Dernière mise à jour le 29/06/2026
ENANTONE LP 3,75 mg, poudre et solvant pour suspension injectable (S.C. ou I.M.) à libération prolongée
Ce médicament n'est ou ne sera bientôt plus disponible sur le marché.
Si vous prenez actuellement ce médicament, il vous est recommandé d'en parler avec votre médecin
ou avec votre pharmacien qui pourra vous orienter vers un autre traitement.
Indications thérapeutiques
ANALOGUE DE L'HORMONE ENTRAINANT LA LIBERATION DE GONADOTROPHINES - code ATC : L02AE02
Ce médicament contient une substance active, la leuproréline, qui a la même action qu'une hormone naturelle (la GnRH). Cette hormone contrôle la production d'autres hormones : la testostérone, les œstrogènes et la progestérone.
L'utilisation sur une longue durée de ce médicament permet d'arrêter la production des hormones sexuelles (testostérone, œstrogène et progestérone) aussi bien chez l'homme que chez la femme et l’enfant.
ENANTONE est donc utilisé :
· pour traiter certains cancers qui ont besoin de ces hormones pour se développer (et notamment le cancer de la prostate chez l'homme, seul ou associé à des rayons (radiothérapie), et le cancer du sein chez la femme) ;
· pour traiter un développement excessif de la muqueuse utérine en dehors de l'utérus (endométriose) ou pour certaines tumeurs bénignes de l'utérus (fibromes utérins) chez la femme ;
· ou pour traiter des pubertés précoces centrales de l'enfant (avant 9 ans chez la fille, avant 10 ans chez le garçon).
Service médical rendu (SMR)
Les libellés affichés ci-dessous ne sont que des résumés ou extraits issus des avis rendus par la Commission de la Transparence. Seul l'avis complet de la Commission de la Transparence fait référence.
Cet avis est consultable à partir du lien `Avis du jj/mm/aaaa` ou encore sur demande auprès de la HAS (plus d'informations dans l'aide). Les avis et synthèses d'avis contiennent un paragraphe sur la place du médicament dans la stratégie thérapeutique.
| Valeur du SMR | Avis | Motif de l'évaluation | Résumé de l'avis |
|---|---|---|---|
| Important | Avis du 05/12/2018 | Renouvellement d'inscription (CT) | Le service médical rendu par ENANTONE reste important dans les indications de l’AMM. |
Amélioration du service médical rendu (ASMR)
Les libellés affichés ci-dessous ne sont que des résumés ou extraits issus des avis rendus par la Commission de la Transparence. Seul l'avis complet de la Commission de la Transparence fait référence.
Cet avis est consultable à partir du lien `Avis du jj/mm/aaaa` ou encore sur demande auprés de la HAS (plus d'informations dans l'aide). Les avis et synthéses d'avis contiennent un paragraphe sur la place du médicament dans la stratégie thérapeutique.
| Valeur de l'ASMR | Avis | Motif de l'évaluation | Résumé de l'avis |
|---|---|---|---|
| V (Inexistant) | Avis du 18/07/2007 | Extension d'indication | ENANTONE LP 3,75 mg et 11,25 mg, n'apportent pas d'amélioration du service médical rendu (niveau V) par rapport aux autres analogues de la GnRH dans le traitement du cancer de la prostate à un stade localement avancé. |
ANSM - Mis à jour le : 10/07/2025
ENANTONE LP 3,75 mg, poudre et solvant pour suspension injectable (S.C. ou I.M.) à libération prolongée
2. COMPOSITION QUALITATIVE ET QUANTITATIVE
Leuproréline........................................................................................................................ 3,75 mg
Pour un flacon.
Pour la liste complète des excipients, voir rubrique 6.1.
Poudre et solvant pour suspension injectable (S.C. ou I.M.) à libération prolongée.
4.1. Indications thérapeutiques
o Traitement du cancer de la prostate localement avancé ou métastatique.
o Traitement concomitant et adjuvant à la radiothérapie externe dans le cancer de la prostate localement avancé (stade T3-T4 de la classification TNM ou stade C de la classification AUA)
· Traitement de la puberté précoce centrale (avant 9 ans chez la fille, avant 10 ans chez le garçon).
· Traitement de l'endométriose à localisation génitale et extragénitale (du stade I au stade IV).
L'expérience clinique relative au traitement de l'endométriose est limitée aux femmes âgées de 18 ans et plus.
Durée du traitement : voir rubrique 4.2.
· Traitement du cancer du sein métastatique hormono-dépendant de la femme pré-ménopausée quand une suppression de la fonction ovarienne est nécessaire.
· Traitement préopératoire des fibromes utérins :
o associés à une anémie (avec un taux d'hémoglobine inférieur ou égal à 8 g/dL),
o dans le cas où une réduction de la taille du fibrome est nécessaire pour faciliter ou modifier la technique opératoire : chirurgie endoscopique, chirurgie transvaginale.
La durée du traitement est limitée à 3 mois.
4.2. Posologie et mode d'administration
Cancer de la prostate
Une injection sous-cutanée qui sera renouvelée toutes les quatre semaines.
Dans l'indication « Traitement concomitant et adjuvant à la radiothérapie externe dans le cancer de la prostate localement avancé (stade T3-T4 de la classification TNM ou stade C de la classification AUA) », il est recommandé de poursuivre le traitement pendant 3 ans.
Chez les patients traités par analogues de la GnRH pour un cancer de la prostate métastatique, le traitement est généralement maintenu en cas de développement d’un cancer de la prostate résistant à la castration. Il convient de tenir compte des recommandations en vigueur.
Puberté précoce centrale
Le traitement des enfants par la leuproréline doit se faire sous la surveillance générale d'un endocrino-pédiatre, d'un pédiatre ou d’un endocrinologue ayant une expertise dans le traitement de la puberté précoce centrale.
Le schéma posologique doit être adapté individuellement.
La dose initiale recommandée dépend du poids corporel.
Enfants d'un poids supérieur ou égal à 20 kg :
La dose administrée sera de 2 mL (3,75 mg d’acétate de leuproréline) de suspension reconstituée à partir des 44,1 mg de microcapsules dans 2 mL de solvant, une fois par mois, en une seule injection sous-cutanée.
Enfants d'un poids inférieur à 20 kg :
Dans ces rares cas, la dose suivante sera administrée en fonction de l’activité clinique de la puberté précoce centrale :
1 mL (1,88 mg d’acétate de leuproréline) soit la moitié du volume de la suspension reconstituée à partir des 44,1 mg de microcapsules dans 2 mL de solvant, une fois par mois, en une seule injection sous-cutanée.
Le reste de la suspension doit être éliminé. La prise de poids de l’enfant doit être surveillée.
Selon l’activité de la puberté précoce centrale, il peut être nécessaire d’augmenter la dose en présence d’une suppression insuffisante (détection clinique par exemple spotting ou suppression gonadotrope insuffisante confirmée par le test LHRH). La dose efficace minimale mensuelle à administrer doit être déterminée par un test LHRH.
Des abcès stériles au site d’injection apparaissent souvent quand la leuproréline est administrée en intramusculaire à des doses supérieures aux doses recommandées. C’est pourquoi, dans ce cas, le médicament doit être administré en sous-cutané (voir rubrique 4.4).
Il est recommandé d’utiliser les volumes les plus faibles possible pour l’injection chez l’enfant pour diminuer les désagréments associés à l’injection intramusculaire/sous-cutanée.
La durée du traitement dépend des paramètres cliniques au début du traitement ou pendant le traitement (prévision de la taille finale, vitesse de la croissance, âge osseux et/ou accélération de l’âge osseux) et est décidée en accord avec le pédiatre et le représentant légal et le cas échéant, l’enfant traité. L’âge osseux doit être surveillé pendant le traitement à 6-12 mois d’intervalle.
Chez les filles ayant un âge osseux supérieur à 12 ans et chez les garçons ayant un âge osseux supérieur à 13 ans, l’arrêt du traitement doit être envisagé en prenant en compte les paramètres cliniques.
Chez les filles, une grossesse doit être exclue avant le début du traitement. La survenue d’une grossesse pendant le traitement ne peut pas être exclue. Dans ce cas, un avis médical doit être demandé.
Note :
L’administration doit se faire à intervalle de 30 ± 2 jours pour prévenir la réapparition des symptômes de la puberté précoce.
Endométriose
Le traitement doit être débuté dans les cinq premiers jours du cycle.
Une injection sous-cutanée ou intramusculaire qui sera renouvelée toutes les quatre semaines.
Durée : l'endométriose, quel que soit le stade, sera traitée au maximum durant 6 mois.
Toutefois dans les cas associés à une symptomatologie pelvienne chronique et en l'absence de désir immédiat de grossesse, la durée de traitement peut être portée à un an en associant ENANTONE à une hormonothérapie de substitution (« add-back therapy ») à partir du 3ème mois.
Le schéma thérapeutique validé est : ENANTONE en association avec du valérate d'œstradiol micronisé 2 mg par jour administré par voie orale et de la promégestone 0,5 mg par jour administrée par voie orale.
Il n'est pas souhaitable d'entreprendre une seconde cure par ENANTONE ou par un autre analogue de la GnRH.
Cancer du sein métastatique
Une injection sous-cutanée ou intramusculaire qui sera renouvelée toutes les quatre semaines.
Traitement préopératoire des fibromes utérins
Une injection sous-cutanée ou intramusculaire qui sera renouvelée toutes les quatre semaines.
Durée : le traitement sera limité à 3 mois.
Ce médicament ne doit pas être prescrit en cas :
·
· Hypersensibilité à la substance active, aux dérivés de la GnRH, aux analogues de la GnRH ou à l'un des excipients mentionnés à la rubrique 6.1
· Grossesse et allaitement
· Chez les filles avec puberté précoce centrale : hémorragie génitale de cause non déterminée
· Dans le cas d'un traitement de l'endométriose associant ENANTONE et une hormonothérapie de substitution, les contre-indications d'utilisation d'oestroprogestatifs doivent être respectées
4.4. Mises en garde spéciales et précautions d'emploi
Il existe un risque accru de survenue de dépression incidente (qui peut être sévère) chez les patients traités par agonistes de la GnRH, tels que la leuproréline. Les patients doivent être informés en conséquence et traités de façon appropriée si des symptômes apparaissent.
Une apoplexie hypophysaire peut survenir de façon très rare lors de la première administration chez des patients présentant un adénome hypophysaire, en particulier gonadotrope. Les symptômes qui peuvent évoquer cette pathologie sont des céphalées et des troubles visuels.
Les traitements par privation androgénique peuvent allonger l’intervalle QT.
Chez les patients ayant un antécédent ou des facteurs de risque d’allongement de l’intervalle QT et chez les patients recevant un traitement concomitant qui risque d’allonger l’intervalle QT (voir rubrique 4.5), le rapport bénéfice risque incluant le risque potentiel de torsade de pointe devra être évalué avant l’instauration du traitement par ENANTONE.
Des cas de convulsions ont été rapportés chez les patients traités par la leuproréline après sa mise sur le marché. Ces convulsions ont été observées aussi bien chez les adultes que chez les enfants, avec ou sans antécédents, troubles, ou facteurs de risque liés aux convulsions.
Des cas d’hypertension intracrânienne idiopathique (méningite séreuse) ont été rapportés chez des patients recevant de la leuproréline. Les patients doivent être avertis de la possibilité de signes et symptômes d’hypertension intracrânienne idiopathique, notamment des céphalées sévères ou récurrentes, de troubles visuels et d’acouphènes. En présence d’une hypertension intracrânienne idiopathique, l’interruption du traitement par leuproréline doit être envisagée.
Réactions indésirables cutanées sévères
Des réactions indésirables cutanées sévères (SCAR), dont le syndrome de Stevens-Johnson (SSJ), et la nécrolyse épidermique toxique (NET, ou syndrome de Lyell), qui peuvent engager le pronostic vital ou être fatals, ont été rapportés en association avec le traitement par leuproréline. Au moment de la prescription, les patients doivent être informés des signes et symptômes, et surveillés étroitement en cas de réactions cutanées graves. En cas d’apparition de signes et symptômes évocateurs de ces réactions, le traitement par leuproréline doit être arrêté immédiatement et un autre traitement doit être envisagé (le cas échéant).
Cancer de la prostate
Mise en route du traitement : des cas isolés d'aggravation, le plus souvent transitoire, des symptômes cliniques (douleurs osseuses en particulier) ont été rapportés lors de la mise en route d'un traitement par les analogues de la GnRH. Ils justifient une surveillance médicale particulièrement attentive durant les premières semaines du traitement chez les patients porteurs d'une obstruction des voies excrétrices et chez les malades présentant des métastases vertébrales (voir rubrique 4.8).
Pour la même raison, la mise en route du traitement chez les sujets présentant des signes prémonitoires de compression médullaire doit être soigneusement pesée.
Une élévation transitoire des phosphatases acides en début de traitement peut être observée.
Cancer de la prostate, endométriose, cancer du sein métastatique et traitement préopératoire des fibromes utérins
Chez l’homme, une privation à long terme d’androgènes par orchidectomie bilatérale ou par administration d’analogues de la GnRH est associée à un risque accru de perte osseuse qui, chez les patients présentant des facteurs de risque additionnels, peut entrainer une ostéoporose et accroître le risque de fracture osseuse (voir rubrique 4.8).
Chez la femme, une privation à long terme d’œstrogènes par ovariectomie bilatérale, une ablation des ovaires, ou l'administration d'analogues de la GnRH, est associée à un risque accru de perte osseuse qui, chez les patients présentant des facteurs de risque additionnels, peut entrainer une ostéoporose et accroître le risque de fracture osseuse (voir rubrique 4.8).
L’inhibition de la production d'hormones sexuelles endogènes, comme lors du traitement de privation d'androgènes (tel qu’identifié à partir de données épidémiologiques) ou de privation en œstrogènes (par exemple chez les femmes ménopausées) est associée à des modifications métaboliques (par exemple réduction de la tolérance au glucose, stéatose hépatique ou aggravation de diabètes préexistants) ainsi qu’à un risque accru de maladies cardiovasculaires.
Cependant, les données prospectives n'ont pas confirmé le lien entre le traitement par des analogues de la GnRH et une augmentation de la mortalité cardiovasculaire. Les patients à risque élevé de maladies métaboliques ou cardiovasculaires doivent faire l’objet d’une surveillance appropriée.
Endométriose, cancer du sein métastatique et traitement pré-opératoire des fibromes utérins
Il est indispensable de vérifier avant toute prescription d'ENANTONE LP 3,75 mg, l'absence de grossesse.
A l’initiation du traitement, une aggravation transitoire de l'état clinique du patient peut survenir. Cependant, cela peut disparaître avec la poursuite du traitement.
Avant l'administration de leuproréline, des saignements vaginaux anormaux doivent être recherchés, le diagnostic doit être confirmé et une prise en charge appropriée doit être mise en place.
Il est conseillé, comme avec tous les autres agonistes de la GnRH, de surveiller les patientes présentant un état ostéoporotique lors de traitement prolongé.
En cas d'association d'une hormonothérapie de substitution au traitement de l'endométriose par ENANTONE, les mises en garde et les précautions d'emploi des oestroprogestatifs doivent être respectées.
Cancer du sein métastatique
Chez les patientes présentant un cancer du sein, comme avec les autres agonistes de la GnRH, il peut y avoir une augmentation possible et transitoire, en début de traitement, des signes et symptômes devant être traités de façon symptomatique.
Précautions d'emploi
Cancer de la prostate
Il peut être utile de vérifier périodiquement la testostéronémie qui ne doit pas être supérieure à 1 ng/mL.
La réponse thérapeutique peut être évaluée au niveau osseux par examen scintigraphique et/ou scannographique; au niveau prostatique, la réponse sera appréciée par (outre l'examen clinique et le toucher rectal) échographie et/ou par examen scannographique.
Endométriose, cancer du sein et traitement pré-opératoire des fibromes utérins
L'administration régulière toutes les quatre semaines d'une injection d'ENANTONE LP 3,75 mg entraîne constamment une aménorrhée hypogonadotrophique.
En dehors du premier mois, la survenue de métrorragies au cours du traitement est anormale et doit conduire à la réalisation d'un dosage du taux d'œstradiol plasmatique. Si celui-ci est inférieur à 50 pg/mL, la recherche d'éventuelles lésions organiques associées doit être effectuée.
En cas de traitement des fibromes utérins, des métrorragies sévères peuvent survenir au cours du traitement en raison d'une dégénérescence aiguë des fibromes. Les patientes doivent être averties de la possibilité de survenue de saignements anormaux ou de douleurs.
Dans le cas d'un traitement de l'endométriose associant ENANTONE et une hormonothérapie de substitution (« add-back therapy »), des métrorragies peuvent se produire, en relation avec le traitement hormonal de substitution.
En cas d'association de l'hormonothérapie de substitution au traitement de l'endométriose par ENANTONE, des précautions particulières sont nécessaires afin d'écarter des patientes présentant une thrombophilie.
En cas d'administration prolongée, il est recommandé de surveiller la masse osseuse afin de mieux prendre en compte le risque d'ostéoporose (voir rubrique 4.8).
Puberté précoce centrale :
Avant de commencer le traitement, un diagnostic précis de puberté précoce centrale idiopathique et/ou neurogénique doit être posé. Chez les filles, une grossesse doit être exclue.
Le traitement est un traitement au long cours, adapté de manière individuelle.
ENANTONE LP 3,75 mg doit être administré dans la mesure du possible de manière régulière chaque mois. Un retard exceptionnel de quelques jours dans la date de l’injection (30 ± 2 jours) n’influence pas les résultats du traitement.
Dans le cas d’un abcès stérile au site d’injection (principalement rapporté en cas d’administration IM de doses supérieures aux doses recommandées), l’absorption de leuproréline peut être diminuée. Dans ce cas les paramètres hormonaux (testostérone, estradiol) doivent être surveillés à intervalle de 2 semaines (voir rubrique 4.2).
Le traitement des enfants avec une tumeur cérébrale évolutive doit faire l’objet d’une évaluation individuelle attentive du rapport bénéfices risques.
Après la première injection chez les filles, des saignements vaginaux, des spotting et des sécrétions peuvent apparaître en signe de privation hormonale. L’apparition de saignements vaginaux au-delà des deux premiers mois de traitement doit être explorée.
Le traitement par les agonistes de la GnRH peut entraîner une diminution de la densité minérale osseuse (DMO). Toutefois, après l'arrêt du traitement, le bilan ultérieur de la masse osseuse est préservé et le pic de croissance de la masse osseuse à la fin de la puberté ne semble pas être affecté par le traitement.
Une épiphysiolyse fémorale peut se produire après l’arrêt du traitement. Il se pourrait que ce soit consécutif à l’affaiblissement du cartilage de conjugaison en raison des faibles concentrations en œstrogène pendant le traitement par les agonistes de la GnRH et à l’augmentation de la vitesse de croissance qui se produit après l’arrêt du traitement et qui faciliterait le déplacement des épiphyses.
Sodium
Ce médicament contient moins de 1 mmol (23 mg) de sodium par dose, c’est-à-dire qu’il est essentiellement « sans sodium ».
4.5. Interactions avec d'autres médicaments et autres formes d'interactions
Les traitements par privation androgénique pouvant allonger l’intervalle QT, l’utilisation concomitante d’ENANTONE avec des médicaments connus pour allonger l’intervalle QT ou avec des médicaments induisant des torsades de pointe tels que les antiarythmiques de classe IA (par exemple quinidine, disopyramide) ou de classe III (par exemple amiodarone, sotalol, dofetilide, ibutilide), méthadone, moxifloxacine, antipsychotiques, etc devra être évaluée avec prudence (voir rubrique 4.4).
4.6. Fertilité, grossesse et allaitement
Grossesse
Les données actuellement disponibles sur les effets de cette classe de produits au cours de a grossesse sont les suivants :
Chez l'animal, les études effectuées n'ont pas mis en évidence d'effet tératogène. En l'absence d'effet tératogène chez l'animal, un effet malformatif dans l'espèce humaine n'est pas attendu.
En effet, à ce jour, les substances responsables de malformations dans l'espèce humaine se sont révélées tératogènes chez l'animal au cours d'études bien conduites sur deux espèces.
En clinique, l'utilisation par mégarde d'analogues de la GnRH, sur des effectifs limités de grossesses exposées, n'a révélé aucun effet malformatif ou fœtotoxique particulier à ce jour.
Toutefois, des études complémentaires sont nécessaires pour vérifier les conséquences d'une exposition en cours de grossesse.
Cependant, par mesure de précaution, ENANTONE ne devra pas être utilisé chez les femmes enceintes.
Les femmes en âge de procréer doivent utiliser une contraception (méthode non hormonale) pendant le traitement par leuproréline et jusqu’à la reprise des règles.
Allaitement
En l'absence de données concernant le passage de ce médicament dans le lait et les effets éventuels sur l'enfant nourri au sein, ENANTONE ne devra pas être utilisé en cas d'allaitement.
4.7. Effets sur l'aptitude à conduire des véhicules et à utiliser des machines
Cancer de la prostate :
Les effets indésirables rapportés avec une fréquence supérieure ou égale à 0,5% chez des patients recevant de la leuproréline sont listés ci-dessous selon la classification MedDRA (par classe organe et de fréquence absolue). Les fréquences sont définies comme suit : très fréquent (> 1/10), fréquent (> 1/100, < 1/10), peu fréquent (> 1/1 000, < 1/100), rare (> 1/10 000, < 1/1 000), très rare (< 1/10 000), indéterminée (ne peut être estimée sur la base des données disponibles).
|
Classes de systèmes d’organes |
Très fréquent |
Fréquent |
Peu fréquent |
Rare |
Très rare |
Indéterminée |
|
Investigations |
Prise de poids |
Augmentation de l’alanine aminotransférase, augmentation de la gamma glutamyl transférase, augmentation de la lactico déshydrogénase |
Augmentation de la phosphatase alcaline |
|
|
|
|
Affections hématologiques et du système lymphatique
|
|
Anémie |
|
|
|
|
|
Affections du système nerveux |
|
Céphalées |
Étourdissement, paresthésie |
|
Apoplexie hypophysaire (après administration initiale chez des patients porteurs d’un adénome hypophysaire) |
Convulsions Hypertension intracrânienne idiopathique (méningite séreuse) (voir rubrique 4.4) |
|
Affections respiratoires, thoraciques et médiastinales |
|
Dyspnée
|
|
|
|
Pneumopathie interstitielle |
|
Affections gastro-intestinales |
|
Nausées, constipation |
Vomissement, diarrhées |
|
|
|
|
Affections du rein et des voies urinaires |
Nycturie, dysurie |
Pollakiurie, hématurie |
|
|
|
|
|
Affections de la peau et du tissu sous-cutané |
Sudation |
Prurit |
Éruption |
Alopécie |
|
Syndrome de Stevens-Johnson (SSJ) / nécrolyse épidermique toxique (NET ou syndrome de Lyell) (voir rubrique 4.4), éruption cutanée toxique, érythème polymorphe |
|
Douleur osseuse, faiblesse musculaire |
Douleur dorsale, arthralgie |
Myalgie, douleur des extrémités |
|
|
|
|
|
Troubles du métabolisme et de la nutrition |
|
Anorexie, diminution de l’appétit |
|
Modification du métabolisme du glucose (diminution ou augmentation) |
|
Intolérance au glucose |
|
Infections et infestations |
|
Infection des voies urinaires |
|
|
|
|
|
Affections cardiaques |
|
|
|
|
|
Allongement de l’intervalle QT (voir rubriques 4.4 et 4.5) |
|
Affections vasculaires |
Bouffées de chaleur |
Lymphœdème |
|
|
|
|
|
Troubles généraux et anomalies au site d'administration |
Fatigue |
Réaction au point d’injection, douleur, œdème périphérique, douleur au point d’injection |
Douleur thoracique, asthénie, œdème au point d’injection |
|
|
|
|
Affections hépatobiliaires |
|
Fonction hépatique anormale |
|
|
|
|
|
Affections des organes de reproduction et du sein |
Impuissance, atrophie testiculaire, trouble testiculaire |
Gynécomastie |
|
|
|
|
|
Affections psychiatriques |
Diminution de la libido |
Altération d’humeur, dépression, lors des traitements à long terme, trouble du sommeil |
Altération d’humeur, dépression, lors des traitements à court terme |
|
|
|
|
Tumeurs bénignes, malignes et non précisées |
|
|
|
|
Adénome hypophysaire |
|
|
Affections du système immunitaire |
|
|
|
|
Réaction anaphylactique (urticaire, angiœdème, choc anaphylactique) |
|
|
Affections oculaires |
|
|
|
|
|
Troubles visuels |
Mise en route du traitement (voir rubrique 4.4) : elle est parfois accompagnée d'une accentuation des signes cliniques et des symptômes (en particulier des douleurs osseuses).
Quelques cas d'aggravation d'une hématurie préexistante ou d'une obstruction urinaire, de sensations de faiblesse ou de paresthésies des membres inférieurs ont été signalés avec les analogues de la GnRH.
Ces manifestations sont habituellement transitoires, disparaissant en 1 à 2 semaines lors de la poursuite du traitement. Néanmoins, la possibilité d'une exacerbation temporaire des symptômes durant les premières semaines de traitement doit être prise en compte chez des malades menacés par la survenue de désordres neurologiques ou chez ceux présentant une obstruction urinaire.
En cours de traitement : Les effets indésirables les plus fréquents liés à l'activité pharmacologique du produit sont : bouffées de chaleur, sueurs, impuissance, diminution de la libido et diminution de la taille des testicules.
Avec d'autres agonistes de la GnRH, d'autres effets indésirables ont été observés : fièvre (de l'ordre de 3 à 4 %), palpitations, troubles de la vue, chute des cheveux, modification de la tolérance au glucose, leucopénie et thrombocytopénie (moins de 1%).
Variations de la densité osseuse : Une diminution de la densité osseuse a été rapportée dans la littérature médicale chez les hommes ayant bénéficié d’une orchidectomie ou ceux traités par un agoniste de la GnRH. Il est probable qu’un traitement à long terme par la leuproréline révèle des signes d’aggravation d’ostéoporose, en ce qui concerne l’augmentation du risque de fracture d’origine ostéoporotique.
Endométriose et traitement préopératoire des fibromes utérins :
Les effets indésirables rapportés avec une fréquence supérieure ou égale à 0,5% chez des patientes recevant de la leuproréline sont listés ci-dessous selon la classification MedDRA (par classe organe et de fréquence absolue). Les fréquences sont définies comme suit : très fréquent (> 1/10), fréquent (> 1/100, < 1/10), peu fréquent (> 1/1 000, < 1/100), rare (> 1/10 000, < 1/1 000), très rare (< 1/10 000), indéterminée (ne peut être estimée sur la base des données disponibles).
|
Classes de systèmes d’organes |
Très fréquent |
Fréquent |
Peu fréquent |
Rare |
Très rare |
Indéterminée |
|
Investigations |
|
Prise de poids, perte de poids |
Augmentation de l’alanine aminotransférase, augmentation de la lactico déshydrogénase, augmentation du phosphore sanguin, augmentation des triglycérides |
|
|
|
|
Affections hématologiques et du système lymphatique
|
|
|
|
|
|
Anémie |
|
Affections cardiaques |
|
|
Palpitations |
|
|
|
|
Affections du système nerveux |
Céphalées |
Étourdissements, paresthésies, hypertonie |
Hypoesthésie |
|
Apoplexie hypophysaire (après administration initiale chez des patients porteurs d’un adénome hypophysaire) |
Convulsions Hypertension intracrânienne idiopathique (méningite séreuse) (voir rubrique 4.4) |
|
Affections oculaires |
|
|
Trouble visuel, amblyopie |
|
|
|
|
Affections gastro-intestinales |
|
Nausées, douleur abdominale, constipation |
Flatulence, bouche sèche, vomissement, diarrhées |
|
|
|
|
Affections hépatobiliaires |
|
|
|
|
|
Fonction hépatique anormale |
|
Affections du rein et des voies urinaires |
|
|
Dysurie |
|
|
|
|
Affections de la peau et du tissu sous-cutané |
|
Sueurs, acné, éruption, sécheresse cutanée |
Séborrhée, alopécie, trouble du cheveu, ecchymose |
|
|
Syndrome de Stevens-Johnson (SSJ) / nécrolyse épidermique toxique (NET ou syndrome de Lyell) (voir rubrique 4.4), éruption cutanée toxique, érythème polymorphe |
|
Affections musculosquelettiques et du tissu conjonctif |
|
Arthralgie, raideur des épaules, douleur dorsale |
Douleur de la nuque, myalgies, raideur de la nuque, arthropathie |
|
|
|
|
Troubles du métabolisme et de la nutrition |
|
|
Hypercholestérolémie, anorexie |
|
|
Intolérance au glucose |
|
Affections vasculaires |
Bouffées de chaleur |
|
|
|
|
|
|
Troubles généraux et anomalies au site d'administration |
|
Asthénie, douleur, œdème périphérique, douleur au point d’injection, induration au point d’injection, douleur thoracique |
Frissons, fatigue, rougeur au point d’injection |
|
|
|
|
Affections des organes de reproduction et du sein |
Vaginite |
Sécheresse vaginale, douleur mammaire |
Douleur pelvienne, atrophie mammaire, leucorrhée |
|
|
Métrorragie |
|
Affections psychiatriques |
Trouble du sommeil |
Altération d’humeur, dépression, lors des traitements à long terme, instabilité émotionnelle, diminution de la libido, nervosité |
Altération d’humeur, dépression, lors des traitements à court terme, anxiété, confusion |
|
|
|
|
Affections respiratoires, thoraciques et médiastinales |
|
|
|
|
|
Pneumopathie interstitielle |
|
Tumeurs bénignes, malignes et non précisées |
|
|
|
|
Adénome hypophysaire |
|
|
Affections du système immunitaire |
|
|
|
|
Réaction anaphylactique (urticaire, angiœdème, choc anaphylactique) |
|
Description d’une sélection d’effets indésirables
Les effets indésirables liés au mode d'action du produit et à l'hypoestrogénie induite sont : bouffées de chaleur, céphalées, modification de la libido, sécheresse vaginale, dyspareunie, troubles de l’humeur, asthénie (ces manifestations sont généralement bien tolérées et ne conduisent que rarement à l’arrêt du traitement), myalgies, diminution du volume mammaire, modification de la densité osseuse (l’utilisation prolongée entraîne une diminution de la densité osseuse, facteur de risque d'une éventuelle ostéoporose, incluant la survenue de fracture).
Dans le cadre d’un traitement de l’endométriose associant ENANTONE à une hormonothérapie de substitution (« add-back therapy »), on note également des métrorragies.
Cancer du sein métastatique :
Les effets indésirables rapportés avec une fréquence supérieure ou égale à 0,5% chez des patientes recevant de la leuproréline sont listés ci-dessous selon la classification MedDRA (par classe organe et de fréquence absolue). Les fréquences sont définies comme suit : très fréquent (> 1/10), fréquent (> 1/100, < 1/10), peu fréquent (> 1/1 000, < 1/100), rare (> 1/10 000, < 1/1 000), très rare (< 1/10 000), indéterminée (ne peut être estimée sur la base des données disponibles).
|
Classes de systèmes d’organes |
Très fréquent |
Fréquent |
Peu fréquent |
Rare |
Très rare |
Indéterminée |
|
Investigations |
|
Prise de poids |
|
|
|
|
|
Affections hématologiques et du système lymphatique |
|
|
|
|
|
Anémie |
|
Affections cardiaques |
|
Palpitations |
|
|
|
|
|
Affections du système nerveux |
Céphalées |
Étourdissements, paresthésie |
|
|
Apoplexie hypophysaire (après administration initiale chez des patients porteurs d’un adénome hypophysaire) |
Convulsions Hypertension intracrânienne idiopathique (méningite séreuse) (voir rubrique 4.4) |
|
Affections oculaires |
|
Troubles visuels |
|
|
|
|
|
Affections gastro-intestinales |
Nausées |
Vomissements, diarrhées |
|
|
|
|
|
Affections hépatobiliaires |
|
|
|
|
|
Fonction hépatique anormale |
|
Affections de la peau et du tissu sous-cutané |
Sueurs |
Alopécie |
|
|
|
Syndrome de Stevens-Johnson (SSJ) / nécrolyse épidermique toxique (NET ou syndrome de Lyell) (voir rubrique 4.4), éruption cutanée toxique, érythème polymorphe |
|
Affections musculosquelettiques et du tissu conjonctif |
Douleur osseuse, faiblesse musculaire |
Raideur des épaules, douleur lombaire |
|
|
|
Myalgie, arthralgie |
|
Troubles du métabolisme et de la nutrition |
Diminution de l’appétit |
|
|
|
|
Intolérance au glucose |
|
Affections vasculaires |
Bouffées de chaleur |
|
|
|
|
|
|
Troubles généraux et anomalies au site d'administration |
Fatigue |
Douleur, réaction au point d’injection, oedème périphérique |
|
|
|
|
|
Affections des organes de reproduction et du sein |
Métrorragie, sécheresse vaginale |
Douleur mammaire, vaginite |
|
|
|
|
|
Affections psychiatriques |
Diminution de la libido |
Altération d’humeur, dépression, lors des traitements à long terme, insomnie |
Altération d’humeur, dépression, lors des traitements à court terme |
|
|
|
|
Affections respiratoires, thoraciques et médiastinales |
|
|
|
|
|
Pneumopathie interstitielle |
|
Tumeurs bénignes, malignes et non précisées |
|
|
|
|
Adénome hypophysaire |
|
|
Affections du système immunitaire |
|
|
|
|
Réaction anaphylactique (urticaire, angiœdème, choc anaphylactique) |
|
Les effets indésirables les plus fréquents sont liés à l'activité pharmacologique du produit : notamment bouffées de chaleur et crises sudorales.
Description d’une sélection d’effets indésirables
Les effets indésirables liés au mode d'action du produit et à l'hypoestrogénie induite sont : bouffées de chaleur, céphalées, modification de la libido, sécheresse vaginale, dyspareunie, troubles de l’humeur, asthénie (ces manifestations sont généralement bien tolérées et ne conduisent que rarement à l’arrêt du traitement), myalgies, diminution du volume mammaire, modification de la densité osseuse (l’utilisation prolongée entraîne une diminution de la densité osseuse, facteur de risque d'une éventuelle ostéoporose, incluant la survenue de fracture).
Rarement, comme avec les autres agonistes de la GnRH, une hypercalcémie peut survenir à la mise sous traitement chez les patientes présentant des métastases osseuses.
Puberté précoce centrale :
A l’initiation du traitement, une augmentation importante de courte durée du taux d’hormones sexuelles apparaît, suivie d’une diminution jusqu’aux valeurs observées lors de la pré-puberté. En raison de ces effets pharmacologiques, des effets indésirables peuvent survenir, particulièrement au début du traitement.
Les effets indésirables rapportés avec une fréquence supérieure ou égale à 0,5% chez des patients recevant de la leuproréline sont listés ci-dessous selon la classification MedDRA (par classe organe et de fréquence absolue). Les fréquences sont définies comme suit : très fréquent (> 1/10), fréquent (>1/100, < 1/10), peu fréquent (> 1/1 000, < 1/100), rare (> 1/10 000, < 1/1 000), très rare (< 1/10 000), indéterminée (ne peut être estimée sur la base des données disponibles).
|
Classes de systèmes d’organes |
Très fréquent |
Fréquent |
Peu fréquent |
Rare |
Très rare |
Indéterminée |
|
Affections du système immunitaire |
|
|
|
|
Réactions allergiques générales (fièvre, rash, démangeaisons, réactions anaphylactiques) |
|
|
Affections du système nerveux |
|
Céphalées |
|
|
Apoplexie hypophysaire (après administration initiale chez des patients porteurs d’un adénome hypophysaire) |
Convulsions Hypertension intracrânienne idiopathique (méningite séreuse) (voir rubrique 4.4) |
|
Affections gastro-intestinales |
|
Douleur abdominale/crampes abdominales, nausées/vomissements |
|
|
|
|
|
Affections de la peau et du tissu sous-cutané |
|
Acné |
Eruption |
|
|
Syndrome de Stevens-Johnson (SSJ) / nécrolyse épidermique toxique (NET ou syndrome de Lyell) (voir rubrique 4.4), éruption cutanée toxique, érythème polymorphe |
|
Affections musculosquelettiques et du tissu conjonctif |
|
|
|
|
|
Myalgie |
|
Affections vasculaires |
|
Bouffées de chaleur |
|
|
|
|
|
Troubles généraux et anomalies au site d'administration |
|
Réaction au point d’injection, inflammation au point d’injection (si celle-ci persiste, le traitement devra être interrompu), douleur au point d’injection |
Induration au point d’injection (si celle-ci persiste, le traitement devra être interrompu) |
|
|
|
|
Affections des organes de reproduction et du sein |
|
Vaginite, métrorragies*, sécrétions vaginales |
Leucorrhées |
|
|
|
|
Affections psychiatriques |
|
Instabilité émotionnelle, altération d’humeur, dépression lors des traitements à long terme |
|
|
|
|
|
Affections respiratoires, thoraciques et médiastinales |
|
|
|
|
|
Pneumopathie interstitielle |
*Note : en général, l’apparition de saignements vaginaux avec un traitement continu (consécutifs à une possible hémorragie de privation dans le premier mois de traitement) doit être évaluée comme un signe de potentiel sous-dosage. La suppression hypophysaire doit ensuite être confirmée par un test LHRH.
La survenue éventuelle de petites hémorragies génitales après la première injection chez les filles ne justifie l’adjonction d’un traitement freinateur que si celles-ci se poursuivent au-delà du premier mois de traitement.
Déclaration des effets indésirables suspectés
La déclaration des effets indésirables suspectés après autorisation du médicament est importante. Elle permet une surveillance continue du rapport bénéfice/risque du médicament. Les professionnels de santé déclarent tout effet indésirable suspecté via le système national de déclaration : Agence nationale de sécurité du médicament et des produits de santé (ANSM) et réseau des Centres Régionaux de Pharmacovigilance - Site internet : https://signalement.social-sante.gouv.fr/.
Sans objet.
5. PROPRIETES PHARMACOLOGIQUES
5.1. Propriétés pharmacodynamiques
Classe pharmacothérapeutique : ANALOGUE DE L'HORMONE ENTRAINANT LA LIBERATION DE GONADOTROPHINES, code ATC : L02AE02
La leuproréline est un nonapeptide de synthèse analogue de la GnRH naturelle. Les études conduites chez l'homme comme chez l'animal ont montré qu'après une stimulation initiale, l'administration prolongée de leuproréline entraîne une diminution de la sécrétion gonadotrope, supprimant par conséquent les fonctions testiculaires chez l'homme, et la sécrétion d'œstradiol gonadique chez la femme.
Elle provoque une involution des tissus soumis à une influence hormonale :
· telle une atrophie endométriale lors d'endométrioses et d'hémorragies endométriales sévères dues à la présence de fibrome utérin,
· ainsi que les tissus tumoraux du cancer de la prostate et du cancer du sein.
Cet effet est réversible à l'arrêt du traitement.
A la suite de certaines études animales, un autre mécanisme d'action a été évoqué : il s'agit d'un effet direct par diminution de la sensibilité des récepteurs gonadotropes.
Chez l'homme, après administration de la première dose, il se produit une élévation des taux sanguins de LH et FSH ce qui a pour corollaire une augmentation initiale des taux de stéroïdes gonadiques (testostérone et dihydrotestostérone chez l'homme et œstradiol chez la femme).
La poursuite du traitement entraîne une diminution des taux de LH et FSH conduisant, dans un délai de 3 à 4 semaines à des taux d'androgènes ou d'œstrogènes équivalents à ceux observés après castration ou après ménopause, aussi longtemps que le produit est administré.
Dans le cadre du traitement de l'endométriose par ENANTONE combiné à une hormonothérapie de substitution (« add-back therapy »), des données cliniques sur une période thérapeutique de un an ont été obtenues avec le schéma posologique comportant du valérate d'œstradiol micronisé 2 mg par jour administré par voie orale et de la promégestone 0,5 mg par jour administrée par voie orale.
Chez l’enfant, l’inhibition réversible de la libération gonadotrope hypophysaire se manifeste par la diminution consécutive des taux d’estradiol (E2) ou de testostérone proches des valeurs observées à la pré-puberté.
La stimulation gonadique initiale peut être responsable de petites hémorragies génitales chez les filles qui ont déjà leurs règles au début du traitement. Une hémorragie de privation peut apparaître en début de traitement. Les saignements s’arrêtent normalement à la poursuite du traitement.
Les effets thérapeutiques suivants peuvent être démontrés :
· suppression des taux de base des gonadotrophines aux taux observés à la pré-puberté
· suppression des taux de l’hormone sexuelle prématurément augmentée aux taux observés à la pré-puberté et arrêt des menstruations prématurées
· arrêt/régression du développement somatique pubertaire (stades de Tanner)
· amélioration/normalisation du ratio de l’âge statural/l’âge osseux
· prévention de l’accélération de l’âge osseux
· baisse de la vitesse de la croissance et sa normalisation
· augmentation de la taille finale
Les résultats du traitement sont la suppression de l’activation pathologique et prématurée de l’axe gonadique hypothalamo hypophysaire avec retour à l’âge pré-pubertaire.
Dans une étude clinique au long cours chez des enfants traités par leuproréline à des doses supérieures à 15 mg par mois pendant plus de 4 ans la reprise de la progression de la puberté a été observée à l’arrêt du traitement. Le suivi de 20 femmes jusqu’à l’âge adulte a montré un cycle menstruel normal chez 80% d’entre elles et 12 grossesses chez 7 des 20 femmes y compris des grossesses multiples pour 4 sujets.
5.2. Propriétés pharmacocinétiques
Chez les enfants :
La Figure 1 présente les concentrations sériques de leuproréline après une administration SC d’acétate de leuproréline à une dose de 30 µg/kg de poids corporel. Les pics sériques sont atteints 60 minutes après administration (7,81 ± 3,59 ng/mL). L’ ASC0-672 est de 105,78 ± 52,40 ng x hr/mL.
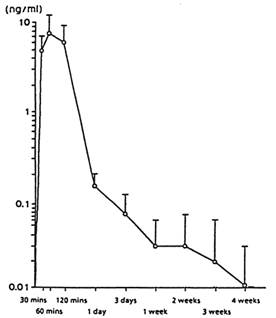
Figure 1 : concentrations sériques de leuproréline après administration SC de 30 µg/kg de poids corporel d’acétate de leuproréline (n=6) (moyenne +/- écart-type)
5.3. Données de sécurité préclinique
Ampoule de solvant : carmellose sodique, D-mannitol, polysorbate 80, eau pour préparations injectables.
3 ans.
6.4. Précautions particulières de conservation
A conserver à température ambiante, à l'abri de la chaleur.
6.5. Nature et contenu de l'emballage extérieur
44,15 mg de poudre en flacon (verre) + 2 mL de solvant en ampoule (verre) ; boîte de 1.
44,15 mg de poudre en flacon (verre) + 2 mL de solvant en ampoule (verre) avec un dispositif d’administration contenant une seringue de 2,5 mL accompagnée de 3 aiguilles (de tailles différentes : 23G (bleue), 21G (verte) et 18G (rose)) ; avec système de sécurité (safe system) ; boîte de 1.
Toutes les présentations peuvent ne pas être commercialisées.
6.6. Précautions particulières d’élimination et de manipulation
· Mettre le pouce sur le point bleu et casser l'ampoule de solvant en exerçant une flexion à ce niveau.
POUR LE CONDITIONNEMENT AVEC LES FLACONS DE POUDRE ET DE SOLVANTS SEULS :
· Soutirer le contenu de l’ampoule de solvant à l’aide d’une seringue et l’introduire dans le flacon de poudre pour reconstituer la suspension. Ce solvant est spécifique d’ENANTONE LP 3,75 mg : il ne devra jamais être remplacé par un autre solvant.
· Bien agiter la préparation.
· Aspirer le mélange obtenu en s’assurant que la totalité de la suspension a bien été soutirée.
· Injecter la suspension immédiatement après la reconstitution à l’aide d’une aiguille S.C. ou I.M.
POUR LE CONDITIONNEMENT AVEC SERINGUES ET AIGUILLES :
· A l’aide de la seringue et de l’aiguille ROSE, soutirer le contenu de l'ampoule de solvant et l'introduire dans le flacon de poudre pour reconstituer la suspension. Ce solvant est spécifique d'ENANTONE LP 3,75 mg : il ne devra jamais être remplacé par un autre solvant.
· Bien agiter la préparation.
· Aspirer le mélange obtenu en s’assurant que la totalité de la suspension a bien été soutirée.
· Changer l’aiguille afin de procéder à l’injection : il conviendra d’utiliser l’aiguille BLEUE pour une injection sous-cutanée et l’aiguille VERTE pour une injection intramusculaire.
· Injecter la suspension immédiatement après la reconstitution.
· La présence du système de sécurité de l’aiguille est destinée à protéger le personnel soignant contre le risque de piqûre accidentelle.
Tout médicament non utilisé ou déchet doit être éliminé conformément à la réglementation en vigueur.
7. TITULAIRE DE L’AUTORISATION DE MISE SUR LE MARCHE
16 PLACE DE L’IRIS
92400 COURBEVOIE
8. NUMERO(S) D’AUTORISATION DE MISE SUR LE MARCHE
· 34009 331 291 2 5 : 44,15 mg de poudre en flacon (verre) + 2 mL de solvant en ampoule (verre) ; boîte de 1.
· 34009 375 752 5 6 : 44,15 mg de poudre en flacon (verre) + 2 mL de solvant en ampoule (verre) avec un dispositif d’administration ; avec système de sécurité (safe system) ; boîte de 1.
9. DATE DE PREMIERE AUTORISATION/DE RENOUVELLEMENT DE L’AUTORISATION
[à compléter ultérieurement par le titulaire]
10. DATE DE MISE A JOUR DU TEXTE
[à compléter ultérieurement par le titulaire]
Sans objet.
12. INSTRUCTIONS POUR LA PREPARATION DES RADIOPHARMACEUTIQUES
Liste I
ANSM - Mis à jour le : 10/07/2025
leuproréline
· Gardez cette notice. Vous pourriez avoir besoin de la relire.
· Si vous avez d’autres questions, interrogez votre médecin, votre pharmacien ou votre infirmier/ère.
· Ce médicament vous a été personnellement prescrit. Ne le donnez pas à d’autres personnes. Il pourrait leur être nocif, même si les signes de leur maladie sont identiques aux vôtres.
· Si vous ressentez un quelconque effet indésirable, parlez-en à votre médecin, votre pharmacien ou votre infirmier/ère. Ceci s’applique aussi à tout effet indésirable qui ne serait pas mentionné dans cette notice. Voir rubrique 4.
1. Qu'est-ce que ENANTONE LP 3,75 mg, poudre et solvant pour suspension injectable (S.C. ou I.M.) à libération prolongée et dans quels cas est-il utilisé ?
2. Quelles sont les informations à connaître avant d'utiliser ENANTONE LP 3,75 mg, poudre et solvant pour suspension injectable (S.C. ou I.M.) à libération prolongée ?
3. Comment utiliser ENANTONE LP 3,75 mg, poudre et solvant pour suspension injectable (S.C. ou I.M.) à libération prolongée ?
4. Quels sont les effets indésirables éventuels ?
5. Comment conserver ENANTONE LP 3,75 mg, poudre et solvant pour suspension injectable (S.C. ou I.M.) à libération prolongée ?
6. Contenu de l’emballage et autres informations.
1. QU’EST-CE QUE ENANTONE LP 3,75 mg, poudre et solvant pour suspension injectable (S.C. ou I.M.) à libération prolongée ET DANS QUELS CAS EST-IL UTILISE ?
ANALOGUE DE L'HORMONE ENTRAINANT LA LIBERATION DE GONADOTROPHINES - code ATC : L02AE02
Ce médicament contient une substance active, la leuproréline, qui a la même action qu'une hormone naturelle (la GnRH). Cette hormone contrôle la production d'autres hormones : la testostérone, les œstrogènes et la progestérone.
L'utilisation sur une longue durée de ce médicament permet d'arrêter la production des hormones sexuelles (testostérone, œstrogène et progestérone) aussi bien chez l'homme que chez la femme et l’enfant.
ENANTONE est donc utilisé :
· pour traiter certains cancers qui ont besoin de ces hormones pour se développer (et notamment le cancer de la prostate chez l'homme, seul ou associé à des rayons (radiothérapie), et le cancer du sein chez la femme) ;
· pour traiter un développement excessif de la muqueuse utérine en dehors de l'utérus (endométriose) ou pour certaines tumeurs bénignes de l'utérus (fibromes utérins) chez la femme ;
· ou pour traiter des pubertés précoces centrales de l'enfant (avant 9 ans chez la fille, avant 10 ans chez le garçon).
2. QUELLES SONT LES INFORMATIONS A CONNAITRE AVANT DE PRENDRE D’UTILISER ENANTONE LP 3,75 mg, poudre et solvant pour suspension injectable (S.C. ou I.M.) à libération prolongée ?
· si vous êtes allergique à la leuproréline, aux dérivés ou aux analogues de la GnRH, ou à l'un des autres composants contenus dans ce médicament, mentionnés dans la rubrique 6 ;
· grossesse ou allaitement ;
· chez la fille avec puberté précoce centrale : saignement vaginal de cause non déterminée ;
· en cas d'association d'une hormonothérapie de substitution au traitement de l'endométriose par ENANTONE, les contre-indications d'utilisation d'oestroprogestatifs doivent être respectées.
Avertissements et précautions
Faites attention avec ENANTONE LP 3,75 mg, poudre et solvant pour suspension injectable (S.C. ou I.M.) à libération prolongée
Des cas de dépression, pouvant être sévère ont été rapportés chez des patients traités par ENANTONE LP 3,75 mg, poudre et solvant pour suspension injectable (S.C. ou I.M.) à libération prolongée.
Si vous prenez ENANTONE LP 3,75 mg, poudre et solvant pour suspension injectable (S.C. ou I.M.) à libération prolongée et si vous ressentez une humeur dépressive, parlez-en à votre médecin.
Veuillez signaler à votre médecin si vous souffrez du cœur ou des vaisseaux, y compris si vous avez des troubles du rythme (arythmies) ou si vous êtes traités par des médicaments pour ces problèmes. Le risque de troubles du rythme cardiaque peut être augmenté avec ENANTONE.
Votre médecin surveillera votre taux sanguin de sucre régulièrement car ENANTONE peut modifier votre métabolisme et donc votre taux sanguin de sucre.
Votre médecin doit s’assurer de la surveillance de votre densité osseuse et peut vous prescrire un traitement approprié car l’utilisation de ce médicament peut entraîner une diminution de la densité osseuse, une ostéoporose (fragilisation des os) et un risque accru de fractures osseuses.
Des cas de convulsions ont été rapportés chez les patients traités par ENANTONE LP 3,75 mg poudre et solvant après sa mise sur le marché. Ces convulsions ont été observées aussi bien chez les adultes que chez les enfants, avec ou sans antécédents, troubles, ou facteurs de risque liés aux convulsions.
Si vous (ou votre enfant) souffrez de maux de tête intenses ou récurrents, de problèmes de vue et de sifflements ou bourdonnements dans les oreilles, contactez votre médecin immédiatement.
Des éruptions cutanées sévères, dont le syndrome de Stevens-Johnson (SSJ) et la nécrolyse épidermique toxique (NET ou syndrome de Lyell) ont été rapportées en association avec la leuproréline. Arrêtez d’utiliser la leuproréline et consultez immédiatement un médecin si vous remarquez l’un des symptômes liés à ces réactions cutanées graves, décrits à la rubrique 4.
Si vous présentez une stéatose hépatique (une maladie dans laquelle un excès de graisse s’accumule dans le foie), contactez votre médecin immédiatement.
Maladie de la prostate :
Lors de la mise en route d'un traitement par ENANTONE, des cas isolés d'aggravation des symptômes cliniques (en particulier des douleurs osseuses), le plus souvent transitoires, ont été observés.
Une surveillance attentive sera effectuée pendant les premières semaines de traitement et une consultation médicale sera demandée rapidement devant toute aggravation des symptômes.
Endométriose, cancer du sein métastatique et traitement pré-opératoire des fibromes utérins :
Une absence de grossesse sera vérifiée avant le traitement.
A l’initiation du traitement, une aggravation transitoire des symptômes cliniques peut survenir. Cependant, cela peut disparaître avec la poursuite du traitement.
En cas d'association d'une hormonothérapie de substitution au traitement de l'endométriose par ENANTONE, les mises en garde et les précautions d'emploi d'utilisation d'oestroprogestatifs doivent être respectées.
Avant le début du traitement, en cas de présence de saignements vaginaux anormaux, la cause de ces saignements doit être recherchée et une prise en charge doit être mise en place.
Demandez conseil à votre médecin ou à votre pharmacien avant de prendre tout médicament.
Prendre des précautions particulières avec ENANTONE LP 3,75 mg, poudre et solvant pour suspension injectable (S.C. ou I.M.) à libération prolongée :
Ce médicament doit être utilisé sous surveillance médicale.
Endométriose, cancer du sein métastatique et traitement pré-opératoire des fibromes utérins :
Dans cette indication, ce médicament entraîne dans tous les cas une absence de règles. En dehors du premier mois de traitement, la survenue de saignements doit conduire à consulter votre médecin.
Si vous présentez un risque d'ostéoporose (fragilisation osseuse), une surveillance de la masse osseuse est conseillée lors de traitement prolongé.
En cas de traitement des fibromes utérins, ce médicament peut entrainer des saignements vaginaux anormaux ou des douleurs au cours du traitement.
Chez l’enfant :
Dans le cas d’un abcès stérile au niveau du site d’injection (principalement rapporté après administration dans le muscle), votre médecin surveillera vos taux d’hormones étant donné que l’absorption de leuproréline pourrait être diminuée à partir du site d’injection.
Si l’enfant a une tumeur cérébrale évolutive, votre médecin décidera si le traitement par ENANTONE LP 3,75 mg est adapté. Chez les filles avec puberté précoce centrale :
L’absence de grossesse sera vérifiée avant le traitement.
Après la première injection, des saignements vaginaux et des sécrétions peuvent apparaître en signe de privation hormonale. L’apparition de saignements vaginaux au-delà des deux premiers mois de traitement doit être explorée.
Le traitement par ENANTONE LP 3,75 mg peut entraîner une diminution de la densité minérale osseuse (DMO). Toutefois, après l'arrêt du traitement, l’augmentation ultérieure de la masse osseuse est préservée et le pic de croissance de la masse osseuse à la fin de la puberté ne semble pas être affecté par le traitement.
L’arrêt du traitement peut conduire à l’apparition soudaine d’un glissement du cartilage de croissance du fémur. Il se pourrait que ce soit consécutif à l’affaiblissement du cartilage de conjugaison en raison des faibles concentrations en hormones sexuelles féminines pendant le traitement.
Autres médicaments et ENANTONE LP 3,75 mg, poudre et solvant pour suspension injectable (S.C. ou I.M.) à libération prolongée
D’autres médicaments et ENANTONE peuvent interagir avec les médicaments utilisés pour traiter les problèmes de rythme cardiaque (par exemple quinidine, procainamide, amiodarone et sotalol) ou peuvent augmenter le risque de troubles du rythme quand ils sont utilisés avec d’autres médicaments (par exemple méthadone (utilisé pour le traitement de la douleur et en désintoxication de l’addiction médicamenteuse), moxifloxacine (un antibiotique), et antipsychotiques (utilisés pour les maladies mentales graves)).
Informez votre médecin ou pharmacien si vous utilisez, avez récemment utilisé ou pourriez utiliser tout autre médicament.
ENANTONE LP 3,75 mg, poudre et solvant pour suspension injectable (S.C. ou I.M.) à libération prolongée avec des aliments, boissons et de l’alcool
Sans objet.
Grossesse, allaitement et fertilité
Grossesse
Ce médicament ne doit pas être utilisé pendant la grossesse. En cas de découverte fortuite d'une grossesse alors que vous preniez ce médicament, avertissez tout de suite votre médecin, qui interrompra alors ce traitement.
Si vous êtes une femme en âge d’avoir des enfants, une contraception non hormonale doit être utilisée pendant le traitement et jusqu’à la reprise de vos règles.
Demandez conseil à votre médecin ou à votre pharmacien avant de prendre tout médicament.
Allaitement
Ce médicament ne doit pas être utilisé en cas d'allaitement.
Demandez conseil à votre médecin ou à votre pharmacien avant de prendre tout médicament.
ENANTONE LP 3,75 mg ne doit pas être administré aux filles enceintes ou qui allaitent (voir aussi N’utilisez jamais ENANTONE LP 3,75 mg, poudre et solvant pour suspension injectable (S.C. ou I.M.) à libération prolongée dans les cas suivants)
Conduite de véhicules et utilisation de machines
Les effets d'ENANTONE LP 3,75 mg sur l'aptitude à conduire des véhicules et à utiliser des machines n'ont pas été étudiés. Des vertiges, des troubles de la vue, une faiblesse des membres inférieurs, une fatigue et une somnolence peuvent survenir au cours de votre maladie ou de votre traitement. Si vous ressentez l'un de ces effets, vous devez être prudent si vous conduisez ou utilisez des machines.
Ce médicament contient moins de 1 mmol (23 mg) de sodium par dose, c’est-à-dire qu’il est essentiellement « sans sodium ».
3. COMMENT UTILISER ENANTONE LP 3,75 mg, poudre et solvant pour suspension injectable (S.C. ou I.M.) à libération prolongée ?
Une injection mensuelle, par voie sous-cutanée ou intramusculaire.
Utilisation chez les enfants
Le traitement des enfants doit se faire sous la surveillance générale d'un endocrino-pédiatre, d'un pédiatre ou d’un endocrinologue ayant une expertise dans le traitement de la puberté précoce centrale.
Le schéma posologique doit être adapté individuellement.
La dose initiale recommandée dépend du poids corporel.
Enfants d'un poids supérieur ou égal à 20 kg :
Sauf prescription contraire, 2 mL d'ENANTONE LP 3,75 mg (3,75 mg d’acétate de leuproréline) sont administrés une fois par mois, en une seule injection, sous la peau par exemple dans l’abdomen, la fesse ou la cuisse.
Enfants d'un poids inférieur à 20 kg :
En prenant en compte l’activité clinique de la puberté précoce centrale, dans ces rares cas, la conduite à tenir est la suivante :
Sauf prescription contraire, 1 mL d'ENANTONE LP 3,75 mg (1,88 mg d’acétate de leuproréline) est administré une fois par mois, en une seule injection, sous la peau par exemple dans l’abdomen, la fesse ou la cuisse. Le reste de la suspension doit être éliminé. Votre médecin surveillera la prise de poids de l’enfant.
Selon l’activité clinique de la puberté précoce centrale, votre médecin pourra augmenter la dose en présence d’une suppression insuffisante (par exemple saignements vaginaux). Votre médecin déterminera la dose efficace minimale à l’aide d’un test sanguin.
La durée du traitement dépend des signes cliniques au début du traitement ou pendant le traitement et est décidée en accord avec le médecin et le représentant légal et le cas échéant, l’enfant traité. Votre médecin déterminera l’âge osseux de l’enfant à intervalles réguliers.
Chez les filles ayant un âge osseux supérieur à 12 ans et chez les garçons ayant un âge osseux supérieur à 13 ans, votre médecin envisagera l’arrêt du traitement en prenant en compte les effets cliniques sur votre enfant.
Chez les filles, une grossesse doit être exclue avant le début du traitement. La survenue d’une grossesse pendant le traitement ne peut pas être exclue. Dans ce cas, parlez-en à votre médecin.
Ce traitement est un traitement au long terme, adapté de manière individuelle. Prévoyez avec votre médecin des administrations mensuelles régulières d’ENANTONE LP 3,75 mg. Un retard exceptionnel de quelques jours dans la date de l’injection (30 ± 2 jours) n’influence pas les résultats du traitement.
Mode et voie d’administration
· Voie sous-cutanée ou intramusculaire dans les indications : maladie de la prostate et des os, endométriose, fibrome utérin, maladie du sein.
· Voie sous-cutanée chez l’enfant dans le traitement de la puberté précoce.
· De fréquents abcès stériles au site d’injection apparaissent quand ENANTONE LP 3,75 mg est administré à des doses supérieures à celles recommandées et quand il est administré dans le muscle.
Votre médecin vous administrera alors le médicament sous la peau par exemple dans l’abdomen, la fesse ou la cuisse.
Mode d’utilisation
ENANTONE LP 3,75 mg doit être uniquement administré par votre médecin ou un(e) infirmier(e). Ce sont eux qui seront chargés de préparer le produit.
Mettre le pouce sur le point bleu et casser l'ampoule de solvant en exerçant une flexion à ce niveau.
· A l’aide de la seringue et de l’aiguille ROSE, soutirer le contenu de l’ampoule de solvant et l’introduire dans le flacon de poudre pour reconstituer la suspension. Ce solvant est spécifique d’ENANTONE LP 3,75 mg : il ne devra jamais être remplacé par un autre solvant.
· Bien agiter la préparation.
· Aspirer le mélange obtenu en s’assurant que la totalité de la suspension a bien été soutirée.
· Changer l’aiguille afin de procéder à l’injection : il conviendra d’utiliser l’aiguille BLEUE pour une injection sous-cutanée et l’aiguille VERTE pour une injection intramusculaire.
· Injecter la suspension immédiatement après la reconstitution.
Après injection :
· Retirer l’aiguille de la peau du patient. Activer immédiatement le système de sécurité de l’aiguille en procédant comme suit :
|
· Positionner le capuchon protecteur sur une surface plane de façon à former avec celle-ci un angle de 45° environ ;
|
|
· Appuyer d’un geste ferme et rapide jusqu’à entendre un « clic » sonore ;
|
|
· S’assurer au bruit et/ou visuellement que le capuchon protecteur recouvre l’aiguille.
|
La présence du système de sécurité de l’aiguille est destinée à protéger le personnel soignant contre le risque de piqûre accidentelle.
Fréquence d'administration
Les injections seront renouvelées toutes les 4 semaines.
Chez la femme (endométriose), le traitement sera limité à 6 mois.
Toutefois, dans les cas associés à une symptomatologie pelvienne chronique et en l'absence de désir immédiat de grossesse, la durée de traitement peut être portée à un an en associant ENANTONE à une hormonothérapie de substitution. Au-delà de cette période, le renouvellement de ce traitement ou d'un autre médicament du même type n'est pas souhaitable.
Durée de traitement
Se conformer strictement à la prescription de votre médecin traitant.
Si vous avez utilisé plus d’ENANTONE LP 3,75 mg, poudre et solvant pour suspension injectable (S.C. ou I.M.) à libération prolongée que vous n’auriez dû :
Consultez immédiatement votre médecin ou votre pharmacien.
Si vous oubliez de prendre ENANTONE LP 3,75 mg, poudre et solvant pour suspension injectable (S.C. ou I.M.) à libération prolongée :
Ne prenez pas de dose double pour compenser la dose simple que vous avez oublié de prendre.
4. QUELS SONT LES EFFETS INDESIRABLES EVENTUELS ?
Chez l’homme :
Dans certains cas rares, il est possible d’observer en début de traitement, une accentuation des troubles qui l’ont motivé : troubles urinaires, douleurs.
Les effets indésirables les plus fréquemment rapportés sont (peuvent affecter 1 personne sur 100 à plus d’1 personne sur 10) : prise de poids, augmentation de certaines enzymes (notamment du foie), anémie (faible nombre de globules rouges), maux de tête, difficulté respiratoire, nausées, constipation, troubles urinaires, sang dans les urines, infection des voies urinaires, sueurs, démangeaisons, douleur osseuse, faiblesse musculaire, douleur du dos, douleurs articulaires, perte d’appétit, diminution de l’appétit, bouffées de chaleur, fatigue, réaction ou douleur au point d’injection, douleur, gonflement localisé (œdème), fonction hépatique anormale, impuissance, diminution de la taille des testicules, trouble testiculaire, développement des seins, diminution de la libido, dépression, altération de l’humeur, trouble du sommeil.
Les effets indésirables suivants sont peu fréquents (peuvent affecter jusqu’à 1 personne sur 100) : étourdissement, paresthésie (fourmillement), vomissement, diarrhées, éruption cutanée, douleur musculaire, douleur des extrémités, douleur thoracique, gonflement localisé (œdème) au point d’injection.
Les effets indésirables suivants sont très rares (peuvent affecter jusqu’à 1 personne sur 10 000) : arrêt de fonctionnement de la glande hypophyse (apoplexie hypophysaire) chez des patients présentant une hypophyse de taille augmentée (si vous avez une lésion hypophysaire préexistante, il peut y avoir une augmentation du risque d’hémorragie dans cette région, pouvant provoquer des maux de tête violents, des troubles visuels et des troubles de la conscience nécessitant une prise en charge en urgence), réactions allergiques incluant urticaire, choc allergique, angiœdème (réaction allergique grave provoquant un gonflement du cou et du visage, et une gêne respiratoire).
Les effets indésirables suivants ont été observés avec une fréquence indéterminée (ne peut être estimée sur la base des données disponibles) : intolérance au glucose (élévation du taux de glucose dans le sang), hypertension intracrânienne idiopathique (augmentation de la pression intracrânienne dans la zone du cerveau caractérisée par des maux de tête, une vision double et autres symptômes visuels et des sifflements ou bourdonnements dans une ou les deux oreilles), modifications de l’ECG (allongement de l’intervalle QT) après une administration prolongée, convulsions, inflammation des poumons, maladie pulmonaire, troubles visuels, rougeur cutanée et éruption cutanée accompagnée de démangeaisons (éruption cutanée toxique), éruption cutanée qui provoque l’apparition de boutons ou de plaques rouges sur la peau, pouvant ressembler à une cible, avec un centre rouge entouré de cercles rouges plus pâles (érythème polymorphe).
Il est probable qu’un traitement à long terme par leuproréline révèle des signes d’aggravation d’ostéoporose (fragilisation osseuse).
Ces phénomènes peuvent survenir même si l’effet du traitement est favorable. Il faut cependant avertir immédiatement votre médecin traitant.
Si vous présentez des plaques rougeâtres non surélevées, en forme de cible ou circulaires sur le tronc, souvent avec des cloques centrales, une desquamation de la peau, des aphtes de la bouche, de la gorge, du nez, des organes génitaux et des yeux. Ces éruptions cutanées graves peuvent être précédées de fièvre et de symptômes pseudo-grippaux (Syndrome de Stevens-Johnson/Nécrolyse épidermique toxique).
Chez la femme :
En début de traitement, il est possible d’observer une accentuation des troubles (douleurs pelviennes) mais qui doivent disparaître en 1 à 2 semaines.
Les effets indésirables les plus fréquemment rapportés sont (peuvent affecter 1 personne sur 100 à plus d’1 personne sur 10) : bouffées de chaleur, prise de poids, perte de poids, maux de tête, étourdissements, trouble de la sensibilité, hypertonie, diminution de l’appétit, nausées, vomissements, diarrhées, chute des cheveux, maux de ventre, constipation, sueurs, acné, éruption cutanée, sécheresse cutanée, douleurs articulaires, raideur des épaules, douleur du dos, faiblesse musculaire, fatigue, douleur, gonflement localisé (œdème), douleur ou induration (zone durcie de la peau) au point d’injection, douleur thoracique, inflammation du vagin, hémorragie vaginale, sécheresse vaginale, douleur mammaire, trouble du sommeil, dépression, altération de l’humeur, instabilité émotionnelle, diminution de la libido, nervosité, palpitations cardiaques, troubles visuels.
Les effets indésirables suivants sont peu fréquents (peuvent affecter jusqu’à 1 personne sur 100) : augmentation de certaines enzymes (notamment du foie), augmentation du phosphore dans le sang, augmentation des graisses, diminution de la sensibilité cutanée, flatulence, bouche sèche, difficultés pour uriner, excès de sébum, trouble du cheveu, ecchymose, douleur de la nuque, douleurs musculaires, raideur de la nuque, affections articulaires, excès de cholestérol dans le sang, frissons, fatigue, rougeur au point d’injection, douleur pelvienne, diminution du volume des seins, pertes vaginales, anxiété, confusion.
Les effets indésirables suivants sont très rares (peuvent affecter jusqu’à 1 personne sur 10 000) : arrêt de fonctionnement de la glande hypophyse (apoplexie hypophysaire) chez des patients présentant une hypophyse de taille augmentée (si vous avez une lésion hypophysaire préexistante, il peut y avoir une augmentation du risque d’hémorragie dans cette région, pouvant provoquer des maux de tête violents, des troubles visuels et des troubles de la conscience nécessitant une prise en charge en urgence), réactions allergiques incluant urticaire, choc allergique, angiœdème (réaction allergique grave provoquant un gonflement du cou et du visage, et une gêne respiratoire).
Les effets indésirables suivants ont été observés avec une fréquence indéterminée (ne peut être estimée sur la base des données disponibles) : intolérance au glucose (élévation du taux de glucose dans le sang), hypertension intracrânienne idiopathique (augmentation de la pression intracrânienne dans la zone du cerveau caractérisée par des maux de tête, une vision double et autres symptômes visuels et des sifflements ou bourdonnements dans une ou les deux oreilles), fonction hépatique anormale, diminution de la masse osseuse avec risque d’ostéoporose (incluant la survenue de fractures), diminution de la quantité de globules rouges (anémie), convulsions, inflammation des poumons, maladie pulmonaire, rougeur cutanée et éruption cutanée accompagnée de démangeaisons (éruption cutanée toxique), éruption cutanée qui provoque l’apparition de boutons ou de plaques rouges sur la peau, pouvant ressembler à une cible, avec un centre rouge entouré de cercles rouges plus pâles (érythème polymorphe).
Dans le cadre d’un traitement de l’endométriose associant ENANTONE à un traitement hormonal complémentaire, on note également des saignements vaginaux.
Si vous présentez des plaques rougeâtres non surélevées, en forme de cible ou circulaires sur le tronc, souvent avec des cloques centrales, une desquamation de la peau, des aphtes de la bouche, de la gorge, du nez, des organes génitaux et des yeux. Ces éruptions cutanées graves peuvent être précédées de fièvre et de symptômes pseudo-grippaux (Syndrome de Stevens-Johnson/Nécrolyse épidermique toxique).Chez l’enfant :
A l’initiation du traitement, une augmentation importante de courte durée du taux d’hormones sexuelles apparaît, suivie d’une diminution jusqu’aux valeurs de la pré-puberté. En raison de ces effets, des effets indésirables peuvent survenir, particulièrement au début du traitement.
Les effets indésirables fréquemment rapportés sont (peuvent affecter 1 personne sur 100 à plus d’1 personne sur 10) : sautes d’humeur, dépression, maux de tête, maux de ventre/crampes abdominales, nausées/vomissements, acné, bouffées de chaleur, réaction au point d’injection, inflammation au point d’injection (si celle-ci persiste, le traitement devra être interrompu), douleur au point d’injection, inflammation du vagin, saignements vaginaux, sécrétions vaginales, instabilité émotionnelle.
Les effets indésirables suivants sont peu fréquents (peuvent affecter jusqu’à 1 personne sur 100) : éruption cutanée, induration (zone durcie de la peau) au point d’injection (si celle-ci persiste, le traitement devra être interrompu), pertes vaginales.
Les effets indésirables suivants sont très rares (peuvent affecter jusqu’à 1 personne sur 10 000) : réactions allergiques générales (fièvre, rash, démangeaisons), réactions allergiques graves pouvant causer des difficultés à respirer ou des vertiges, un gonflement du visage et des lèvres et comme les autres médicaments de cette classe : si vous avez une lésion hypophysaire préexistante, il peut y avoir une augmentation du risque d’hémorragie dans cette région, pouvant provoquer des maux de tête violents, des troubles visuels et des troubles de la conscience nécessitant une prise en charge en urgence.
Les effets indésirables suivants ont été observés avec une fréquence indéterminée (ne peut être estimée sur la base des données disponibles) : convulsions, hypertension intracrânienne idiopathique (augmentation de la pression intracrânienne dans la zone du cerveau caractérisée par des maux de tête, une vision double et autres symptômes visuels et des sifflements ou bourdonnements dans une ou les deux oreilles), inflammation des poumons, douleur musculaire, maladie pulmonaire, rougeur cutanée et éruption cutanée accompagnée de démangeaisons (éruption cutanée toxique), éruption cutanée qui provoque l’apparition de boutons ou de plaques rouges sur la peau, pouvant ressembler à une cible, avec un centre rouge entouré de cercles rouges plus pâles (érythème polymorphe).
Quelques pertes sanguines peuvent être observées chez la fille lors de la première semaine de traitement. Elles peuvent justifier un traitement complémentaire de courte durée.
Note : en général, si des saignements vaginaux apparaissent avec un traitement continu (après une possible hémorragie de privation dans le premier mois de traitement), cela peut être un signe de potentiel sous-dosage. Prévenez votre médecin si des saignements vaginaux apparaissent.
Si vous présentez des plaques rougeâtres non surélevées, en forme de cible ou circulaires sur le tronc, souvent avec des cloques centrales, une desquamation de la peau, des aphtes de la bouche, de la gorge, du nez, des organes génitaux et des yeux. Ces éruptions cutanées graves peuvent être précédées de fièvre et de symptômes pseudo-grippaux (Syndrome de Stevens-Johnson/Nécrolyse épidermique toxique).
Déclaration des effets secondaires
Si vous ressentez un quelconque effet indésirable, parlez-en à votre médecin, votre pharmacien ou à votre infirmier/ère. Ceci s’applique aussi à tout effet indésirable qui ne serait pas mentionné dans cette notice. Vous pouvez également déclarer les effets indésirables directement via le système national de déclaration : Agence nationale de sécurité du médicament et des produits de santé (ANSM) et réseau des Centres Régionaux de Pharmacovigilance - Site internet : https://signalement.social-sante.gouv.fr/.
En signalant les effets indésirables, vous contribuez à fournir davantage d’informations sur la sécurité du médicament.
5. COMMENT CONSERVER ENANTONE LP 3,75 mg, poudre et solvant pour suspension injectable (S.C. ou I.M.) à libération prolongée ?
Tenir ce médicament hors de la vue et de la portée des enfants.
N’utilisez pas ce médicament après la date de péremption indiquée sur l’emballage. La date de péremption fait référence au dernier jour de ce mois.
A conserver à température ambiante, à l’abri de la chaleur.
Ne jetez aucun médicament au tout-à-l’égout ou avec les ordures ménagères. Demandez à votre pharmacien d’éliminer les médicaments que vous n’utilisez plus. Ces mesures contribueront à protéger l’environnement.
6. CONTENU DE L’EMBALLAGE ET AUTRES INFORMATIONS
· La substance active est :
leuproréline................................................................................................................... 3,75 mg
Pour un flacon
· Les autres composants sont :
Flacon de poudre : gélatine, copolymère d'acide DL lactique et d'acide glycolique (75/25 mol pour cent), D-mannitol
Ampoule de solvant : carmellose sodique, D-mannitol, polysorbate 80, eau pour préparations injectables.
Poudre et solvant pour suspension injectable (S.C. ou I.M.) à libération prolongée
Boîte de 1 flacon et de 1 ampoule avec dispositif d’administration (1 seringue de 2,5 mL accompagnée de 3 aiguilles (de tailles différentes : 23G (bleue), 21G (verte) et 18G (rose)).
Titulaire de l’autorisation de mise sur le marché
16 PLACE DE L’IRIS
92400 COURBEVOIE
Exploitant de l’autorisation de mise sur le marché
TAKEDA FRANCE SAS
16 PLACE DE L’IRIS
92400 COURBEVOIE
VIA CROSA, 86
28065 CERANO (NO)
ITALIE
Noms du médicament dans les Etats membres de l'Espace Economique Européen
Sans objet.
La dernière date à laquelle cette notice a été révisée est :
[à compléter ultérieurement par le titulaire]
Sans objet.
Des informations détaillées sur ce médicament sont disponibles sur le site Internet de l’ANSM (France).