Dernière mise à jour le 03/08/2026
DIMETHYL FUMARATE BIOGARAN 120 mg, gélule gastro-résistante
Ce médicament n'est ou ne sera bientôt plus disponible sur le marché.
Si vous prenez actuellement ce médicament, il vous est recommandé d'en parler avec votre médecin
ou avec votre pharmacien qui pourra vous orienter vers un autre traitement.
Indications thérapeutiques
Classe pharmacothérapeutique - Immunosuppresseurs, autres immunosuppresseurs, code ATC : L04AX07.
Qu’est-ce que DIMETHYL FUMARATE BIOGARAN ?
DIMETHYL FUMARATE BIOGARAN est un médicament dont la substance active est le fumarate de diméthyle.
Dans quel cas DIMETHYL FUMARATE BIOGARAN est-il utilisé ?
DIMETHYL FUMARATE BIOGARAN est utilisé pour traiter la sclérose en plaques (SEP) récurrente rémittente chez les patients âgés de 13 ans et plus.
La sclérose en plaques est une maladie chronique qui affecte le système nerveux central comprenant le cerveau et la moelle épinière. La sclérose en plaques récurrente rémittente se caractérise par l’apparition répétée de symptômes neurologiques (poussées). Les symptômes varient d'un patient à un autre mais comportent de façon caractéristique des difficultés pour marcher, une sensation de perte de l'équilibre ainsi que des troubles visuels (par exemple vision floue ou double). Ces symptômes peuvent disparaître complètement lorsque la poussée est terminée mais certains problèmes peuvent persister.
Comment agit DIMETHYL FUMARATE BIOGARAN ?
DIMETHYL FUMARATE BIOGARAN semblerait agir en bloquant les systèmes de défense de l'organisme qui peuvent léser le cerveau et la moelle épinière. Cette possible action pourrait aussi retarder l'évolution future de votre SEP.
Service médical rendu (SMR)
Pas de SMR disponible pour ce médicament ( plus d'informations dans l'aide )
Amélioration du service médical rendu (ASMR)
Pas d'ASMR disponible pour ce médicament (plus d'informations dans l'aide )
ANSM - Mis à jour le : 28/04/2023
DIMETHYL FUMARATE BIOGARAN 120 mg, gélule gastro-résistante
2. COMPOSITION QUALITATIVE ET QUANTITATIVE
Fumarate de diméthyle.......................................................................................................... 120 mg
Pour une gélule gastro-résistante.
Pour la liste complète des excipients, voir rubrique 6.1.
Gélule gastro-résistante de longueur : 19,4 mm ± 0,3 mm, avec un corps blanc et une tête verte clair, portant l’inscription « 120 mg ».
4.1. Indications thérapeutiques
4.2. Posologie et mode d'administration
Posologie
La dose initiale est de 120 mg deux fois par jour. Après 7 jours de traitement, la dose doit être augmentée à la dose d’entretien recommandée de 240 mg deux fois par jour (voir rubrique 4.4).
En cas d’oubli d’une dose, le patient ne doit pas prendre de dose double. Il ne peut prendre la dose oubliée qu’en respectant un intervalle de 4 heures entre les doses. Sinon, le patient doit attendre et prendre la dose suivante au moment habituel.
Une réduction temporaire de la dose à 120 mg deux fois par jour peut permettre de réduire la fréquence des bouffées congestives et des effets indésirables gastro-intestinaux. Il convient de revenir à la dose d’entretien recommandée de 240 mg deux fois par jour au cours du mois suivant.
DIMETHYL FUMARATE BIOGARAN doit être pris au moment des repas (voir rubrique 5.2). Chez les patients présentant des effets indésirables gastro-intestinaux ou des bouffées congestives, la prise de DIMETHYL FUMARATE BIOGARAN au moment des repas peut améliorer la tolérance (voir rubriques 4.4, 4.5 et 4.8).
Populations particulières Sujets âgés
Les études cliniques réalisées avec le fumarate de diméthyle ont concerné un nombre limité de patients âgés de 55 ans et plus ainsi qu’un nombre insuffisant de patients âgés de 65 ans et plus ce qui n’a pas permis de déterminer si cette population de patients répondait différemment à ce médicament par comparaison à des patients plus jeunes (voir rubrique 5.2). Compte tenu du mécanisme d'action de cette substance active, il n'y a théoriquement aucune raison de modifier la posologie chez le sujet âgé.
Insuffisants rénaux et hépatiques
Le fumarate de diméthyle n’a pas été étudié chez les patients insuffisants rénaux ou hépatiques. Selon les études de pharmacologie clinique, aucune adaptation posologique n’est nécessaire (voir rubrique 5.2). Le traitement de patients présentant une insuffisance rénale sévère ou hépatique sévère doit être instauré avec prudence (voir rubrique 4.4).
Population pédiatrique
La posologie est la même chez les adultes et les enfants âgés de 13 ans et plus.
Les données actuellement disponibles sont décrites aux rubriques 4.4, 4.8, 5.1 et 5.2.
Les données disponibles chez les enfants âgés de 10 à 12 ans sont limitées.
La sécurité et l’efficacité du fumarate de diméthyle chez les enfants âgés de moins de 10 ans n’ont pas encore été établies.
Mode d’administration
Voie orale.
La gélule doit être avalée entière. Ne pas écraser, ouvrir, dissoudre, sucer ou mâcher la gélule ou son contenu car le pelliculage gastro-résistant des granulés évite les effets irritants intestinaux.
· Hypersensibilité à la substance active ou à l’un des excipients mentionnés à la rubrique 6.1.
· Leucoencéphalopathie multifocale progressive (LEMP) suspectée ou confirmée.
4.4. Mises en garde spéciales et précautions d'emploi
Des modifications des résultats des analyses biologiques rénales ont été observées chez des patients ayant été traités par le fumarate de diméthyle dans les essais cliniques (voir rubrique 4.8). Les conséquences cliniques de ces modifications ne sont pas connues. Il est recommandé d’évaluer la fonction rénale (par exemple créatinine, urée et analyse d’urines) avant d’instaurer le traitement puis à 3 mois, 6 mois de traitement, ensuite tous les 6 à 12 mois et également en présence de signes cliniques.
Le traitement par le fumarate de diméthyle peut provoquer une atteinte hépatique médicamenteuse, incluant une augmentation des taux d’enzymes hépatiques (≥ 3 fois la limite supérieure de la normale [LSN]) et de bilirubine totale (≥ 2 x LSN). Le délai d’apparition peut être immédiat, de plusieurs semaines ou plus long. Une résolution des effets indésirables a été observée après l’arrêt du traitement. Il est recommandé de contrôler les taux de transaminases sériques (par exemple alanine aminotransférase [ALAT], aspartate aminotransférase [ASAT]) et de bilirubine totale avant l’instauration du traitement et pendant le traitement si le tableau clinique le justifie.
Les patients traités par DIMETHYL FUMARATE BIOGARAN peuvent développer une lymphopénie (voir rubrique 4.8). Avant d’instaurer un traitement par DIMETHYL FUMARATE BIOGARAN, une numération formule sanguine (NFS) complète incluant une numération des lymphocytes, doit être effectuée.
Si le taux des lymphocytes est inférieur à la limite de la normale, il est nécessaire de rechercher de façon approfondie les causes possibles avant l’instauration du traitement par DIMETHYL FUMARATE BIOGARAN. Le fumarate de diméthyle n’a pas été étudié chez des patients présentant un faible taux de lymphocytes pré-existant ; la prudence s’impose donc lors du traitement de ces patients. DIMETHYL FUMARATE BIOGARAN ne doit pas être instauré chez les patients présentant une lymphopénie sévère (nombre de lymphocytes < 0,5 × 109/L).
Après le début du traitement, une NFS complète incluant une numération des lymphocytes doit être effectuée tous les 3 mois.
Une vigilance accrue en raison d’un risque majoré de leucoencéphalopathie multifocale progressive (LEMP) est recommandée chez les patients présentant une lymphopénie comme suit :
· Le traitement par DIMETHYL FUMARATE BIOGARAN doit être interrompu chez les patients présentant une lymphopénie sévère et prolongée (nombre de lymphocytes < 0,5 × 109/L) persistant pendant plus de 6 mois.
· Chez les patients présentant des réductions modérées et durables du nombre de lymphocytes ≥ 0,5 × 109/L à < 0,8 × 109/L pendant plus de 6 mois, le rapport bénéfice/risque du traitement par DIMETHYL FUMARATE BIOGARAN doit être réévalué.
· Chez les patients dont le nombre de lymphocytes est inférieur à la limite inférieure de la normale (LIN) telle que définie par l’intervalle de référence du laboratoire local, une surveillance régulière du nombre absolu de lymphocytes est recommandée. D'autres facteurs susceptibles d'augmenter davantage le risque individuel de LEMP doivent être pris en compte (voir la sous-rubrique sur la LEMP ci-dessous).
Le nombre de lymphocytes doit être surveillé jusqu’à normalisation (voir rubrique 5.1). Lors du retour à la normale et en l’absence d’alternatives thérapeutiques, la décision concernant la reprise ou non du traitement par DIMETHYL FUMARATE BIOGARAN après l’arrêt devra alors reposer sur le jugement clinique.
Imagerie par résonance magnétique (IRM)
Avant l’instauration d’un traitement par DIMETHYL FUMARATE BIOGARAN, une IRM initiale de référence doit être disponible (datant de moins de 3 mois). La surveillance par des examens d’IRM supplémentaires doit être conforme aux recommandations nationales et locales. Envisager la réalisation d’une IRM dans le contexte d’un suivi renforcé chez les patients à risque plus élevé de LEMP. En cas de suspicion clinique de LEMP, une IRM devra être réalisée immédiatement à des fins diagnostiques.
Leucoencéphalopathie multifocale progressive (LEMP)
Des cas de LEMP ont été rapportés chez des patients traités par DIMETHYL FUMARATE BIOGARAN (voir rubrique 4.8). La LEMP est une infection opportuniste causée par le virus de John Cunningham (JCV) qui peut avoir une issue fatale ou entraîner un handicap sévère.
Des cas de LEMP sont survenus avec le fumarate de diméthyle et d’autres produits contenant des fumarates dans le cadre de lymphopénies (nombre de lymphocytes inférieur à la LIN). La lymphopénie modérée à sévère prolongée semble augmenter le risque de LEMP avec DIMETHYL FUMARATE BIOGARAN ; cependant, le risque ne peut être exclu chez les patients présentant une lymphopénie légère.
D'autres facteurs pouvant contribuer à augmenter le risque de LEMP dans le cadre d'une lymphopénie sont :
· durée du traitement par DIMETHYL FUMARATE BIOGARAN. Des cas de LEMP sont apparus après environ 1 à 5 ans de traitement, bien que le lien exact avec la durée du traitement ne soit pas connu.
· une diminution importante du nombre de lymphocytes T CD4+ et surtout CD8+, qui sont importants pour la défense immunologique (voir rubrique 4.8), et
· un traitement immunosuppresseur ou immunomodulateur préalable (voir ci-dessous).
Les médecins doivent évaluer leurs patients afin de déterminer si les symptômes indiquent un dysfonctionnement neurologique et, si c’est le cas, si ces symptômes sont typiques de la SEP ou s’ils peuvent évoquer une LEMP.
Au premier signe ou symptôme évocateur d’une LEMP, le traitement par DIMETHYL FUMARATE BIOGARAN doit être interrompu et les examens appropriés, y compris la détection de l’ADN du JCV dans le liquide céphalorachidien (LCR) par la méthode quantitative d’amplification en chaîne par polymérase (PCR), doivent être réalisés. Les symptômes de la LEMP peuvent être similaires à ceux d’une poussée de SEP. Les symptômes caractéristiques de la LEMP sont divers tels qu’une faiblesse progressive d’un côté du corps ou un manque de coordination des membres, des troubles visuels et des troubles de la pensée, de la mémoire et de l’orientation entraînant une confusion et des modifications de la personnalité. La progression de ces symptômes évolue sur une durée allant de quelques jours à quelques semaines. Les médecins doivent être particulièrement attentifs aux symptômes évocateurs de la LEMP que le patient peut ne pas remarquer. Il faut également conseiller aux patients d’informer leur partenaire ou leurs soignants de leur traitement, car ils peuvent remarquer des symptômes dont le patient n’est pas conscient.
La LEMP ne peut survenir qu’en présence d’une infection causée par le virus JC. Il faut considérer que l’influence de la lymphopénie sur la précision du test de détection des anticorps anti-JCV dans le sérum n’a pas été étudiée chez les patients traités par le fumarate de diméthyle. Il faut également noter qu’un test de détection des anticorps anti-JCV négatif (en présence de taux normaux de lymphocytes) n’exclut pas la possibilité d’une infection ultérieure par le JCV.
Si un patient développe une LEMP, DIMETHYL FUMARATE BIOGARAN doit être arrêté définitivement.
Traitement antérieur par immunosuppresseurs ou immunomodulateurs
Il n’y a pas d’études évaluant l’efficacité et la tolérance de DIMETHYL FUMARATE BIOGARAN en relais à d’autres traitements de fond de la maladie. La contribution d’un traitement immunosuppresseur antérieur sur le développement de la LEMP chez les patients traités par le fumarate de diméthyle est possible.
Des cas de LEMP sont survenus chez des patients ayant auparavant été traités par le natalizumab, pour lequel la LEMP est un risque établi. Les médecins doivent savoir que les cas de LEMP qui surviennent après l’arrêt récent du natalizumab peuvent ne pas présenter de lymphopénie.
En outre, la majorité des cas de LEMP confirmés avec le fumarate de diméthyle sont survenus chez des patients ayant reçu un traitement immunomodulateur antérieur.
En cas de relais d’un autre traitement de fond de la sclérose en plaques par DIMETHYL FUMARATE BIOGARAN, la demi-vie et le mode d’action de l’autre traitement doivent être pris en compte afin d’éviter un effet additif sur le système immunitaire et de minimiser le risque de réactivation de la maladie. Une NFS complète est recommandée avant d’instaurer DIMETHYL FUMARATE BIOGARAN et régulièrement durant le traitement (voir Analyses de sang/biologiques ci-dessus).
Insuffisances rénale et hépatique sévères
DIMETHYL FUMARATE BIOGARAN n’a pas été étudié chez des patients présentant une insuffisance rénale ou hépatique sévère ; le traitement doit être instauré avec prudence chez ces patients (voir rubrique 4.2).
Pathologie gastro-intestinale active sévère
DIMETHYL FUMARATE BIOGARAN n’a pas été étudié chez les patients présentant une pathologie gastro-intestinale active sévère ; par conséquent, le traitement doit être instauré avec prudence chez ces patients.
Bouffées congestives
Lors des essais cliniques, 34 % des patients sous fumarate de diméthyle ont présenté des bouffées congestives. Pour la majorité des patients présentant des bouffées congestives, ces dernières étaient de sévérité légère ou modérée. Les données issues d’études menées chez des volontaires sains montrent que les bouffées congestives associées au fumarate de diméthyle sont probablement médiées par les prostaglandines. Un traitement court par de l’acide acétylsalicylique 75 mg en formulation non gastro-résistante peut être bénéfique pour les patients souffrant de bouffées congestives insupportables (voir rubrique 4.5). Ce traitement a réduit la fréquence et la sévérité des bouffées congestives dans deux études menées chez le volontaire sain.
Lors des essais cliniques, 3 patients sur un total de 2 560 patients sous fumarate de diméthyle ont présenté des symptômes de bouffées congestives graves probablement dues à une hypersensibilité ou à des réactions anaphylactoïdes. Sans engager le pronostic vital, ces effets ont nécessité une hospitalisation. Les prescripteurs et les patients devront être alertés de cette éventualité en cas de réactions graves avec bouffées congestives (voir rubriques 4.2, 4.5 et 4.8).
Réactions anaphylactiques
Des cas d’anaphylaxie/réaction anaphylactoïde ont été rapportés après l’administration de dimethyl fumarate depuis la commercialisation. Les symptômes peuvent être : dyspnée, hypoxie, hypotension, angiœdème, rash ou urticaire. Le mécanisme de l’anaphylaxie induite par le fumarate de diméthyle n’est pas connu. Ces réactions apparaissent généralement après la première dose, mais peuvent également survenir à tout moment au cours du traitement et peuvent être graves et engager le pronostic vital. Les patients doivent être informés qu’ils doivent arrêter de prendre DIMETHYL FUMARATE BIOGARAN et consulter immédiatement un médecin s’ils présentent des signes ou symptômes d’anaphylaxie. Le traitement ne doit pas être repris (voir rubrique 4.8).
Infections
Lors des essais de phase III contrôlés versus placebo, l'incidence des infections (60 % vs 58 %) et des infections graves (2 % vs 2 %) était similaire chez les patients sous fumarate de diméthyle ou sous placebo, respectivement. Cependant, du fait des propriétés immunomodulatrices de DIMETHYL FUMARATE BIOGARAN (voir rubrique 5.1), si un patient développe une infection grave, l’interruption du traitement par DIMETHYL FUMARATE BIOGARAN doit être envisagée et les bénéfices et les risques doivent être réévalués avant la reprise du traitement. Les patients traités par DIMETHYL FUMARATE BIOGARAN doivent être avertis de la nécessité de signaler les symptômes d’infection à un médecin. Chez les patients présentant des infections graves, le traitement par DIMETHYL FUMARATE BIOGARAN ne doit débuter qu’après la résolution de la ou des infection(s).
Il n’a pas été observé d’augmentation de l’incidence d’infections graves chez les patients ayant un taux de lymphocytes < 0,8 x 109/L ou < 0,5 x 109/L (voir rubrique 4.8). Si le traitement est poursuivi en présence d’une lymphopénie prolongée, modérée à sévère, le risque d’infection opportuniste, y compris de leucoencéphalopathie multifocale progressive (LEMP) ne peut être exclu (voir rubrique 4.4, sous-rubrique LEMP).
Infections zostériennes (zona)
Des cas de zona ont été rapportés avec le fumarate de diméthyle. La majorité des cas étaient sans gravité, cependant des cas graves incluant zona disséminé, zona ophtalmique, zona otitique, infection neurologique zostérienne, méningoencéphalite zostérienne et méningomyélite zostérienne ont été rapportés. Ces événements peuvent survenir à tout moment au cours du traitement. Les patients sous DIMETHYL FUMARATE BIOGARAN doivent faire l’objet d’une surveillance afin de détecter tout signe ou symptôme du zona, surtout lorsqu’une lymphocytopénie concomitante est rapportée. En cas de survenue d’un zona, un traitement approprié contre le zona doit être administré. L’interruption du traitement par DIMETHYL FUMARATE BIOGARAN doit être envisagée chez les patients atteints d’infections graves jusqu’à celles-ci soient résolues (voir rubrique 4.8).
Instauration du traitement
Le traitement par DIMETHYL FUMARATE BIOGARAN doit être débuté progressivement pour réduire la fréquence des bouffées congestives et des effets indésirables gastro-intestinaux (voir rubrique 4.2).
Syndrome de Fanconi
Des cas de syndrome de Fanconi ont été rapportés avec un médicament contenant du fumarate de diméthyle associé à d’autres esters de l’acide fumarique. Le diagnostic précoce du syndrome de Fanconi et l’arrêt du traitement par fumarate de diméthyle sont primordiaux afin de prévenir l’apparition d’une insuffisance rénale et d’une ostéomalacie, car le syndrome est généralement réversible. Les signes les plus importants sont les suivants : protéinurie, glycosurie (avec glycémie normale), hyperaminoacidurie et phosphaturie (éventuellement associée à une hypophosphatémie). La progression peut impliquer des symptômes tels que polyurie, polydipsie et faiblesse musculaire proximale. Dans de rares cas, une ostéomalacie hypophosphatémique accompagnée de douleurs osseuses non localisées, une phosphatase alcaline sérique élevée et des fractures de fatigue peuvent survenir. Il est important de noter que le syndrome de Fanconi peut survenir sans élévation des taux de créatinine ou sans diminution du débit de filtration glomérulaire.
En cas de symptômes flous, le syndrome de Fanconi doit être envisagé et des examens appropriés doivent être effectués.
Population pédiatrique
Le profil de sécurité des patients pédiatriques est qualitativement similaire à celui des adultes. En conséquence, les mises en garde et les précautions d’emploi pour les adultes s’appliquent également à la population pédiatrique. Pour les différences quantitatives du profil de sécurité, voir la rubrique 4.8.
La sécurité à long terme du fumarate de diméthyle chez les enfants et les adolescents n’a pas encore été établie.
Ce médicament contient moins de 1 mmol (23 mg) de sodium par gélule, c’est-à-dire qu’il est essentiellement « sans sodium ».
4.5. Interactions avec d'autres médicaments et autres formes d'interactions
DIMETHYL FUMARATE BIOGARAN n'a pas été étudié en association avec des traitements anticancéreux ou immunosuppresseurs. Par conséquent, la prudence s’impose lorsque ces médicaments sont administrés de façon concomitante. Dans les études cliniques dans la sclérose en plaques, un traitement concomitant des poussées par des corticostéroïdes intraveineux en cure de courte durée n'a pas été associé à une augmentation du nombre d’infections.
L’administration concomitante de vaccins non vivants conformément aux programmes de vaccination nationaux peut être envisagée pendant le traitement par DIMETHYL FUMARATE BIOGARAN. Dans une étude clinique menée chez 71 patients au total atteints de sclérose en plaques récurrente-rémittente, les patients recevant du fumarate de diméthyle 240 mg deux fois par jour pendant au moins 6 mois (n = 38) ou un interféron non pégylé pendant au moins 3 mois (n = 33) ont développé une réponse immunitaire comparable (définie comme une augmentation ≥ 2 fois des titres post-vaccination par rapport à la valeur avant la vaccination) à l’anatoxine tétanique (antigène de rappel) et à un vaccin méningococcique C polysaccharidique conjugué (néoantigène), tandis que la réponse immunitaire aux différents sérotypes d’un vaccin pneumococcique polysaccharidique 23-valent non conjugué (antigène indépendant des cellules T) a varié dans les deux groupes de traitement. Une réponse immunitaire positive aux trois vaccins, définie comme une augmentation ≥ 4 fois des titres d’anticorps, a été atteinte chez un nombre moindre de patients dans les deux groupes de traitement. De faibles différences numériques dans la réponse à l’anatoxine tétanique et au polysaccharide du pneumocoque de sérotype 3 ont été observées en faveur de l’interféron non pégylé.
Il n’existe pas de données cliniques concernant l’efficacité et la sécurité des vaccins vivants atténués chez les patients traités par du fumarate de diméthyle. Il est possible que les vaccins vivants comportent un plus grand risque infectieux et ils ne doivent pas être administrés aux patients sous DIMETHYL FUMARATE BIOGARAN sauf, dans des cas exceptionnels, par exemple si l'on considère que ce risque potentiel est inférieur au risque lié à l'absence de vaccination.
Au cours du traitement par DIMETHYL FUMARATE BIOGARAN, il convient d’éviter d’utiliser simultanément d’autres dérivés de l’acide fumarique (topiques ou systémiques).
Chez l’homme, le fumarate de diméthyle est essentiellement métabolisé par les estérases avant d’atteindre la circulation systémique, puis son métabolisme fait intervenir le cycle de l'acide tricarboxylique, sans aucune intervention du cytochrome P450 (CYP). Aucun risque d’éventuelles interactions médicamenteuses n’a été identifié au cours des études suivantes : études in vitro d'inhibition et d’induction du CYP, étude sur la glycoprotéine-p, ou études sur la liaison aux protéines du fumarate de diméthyle et du fumarate de monométhyle (un métabolite primaire du fumarate de diméthyle).
Des médicaments fréquemment utilisés chez les patients atteints de sclérose en plaques, comme l’interféron bêta 1a en intramusculaire et l’acétate de glatiramère, ont été évalués cliniquement afin de détecter une interaction éventuelle avec le fumarate de diméthyle. Le profil pharmacocinétique du fumarate de diméthyle n’a pas été modifié par ces médicaments.
Les données d’études menées chez des volontaires sains semblent indiquer que les bouffées congestives associées au fumarate de diméthyle sont probablement médiées par les prostaglandines. Dans deux études chez des volontaires sains, l’administration de 325 mg (ou équivalent) d’acide acétylsalicylique non gastro-résistant 30 minutes avant le fumarate de diméthyle, pendant 4 jours et pendant 4 semaines respectivement n’a pas modifié le profil pharmacocinétique du fumarate de diméthyle. Les risques potentiels associés au traitement par l’acide acétylsalicylique doivent être pris en compte avant l’administration concomitante avec DIMETHYL FUMARATE BIOGARAN chez les patients atteints de SEP récurrente-rémittente. L’utilisation continue à long terme (plus de 4 semaines) d’acide acétylsalicylique n’a pas été étudiée (voir rubriques 4.4 et 4.8).
Un traitement concomitant par des médicaments néphrotoxiques (tels que les aminoglycosides, les diurétiques, les anti-inflammatoires non stéroïdiens ou le lithium) peut augmenter le risque de survenue d’effets indésirables rénaux (par exemple protéinurie, voir rubrique 4.8) chez les patients traités par DIMETHYL FUMARATE BIOGARAN (voir rubrique 4.4 Analyses de sang/biologiques).
La consommation modérée d'alcool n'a pas modifié l'exposition au fumarate de diméthyle et n'a pas été associée à un nombre plus élevé de réactions indésirables. La consommation d'une grande quantité de boissons fortement alcoolisées (taux d'alcool supérieur à 30 %) doit être évitée dans l’heure suivant la prise de DIMETHYL FUMARATE BIOGARAN car l’alcool peut entraîner une augmentation de la fréquence des effets indésirables gastro-intestinaux.
Les essais in vitro d’induction du CYP n'ont pas révélé d'interaction entre le fumarate de diméthyle et les contraceptifs oraux. Dans une étude in vivo, l’administration concomitante de fumarate de diméthyle avec un contraceptif oral combiné (norgestimate et éthinylestradiol) n’a pas induit de modification significative de l’exposition au contraceptif oral. Il n’a pas été réalisé d’études d’interactions avec les contraceptifs oraux contenant d’autres progestatifs ; cependant, aucun effet de DIMETHYL FUMARATE BIOGARAN sur l’exposition à ces médicaments n’est attendu.
Population pédiatrique
Les études d’interaction n’ont été réalisées que chez l’adulte.
4.6. Fertilité, grossesse et allaitement
Grossesse
Il n’existe pas de données ou il existe des données limitées sur l’utilisation du fumarate de diméthyle chez la femme enceinte. Les études effectuées chez l’animal ont mis en évidence une toxicité sur la reproduction (voir rubrique 5.3). DIMETHYL FUMARATE BIOGARAN n’est pas recommandé pendant la grossesse et chez les femmes en âge de procréer n’utilisant pas de méthode appropriée de contraception (voir rubrique 4.5). DIMETHYL FUMARATE BIOGARAN ne doit être utilisé pendant la grossesse qu'en cas de nécessité absolue et uniquement si le bénéfice éventuel est supérieur au risque potentiel pour le fœtus.
Allaitement
On ne sait pas si le fumarate de diméthyle ou ses métabolites sont excrétés dans le lait maternel. Un risque pour les nouveau-nés/nourrissons ne peut être exclu. Une décision doit être prise soit d’interrompre l’allaitement soit d’interrompre le traitement avec DIMETHYL FUMARATE BIOGARAN, en prenant en compte le bénéfice de l’allaitement pour l’enfant au regard du bénéfice du traitement pour la femme.
Fertilité
Il n’existe pas de données relatives aux effets du fumarate de diméthyle sur la fertilité humaine. Les données issues des études précliniques ne suggèrent pas que le fumarate de diméthyle soit associé à un risque accru de diminution de la fertilité (voir rubrique 5.3).
4.7. Effets sur l'aptitude à conduire des véhicules et à utiliser des machines
Synthèse du profil de sécurité
Les effets indésirables les plus fréquents (incidence ≥ 10 %) rapportés chez les patients traités par le fumarate de diméthyle sont les bouffées congestives et les effets gastro-intestinaux (c’est-à-dire, diarrhées, nausées, douleurs abdominales, douleurs abdominales hautes). Les bouffées congestives et les effets gastro-intestinaux ont tendance à survenir en début de traitement (principalement au cours du premier mois) et chez les patients présentant des bouffées congestives et troubles gastrointestinaux, ces troubles peuvent éventuellement continuer de manière intermittente pendant le traitement par DIMETHYL FUMARATE BIOGARAN. Les effets indésirables rapportés le plus fréquemment et ayant entraîné l’arrêt du traitement (incidence >1 %) chez les patients traités par fumarate de diméthyle sont les bouffées congestives (3 %) et les effets gastro-intestinaux (4 %).
Dans le cadre des études cliniques contrôlées contre placebo et non contrôlées, 2 études au total, 2 513 patients ont reçu du fumarate de diméthyle pendant une durée allant jusqu’à 12 ans, avec une exposition globale au produit équivalente à 11 318 patient-années. Au total, 1 169 patients ont été traités par fumarate de diméthyle pendant moins 5 ans et 426 patients pendant au moins 10 ans. L’expérience au cours des essais cliniques non contrôlés est comparable à celle des essais cliniques contrôlés contre placebo.
Liste tabulée des effets indésirables
Les effets indésirables rapportés au cours des études cliniques, des études de sécurité post-autorisation et des déclarations spontanées sont présentés dans le tableau ci-dessous.
Les effets indésirables sont présentés selon les termes préférentiels de la base de données MedDRA et les classes de systèmes d’organes. L’incidence des effets indésirables ci-dessous est exprimée en fonction des catégories suivantes :
· Très fréquent (≥ 1/10)
· Fréquent (≥ 1/100, < 1/10)
· Peu fréquent (≥ 1/1000, < 1/100)
· Rare (≥ 1/10 000, < 1/1 000)
· Très rare (< 1/10 000)
· Fréquence indéterminée (ne peut être estimée sur la base des données disponibles).
|
Base de données MedDRA des classes de systèmes d’organes |
Effet indésirable |
Catégorie de fréquence |
|
Infections et infestations |
Gastro-entérite |
Fréquent |
|
Leucoencéphalopathie multifocale progressive (LEMP) |
Fréquence indéterminée |
|
|
Zona |
Fréquence indéterminée |
|
|
Affections hématologiques et du système lymphatique |
Lymphopénie |
Fréquent |
|
Leucopénie |
Fréquent |
|
|
Thrombocytopénie |
Peu fréquent |
|
|
Affections du système immunitaire |
Hypersensibilité |
Peu fréquent |
|
Anaphylaxie |
Fréquence indéterminée |
|
|
Dyspnée |
Fréquence indéterminée |
|
|
Hypoxie |
Fréquence indéterminée |
|
|
Hypotension |
Fréquence indéterminée |
|
|
Angiœdème |
Fréquence indéterminée |
|
|
Affections du système nerveux |
Sensation de brûlures |
Fréquent |
|
Affections vasculaires |
Bouffées congestives |
Très fréquent |
|
Bouffées de chaleur |
Fréquent |
|
|
Affections respiratoires, thoraciques et médiastinales |
Rhinorrhée |
Fréquence indéterminée |
|
Affections gastro-intestinales |
Diarrhées |
Très fréquent |
|
Nausées |
Très fréquent |
|
|
Douleurs abdominales hautes |
Très fréquent |
|
|
Douleurs abdominales |
Très fréquent |
|
|
Vomissements |
Fréquent |
|
|
Dyspepsie |
Fréquent |
|
|
Gastrite |
Fréquent |
|
|
Troubles gastro-intestinaux |
Fréquent |
|
|
Affections hépatobiliaires |
Augmentation de l’aspartate aminotransférase |
Fréquent |
|
Augmentation de l’alanine aminotransférase |
Fréquent |
|
|
Atteinte hépatique médicamenteuse |
Fréquence indéterminée |
|
|
Affections de la peau et du tissu sous-cutané |
Prurit |
Fréquent |
|
Rash |
Fréquent |
|
|
Érythème |
Fréquent |
|
|
Alopécie |
Fréquent |
|
|
Affections du rein et des voies urinaires |
Protéinurie |
Fréquent |
|
Troubles généraux et anomalies au site d’administration |
Sensation de chaleur |
Fréquent |
|
Investigations |
Présence de cétones dans les urines |
Très fréquent |
|
Présence d’albumine dans les urines |
Fréquent |
|
|
Diminution du nombre de globules blancs |
Fréquent |
Description de certaines réactions indésirables
Bouffées congestives
Dans les études contre placebo, l’incidence des bouffées congestives (34 % versus 4 %) et des bouffées de chaleur (7 % versus 2 %) était respectivement plus élevée chez les patients traités par fumarate de diméthyle que chez ceux recevant le placebo. Les bouffées congestives étaient habituellement décrites comme des bouffées congestives ou de chaleur, mais elles pouvaient également comprendre d’autres effets (chaleur, rougeur, démangeaisons ou sensation de brûlure, par exemple). Les bouffées congestives tendaient à survenir en début de traitement (principalement pendant le premier mois) et chez les patients qui les présentaient, ces effets pouvaient se manifester de manière intermittente pendant tout le traitement par fumarate de diméthyle. Dans la majorité des cas, ces bouffées congestives étaient d'une sévérité légère à modérée. Au total, 3 % des patients traités par du fumarate de diméthyle ont arrêté le traitement en raison de bouffées congestives. L'incidence des bouffées congestives graves pouvant se caractériser par un érythème généralisé, un rash et/ou un prurit, a été observée chez moins de 1 % des patients traités par du fumarate de diméthyle (voir rubriques 4.2, 4.4 et 4.5).
Effets gastro-intestinaux
L’incidence des effets gastro-intestinaux (tels que diarrhées [14 % versus 10 %], nausées [12 % versus 9 %], douleurs abdominales hautes [10 % versus 6 %], douleurs abdominales [9 % versus 4 %], vomissements [8 % versus 5 %] et dyspepsie [5 % versus 3 %]) était respectivement plus élevée chez les patients traités par fumarate de diméthyle que chez les patients sous placebo. L’incidence des effets gastro-intestinaux était plus élevée en début de traitement (principalement durant le premier mois) et chez les patients présentant des troubles gastrointestinaux, ces troubles peuvent éventuellement continuer de manière intermittente pendant le traitement par DIMETHYL FUMARATE BIOGARAN. Pour la majorité des patients présentant des troubles gastro-intestinaux, ces derniers étaient légers ou modérés. Quatre pour cent (4 %) des patients traités par du fumarate de diméthyle ont dû arrêter leur traitement à cause d’effets gastro-intestinaux. L’incidence des effets gastro-intestinaux graves, notamment des gastro-entérites et des gastrites, a été observée chez moins de 1 % des patients traités par du fumarate de diméthyle (voir rubrique 4.2).
Fonction hépatique
Sur la base des données des études contrôlées contre placebo, chez la majorité des patients présentant des augmentations des transaminases hépatiques, ces augmentations étaient < 3 fois la limite supérieure de la normale (LSN). L’incidence accrue d’une augmentation du taux des transaminases hépatiques chez les patients traités par du fumarate de diméthyle, en comparaison au placebo, était principalement observée durant les 6 premiers mois de traitement. Une augmentation du taux d'alanine aminotransférase et d’aspartate aminotransférase ≥ 3 fois la LSN a été observée respectivement chez 5 % et 2 % des patients sous placebo et chez 6 % et 2 % des patients traités par du fumarate de diméthyle. Les arrêts de traitement dus à un taux élevé de transaminases hépatiques ont été < 1 % et comparables chez les patients traités par du fumarate de diméthyle et chez ceux sous placebo. Il n’a pas été observé d’augmentations des taux de transaminases ≥ 3 fois la LSN accompagnées d’augmentations du taux de bilirubine totale > 2 fois la LSN dans les études contrôlées contre placebo.
Des cas d’augmentation des enzymes hépatiques et d’atteinte hépatique médicamenteuse (élévations des transaminases ≥ 3 fois la LSN accompagnées d’élévations de la bilirubine totale > 2 fois la LSN) après l’administration de fumarate de diméthyle ont été rapportés depuis la commercialisation ; ils se sont résolus après l’arrêt du traitement.
Lymphopénie
Dans les études contrôlées contre placebo, la majorité des patients (> 98 %) présentait avant l’instauration du traitement des taux normaux de lymphocytes. Après le traitement par fumarate de diméthyle, le nombre moyen de lymphocytes a diminué au cours de la première année puis a atteint un plateau. En moyenne, le nombre de lymphocytes a diminué d’environ 30 % par rapport à la valeur initiale. Les nombres moyen et médian de lymphocytes sont restés dans les limites de la normale. Un nombre de lymphocytes < 0,5 × 109/L a été observé chez < 1 % des patients sous placebo et chez 6 % de ceux traités par du fumarate de diméthyle. Un nombre de lymphocytes < 0,2 × 109/L a été observé chez 1 patient traité par du fumarate de diméthyle contre aucun patient sous placebo.
Dans les études cliniques (contrôlées et non contrôlées), 41 % des patients traités par du fumarate de diméthyle présentaient une lymphopénie (définie dans ces études comme < 0,91 × 109/L). Une lymphopénie légère (taux ≥ 0,8 × 109/L et < 0,91 × 109/L) a été observée chez 28 % des patients ; une lymphopénie modérée (taux ≥ 0,5 × 109/L et < 0,8 × 109/L) persistant pendant au moins six mois a été observée chez 10 % des patients ; une lymphopénie sévère (taux < 0,5 × 109/L) persistant pendant au moins six mois a été observée chez 2 % des patients. Dans le groupe présentant une lymphopénie sévère, les taux de lymphocytes sont restés < 0,5 × 109/L avec la poursuite du traitement chez la majorité des patients.
Infections, y compris LEMP et infections opportunistes
Des cas d’infections par le virus de John Cunningham (JCV) provoquant une leucoencéphalopathie multifocale progressive (LEMP) ont été rapportés avec fumarate de diméthyle (voir rubrique 4.4). La LEMP peut avoir une issue fatale ou entraîner un handicap sévère. Dans l’un des essais cliniques, un patient prenant du fumarate de diméthyle a développé une LEMP dans le cadre d’une lymphopénie sévère et prolongée (nombre de lymphocytes principalement < 0,5 × 109/L pendant 3,5 ans), avec une issue fatale. Dans le cadre de la post-commercialisation, la LEMP est également survenue en présence d’une lymphopénie modérée et légère (> 0,5 × 109/L à < LIN, telle que définie par l’intervalle de référence du laboratoire local).
Dans plusieurs cas de LEMP avec détermination des sous-types de lymphocytes T au moment du diagnostic de la LEMP, on a constaté que le nombre de lymphocytes T CD8+ était réduit à <0,1×109/L, alors que les réductions du nombre de lymphocytes T CD4+ étaient variables (allant de <0,05 à 0,5×109/L) et étaient davantage corrélées avec la sévérité globale de la lymphopénie (<0,5 x109/L à Une lymphopénie modérée à sévère prolongée semble augmenter le risque de LEMP avec du fumarate de diméthyle ; cependant, la LEMP est également survenue chez des patients présentant une lymphopénie légère. En outre, la majorité des cas de LEMP dans le cadre de la post-commercialisation sont survenus chez des patients > 50 ans. Des infections zostériennes (zona) ont été rapportées lors de l’utilisation de fumarate de diméthyle. Dans une étude d’extension à long terme en cours, environ 5 % des 1 736 patients atteints de SEP traités par du fumarate de diméthyle ont présenté un ou plusieurs événements de type zona, en majorité d’intensité légère à modérée. La plupart des patients, notamment ceux ayant présenté une infection zostérienne grave, avaient un nombre de lymphocytes supérieur à la limite inférieure de la normale. Chez une majorité de sujets dont le nombre de lymphocytes concomitant était inférieur à la LIN, la lymphopénie a été jugée modérée ou sévère. Depuis la commercialisation, la plupart des cas d’infection zostérienne (zona) étaient sans gravité et ont disparu après traitement. Les données disponibles concernant le nombre absolu de lymphocytes (NAL) chez les patients atteints d’infection herpétique depuis la commercialisation sont limitées. Toutefois, la plupart des patients chez qui le NAL a été rapporté ont présenté une lymphopénie modérée (≥ 0,5 × 109/L à < 0,8 × 109/L) ou sévère (< 0,5 × 109/L à 0,2 × 109/L) (voir rubrique 4.4). Anomalies biologiques Dans les études contrôlées contre placebo, le taux de cétones urinaires (1+ ou plus) était plus élevé chez les patients traités par du fumarate de diméthyle (45 %) que chez ceux sous placebo (10 %), sans qu’aucune conséquence clinique négative n’ait été observée pendant les essais cliniques. Le taux de 1,25-dihydroxy-vitamine D a diminué chez les patients traités par du fumarate de diméthyle, comparé à ceux sous placebo (diminution du pourcentage médian, par rapport au pourcentage initial, à 2 ans respectivement de 25 % versus 15 %), alors que le taux d’hormone parathyroïdienne (PTH) a augmenté chez les patients traités par du fumarate de diméthyle, par rapport à ceux sous placebo (augmentation du pourcentage médian, par rapport au pourcentage initial, à 2 ans respectivement de 29 % versus 15 %,). Les valeurs moyennes de ces deux paramètres sont restées dans les limites de la normale. Une augmentation transitoire du nombre moyen d’éosinophiles a été observée durant les deux premiers mois de traitement. Population pédiatrique Dans une étude en ouvert randomisée, contrôlée versus comparateur actif d’une durée de 96 semaines menée chez des enfants et des adolescents atteints de SEP-RR âgés de 10 à moins de 18 ans (dose de 120 mg deux fois par jour pendant 7 jours puis 240 mg deux fois par jour pendant le reste de la période de traitement, population d’analyse de la sécurité, n = 78), le profil de sécurité chez ces patients était comparable à celui précédemment observé chez les patients adultes. Le plan expérimental de l’étude clinique pédiatrique était différent de celui des études cliniques contrôlées versus placebo menées chez des adultes. Par conséquent, une contribution du plan expérimental de l’étude aux différences numériques des effets indésirables entre les populations pédiatrique et adulte ne peut être exclue. Les effets indésirables suivants ont été rapportés plus fréquemment (fréquence ≥ 10 %) dans la population pédiatrique que dans la population adulte : · Des céphalées ont été rapportées chez 28 % des patients traités par fumarate de diméthyle versus 36 % des patients traités par l’interféron bêta-1a. · Des affections gastro-intestinales ont été rapportées chez 74 % des patients traités par fumarate de diméthyle versus 31 % des patients traités par l’interféron bêta-1a. Parmi celles-ci, les plus fréquemment rapportées avec fumarate de diméthyle étaient des douleurs abdominales et des vomissements. · Des affections respiratoires, thoraciques et médiastinales ont été rapportées chez 32 % des patients traités par fumarate de diméthyle versus 11 % des patients traités par l’interféron bêta-1a. Parmi celles-ci, les plus fréquemment rapportées avec fumarate de diméthyle étaient des douleurs oropharyngées et une toux. · Des dysménorrhées ont été rapportées chez 17 % des patientes traitées par fumarate de diméthyle versus 7 % des patientes traitées par l’interféron bêta-1a. Dans une petite étude en ouvert non contrôlée d’une durée de 24 semaines menée chez des enfants et des adolescents atteints de SEP-RR âgés de 13 à 17 ans (dose de 120 mg deux fois par jour pendant 7 jours puis 240 mg deux fois par jour pendant le reste de la période de traitement ; population d’analyse de la sécurité, n = 22), suivie d’une étude d’extension de 96 semaines (dose de 240 mg deux fois par jour ; population d’analyse de la sécurité, n = 20), le profil de sécurité était comparable à celui observé chez les patients adultes. Les données chez les enfants âgés de 10 à 12 ans sont limitées. La sécurité et l’efficacité de fumarate de diméthyle chez les enfants âgés de moins de 10 ans n’ont pas encore été établies. Déclaration des effets indésirables suspectés La déclaration des effets indésirables suspectés après autorisation du médicament est importante. Elle permet une surveillance continue du rapport bénéfice/risque du médicament. Les professionnels de santé déclarent tout effet indésirable suspecté via le système national de déclaration : Agence nationale de sécurité du médicament et des produits de santé (ANSM) et réseau des Centres Régionaux de Pharmacovigilance - Site internet : https://signalement.social-sante.gouv.fr/ 5. PROPRIETES PHARMACOLOGIQUES 5.1. Propriétés pharmacodynamiques Classe pharmacothérapeutique : Immunosuppresseurs, autres immunosuppresseurs, code ATC : L04AX07. Mécanisme d’action Le mécanisme par lequel le fumarate de diméthyle exerce ses effets thérapeutiques chez les patients atteints de SEP n’est pas entièrement connu. Les études précliniques indiquent que les réponses pharmacodynamiques au fumarate de diméthyle semblent être principalement médiées par l’activation de la voie transcriptionnelle du facteur nucléaire NRF2 (erythroid-derived 2-like 2). Il a été montré chez des patients que le fumarate de diméthyle augmente l’expression des gènes antioxydants NRF2- dépendants (par exemple NAD(P)H déshydrogénase, quinone 1; [NQO1]). Effets pharmacodynamiques Effets sur le système immunitaire Dans les études précliniques et cliniques, le fumarate de diméthyle a démontré des propriétés anti- inflammatoires et immunomodulatrices. Dans des modèles précliniques, le fumarate de diméthyle et le monométhyl fumarate, métabolite primaire du fumarate de diméthyle, ont réduit significativement l’activation des cellules immunitaires et la libération ultérieure de cytokines pro-inflammatoires en réponse aux stimuli inflammatoires. Durant les essais cliniques chez les patients psoriasiques, le fumarate de diméthyle a affecté les phénotypes des lymphocytes en réduisant le profil des cytokines pro- inflammatoires (TH1, TH17), et a favorisé la production de cytokines anti-inflammatoires (TH2). Le fumarate de diméthyle a démontré une activité thérapeutique dans de multiples modèles de lésion inflammatoire et neuro-inflammatoire. Dans les études de phase III menées chez des patients atteints de SEP (DEFINE, CONFIRM et ENDORSE), lors du traitement par fumarate de diméthyle, le nombre moyen de lymphocytes a diminué en moyenne d’environ 30 % par rapport au nombre initial au cours de la première année puis est resté stable. Dans ces études, les patients qui arrêtaient le traitement par fumarate de diméthyle avec un taux de lymphocytes inférieur à la limite inférieure de la normale (LIN, 910/mm3) étaient suivis afin que le retour à la normale puisse être surveillé. La figure 1 présente l’estimation selon la méthode de Kaplan-Meier du pourcentage de patients chez lesquels le taux de lymphocytes est revenu à la LIN sans lymphopénie sévère prolongée. La valeur de référence pour la normalisation (VRN) était définie comme le dernier nombre absolu de lymphocytes sous traitement avant l’arrêt de fumarate de diméthyle. Les pourcentages estimés de patients qui présentaient une lymphopénie légère, modérée ou sévère à la VRN et qui avaient un retour du taux de lymphocytes à la LIN (NAL ≥ 0,9 × 109/L) à la semaine 12 et à la semaine 24 sont présentés avec les intervalles de confiance à 95 % ponctuels dans les tableaux 1, 2 et 3. L’erreur standard de l’estimateur de Kaplan-Meier de la fonction de survie est calculée selon la formule de Greenwood. Figure 1 : Méthode de Kaplan-Meier ; pourcentage de patients présentant un retour du taux de lymphocytes à ≥ 910/mm3 (LIN) par rapport à la valeur de référence pour la normalisation (VRN)
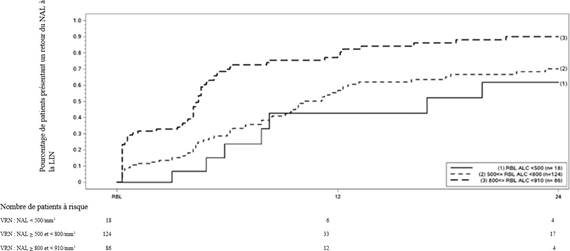
Tableau 1 : Méthode de Kaplan-Meier ; estimation du pourcentage de patients présentant une lymphopénie légère par rapport à la valeur de référence pour la normalisation (VRN) ayant obtenu un retour du taux de lymphocytes à la LIN, à l’exclusion des patients présentant une lymphopénie sévère prolongée
|
Nombre de patients présentant une lymphopénie légèrea à risque |
Inclusion N = 86 |
Semaine 12 N = 12 |
Semaine 24 N = 4 |
|
Pourcentage de patients dont le taux de lymphocytes est revenu à la LIN (IC à 95 %) |
|
0,81 (0,71 ; 0,89) |
0,90 (0,81 ; 0,96) |
a Patients ayant un NAL < 910 et ≥ 800/mm3 à la VRN, à l’exclusion des patients présentant une lymphopénie sévère prolongée.
Tableau 2 : Méthode de Kaplan-Meier ; estimation du pourcentage de patients présentant une lymphopénie modérée par rapport la valeur de référence pour la normalisation (VRN) ayant obtenu un retour du taux de lymphocytes à la LIN, à l’exclusion des patients présentant une lymphopénie sévère prolongée
|
Nombre de patients présentant une lymphopénie modéréea à risque |
Inclusion N = 124 |
Semaine 12 N = 33 |
Semaine 24 N = 17 |
|
Pourcentage de patients dont le taux de lymphocytes est revenu à la LIN (IC à 95 %) |
|
0,57 (0,46 ; 0,67) |
0,70 (0,60 ; 0,80) |
a Patients ayant un NAL < 800 et ≥ 500/mm3 à la VRN, à l’exclusion des patients présentant une lymphopénie sévère prolongée.
Tableau 3 : Méthode de Kaplan-Meier ; estimation du pourcentage de patients présentant une lymphopénie sévère par rapport la valeur de référence pour la normalisation (VRN) ayant obtenu un retour du taux de lymphocytes à la LIN, à l’exclusion des patients présentant une lymphopénie sévère prolongée
|
Nombre de patients présentant une lymphopénie sévèrea à risque |
Inclusion N = 18 |
Semaine 12 N = 6 |
Semaine 24 N = 4 |
|
Pourcentage de patients dont le taux de lymphocytes est revenu à la LIN (IC à 95 %) |
|
0,43 (0,20 ; 0,75) |
0,62 (0,35 ; 0,88) |
a Patients ayant un NAL < 500 à la VRN, à l’exclusion des patients présentant une lymphopénie sévère prolongée.
Efficacité et sécurité cliniques
Dans l’étude 1 DEFINE, les valeurs médianes des caractéristiques initiales des patients étaient les suivantes : âge 39 ans ; durée de la maladie 7,0 ans ; score EDSS 2,0. Par ailleurs, 16 % des patients avaient un score EDSS > 3,5 ; 28 % avaient présenté au moins 2 poussées au cours de l'année précédente et 42 % avaient déjà reçu un traitement de la SEP. Dans la cohorte IRM, 36 % des patients entrant dans l'étude présentaient initialement des lésions Gd+ (nombre moyen de lésions Gd+ : 1,4).
Dans l'étude 2 CONFIRM, les valeurs médianes des caractéristiques initiales des patients étaient les suivantes : âge 37 ans, durée de la maladie 6,0 ans, score EDSS 2,5. Par ailleurs, 17 % des patients avaient un score EDSS > 3,5 ; 32 % avaient présenté au moins 2 poussées au cours de l'année précédente et 30 % avaient déjà reçu un traitement de la SEP. Dans la cohorte IRM, 45 % des patients entrant dans l'étude présentaient initialement des lésions Gd+ (nombre moyen de lésions Gd+ : 2,4).
Comparativement au placebo, les groupes de patients traités par du fumarate de diméthyle ont présenté une réduction cliniquement et statistiquement significative du pourcentage de patients ayant eu au moins une poussée à 2 ans (critère d’évaluation principal dans l’étude 1 DEFINE), et du taux annualisé de poussées (TAP) à 2 ans (critère d’évaluation principal dans l’étude 2 CONFIRM).
Le TAP dans les groupes traités par l'acétate de glatiramère et le placebo, était respectivement de 0,286 et 0,401 dans l’étude 2 CONFIRM, ce qui correspond à une réduction de 29 % (p = 0,013), pourcentage comparable aux données figurant dans le RCP de ce médicament.
|
|
DEFINE |
CONFIRM |
|||
|
|
Placebo |
Fumarate de diméthyle 240 mg 2 fois/jour |
Placebo |
Fumarate de diméthyle 240 mg 2 fois/par jour |
Acétate de glatiramère |
|
Critères d’évaluation cliniquea |
|||||
|
Nombre de patients |
408 |
410 |
363 |
359 |
350 |
|
|
DEFINE |
CONFIRM |
|||
|
|
Placebo |
Fumarate de diméthyle 240 mg 2 fois/jour |
Placebo |
Fumarate de diméthyle 240 mg 2 fois/par jour |
Acétate de glatiramère |
|
Taux annualisé de poussées |
0,364 |
0,172*** |
0,401 |
0,224*** |
0,286* |
|
Rapport des taux de poussées (IC à 95 %) |
|
0,47 (0,37 ; 0,61) |
|
0,56 (0,42 ; 0,74) |
0,71 (0,55 ; 0,93) |
|
Pourcentage de patients ayant présenté au moins une poussée |
0,461 |
0,270*** |
0,410 |
0,291** |
0,321** |
|
Rapport de risques (IC à 95 %) |
|
0,51 (0,40 ; 0,66) |
|
0,66 (0,51 ; 0,86) |
0,71 (0,55 ; 0,92) |
|
Pourcentage de patients présentant une progression du handicap confirmée à 12 semaines |
0,271 |
0,164** |
0,169 |
0,128# |
0,156# |
|
Rapport de risques (IC à 95 %) |
|
0,62 (0,44 ; 0,87) |
|
0,79 (0,52 ; 1,19) |
0,93 (0,63 ; 1,37) |
|
Pourcentage de patients présentant une progression du handicap confirmée à 24 semaines |
0,169 |
0,128# |
0,125 |
0,078# |
0,108# |
|
Rapport de risques (IC à 95 %) |
|
0,77 (0,52 ; 1,14) |
|
0,62 (0,37 ; 1,03) |
0,87 (0,55 ; 1,38) |
|
Critères d’évaluation IRMb |
|
||||
|
Nombre de patients |
165 |
152 |
144 |
147 |
161 |
|
Nombre de nouvelles lésions T2 ou de lésions élargies en T2 sur 2 ans ; moyenne (médiane) |
16,5 (7,0) |
3,2 (1,0)*** |
19,9 (11,0) |
5,7 (2,0)*** |
9,6 (3,0)*** |
|
Rapport du nombre moyen de lésions (IC à 95 %) |
|
0,15 (0,10 ; 0,23) |
|
0,29 (0,21 ; 0,41) |
0,46 (0,33 ; 0,63) |
|
Nombre de lésions rehaussées par le Gd à 2 ans ; moyenne (médiane) |
1,8 (0) |
0,1 (0)*** |
2,0 (0,0) |
0,5 (0,0)*** |
0,7 (0,0)** |
|
Odds ratio (IC à 95 %) |
|
0,10 (0,05 ; 0,22) |
|
0,26 (0,15 ; 0,46) |
0,39 (0,24 ; 0,65) |
|
Nombre de nouvelles lésions T1 hypo-intenses sur 2 ans ; moyenne (médiane) |
5,7 (2,0) |
2,0 (1,0)*** |
8,1 (4,0) |
3,8 (1,0)*** |
4,5 (2,0)** |
|
Rapport du nombre moyen de lésions (IC à 95 %) |
|
0,28 (0,20 ; 0,39) |
|
0,43 (0,30 ; 0,61) |
0,59 (0,42 ; 0,82) |
a Toutes les analyses des critères d’évaluation cliniques étaient en intention de traiter.
bL’analyse IRM a été réalisée sur une cohorte IRM.
*Valeur p < 0,05; **Valeur p < 0,01 ; ***Valeur p < 0,0001 ; # non statistiquement significatif.
L’objectif principal de l’étude était d’évaluer la sécurité à long terme de fumarate de diméthyle chez les patients atteints de SEP-RR. Sur les 1 736 patients, environ la moitié (909 ; 52 %) ont été traités pendant 6 ans ou plus. Cinq-cent-un (501) patients ont été traités en continu par fumarate de diméthyle 240 mg deux fois par jour dans les trois études, et 249 patients qui avaient reçu précédemment le placebo dans les études DEFINE et CONFIRM ont reçu du fumarate de diméthyle 240 mg deux fois par jour dans l’étude ENDORSE. Les patients qui recevaient du fumarate de diméthyle deux fois par jour en continu ont été traités pendant une durée allant jusqu’à 12 ans.
Pendant l’étude ENDORSE, plus de la moitié des patients traités par fumarate de diméthyle 240 mg deux fois par jour n’ont pas présenté de poussée. Les TAP ajustés chez les patients recevant le traitement deux fois par jour en continu dans les trois études étaient de 0,187 (IC à 95 % : 0,156 ; 0,224) dans les études DEFINE et CONFIRM et de 0,141 (IC à 95 % : 0,119 ; 0,167) dans l’étude ENDORSE. Chez les patients ayant reçu précédemment le placebo, le TAP ajusté a diminué de 0,330 (IC à 95 % : 0,266 ; 0,408) dans les études DEFINE et CONFIRM à 0,149 (IC à 95 % : 0,116 ; 0,190) dans l’étude ENDORSE.
Dans l’étude ENDORSE, la majorité des patients (> 75%) n’a pas présenté de progression du handicap confirmée (mesurée comme une progression du handicap maintenue à 6 mois). Les résultats combinés des trois études ont montré des taux de progression du handicap confirmée uniformes et faibles chez les patients traités par fumarate de diméthyle, avec une légère augmentation des scores EDSS moyens dans l’étude ENDORSE. Les analyses des IRM (jusqu’à l’année 6, portant sur 752 patients qui avaient été inclus précédemment dans la cohorte IRM des études DEFINE et CONFIRM) ont montré l’absence de lésions rehaussées par le Gd+ chez la majorité des patients (environ 90%). Sur les 6 ans, le nombre moyen annuel ajusté de lésions nouvelles ou élargies en T2 et de nouvelles lésions en T1 est resté faible.
Efficacité chez les patients présentant une maladie très active :
Dans les études DEFINE et CONFIRM, un effet thérapeutique cohérent sur les poussées a été observé dans un sous-groupe de patients présentant une maladie très active alors que l'effet sur la progression du handicap à 3 mois n'a pas été clairement établi. Dans ces études, la définition d’une maladie très active était la suivante :
· Patients ayant présenté au moins 2 poussées en un an et présentant une ou plusieurs lésions rehaussées par le gadolinium sur l'IRM cérébrale (n=42 dans DEFINE ; n=51 dans CONFIRM) ou;
· Patients n'ayant pas répondu à un traitement médicamenteux complet et bien conduit par interféron bêta (d’une durée d’au moins un an), ayant présenté au moins 1 poussée durant l'année précédente sous ce traitement, et au moins 9 lésions T2-hyper intenses sur l'IRM cérébrale ou au moins 1 lésion rehaussée par Gd, ou des patients dont le taux de poussées n’a pas changé ou a augmenté pendant l'année précédente par rapport aux 2 années antérieures (n=177 dans l’étude DEFINE; n=141 dans l’étude CONFIRM).
Population pédiatrique
Les pourcentages de patients de la population ITT ne présentant pas de lésions nouvelles ou élargies en T2 sur l’IRM à la semaine 96 par rapport à l’inclusion étaient de 12,8 % dans le groupe fumarate de diméthyle versus 2,8 % dans le groupe interféron bêta-1a. Les nombres moyens de lésions nouvelles ou élargies en T2 à la semaine 96 par rapport à l’inclusion, ajustés pour le nombre initial de lésions T2 et l’âge (population ITT dont étaient exclus les patients pour lesquels des mesures IRM n’étaient pas disponibles), étaient de 12,4 dans le groupe fumarate de diméthyle et de 32,6 dans le groupe interféron bêta-1a.
La probabilité de poussée clinique était de 34 % dans le groupe fumarate de diméthyle et de 48 % dans le groupe interféron bêta-1a à la fin de la période d’étude en ouvert de 96 semaines. Le profil de sécurité chez les enfants et adolescents (âgés de 13 à moins de 18 ans) traités par fumarate de diméthyle était qualitativement conforme à celui précédemment observé chez les patients adultes (voir rubrique 4.8).
5.2. Propriétés pharmacocinétiques
Absorption
Le Tmax du monométhyl fumarate est de 2 à 2,5 heures. Les gélules gastro-résistantes de fumarate de diméthyle contiennent des granulés, protégés par un pelliculage entérique ; l’absorption ne débute donc que lorsque les gélules ont quitté l’estomac (généralement en moins d’1 heure). Après administration de 240 mg deux fois par jour au moment des repas, la concentration maximale médiane (Cmax) atteint 1,72 mg/L et l’exposition globale (aire sous la courbe [ASC]) est de 8,02 h.mg/L chez les patients ayant une sclérose en plaques. Dans l’ensemble, la Cmax et l’ASC augmentent approximativement de façon proportionnelle à la dose dans l’intervalle de doses étudiées (120 mg à 360 mg). Chez des patients atteints de SEP, deux doses de 240 mg ont été administrées 3 fois par jour à 4 heures d'intervalle. Une accumulation minimale de l’exposition a été observée conduisant à une augmentation de la Cmax médiane de 12 %, par comparaison à une administration 2 fois par jour (1,72 mg/L après administration 2 fois par jour, et 1,93 mg/L après administration 3 fois par jour), sans modification de la tolérance.
La prise d’aliments ne modifie pas de manière cliniquement significative l’exposition au fumarate de diméthyle. Cependant, le fumarate de diméthyle doit être pris au moment des repas pour une meilleure tolérance et pour diminuer les effets indésirables à type de bouffées congestives ou d’effets gastro-intestinaux (voir rubrique 4.2).
Distribution
Le volume apparent de distribution après administration orale de 240 mg de fumarate de diméthyle varie de 60 à 90 litres. La liaison aux protéines plasmatiques humaines varie, en général, de 27 % à 40 %.
Biotransformation
Chez l’homme, le fumarate de diméthyle est essentiellement métabolisé, moins de 0,1 % de la dose étant excrétée sous forme de fumarate de diméthyle inchangé dans les urines. Il est tout d’abord métabolisé par les estérases, présentes dans le tube digestif, le sang et les tissus, avant d’atteindre la circulation systémique. Son métabolisme est ensuite effectué par la voie du cycle de l'acide tricarboxylique, sans participation du cytochrome P450 (CYP). Une étude à dose unique de 240 mg de fumarate de diméthyle marquée au 14C a montré que le glucose est le métabolite prédominant dans le plasma humain. Parmi les autres métabolites présents dans la circulation se trouvaient l'acide fumarique, l'acide citrique et le monométhyl fumarate. L’acide fumarique est ensuite métabolisé par l’intermédiaire du cycle de l’acide tricarboxylique et éliminé principalement dans l’air expiré sous forme de CO2.
Élimination
La libération de CO2 est la principale voie d’élimination du fumarate de diméthyle et permetd’éliminer 60 % de la dose. L’élimination par voie rénale et fécale est secondaire, correspondant respectivement à 15,5 % et 0,9 % de la dose.
La demi-vie terminale du monométhyl fumarate est courte (approximativement 1 heure) et il n’est pas retrouvé dans la circulation après 24 heures chez la majorité des sujets. Il n’y a pas d’accumulation de fumarate de diméthyle ou de monométhyl fumarate après administration de doses répétées à la posologie recommandée.
Linéarité/non-linéarité
L’exposition au fumarate de diméthyle augmente de façon quasi-proportionnelle à la dose après administrations répétées de doses multiples comprises entre 120 mg et 360 mg.
Pharmacocinétique dans des groupes particuliers de patients
Selon les résultats de l’analyse de variance (ANCOVA), le poids corporel est la principale covariable de l’exposition (Cmax et ASC) chez les patients atteints de SEP-RR, cependant il n’a pas d’influence sur la tolérance et l’efficacité évaluées dans le cadre des études cliniques.
Le sexe et l’âge n’ont pas eu d’effet cliniquement significatif sur la pharmacocinétique du fumarate de diméthyle. La pharmacocinétique chez les patients d’âge supérieur ou égal à 65 ans n’a pas encore été étudiée.
Population pédiatrique
Le profil pharmacocinétique du fumarate de diméthyle 240 mg deux fois par jour a été évalué dans une petite étude non contrôlée en ouvert menée chez des patients atteints de SEP-RR âgés de 13 à 17 ans (n = 21). La pharmacocinétique du fumarate de diméthyle chez ces patients adolescents concordait avec celle observée antérieurement chez des patients adultes (Cmax : 2,00 ± 1,29 mg/L ; ASC0-12 h :3,62 ± 1,16 h.mg/L, soit une ASC quotidienne globale de 7,24 h.mg/L).
Insuffisance rénale
Sachant que la voie rénale est une voie d’élimination secondaire du fumarate de diméthyle (moins de 16 % de la dose administrée sont éliminés par cette voie), la pharmacocinétique n’a pas encore été évaluée chez les patients présentant une insuffisance rénale.
Insuffisance hépatique
Compte tenu que le fumarate de diméthyle et le monométhyl fumarate sont métabolisés par des estérases, sans implication du CYP450, la pharmacocinétique n’a pas encore été évaluée chez les patients présentant une insuffisance hépatique.
5.3. Données de sécurité préclinique
Génotoxicité
Le fumarate de diméthyle et le monométhyl fumarate ont donné des résultats négatifs dans une batterie de tests in vitro (test d’Ames, essai d’aberration chromosomique sur cellules de mammifères). Le fumarate de diméthyle a également donné un résultat négatif dans le test du micronoyau in vivo chez le rat.
Cancérogenèse
Des études de cancérogénicité ont été réalisées avec du fumarate de diméthyle sur une durée de 2 ans chez la souris et le rat. Le fumarate de diméthyle était administré par voie orale à des doses de 25, 75, 200 et 400 mg/kg/jour chez la souris, et à des doses de 25, 50, 100 et 150 mg/kg/jour chez le rat.
L’incidence des papillomes et des carcinomes à cellules squameuses dans l'estomac non glandulaire (secteur gastrique antérieur) a augmenté chez la souris lors d'une exposition équivalente à celle obtenue avec la dose recommandée chez l’homme et chez le rat lors d'une exposition inférieure à celle correspondant à la posologie recommandée chez l’homme (sur la base de l’aire sous la courbe). Le tube digestif chez l’homme ne comporte pas de partie équivalente à la section antérieure de l’estomac de la souris et du rat.
Toxicologie
Les études non cliniques ont été réalisées chez des rongeurs, des lapins et des singes par gavage oral d’une suspension de fumarate de diméthyle (fumarate de diméthyle dans de l’hydroxypropylméthylcellulose à 0,8 %). L’étude chronique chez le chien a été réalisée par administration orale de gélules de fumarate de diméthyle.
Des effets ont été observés au niveau des reins après administration orale répétée du fumarate de diméthyle chez la souris, le rat, le chien et le singe. Une régénérescence épithéliale des tubules rénaux, évoquant la présence de lésions, a été observée dans toutes ces espèces. Une hyperplasie des tubules rénaux a été observée chez le rat après administration sur le long-terme (2 ans). Après administration de doses orales quotidiennes de fumarate de diméthyle pendant 11 mois chez le chien, la marge calculée pour l’atrophie corticale a été observée à une exposition correspondant à 3 fois la dose recommandée chez l’homme sur la base de l’ASC. Après administration de doses orales quotidiennes de fumarate de diméthyle pendant 12 mois chez le singe, une nécrose monocellulaire a été observée à une exposition correspondant à 2 fois la dose recommandée chez l’homme sur la base de l’ASC. Une fibrose interstitielle et une atrophie corticale ont été observées à une exposition correspondant à 6 fois la dose recommandée chez l’homme sur la base de l’ASC. La pertinence de ces résultats pour l'homme n'est pas connue.
Dans les testicules, une dégénérescence de l'épithélium séminifère a été observée chez le rat et le chien. Ces effets ont été observés chez le rat à une exposition correspondant à environ la dose recommandée chez l’homme et chez le chien à une exposition correspondant à 3 fois la dose recommandée chez l’homme (sur la base des ASC). La pertinence de ces résultats pour l'homme n’est pas connue.
Dans la section antérieure de l'estomac de la souris et du rat, la présence d'une hyperplasie et une hyperkératose des cellules épithéliales squameuses, une inflammation, un papillome à cellules squameuses et un carcinome ont été observés dans des études d’une durée supérieure ou égale à 3 mois. Le tube digestif chez l’homme ne comporte pas de partie équivalente à la section antérieure de l’estomac de la souris et du rat.
Toxicité sur la reproduction
L’administration orale de fumarate de diméthyle à des rats mâles, à des doses de 75, 250 et 375 mg/kg/jour, avant et pendant l’accouplement n’a eu aucun effet sur la fertilité des mâles, y compris à la dose testée la plus élevée (correspondant à une exposition, basée sur l’ASC, d’au moins 2 fois celle obtenue avec la dose recommandée chez l’homme).
L’administration orale de fumarate de diméthyle à des rates, à des doses de 25, 100 et 250 mg/kg/jour, avant, pendant l’accouplement et jusqu’au 7ème jour de la gestation, a réduit de 14 jours le nombre de stades d’œstrus et entraîné une augmentation du nombre d'animaux présentant un dioestrus prolongé à la dose testée la plus élevée (correspondant à une exposition, basée sur l'ASC, de 11 fois la dose recommandée chez l’homme).
Toutefois, ces effets n’ont pas affecté la fertilité ou le nombre de fœtus viables engendrés.
Les études ont montré que le fumarate de diméthyle traverse la membrane placentaire et pénètre dans le sang fœtal chez le rat et le lapin, les rapports entre les concentrations plasmatiques du fœtus et de la mère étant respectivement de 0,48 à 0,64 et 0,1.
Aucune malformation n’a été observée, quelle que soit la dose de fumarate de diméthyle administrée au rat ou au lapin. L’administration du fumarate de diméthyle par voie orale, à des doses de 25, 100 et 250 mg/kg/jour, à des rates gravides durant la période d’organogenèse a entraîné des effets indésirables à une exposition des mères (basée sur l’ASC) de 4 fois la dose recommandée chez l’homme ainsi qu’un faible poids fœtal et un retard de l’ossification (métatarses et phalanges de la patte arrière) à 11 fois la dose recommandée chez l’homme. Il a été considéré que le poids fœtal plus faible et le retard de l'ossification étaient secondaires à la toxicité maternelle (réduction du poids corporel et de la consommation d'aliments).
L’administration orale du fumarate de diméthyle à raison de 25, 75 et 150 mg/kg/jour à des lapines gravides pendant la période d’organogenèse n’a eu aucun effet sur le développement fœto- embryonnaire et a entraîné une réduction du poids corporel de la mère à une exposition correspondant à 7 fois la dose recommandée chez l’homme et un taux d'avortement accru à 16 fois la dose recommandée (basée sur l'ASC).
L’administration orale du fumarate de diméthyle à raison de 25, 100 et 250 mg/kg/jour à des rates durant la gestation et la période d’allaitement a entraîné une réduction du poids corporel de la progéniture F1, ainsi qu’un retard de la maturation sexuelle chez les mâles F1 à une exposition (basées sur l’ASC) de 11 fois la dose recommandée chez l’homme. La fertilité de la progéniture F1 n’a pas été affectée. La réduction du poids corporel de la progéniture a été considéré comme secondaire à la toxicité maternelle.
Croscarmellose sodique
Silice colloïdale anhydre
Fumarate de stéaryle sodique
Copolymère d'acide méthacrylique et de méthacrylate de méthyle (1:1)
Copolymère d'acide méthacrylique et d'acrylate d'éthyle (1:1) dispersion à 30%Talc
Citrate de triéthyle
Polysorbate 80
Monostéarate de glycérol
Enveloppe de la gélule
Gélatine
Dioxyde de titane (E171)
Oxyde de fer jaune (E172)
Laque aluminique de bleu brillant FCF (E133)
Impression de la gélule
Shellac gomme 45% (20 % estérifiée)
Oxyde de fer noir (E172)
Propylèneglycol (E1520)
Solution d’hydroxyde d’ammonium 28%
3 ans.
6.4. Précautions particulières de conservation
A conserver à une température ne dépassant pas 30°C.
6.5. Nature et contenu de l'emballage extérieur
Plaquette (Aluminium//PVC/PVDC). Boites de 14 ou 56 gélules.
Toutes les présentations peuvent ne pas être commercialisées.
6.6. Précautions particulières d’élimination et de manipulation
Pas d’exigences particulières pour l’élimination.
7. TITULAIRE DE L’AUTORISATION DE MISE SUR LE MARCHE
15, BOULEVARD CHARLES DE GAULLE
92700 COLOMBES
8. NUMERO(S) D’AUTORISATION DE MISE SUR LE MARCHE
· 34009 302 647 8 2 : 14 gélules sous plaquettes (Aluminium/PVC/PVDC).
· 34009 302 647 9 9 : 56 gélules sous plaquettes (Aluminium/PVC/PVDC).
9. DATE DE PREMIERE AUTORISATION/DE RENOUVELLEMENT DE L’AUTORISATION
[à compléter ultérieurement par le titulaire]
10. DATE DE MISE A JOUR DU TEXTE
[à compléter ultérieurement par le titulaire]
Sans objet.
12. INSTRUCTIONS POUR LA PREPARATION DES RADIOPHARMACEUTIQUES
Liste I.
Prescription réservée aux spécialistes en neurologie ou en neuropédiatrie.
Médicament nécessitant une surveillance particulière pendant le traitement.
ANSM - Mis à jour le : 28/04/2023
DIMETHYL FUMARATE BIOGARAN 120 mg, gélule gastro-résistante
Fumarate de diméthyle
· Gardez cette notice. Vous pourriez avoir besoin de la relire.
· Si vous avez d’autres questions, interrogez votre médecin ou votre pharmacien.
· Ce médicament vous a été personnellement prescrit. Ne le donnez pas à d’autres personnes. Il pourrait leur être nocif, même si les signes de leur maladie sont identiques aux vôtres.
· Si vous ressentez un quelconque effet indésirable, parlez-en à votre médecin ou votre pharmacien. Ceci s’applique aussi à tout effet indésirable qui ne serait pas mentionné dans cette notice. Voir rubrique 4.
1. Qu'est-ce que DIMETHYL FUMARATE BIOGARAN 120 mg, gélule gastro-résistante et dans quels cas est-il utilisé ?
2. Quelles sont les informations à connaître avant de prendre DIMETHYL FUMARATE BIOGARAN 120 mg, gélule gastro-résistante ?
3. Comment prendre DIMETHYL FUMARATE BIOGARAN 120 mg, gélule gastro-résistante ?
4. Quels sont les effets indésirables éventuels ?
5. Comment conserver DIMETHYL FUMARATE BIOGARAN 120 mg, gélule gastro-résistante ?
6. Contenu de l’emballage et autres informations.
1. QU’EST-CE QUE DIMETHYL FUMARATE BIOGARAN 120 mg, gélule gastro-résistante ET DANS QUELS CAS EST-IL UTILISE ?
Classe pharmacothérapeutique - Immunosuppresseurs, autres immunosuppresseurs, code ATC : L04AX07.
Qu’est-ce que DIMETHYL FUMARATE BIOGARAN ?
DIMETHYL FUMARATE BIOGARAN est un médicament dont la substance active est le fumarate de diméthyle.
Dans quel cas DIMETHYL FUMARATE BIOGARAN est-il utilisé ?
DIMETHYL FUMARATE BIOGARAN est utilisé pour traiter la sclérose en plaques (SEP) récurrente rémittente chez les patients âgés de 13 ans et plus.
La sclérose en plaques est une maladie chronique qui affecte le système nerveux central comprenant le cerveau et la moelle épinière. La sclérose en plaques récurrente rémittente se caractérise par l’apparition répétée de symptômes neurologiques (poussées). Les symptômes varient d'un patient à un autre mais comportent de façon caractéristique des difficultés pour marcher, une sensation de perte de l'équilibre ainsi que des troubles visuels (par exemple vision floue ou double). Ces symptômes peuvent disparaître complètement lorsque la poussée est terminée mais certains problèmes peuvent persister.
Comment agit DIMETHYL FUMARATE BIOGARAN ?
DIMETHYL FUMARATE BIOGARAN semblerait agir en bloquant les systèmes de défense de l'organisme qui peuvent léser le cerveau et la moelle épinière. Cette possible action pourrait aussi retarder l'évolution future de votre SEP.
2. QUELLES SONT LES INFORMATIONS A CONNAITRE AVANT DE PRENDRE DIMETHYL FUMARATE BIOGARAN 120 mg, gélule gastro-résistante ?
Ne prenez jamais DIMETHYL FUMARATE BIOGARAN 120 mg, gélule gastro-résistante :
· si vous êtes allergique au fumarate de diméthyle ou à l’un des autres composants contenus dans ce médicament, mentionnés dans la rubrique 6.
· si vous êtes suspecté de souffrir d'une infection cérébrale rare appelée leucoencéphalopathie multifocale progressive (LEMP) ou si la LEMP a été confirmée.
Avertissements et précautions
Adressez-vous à votre médecin ou pharmacien avant de prendre DIMETHYL FUMARATE BIOGARAN.
DIMETHYL FUMARATE BIOGARAN peut agir sur le nombre de globules blancs, sur les reins et sur le foie. Avant de commencer le traitement par DIMETHYL FUMARATE BIOGARAN, votre médecin réalisera une analyse de sang pour vérifier votre nombre de globules blancs et voir si vos reins et votre foie fonctionnent correctement. Votre médecin réalisera régulièrement ces analyses durant le traitement. Si votre nombre de globules blancs diminue durant le traitement, votre médecin pourra envisager des mesures analytiques supplémentaires ou d’interrompre votre traitement.
Signalez à votre médecin avant de prendre DIMETHYL FUMARATE BIOGARAN si vous souffrez :
· d’une maladie rénale sévère.
· d’une maladie hépatique sévère.
· d’une maladie de l’estomac ou de l’intestin.
· d'une infection grave (pneumonie par exemple).
Un zona peut survenir au cours du traitement par DIMETHYL FUMARATE BIOGARAN. Dans certains cas, de graves complications sont apparues. Vous devez informer votre médecin immédiatement si vous pensez présenter des symptômes du zona.
Si vous pensez que votre SEP s'aggrave (par exemple, faiblesse ou changements visuels) ou si vous remarquez de nouveaux symptômes, parlez-en immédiatement à votre médecin car il peut s'agir des symptômes d'une infection cérébrale rare appelée leucoencéphalopathie multifocale progressive (LEMP). La LEMP est une affection grave qui peut entraîner une invalidité sévère ou le décès.
Un trouble rénal rare mais grave (syndrome de Fanconi) a été rapporté avec un médicament contenant du fumarate de diméthyle associé à d’autres esters de l’acide fumarique, utilisé pour traiter le psoriasis (une maladie de la peau). Si vous remarquez que vous urinez davantage, que vous avez plus soif et que vous buvez plus que d’habitude, que vos muscles semblent plus faibles, que vous vous cassez un os ou que vous ressentez simplement des douleurs, parlez-en à votre médecin le plus tôt possible afin qu’il puisse faire des examens complémentaires.
Enfants et adolescents
Autres médicaments et DIMETHYL FUMARATE BIOGARAN 120 mg, gélule gastro-résistante
Informez votre médecin ou votre pharmacien si vous prenez, avez récemment pris ou pourriez prendre tout autre médicament, notamment :
· des médicaments qui contiennent des esters de l'acide fumarique (fumarates) utilisés pour traiter le psoriasis.
· des médicaments ayant une action sur le système immunitaire, y compris d’autres médicaments utilisés pour traiter la SEP, tels que le fingolimod, le natalizumab, le tériflunomide, l’alemtuzumab, l’ocrelizumab ou la cladribine ou certains autres traitements anticancéreux fréquemment utilisés (rituximab ou mitoxantrone).
· des médicaments ayant une action sur les reins, y compris certains antibiotiques (utilisés pour traiter les infections), les diurétiques, certains antalgiques (comme l'ibuprofène et d’autres anti-inflammatoires courants y compris des médicaments achetés sans ordonnance) et des médicaments contenant du lithium.
· la prise de DIMETHYL FUMARATE BIOGARAN avec certains groupes de vaccins (vaccins vivants) peut provoquer une infection et doit donc être évitée. Votre médecin vous indiquera si d’autres types de vaccins (vaccins non vivants) doivent être prescrits.
DIMETHYL FUMARATE BIOGARAN 120 mg, gélule gastro-résistante avec de l’alcool
Grossesse et allaitement
Si vous êtes enceinte ou que vous allaitez, si vous pensez être enceinte ou planifiez une grossesse, demandez conseil à votre médecin ou pharmacien avant de prendre ce médicament.
Grossesse
Ne prenez pas DIMETHYL FUMARATE BIOGARAN si vous êtes enceinte, sauf si vous en avez discuté avec votre médecin.
Allaitement
On ne sait pas si la substance active de DIMETHYL FUMARATE BIOGARAN passe dans le lait maternel. Ne prenez pas DIMETHYL FUMARATE BIOGARAN si vous allaitez. Votre médecin vous aidera à décider soit d’arrêter d'allaiter soit d’arrêter le traitement par DIMETHYL FUMARATE BIOGARAN, en mettant en balance les bénéfices de l’allaitement et les bénéfices de votre traitement.
Conduite de véhicules et utilisation de machines
On ne connaît pas l'effet de DIMETHYL FUMARATE BIOGARAN sur l’aptitude à conduire ou à utiliser des machines. DIMETHYL FUMARATE BIOGARAN ne devrait pas avoir d’effet sur votre aptitude à conduire des véhicules et à utiliser des machines.
DIMETHYL FUMARATE BIOGARAN 120 mg, gélule gastro-résistante contient du sodium
3. COMMENT PRENDRE DIMETHYL FUMARATE BIOGARAN 120 mg, gélule gastro-résistante ?
Dose initiale
120 mg deux fois par jour.
Prenez cette dose initiale pendant les 7 premiers jours, puis passez à la dose habituelle.
Dose habituelle
240 mg deux fois par jour.
DIMETHYL FUMARATE BIOGARAN est pris par voie orale.
Avalez chaque gélule entière, avec un peu d’eau. N’ouvrez pas, n’écrasez pas, ne dissolvez pas, ne sucez pas ou ne mâchez pas la gélule car ceci risque d'augmenter certains effets indésirables.
Prenez DIMETHYL FUMARATE BIOGARAN au moment des repas – cela peut aider à réduire les effets indésirables très fréquents (voir rubrique 4).
Si vous avez pris plus de DIMETHYL FUMARATE BIOGARAN 120 mg, gélule gastro-résistante que vous n’auriez dû
Si vous avez pris trop de gélules, informez-en immédiatement votre médecin. Vous pourriez ressentir des effets indésirables similaires à ceux décrits ci-dessous à la rubrique 4.
Si vous oubliez de prendre DIMETHYL FUMARATE BIOGARAN 120 mg, gélule gastro-résistante
Vous pouvez prendre la dose oubliée si vous espacez d’au moins 4 heures les 2 doses. Sinon, prenez la prochaine dose au moment prévu.
Si vous arrêtez de prendre DIMETHYL FUMARATE BIOGARAN 120 mg, gélule gastro-résistante
Sans objet.
Si vous avez d’autres questions sur l’utilisation de ce médicament, demandez plus d’informations à votre médecin ou à votre pharmacien.
4. QUELS SONT LES EFFETS INDESIRABLES EVENTUELS ?
Effets indésirables graves
DIMETHYL FUMARATE BIOGARAN peut diminuer le nombre de lymphocytes (un type de globule blanc). Le fait d’avoir un faible nombre de globules blancs peut augmenter votre risque d’infection, y compris vous exposer au risque d’une infection rare du cerveau appelée leucoencéphalopathie multifocale progressive (LEMP). La LEMP peut entraîner un handicap sévère ou le décès. La LEMP est apparue après 1 à 5 ans de traitement. Votre médecin doit donc continuer à surveiller vos globules blancs tout au long de votre traitement, et vous devez rester attentif à tout symptôme potentiel de LEMP comme décrit ci-dessous. Le risque de LEMP peut être plus élevé si vous avez déjà pris un médicament altérant le fonctionnement de votre système immunitaire.
Les symptômes de la LEMP peuvent être similaires à une poussée de SEP. Ils peuvent inclure l’apparition ou l’aggravation d’un affaiblissement d’un côté du corps ; de la maladresse, des troubles visuels, des troubles de la pensée ou de la mémoire ; une confusion ou des changements dans la personnalité, ou des difficultés d’élocution et de communication pendant quelques jours ou plus. Par conséquent, si vous pensez que votre SEP s’aggrave ou si vous remarquez de nouveaux symptômes pendant votre traitement par DIMETHYL FUMARATE BIOGARAN, il est très important que vous en parliez à votre médecin dès que possible. Parlez-en également à votre partenaire ou aux personnes qui s’occupent de vous et informez-les de votre traitement. Il se peut que des symptômes apparaissent et que vous ne vous en rendiez pas compte par vous-même.
Appelez immédiatement votre médecin si vous présentez l’un quelconque de ces symptômes.
Réactions allergiques sévères
La fréquence des réactions allergiques sévères ne peut pas être estimée à partir des données disponibles (fréquence indéterminée).
L'apparition de rougeurs sur le visage ou le corps (bouffées congestives) est un effet indésirable très fréquent. Toutefois, si les bouffées congestives sont accompagnées d’une éruption cutanée rouge ou d’urticaire et que vous présentez l’un des symptômes suivants :
· gonflement du visage, des lèvres, de la bouche ou de la langue (angiœdème).
· respiration sifflante, difficulté à respirer ou essoufflement (dyspnée, hypoxie).
· sensations vertigineuses ou perte de conscience (hypotension).
Cela peut alors représenter une réaction allergique sévère (anaphylaxie).
Arrêtez immédiatement de prendre DIMETHYL FUMARATE BIOGARAN et appelez immédiatement un médecin.
Effets indésirables très fréquents
Ceux-ci peuvent affecter plus d’1 personne sur 10 :
· rougeurs sur le visage ou le corps, chaleurs, sensations de brûlures ou de démangeaisons (bouffées congestives).
· selles molles (diarrhée).
· envie de vomir (nausée).
· douleurs ou crampes au niveau de l’estomac.
Le fait de prendre votre médicament au moment des repas peut aider à réduire les effets indésirables mentionnés ci-dessus.
Les substances nommées cétones, naturellement produites dans le corps, apparaissent fréquemment dans les analyses d'urine pendant la prise de DIMETHYL FUMARATE BIOGARAN.
Discutez avec votre médecin de la façon de prendre en charge ces effets indésirables. Votre médecin réduira peut-être la dose. Ne réduisez votre dose que si votre médecin vous l’a prescrit.
Effets indésirables fréquents
Ceux-ci peuvent affecter 1 personne sur 10 au maximum :
· inflammation de la paroi intestinale (gastroentérite).
· nausées (vomissements).
· indigestion (dyspepsie).
· inflammation de la paroi de l'estomac (gastrite).
· troubles gastro-intestinaux.
· sensation de brûlures.
· bouffées de chaleur, sensation de chaleur.
· démangeaisons cutanées (prurit).
· éruption cutanée.
· taches cutanées roses ou rouges (érythème).
· perte de cheveux (alopécie).
Effets indésirables qui peuvent être détectés dans les analyses de sang ou d'urine :
· faible nombre de globules blancs (lymphopénie, leucopénie) dans le sang. Un nombre réduit de globules blancs peut signifier que votre corps est moins apte à lutter contre une infection. Si vous avez une infection grave (pneumonie par exemple), consultez immédiatement votre médecin.
· protéines (albumine) dans l’urine.
· augmentation du taux d’enzymes hépatiques (ALAT, ASAT) dans le sang.
Effets indésirables peu fréquents
Ceux-ci peuvent affecter jusqu’à 1 personne sur 100 :
· réactions allergiques (hypersensibilité).
· réduction des plaquettes sanguines.
Fréquence indéterminée (ne peut être estimée sur la base des données disponibles)
· inflammation du foie et augmentation des taux d’enzymes hépatiques (ALAT ou ASAT en association avec la bilirubine).
· zona avec symptômes tels que vésicules, brûlures, démangeaisons ou douleurs au niveau de la peau, généralement sur un côté du haut du corps ou du visage, et d’autres symptômes, tels que fièvre et faiblesse dès les premiers stades de l’infection, suivis d’engourdissements, de démangeaisons ou de plaques rouges s’accompagnant d’une douleur intense.
· écoulement nasal (rhinorrhée).
Enfants et adolescents (âgés de 13 ans et plus)
Les effets indésirables répertoriés ci-dessus s’appliquent également aux enfants et aux adolescents. Certains effets indésirables ont été plus fréquemment rapportés chez les enfants et adolescents que chez les adultes, par exemple : maux de tête, douleurs abdominales ou crampes d’estomac, vomissements, mal de gorge, toux et règles douloureuses.
Déclaration des effets secondaires
Si vous ressentez un quelconque effet indésirable, parlez-en à votre médecin ou votre pharmacien. Ceci s’applique aussi à tout effet indésirable qui ne serait pas mentionné dans cette notice. Vous pouvez également déclarer les effets indésirables directement via le système national de déclaration : Agence nationale de sécurité du médicament et des produits de santé (ANSM) et réseau des Centres Régionaux de Pharmacovigilance - Site internet : https://signalement.social-sante.gouv.fr/
En signalant les effets indésirables, vous contribuez à fournir davantage d’informations sur la sécurité du médicament.
5. COMMENT CONSERVER DIMETHYL FUMARATE BIOGARAN 120 mg, gélule gastro-résistante ?
Tenir ce médicament hors de la vue et de la portée des enfants.
N’utilisez pas ce médicament après la date de péremption indiquée sur l’emballage. La date de péremption fait référence au dernier jour de ce mois.
A conserver à une température ne dépassant pas 30°C.
Ne jetez aucun médicament au tout-à-l’égout ou avec les ordures ménagères. Demandez à votre pharmacien d’éliminer les médicaments que vous n’utilisez plus. Ces mesures contribueront à protéger l’environnement.
6. CONTENU DE L’EMBALLAGE ET AUTRES INFORMATIONS
Ce que contient DIMETHYL FUMARATE BIOGARAN 120 mg, gélule gastro-résistante
· La substance active est :
Fumarate de diméthyle.................................................................................................... 120 mg
Pour une gélule gastro-résistante.
· Les autres composants sont :
Enveloppe de la gélule : gélatine, dioxyde de titane (E171), oxyde de fer jaune (E172), laque aluminique de bleu brillant FCF (E133).
Impression de la gélule : shellac gomme 45% (20 % estérifiée), oxyde de fer noir (E172), propylèneglycol (E1520), solution d’hydroxyde d’ammonium 28%.
Gélule gastro-résistante, de longueur : 19,4 mm ± 0,3 mm, avec un corps blanc et une tête verte clair, portant l’inscription « 120 mg ».
Disponible en boite de 14 ou 56 gélules.
Toutes les présentations peuvent ne pas être commercialisées.
Titulaire de l’autorisation de mise sur le marché
15, BOULEVARD CHARLES DE GAULLE
92700 COLOMBES
Exploitant de l’autorisation de mise sur le marché
15, BOULEVARD CHARLES DE GAULLE
92700 COLOMBES
ZAKLADY FARMACEUTYCZNE POLPHARMA SA
2 METALOWCA STREET
39460 NOWA DEBA
POLOGNE
Noms du médicament dans les Etats membres de l'Espace Economique Européen
Ce médicament est autorisé dans les Etats membres de l'Espace Economique Européen sous les noms suivants : Conformément à la réglementation en vigueur.
[À compléter ultérieurement par le titulaire]
La dernière date à laquelle cette notice a été révisée est :
[à compléter ultérieurement par le titulaire]
Des informations détaillées sur ce médicament sont disponibles sur le site Internet de l’ANSM (France).