Dernière mise à jour le 01/06/2026
OCTAFIX 100 UI/ml, poudre et solvant pour solution injectable
Indications thérapeutiques
OCTAFIX appartient à un groupe de médicaments appelés facteurs de coagulation et contient le facteur IX de coagulation humain. C’est une protéine spéciale qui augmente la capacité du sang à coaguler.
OCTAFIX est utilisé dans le traitement et la prévention des saignements chez les patients présentant un trouble hémorragique (hémophilie B). Ceci est un trouble au cours duquel un saignement peut continuer plus longtemps que prévu. Il est dû à un déficit congénital en facteur IX dans le sang.
OCTAFIX est fourni en tant que poudre et solvant pour solution injectable. Après reconstitution avec le solvant fourni, OCTAFIX est administré par voie intraveineuse.
Présentations
> 1 flacon(s) poudre en verre de 1000 UI - 1 flacon(s) solvant en verre de 10 ml avec 1 boîte de matériel pour injection intraveineuse (avec 1 dispositif de transfert avec 1 kit de perfusion avec 1 seringue jetable) avec 2 tampons alcoolisés
Code CIP : 563 415-2 ou 34009 563 415 2 8
Déclaration de commercialisation : 03/11/2003
Cette présentation est agréée aux collectivités
Inscription sur la liste de rétrocession au titre de son AMM, selon les conditions précisées au Journal Officiel. Prix de cession publié au Journal Officiel.
Service médical rendu (SMR)
Les libellés affichés ci-dessous ne sont que des résumés ou extraits issus des avis rendus par la Commission de la Transparence. Seul l'avis complet de la Commission de la Transparence fait référence.
Cet avis est consultable à partir du lien `Avis du jj/mm/aaaa` ou encore sur demande auprès de la HAS (plus d'informations dans l'aide). Les avis et synthèses d'avis contiennent un paragraphe sur la place du médicament dans la stratégie thérapeutique.
| Valeur du SMR | Avis | Motif de l'évaluation | Résumé de l'avis |
|---|---|---|---|
| Important | Avis du 03/09/2008 | Extension d'indication | Le service médical rendu par cette spécialités est important dans ses indications. |
| Important | Avis du 07/05/2003 | Inscription (CT) | Le niveau de service médical rendu par cette spécialité est important dans le traitement et la prophylaxie des hémorragies chez les patients atteints d’hémophilie B (déficit congénital en facteur IX). |
Amélioration du service médical rendu (ASMR)
Les libellés affichés ci-dessous ne sont que des résumés ou extraits issus des avis rendus par la Commission de la Transparence. Seul l'avis complet de la Commission de la Transparence fait référence.
Cet avis est consultable à partir du lien `Avis du jj/mm/aaaa` ou encore sur demande auprés de la HAS (plus d'informations dans l'aide). Les avis et synthéses d'avis contiennent un paragraphe sur la place du médicament dans la stratégie thérapeutique.
| Valeur de l'ASMR | Avis | Motif de l'évaluation | Résumé de l'avis |
|---|---|---|---|
| V (Inexistant) | Avis du 03/09/2008 | Extension d'indication | Cette spécialité n'apporte pas d'amélioration du service médical rendu (ASMR V) par rapport aux autres spécialités à base de facteur IX de coagulation humain dont l'utilisation est autorisée chez l'enfant de moins de 6 ans. |
| V (Inexistant) | Avis du 07/05/2003 | Inscription (CT) | Cette spécialité n'apporte pas d'amélioration du service médical rendu (ASMR V) par rapport aux produits disponibles. |
Autres informations
- Titulaire de l'autorisation : OCTAPHARMA France
- Conditions de prescription et de délivrance :
- délivrance réservée aux ETS pour les malades qui y sont traités
- délivrance réservée aux pharmacies à usage intérieur
- liste I
- prescription initiale hospitalière semestrielle
- Statut de l'autorisation : Valide
- Type de procédure : Procédure de reconnaissance mutuelle
- Code CIS : 6 912 824 4
ANSM - Mis à jour le : 16/03/2021
OCTAFIX 100 UI / ml, poudre et solvant pour solution injectable
2. COMPOSITION QUALITATIVE ET QUANTITATIVE
OCTAFIX se présente sous forme de poudre et solvant pour solution injectable, contenant nominalement
Soit 500 UI de facteur IX de coagulation humain par flacon.
Le produit contient environ 100 Ul/ml de facteur IX de coagulation humain après reconstitution avec 5 ml d'eau pour préparations injectables (Ph. Eur.)
Soit 1000 UI de facteur IX de coagulation humain par flacon.
Le produit contient environ 100 Ul/ml de facteur IX de coagulation humain après reconstitution avec 10 ml d'eau pour préparations injectables (Ph. Eur.)
OCTAFIX est produit à partir de plasma de donneurs humain.
L’activité (UI) est déterminée en utilisant le test de coagulation en un temps de la Pharmacopée européenne, en comparaison avec une référence internationale de l’Organisation Mondiale de la Santé (OMS). L'activité spécifique d'OCTAFIX est d'environ 100 UI/mg de protéine.
Excipients à effet notoire
Ce médicament contient jusqu’à 69 mg de sodium pour 1 flacon d’OCTAFIX 500 UI et jusqu’à 138 mg de sodium pour 1 flacon d’OCTAFIX 1000 UI par dose.
Pour la liste complète des excipients, voir rubrique 6.1
Poudre et solvant pour solution injectable
La poudre est blanche ou jaune pâle, sous forme d'un solide friable.
4.1. Indications thérapeutiques
4.2. Posologie et mode d'administration
Le traitement doit être initié sous la surveillance d'un médecin spécialiste de l'hémophilie.
Patients non préalablement traités
La sécurité et l’efficacité d’OCTAFIX chez les patients non préalablement traités n’ont pas encore été établies.
Contrôle du traitement
Au cours du traitement, une détermination appropriée des taux de facteurs IX est conseillée afin d’évaluer la dose à administrer et la fréquence du renouvellement des perfusions. La réponse au traitement par le facteur IX peut varier selon les individus, notamment la demi-vie et le taux de récupération in vivo. La dose basée sur le poids doit être ajustée en fonction des patients. En cas d’interventions chirurgicales majeures, un contrôle précis du traitement substitutif au moyen de tests de coagulation (activité du facteur IX plasmatique) est indispensable.
Posologie
La posologie et la durée du traitement de substitution dépendent de la sévérité du déficit en facteur IX, de la localisation et de l'intensité de l'accident hémorragique, ainsi que de l'état clinique du patient.
Le nombre d'unités de facteur IX administré s'exprime en unités internationales (UI) calculées par rapport à l'étalon OMS pour le facteur IX. L'activité plasmatique du facteur IX est exprimée soit en pourcentage de l'activité normale du plasma humain, soit en unités internationales (selon l'étalon international pour le facteur IX plasmatique).
Une unité internationale (UI) de facteur IX de coagulation humain correspond à la quantité de facteur IX contenue dans un ml de plasma humain normal.
Traitement à la demande :
Le calcul de la dose nécessaire de facteur IX est basé sur le résultat empirique qu'une UI de facteur IX de coagulation humain par kg de poids corporel augmente l'activité plasmatique du facteur IX de 1 % de la normale. Cette dose est déterminée à l'aide de la formule suivante:
Nombre d'Unités à administrer = poids corporel (kg) x augmentation souhaitée en facteur IX (%) (Ul/dl) x 0.8
La quantité à administrer et la fréquence des administrations doivent toujours être adaptées en fonction de l’efficacité clinique individuelle.
En cas de survenue des événements hémorragiques suivants, l’activité du facteur IX ne doit pas descendre en dessous du taux d’activité plasmatique indiqué (en % de la normale) pendant la durée mentionnée.
Le tableau ci-dessous peut être utilisé comme guide pour les posologies lors d’épisodes hémorragiques et de chirurgies:
|
Degré de l’hémorragie/Type d’intervention chirurgicale |
Taux de facteur FIX nécessaire (%) (UI/dl) |
Fréquence des doses (heures)/Durée de traitement (jours) |
|
Hémorragie |
||
|
Début d’hémarthrose, saignement musculaire ou buccal |
20-40 |
Renouveler toutes les 24 heures. Au moins 1 jour, jusqu’à ce que l’épisode hémorragique soit résolu comme indiqué par la douleur ou que la cicatrisation soit obtenue. |
|
Hémarthrose plus étendue, hémorragie musculaire ou hématome |
30-60 |
Renouveler la perfusion toutes les 24 heures pendant 3 à 4 jours ou plus jusqu’à ce que la douleur et le handicap sévère disparaissent. |
|
Hémarthrose mettant en jeu le pronostic vital |
60-100 |
Renouveler la perfusion toutes les 8 à 24 heures, jusqu’à disparition du risque vital. |
|
Chirurgie |
||
|
Chirurgie mineure Dont extraction dentaire |
30-60 |
Toutes les 24 heures, au moins 1 jour, jusqu’à ce que la cicatrisation soit obtenue. |
|
Chirurgie majeure |
80-100 (pré- et post - opératoire) |
Renouveler la perfusion toutes les 8 à 24 heures, jusqu’à cicatrisation suffisante de la plaie, puis poursuivre le traitement pendant au moins 7 jours supplémentaires pour maintenir l’activité du FIX entre 30% et 60% (UI/dl). |
Prophylaxie
Dans le traitement prophylactique au long cours des épisodes hémorragiques chez les patients atteints d'hémophilie B sévère, les doses habituelles sont 20 à 40 UI de facteur IX de coagulation humain par kg de poids corporel tous les 3 à 4 jours.
Dans certains cas, particulièrement chez les patients plus jeunes, des intervalles d’administration plus courts ou des posologies plus élevées peuvent être nécessaires.
Perfusion continue
Il n’y a pas de données cliniques suffisantes pour recommander l'utilisation d'OCTAFIX en perfusion continue lors d'interventions chirurgicales.
Population pédiatrique
Dans une étude clinique réalisée sur 25 enfants âgés de moins de 6 ans, la dose médiane administrée par jour d’exposition était similaire dans la prophylaxie et le traitement des hémorragies, soit une dose comprise entre 35 et 40 UI/kg de poids corporel.
Mode d’administration
Voie intraveineuse
Il est recommandé de ne pas administrer plus de 2 à 3 ml par minute. Pour les instructions concernant la reconstitution du médicament avant administration, voir la rubrique 6.6.
Hypersensibilité à la substance active ou à l'un des excipients mentionnés à la rubrique 6.1.
Diminution des thrombocytes liée à une allergie connue pendant un traitement par héparine (Thrombocytopénie induite par l'héparine [TIH] type II).
4.4. Mises en garde spéciales et précautions d'emploi
Afin d'améliorer la traçabilité des médicaments biologiques, le nom et le numéro de lot du produit administré doivent être clairement enregistrés.
Hypersensibilité
Des réactions d’hypersensibilité de type allergique peuvent apparaître avec OCTAFIX. Le produit contient des traces de protéine humaine autre que le facteur IX et l’héparine. Si ces symptômes apparaissent, les patients doivent être prévenus de la nécessité d’interrompre immédiatement le traitement et de contacter leur médecin. Les patients doivent être informés des signes précoces des réactions d’hypersensibilité, y compris démangeaisons, urticaire généralisée, oppression thoracique, respiration sifflante, hypotension et anaphylaxie.
En cas de choc, le traitement médical standard relatif à l’état de choc devra être instauré.
Inhibiteurs
Chez les patients recevant régulièrement des produits à base de facteur IX de coagulation humain, la recherche d'anticorps neutralisants (inhibiteurs) doit être effectuée au moyen de tests biologiques appropriés avec quantification de l'inhibiteur en unités Bethesda (BU).
Il a été rapporté dans la littérature une corrélation entre la survenue d’un inhibiteur anti-facteur IX et des réactions allergiques. Ainsi, en cas d’apparition de réaction allergique, la présence d’inhibiteurs doit être recherchée chez le patient. Les patients présentant des inhibiteurs anti-facteur IX peuvent présenter un risque anaphylactique accru lors d’un traitement ultérieur avec le facteur IX.
En raison du risque d'allergie lors de l'administration de produits contenant du facteur IX, les premières administrations de facteur IX doivent, selon l’avis du médecin traitant, être effectuées sous surveillance médicale permettant de fournir un traitement médical approprié en cas de réactions allergiques.
Thromboembolie
En raison du risque potentiel de complications thrombotiques, une surveillance clinique et la réalisation de tests biologiques devront être instaurées pour rechercher les marqueurs précoces d'une thrombose ou d'une coagulopathie de consommation chez les patients atteints de maladies hépatiques, en période post-opératoire, chez les nouveau-nés ou chez les patients présentant un risque thrombotique ou de coagulation intravasculaire disséminée. Dans chacune de ces situations, le bénéfice potentiel du traitement par OCTAFIX doit être évalué par rapport aux risques de complication.
Événements cardiovasculaires
Chez les patients présentant des facteurs de risque de maladies cardiovasculaires, un traitement substitutif par facteur IX peut augmenter le risque cardiovasculaire.
Complications liées au cathéter
Si un dispositif d’accès veineux central (DAVC) est nécessaire, il existe un risque d’infections locales, de bactériémie et de thrombose sur le site d’insertion du cathéter.
Agents transmissibles
Les mesures habituelles de prévention du risque de transmission d’agents infectieux par les médicaments préparés à partir de sang ou de plasma humain comprennent la sélection clinique des donneurs, la recherche des marqueurs spécifiques d’infection sur chaque don et sur les mélanges de plasma ainsi que la mise en œuvre dans le procédé de fabrication d’étapes efficaces pour l’inactivation/élimination virale. Cependant, lorsque des médicaments préparés à partir de sang ou de plasma humain sont administrés, le risque de transmission d’agents infectieux ne peut pas être totalement exclu. Ceci s’applique également aux virus inconnus ou émergents ou autres types d’agents infectieux. Les mesures prises sont considérées comme efficaces vis-à-vis des virus enveloppés tels que le virus de l’immunodéficience humaine (VIH), le virus de l’hépatite B (VHB) et le virus de l’hépatite C (VHC), et vis-à-vis du virus non-enveloppé de l’hépatite A (VHA)
Les mesures prises peuvent être d’efficacité limitée vis-à-vis des virus non-enveloppés tels que le parvovirus B19. L’infection par le parvovirus B19 peut être sévère chez la femme enceinte (infection du fœtus) et chez les personnes atteintes de déficit immunitaires ou d’une augmentation de la production d’hématies (par exemple, l’anémie hémolytique).
Une vaccination appropriée (hépatite A et hépatite B) des patients recevant de façon répétée/régulière des concentrés de facteur IX dérivé du plasma est recommandée.
Patients sous régime contrôlé en sel
Ce médicament contient jusqu’à 69 mg de sodium pour 1 flacon OCTAFIX 500 UI, ce qui équivaut à 3,45 % de l’apport alimentaire maximal quotidien recommandé par l'’OMS de 2g sodium par adulte et jusqu’à 138 mg de sodium pour 1 flacon d’OCTAFIX 1000 UI, ce qui équivaut à 6,9 % de l’apport alimentaire maximal quotidien recommandé par l'’OMS de 2g sodium par adulte.
Cette information doit être prise en compte chez les patients suivant un régime contrôlé en sodium.
Population pédiatrique
Les mises en gardes et précautions d'emploi indiquées concernent aussi bien les adultes que les enfants.
4.5. Interactions avec d'autres médicaments et autres formes d'interactions
Aucune interaction des préparations de facteur IX de coagulation humain avec d'autres spécialités pharmaceutiques n'a été rapportée à ce jour.
4.6. Fertilité, grossesse et allaitement
Aucune étude sur la reproduction animale n'a été menée avec le facteur IX. Etant donné la rareté de l'hémophilie B chez la femme, aucune donnée concernant l'utilisation du facteur IX pendant la grossesse et l'allaitement n'est disponible. Par conséquent, le facteur IX ne doit être administré qu'en cas de nécessité absolue au cours de la grossesse et de l'allaitement.
4.7. Effets sur l'aptitude à conduire des véhicules et à utiliser des machines
OCTAFIX n’a aucun effet sur l'aptitude à conduire des véhicules et à utiliser des machines.
Une hypersensibilité ou des réactions allergiques (telles qu'angio-œdème, inflammation et sensation de brûlure au site d'injection, frissons, rougeurs, urticaire généralisée, céphalées, démangeaisons, hypotension, léthargie, nausée, agitation, tachycardie, oppression thoracique, fourmillements, vomissement, respiration sifflante) ont été observés de façon peu fréquente chez les patients traités par des préparations de facteur IX de coagulation humain et peuvent dans certains cas évoluer en anaphylaxie sévère (avec choc). Dans certains cas, ces réactions ont abouti à une réaction anaphylactique sévère associée à l'apparition simultanée d'inhibiteurs du facteur IX (voir aussi la rubrique 4.4). Des cas de syndrome néphrotique ont été rapportés suite à une induction de tolérance immune chez des patients atteints d’hémophilie B avec des inhibiteurs anti-facteur IX et des antécédents de réactions allergiques.
À de rares occasions, de la fièvre a été observée.
Les patients atteints d'hémophilie B peuvent développer des anticorps neutralisants (inhibiteurs) anti-facteur IX. Si de tels inhibiteurs apparaissent, la réaction se manifeste sous forme d’une réponse clinique insuffisante. Dans ce cas, il est recommandé de contacter un centre spécialisé en hémophilie. Dans une étude clinique 25 enfants atteints d’hémophilie B, dont 6 patients non préalablement traités ont été traité par OCTAFIX pendant une médiane de 38 JCPA (entre 8 et 90 JCPA). Tous les patients avaient un taux de base en inhibiteur de facteur IX inférieur à 0.4 BU. Aucune apparition d’anticorps inhibiteur n’est survenue pendant l’étude.
L'administration de produits contenant le facteur IX comprend un risque potentiel de complications thromboemboliques, avec un risque accru pour des préparations de faible pureté. L’utilisation de préparation de facteur IX de faible pureté a été associée à la survenue d'infarctus du myocarde, de coagulation intravasculaire disséminée, de thrombose veineuse et d'embolie pulmonaire. L'administration de facteur IX humain hautement purifié est rarement associée à de tels effets secondaires.
Pour toute information sur la sécurité virale, voir la rubrique 4.4.
Tableau des effets indésirables
Le tableau suivant est conforme à la classification des systèmes d’organes selon le système organe classe MedDRA (et terme privilégié).
Les fréquences ont été évaluées selon le classement suivant : très fréquent (≥ 1/10); fréquent (≥ 1/100, <1/10); peu fréquent (≥ 1/1 000, <1/100); rare (≥ 1/10 000, <1/1 000); très rare (<1/10 000), fréquence indéterminée (ne peut être estimée sur la base des données disponibles).
|
Classes de systèmes d’organes |
Rares |
Très rares |
|
Affections du système immunitaire |
Réaction allergique |
Choc anaphylactique |
|
Affections vasculaires |
|
Evènement thromboembolique* |
|
Affections du rein et des voies urinaires |
|
Syndrome néphrotique |
|
Troubles généraux et anomalies au site d’administration |
|
Thrombocytopénie induite par l’héparine Fièvre |
|
Investigations |
|
Anticorps anti-facteur IX positif |
*Terme MEDRA de plus bas niveau (LLT)
Description des effets indésirables sélectionnés
Dans de rares cas, une thrombocytopénie de type II liée à la présence d'héparine dans la préparation d’OCTAFIX peut survenir. Elle conduit à une diminution de la numération des plaquettes inférieure à 100 000/µl ou à 50% de la numération initiale. Cette thrombocytopénie d’origine allergique peut survenir quelques heures après le traitement chez les patients ayant des antécédents d'hypersensibilité à l'héparine, et jusqu'à 6 à 14 jours après le début du traitement chez les patients sans antécédents d'hypersensibilité à l'héparine.
Cette forme sévère de thrombocytopénie peut s'accompagner de/ou occasionner une thrombose artérielle et veineuse, un thrombo-embolisme, des troubles sévères de la coagulation (coagulopathie de consommation), une nécrose cutanée au site d'injection, une hémorragie de type pétéchiale, un purpura et des selles goudronneuses. Si de telles manifestations allergiques surviennent, l'injection d’OCTAFIX doit être interrompue immédiatement. Le patient traité devra éviter d'utiliser des médicaments contenant de l'héparine à l'avenir. En raison de cet effet rare induit par l'héparine sur les plaquettes sanguines, la numération plaquettaire doit être surveillée attentivement, particulièrement en début de traitement.
Population pédiatrique
La fréquence, le type et la sévérité des effets indésirables chez l’enfant sont présumés identiques à ceux chez l’adulte.
Déclaration des effets indésirables suspectés
La déclaration des effets indésirables suspectés après autorisation du médicament est importante. Elle permet une surveillance continue du rapport bénéfice/risque du médicament. Les professionnels de santé déclarent tout effet indésirable suspecté via le système national de déclaration : Agence nationale de sécurité du médicament et des produits de santé (ANSM) et réseau des Centres Régionaux de Pharmacovigilance - Site internet : www.signalement-sante.gouv.fr.
Aucun symptôme lié à un surdosage par OCTAFIX n’a été rapporté.
5. PROPRIETES PHARMACOLOGIQUES
5.1. Propriétés pharmacodynamiques
Le facteur IX est une glycoprotéine monocaténaire d'un poids moléculaire d'environ
68 000 dalton. C'est un facteur de coagulation vitamine-K dépendant, synthétisé par le foie. Le facteur IX est activé par le facteur XIa dans la voie endogène de la coagulation et par le complexe facteur VII/facteur tissulaire dans la voie exogène. Le facteur IX activé, associé au facteur VIII activé, active le facteur X. Le facteur X activé convertit la prothrombine en thrombine. La thrombine convertit ensuite le fibrinogène en fibrine, ce qui aboutit à la formation du caillot.
L’hémophilie B est un déficit de la coagulation sanguine héréditaire et lié au sexe, dû à une diminution des taux du facteur IX, provoquant des accidents hémorragiques profus au niveau des articulations, des muscles, ou des organes internes, soit spontanément ou résultant de traumatismes accidentels ou chirurgicaux. A l’aide d’un traitement substitutif, les taux plasmatiques du facteur IX sont augmentés, permettant ainsi une correction temporaire du déficit en ce facteur et une correction des tendances hémorragiques.
Population pédiatrique
Une étude clinique incluant 25 enfants de moins de 6 ans a été réalisée incluant 6 patients non préalablement traités (naïfs). Le taux de récupération après administration de plus de 25 UI/kg de poids corporel d’OCTAFIX a été évalué pendant les trois premiers mois de traitement et après 12-24 mois. Le taux de récupération a été évalué à 0.8±1.4 et 0.9±1.3%/UI/kg respectivement dans la première et la deuxième évaluation.
5.2. Propriétés pharmacocinétiques
|
N = 13 |
Médiane |
Moyenne |
Ecart Type |
Minimum |
Maximum |
|
Taux de récupération |
1,2 |
1,3 |
0,5 |
0,8 |
2,4 |
|
Aire sous la courbenorm |
32,4 |
37,7 |
13,0 |
24,5 |
64,0 |
|
Demi-vie (h) |
27,8 |
29,1 |
5,2 |
22,0 |
36,8 |
|
Temps de résidence moyen (h) |
39,4 |
40,0 |
7,3 |
30,2 |
51,6 |
|
Clairance (ml x h-1 x kg) |
3,1 |
2,9 |
0,9 |
1,6 |
4,1 |
Le taux de récupération a également été évalué lors d'une seconde étude. La méta-analyse de toutes les évaluations de récupération (n=19) donne une récupération moyenne de 1,1 [UI/dl]/[UI/kg].
5.3. Données de sécurité préclinique
Les études réalisées chez l’animal sont limitées et ne mettent pas en évidence de risques supplémentaires par rapports aux effets déjà décrits dans les autres rubriques.
Héparine
Chlorure de sodium
Citrate de sodium
Chlorhydrate d'arginine
Chlorhydrate de lysine.
Solvant:
Eau pour préparations injectables.
Seuls les dispositifs d'injection/perfusion fournis doivent être utilisés car un échec du traitement peut se produire comme résultant de l'adsorption du facteur IX de coagulation humain sur la surface interne de certains dispositifs d'injection/perfusion.
La stabilité biochimique et physique après reconstitution a été démontrée pendant 72 heures à 25 °C. D’un point de vue microbiologique, le produit reconstitué doit être utilisé immédiatement. S’il n’est pas utilisé immédiatement, le temps et les conditions de conservation avant emploi relèvent de la responsabilité des utilisateurs et il est recommandé que la conservation ne dépasse pas 8 heures à température ambiante (25 °C).
6.4. Précautions particulières de conservation
A conserver à une température ne dépassant pas 25 °C.
Ne pas congeler.
Conserver les flacons dans l’emballage extérieur, à l’abri de la lumière.
Pour les conditions de conservation du médicament après reconstitution, voir la rubrique 6.3.
6.5. Nature et contenu de l'emballage extérieur
OCTAFIX se présente sous forme d’un emballage combiné composé de deux boîtes maintenues ensemble par un film plastique.
· OCTAFIX 500 UI :
Boîte 1 : poudre en flacon de 30 ml (verre type I) muni d'un bouchon (caoutchouc bromobutyle) et d'un capuchon amovible (aluminium) + une notice
Boîte 2 : 5 ml de solvant (eau pour préparations injectables) en flacon (verre type I ou type II) muni d'un bouchon (caoutchouc chlorobutyle ou bromobutyle) et d'un capuchon amovible (aluminium).
· OCTAFIX 1000 UI :
Boîte 1 : poudre en flacon de 30 ml (verre type I) muni d'un bouchon (caoutchouc bromobutyle) et d'un capuchon amovible (aluminium) + une notice
Boîte 2 : 10 ml de solvant (eau pour préparations injectables) en flacon (verre type I ou type II) muni d'un bouchon (caoutchouc chlorobutyle ou bromobutyle) et d'un capuchon amovible (aluminium).
La boîte 2 contient également les dispositifs médicaux suivants :
· 1 boîte de matériel pour injection intraveineuse (1 dispositif de transfert, 1 kit de perfusion, 1 seringue jetable),
· 2 tampons d'alcool.
6.6. Précautions particulières d’élimination et de manipulation
Lire attentivement les instructions et les suivre soigneusement !
N’utilisez pas OCTAFIX après la date de péremption mentionnée sur l'étiquette et l’emballage.
Durant la procédure décrite ci-dessous, la stérilité doit être maintenue.
Avant administration, la solution reconstituée doit être examinée visuellement pour déceler une décoloration et la présence de particules étrangères.
La solution doit être limpide ou légèrement opalescente. N'utilisez pas de solutions troubles ou présentant des dépôts.
Utilisez immédiatement la solution préparée afin de prévenir toute contamination microbienne.
Pour la reconstitution, utilisez seulement le kit de perfusion fourni. L'utilisation d'un autre dispositif d'injection/perfusion peut induire des risques supplémentaires et un échec du traitement.
Instructions pour la préparation de la solution:
|
· N'utilisez pas le produit directement à la sortie du réfrigérateur. Laissez le solvant et la poudre contenus dans les flacons fermés revenir à température ambiante. |
|
|||||||||||||
|
· Retirez les opercules des deux flacons et nettoyez les bouchons en caoutchouc avec l'un des tampons imbibés d'alcool fournis. |
|
|||||||||||||
|
· Le dispositif de transfert est représenté à la Fig. 1. Placez le flacon de solvant sur une surface plane et tenez-le fermement. Prenez le dispositif de transfert et retournez-le. Placez la partie bleue du dispositif de transfert sur le dessus du flacon de solvant et appuyez fermement jusqu'à ce qu'il s'enclenche (Fig. 2 + 3). Ne le faites pas tourner au moment de le fixer.
Fig. 3 Fig. 2
|
|
|||||||||||||
|
· Placez le flacon de poudre sur une surface plane et tenez-le fermement. Prenez le flacon de solvant avec le dispositif de transfert fixé et retournez-le. Placez la partie blanche sur le dessus du flacon de poudre et appuyez fermement jusqu'à ce qu'il s'enclenche (Fig. 4). Ne le faites pas tourner au moment de le fixer. Le solvant s'écoule automatiquement dans le flacon de poudre.
|
|
|
||||||||||||
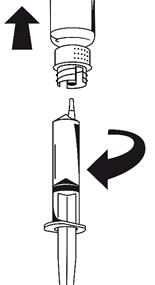
· Les deux flacons toujours fixés, tournez doucement le flacon de poudre jusqu'à ce que le produit soit dissous. La dissolution est terminée en moins de 10 minutes à température ambiante. Il peut se produire une légère formation de mousse pendant la préparation. Dévissez le dispositif de transfert en deux parties (Fig. 5). La mousse va disparaître. Eliminez le flacon de solvant vide avec la partie bleue du dispositif de transfert.
|
|
|||||||||||||
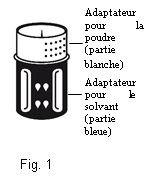
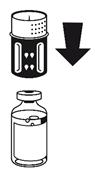
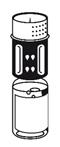
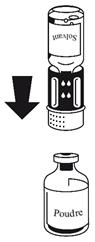
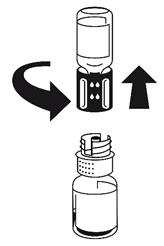
Fig. 5
Instructions pour l’injection :
À titre de précaution, vous devez mesurer votre pouls avant et pendant l'injection. S'il se produit une forte augmentation de votre fréquence cardiaque, réduisez la vitesse d'injection ou interrompez l'administration pendant un court moment.
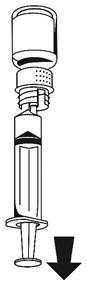
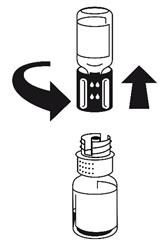
· Fixez la seringue à la partie blanche du dispositif de transfert. Retournez le flacon et prélevez la solution dans la seringue (Fig. 6).
La solution doit être limpide ou légèrement opalescente.
Dès que la solution a été transférée, tenez fermement le piston de la seringue (en la tenant tournée vers le bas) et retirez la seringue du dispositif de transfert (Fig. 7). Eliminez le flacon vide, ainsi que la partie blanche du dispositif de transfert.
![]()
![]()
|
|||
|
|||
· Nettoyez le site d'injection choisi avec l'un des tampons imbibés d'alcool fournis.
· Fixez le kit de perfusion fourni à la seringue.
· Introduisez l'aiguille pour injection dans la veine choisie. Si vous avez utilisé un garrot pour rendre la veine plus facile à voir, ce garrot doit être relâché avant de commencer à injecter OCTAFIX.
Du sang ne doit pas pénétrer dans la seringue, en raison du risque de formation de caillots de fibrine.
· Injectez la solution dans la veine lentement, pas plus vite que 2 à 3 ml par minute.
Si vous utilisez plus d'un flacon de poudre d’OCTAFIX pour un traitement, vous pouvez réutiliser la même aiguille pour injection et la même seringue. Le dispositif de transfert est réservé à un usage unique.
Tout produit non utilisé ou déchet doit être éliminé conformément à la réglementation en vigueur.
7. TITULAIRE DE L’AUTORISATION DE MISE SUR LE MARCHE
62 BIS AVENUE ANDRE MORIZET
92100 BOULOGNE BILLANCOURT
France
8. NUMERO(S) D’AUTORISATION DE MISE SUR LE MARCHE
· 563 415-2 ou 34009 563 415 2 8 : OCTAFIX 1000 UI : poudre en flacon de 30 ml (verre type I) muni d'un bouchon (bromobutyle) et d'un opercule détachable (aluminium) + 10 ml de solvant en flacon (verre type I) muni d'un bouchon (chlorobutyle ou bromobutyle) et d'un opercule détachable ; boîte de 1 flacon de poudre et 1 flacon de solvant.
9. DATE DE PREMIERE AUTORISATION/DE RENOUVELLEMENT DE L’AUTORISATION
[à compléter ultérieurement par le titulaire]
10. DATE DE MISE A JOUR DU TEXTE
[à compléter ultérieurement par le titulaire]
Sans objet.
12. INSTRUCTIONS POUR LA PREPARATION DES RADIOPHARMACEUTIQUES
Liste I.
Prescription initiale hospitalière de six mois (les établissements de transfusion sanguine autorisés à dispenser des médicaments dérivés du sang qui y sont traités, inclus).
Délivrance réservée aux pharmacies à usage intérieur des établissements de santé ou des établissements de transfusion sanguine pour les malades qui y sont traités.
ANSM - Mis à jour le : 16/03/2021
OCTAFIX 100 UI/ml, poudre et solvant pour solution injectable
Facteur IX de coagulation humain
Veuillez lire attentivement cette notice avant d’utiliser ce médicament car elle contient des informations importantes pour vous.
· Gardez cette notice. Vous pourriez avoir besoin de la relire.
· Si vous avez d’autres questions, interrogez votre médecin, votre pharmacien ou votre infirmier/ère.
· Ce médicament vous a été personnellement prescrit. Ne le donnez pas à d’autres personnes. Il pourrait leur être nocif, même si les signes de leur maladie sont identiques aux vôtres.
· Si vous ressentez un quelconque effet indésirable, parlez-en à votre médecin, votre pharmacien ou votre infirmier/ère. Ceci s’applique aussi à tout effet indésirable qui ne serait pas mentionné dans cette notice. Voir rubrique 4.
1. Qu'est-ce que OCTAFIX 100 UI/ml, poudre et solvant pour solution injectable et dans quels cas est-il utilisé ?
2. Quelles sont les informations à connaître avant d'utiliser OCTAFIX 100 UI/ml, poudre et solvant pour solution injectable ?
3. Comment utiliser OCTAFIX 100 UI/ml, poudre et solvant pour solution injectable ?
4. Quels sont les effets indésirables éventuels ?
5. Comment conserver OCTAFIX 100 UI/ml, poudre et solvant pour solution injectable ?
6. Contenu de l’emballage et autres informations.
1. QU’EST-CE QUE OCTAFIX 100 UI/ml, poudre et solvant pour solution injectable ET DANS QUELS CAS EST-IL UTILISE ?
OCTAFIX appartient à un groupe de médicaments appelés facteurs de coagulation et contient le facteur IX de coagulation humain. C’est une protéine spéciale qui augmente la capacité du sang à coaguler.
OCTAFIX est utilisé dans le traitement et la prévention des saignements chez les patients présentant un trouble hémorragique (hémophilie B). Ceci est un trouble au cours duquel un saignement peut continuer plus longtemps que prévu. Il est dû à un déficit congénital en facteur IX dans le sang.
OCTAFIX est fourni en tant que poudre et solvant pour solution injectable. Après reconstitution avec le solvant fourni, OCTAFIX est administré par voie intraveineuse.
2. QUELLES SONT LES INFORMATIONS A CONNAITRE AVANT D’UTILISER OCTAFIX 100 UI/ml, poudre et solvant pour solution injectable ?
N’utilisez jamais OCTAFIX 100 UI/ml, poudre et solvant pour solution injectable :
· si vous êtes allergique (hypersensible) au facteur IX de coagulation humain ou à l’un des autres composants contenus dans ce médicament, mentionnés dans la rubrique 6.
· si vous avez déjà présenté une thrombocytopénie induite par l'héparine (type II), qui correspond à la chute du nombre de plaquettes sanguines après administration d’héparine. Les plaquettes sont des cellules sanguines qui aident à stopper le saignement. L’héparine est un médicament utilisé pour prévenir la coagulation sanguine.
Avertissements et précautions
Adressez-vous à votre médecin ou pharmacien avant d’utiliser OCTAFIX 100 UI/ml, poudre et solvant pour solution injectable
Comme pour tout médicament contenant des protéines et administré par voie intraveineuse, des réactions allergiques type hypersensibilité peuvent survenir. OCTAFIX contient des traces de protéines humaines autres que le facteur IX et de l'héparine.
Les signes annonciateurs d'une réaction d'hypersensibilité sont :
· démangeaisons,
· urticaire généralisée,
· oppression thoracique,
· respiration sifflante,
· tension artérielle basse,
· allergie généralisée sévère (anaphylaxie, quand tout ou partie des signes décrits ci-dessus se développent rapidement et s’intensifient).
En cas de survenue de ces signes, le traitement doit être arrêté immédiatement et vous devez prévenir votre médecin.
En cas de choc anaphylactique, le traitement approprié devra être instauré dès que possible.
Votre médecin peut vous recommander la vaccination contre l’hépatite A et l’hépatite B si vous recevez régulièrement/de façon répétée des produits à base de facteur IX de coagulation humain.
La formation d’inhibiteurs (anticorps neutralisants) du facteur IX, produits par les cellules immunitaires, est une complication connue du traitement de l’hémophilie B. Les inhibiteurs peuvent augmenter le risque de choc anaphylactique (réactions allergiques sévères). C’est pourquoi, si vous présentez une réaction allergique, un test biologique devra être effectué afin de rechercher la présence d’inhibiteurs. Les patients présentant un inhibiteur du facteur IX peuvent présenter un risque accru de choc anaphylactique s’ils sont traités par du facteur IX. Votre médecin pourra donc décider de faire la première perfusion de facteur IX sous surveillance médicale, avec possibilité de traitement immédiat des réactions allergiques en cas de besoin.
Les concentrés de facteur IX peuvent provoquer l’obstruction des vaisseaux sanguins par un caillot. En raison de ce risque, accru avec les préparations de faible pureté, une surveillance biologique devra être instaurée pour rechercher les marqueurs précoces de ces complications après administration de produits à base de facteur IX si :
· vous présentez des signes de fibrinolyse (désagrégation des caillots sanguins),
· vous présentez une coagulation intra vasculaire disséminée (formation de multiples caillots dans la circulation sanguine)
· on vous a fait un diagnostic de maladie hépatique,
· vous présentez des facteurs de risques cardiovasculaires,
· vous avez récemment subit une opération de chirurgie,
· vous présentez un risque fort de formation de caillot ou de coagulation intra vasculaire disséminée.
Si vous présentez l’une des complications évoquées ci-dessus, votre médecin ne vous donnera OCTAFIX que si les bénéfices attendus sont supérieurs aux risques.
Chez les patients recevant régulièrement des produits à base de facteur IX de coagulation humain, la recherche d’anticorps neutralisants (inhibiteurs) devra être effectuée au moyen de tests biologiques appropriés avec quantification en unités Bethesda (UB).
Données de Sécurité virale
Lorsque des médicaments préparés à partir de sang ou de plasma humain sont administrés, des mesures sont mises en place afin de prévenir le risque de transmission d’infection aux patients. Ceci implique la sélection rigoureuse des donneurs de sang et plasma afin d’exclure tout risque d’infection et la réalisation de tests de dépistage des principaux marqueurs viraux sur chaque don et chaque mélange de plasma. La fabrication de ces produits inclut également des étapes d'élimination et/ou d'inactivation des virus dans le sang ou le plasma.
Malgré ces mesures, lorsque des médicaments préparés à partir de sang ou de plasma humain sont administrés, le risque de transmission d’agents infectieux ne peut être totalement exclu. Ceci s’applique également à des agents qui pourraient engendrer une maladie et dont la nature est jusqu’ici inconnue.
Les mesures prises sont considérées comme efficaces vis-à-vis des virus enveloppés tels que le virus de l’immunodéficience humaine (VIH), le virus de l’hépatite B (VHB) et le virus de l’hépatite C (VHC), et vis-à-vis du virus non enveloppé VHA.
Les mesures prises peuvent être d’efficacité limitée vis-à-vis des virus non enveloppés tels que le parvovirus B19. L’infection par le parvovirus B19 peut être sévère chez la femme enceinte (avec infection du fœtus) et chez les personnes dont le système immunitaire est déficient ou qui ont certains types d’anémie (par exemple l’anémie hémolytique).
Enfants et adolescents
Si OCTAFIX est administré au nouveau-né, l’enfant devra être étroitement surveillé afin de prévenir les signes de coagulation intravasculaire disséminée.
Autres médicaments et OCTAFIX 100 UI/ml, poudre et solvant pour solution injectable
Aucune interaction des concentrés du facteur IX de coagulation humain avec d'autres spécialités pharmaceutiques n'a été rapportée à ce jour.
Informez votre médecin ou pharmacien si vous prenez, avez récemment pris ou pourriez prendre tout autre médicament.
OCTAFIX 100 UI/ml, poudre et solvant pour solution injectable avec des aliments, boissons et de l’alcool
Sans objet.
Grossesse, allaitement et fertilité
Aucune étude sur l’utilisation de facteur IX pendant la grossesse et l’allaitement n’a été menée.
OCTAFIX ne doit donc être administré qu'en cas d'indication absolue au cours de la grossesse et de l'allaitement.
Si vous êtes enceinte ou que vous allaitez, si vous pensez être enceinte ou planifiez une grossesse, demandez conseil à votre médecin avant de prendre ce médicament.
Conduite de véhicules et utilisation de machines
Aucun effet sur l'aptitude à conduire des véhicules et à utiliser des machines n'a été observé.
OCTAFIX contient du sodium
Ce médicament contient jusqu’à 69 mg de sodium (composant principal du sel de cuisine/table) pour 1 flacon d’OCTAFIX 500 UI, ce qui équivaut à 3,45 % de l’apport alimentaire maximal quotidien recommandé de sodium par adulte et
jusqu’à 138 mg de sodium pour 1 flacon d’OCTAFIX 1000 UI, ce qui équivaut à 6,9 % de l’apport alimentaire maximal quotidien recommandé de sodium par adulte.
Cette information doit être prise en compte chez les patients suivant un régime contrôlé en sodium.
3. COMMENT UTILISER OCTAFIX 100 UI/ml, poudre et solvant pour solution injectable ?
OCTAFIX doit être administré par voie intraveineuse (dans la veine) après reconstitution avec le solvant fourni.
Seul le dispositif de perfusion fourni doit être utilisé. L’utilisation de tout autre set d’injection/perfusion peut provoquer des risques supplémentaires et un échec du traitement.
Le traitement doit être initié sous la surveillance d'un médecin spécialiste de l'hémophilie. La posologie et la durée du traitement de substitution dépendent de la sévérité de votre déficit en facteur IX, de la localisation et de l'intensité de l'accident hémorragique, ainsi que de votre état clinique.
Posologie
Votre médecin vous indiquera la fréquence et la quantité d’OCTAFIX qui doit vous être injecté.
La dose de facteur IX s'exprime en unités internationales (UI). L'activité du facteur IX dans le plasma se réfère à la quantité de facteur IX présent dans le plasma. Elle est exprimée soit en pourcentage (relative à l'activité normale du plasma sanguin humain), soit en unités internationales (relative au standard international pour le facteur IX dans le plasma sanguin).
Une unité internationale (UI) de facteur IX correspond à la quantité de facteur IX contenue dans un ml de plasma sanguin humain normal. Le calcul de la dose nécessaire repose sur l'observation que 1 UI de facteur IX par kg de poids corporel augmente l'activité du facteur IX dans le plasma humain de 1 % par rapport à l’activité normale. Afin de calculer la dose requise, le niveau d’activité du facteur IX dans le plasma est mesuré. Celui-ci indiquera de combien l’activité doit être augmentée.
La dose requise est calculée selon la formule :
Unités requises = poids corporel (kg) x augmentation souhaitée en facteur IX (%) (Ul/dl) x 0.8
La dose et la fréquence des injections vont dépendre de votre réponse au traitement et seront adaptées par votre médecin. Les produits à base de facteur IX doivent rarement être injectés plus d'une fois par jour.
Votre réponse aux produits à base de facteur IX peut varier. C’est pourquoi votre taux de facteur IX devra être mesuré pendant le traitement afin de calculer la dose correcte et la fréquence de l’injection. Plus spécifiquement en cas d’intervention chirurgicale, votre médecin utilisera des tests sanguins (activité du facteur IX dans le plasma sanguin) afin de surveiller étroitement le traitement de substitution.
Prévention des saignements
Si vous souffrez d’hémophilie B sévère, l’injection sera de 20 à 40 UI de facteur IX par kg de poids corporel. Cette dose sera administrée deux fois par semaine pour le traitement prophylactique au long cours. La dose sera adaptée en fonction de votre réponse. Dans certains cas, spécialement chez les sujets jeunes, il peut être nécessaire de raccourcir l'intervalle entre deux injections ou d'augmenter la posologie.
Utilisation chez les enfants
Dans une étude menée chez des enfants de moins de 6 ans, la dose moyenne administrée par jour d’exposition était de 40 UI/kg de poids corporel.
Si votre saignement ne peut être stoppé à cause des inhibiteurs
Si l’activité attendue de facteur IX n’est pas atteinte après une injection, ou si le saignement n’est pas stoppé après une dose correcte, informez en votre médecin. Il examinera votre plasma sanguin afin de voir si vous avez développé des inhibiteurs (anticorps) contre la protéine du facteur IX. Ces inhibiteurs peuvent réduire l’activité du facteur IX. Dans ce cas, il peut devenir nécessaire de choisir un traitement différent. Votre médecin discutera de cela avec vous et vous recommandera un traitement différent si nécessaire.
Mode d’administration
Lire attentivement les instructions et les suivre soigneusement !
N’utilisez pas OCTAFIX après la date de péremption mentionnée sur l'étiquette et l’emballage.
Durant la procédure décrite ci-dessous, la stérilité doit être maintenue.
Avant administration, la solution reconstituée doit être examinée visuellement pour déceler une décoloration et la présence de particules étrangères.
La solution doit être limpide ou légèrement opalescente. N'utilisez pas de solutions troubles ou présentant des dépôts.
Utilisez immédiatement la solution préparée afin de prévenir toute contamination microbienne.
Pour la reconstitution, utilisez seulement le kit de perfusion fourni. L'utilisation d'un autre dispositif d'injection/perfusion peut induire des risques supplémentaires et un échec du traitement.
Instructions pour la préparation de la solution :
|
1. N'utilisez pas le produit directement à la sortie du réfrigérateur. Laissez le solvant et la poudre contenus dans les flacons fermés revenir à température ambiante. |
|
|||||||||||||||
|
2. Retirez les opercules des deux flacons et nettoyez les bouchons en caoutchouc avec l'un des tampons imbibés d'alcool fournis. |
|
|||||||||||||||
|
3. Le dispositif de transfert est représenté à la Fig. 1. Placez le flacon de solvant sur une surface plane et tenez-le fermement. Prenez le dispositif de transfert et retournez-le. Placez la partie bleue du dispositif de transfert sur le dessus du flacon de solvant et appuyez fermement jusqu'à ce qu'il s'enclenche (Fig. 2 + 3). Ne le faites pas tourner au moment de le fixer.
Fig. 3 Fig. 2
|
|
|||||||||||||||
|
4. Placez le flacon de poudre sur une surface plane et tenez-le fermement. Prenez le flacon de solvant avec le dispositif de transfert fixé et retournez-le. Placez la partie blanche sur le dessus du flacon de poudre et appuyez fermement jusqu'à ce qu'il s'enclenche (Fig. 4). Ne le faites pas tourner au moment de le fixer. Le solvant s'écoule automatiquement dans le flacon de poudre.
|
|
|
||||||||||||||
5. Les deux flacons toujours fixés, tournez doucement le flacon de poudre jusqu'à ce que le produit soit dissous. La dissolution est terminée en moins de 10 minutes à température ambiante. Il peut se produire une légère formation de mousse pendant la préparation. Dévissez le dispositif de transfert en deux parties (Fig. 5). La mousse va disparaître. Eliminez le flacon de solvant vide avec la partie bleue du dispositif de transfert. |
|
|||||||||||||||
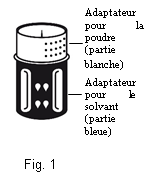
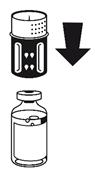
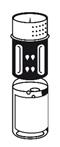
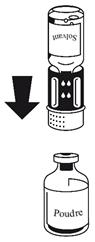
Fig. 5![]()
![]()
Instructions pour l’injection :
À titre de précaution, vous devez mesurer votre pouls avant et pendant l'injection. S'il se produit une forte augmentation de votre fréquence cardiaque, réduisez la vitesse d'injection ou interrompez l'administration pendant un court moment.
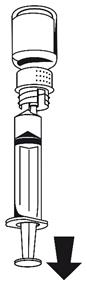
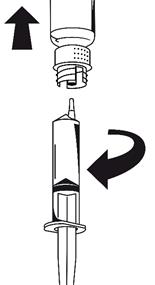
1. Fixez la seringue à la partie blanche du dispositif de transfert. Retournez le flacon et prélevez la solution dans la seringue (Fig. 6).
La solution doit être limpide ou légèrement opalescente.
Dès que la solution a été transférée, tenez fermement le piston de la seringue (en la tenant tournée vers le bas) et retirez la seringue du dispositif de transfert (Fig. 7). Eliminez le flacon vide, ainsi que la partie blanche du dispositif de transfert.
![]()
![]()
|
|||
|
|||
2. Nettoyez le site d'injection choisi avec l'un des tampons imbibés d'alcool fournis.
3. Fixez le kit de perfusion fourni à la seringue.
4. Introduisez l'aiguille pour injection dans la veine choisie. Si vous avez utilisé un garrot pour rendre la veine plus facile à voir, ce garrot doit être relâché avant de commencer à injecter OCTAFIX.
Du sang ne doit pas pénétrer dans la seringue, en raison du risque de formation de caillots de fibrine.
5. Injectez la solution dans la veine lentement, pas plus vite que 2 à 3 ml par minute.
Si vous utilisez plus d'un flacon de poudre d’OCTAFIX poudre pour un traitement, vous pouvez réutilisez la même aiguille pour injection et la même seringue. Le dispositif de transfert est réservé à un usage unique.
Tout produit non utilisé ou déchet doit être éliminé conformément à la réglementation en vigueur.
Si vous avez utilisé plus de OCTAFIX 100 UI/ml, poudre et solvant pour solution injectable que vous n’auriez dû :
Aucun symptôme lié à un surdosage par facteur IX de coagulation humain n’a été rapporté.
Cependant, la dose recommandée ne devra pas être dépassée.
Votre traitement doit être initié sous la surveillance d'un médecin spécialiste de l'hémophilie.
Si vous oubliez d’utiliser OCTAFIX 100 UI/ml, poudre et solvant pour solution injectable :
Ne prenez pas de dose double pour compenser la dose que vous avez oublié de prendre.
Si vous arrêtez d’utiliser OCTAFIX 100 UI/ml, poudre et solvant pour solution injectable :
Sans objet.
4. QUELS SONT LES EFFETS INDESIRABLES EVENTUELS ?
Une hypersensibilité ou des réactions allergiques ont été observées de façon peu fréquente chez des patients traités par des produits contenant du facteur IX. Ces réactions peuvent inclure :
· contraction involontaire des vaisseaux sanguins (spasmes) avec gonflement du cou, de la bouche et du visage,
· inflammation et sensation de brûlure au site d’injection,
· frissons,
· rougeurs,
· urticaire généralisée,
· maux de tête,
· démangeaisons,
· tension artérielle basse,
· fatigue,
· sensation de mal être,
· agitation,
· accélération du rythme cardiaque,
· oppression thoracique,
· fourmillements,
· vomissements,
· respiration sifflante.
Dans certains cas, ces réactions allergiques ont abouti à une réaction allergique généralisée sévère appelée anaphylaxie, pouvant inclure un choc. Ces réactions sont le plus souvent associées à l'apparition simultanée d'inhibiteurs du facteur IX.
Si vous présentez l’un des signes ci-dessus, contactez votre médecin.
Si vous êtes atteint d'hémophilie B, vous pouvez développer des anticorps neutralisants (inhibiteurs) du facteur IX. Ces anticorps peuvent empêcher votre médicament de fonctionner correctement. Votre médecin discutera de cela avec vous et vous recommandera un autre traitement si nécessaire.
Une étude clinique a été menée chez 25 enfants atteints d’hémophilie B dont 6 patients non préalablement traités. Aucun inhibiteur n’a été observé pendant cette étude. La tolérance de toutes les injections a été évaluée comme « très bonne » ou « bonne ».
Certains patients atteints d’hémophilie B ayant développé une tolérance immune avec présence d’inhibiteurs du facteur IX et des antécédents de réactions allergiques, ont présenté un syndrome néphrotique (état rénal sévère).
Une fièvre peut apparaître dans de rares cas.
Des produits de faible pureté contenant du facteur IX peuvent provoquer, dans de rares cas, la formation d’un caillot de sang dans un vaisseau sanguin. Ceci peut conduire aux complications suivantes :
· attaque cardiaque,
· formation de multiples caillots dans la circulation sanguine (coagulation intravasculaire disséminée),
· formation de caillot de sang dans les veines (thrombose veineuse),
· formation de caillot de sang dans les poumons (embolie pulmonaire).
Ces effets secondaires sont plus fréquents si vous utilisez des préparations de facteur IX de faible pureté. L'administration de facteur IX humain hautement purifié, tels qu’OCTAFIX, est rarement associée à de tels effets secondaires.
La présence d’héparine dans la préparation peut provoquer une diminution soudaine du nombre de plaquettes dans le sang à moins de 100 000 par microlitre ou moins de 50% de la numération initiale. C’est une réaction allergique appelée « thrombocytopénie de type II induite par l’héparine ». Dans de rares cas, chez des patients sans antécédents d'hypersensibilité à l'héparine, la diminution du nombre de plaquettes peut survenir 6 à 14 jours après le début du traitement. Chez les patients ayant des antécédents d'hypersensibilité à l'héparine, cette diminution peut survenir quelques heures après le traitement. Cette forme sévère de diminution du nombre de plaquettes peut s'accompagner de/ou occasionner :
· formation d’un caillot de sang dans les artères et les veines,
· obstruction d’un vaisseau sanguin par un caillot d’un autre endroit,
· troubles sévères de la coagulation sanguine (coagulopathie de consommation),
· nécrose de la peau au site d'injection,
· hémorragie avec petites taches rouges violacées sur la peau,
· purpura (bleu ou petites taches rouges),
· selles goudronneuses.
Si de telles réactions allergiques surviennent, les injections d’OctaFIX doivent être interrompues immédiatement et vous ne devrez plus utiliser des médicaments contenant de l'héparine à l'avenir. En raison de cet effet rare sur les plaquettes sanguines, votre médecin devra surveiller étroitement la numération des plaquettes sanguines, particulièrement en début de traitement.
Pour la sécurité virale, veuillez-vous référez à la section 2.
Déclaration des effets secondaires
Si vous ressentez un quelconque effet indésirable, parlez-en à votre médecin, votre pharmacien ou à votre infirmier/ère. Ceci s’applique aussi à tout effet indésirable qui ne serait pas mentionné dans cette notice. Vous pouvez également déclarer les effets indésirables directement via le système national de déclaration : Agence nationale de sécurité du médicament et des produits de santé (ANSM) et réseau des Centres Régionaux de Pharmacovigilance - Site internet: www.signalement-sante.gouv.fr
En signalant les effets indésirables, vous contribuez à fournir davantage d’informations sur la sécurité du médicament.
5. COMMENT CONSERVER OCTAFIX 100 UI/ml, poudre et solvant pour solution injectable ?
Tenir ce médicament hors de la vue et de la portée des enfants.
A conserver à une température ne dépassant pas 25°C.
Ne pas congeler.
Conserver les flacons dans l'emballage extérieur à l'abri de la lumière.
N’utilisez pas ce médicament après la date de péremption indiquée sur l’étiquette et le flacon. La date de péremption fait référence au dernier jour de ce mois. Le produit reconstitué doit être utilisé immédiatement et la conservation ne doit pas dépasser 8 heures à température ambiante (25 °C).
Utilisez OCTAFIX en une seule fois.
N’utilisez pas ce médicament si vous remarquez des solutions troubles ou non dissoutes complètement.
Ne jetez aucun médicament au tout-à-l’égout ni avec les ordures ménagères. Demandez à votre pharmacien d’éliminer les médicaments que vous n’utilisez plus. Ces mesures contribueront à protéger l’environnement.
6. CONTENU DE L’EMBALLAGE ET AUTRES INFORMATIONS
Ce que contient OCTAFIX 100 UI/ml, poudre et solvant pour solution injectable
· La substance active est :
le facteur IX de coagulation humain.
· Les autres composants sont :
l’héparine, le chlorure de sodium, le citrate de sodium, le chlorhydrate d'arginine et le chlorhydrate de lysine.
OCTAFIX se présente sous forme de poudre et solvant pour solution injectable et existe sous la forme de 2 présentations contenant nominalement :
· Soit 500 UI de facteur IX de coagulation humain par flacon.
Le produit contient environ 100 UI/ml de facteur IX de coagulation humain après reconstitution avec les 5 ml d’eau pour préparations injectables.
· Soit 1000 UI de facteur IX de coagulation humain par flacon.
Le produit contient environ 100 UI/ml de facteur IX de coagulation humain après reconstitution avec les 10 ml d’eau pour préparations injectables.
OCTAFIX est produit à partir de dons de plasma humain.
L’activité (UI) est déterminée en utilisant le test de coagulation en un temps de la Pharmacopée européenne, en comparaison avec une référence internationale de l’Organisation Mondiale de la Santé (OMS). L'activité spécifique d'OCTAFIX est d'environ 100 Ul/mg de protéine.
Description de l'emballage
OCTAFIX est vendu sous forme d’emballage combiné, composé en deux emballages maintenus ensemble par un film plastique.
Une des boîtes contient le flacon de poudre pour solution injectable (500 UI ou 1000 UI) ainsi que la notice.
L’autre boîte contient le flacon de solvant (eau pour préparations injectables); 5 ml pour OCTAFIX 500 UI ou 10 ml pour OCTAFIX 1000 UI. Cette boîte contient aussi les dispositifs médicaux suivants :
· 1 boîte de matériel pour injection intraveineuse (1 dispositif de transfert, 1 kit de perfusion, 1 seringue jetable),
· 2 tampons d'alcool.
Titulaire de l’autorisation de mise sur le marché
62 BIS AVENUE ANDRE MORIZET
92100 BOULOGNE BILLANCOURT
FRANCE
Exploitant de l’autorisation de mise sur le marché
62 BIS AVENUE ANDRE MORIZET
92100 BOULOGNE BILLANCOURT
FRANCE
OCTAPHARMA PHARMAZEUTIKA Produktionsges M.B.H
OBERLAAER STRASSE 235
1100 VIENNE
AUTRICHE
ou
OCTAPHARMA S.A.S.
70-72 RUE DU MARECHAL FOCH
67380 LINGOLSHEIM
FRANCE
Noms du médicament dans les Etats membres de l'Espace Economique Européen
Ce médicament est autorisé dans les Etats membres de l'Espace Economique Européen sous les noms suivants : Conformément à la réglementation en vigueur.
[à compléter ultérieurement par le titulaire]
La dernière date à laquelle cette notice a été révisée est :
[à compléter ultérieurement par le titulaire]
Sans objet.