces signes, il doit être conseillé aux patients de contacter leur médecin.
Les patients traités avec des médicaments opioïdes doivent être surveillés pour détecter des signes de TUO, tels que les comportements de recherche de médicaments (par exemple, demandes de renouvellement trop précoces), en particulier chez les patients à risque accru. Cela inclut l'examen des traitements concomitants par opioïdes et médicaments psychoactifs (comme les benzodiazépines). Pour les patients présentant des signes et des symptômes de TUO, il convient d'envisager une consultation avec un spécialiste des dépendances. En cas d'arrêt du traitement aux opioïdes, voir rubrique 4.4.
Troubles du Système Nerveux Central incluant l’augmentation de la pression intracrânienne
MATRIFEN doit être utilisé avec prudence chez les patients qui peuvent être particulièrement sensibles aux effets intracrâniens de la rétention de CO2, par exemple les patients présentant des signes d’augmentation de la pression intracrânienne, une altération de la conscience ou les patients dans le coma. MATRIFEN doit être utilisé avec prudence chez les patients présentant des tumeurs cérébrales.
Maladies cardiaques
Le fentanyl peut induire une bradycardie et doit donc être utilisé avec prudence chez les patients présentant des bradyarythmies.
Hypotension
Les opioïdes peuvent provoquer une hypotension, en particulier chez les patients présentant une hypovolémie sévère. Une hypotension et/ou une hypovolémie symptomatique sous-jacente doivent être corrigées avant l’initiation d’un traitement par fentanyl sous forme de patch transdermique.
Insuffisance hépatique
Le fentanyl est métabolisé en métabolites inactifs dans le foie, l’insuffisance hépatique peut retarder son élimination. Les patients présentant une insuffisance hépatique qui reçoivent MATRIFEN doivent être surveillés attentivement pour détecter des signes de toxicité du fentanyl et la dose de MATRIFEN doit être réduite si nécessaire (voir rubrique 5.2).
Insuffisance rénale
Bien qu’il ne soit pas attendu que l’insuffisance rénale modifie l’élimination du fentanyl de façon cliniquement significative, la prudence est recommandée car la pharmacocinétique du fentanyl n’a pas été évaluée chez cette population de patients (voir rubrique 5.2).
Le traitement ne doit être envisagé que si les avantages l’emportent sur les risques. Les patients présentant une insuffisance rénale qui reçoivent MATRIFEN doivent être surveillés attentivement pour détecter des signes de toxicité du fentanyl et la dose de MATRIFEN doit être réduite si nécessaire. Des restrictions supplémentaires s’appliquent aux patients naïfs d’opioïdes présentant une insuffisance rénale (voir rubrique 4.2).
Fièvre/Exposition à la chaleur extérieure
Les concentrations du fentanyl peuvent augmenter si la température cutanée augmente (voir rubrique 5.2).
En conséquence, les patients fébriles doivent être surveillés à la recherche d’effets indésirables des opioïdes et la dose de MATRIFEN doit être adaptée si nécessaire. Il est possible que la libération du fentanyl à partir du dispositif soit augmentée par la température pouvant possiblement entraîner un surdosage et un décès.
Tous les patients doivent être informés d’éviter d’exposer le site d’application de MATRIFEN à des sources de chaleur extérieures telles que : coussins chauffants, couvertures chauffantes, matelas d’eau chauffée, lampes solaires ou lampes bronzantes, bains de soleil, bouillotes, bains chauds prolongés, saunas et bains chauds à remous.
Syndrome sérotoninergique
La prudence est conseillée lorsque MATRIFEN est co-administré avec des médicaments affectant les systèmes de neurotransmission sérotoninergique.
Le développement d’un syndrome sérotoninergique pouvant mettre en jeu le pronostic vital peut survenir lors de l’utilisation concomitante de substances actives sérotoninergiques telles que les Inhibiteurs Sélectifs de la Recapture de la Sérotonine (ISRS), les Inhibiteurs de la Recapture de la Sérotonine et de la Noradrénaline (IRSN), et avec les substances actives altérant le métabolisme de la sérotonine (incluant les Inhibiteurs de la Monoamine Oxydase [IMAO]). Cela peut se produire à la posologie recommandée (voir rubrique 4.5).
Le syndrome sérotoninergique peut inclure des modifications de l’état mental (par exemple : agitation, hallucinations, coma), une instabilité du système nerveux autonome (par exemple : tachycardie, pression artérielle instable, hyperthermie), des anomalies neuromusculaires (par exemple : hyperréflexie, incoordination, rigidité) et/ou des symptômes gastro-intestinaux (par exemple : nausées, vomissement, diarrhée).
Si un syndrome sérotoninergique est suspecté, le traitement par MATRIFEN doit être arrêté.
Interactions avec d’autres médicaments
Inhibiteurs du CYP3A4
L’utilisation concomitante de MATRIFEN avec des inhibiteurs du cytochrome P450 3A4 (CYP 3A4) peut entraîner une augmentation des concentrations plasmatiques de fentanyl, ce qui peut augmenter ou prolonger à la fois les effets thérapeutiques et les effets indésirables, et provoquer une dépression respiratoire grave. Par conséquent, l’utilisation concomitante de MATRIFEN avec des inhibiteurs du CYP3A4 n’est pas recommandée à moins que les bénéfices soient supérieurs au risque accru d’effets indésirables. En général, un patient doit attendre 2 jours après l’arrêt d’un traitement par un inhibiteur du CYP3A4 avant d’appliquer le premier patch de MATRIFEN. Cependant, la durée de l’inhibition varie et pour certains inhibiteurs du CYP3A4 avec une longue demi-vie d’élimination, tels que l’amiodarone, ou pour les inhibiteurs temps-dépendants tels que l’érythromycine, l’idélalisib, la nicardipine et le ritonavir, il peut être nécessaire d’attendre plus longtemps. Par conséquent, il est nécessaire de consulter l’information produit de l’inhibiteur du CYP3A4 concernant la demi-vie de la substance active et la durée de l’effet inhibiteur avant d’appliquer le premier patch de MATRIFEN. Un patient traité par MATRIFEN doit attendre au moins 1 semaine après le retrait du dernier patch avant d’initier un traitement par un inhibiteur du CYP3A4. Si l’utilisation concomitante de MATRIFEN avec un inhibiteur du CYP3A4 ne peut être évitée, il est conseillé de surveiller attentivement l’apparition des signes ou symptômes d’une augmentation ou d’une prolongation des effets thérapeutiques et des effets indésirables du fentanyl (notamment une dépression respiratoire), et si nécessaire la dose de MATRIFEN doit être réduite ou le traitement interrompu (voir rubrique 4.5).
Exposition accidentelle par transfert du patch
Un transfert accidentel du patch de fentanyl sur la peau d’une tierce personne non traitée (plus particulièrement un enfant) au cours du sommeil, ou lors de contacts physiques rapprochés avec une personne traitée peut entraîner un surdosage en opioïde chez cette personne non-traitée.
Les patients doivent être informés qu’en cas d’exposition accidentelle, il est nécessaire de retirer immédiatement le patch transféré de la peau de la personne non traitée (voir rubrique 4.9).
Utilisation chez les personnes âgées
Les résultats des études menées après l’administration intraveineuse du fentanyl suggèrent que la clairance du produit peut être réduite et sa demi-vie prolongée chez les personnes âgées. En outre, les patients âgés risquent d’être plus sensibles à la substance active que les patients plus jeunes. Les patients âgés qui reçoivent MATRIFEN doivent être surveillés attentivement pour détecter des signes de toxicité du fentanyl et la dose de MATRIFEN doit être réduite, si nécessaire (voir rubrique 5.2).
Tractus gastro-intestinal
Les opioïdes augmentent le tonus et diminuent les contractions propulsives des muscles lisses du tractus gastro-intestinal. L’augmentation du temps du transit gastro-intestinal qui en résulte peut être responsable de l’effet de constipation du fentanyl. Les patients doivent être informés des mesures permettant de prévenir la constipation et un traitement laxatif prophylactique doit être envisagé. La plus grande prudence s’impose chez les patients souffrant de constipation chronique. Si un iléus paralytique est présent ou suspecté, le traitement par MATRIFEN doit être arrêté.
Patients présentant une myasthénie grave
Des réactions (myo)cloniques non épileptiques peuvent survenir. La prudence s’impose lors du traitement des patients présentant une myasthénie grave.
Utilisation concomitante d’un mélange d’agonistes/antagonistes des opioïdes
L’utilisation concomitante de buprénorphine, nalbuphine ou pentazocine n’est pas recommandée (voir aussi rubrique 4.5).
Population pédiatrique
MATRIFEN ne doit pas être administré chez les enfants naïfs de traitement opioïde (voir rubrique 4.2). Il existe un risque d'hypoventilation grave ou potentiellement fatale quelle que soit la dose de MATRIFEN dispositif transdermique administrée.
MATRIFEN n'a pas été étudié chez l'enfant de moins de 2 ans. MATRIFEN doit être administré uniquement aux enfants de 2 ans ou plus tolérants aux opioïdes (voir rubrique 4.2).
Afin de prévenir une ingestion accidentelle par l'enfant, choisir avec prudence le site d'application de MATRIFEN (voir rubrique 4.2 et 6.6) et vérifier attentivement la bonne adhésion du patch.
Hyperalgésie induite par les opioïdes
L’hyperalgésie induite par les opioïdes (HIO) est une réponse paradoxale à un opioïde, dans laquelle la perception de la douleur est accrue, malgré une exposition stable ou croissante à l’opioïde. Elle se distingue de la tolérance, pour laquelle des doses plus importantes d’opioïdes sont nécessaires pour atteindre un effet analgésique identique ou pour traiter une douleur récurrente. L’HIO peut se manifester sous la forme d’une augmentation de la douleur, d’une douleur plus généralisée (c.-à-d. moins topique) ou d’une douleur provoquée par des stimuli ordinaires (c.-à-d. non douloureux ; allodynie) sans signe d’une progression de la maladie. En cas de suspicion d’HIO, la dose d’opioïde devra être réduite ou progressivement diminuée, si possible.
4.5. Interactions avec d'autres médicaments et autres formes d'interactions
Interactions liées à la pharmacodynamique
Médicaments à action centrale/dépresseurs du système nerveux central (SNC), incluant l’alcool et les médicaments dépresseurs du SNC.
L’utilisation concomitante de MATRIFEN avec d’autres dépresseurs du système nerveux central (incluant benzodiazépines et autres sédatifs/hypnotique, opioïdes, anesthésiques généraux, phénothiazines, tranquillisants, anti-histaminiques sédatifs, alcool et médicaments narcotiques dépresseurs du SNC), des relaxants musculo-squelettiques et des gabapentinoïdes (gabapentine et prégabaline) peut entrainer une dépression respiratoire, une hypotension, une sédation profonde, un coma ou un décès. La prescription concomitante de dépresseurs du SNC et de MATRIFEN doit être réservée aux patients pour lesquels d'autres options de traitement ne sont pas possibles. L’utilisation concomitante d’un de ces médicaments avec MATRIFEN nécessite une surveillance et une observation étroite du patient.. Par conséquent, l’utilisation concomitante d’un de ces médicaments avec MATRIFEN nécessite des soins particuliers et une observation du patient.
La dose et la durée de l’utilisation concomitante doivent être limitées (voir rubrique 4.4).
Inhibiteurs de la Monoamine Oxydase
L’utilisation de MATRIFEN n’est pas recommandée chez les patients nécessitant une administration concomitante d’un IMAO. Des interactions sévères et imprévisibles avec les IMAO, comportant une potentialisation des effets opiacés ou des effets sérotoninergiques, ont été rapportées. Ainsi, MATRIFEN ne doit pas être utilisé dans les 14 jours suivant l’arrêt d’un traitement par IMAO.
Médicaments sérotoninergiques
La co-administration de fentanyl avec un médicament sérotoninergique, tel qu’un Inhibiteur Sélectif de la Recapture de la Sérotonine (ISRS), un Inhibiteur de la Recapture de la Sérotonine et de la Noradrénaline (IRSN) ou un Inhibiteur de la Monoamine Oxydase (IMAO) peut augmenter le risque de syndrome sérotoninergique, pouvant mettre en jeu le pronostic vital.. A utiliser avec prudence de manière concomitante. Observer attentivement le patient, en particulier lors de l'instauration du traitement et de l'ajustement de la dose (voir rubrique 4.4).
Utilisation concomitante d’un mélange d’agonistes/antagonistes des opioïdes
L’utilisation concomitante de buprénorphine, nalbuphine ou pentazocine n’est pas recommandée. Ces derniers présentent une forte affinité pour les récepteurs aux opioïdes avec une activité intrinsèque relativement faible. Par conséquent, ils antagonisent partiellement l’effet analgésique du fentanyl et peuvent induire des symptômes de sevrage chez les patients dépendants des opioïdes (voir rubrique 4.4).
Interactions liées à la pharmacocinétique
Inhibiteurs du CYP3A4
Le fentanyl, substance active ayant une clairance élevée, est rapidement et largement métabolisé, essentiellement par le CYP3A4.
L’utilisation concomitante de MATRIFEN avec des inhibiteurs du cytochrome P450 3A4 (CYP3A4) peut entraîner une augmentation des concentrations plasmatiques du fentanyl, ce qui peut augmenter ou prolonger à la fois ses effets thérapeutiques et ses effets indésirables et provoquer une dépression respiratoire grave. Il est attendu que l’interaction avec les inhibiteurs puissants du CYP3A4 soit supérieure à l’interaction avec les inhibiteurs faibles ou modérés du CYP3A4. Des cas de dépression respiratoire grave après co-administration d’inhibiteurs du CYP3A4 avec du fentanyl transdermique ont été rapportés, incluant un cas mortel après une co-administration avec un inhibiteur modéré du CYP3A4. L’utilisation concomitante de MATRIFEN avec des inhibiteurs du CYP3A4 n’est pas recommandée à moins que le patient soit étroitement surveillé (voir rubrique 4.4). Des exemples de substances actives pouvant augmenter les concentrations de fentanyl incluent : amiodarone, cimétidine, clarithromycine, diltiazem, érythromycine, fluconazole, itraconazole, kétoconazole, néfazodone, ritonavir, vérapamil et voriconazole (cette liste n’est pas exhaustive). Après la co-administration d’inhibiteurs faibles, modérés ou sévères du CYP3A4 avec du fentanyl par voie intraveineuse administré sur une courte durée, les diminutions de la clairance du fentanyl étaient généralement ≤ 25 %. Cependant, avec le ritonavir (inhibiteur puissant du CYP3A4), la clairance du fentanyl a diminué en moyenne de 67 %. L’importance des interactions des inhibiteurs du CYP3A4 en cas d’administration à long terme du fentanyl transdermique est inconnue, mais elle peut être supérieure à celle d’une administration intraveineuse de courte durée.
Inducteurs du CYP3A4
L’utilisation concomitante du fentanyl transdermique avec des inducteurs du CYP3A4 peut entraîner une diminution des concentrations plasmatiques du fentanyl et une diminution de l’effet thérapeutique. La prudence est conseillée lors d’une utilisation concomitante d’inducteurs du CYP3A4 et de MATRIFEN. Il peut être nécessaire d’augmenter la dose de MATRIFEN ou de changer pour une autre substance active analgésique. Une diminution de la dose de fentanyl et une surveillance particulière sont nécessaires en prévision de l’arrêt du traitement concomitant par un inducteur du CYP3A4. Les effets de l’inducteur diminuent progressivement et peuvent entraîner une augmentation des concentrations plasmatiques du fentanyl, ce qui peut augmenter ou prolonger à la fois ses effets thérapeutiques et ses effets indésirables, et provoquer une dépression respiratoire grave. Une surveillance étroite doit être maintenue jusqu’à ce que les effets du médicament soient stabilisés. Parmi les exemples de substances actives pouvant diminuer les concentrations plasmatiques du fentanyl : carbamazépine, phénobarbital, phénytoïne et rifampicine (cette liste n’est pas exhaustive).
Population pédiatrique
Les études d’interaction n’ont été réalisées que chez l’adulte.
4.6. Fertilité, grossesse et allaitement
Grossesse
Il n’existe pas de données suffisantes sur l’utilisation de MATRIFEN chez la femme enceinte. Les études chez l’animal ont mis en évidence une toxicité sur la reproduction (voir rubrique 5.3). Le risque potentiel chez l’Homme n’est pas bien connu, bien qu’il a été constaté que le fentanyl utilisé en tant qu’anesthésique par voie intraveineuse franchit la barrière placentaire chez les femmes enceintes. Un syndrome de sevrage néonatal a été rapporté chez des nouveau-nés dont les mères avaient reçu un traitement prolongé par MATRIFEN durant la grossesse. MATRIFEN ne doit pas être utilisé pendant la grossesse sauf en cas de nécessité absolue.
L’utilisation du MATRIFEN pendant l’accouchement n’est pas recommandé car il ne doit pas être utilisé dans le traitement de la douleur aiguë ou post-opératoire (voir rubrique 4.3). De plus, comme le fentanyl franchit la barrière placentaire, l’utilisation de MATRIFEN pendant l’accouchement pourrait provoquer une dépression respiratoire chez le nouveau-né.
Allaitement
Le fentanyl est excrété dans le lait maternel et peut provoquer une sédation/ dépression respiratoire chez le nourrisson allaité. L’allaitement doit donc être interrompu au cours du traitement par MATRIFEN et pendant au moins 72 heures après le retrait du patch.
Fertilité
Il n’existe pas de données cliniques concernant les effets du fentanyl sur la fertilité. Des études chez le rat ont révélé une diminution de la fertilité et une augmentation de la mortalité embryonnaire à des doses toxiques pour la mère (voir rubrique 5.3).
4.7. Effets sur l'aptitude à conduire des véhicules et à utiliser des machines
Les effets indésirables rapportés lors de l’utilisation par des patchs de fentanyl transdermique à partir de ces études cliniques, incluant les effets indésirables mentionnées ci-dessus et ceux signalés après la commercialisation sont présentés dans le tableau 5.
Les catégories de fréquence sont définies de la manière suivante : Très fréquent (≥ 1/10), fréquent (≥ 1/100, < 1/10), peu fréquent (≥ 1/1000, < 1/100), rare (≥ 1/10 000, < 1/1000), très rares < 1/10 000, et fréquence indéterminée (ne peut être estimée sur la base des données cliniques disponibles). Les effets indésirables sont listés par classe de système organe suivant un ordre décroissant de gravité dans chaque catégorie de fréquence.
Tableau 5 : Effets indésirables chez les patients adultes et enfants
|
Classe de système organe |
Effets indésirables |
||||
|
Catégorie de fréquence |
|||||
|
Très fréquent |
Fréquent |
Peu fréquent |
Rare |
Fréquence indéterminée |
|
|
Affections du système immunitaire |
|
Hypersensibilité |
|
|
Choc anaphylactique, réaction anaphylactique, réaction anaphylactoïde |
|
Affections endocriniennes |
|
|
|
|
Déficience androgénique |
|
Troubles du métabolisme et de la nutrition |
|
Anorexie |
|
|
|
|
Affections psychiatriques |
|
Insomnie, dépression, anxiété, confusion, hallucination |
Agitation, désorientation, humeur euphorique |
|
Délire, dépendance |
|
Affections du système nerveux |
Somnolence, sensations vertigineuses, maux de tête |
Tremblement, paresthésie |
Hypoesthésie, convulsion (incluant les convulsions cloniques et le grand mal convulsif), amnésie, diminution du niveau de conscience, perte de conscience |
|
|
|
Affections oculaires |
|
|
Vision trouble |
Myosis |
|
|
Affections de l’oreille et du labyrinthe |
|
Vertiges |
|
|
|
|
Affections cardiaques |
|
Palpitations, tachycardie |
Bradycardie, cyanose |
|
|
|
Affections vasculaires |
|
Hypertension |
Hypotension |
|
|
|
Affections respiratoires, thoraciques et médiastinales |
|
Dyspnée |
Dépression respiratoire, détresse respiratoire |
Apnée, hypoventilation |
Bradypnée |
|
Affections gastro-intestinales |
Nausées, vomissements, constipation |
Diarrhées, bouche sèche, douleur abdominale, douleur abdominale haute, dyspepsie |
Ileus, dysphagie |
Sub-ileus |
|
|
Affections de la peau et du tissu sous-cutané |
|
Hyperhidrose, prurit, rash, érythème |
Eczéma, dermatite allergique, troubles de la peau, dermatite, dermatite de contact |
|
|
|
Affections musculo-squelettiques et systémiques |
|
Contractures musculaires |
Contractions fasciculaires |
|
|
|
Affections du rein et des voies urinaires |
|
Rétention urinaire |
|
|
|
|
Affections des organes de reproduction et du sein |
|
|
Dysfonction érectile, dysfonction sexuelle |
|
|
|
Troubles généraux et anomalies au site d’administration |
|
Fatigue, œdème périphérique, asthénie, malaise, sensation de froid |
Réaction au site d’application, syndrome grippal, sensation de modification de la température corporelle, hypersensibilité au site d’application, syndrome de sevrage de drogue ou de médicament, pyrexie* |
Dermatite au site d’application, eczéma au site d’application |
Tolérance au médicament |
*la fréquence assignée (peu fréquent) est fonction des analyses de fréquence incluant seulement les sujets adultes et enfants des études cliniques présentant une douleur d’origine non-cancéreuse.
Population pédiatrique
La sécurité d’emploi des patchs de fentanyl transdermique a été évaluée chez 289 enfants (<18 ans) ayant participé à 3 études cliniques relatives au traitement de la douleur chronique ou continue d’origine cancéreuse ou non-cancéreuse. Ces sujets ont reçu au moins une dose de fentanyl transdermique et ont permis de fournir des données de sécurité (voir rubrique 5.1).
Le profil de sécurité d’emploi chez les enfants et les adolescents traités par des patchs de fentanyl transdermique était similaire à celui observé chez les adultes. Chez l’enfant, il n’a pas été identifié de risque supérieur à celui attendu lors de l’utilisation des opioïdes dans le traitement des douleurs associées à une maladie grave et il ne semble pas qu’il y ait de risque pédiatrique spécifique associé à l’utilisation des patchs de fentanyl transdermique chez l’enfant à partir de l’âge de 2 ans dès lors qu’il est utilisé selon les recommandations définies.
Sur la base des données de sécurité combinées à partir de ces 3 essais cliniques chez les enfants, les effets indésirables les plus fréquemment rapportés (fréquence ≥10 %) étaient : vomissements (33,9 %), nausées (23,5 %), céphalées (16,3 %), constipation (13,5 %), diarrhées (12,8 %), et prurit (12,8 %).
Tolérance
La tolérance peut se développer lors d’une utilisation répétée.
Pharmacodépendance
L’utilisation répétée du MATRIFEN peut entraîner une dépendance aux médicaments (pharmacodépendance), même à des doses thérapeutiques. Le risque de dépendance aux médicaments peut varier en fonction des facteurs de risque, de la posologie et de la durée du traitement par opioïdes du patient (voir rubrique 4.4).
Après le relais d’autres analgésiques opioïdes par des patchs de fentanyl transdermique ou après l’arrêt brutal du traitement, certains patients peuvent présenter des symptômes de sevrage aux opioïdes (tels que nausées, vomissements, diarrhée, anxiété et frissons) (voir rubriques 4.2 et 4.4).
Il y a eu de très rares cas de syndrome de sevrage néonatal chez des nouveau-nés dont les mères avaient reçu un traitement prolongé par des patchs de fentanyl transdermique durant la grossesse (voir rubrique 4.6).
Des cas de syndrome sérotoninergique ont été rapportés avec le fentanyl administré de façon concomitante avec des médicaments sérotoninergiques puissants (voir rubriques 4.4 et 4.5).
Déclaration des effets indésirables suspectés
La déclaration des effets indésirables suspectés après autorisation du médicament est importante. Elle permet une surveillance continue du rapport bénéfice/risque du médicament. Les professionnels de santé déclarent tout effet indésirable suspecté via le système national de déclaration : Agence nationale de sécurité du médicament et des produits de santé (ANSM) et réseau des Centres Régionaux de Pharmacovigilance - Site internet : https://signalement.social-sante.gouv.fr/
Les manifestations d’un surdosage en fentanyl consistent en une accentuation de ses effets pharmacologiques, l’effet le plus grave étant la dépression respiratoire. Une leucoencéphalopathie toxique a également été observée lors d’un surdosage de fentanyl.
Traitement
Pour la prise en charge de la dépression respiratoire, les mesures immédiates à prendre comprennent le retrait du patch de MATRIFEN et la stimulation physique ou verbale du patient. Ces mesures peuvent être suivies par l’administration d’un antagoniste spécifique des opioïdes tel que la naloxone.
La dépression respiratoire consécutive à un surdosage peut persister plus longtemps que l’effet de l’antagoniste des opioïdes. L’intervalle entre les doses d’antagoniste par voie IV doit être soigneusement choisi en raison de la possibilité de re-narcotisation après le retrait du patch ; une administration répétée ou une perfusion continue de naloxone peuvent être nécessaires. La neutralisation de l’effet narcotique peut provoquer l’apparition d’une douleur aigüe et une libération de catécholamines.
Si la situation clinique le justifie, la perméabilité des voies respiratoires doit être assurée et maintenue, éventuellement à l’aide d’une canule oropharyngée ou d’une sonde endotrachéale, de l’oxygène doit être administré et si besoin, la ventilation doit être assistée ou contrôlée. Une température corporelle adéquate et un apport hydrique doivent être maintenus.
En cas de survenue d’une hypotension sévère ou persistante, une hypovolémie doit être envisagée et l’état doit être géré par l’administration appropriée de liquides par voie parentérale.
5. PROPRIETES PHARMACOLOGIQUES
5.1. Propriétés pharmacodynamiques
Mécanisme d’action
Le fentanyl est un analgésique opioïde qui interagit principalement sur les récepteurs aux opioïdes μ. Ses principaux effets thérapeutiques sont une analgésie et une sédation.
Population pédiatrique
La sécurité d’emploi des patchs de fentanyl transdermique a été évaluée au cours de 3 études en ouvert chez 289 enfants âgés de 2 ans à 17 ans inclus, présentant des douleurs chroniques, parmi lesquels 80 enfants étaient âgés de 2 à 6 ans inclus. Sur les 289 patients inclus dans ces 3 études, 110 ont débuté un traitement par des patchs de fentanyl transdermique à la dose de 12 microgrammes/heure. Sur ces 110 patients, 23 (20,9 %) avaient précédemment reçu une dose équivalente à < 30 mg de morphine orale par jour, 66 (60,0 %) avaient reçu une dose équivalente à 30 à 44 mg de morphine orale par jour et 12 (10,9 %) avaient reçu une dose équivalente à au moins 45 mg de morphine orale par jour (données non disponibles pour 9 [8,2 %] sujets). Des doses d’initiation de 25 microgrammes/heure et plus ont été utilisées chez les 179 sujets restants dont 174 (97,2 %) qui ont reçu des doses d’opioïdes équivalentes à au moins 45 mg de morphine orale par jour. Parmi les 5 patients restants ayant reçu une dose d’initiation d’au moins 25 microgrammes/heure avec des doses d’opioïdes précédentes équivalentes à < 45 mg de morphine orale par jour, 1 (0,6 %) avait précédemment reçu une dose équivalente à < 30 mg de morphine orale par jour et 4 (2,2 %) avaient reçu une dose équivalente à 30 à 44 mg de morphine orale par jour (voir rubrique 4.8).
5.2. Propriétés pharmacocinétiques
Les patchs de fentanyl transdermique permettent une libération systémique continue de fentanyl pendant l’application de 72 heures. Après l’application des patchs, le fentanyl est absorbé par la peau située sous le dispositif et un dépôt de fentanyl se concentre dans les couches supérieures de la peau. Le fentanyl devient ensuite accessible à la circulation systémique. Grâce à la matrice de polymère et à la diffusion du fentanyl au travers des couches cutanées, la vitesse de libération reste relativement constante. Le gradient de concentration qui existe entre le dispositif et la concentration la plus basse dans la peau entraîne la libération du médicament. La biodisponibilité moyenne du fentanyl après l’application du patch transdermique est de 92 %.
Après la première application des patchs, les concentrations sériques de fentanyl augmentent progressivement et atteignent généralement un plateau au bout de 12 à 24 heures, après quoi elles restent relativement stables pendant le reste des 72 heures d’application. La concentration sérique d’équilibre est atteinte avant la fin de la deuxième application de 72 heures et se maintient pendant les applications ultérieures d’un patch de même taille. Du fait de l’accumulation, les valeurs de l’ASC et de la Cmax entre deux doses sont, à l’équilibre, environ 40 % plus élevées qu’après une application unique. Les patients atteignent et maintiennent une concentration sérique d’équilibre qui est déterminée par les variations individuelles de la perméabilité de la peau et la clairance corporelle du fentanyl. Une importante variabilité inter-individuelle dans les concentrations plasmatiques a été observée.
Un modèle pharmacocinétique a indiqué que les concentrations sériques de fentanyl pouvaient augmenter de 14 % (intervalle de 0 à 26 %) si un nouveau patch était appliqué après un délai de 24 heures plutôt qu’après le délai d’application recommandé de 72 heures.
Une augmentation de la température cutanée peut accroître l’absorption du fentanyl appliqué par voie transdermique (voir rubrique 4.4). L’élévation de la température cutanée par l’application d’un coussin chauffant réglé à basse température sur le patch de MATRIFEN pendant les 10 premières heures lors d’une application unique a multiplié la valeur moyenne de l’ASC du fentanyl par 2,2 et a augmenté de 61 % la concentration moyenne à la fin de l’exposition à la chaleur.
Distribution
Le fentanyl est rapidement distribué dans divers tissus et organes comme l’indique l’important volume de distribution (3 à 10 L/kg après administration intraveineuse chez des patients). Le fentanyl s’accumule dans le muscle squelettique et les graisses et est lentement libéré dans le sang.
Dans une étude chez des patients atteints d’un cancer et traités par du fentanyl transdermique, le taux moyen de liaison aux protéines plasmatiques était de 95 % (intervalle de 77 à 100 %). Le fentanyl traverse facilement la barrière hémato-encéphalique. Le fentanyl traverse également le placenta et est excrété dans le lait maternel.
Biotransformation
Le fentanyl est une substance active avec une clairance élevée et est rapidement et largement métabolisé, principalement par le CYP3A4, dans le foie. Le métabolite principal, le norfentanyl, et les autres métabolites sont inactifs. Le fentanyl délivré par voie transdermique ne semble pas être métabolisé au niveau de la peau.
Cette observation a été établie par un test sur des kératinocytes humains et par des études cliniques dans lesquelles 92 % de la dose libérée par le dispositif l’était sous forme de fentanyl inchangé, retrouvé dans la circulation systémique.
Élimination
Après une application du patch pendant 72 heures, la demi-vie moyenne du fentanyl varie de 20 à 27 heures. En raison de l’absorption continue du fentanyl à partir du dépôt cutané après le retrait du patch, la demi-vie du fentanyl après administration transdermique est d’environ 2 à 3 fois plus longue qu’après administration intraveineuse.
Après administration intraveineuse, la clairance totale moyenne du fentanyl dans les études varie généralement entre 34 et 66 L/h.
Dans les 72 heures suivant une administration intraveineuse de fentanyl, environ 75 % de la dose est excrété dans les urines et environ 9 % de la dose est retrouvé dans les selles. L’excrétion a principalement lieu sous la forme de métabolites, avec moins de 10 % de la dose excrété sous la forme active inchangée.
Linéarité/non-linéarité
Les concentrations sériques de fentanyl atteintes sont proportionnelles à la taille du patch de fentanyl transdermique. La pharmacocinétique du fentanyl transdermique n’est pas modifiée par des applications répétées.
Relations pharmacocinétique/pharmacodynamique
Il existe une importante variabilité inter-individuelle dans la pharmacocinétique du fentanyl, dans les relations entre les concentrations de fentanyl, les effets thérapeutiques et les effets indésirables, et dans la tolérance aux opioïdes. La concentration minimale efficace du fentanyl dépend de l’intensité de la douleur et de l’utilisation antérieure de traitement opioïde. La concentration minimale efficace et la concentration toxique augmentent avec la tolérance. Il n’est donc pas possible d’établir un intervalle de concentration thérapeutique optimale en fentanyl. L’adaptation de la dose individuelle en fentanyl doit se baser sur la réponse du patient et le niveau de tolérance. Après l’application du premier patch et après une augmentation de la dose, un délai de 12 à 24 heures doit être pris en compte.
Populations particulières
Personnes âgées
Les données des études avec le fentanyl administré par voie intraveineuse suggèrent que la clairance peut être réduite et la demi-vie prolongée chez les personnes âgées. En outre, les patients âgés risquent d’être plus sensibles au médicament que les patients plus jeunes. Dans une étude conduite avec des patchs de fentanyl transdermique chez des sujets âgés sains la pharmacocinétique du fentanyl ne différait pas de façon significative de celle observée chez les sujets jeunes sains bien que le pic des concentrations sériques tendait à être inférieur et que les valeurs moyennes de demi-vie étaient augmentées jusqu’à environ 34 heures. Les patients âgés doivent être étroitement surveillés à la recherche de signes de toxicité du fentanyl et la dose de MATRIFEN doit être réduite si nécessaire (voir rubrique 4.4).
Insuffisance rénale
L’effet d’une insuffisance rénale sur la pharmacocinétique du fentanyl devrait être limité car l’excrétion urinaire du fentanyl sous forme inchangée est inférieure à 10 % et il n’existe pas de métabolites actifs connus éliminés par le rein. Cependant, comme l’effet de l’insuffisance rénale sur la pharmacocinétique du fentanyl n’a pas été évalué, la prudence est de rigueur (voir rubriques 4.2 et 4.4).
Insuffisance hépatique
Les patients présentant une insuffisance hépatique doivent être étroitement surveillés à la recherche de signes de toxicité du fentanyl et la dose des patchs de fentanyl transdermique doit être réduite si nécessaire (voir rubrique 4.4). Des données chez des patients cirrhotiques et des données de simulation chez des patients présentant différents grades d’insuffisance hépatique traités par du fentanyl transdermique indiquent que les concentrations de fentanyl peuvent être augmentées et que la clairance du fentanyl peut être diminuée en comparaison avec les sujets présentant une fonction hépatique normale. Les simulations indiquent que l’ASC à l’équilibre des patients atteints d’une insuffisance hépatique de grade B selon le score de Child-Pugh (score de Child-Pugh = 8) serait 1,36 fois plus importante que chez des patients présentant une fonction hépatique normale (grade A ; score de Child-Pugh = 5,5).
Pour les patients présentant une insuffisance hépatique de grade C (score de Child-Pugh = 12,5), les résultats indiquent que la concentration du fentanyl s’accumule à chaque administration, conduisant chez ces patients à une ASC à l’équilibre 3,72 fois plus grande.
Population pédiatrique
Les concentrations de fentanyl ont été mesurées chez plus de 250 enfants âgés de 2 à 17 ans ayant reçu du fentanyl par voie transdermique à une dose allant de 12,5 à 300 microgrammes/heure. La clairance ajustée au poids corporel (L/h/kg) s’avère être plus élevée d’environ 80 % chez les enfants âgés de 2 à 5 ans, et d’environ 25 % chez les enfants de 6 à 10 ans, comparativement à celle observée chez les enfants de 11 à 16 ans dont la clairance devrait être similaire à celle des adultes. Ces résultats ont été pris en compte pour déterminer les recommandations posologiques chez les enfants (voir rubriques 4.2 et 4.4).
5.3. Données de sécurité préclinique
Des études de toxicité standards sur la reproduction et le développement ont été conduites avec le fentanyl administré par voie parentérale. Dans une étude chez le rat, le fentanyl n’a pas eu d’effet sur la fertilité chez le mâle. Des études chez les rats femelles ont montré une réduction de la fertilité et une augmentation de la mortalité embryonnaire.
Les effets sur l’embryon étaient dus à la toxicité maternelle et non aux effets directs de la substance sur le développement embryonnaire. Il n’a pas été noté d’effet tératogène dans les études effectuées chez deux espèces (rat et lapin). Dans une étude sur le développement pré- et post-natal, le taux de survie de la progéniture a été significativement réduit aux doses qui entraînaient une légère diminution du poids des mères. Cet effet pourrait être dû soit à une modification des soins maternels, soit à un effet direct du fentanyl sur les petits. Il n’a pas été observé d’effets sur le développement somatique et sur le comportement de la progéniture.
Les tests de mutagénicité sur bactéries et chez les rongeurs ont donné des résultats négatifs. In vitro, le fentanyl a induit des effets mutagènes dans des cellules de mammifères comparables à ceux des autres analgésiques opioïdes. Un risque mutagène en cas d’utilisation à des doses thérapeutiques semble peu probable puisque ces effets ne sont apparus qu’à des concentrations élevées.
Une étude de cancérogenèse (injections sous-cutanées quotidiennes de chlorhydate de fentanyl hydrochloride pendant deux ans chez des rats Sprague Dawley) n’a donné aucun résultat indiquant un potentiel oncogène.
6.4. Précautions particulières de conservation
Pas de conditions particulières de conservation.
6.5. Nature et contenu de l'emballage extérieur
Chaque patch est conditionné dans un sachet thermoscellé avec sécurité enfant, composé de papier, d’aluminium et de polyéthylène téréphtalate (PET).
Toutes les présentations peuvent ne pas être commercialisées.
6.6. Précautions particulières d’élimination et de manipulation
Instructions pour l’élimination :
Les patchs usagés doivent être pliés de telle façon que la face adhésive du patch soit repliée sur elle-même et ils doivent ensuite être éliminés en toute sécurité. Tout médicament non utilisé ou déchet doit être éliminé conformément à la réglementation en vigueur.
Se laver les mains à l’eau après application et retrait du patch.
7. TITULAIRE DE L’AUTORISATION DE MISE SUR LE MARCHE
ISTITUTO GENTILI S.R.L.
VIA SAN GIUSEPPE COTTOLENGO 15
20143 MILANO
ITALIE
8. NUMERO(S) D’AUTORISATION DE MISE SUR LE MARCHE
· 34009 386 506 0 0 : dispositif transdermique en sachet (Papier/Aluminium/Polycrylonitrile); boîte de 2.
· 34009 386 507 7 8 : dispositif transdermique en sachet (Papier/Aluminium/Polycrylonitrile); boîte de 3.
· 34009 386 508 3 9 : dispositif transdermique en sachet (Papier/Aluminium/Polycrylonitrile); boîte de 4.
· 34009 386 510 8 9 : dispositif transdermique en sachet (Papier/Aluminium/Polycrylonitrile); boîte de 5.
· 34009 573 053 6 9 : dispositif transdermique en sachet (Papier/Aluminium/Polycrylonitrile); boîte de 8.
· 34009 573 054 2 0 : dispositif transdermique en sachet (Papier/Aluminium/Polycrylonitrile); boîte de 10.
· 34009 573 078 9 9 : dispositif transdermique en sachet (Papier/Aluminium/Polycrylonitrile); boîte de 16.
· 34009 573 055 9 8 : dispositif transdermique en sachet (Papier/Aluminium/Polycrylonitrile); boîte de 20.
9. DATE DE PREMIERE AUTORISATION/DE RENOUVELLEMENT DE L’AUTORISATION
[à compléter ultérieurement par le titulaire]
10. DATE DE MISE A JOUR DU TEXTE
[à compléter ultérieurement par le titulaire]
12. INSTRUCTIONS POUR LA PREPARATION DES RADIOPHARMACEUTIQUES
Stupéfiant : prescription limitée à 28 jours.
Délivrance fractionnée de 14 jours.
Prescription sur ordonnance répondant aux spécifications fixées par l’arrêté du 31 mars 1999
ANSM - Mis à jour le : 19/05/2025
MATRIFEN 25 microgrammes/heure, dispositif transdermique
Fentanyl
Veuillez lire attentivement cette notice avant d'utiliser ce médicament car elle contient des informations importantes pour vous.
· Gardez cette notice. Vous pourriez avoir besoin de la relire.
· Si vous avez d’autres questions, interrogez votre médecin, votre pharmacien ou votre infirmier/ère.
· Ce médicament vous a été personnellement prescrit (ou à votre enfant). Ne le donnez pas à d'autres personnes. Il pourrait leur être nocif, même si les signes de leur maladie sont identiques aux vôtres.
· Si vous ressentez un quelconque effet indésirable, parlez-en à votre médecin, votre pharmacien ou votre infirmier/ère. Ceci s’applique aussi à tout effet indésirable qui ne serait pas mentionné dans cette notice. Voir rubrique 4.
2. Quelles sont les informations à connaître avant d'utiliser MATRIFEN 25 microgrammes/heure, dispositif transdermique ?
3. Comment utiliser MATRIFEN 25 microgrammes/heure, dispositif transdermique ?
4. Quels sont les effets indésirables éventuels ?
5. Comment conserver MATRIFEN 25 microgrammes/heure, dispositif transdermique ?
6. Contenu de l’emballage et autres informations.
1. QU’EST-CE QUE MATRIFEN 25 microgrammes/heure, dispositif transdermique ET DANS QUELS CAS EST-IL UTILISE ?
Le nom de votre médicament est MATRIFEN.
Les patchs aident à soulager les douleurs très fortes et de longue durée :
· chez les adultes qui ont besoin d’un traitement continu de la douleur
· chez les enfants à partir de l’âge de 2 ans qui ont déjà reçu des médicaments opioïdes et qui ont besoin d’un traitement continu de la douleur.
MATRIFEN contient un médicament appelé fentanyl. Il appartient à la classe des opioïdes, des anti-douleurs puissants.
2. QUELLES SONT LES INFORMATIONS A CONNAITRE AVANT D’UTILISER MATRIFEN 25 microgrammes/heure, dispositif transdermique ?
N’utilisez jamais MATRIFEN 25 microgrammes/heure, dispositif transdermique :
· si vous êtes allergique au fentanyl ou à l'un des autres composants contenus dans ce médicament, mentionnés dans la rubrique 6.
· si vous souffrez d’une douleur de courte durée, telle qu’une douleur soudaine ou une douleur survenant après une intervention chirurgicale.
· si vous avez des difficultés à respirer, avec une respiration lente ou peu profonde.
N’utilisez pas ce médicament si l’une des situations ci-dessus s’applique à vous ou à votre enfant. En cas de doute, parlez-en avec votre médecin ou votre pharmacien avant d’utiliser MATRIFEN.
Avertissements et précautions
|
· MATRIFEN peut avoir des effets indésirables pouvant mettre la vie en danger chez les personnes qui n’utilisent pas déjà régulièrement des médicaments opioïdes sur ordonnance. · MATRIFEN est un médicament qui peut mettre la vie des enfants en danger, même si les patchs ont déjà servi. Gardez à l’esprit qu'un patch adhésif (utilisé ou non) peut être attractif pour un enfant ; si le patch se colle sur sa peau ou si l’enfant le porte à la bouche, cela peut être fatal. · Conserver ce médicament dans un endroit sûr et sécurisé, où d’autres personnes n’y ont pas accès – voir rubrique 5 pour de plus amples informations. |
Transfert de patch sur la peau d’une tierce personne
Le patch doit être appliqué exclusivement sur la peau de la personne pour laquelle il a été prescrit. Des cas ont été rapportés de transfert accidentel de patch sur la peau d’un membre de la famille du patient traité, lors de contacts physiques rapprochés ou en cas de partage du même lit. Le transfert accidentel du patch sur la peau d’une tierce personne (notamment un enfant) peut entraîner le passage du médicament contenu dans le patch à travers sa peau et provoquer des effets indésirables graves qui peuvent être fatals, telles que des difficultés respiratoires, avec une respiration lente ou peu profonde. Dans le cas où le patch se serait collé à la peau d’une autre personne, retirez-le immédiatement et consultez un médecin.
Soyez extrêmement prudent(e) avec MATRIFEN
Adressez-vous à votre médecin ou votre pharmacien avant d'utiliser ce médicament si l'un des cas suivants vous concerne, votre médecin devra peut-être vous surveiller plus étroitement si :
· Vous avez déjà eu des problèmes aux poumons ou de respiration
· Vous avez déjà eu des problèmes au cœur, au foie, aux reins, ou de basse pression artérielle
· Vous avez déjà eu une tumeur cérébrale
· Vous avez déjà eu des maux de tête persistants ou un traumatisme crânien
· Vous êtes âgé : vous pouvez être plus sensible aux effets de ce médicament
· Vous souffrez d’une maladie appelée « myasthénie grave », qui provoque une faiblesse musculaire et une fatigue
Si l'une des situations décrites ci-dessus vous concerne (ou en cas de doute), parlez-en à votre médecin ou votre pharmacien avant d'utiliser MATRIFEN.
Pendant que vous utilisez le patch, prévenez votre médecin si vous présentez des problèmes respiratoires pendant le sommeil.
Les opioïdes tels MATRIFEN peuvent provoquer des troubles respiratoires du sommeil tels que l’apnée du sommeil (pauses de la respiration pendant le sommeil) et une hypoxie du sommeil (faible taux d’oxygène dans le sang). Informez votre médecin si vous, votre partenaire ou votre aidant remarquez l’un des symptômes suivants :
· Pauses de la respiration pendant le sommeil
· Réveil nocturne en raison d’un essoufflement
· Difficultés à rester endormi(e)
· Endormissement excessif pendant la journée
Votre médecin pourrait décider de modifier votre dose.
Pendant que vous utilisez le patch, prévenez votre médecin si vous remarquez un changement dans la douleur que vous ressentez. Si vous sentez :
· que votre douleur n’est plus soulagée par le patch ;
· une augmentation de la douleur ;
· une modification de la manière dont vous ressentez la douleur (par exemple, vous ressentez la douleur dans une autre partie de votre corps) ;
· une douleur lorsque vous touchez quelque chose qui ne devrait pas entraîner de douleur ;
Ne modifiez pas la posologie vous-même. Votre médecin décidera lui-même s’il faut changer votre dose ou votre traitement.
Effets indésirables et MATRIFEN
· MATRIFEN peut provoquer une somnolence inhabituelle et être à l'origine d'une respiration plus lente ou peu profonde. Très rarement, ces problèmes respiratoires peuvent mettre la vie en danger, voire être mortels, en particulier chez les personnes n'ayant pas utilisé auparavant des anti-douleurs opioïdes puissants (comme MATRIFEN ou la morphine). Si vous, votre partenaire ou votre personnel soignant remarquez que la personne portant le patch est inhabituellement somnolente et a une respiration lente ou peu profonde :
o Retirez le patch
o Appelez un médecin ou rendez-vous immédiatement à l'hôpital le plus proche
o Maintenez la personne animée et faites-la parler autant que possible
· Si vous avez de la fièvre pendant l'utilisation de MATRIFEN, parlez-en à votre médecin ; cela peut augmenter la quantité de médicament qui passe à travers votre peau.
· MATRIFEN peut provoquer une constipation, demandez conseil auprès de votre médecin ou de votre pharmacien sur les moyens de prévenir ou de soulager la constipation.
Veuillez-vous reporter à la rubrique 4 pour la liste complète des effets indésirables éventuels.
Lorsque vous portez le patch vous devez éviter de l’exposer à des sources directes de chaleur, comme des coussins chauffants, couvertures électriques, bouillottes, lits chauffants ou lampes chauffantes ou lampes à bronzer. Ne prenez pas de bains de soleil, ni de bains chauds prolongés, de saunas ou de bains bouillonnants chauds, car vous risqueriez d'augmenter la quantité de médicament libérée par le patch.
Utilisation à long terme et tolérance
Ce médicament contient du fentanyl, qui est un opioïde. L’utilisation répétée d’analgésiques opioïdes peut entraîner une diminution de l’efficacité du médicament (vous vous y habituez, ce que l’on appelle la tolérance aux médicaments). Vous pouvez également devenir plus sensible à la douleur lors de l’utilisation du MATRIFEN. C’est ce que l’on appelle l’hyperalgésie. L’augmentation de la dose de vos dispositifs peut vous aider à réduire davantage vos douleurs pendant un certain temps, mais elle peut également être nocive. Si vous remarquez que votre médicament devient moins efficace, parlez-en à votre médecin. Votre médecin décidera s’il est préférable pour vous d’augmenter la dose ou de diminuer progressivement votre utilisation du MATRIFEN.
Dépendance et addiction
|
Ce médicament contient du fentanyl, qui est un médicament opioïde. Il peut entraîner une dépendance et/ou une addiction. |
L’utilisation répétée de MATRIFEN peut également entraîner une dépendance, des abus et une addiction, qui peuvent aboutir à un surdosage mettant la vie en danger. Le risque de ces effets indésirables peut augmenter avec une dose plus élevée et une durée d’utilisation plus longue. La dépendance ou l’addiction peut vous donner l’impression que vous ne contrôlez plus la quantité de médicament que vous devez utiliser ou la fréquence à laquelle vous devez l’utiliser. Vous pouvez avoir l’impression que vous devez continuer à prendre votre médicament, même s’il ne vous aide pas à soulager votre douleur.
Le risque de développer une dépendance ou une addiction varie d’une personne à l’autre. Vous pouvez présenter un risque plus élevé de développer une dépendance ou une addiction au MATRIFEN :
si vous ou un membre de votre famille avez déjà consommé de façon abusive ou été dépendant(e) de l’alcool, à des médicaments sur ordonnance ou à des substances illicites («addiction»);
si vous fumez;
si vous avez déjà présenté des troubles de l’humeur (dépression, anxiété ou trouble de la personnalité) ou avez été traité(e) par un psychiatre pour d’autres maladies mentales;
Si vous remarquez l’un des signes suivants lors de l’utilisation du MATRIFEN, il pourrait s’agir d’un signe que vous avez développé une dépendance ou une addiction ;
vous devez utiliser le médicament pendant une durée plus longue que celle indiquée par votre médecin;
vous devez utiliser une dose supérieure à la dose recommandée;
vous utilisez le médicament pour des raisons autres que celles qui vous ont été prescrites, par exemple «pour rester calme» ou «pour vous aider à dormir»;
vous avez tenté à plusieurs reprises et sans succès d’arrêter d’utiliser le médicament ou de contrôler son utilisation;
lorsque vous arrêtez de prendre le médicament, vous vous sentez mal, et vous vous sentez mieux lorsque vous reprenez le médicament («effets de sevrage»).
Si vous remarquez l’un de ces signes, parlez-en à votre médecin pour discuter de la meilleure voie de traitement pour vous, y compris le moment opportun pour arrêter et la façon d’arrêter en toute sécurité.
Symptômes de sevrage à l’arrêt de MATRIFEN
N’arrêtez pas brusquement de prendre ce médicament. Des symptômes de sevrage comme de l’impatience, des difficultés à dormir, de l’irritabilité, de l’agitation, de l’anxiété, le fait de sentir son cœur battre (palpitations), une tension artérielle élevée, des nausées ou des vomissements, de la diarrhée, une perte d’appétit, des tremblements, des frissonnements ou de la transpiration pourraient se manifester. Si vous souhaitez arrêter de prendre ce médicament, parlez-en d’abord avec votre médecin. Il vous expliquera comme procéder, habituellement en réduisant progressivement la dose de manière à maintenir au minimum tout effet désagréable de sevrage.
Autres médicaments et MATRIFEN 25 microgrammes/heure, dispositif transdermique
Informez votre médecin ou pharmacien si vous prenez, avez récemment pris ou pourriez prendre tout autre médicament. Ceci inclut les médicaments achetés sans ordonnance ou les médicaments à base de plantes. Vous devez également signaler à votre pharmacien que vous utilisez MATRIFEN si vous achetez un médicament à la pharmacie.
Votre médecin vous indiquera les médicaments que vous pouvez prendre en toute sécurité avec MATRIFEN. Vous pouvez avoir besoin d’une surveillance étroite si vous prenez ou arrêtez de prendre certains types de médicaments énumérés ci-dessous ; en effet, ils sont susceptibles de modifier la dose de MATRIFEN qui vous est nécessaire.
Vous devez en particulier informer votre médecin ou votre pharmacien si vous prenez :
· D’autres médicaments contre la douleur, par exemple d'autres anti-douleurs opioïdes (tels que buprénorphine, nalbuphine ou pentazocine) et certains anti-douleurs pour traiter les douleurs neuropathiques (gabapentine et prégabaline)
· Des médicaments utilisés pour traiter les troubles du sommeil (tels que témazépam, zaleplon ou zolpidem)
· Des médicaments utilisés pour traiter l’anxiété (tranquillisants, tels que alprazolam, clonazépam, diazépam, hydroxyzine ou lorazépam) et des médicaments utilisés pour traiter les troubles psychiques (anti-psychotiques, tels que aripiprazole, halopéridol, olanzapine, rispéridone ou phénothiazines)
· Des médicaments utilisés pour détendre les muscles (tels que cyclobenzaprine ou diazépam)
· Certains médicaments utilisés pour traiter la dépression, appelés ISRS ou IRSNa (tels que citalopram, duloxétine, escitalopram, fluoxétine, fluvoxamine, paroxétine, sertraline ou venlafaxine) – voir ci-dessous pour plus d'informations
· Certains médicaments utilisés pour traiter la dépression ou la maladie de Parkinson appelés IMAO (par exemple isocarboxazide, phénelzine, sélégiline ou tranylcypromine). Vous ne devez pas utiliser MATRIFEN au cours des 14 jours suivant l'arrêt de ces médicaments – voir ci-dessous pour plus d'informations
· Certains antihistaminiques, en particulier ceux qui font dormir (tels que chlorphéniramine, clémastine, cyproheptadine, diphénhydramine ou hydroxyzine)
· Certains antibiotiques utilisés pour traiter les infections (tels que érythromycine ou clarithromycine)
· Des médicaments utilisés pour traiter les infections fongiques (tels que itraconazole, kétoconazole, fluconazole ou voriconazole)
· Des médicaments utilisés pour traiter l'infection à VIH (tels que ritonavir)
· Des médicaments utilisés pour traiter des troubles du rythme cardiaque (tels que amiodarone, diltiazem ou vérapamil)
· Des médicaments contre la tuberculose (tels que rifampicine)
· Certains médicaments antiépileptiques (tels que carbamazépine, phénobarbital ou phénytoïne)
· Certains médicaments utilisés pour traiter les nausées ou le mal des transports (tels que phénothiazines)
· Certains médicaments utilisés pour traiter les brûlures d'estomac ou les ulcères (tels que cimétidine).
· Certains médicaments utilisés pour traiter l’angine de poitrine (douleur à la poitrine) ou une pression sanguine élevée (tels que la nicardipine).
· Certains médicaments utilisés pour traiter le cancer du sang (tels que idélalisib).
MATRIFEN avec les antidépresseurs
Le risque d'effets indésirables augmente si vous prenez des médicaments tels que certains antidépresseurs. MATRIFEN est susceptible d'interagir avec ces médicaments, et vous pourriez présenter des modifications de votre état mental se manifestant par une agitation, la perception d'informations visuelles, tactiles, auditives ou olfactives qui ne sont pas réelles (hallucinations), ainsi que d'autres effets comme des modifications de la pression artérielle, des battements cardiaques rapides, une augmentation de la température corporelle, des réflexes hyperactifs, un manque de coordination, une raideur musculaire, des nausées, des vomissements et une diarrhée (il pourrait s'agir de signes du syndrome sérotoninergique). En cas d'utilisation conjointe, votre médecin peut vouloir vous surveiller étroitement pour détecter ces effets secondaires, en particulier au début du traitement ou lors d'une modification de la dose de votre médicament.
Utilisation avec les dépresseurs du système nerveux central, incluant l'alcool et certains médicaments narcotiques
L’utilisation concomitante de MATRIFEN et de médicaments sédatifs tels que les benzodiazépines ou médicaments apparentés augmente le risque de somnolence, de difficultés respiratoires (dépression respiratoire), de coma et peut mettre la vie du patient en danger. Pour cette raison, l’utilisation concomitante ne doit être envisagée que lorsque d’autres traitements ne sont pas possibles.
Toutefois, si votre médecin vous prescrit MATRIFEN avec des médicaments sédatifs, la posologie et la durée du traitement concomitant doivent être limitées par votre médecin.
Informez votre médecin de tous les médicaments sédatifs que vous prenez et suivez attentivement les recommandations de votre médecin concernant la posologie. Il pourrait être utile d’informer vos amis ou votre famille des signes et symptômes indiqués ci-dessus. Contactez votre médecin si vous ressentez de tels symptômes.
Interventions chirurgicales
Si vous pensez que vous allez recevoir une anesthésie, prévenez le médecin ou le dentiste que vous utilisez MATRIFEN.
MATRIFEN avec de l’alcool
Ne buvez pas d'alcool pendant l'utilisation de MATRIFEN sans en avoir préalablement discuté avec votre médecin.
MATRIFEN est susceptible de provoquer une somnolence et un ralentissement de la respiration. La consommation d'alcool peut aggraver ces effets.
Si vous êtes enceinte ou que vous allaitez, si vous pensez être enceinte ou planifiez une grossesse, demandez conseil à votre médecin ou pharmacien avant de prendre ce médicament.
MATRIFEN ne doit pas être utilisé pendant la grossesse sans en avoir discuté avec votre médecin.
MATRIFEN ne doit pas être utilisé pendant l'accouchement car le médicament peut perturber la respiration du nouveau-né.
Vous ne devez pas utiliser MATRIFEN si vous allaitez. Vous ne devez pas allaiter pendant les 3 jours qui suivent le retrait du patch de MATRIFEN. Ces précautions sont nécessaires car le médicament peut passer dans le lait maternel.
Conduite de véhicules et utilisation de machines
Discutez avec votre médecin ou votre pharmacien si vous n’êtes pas sûr(e) de pouvoir conduire en toute sécurité pendant l’utilisation de ce médicament.
Lisez toutes les informations contenues dans cette notice pour vous guider.
Si vous n'êtes pas sûr, parlez-en à votre médecin ou à votre pharmacien.
3. COMMENT UTILISER MATRIFEN 25 microgrammes/heure, dispositif transdermique ?
Votre médecin déterminera le dosage de MATRIFEN le mieux adapté pour vous, en tenant compte de l’intensité de votre douleur, de votre état général et du type de traitement anti-douleur que vous avez reçu jusque-là.
Avant de commencer le traitement et régulièrement pendant le traitement, votre médecin discutera également avec vous de ce que vous pouvez attendre de l’utilisation du MATRIFEN, du moment et de la durée du traitement, du moment auquel vous devez contacter votre médecin et du moment où vous devez l’arrêter (voir également rubrique 2, symptômes de sevrage lors de l’arrêt du MATRIFEN).
Application et changement de patch
· La quantité de médicament dans chaque patch est suffisante pour une durée de 3 jours (72 heures).
· Vous devez donc changer de patch tous les trois jours, sauf indication contraire du médecin.
· Veillez à toujours retirer l’ancien patch avant d’appliquer le nouveau.
· Effectuez toujours le changement de patch à la même heure tous les 3 jours (72 heures).
· Si vous utilisez plusieurs patchs, changez-les tous en même temps.
· Notez le jour, la date et l’heure de l’application du patch, pour vous rappeler quand vous aurez besoin de remplacer le patch.
· Le tableau suivant est fourni à titre indicatif comme aide pour les changements de patch :
|
Application du patch le |
Changement du patch le |
|
Lundi |
Jeudi |
|
Mardi |
Vendredi |
|
Mercredi |
Samedi |
|
Jeudi |
Dimanche |
|
Vendredi |
Lundi |
|
Samedi |
Mardi |
|
Dimanche |
Mercredi |
Où appliquer le patch ?
Adultes
· Appliquer le patch sur une partie plane du haut du corps ou des bras (pas sur les articulations).
Enfants
· Appliquer toujours le patch en haut du dos pour que votre enfant ne puisse pas l’atteindre et l’enlever.
· Même dans ce cas, vérifiez régulièrement que le patch est bien resté en place sur la peau
· Il est primordial que votre enfant ne retire pas le patch pour le mettre dans sa bouche ; cela pourrait mettre sa vie en danger, voir lui être fatal.
· Vous devez surveiller très attentivement votre enfant pendant 48 heures après :
o L’application du premier patch
o L’application d’un patch à plus fort dosage
· L'effet maximal du premier patch peut être un peu long à obtenir. Votre enfant aura donc peut-être besoin d'autres anti-douleurs en attendant que le patch devienne efficace. Votre médecin en discutera avec vous.
Adultes et enfants :
Ne pas appliquer le patch sur
· Le même endroit deux fois de suite
· Les régions du corps qui bougent beaucoup (articulations), des zones de peau irritée ou coupée
· Une zone de peau très poilue. Si la peau est poilue, ne la rasez pas (le rasage irrite la peau). Coupez plutôt les poils le plus près possible de la peau.
Application du patch
Etape 1 : Préparation de la peau
· Vérifiez que la peau est complètement sèche, propre et fraîche avant l’application du patch.
· Si la peau a besoin d’être lavée, lavez-la uniquement à l’eau froide.
· N’utilisez pas de savons ou autres détergents, crèmes, crèmes hydratantes, huiles ou talc avant l’application du patch.
· Evitez d’appliquer le patch juste après un bain ou une douche chaude.
Etape 2 : Ouverture du sachet
· Chaque patch est scellé dans son propre sachet.
· Découpez le sachet le long de la ligne pointillée à l’aide de ciseaux.
· Coupez délicatement et complètement le bord du sachet pour éviter d’endommager le patch.
· Prenez les deux côtés de l’ouverture du sachet et écartez-les.
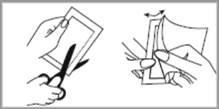
· Sortez le patch de l’emballage et utilisez-le immédiatement
· Gardez le sachet vide pour y jeter ultérieurement le patch usagé.
· Chaque patch ne doit être utilisé qu’une seule fois.
· Ne sortez pas le patch de son sachet avant d’être prêt(e) à l’utiliser.
· Vérifiez visuellement que le patch n’a pas été endommagé.
· N’utilisez pas le patch s’il a été divisé, coupé ou s’il semble endommagé.
· Ne divisez pas ou ne coupez pas le patch.
Etape 3 : Retrait de la pellicule de protection et application
· Assurez-vous que le patch sera recouvert de vêtements amples et ne le recouvrez pas d’une bande serrée ou élastique.
· Enlevez doucement la moitié de la pellicule de plastique brillante à partir du centre du patch. Essayez de ne pas toucher la partie adhésive du patch.
· Appliquer la partie adhésive du patch sur la peau.
· Retirez l’autre moitié de la pellicule de protection et appliquez la totalité du patch sur la peau en appuyant avec la paume de la main.
· Maintenez pendant au moins 30 secondes. Assurez-vous que le patch colle bien, notamment sur les bords.
Etape 4 : Elimination du patch
· Dès que vous retirez un patch, pliez-le fermement en deux de telle façon que la face adhésive se replie sur elle-même.
· Replacez-le ensuite dans le système de récupération fourni et rapportez-le à votre pharmacien.
· Veillez à tenir les patchs hors de la vue et de la portée des enfants ; en effet, même usagés, les patchs contiennent une quantité de médicament qui peut être nocive, voire fatale, pour les enfants.
Etape 5 : Lavage des mains
· Après avoir manipulé le patch, lavez-vous toujours les mains à l'eau propre uniquement.
Informations supplémentaires sur l'utilisation de MATRIFEN
Activités quotidiennes pendant l'utilisation des patchs
· Les patchs sont imperméables.
· Vous pouvez donc vous doucher ou prendre un bain pendant que vous portez un patch, mais vous ne devez pas frotter le patch lui-même.
· Si votre médecin l'accepte, vous pouvez faire de l'exercice ou du sport pendant le port du patch.
· Vous pouvez également pratiquer la natation pendant le port du patch mais :
o vous ne devez pas utiliser de bains bouillonnants chauds
o vous ne devez pas mettre de bande serrée ou élastique au-dessus du patch.
· Pendant le port du patch, vous devez éviter d’exposer le patch à des sources directes de chaleur, comme des coussins chauffants, couvertures électriques, bouillottes, lits chauffants ou lampes chauffantes ou lampes à bronzer. Ne prenez pas de bains de soleil, ni de bains chauds prolongés ou de saunas car vous risqueriez d'augmenter la quantité de médicament libérée par le patch.
En combien de temps le patch agit-il ?
· L'effet maximal du premier patch peut être un peu long à obtenir.
· Votre médecin pourra aussi vous donner d'autres anti-douleurs pour le premier jour et les autres jours si besoin.
· Ensuite, le patch doit permettre de soulager la douleur de façon continue, si bien que vous pourrez arrêter de prendre les autres anti-douleurs. Toutefois, votre médecin pourra encore vous prescrire des anti-douleurs supplémentaires de temps en temps.
Quelle sera la durée d’utilisation des patchs ?
· Les patchs de MATRIFEN sont indiqués pour les douleurs de longue durée. Votre médecin vous indiquera la durée prévue d’utilisation des patchs.
Si votre douleur devient plus intense
· Si votre douleur devient plus intense soudainement après l’application de votre dernier patch, vous devez vérifier votre patch. S'il n’adhère plus correctement ou s'il s’est décollé, remplacez-le (voir également la rubrique Si un patch se détache).
· Si votre douleur devient plus intense au fil du temps pendant l’utilisation de ces patchs, votre médecin pourra essayer un patch à plus fort dosage ou vous prescrire des antidouleurs supplémentaires (ou les deux).
· Si l’augmentation du dosage du patch ne vous aide pas, votre médecin pourra décider d’arrêter l’utilisation des patchs.
Si vous avez utilisé plus de MATRIFEN 25 microgrammes/heure, dispositif transdermique que vous n’auriez dû ou un patch avec un dosage erroné
Si vous avez appliqué trop de patchs ou un patch avec un mauvais dosage, retirez les patchs et contactez immédiatement un médecin.
Les signes de surdosage se manifestent notamment par des troubles respiratoires ou une respiration peu profonde, une fatigue, une somnolence extrême, une incapacité à penser clairement, à marcher ou à parler normalement et une sensation d'étourdissement, de vertige ou de confusion. Un surdosage peut également entraîner un trouble cérébral connu sous le nom de leucoencéphalopathie toxique.
Si vous oubliez de changer MATRIFEN 25 microgrammes/heure, dispositif transdermique
· Si vous avez oublié de changer votre patch, remplacez-le dès que vous constatez l'oubli et notez le jour et l'heure du remplacement de patch. Changez à nouveau le patch au bout de 3 jours (72 heures) comme d'habitude.
· Si vous avez beaucoup de retard pour changer votre patch, vous devez en discuter avec votre médecin car vous aurez peut-être besoin d'anti-douleurs supplémentaires mais n'appliquez pas de patch supplémentaire.
Si un patch se détache
· Si un patch se détache avant son changement prévu, appliquez immédiatement un nouveau patch et notez le jour et l'heure. Appliquez le patch sur une nouvelle zone de peau située sur :
o la partie supérieure du corps ou du bras
o la partie supérieure du dos de votre enfant.
· Signalez à votre médecin que le patch s'est détaché et laissez le nouveau patch en place pendant 3 jours (72 heures) ou en fonction des instructions de votre médecin, avant de le remplacer comme d'habitude.
· Si vos patchs continuent de se détacher, parlez-en à votre médecin, votre pharmacien ou à votre infirmier/ère.
Si vous arrêtez d’utiliser MATRIFEN 25 microgrammes/heure, dispositif transdermique
· Si vous arrêtez l'utilisation des patchs, ne recommencez pas le traitement sans en discuter au préalable avec votre médecin. En effet, la reprise du traitement peut nécessiter un autre dosage de patch.
· Si vous avez d’autres questions sur l’utilisation de ce médicament, demandez plus d’informations à votre médecin ou à votre pharmacien.
4. QUELS SONT LES EFFETS INDESIRABLES EVENTUELS ?
Si vous, ou votre partenaire ou votre personnel soignant remarquez que la personne portant le patch présente les symptômes suivants, retirez le patch et contactez un médecin ou rendez-vous immédiatement à l'hôpital le plus proche. Un traitement médical d'urgence peut être nécessaire.
· sensation de somnolence inhabituelle, respiration plus lente ou superficielle que d’habitude. Suivez les conseils ci-dessus et maintenez la personne portant le patch animée et faites-la parler autant que possible. Très rarement, ces difficultés respiratoires peuvent mettre la vie en danger, voire être fatales, notamment chez les personnes n'ayant pas utilisé auparavant des anti-douleurs opioïdes forts (comme MATRIFEN ou la morphine). (Peu fréquent, pouvant affecter jusqu'à 1 personne sur 100)
· gonflement soudain du visage ou de la gorge, irritation sévère, rougeur de la peau ou formation de cloques sur la peau. Il peut s’agir des signes d’une réaction allergique sévère. (Fréquence ne pouvant être estimée sur la base des données disponibles).
· convulsions (crises épileptiques). (Peu fréquent, pouvant affecter jusqu’à 1 personne sur 100).
· diminution de la conscience ou perte de conscience. (Peu fréquent, pouvant affecter jusqu’à 1 personne sur 100).
Les effets secondaires suivants ont également été signalés :
Très fréquents (pouvant affecter plus de 1 personne sur 10)
· Nausées, vomissements, constipation
· Envie de dormir (somnolence)
· Sensations vertigineuses
· Maux de tête
Fréquents (pouvant affecter jusqu’à 1 personne sur 10)
· Réaction allergique
· Perte d'appétit
· Difficultés pour dormir
· Dépression
· Sensation d'anxiété ou de confusion
· Perception d'informations visuelles, tactiles, auditives ou olfactives qui ne sont pas réelles (hallucinations)
· Tremblements ou spasmes musculaires
· Sensation inhabituelle au niveau de la peau, comme des picotements ou des fourmillements (paresthésies)
· Sensation de tournis (vertige)
· Battements cardiaques rapides ou irréguliers (palpitations, tachycardie)
· Pression artérielle élevée
· Essoufflement (dyspnée)
· Diarrhée
· Sécheresse buccale
· Douleur au niveau de l'estomac ou troubles de la digestion
· Transpiration excessive
· Démangeaisons, éruption cutanée ou rougeur de la peau
· Incapacité à uriner ou à vider complètement la vessie
· Sensation de fatigue extrême, de faiblesse ou de mal-être général
· Sensation de froid
· Gonflement des mains, des chevilles ou des pieds (œdèmes périphériques).
Peu fréquents (pouvant affecter jusqu’à 1 personne sur 100)
· Sensation d'agitation ou de désorientation
· Sensation de bonheur extrême (euphorie)
· Diminution de la sensibilité, notamment au niveau de la peau (hypoesthésie)
· Perte de mémoire
· Vision trouble
· Battements cardiaques lents (bradycardie) ou pression artérielle basse
· Coloration bleue de la peau due à un faible taux d'oxygène dans le sang (cyanose)
· Perte des contractions intestinales (iléus)
· Éruption cutanée associée à des démangeaisons (eczéma), réaction allergique ou autres troubles cutanés sur le site d'application du patch
· Syndrome pseudo-grippal
· Sensation de modification de la température corporelle
· Fièvre
· Contractions musculaires
· Troubles à obtenir et maintenir une érection (impuissance) ou problèmes lors des rapports sexuels
· Difficultés à avaler.
Rares (pouvant affecter jusqu’à 1 personne sur 1000)
· Constriction des pupilles (myosis)
· Interruptions ponctuelles de la respiration (apnée).
Les effets indésirables suivants ont aussi été rapportés, mais leur fréquence exacte est inconnue :
· Manque d’hormones sexuelles masculines (déficience androgénique)
· Délire (les symptômes peuvent inclure une combinaison d’agitation, d’impatience, de désorientation, de confusion, de peur, de troubles du sommeil et de cauchemars, ou le fait d’avoir des visions ou d’entendre des choses
· Vous pouvez développer une dépendance au MATRIFEN (voir rubrique 2).
Une éruption cutanée, une rougeur ou de légères démangeaisons peuvent apparaître sur le site d'application du patch. Ces effets sont généralement bénins et disparaissent après le retrait du patch. Dans le cas contraire, ou si le patch irrite trop votre peau, signalez-le à votre médecin.
L'utilisation répétée des dispositifs peut réduire l'efficacité du médicament (vous vous y habituez ou vous pouvez devenir plus sensible à la douleur) ou vous pouvez développer une dépendance à celui-ci.
Si vous passez d'un autre anti-douleur à MATRIFEN ou si vous arrêtez brutalement le traitement par MATRIFEN, des symptômes de sevrage pourront apparaître ; tels que des nausées, des vomissements, une diarrhée, une anxiété et des frissons. Si vous présentez l'un de ces effets, quel qu'il soit, signalez-le à votre médecin.
Des cas de symptômes de sevrage chez le nouveau-né ont également été rapportés lorsque les mères avaient utilisé MATRIFEN de façon prolongée pendant la grossesse.
Déclaration des effets secondaires
Si vous ressentez un quelconque effet indésirable, parlez-en à votre médecin ou votre pharmacien ou à votre infirmier/ère. Ceci s’applique aussi à tout effet indésirable qui ne serait pas mentionné dans cette notice. Vous pouvez également déclarer les effets indésirables directement via le système national de déclaration : Agence nationale de sécurité du médicament et des produits de santé (ANSM) et réseau des Centres Régionaux de Pharmacovigilance - Site internet : https://signalement.social-sante.gouv.fr/
En signalant les effets indésirables, vous contribuez à fournir davantage d’informations sur la sécurité du médicament.
5. COMMENT CONSERVER MATRIFEN 25 microgrammes/heure, dispositif transdermique ?
Lieu de conservation des patchs
Tenir les patchs (usagés et non utilisés) hors de la vue et de la portée des enfants.
Conservez ce médicament dans un endroit sûr et sécurisé, où d’autres personnes ne peuvent pas y accéder. Il peut causer des dommages graves et être fatal pour les personnes qui peuvent prendre ce médicament par accident, ou intentionnellement lorsqu’il n’a pas été prescrit pour elles.
Durée de conservation de MATRIFEN
N’utilisez pas ce médicament après la date de péremption indiquée sur l'emballage et le sachet après EXP. La date de péremption fait référence au dernier jour de ce mois. Si les patchs sont périmés, les rapporter à la pharmacie.
Comment éliminer les patchs usagés ou les patchs qui ne sont plus utilisés ?
Une exposition accidentelle à des patchs usagés ou non utilisés, en particulier chez l’enfant, peut entraîner la mort.
Les patchs usagés doivent être fermement pliés en deux de façon à ce que la face adhésive colle sur elle-même, puis placés dans le système de récupération fourni.
Le système sera ensuite replié et refermé par la languette et placé hors de la vue et de la portée d’autres personnes, particulièrement des enfants, jusqu’à leur élimination sécurisée.
Demandez à votre pharmacien d’éliminer les médicaments que vous n’utilisez plus.
Ne jetez aucun médicament au tout-à-l’égout ou avec les ordures ménagères. Ces mesures contribueront à protéger l’environnement.
6. CONTENU DE L’EMBALLAGE ET AUTRES INFORMATIONS
Ce que contient MATRIFEN 25 microgrammes/heure, dispositif transdermique
· La substance active est :
Fentanyl ................................................................................................................................2,75 mg
Pour un dispositif transdermique de 8,4 cm2 libérant 25 microgrammes de fentanyl par heure.
· Les autres composants sont :
Dipropylène glycol, hydroxypropyl cellulose, diméticone, adhésif siliconé (amino-résistant), acétate d'éthyle vinyle (EVA, membrane libératrice), film polyéthylène téréphtalate (PET, film externe), film polyester recouvert de fluoropolymère (film protecteur), encre d'impression.
MATRIFEN est un dispositif transdermique transparent et rectangulaire. Chaque dispositif est conditionné dans un sachet thermoscellé avec sécurité enfant, composé de papier, d’aluminium et de polyéthylène téréphtalate (PET).
Les dispositifs transdermiques portent le nom de marque, la substance active et le dosage imprimés à l’aide d’une encre colorée : encre rouge.
Boîte de 1, 2, 3, 4, 5, 8, 10, 16 ou 20 dispositifs transdermiques.
Toutes les présentations peuvent ne pas être commercialisées.
Titulaire de l’autorisation de mise sur le marché
VIA SAN GIUSEPPE COTTOLENGO 15
20143 MILANO
ITALIE
Exploitant de l’autorisation de mise sur le marché
LABORATOIRES LEURQUIN MEDIOLANUM S.A.S.
86/88 RUE LOUIS AMPERE
ZI DES CHANOUX
93330 NEUILLY SUR MARNE
LTS LOHMANN THERAPIE SYSTEME AG
LOHMANNSTRASSE 2
D – 56626 ANDERNACH
ALLEMAGNE
Noms du médicament dans les Etats membres de l'Espace Economique Européen
Ce médicament est autorisé dans les Etats membres de l'Espace Economique Européen sous les noms suivants : Conformément à la réglementation en vigueur.
[À compléter ultérieurement par le titulaire]
La dernière date à laquelle cette notice a été révisée est :
[à compléter ultérieurement par le titulaire]
{mois AAAA}.
Des informations détaillées sur ce médicament sont disponibles sur le site Internet de l’ANSM (France).