Dernière mise à jour le 03/08/2026
OXYBATE DE SODIUM REIG JOFRE 500 mg/mL, solution buvable
Indications thérapeutiques
OXYBATE DE SODIUM REIG JOFRE est un médicament utilisé dans le traitement de la narcolepsie chez les adultes, les adolescents et les enfants à partir de 7 ans, présentant une cataplexie.
La narcolepsie se caractérise par des troubles du sommeil pouvant comporter une somnolence diurne, ainsi qu’une cataplexie, des paralysies du sommeil, des hallucinations et un sommeil de mauvaise qualité. La cataplexie se caractérise par une brutale faiblesse musculaire ou une paralysie sans perte de conscience déclenchée par une réaction émotionnelle soudaine telle que colère, peur, joie, éclats de rire ou surprise.
Présentations
> 1 flacon(s) polytéréphtalate (PET) jaune(brun) de 180 ml avec fermeture de sécurité enfant avec adaptateur(s) pour flacon avec seringue(s) doseuse(s) avec 2 gobelet(s) doseur(s)
Code CIP : 34009 301 870 6 7
Déclaration de commercialisation : 08/12/2020
Cette présentation est agréée aux collectivités
Service médical rendu (SMR)
Les libellés affichés ci-dessous ne sont que des résumés ou extraits issus des avis rendus par la Commission de la Transparence. Seul l'avis complet de la Commission de la Transparence fait référence.
Cet avis est consultable à partir du lien `Avis du jj/mm/aaaa` ou encore sur demande auprès de la HAS (plus d'informations dans l'aide). Les avis et synthèses d'avis contiennent un paragraphe sur la place du médicament dans la stratégie thérapeutique.
| Valeur du SMR | Avis | Motif de l'évaluation | Résumé de l'avis |
|---|---|---|---|
| Important | Avis du 08/01/2020 | Inscription (CT) | Le service médical rendu par OXYBATE DE SODIUM REIG JOFRE 500 mg/ml, solution buvable, est important dans l’indication de l’AMM. |
Amélioration du service médical rendu (ASMR)
Les libellés affichés ci-dessous ne sont que des résumés ou extraits issus des avis rendus par la Commission de la Transparence. Seul l'avis complet de la Commission de la Transparence fait référence.
Cet avis est consultable à partir du lien `Avis du jj/mm/aaaa` ou encore sur demande auprés de la HAS (plus d'informations dans l'aide). Les avis et synthéses d'avis contiennent un paragraphe sur la place du médicament dans la stratégie thérapeutique.
| Valeur de l'ASMR | Avis | Motif de l'évaluation | Résumé de l'avis |
|---|---|---|---|
| V (Inexistant) | Avis du 08/01/2020 | Inscription (CT) | Cette spécialité est un générique qui n’apporte pas d’amélioration du service médical rendu (ASMR V) par rapport au princeps XYREM 500 mg/ml, solution buvable (oxybate de sodium). |
Autres informations
- Titulaire de l'autorisation : LABORATORIO REIG JOFRE SA
- Conditions de prescription et de délivrance :
- prescription en toutes lettres sur ordonnance sécurisée
- prescription initiale annuelle réservée à certains spécialistes
- prescription limitée à 4 semaines
- prescription réservée aux médecins exerçant dans les centres du sommeil
- prescription réservée aux spécialistes et services NEUROLOGIE
- renouvellement non restreint
- stupéfiants
- Statut de l'autorisation : Valide
- Type de procédure : Procédure décentralisée
- Code CIS : 6 909 685 7
ANSM - Mis à jour le : 23/05/2022
OXYBATE DE SODIUM REIG JOFRE 500 mg/mL, solution buvable
2. COMPOSITION QUALITATIVE ET QUANTITATIVE
Oxybate de sodium............................................................................................................. 500 mg
Pour 1 mL de solution buvable
Excipient(s) à effet notoire : sodium (91 mg/mL)
Pour la liste complète des excipients, voir rubrique 6.1.
La solution buvable est claire et incolore.
4.1. Indications thérapeutiques
4.2. Posologie et mode d'administration
Posologie
Adulte
La dose initiale recommandée est de 4,5 g/jour d’oxybate de sodium à fractionner en deux prises de 2,25 g/dose. La posologie doit être individualisée en fonction de l’efficacité et de la tolérance (voir rubrique 4.4) jusqu’à une posologie maximale de 9 g/jour à fractionner en deux prises identiques de 4,5 g/dose ; la posologie doit être adaptée par paliers de 1,5 g par jour (soit 0,75 g/dose). Un minimum de 1 à 2 semaines est recommandé entre chaque augmentation de dose. La posologie de 9 g/jour ne doit pas être dépassée en raison de la possible survenue de symptômes sévères à des doses de 18 g/jour ou plus (voir rubrique 4.4).
Des doses uniques de 4,5 g ne doivent pas être administrées sauf si le patient a atteint cette dose après une période d’adaptation posologique.
Arrêt de OXYBATE DE SODIUM REIG JOFRE
Les effets à l’arrêt du traitement par l’oxybate de sodium n’ont pas été évalués de façon systématique lors d’essais cliniques contrôlés (voir rubrique 4.4).
Si le patient arrête de prendre le médicament pendant plus de 14 jours consécutifs, le traitement doit être réinitialisé à la posologie la plus faible.
Populations particulières
Patients âgés
L’apparition de troubles des fonctions motrices ou cognitives doit être recherchée chez les sujets âgés traités par l’oxybate de sodium (voir rubrique 4.4).
Insuffisants hépatiques
La posologie initiale doit être réduite de moitié chez tous les patients insuffisants hépatiques et les effets de chaque augmentation posologique devront être surveillés avec attention (voir rubrique 4.4 et 5.2).
Insuffisants rénaux
Tous les patients insuffisants rénaux devront suivre des recommandations afin de réduire leur consommation de sodium (voir rubrique 4.4).
Population pédiatrique
Adolescents et enfants à partir de 7 ans et avec un poids corporel minimum de 15 kg:
OXYBATE DE SODIUM REIG JOFRE est administré par voie orale deux fois par nuit. Les recommandations posologiques sont présentées dans le Tableau 1.
Tableau 1 Oxybate de sodium Dose initiale et schéma de titration recommandés pour les patients pédiatriques
|
Poids du patient |
Dose journalière totale initiale (prise en 2 doses fractionnées)* |
Schéma de titration (jusqu’à effet clinique) |
Dose journalière maximale recommandée |
|
15kg - <20kg |
≤ 1g/jour |
≤ 0.5 g/jour/semaine |
0.2g/kg/jour |
|
20kg - <30kg |
≤ 2g/jour |
≤ 1 g/jour/semaine |
|
|
30kg - <45kg |
≤ 3g/jour |
≤ 1 g/jour/semaine |
|
|
≥45kg |
≤ 4.5g/jour |
≤ 1.5 g/jour/semaine |
9g/jour |
*Au coucher et 2,5 à 4 heures plus tard. Pour les enfants qui dorment plus de 8 heures par nuit, l’oxybate de sodium peut être administré après le coucher, tandis que l’enfant est au lit, en deux doses fractionnées égales à 2,5 à 4 heures d’intervalle.
La dose doit être progressivement ajustée jusqu’à l’effet recherché en tenant compte de l’efficacité et de la tolérance (voir rubrique 4.4). Un minimum d’une à deux semaines est recommandé entre les paliers d’adaptation posologique. Les recommandations posologiques de l’oxybate de sodium pour les patients pédiatriques (dose initiale, schéma de titration et dose maximale) sont basées sur le poids. Par conséquent, les patients doivent vérifier leur poids à intervalles réguliers, en particulier au cours de la titration pour s’assurer que la dose d’oxybate de sodium appropriée est administrée.
La dose journalière maximale recommandée est de 0,2 g/kg/jour chez les patients pédiatriques pesant moins de 45 kg. Pour les patients pédiatriques pesant 45 kg ou plus, la dose journalière totale maximale est 9 g/jour.
Si l’oxybate de sodium et le valproate sont utilisés de façon concomitante (voir rubrique 4.5), une diminution de 20 % de la dose d’oxybate de sodium est recommandée, par ex. 4,8 g/jour au lieu de 6 g/jour.
La sécurité et l’efficacité de l’oxybate de sodium chez les enfants de moins de 7 ans n’ont pas encore été établies et, par conséquent, l’oxybate de sodium n’est pas recommandé en dessous de 7 ans. Les enfants de moins de 15 kg ne doivent pas recevoir d’oxybate de sodium.
Mode d’administration
OXYBATE DE SODIUM REIG JOFRE doit être absorbé par voie orale au moment du coucher puis de nouveau 2,5 à 4 heures plus tard. Il est recommandé de préparer simultanément les deux doses de OXYBATE DE SODIUM REIG JOFRE au moment du coucher. Une seringue graduée ainsi que deux godets doseurs de 90 mL et deux bouchons de sécurité enfant sont fournis avec OXYBATE DE SODIUM REIG JOFRE. Chaque dose mesurée de OXYBATE DE SODIUM REIG JOFRE doit être versée dans un godet doseur et diluée dans 60 mL d’eau avant absorption. L’alimentation réduisant significativement la biodisponibilité de l’oxybate de sodium, les patients tant adultes que pédiatriques doivent prendre leur repas au minimum plusieurs (2-3) heures avant la première prise de OXYBATE DE SODIUM REIG JOFRE. Les patients adultes et pédiatriques doivent toujours observer le même délai entre la prise du traitement et le repas. Les doses doivent être utilisées dans les 24 heures suivant leur préparation ou être jetées.
· Hypersensibilité à la substance active ou à l’un des excipients mentionnés à la rubrique 6.1.
· Patients présentant une dépression majeure.
· Patients ayant un déficit en succinate-semi-aldéhyde déshydrogénase.
· Patients traités par des opiacés ou des barbituriques.
4.4. Mises en garde spéciales et précautions d'emploi
Dépression respiratoire et du système nerveux central (SNC)
L’oxybate de sodium peut provoquer une dépression respiratoire. Avant le traitement, il convient d’évaluer le risque d’apnée du sommeil chez les patients et d’envisager le traitement avec prudence. Un épisode apnéique et une dépression respiratoire ont été observés chez un volontaire sain à jeun après une prise unique de 4,5 g (le double de la dose initiale recommandée). Dans le cadre de la surveillance après commercialisation, il a été observé que l’utilisation de l’oxybate de sodium pouvait prédisposer les patients à une sensation d’étouffement pendant le sommeil. Les patients doivent être interrogés sur les signes associés à une dépression du système nerveux central ou de l’appareil respiratoire. Une attention particulière devra être portée aux patients ayant une atteinte respiratoire sous-jacente. Pendant le traitement, les patients doivent être surveillés pour détecter d’éventuels signes de dépression respiratoire. En raison d’un risque plus élevé d’apnée du sommeil, les patients traités par l’oxybate de sodium ayant un IMC ≥ 40 kg/m2 doivent être étroitement surveillés.
Au cours des essais cliniques, environ 80 % des patients traités par l’oxybate de sodium ont continué à prendre un stimulant du SNC. L’effet sur la respiration pendant la nuit n’est cependant pas connu. Avant d’augmenter la dose d’oxybate de sodium (voir rubrique 4.2), les prescripteurs doivent être avertis que des apnées du sommeil surviennent chez 50 % des patients narcoleptiques.
· Benzodiazépines
Compte tenu de la possibilité d’augmentation du risque de dépression respiratoire, l’utilisation concomitante de benzodiazépines et d’oxybate de sodium doit être évitée.
· Alcool et dépresseurs du SNC
L’utilisation concomitante d’alcool ou de tout médicament dépresseur du SNC et d’oxybate de sodium peut entraîner une potentialisation des effets dépresseurs centraux de l’oxybate de sodium, ainsi qu’une augmentation du risque de dépression respiratoire. Par conséquent, les patients devront être mis en garde contre la prise d’alcool avec l’oxybate de sodium.
· Inhibiteurs de la Gamma hydroxybutyrate (GHB) déshydrogénase
Des précautions sont requises pour les patients traités de façon concomitante par valproate ou par d’autres inhibiteurs de la GHB déshydrogénase car des interactions pharmacocinétiques et pharmacodynamiques ont été observées quand l’oxybate de sodium est co-administré avec le valproate (voir rubrique 4.5). Si l’utilisation concomitante est justifiée, un ajustement de la posologie doit être envisagé (voir rubrique 4.2). De plus, la réponse du patient et la tolérance devront être suivies avec attention et la posologie devra être adaptée en conséquence.
· Topiramate
Coma et augmentation de la concentration plasmatique de GHB ont été observés après coadministration d'oxybate de sodium et de topiramate. Par conséquent, les patients doivent être mis en garde contre l'utilisation du topiramate avec l'oxybate de sodium (rubrique 4.5).
Risque d’abus et de dépendance
L’oxybate de sodium, qui est le sel de sodium du gamma hydroxybutyrate (GHB), est un dépresseur du système nerveux central (SNC) présentant un risque d’abus bien connu. Avant traitement, les médecins devront rechercher chez les patients des antécédents ou une prédisposition à l’abus médicamenteux. Les patients devront être régulièrement surveillés et en cas de suspicion d’abus, le traitement par l’oxybate de sodium devra être arrêté.
Des cas de dépendance après utilisation illicite de GHB à doses répétées et fréquentes (18 à 250 g/jour) supérieures aux doses thérapeutiques, ont été rapportés. Bien que l’apparition d’une dépendance chez les patients traités par l’oxybate de sodium aux doses thérapeutiques n’ait pas été clairement démontrée, cette possibilité ne peut être exclue.
Porphyrie
Il a été démontré un effet porphyrogénique de l’oxybate de sodium chez l’animal ou dans des modèles in vitro ; par conséquent, son utilisation est considérée comme dangereuse chez les patients ayant une porphyrie.
Effets neuropsychiatriques
Les patients traités par l’oxybate de sodium peuvent présenter une confusion. Dans ce cas, ces patients devront être évalués de façon approfondie et une surveillance appropriée sera effectuée individuellement. D’autres manifestations neuropsychiatriques telles qu’anxiété, psychose, paranoïa, hallucinations et agitation peuvent survenir. La survenue de troubles de la pensée y compris pensées de commettre des actes violents (y compris atteinte à autrui) et/ou d’anomalies du comportement lors d’un traitement par l’oxybate de sodium nécessite une évaluation minutieuse et immédiate.
La survenue d’une dépression lors d’un traitement par l’oxybate de sodium nécessite une évaluation minutieuse et immédiate. Chez les patients ayant des antécédents de troubles affectifs (notamment dépression, anxiété et trouble bipolaire), de tentative de suicide et de psychose, il faudra surveiller particulièrement et avec attention l’apparition de symptômes dépressifs et/ou d’idées suicidaires au cours du traitement par l’oxybate de sodium. L’utilisation de OXYBATE DE SODIUM REIG JOFRE est contre-indiquée en cas de dépression majeure (voir rubrique 4.3).
En cas d’incontinence urinaire ou fécale chez un patient traité par l’oxybate de sodium, le prescripteur devra poursuivre les investigations afin d’éliminer des étiologies sous-jacentes.
Des cas de somnambulisme ont été rapportés au cours des essais cliniques avec l’oxybate de sodium. Il n’est pas possible de déterminer si tous les épisodes ou seulement certains d’entre eux correspondent à un somnambulisme vrai (parasomnie survenant pendant le sommeil non paradoxal) ou à un autre trouble médical spécifique. Le risque de blessure ou d’automutilation doit être pris en compte chez tout patient somnambule. C’est pourquoi, les épisodes de somnambulisme doivent être minutieusement évalués et des actions appropriées envisagées.
Population pédiatrique:
Suivi pendant la phase de titration
La tolérance du patient, en particulier en ce qui concerne les signes potentiels de dépression du système nerveux central et de dépression respiratoire, doit être attentivement surveillée lors de chaque augmentation de dose au cours de la titration. Une surveillance attentive signifie que le parent/soignant observe la respiration de l’enfant après la prise d’oxybate de sodium pour évaluer toute anomalie de la respiration pendant les deux premières heures, par exemple respiration difficile, apnée du sommeil, cyanose des lèvres/du visage. Si une anomalie de la respiration est observée, une aide médicale doit être recherchée. Si une anomalie est observée après la première dose, la deuxième dose ne doit pas être administrée. Si aucune anomalie n’est observée, la deuxième dose peut être administrée. La deuxième dose ne doit pas être administrée plus tôt que 2,5 heures ou plus tard que 4 heures après la première dose. Dans chaque cas, par exemple en cas de doute sur la capacité des parents/soignants à gérer une surveillance attentive comme décrit, l’oxybate de sodium n’est pas recommandé, à moins qu’une surveillance médicale du traitement puisse être organisée.
En cas de doute à propos de l'administration d'une dose, ne pas administrer une nouvelle dose pour réduire le risque de surdosage.
Perte de poids
Une perte de poids est fréquente chez les patients traités par l’oxybate de sodium (voir rubrique 4.8).
Pour les patients pédiatriques, il est important de vérifier leur poids à intervalles réguliers, en particulier pendant la titration de la dose, pour s’assurer que la dose appropriée d’oxybate de sodium est administrée (voir rubrique 4.2).
Effets neuropsychiatriques
Pour les enfants et les adolescents, des précautions supplémentaires doivent être prises pour évaluer tout état suicidaire ou dépressif potentiel avant de commencer le traitement par l’oxybate de sodium (voir rubrique 4.8) et pour surveiller tout événement survenant lors du traitement.
L’alcool et les dépresseurs du SNC
Étant donné le risque de consommation d’alcool chez les adolescents, il convient de noter que l’alcool peut encore amplifier les effets dépresseurs respiratoires et sur le SNC de l’oxybate de sodium chez les enfants – adolescents prenant de l’oxybate de sodium (voir rubrique 4.5).
Apport en sodium
Ce médicament contient 182,24 mg de sodium par gramme de dose d’oxybate de sodium, ce qui équivaut à 9,11 % de l’apport alimentaire quotidien maximal recommandé par l’OMS de 2 g de sodium par adulte.
La dose quotidienne maximale de ce produit équivaut à 82 % de l’apport alimentaire quotidien maximal recommandé par l’OMS pour le sodium.
OXYBATE DE SODIUM REIG JOFRE est considéré comme riche en sodium. Il faut en tenir compte, en particulier chez les patients sous régime à faible teneur en sodium.
Des recommandations pour réduire l’apport sodé doivent être données aux patients ayant une insuffisance cardiaque, une hypertension artérielle ou une altération de la fonction rénale (voir rubriques 4.2 et 4.9). Patients âgés
Les données cliniques chez le sujet âgé sont très limitées. Par conséquent, les patients âgés doivent être suivis avec attention lors d’un traitement par l’oxybate de sodium pour dépister des déficiences des fonctions motrices et/ou cognitives.
Patients épileptiques
Des crises convulsives ont été observées chez des patients traités par l’oxybate de sodium. Chez les patients épileptiques, la sécurité et l’efficacité de l’oxybate de sodium n’ont pas été établies, son utilisation n’est donc pas recommandée.
Effet rebond et syndrome de sevrage
Les effets à l’arrêt du traitement par l’oxybate de sodium n’ont pas été évalués systématiquement au cours des essais cliniques contrôlés. Chez certains patients, la cataplexie peut réapparaître à une fréquence plus élevée à l’arrêt du traitement par l’oxybate de sodium ; cependant, cela peut être dû à la variabilité normale de la maladie. Bien que l’expérience acquise lors des études cliniques avec l’oxybate de sodium aux doses thérapeutiques chez des patients atteints de narcolepsie/cataplexie n’ait pas mis clairement en évidence de syndrome de sevrage, dans de rares cas, des évènements tels qu’insomnie, céphalées, anxiété, sensations vertigineuses, troubles du sommeil, somnolence, hallucinations et troubles psychotiques ont été observés à l’arrêt du GHB.
Matériel éducationnel
Afin d’aider les prescripteurs et les patients/soignants à connaitre les informations importantes sur OXYBATE DE SODIUM REIG JOFRE, du matériel éducationnel leur sera fourni. Ce matériel insistera notamment sur le fait que, pour les patients pédiatriques, une évaluation initiale du patient doit être réalisée en termes de croissance et de capacité d’apprentissage, et qu’en plus de tout effet indésirable, tout changement comportemental (social et apprentissage) doit être signalé à la personne en charge des soins de santé de l’enfant.
4.5. Interactions avec d'autres médicaments et autres formes d'interactions
L’oxybate de sodium ne doit pas être utilisé en association avec des hypnotiques sédatifs ou avec d’autres dépresseurs du SNC.
Hypnotiques sédatifs
Les études d’interactions médicamenteuses chez des adultes volontaires sains traités par oxybate de sodium (dose unique de 2,25 g) et lorazépam (dose unique de 2 mg) et tartrate de zolpidem (dose unique de 5 mg) n’ont pas montré d’interactions pharmacocinétiques. Une augmentation de la somnolence a été observée lors de l’administration concomitante d’oxybate de sodium (2,25 g) et de lorazépam (2 mg). L’interaction pharmacodynamique avec le zolpidem n’a pas été évaluée. Lors de l’association de doses plus élevées d’oxybate de sodium, jusqu’à 9 g/jour, et de doses plus élevées d’hypnotiques (dans l’intervalle de doses recommandé), des interactions pharmacodynamiques avec symptômes de dépression du système nerveux central et/ou de dépression respiratoire ne peuvent être exclues (voir rubrique 4.3).
Tramadol
Une étude d’interaction médicamenteuse chez des adultes volontaires sains traités par oxybate de sodium (dose unique de 2,25 g) et tramadol (dose unique de 100 mg) n’a pas montré d’interaction pharmacocinétique/pharmacodynamique. Lors de l’association de doses plus élevées d’oxybate de sodium, jusqu’à 9 g, et de doses plus élevées d’opiacés (dans l’intervalle de doses recommandé), des interactions pharmacodynamiques avec symptômes de dépression du système nerveux central et/ou de dépression respiratoire ne peuvent être exclues (voir rubrique 4.3).
Antidépresseurs
Les études d’interaction médicamenteuse chez des adultes volontaires sains traités par oxybate de sodium (dose unique de 2,25 g) et antidépresseurs, chlorhydrate de protriptyline (dose unique de 10 mg) et duloxétine (60 mg à l’état d’équilibre), n’ont pas montré d’interaction pharmacocinétique. Il n’a pas été observé d’effet additionnel sur la somnolence lors de la comparaison de doses uniques d’oxybate de sodium seul (2,25 g), et de doses d’oxybate de sodium (2,25 g) en association avec la duloxétine (60 mg à l’état d’équilibre). Les antidépresseurs ont été utilisés dans le traitement de la cataplexie. Un possible effet additif des antidépresseurs et de l’oxybate de sodium ne peut pas être exclu. La fréquence des effets indésirables a été augmentée lors de la co-administration d’oxybate de sodium et d’antidépresseurs tricycliques.
Modafinil
Une étude d’interaction médicamenteuse chez des adultes volontaires sains traités par oxybate de sodium (dose unique de 4,5 g) et modafinil (dose unique de 200 mg) n’a pas montré d’interaction pharmacocinétique. L’oxybate de sodium a été administré de façon concomitante avec des médicaments stimulants du SNC chez environ 80 % des patients au cours des études cliniques dans la narcolepsie. L’effet sur la respiration pendant la nuit n’est cependant pas connu.
Oméprazole
La co-administration d’oméprazole n’a pas d’effet cliniquement significatif sur la pharmacocinétique de l’oxybate de sodium. Il n’est donc pas nécessaire de modifier la dose d’oxybate de sodium lors de l’administration concomitante d’inhibiteurs de la pompe à protons.
Ibuprofène
Des études d’interaction chez le volontaire sain ont démontré l’absence d’interaction pharmacocinétique entre l’oxybate de sodium et l’ibuprofène.
Diclofénac
Des études d’interaction chez le volontaire sain ont démontré l’absence d’interaction pharmacocinétique entre l’oxybate de sodium et le diclofénac. La co-administration d’oxybate de sodium et de diclofénac chez le volontaire sain a réduit les troubles de l’attention dus à l’administration d’oxybate de sodium seul, selon les résultats des tests psychométriques.
Inhibiteurs de la GHB déshydrogénase
L’oxybate de sodium étant métabolisé par la GHB déshydrogénase, il existe un risque potentiel d’interaction avec les médicaments stimulant ou inhibant cette enzyme (ex : valproate, phénytoïne ou éthosuximide) (voir rubrique 4.4).
La co-administration d’oxybate de sodium (6 g par jour) et de valproate (1 250 mg par jour) a entraîné une augmentation de l’exposition systémique à l’oxybate de sodium d’environ 25 % et n’a pas entraîné de modification significative de la Cmax. Aucun effet sur la pharmacocinétique du valproate n’a été observé. Les effets pharmacodynamiques en résultant, y compris l’augmentation des troubles cognitifs et de la somnolence, ont été supérieurs lors de la co-administration par rapport à ceux observés lors de l’administration de chaque médicament seul. Si l’administration concomitante est justifiée, la réponse du patient et la tolérance devront être suivies et la posologie adaptée si nécessaire.
Topiramate
De possibles interactions pharmacodynamiques et pharmacocinétiques ne peuvent être exclues lors de l’utilisation concomitante d'oxybate de sodium et de topiramate, car un coma et une augmentation de la concentration plasmatique de GHB ont été signalés chez des patients utilisant de l’oxybate de sodium et du topiramate de façon concomitante (rubrique 4.4).
Des études in vitro réalisées sur microsomes hépatiques humains poolés montrent que l’oxybate de sodium n’inhibe pas significativement l’activité des isoenzymes humaines (voir rubrique 5.2).
4.6. Fertilité, grossesse et allaitement
Grossesse
Les études effectuées chez l’animal n’ont pas mis en évidence d’effet tératogène mais un effet embryoléthal a été observé au cours des études chez le rat et le lapin (voir rubrique 5.3).
Des données provenant d’un nombre limité de femmes enceintes exposées durant le premier trimestre de la grossesse indiquent la possibilité d’un risque accru d’avortement spontané. A ce jour, il n’y a pas d’autres données épidémiologiques pertinentes disponibles. Les données limitées chez les femmes enceintes pendant les deuxième et troisième trimestres n’ont pas mis en évidence d’effet malformatif, ni de toxicité fœtale/néonatale de l’oxybate de sodium.
L’oxybate de sodium n’est pas recommandé pendant la grossesse.
L'oxybate de sodium et / ou ses métabolites sont excrétés dans le lait maternel. Des modifications de la structure du sommeil ont été observées chez les nourrissons allaités de mères exposées, ce qui pourrait être compatible avec les effets de l'oxybate de sodium sur le système nerveux.
L'oxybate de sodium ne doit pas être utilisé pendant l'allaitement.
Fertilité
Il n’existe pas de données sur les effets de l’oxybate de sodium sur la fertilité. Les études chez les rats mâles et femelles à des doses de GHB allant jusqu'à 1 000 mg/kg/jour, n'ont pas mis en évidence d’effet indésirable sur la fertilité.
4.7. Effets sur l'aptitude à conduire des véhicules et à utiliser des machines
Quand les patients commencent à prendre de l’oxybate de sodium et jusqu’à ce qu’ils sachent si le médicament a encore des effets le jour suivant, ils devront être très prudents lors de la conduite d’un véhicule, la manipulation d’une machine lourde ou la réalisation d’une tâche pouvant être dangereuse ou nécessitant une vigilance complète.
Pour les patients pédiatriques, les médecins et les parents ou soignants doivent être informés que si le rapport de la dose journalière au poids corporel dépasse 0,1 g/kg/jour, le délai d’attente peut être supérieur à 6 heures en fonction de la sensibilité individuelle de chaque patient.
Résumé du profil de sécurité
Études cliniques
Le profil de sécurité était qualitativement le même dans des études chez des patients adultes et pédiatriques.
Chez l’adulte, les effets indésirables les plus fréquemment rapportés sont sensations vertigineuses, nausées et céphalées, survenant chez 10 à 20 % des patients. Les effets indésirables les plus graves sont tentative de suicide, psychose, dépression respiratoire et convulsion.
Chez l’adulte, l’efficacité et la sécurité et l’efficacité de l’oxybate de sodium dans le traitement des symptômes de la narcolepsie ont été établies au cours de quatre études multicentriques, randomisées, en double aveugle, contrôlées versus placebo en groupes parallèles chez des patients narcoleptiques ayant une cataplexie sauf pour une étude où la cataplexie n’était pas un critère d’inclusion. Deux études de phase 3 et une étude de phase 2, en double aveugle, contrôlées versus placebo en groupes parallèles ont été réalisées pour évaluer l’oxybate de sodium dans l’indication fibromyalgie chez l’adulte. En outre, des études croisées d'interaction médicamenteuse avec de l'ibuprofène, du diclofénac et du valproate, en double aveugle, contrôlées versus placebo, randomisées, ont été réalisées chez des sujets sains adultes et sont résumées dans la rubrique 4.5.
Expérience post-commercialisation
En plus des effets indésirables rapportés au cours des études cliniques, des effets indésirables ont été rapportés depuis la commercialisation. Il n'est pas toujours possible d'estimer de manière fiable la fréquence de leur incidence dans la population traitée.
Tableau des effets indésirables
Les effets indésirables sont listés selon les classes de systèmes d’organes MedDRA.
Estimation de la fréquence : très fréquent (≥ 1/10) ; fréquent (≥ 1/100, < 1/10) ; peu fréquent (≥ 1/1 000, < 1/100) ; rare (≥ 1/10 000, < 1/1 000) ; très rare (< 1/10 000) ; fréquence indéterminée (ne peut être estimée sur la base des données disponibles).
Dans chaque groupe de fréquence, les effets indésirables sont présentés par ordre décroissant de gravité.
Infections et infestations
Fréquent: rhinopharyngite, sinusite
Affections du système immunitaire
Peu fréquent: hypersensibilité
Troubles du métabolisme et de la nutrition
Fréquent: anorexie, diminution de l’appétit
Fréquence indéterminée: déshydratation, augmentation de l’appétit
Affections psychiatriques
Fréquent: dépression, cataplexie, anxiété, rêves anormaux, état confusionnel, désorientation, cauchemars, somnambulisme, troubles du sommeil, insomnie, insomnie de milieu de nuit, nervosité
Peu fréquent: tentative de suicide, psychose, paranoïa, hallucinations, troubles de la pensée, agitation, insomnie d’endormissement
Fréquence indéterminée: idées suicidaires, idée d’homicide, agression, humeur euphorique, troubles des conduites alimentaires liés au sommeil, attaque de panique, manie/trouble bipolaire, idée délirante, bruxisme, irritabilité et libido augmentée
Affections du système nerveux
Très fréquent: sensations vertigineuses, céphalées
Fréquent: paralysie du sommeil, somnolence, tremblements, troubles de l’équilibre, troubles de l’attention, hypoesthésie, paresthésie, sédation, dysgueusie
Peu fréquent: myoclonies, amnésie, syndrome des jambes sans repos
Fréquence indéterminée: convulsions, perte de connaissance, dyskinésie
Affections oculaires
Fréquent: vision trouble
Affections de l’oreille et du labyrinthe
Fréquent: vertige
Fréquence indéterminée: acouphènes
Affections cardiaques
Fréquent: palpitations
Affections vasculaires
Fréquent: hypertension
Affections respiratoires, thoraciques et médiastinales
Fréquent: dyspnée, ronflement, congestion nasale
Fréquence indéterminée: dépression respiratoire, apnée du sommeil, sensation d’étouffement
Affections gastro-intestinales
Très fréquent: nausées (la fréquence des nausées est plus importante chez la femme que chez l’homme)
Fréquent: vomissements, diarrhée, douleur abdominale haute
Peu fréquent: incontinence fécale
Fréquence indéterminée: sécheresse de la bouche
Affections de la peau et du tissu sous-cutané
Fréquent: hyperhidrose, rash
Fréquence indéterminée: urticaire, angiœdème, séborrhée
Affections musculo-squelettiques et du tissu conjonctif
Fréquent: arthralgies, spasmes musculaires, douleur dorsale
Affections du rein et des voies urinaires
Fréquent: énurésie nocturne, incontinence urinaire
Fréquence indéterminée: pollakiurie/impériosité mictionnelle, nycturie
Troubles généraux et anomalies au site d’administration
Fréquent: asthénie, fatigue, sensation d’ébriété, œdème périphérique
Investigations
Fréquent: augmentation de la pression artérielle, perte de poids
Lésions, intoxications et complications liées aux procédures
Fréquent: chute
Description de certains effets indésirables
Chez certains patients, les accès de cataplexie peuvent réapparaître à une fréquence plus élevée à l’arrêt du traitement par l’oxybate de sodium, cependant, cela peut être dû à la variabilité normale de la maladie. Bien que l’expérience acquise lors des études cliniques avec l’oxybate de sodium aux doses thérapeutiques chez des patients narcoleptiques/cataplexiques n’ait pas mis clairement en évidence de syndrome de sevrage, dans de rares cas, des effets indésirables tels qu’insomnie, céphalées, anxiété, sensations vertigineuses, troubles du sommeil, somnolence, hallucinations et troubles psychotiques ont été observés à l’arrêt du GHB.
Populations particulières
Population pédiatrique
Dans la population pédiatrique, l’efficacité et la sécurité d’emploi de l’oxybate de sodium pour le traitement de la narcolepsie avec symptômes de cataplexie ont été établies dans une étude de retrait du traitement, de phase 2/3, multicentrique, randomisée, en double aveugle, contrôlée contre placebo.
Dans une étude chez des enfants et des adolescents, les effets indésirables survenus lors du traitement les plus fréquemment rapportés étaient une énurésie (18,3 %), des nausées (12,5 %), des vomissements (8,7 %), une perte de poids (8,7 %), une diminution de l’appétit (6,7 %), des maux de tête (5,8 %) et des sensations vertigineuses (5,8 %). Des effets indésirables au médicament de type pensées suicidaires (1 %) et psychose aiguë (1 %) ont également été rapportés (voir rubrique 4.4 et rubrique 6).
Chez certains enfants entre 7 et < 18 ans, la surveillance post-commercialisation a montré que l’oxybate de sodium a été arrêté en raison d’un comportement anormal, d’agressivité et d’altération de l’humeur.
Déclaration des effets indésirables suspectés
La déclaration des effets indésirables suspectés après autorisation du médicament est importante. Elle permet une surveillance continue du rapport bénéfice/risque du médicament. Les professionnels de santé déclarent tout effet indésirable suspecté via le système national de déclaration : Agence nationale de sécurité du médicament et des produits de santé (ANSM) et réseau des Centres Régionaux de Pharmacovigilance - Site internet : www.signalement-sante.gouv.fr.
Les données sur les signes et les symptômes associés à un surdosage avec l’oxybate de sodium sont limitées. La plupart des données proviennent de l’utilisation illicite du GHB. L’oxybate de sodium est le sel sodique du GHB. Les manifestations associées au syndrome de sevrage ont été observées en dehors des doses thérapeutiques.
Symptômes
Les patients ont présenté des degrés divers d’altération de la conscience pouvant fluctuer rapidement entre un état confusionnel agité, agressif avec ataxie et un coma. Des vomissements (même avec une conscience altérée), des sueurs, des céphalées et une altération des capacités psychomotrices peuvent être observés. Une vision trouble a été rapportée. Un coma profond a été observé à fortes doses, ainsi qu’une acidose. Des myoclonies et des crises tonico-cloniques ont été rapportées. Des cas d’altération du rythme et de l’amplitude de la respiration et de dépression respiratoire mettant en jeu le pronostic vital, nécessitant intubation et ventilation, ont été rapportés. Respiration de Cheyne-Stokes et apnée ont été observées.
Bradycardie et hypothermie peuvent accompagner une perte de conscience ainsi qu’une hypotonie musculaire sans perte des réflexes ostéotendineux. La bradycardie a bien répondu à une administration intraveineuse d’atropine. Des cas d’hypernatrémie avec alcalose métabolique ont été rapportés dans le cadre d’une administration concomitante de NaCl par perfusion.
Prise en charge
Un lavage gastrique peut être envisagé si l’ingestion d’autres produits est suspectée. En raison de la possibilité de vomissements en présence d’une altération de la conscience, la mise en position de sécurité (en décubitus latéral gauche) et une protection des voies aériennes par intubation peuvent être justifiées. Malgré la possibilité d’absence de réflexe pharyngé chez les patients en coma profond, les patients inconscients peuvent devenir agressifs au moment de l’intubation, une séquence d’induction rapide (sans l’usage de sédatifs) doit être envisagée.
Aucune régression des effets dépresseurs centraux de l’oxybate de sodium ne peut être attendue avec l’administration de flumazénil. Les données sont insuffisantes pour recommander l’utilisation de naloxone dans le traitement d’un surdosage avec le GHB. L’utilisation de l’hémodialyse ou de toute autre forme d’épuration extracorporelle n’a pas été étudiée en cas de surdosage par l’oxybate de sodium, mais a été rapportée dans des cas d’acidose induite par un surdosage avec le GHB. Cependant, en raison du métabolisme rapide de l’oxybate de sodium, ces mesures ne sont pas justifiées.
5. PROPRIETES PHARMACOLOGIQUES
5.1. Propriétés pharmacodynamiques
Classe pharmacothérapeutique : autres médicaments du système nerveux, code ATC : N07XX04.
Mécanisme d'action
L’oxybate de sodium est un dépresseur du système nerveux central qui réduit la somnolence diurne excessive et la cataplexie chez les patients narcoleptiques et modifie l’architecture du sommeil en réduisant le sommeil de nuit fragmenté. Le mécanisme d’action précis de l’oxybate de sodium n’est pas connu, cependant l’oxybate de sodium agirait en favorisant le sommeil à ondes lentes (delta) et en consolidant la durée du sommeil nocturne. L’oxybate de sodium administré avant le sommeil nocturne augmente la durée du sommeil des stades 3 et 4 ainsi que la latence du sommeil tandis qu’il réduit la fréquence des épisodes d’endormissement en sommeil paradoxal (SOREMPs). D’autres mécanismes restant à élucider pourraient également être impliqués. Selon les données des essais cliniques, plus de 80 % des patients ont maintenu l’utilisation d’un stimulant en association.
Adultes
L’efficacité de l’oxybate de sodium dans le traitement des symptômes de la narcolepsie a été établie au cours de quatre essais multicentriques, randomisées, en double aveugle, contrôlées versus placebo en groupes parallèles (essais 1, 2, 3 et 4) chez des patients narcoleptiques ayant une cataplexie sauf pour l’essai 2 où la cataplexie n’était pas un critère d’inclusion. L’association d’un stimulant était autorisée dans toutes les études (sauf pour la phase de traitement actif de l’essai 2) ; les antidépresseurs étaient supprimés avant le traitement actif dans tous les essais à l’exception de l’essai 2. Dans chaque étude, la dose quotidienne était divisée en 2 doses égales. Chaque nuit, la première dose était prise au moment du coucher et la deuxième dose 2,5 à 4 heures plus tard.
Tableau 2 Résumé des essais cliniques réalisés avec l’oxybate de sodium pour le traitement de la narcolepsie
|
Essai |
Efficacité primaire |
N |
Efficacité secondaire |
Durée |
Traitement actif et dose (g/j) |
|
Essai 1 |
EDS (ESS) ; CGIc |
246 |
MWT/architecture du sommeil/cataplexie/sommes/FOSQ |
8 semaines |
Oxybate de sodium 4,5-9 |
|
Essai 2 |
EDS (MWT) |
231 |
Architecture du sommeil/ESS/CGIc/sommes |
8 semaines |
Oxybate de sodium 6-9 Modafinil 200-600 mg |
|
Essai 3 |
cataplexie |
136 |
EDS (ESS)/CGIc/sommes |
4 semaines |
Oxybate de sodium 3-9 |
|
Essai 4 |
cataplexie |
55 |
aucun |
4 semaines |
Oxybate de sodium 3-9 |
EDS : somnolence diurne excessive ; ESS : Echelle de sommeil d’Epworth ; MWT : test du maintien d’éveil ; sommes : nombre de sommes inattendus pendant la journée ; CGIc : impression clinique globale de changement ; FOSQ : résultats fonctionnels du questionnaire de sommeil.
L’essai 1 a inclus 246 patients narcoleptiques avec une période d’une semaine de titration. Les mesures primaires de l’efficacité étaient des modifications de la somnolence diurne excessive mesurées par l’échelle de sommeil d’Epworth (ESS) et la modification de la sévérité globale des symptômes de narcolepsie du patient évaluée par l’investigateur utilisant l’impression clinique globale de changement (CGIc).
Tableau 3 Résumé de l’ESS dans l’Essai 1
|
Echelle de sommeil d’Epworth (ESS ; de 0 à 24) |
||||
|
Groupe de dose [g/j (n)] |
Valeur de référence |
Endpoint |
Changement médian par rapport à la valeur de référence |
Changement par rapport à la valeur de référence comparativement au placebo (p-valeur) |
|
Placebo (60) |
17,3 |
16,7 |
-0,5 |
- |
|
4,5 (68) |
17,5 |
15,7 |
-1,0 |
0,119 |
|
6 (63) |
17,9 |
15,3 |
-2,0 |
0,001 |
|
9 (55) |
17,9 |
13,1 |
-2,0 |
< 0,001 |
Tableau 4 Résumé de la CGIc de l’Essai 1
|
Impression clinique globale de changement (CGIc) |
||
|
Groupe de dose ([g/j (n)] |
Répondeurs* N (%) |
Changement par rapport à la valeur de référence comparativement au placebo (p-valeur) |
|
Placebo (60) |
13 (21,7) |
- |
|
4,5 (68) |
32 (47,1) |
0,002 |
|
6 (63) |
30 (47,6) |
< 0,001 |
|
9 (55) |
30 (54,4) |
< 0,001 |
*Les données de la CGIc ont été analysées en définissant les répondeurs comme étant les patients très améliorés ou bien améliorés.
L’essai 2 a comparé les effets de l’administration orale de l’oxybate de sodium, du modafinil et de l’association oxybate de sodium + modafinil au placebo dans le traitement de la somnolence diurne de la narcolepsie. Durant les 8 semaines de double aveugle, les patients prenaient du modafinil à une dose déterminée ou l’équivalent placebo. La dose d’oxybate de sodium ou de placebo était de 6 g/jour pendant les 4 premières semaines et était augmentée à 9 g/jour pendant les 4 semaines restantes. Le critère primaire d’efficacité était la somnolence diurne excessive mesurée par une réponse objective au test du maintien d’éveil (MWT).
Tableau 5 Résumé du MWT de l’Essai 2
|
ESSAI 2 |
||||
|
Groupe de dose |
Valeur de référence |
Endpoint |
Changement moyen par rapport à la valeur de référence |
Endpoint comparé au placebo |
|
Placebo (56) |
9,9 |
6,9 |
-2,7 |
- |
|
Oxybate de sodium (55) |
11,5 |
11,3 |
0,16 |
< 0,001 |
|
Modafinil (63) |
10,5 |
9,8 |
-0,6 |
0,004 |
|
Oxybate de sodium + modafinil (57) |
10,4 |
12,7 |
2,3 |
< 0,001 |
L’essai 3 a inclus 136 patients narcoleptiques présentant une cataplexie modérée à sévère (médiane de 21 attaques de cataplexie par semaine) à la valeur de référence. Le critère d’efficacité primaire dans cet essai était la fréquence des attaques de cataplexie.
Tableau 6 Résumé des résultats de l’Essai 3
|
Dose |
Nombre de sujets |
Attaques de cataplexie |
||
|
Essai 3 |
Valeur de référence |
Changement médian par rapport à la valeur de référence |
Changement par rapport à la valeur de référence comparé au placebo (p-valeur) |
|
|
|
Valeur médiane des attaques/semaine |
|||
|
Placebo |
33 |
20,5 |
-4 |
- |
|
3,0 g/jour |
33 |
20,0 |
-7 |
0,5235 |
|
6,0 g/jour |
31 |
23,0 |
-10 |
0,0529 |
|
9,0 g/jour |
33 |
23,5 |
-16 |
0,0008 |
L’essai 4 a inclus 55 patients narcoleptiques qui ont pris de l’oxybate de sodium en ouvert pendant 7 à 44 mois. Les patients étaient randomisés pour poursuivre le traitement par l’oxybate de sodium à dose stable ou par du placebo. L’essai 4 était destiné à évaluer spécifiquement l’efficacité en continu de l’oxybate de sodium après une longue utilisation. Le critère primaire d’efficacité dans cet essai était la fréquence des attaques de cataplexie.
Tableau 7 Résumé des résultats de l’Essai 4
|
Groupe |
Nombre de sujets |
Attaques de cataplexie |
||
|
Essai 4 |
Valeur de référence |
Changement médian par rapport à la valeur de référence |
Changement par rapport à la valeur de référence comparé au placebo (p-valeur) |
|
|
|
Valeur médiane des attaques/2 semaines |
|||
|
Placebo |
29 |
4,0 |
21,0 |
- |
|
Oxybate de sodium |
26 |
1,9 |
0 |
p < 0,001 |
Dans l’essai 4, la réponse était similaire en nombre pour les patients traités aux doses de 6 à 9 g/jour, mais aucun effet n’a été mis en évidence chez les patients traités à des doses inférieures à 6 g/jour.
Population pédiatrique
L’efficacité du traitement par l’oxybate de sodium chez les patients pédiatriques atteints de narcolepsie avec cataplexie a été établie dans un essai de retrait du traitement en double aveugle, contrôlé contre placebo, randomisé, multicentrique.
Cette étude a démontré l’efficacité clinique de l’oxybate de sodium dans le traitement de la cataplexie et de la somnolence diurne excessive (SDE) dans le cadre de la narcolepsie chez les sujets pédiatriques.
63 patients ont été randomisés dans la population d'efficacité où le critère d’évaluation primaire de l’efficacité dans cet essai était le changement du nombre de crises de cataplexie par semaine entre les deux dernières semaines de la période à dose stable et de la période en double aveugle.
Pendant la période en double aveugle, la variation médiane (Q1, Q3) par rapport à la valeur de référence (c.-à-d. les 2 dernières semaines de la période à dose stable) du nombre d’attaques de cataplexie hebdomadaires était 12,71 (3,44 ; 19,77) pour les patients randomisés sous placebo et de 0,27 (-1,00 , 2,50) pour les patients randomisés sous oxybate de sodium.
Tableau 8 Résumé des résultats dans l’étude 13-005 chez les enfants / adolescents
|
Groupe de traitement |
Nombre de patients |
Nombre de crises de cataplexie hebdomadaires (médiane) |
||
|
|
|
Valeur de Référence (c.-àd. 2 dernières semaines de la période à dose stable) |
Période en double aveugle |
Variation par rapport à la valeur de référence |
|
Placebo |
32 |
4.67 |
21.25 |
12.71 |
|
Oxybate de sodium |
31 |
3.50 |
3.77 |
0.27 |
|
Valeur p |
< 0.0001 |
|||
Lors des analyses en sous-groupes par groupe d’âge (7 - 11 ans et 12 - 17 ans) menées pour le critère d’évaluation principal, des résultats similaires ont été observés. Pendant la période de traitement en double aveugle, chez les sujets âgés de 7 à 11 ans, la variation médiane (Q1, Q3) par rapport à la valeur de référence du nombre de crises de cataplexie hebdomadaires était de 18,32 (7,58 ; 35,75) pour les sujets randomisés sous placebo et de 0,13 (-1,15 , 2,05) pour les sujets randomisés sous oxybate de sodium (p < 0,0001). Pendant la période de traitement en double aveugle, chez les sujets âgés de 12 à 17 ans, la variation médiane (Q1, Q3) par rapport à la valeur de référence du nombre de crises de cataplexie hebdomadaires était de 9,39 (1,08 ; 16,12) pour les sujets randomisés sous placebo et de 0,58 (-0,88 ; 2,58) pour les sujets randomisés sous oxybate de sodium (p = 0,0044).
Pendant la période de traitement en double aveugle, la variation médiane (Q1, Q3) du critère d’évaluation secondaire (changement dans les scores ESS) par rapport à la valeur de référence(Visite 3 – fin de la Période à dose stable) sur le score de l’échelle de Sommeil d’Epworth pour les enfants et les adolescents (ESS-CHAD) était de 3,0 (1,0 ; 5,0) pour les sujets randomisés sous placebo et de 0,0 (-1,0 ; 2,0) pour les sujets randomisés sous oxybate de sodium. La comparaison de la variation de classement entre les traitements par rapport à la valeur de référence était statistiquement significative (p = 0,0004) lors d’une analyse par modélisation ANCOVA avec le traitement comme facteur et la valeur de référence du classement comme covariable. Les sujets randomisés sous placebo ont présenté, en moyenne, des scores ESS (CHAD) plus élevés pour la valeur de référence, par rapport à ceux sous oxybate de sodium.
Tableau 9 Résumé du score ESS (CHAD) pendant la période de traitement en double aveugle (population d’efficacité)
|
Groupe de traitement |
Nombre de patients |
Changement du score ESS (CHAD) (médiane) |
||
|
|
|
Valeur de Référence (Visite 3 - Fin de la Période à dose stable) |
Fin de la période de traitement en double aveugle (Visite 4) |
Variation par rapport à la valeur de référence |
|
Placebo |
32 |
11,0 |
12,0 |
3,0 |
|
Oxybate de sodium |
31 |
8,0 |
9,0 |
0,0 |
|
Valeur p |
0,0004 |
|||
Abréviations : ESS (CHAD) = Échelle du Sommeil d'Epworth pour les enfants et les adolescents
(Children and Adolescent)
5.2. Propriétés pharmacocinétiques
Absorption
L’oxybate de sodium est absorbé rapidement après administration orale avec une biodisponibilité absolue d’environ 88 %. Les pics moyens de concentrations plasmatiques (1er et 2ème pics) ont été respectivement de 78 et 142 µg/ mL, après administration d’une dose quotidienne de 9 g répartie en deux prises équivalentes à 4 heures d’intervalle. Dans 8 études pharmacocinétiques, le temps moyen d’atteinte du pic plasmatique (Tmax) a varié de de 0,5 à 2 heures. Après administration orale, les taux plasmatiques en oxybate de sodium augmentent plus que proportionnellement avec la dose. Des doses uniques supérieures à 4,5 g n’ont pas été étudiées. L’administration d’oxybate de sodium immédiatement après un repas riche en graisses entraîne un retard d’absorption (augmentation du Tmax moyen de 0,75 h à 2,0 h) ainsi qu’une réduction du pic plasmatique (Cmax) de 58 % en moyenne et de l’exposition systémique (AUC) de 37 %.
Distribution
L’oxybate de sodium est un composé hydrophile avec un volume apparent de distribution moyen de 190-384 mL/kg. Pour des concentrations en oxybate de sodium allant de 3 à 300 µg/mL, moins de 1 % est lié aux protéines plasmatiques.
Biotransformation
Les études chez l’animal montrent que le métabolisme est la principale voie d’élimination de l’oxybate de sodium ; avec production de dioxyde de carbone et d’eau par le cycle de l’acide tricarboxylique (Krebs) et secondairement par β-oxydation. La voie principale fait intervenir une enzyme cytosolique liée au NADP+, la GHB déshydrogénase catalysant la transformation de l’oxybate de sodium en semialdéhyde succinique qui est métabolisé ensuite en acide succinique par la déshydrogénase semialdéhyde succinique. L’acide succinique entre dans le cycle de Krebs où il est métabolisé en dioxyde de carbone et en eau. Une seconde enzyme oxydoréductase mitochondriale, une transhydrogénase, catalyse également la transformation de l’oxybate de sodium en semi-aldéhyde succinique en présence d’α-cétoglutarate. Une autre voie fait intervenir une β-oxydation et le 3,4-dihydroxybutyrate pour conduire à la formation d’acétyl-CoA ; l’acétyl-Co-A entre dans le cycle de l’acide citrique pour conduire à la formation de gaz carbonique et d’eau. Aucun métabolite actif n’a été identifié.
Des études in vitro sur des microsomes hépatiques humains poolés ont montré que l’oxybate de sodium n’inhibe pas de façon significative l’activité des isoenzymes humaines CYP1A2, CYP2C9, CYP2C19, CYP2D6, CYP2E1 ou CYP3A jusqu’à une concentration de 3 mM (378 µg/mL). Ces taux sont bien supérieurs aux taux atteints avec des doses thérapeutiques.
Élimination
L’élimination de l’oxybate de sodium se fait presque entièrement par biotransformation en dioxyde de carbone, éliminé ensuite par expiration. En moyenne, moins de 5 % sont éliminés sous forme inchangée dans l’urine 6 à 8 heures après administration. L’excrétion fécale est négligeable.
Populations particulières
Patients âgés
La pharmacocinétique de l’oxybate de sodium étudiée chez un nombre limité de patients âgés de plus de 65 ans n’a pas été différente de celle observée chez des patients âgés de moins de 65 ans.
Population pédiatrique
Les principales caractéristiques pharmacocinétiques de l’oxybate de sodium chez des sujets pédiatriques sont les mêmes que celles rapportées dans les études pharmacocinétiques du traitement par oxybate de sodium chez les adultes.
Des sujets pédiatriques et adultes recevant la même dose en mg/kg ont des profils similaires de concentration plasmatique-temps (voir rubrique 4.2).Insuffisance rénale
Le rein n’intervenant pas de façon significative dans l’excrétion de l’oxybate de sodium, aucune étude pharmacocinétique n’a été menée chez des patients présentant une atteinte rénale ; aucun effet de la fonction rénale sur la pharmacocinétique de l’oxybate de sodium n’est attendu.
Insuffisance hépatique
L’oxybate de sodium subit un métabolisme pré-systémique significatif (premier passage hépatique). Après administration d’une dose orale unique de 25 mg/kg, l’AUC est multipliée par deux chez le patient cirrhotique, la clairance orale apparente passe de 9,1 chez le volontaire sain à 4,5 ml/min/kg chez le patients de classe A (sans ascite) et à 4,1 ml/min/kg chez le patient de classe C (avec ascite). La demi-vie d’élimination est significativement augmentée chez les patients de classe C et de classe A comparativement aux sujets témoins (t1/2 moyenne de 59 et 32 versus 22 minutes). Chez les patients présentant une dysfonction hépatique, la dose initiale d’oxybate de sodium sera réduite de moitié et les réponses aux augmentations de dose seront suivies avec attention (voir rubrique 4.2).
Ethnie
Les effets du métabolisme de l’oxybate de sodium dans les différents groupes ethniques n’ont pas été évalués.
5.3. Données de sécurité préclinique
La gamma butyrolactone, un des précurseurs du GHB testé chez le rat à des doses équivalentes à celles attendues chez l’homme (1,21-1,64 fois), a été considérée (classification NTP) comme noncarcinogène chez le rat et possiblement carcinogène chez la souris, en raison d’une légère augmentation du nombre des phéochromocytomes difficilement interprétable du fait d’une mortalité importante dans le groupe exposé aux fortes doses. Dans une étude de carcinogénicité chez le rat aucune tumeur attribuée à l’oxybate de sodium n’a été mise en évidence.
Le GHB n’a pas eu d’effet sur l’accouplement, la fertilité et sur le spermogramme ; il n’a pas provoqué de toxicité embryofoetale chez des rates exposées à des doses allant jusqu’à 1 000 mg/kg/jour de GHB (1,64 fois l’exposition humaine calculée chez des rates non gestantes). La mortalité périnatale a été augmentée et le poids moyen des petits exposés à la plus forte dose durant la lactation a été diminué. La relation entre ces effets sur le développement et une toxicité chez la mère n’a pas pu être établie. Chez le lapin, une légère fœtotoxicité a été observée.
Dans une étude de toxicité à dose répétée de 10 semaines menée chez de jeunes rats traités des jours 21 à 90 après la naissance, l’oxybate de sodium a produit des effets indésirables, y compris des mortalités pendant la première semaine de traitement, lorsque les animaux étaient âgés de 21 à 27 jours, ce qui correspond à un âge approximatif de 3 à 4 ans chez les enfants. La toxicité aiguë est apparue lors d’expositions inférieures à celles attendues chez les patients pédiatriques et la mortalité était précédée de signes cliniques (bradypnée, respiration profonde, diminution de l’activité, perte de coordination de la démarche, altération du réflexe de redressement), liés à l’oxybate de sodium et en ligne avec ses propriétés pharmacologiques. La raison de cette toxicité relativement plus forte pendant la première semaine de traitement n’est pas entièrement établie. Elle pourrait être liée au fait que les jeunes animaux semblent présenter une exposition systémique plus élevée que les rats juvéniles plus âgés. Elle pourrait également être due à une sensibilité des nouveau-nés à l’oxybate de sodium plus élevée par rapport à des rats juvéniles plus âgés et des rats adultes et/ou à un phénomène de développement de tolérance. Une diminution du poids et de la consommation de nourriture similaire aux adultes a également été observée, avec des signes respiratoires supplémentaires (respiration profonde et ralentie). L’oxybate de sodium n’a pas produit d’effets indésirables sur la croissance et le développement jusqu’à des niveaux d’exposition 2 à 4 fois plus élevés que l’exposition prévue à la dose maximale recommandée chez des sujets pédiatriques (200 mg/kg/jour chez les patients pédiatriques ayant un poids corporel inférieur à 45 kg ou 9 g/jour pour les patients pédiatriques dont le poids corporel est ≥45 kg).
Des études ont montré que le GHB induit une sensation qui ressemble à celle produite par l’alcool, la morphine et certains médicaments GABA-mimétiques. Les études d’auto-administration chez le rat, la souris et le singe ont donné des résultats contradictoires, alors que la tolérance au GHB ainsi que la tolérance croisée à l’alcool et au baclofène ont été clairement établies chez les rongeurs.
· Acide malique (pour ajustement du pH)
· Hydroxyde de sodium (pour ajustement du pH)
Ce médicament ne doit pas être mélangé avec d’autres médicaments.
5 ans
Après la première ouverture : 90 jours.
Après dilution dans les godets doseurs, la préparation doit être utilisée dans les 24 heures.
6.4. Précautions particulières de conservation
Ce médicament ne nécessite pas de précautions particulières de conservation.
Pour les conditions de conservation du médicament après première ouverture, voir la rubrique 6.3.
Pour les conditions de conservation après dilution du médicament, voir rubrique 6.3.
6.5. Nature et contenu de l'emballage extérieur
180 mL de solution dans un flacon ovale ambré en PET de 200 mL et fermé avec un bouchon de sécurité enfant en polyéthylène.
Chaque boîte contient une seringue graduée en polypropylène et en polyéthylène, un adaptateur en polyéthylène et deux godets doseurs en polypropylène et deux bouchons sécurité enfant à vis en polyéthylène.
6.6. Précautions particulières d’élimination et de manipulation
Pas d’exigence particulière.
7. TITULAIRE DE L’AUTORISATION DE MISE SUR LE MARCHE
GRAN CAPITAN 10
08970 SANT JOAN DESPÍ (BARCELONA)
ESPAGNE
8. NUMERO(S) D’AUTORISATION DE MISE SUR LE MARCHE
9. DATE DE PREMIERE AUTORISATION/DE RENOUVELLEMENT DE L’AUTORISATION
[à compléter ultérieurement par le titulaire]
10. DATE DE MISE A JOUR DU TEXTE
[à compléter ultérieurement par le titulaire]
Sans objet.
12. INSTRUCTIONS POUR LA PREPARATION DES RADIOPHARMACEUTIQUES
Stupéfiant : prescription limitée à 28 jours. Prescription sur ordonnance répondant aux spécifications fixées par l’arrêté du 31 mars 1999
Médicament soumis à prescription initiale annuelle réservée aux spécialistes en neurologie et aux médecins exerçant dans les centres du sommeil. Renouvellement non restreint.
Respecter les doses prescrites
Prescription en toutes lettres sur ordonnance sécurisée
Prescription limitée à 4 semaines
ANSM - Mis à jour le : 23/05/2022
OXYBATE DE SODIUM REIG JOFRE 500 mg/mL, solution buvable
oxybate de sodium
· Gardez cette notice. Vous pourriez avoir besoin de la relire.
· Si vous avez d’autres questions, interrogez votre médecin ou votre pharmacien.
· Ce médicament vous a été personnellement prescrit. Ne le donnez pas à d’autres personnes. Il pourrait leur être nocif, même si les signes de leur maladie sont identiques aux vôtres.
· Si vous ressentez un quelconque effet indésirable, parlez-en à votre médecin, ou votre pharmacien. Ceci s’applique aussi à tout effet indésirable qui ne serait pas mentionné dans cette notice. Voir rubrique 4.
1. Qu'est-ce que OXYBATE DE SODIUM REIG JOFRE 500 mg/mL, solution buvable et dans quels cas est-il utilisé ?
2. Quelles sont les informations à connaître avant de prendre OXYBATE DE SODIUM REIG JOFRE 500 mg/mL, solution buvable ?
3. Comment prendre OXYBATE DE SODIUM REIG JOFRE 500 mg/mL, solution buvable ?
4. Quels sont les effets indésirables éventuels ?
5. Comment conserver OXYBATE DE SODIUM REIG JOFRE 500 mg/mL, solution buvable ?
6. Contenu de l’emballage et autres informations.
1. QU’EST-CE QUE OXYBATE DE SODIUM REIG JOFRE 500 mg/mL, solution buvable ET DANS QUELS CAS EST-IL UTILISE ?
OXYBATE DE SODIUM REIG JOFRE est un médicament utilisé dans le traitement de la narcolepsie chez les adultes, les adolescents et les enfants à partir de 7 ans, présentant une cataplexie.
La narcolepsie se caractérise par des troubles du sommeil pouvant comporter une somnolence diurne, ainsi qu’une cataplexie, des paralysies du sommeil, des hallucinations et un sommeil de mauvaise qualité. La cataplexie se caractérise par une brutale faiblesse musculaire ou une paralysie sans perte de conscience déclenchée par une réaction émotionnelle soudaine telle que colère, peur, joie, éclats de rire ou surprise.
2. QUELLES SONT LES INFORMATIONS A CONNAITRE AVANT DE PRENDRE OXYBATE DE SODIUM REIG JOFRE 500 mg/mL, solution buvable ?
Ne prenez jamais OXYBATE DE SODIUM REIG JOFRE
· si vous êtes allergique à l’oxybate de sodium ou à l’un des autres composants contenus dans ce médicament, mentionnés dans la rubrique 6.
· Si vous avez un déficit en succinate-semi-aldéhyde déshydrogénase (maladie métabolique rare).
· Si vous souffrez de dépression grave.
· Si vous êtes traité par des médicaments opiacés ou des barbituriques.
Avertissements et précautions
Adressez-vous à votre médecin ou votre pharmacien avant de prendre OXYBATE DE SODIUM REIG JOFRE.
· si vous avez des problèmes respiratoires ou pulmonaires (particulièrement si vous êtes obèse), car OXYBATE DE SODIUM REIG JOFRE peut causer des difficultés pour respirer;
· si vous souffrez ou avez souffert d’une maladie dépressive, de pensées suicidaires, d’anxiété, de psychose (un trouble mental qui peut impliquer des hallucinations, une élocution incohérente, ou un comportement désorganisé et agité) ou d’un trouble bipolaire;
· si vous avez une insuffisance cardiaque, de l’hypertension (pression sanguine élevée), des problèmes hépatiques ou rénaux, votre dose devant peut-être être ajustée;
· si vous avez déjà abusé de médicaments;
· si vous êtes épileptique, l’utilisation de OXYBATE DE SODIUM REIG JOFRE n’étant pas recommandée dans ce cas;
· si vous souffrez de porphyrie (maladie métabolique peu fréquente).
Si vous êtes concerné par l’une de ces situations, veuillez informer votre médecin avant de prendre OXYBATE DE SODIUM REIG JOFRE.
Pendant le traitement par OXYBATE DE SODIUM REIG JOFRE, si vous souffrez d’incontinence (urinaire ou fécale), de confusion, d’hallucinations, d’épisodes de somnambulisme ou de troubles de la pensée, vous devez informer votre médecin immédiatement. Bien que ces effets soient peu fréquents, s’ils surviennent, ils sont habituellement légers à modérés.
Si vous êtes âgé, votre médecin vous surveillera avec attention, afin de contrôler les effets de ce médicament.
OXYBATE DE SODIUM REIG JOFRE a un risque d’abus bien connu. Des cas de dépendance sont apparus après utilisation illicite de l’oxybate de sodium.
Votre médecin vous demandera si vous avez un jour abusé de médicaments avant que vous ne commenciez à prendre ce médicament et pendant votre traitement.
Enfants et adolescents
OXYBATE DE SODIUM REIG JOFRE peut être pris par les adolescents et les enfants à partir de 7 ans quand ils ont un poids supérieur à 15 kg.
OXYBATE DE SODIUM REIG JOFRE ne peut pas être pris par les enfants de moins de 7 ans ou d’un poids inférieur à 15 kg.
Si vous êtes un enfant ou un adolescent, votre médecin surveillera votre poids régulièrement.
Pendant que le médecin ajuste la dose, ce qui peut prendre un certain nombre de semaines, les parents/soignants doivent surveiller étroitement la respiration de l’enfant pendant les 2 premières heures après la prise d’oxybate de sodium pour repérer toute anomalie de la respiration, par exemple un arrêt de la respiration pendant de courtes périodes durant le sommeil, une respiration bruyante et une coloration bleuâtre des lèvres et du visage. Si une anomalie de la respiration est observée, une aide médicale doit être recherchée, et le médecin doit être informé le plus vite possible. Si une anomalie est observée après la première dose, la deuxième dose ne doit pas être administrée. Si aucune anomalie n’est observée, la deuxième dose peut être administrée. La deuxième dose ne doit pas être administrée plus tôt que 2,5 heures ou plus tard que 4 heures après la première dose.
Si vous avez ressenti ou si vous ressentez un sentiment d’abattement, en particulier si vous vous sentez très triste ou que vous avez perdu l’intérêt à la vie, il est important que vous informiez le médecin ou le soignant. Autres médicaments et OXYBATE DE SODIUM REIG JOFRE
Informez votre médecin ou votre pharmacien si vous prenez, avez récemment pris ou pourriez prendre tout autre médicament.
En particulier, OXYBATE DE SODIUM REIG JOFRE ne doit pas être pris en même temps que des médicaments contre l’insomnie ou des médicaments réduisant l’activité du système nerveux central (le système nerveux central est composé du cerveau et la moelle épinière).
Vous devez également prévenir votre médecin ou votre pharmacien si vous prenez les catégories de médicaments suivantes :
· médicaments qui augmentent l’activité du système nerveux central
· antidépresseurs
· médicaments qui sont transformés par des voies similaires par l’organisme (par ex. valproate, phénytoïne ou éthosuximide qui sont utilisés pour le traitement des crises d’épilepsie).
· topiramate (utilisé pour le traitement de l’épilepsie)
· si vous prenez du valproate, votre dose quotidienne d’OXYBATE DE SODIUM REIG JOFRE 500 mg/mL, solution buvable devra être ajustée car ceci pourrait conduire à des interactions.
OXYBATE DE SODIUM REIG JOFRE avec des aliments, boissons et de l’alcool
Vous ne devez pas boire d’alcool pendant votre traitement par ce médicament, car les effets de ce médicament peuvent être augmentés.
Si vous êtes enceinte ou que vous allaitez, si vous pensez être enceinte ou planifiez une grossesse, demandez conseil à votre médecin avant de prendre ce médicament.
Très peu de femmes ont pris Oxybate de sodium pendant leur grossesse et quelques-unes d’entre elles ont eu un avortement spontané. Le risque associé à la prise de OXYBATE DE SODIUM REIG JOFRE pendant la grossesse n’est pas connu et l’utilisation de ce médicament chez la femme enceinte ou essayant de l’être n’est donc pas recommandé.
Les patientes prenant OXYBATE DE SODIUM REIG JOFRE ne doivent pas allaiter car OXYBATE DE SODIUM REIG JOFRE passe dans le lait maternel. Des modifications de la structure du sommeil ont été observées chez les nourrissons allaités de mères exposées.
Conduite de véhicules et utilisation de machines
La prise de OXYBATE DE SODIUM REIG JOFRE a des conséquences sur la conduite de véhicules et l’utilisation de machines. Ne conduisez pas, n’utilisez pas de machines ou d’outils, ne démarrez pas une activité dangereuse ou nécessitant une vigilance particulière pendant au moins 6 heures après la prise de OXYBATE DE SODIUM REIG JOFRE. Lorsque vous prenez OXYBATE DE SODIUM REIG JOFRE pour la première fois, jusqu’à ce que vous sachiez si ce médicament vous fait dormir le lendemain, soyez très prudent quand vous conduirez un véhicule, utiliserez une machine ou ferez toute activité potentiellement dangereuse ou nécessitant une vigilance particulière.
Pour les patients pédiatriques, les médecins, les parents ou soignants doivent être informés que le temps d'attente pour l’accomplissement des activités nécessitant de la vigilance, de la coordination motrice ou toutes les activités qui peuvent entrainer un risque physique pourrait devoir être de plus de 6 heures, en fonction de la sensibilité individuelle de chaque patient.
OXYBATE DE SODIUM REIG JOFRE contient du sodium
Ce médicament contient 182,24 mg de sodium (composant principal du sel de cuisine/table) par gramme. Cela équivaut à 9,11 % de l’apport alimentaire quotidien maximal recommandé de sodium pour un adulte. Parlez-en à votre médecin ou votre pharmacien si vous avez besoin de 2 g ou plus d’oxybate de sodium quotidiennement pendant une période prolongée, surtout si vous devez suivre un régime à faible teneur en sel (sodium).
3. COMMENT PRENDRE OXYBATE DE SODIUM REIG JOFRE 500 mg/mL, solution buvable ?
Il est important que vous n’utilisiez que la seringue fournie dans la boite pour la préparation des doses de OXYBATE DE SODIUM REIG JOFRE.
Adultes – prise de OXYBATE DE SODIUM REIG JOFRE seul
· Pour les adultes, la dose initiale recommandée est de 4,5 g par jour, à répartir en 2 prises de 2,25 g.
· Votre médecin peut augmenter progressivement votre dose jusqu’à une dose maximale de 9 g par jour à répartir en deux prises de 4,5 g.
· Prenez OXYBATE DE SODIUM REIG JOFRE par voie orale, 2 fois chaque nuit :
o La première dose doit être prise juste avant le coucher et la deuxième 2h30 à 4 heures plus tard. Vous pouvez utiliser un réveil pour être sûr de vous réveiller pour prendre la seconde dose.
o La nourriture diminue la quantité de OXYBATE DE SODIUM REIG JOFRE absorbée par votre organisme. C’est pourquoi, il est préférable de prendre OXYBATE DE SODIUM REIG JOFRE à un horaire fixe 2 à 3 heures après un repas.
o Préparez les deux doses avant de vous coucher.
o Prenez les doses dans les 24 heures suivant leur préparation.
Adolescents et enfants âgés de 7 ans et plus, qui pèsent 15 kg ou plus - prise de OXYBATE DE SODIUM REIG JOFRE seul
Pour les patients âgés de 7 ans et plus, qui pèsent 15 kg ou plus, un médecin déterminera la bonne dose selon votre poids.
Votre médecin déterminera la dose adéquate pour vous. Ne pas dépasser la dose qui vous est prescrite.
Problèmes rénaux ou hépatiques
· Si vous avez des problèmes rénaux, vous devrez envisager des mesures diététiques afin de réduire votre consommation de sodium (sel).
· Si vous avez des problèmes hépatiques, la dose initiale doit être réduite de moitié. Votre médecin pourra augmenter progressivement votre dose.
Instructions pour la préparation de la dilution de OXYBATE DE SODIUM REIG JOFRE
Les instructions suivantes expliquent la préparation de OXYBATE DE SODIUM REIG JOFRE. Merci de les lire avec attention et de les suivre étape par étape. N’autorisez pas un enfant à préparer OXYBATE DE SODIUM REIG JOFRE.
Pour vous aider, la boîte de OXYBATE DE SODIUM REIG JOFRE contient 1 flacon de médicament, une seringue doseuse et 2 godets doseurs avec bouchons de sécurité enfant.
Étape 1
· Enlevez le bouchon du flacon en appuyant dessus, tout en tournant dans le sens contraire des aiguilles d’une montre (vers la gauche).
· Ensuite, posez le flacon en position verticale sur une table.
· Puis, en tenant toujours le flacon en position verticale, insérez l’adaptateur sur le goulot. Cette opération n’est réalisée qu’une seule fois, lors de la première utilisation. L’adaptateur reste ensuite sur le flacon pour les utilisations suivantes.
Étape 2
· Introduisez la pointe de la seringue doseuse au centre de l’ouverture et appuyez fermement (voir figure 1).
|
|
|
Figure 1 |
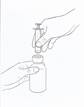
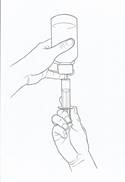
· Tenez le flacon et la seringue d’une main et, de l’autre main, retournez le flacon et tirez sur le piston jusqu’à la graduation correspondant à la dose prescrite. Remarque : la solution ne pourra être prélevée que si le flacon est maintenu en position inversée (voir figure 2).
|
|
|
Figure 2 |
Étape 3
· Posez la bouteille en position verticale.
· Retirez la seringue du flacon.
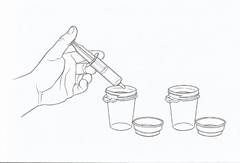
· Videz le contenu de la seringue dans un des deux godets doseurs en appuyant sur le piston (voir figure 3). Répétez cette étape pour le second godet doseur.
· Puis ajoutez environ 60 mL d’eau (soit environ 4 cuillères à soupe) dans chaque godet doseur.
|
|
|
Figure 3 |
Étape 4
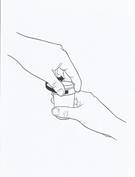
· Fermez les deux godets doseurs à l’aide des bouchons de sécurité enfant fournis, en tournant dans le sens des aiguilles d’une montre (voir figure 4).
· Rincez la seringue avec de l’eau.
|
|
|
Figure 4 |
· Juste avant d’aller vous coucher:
- Les patients adultes placent la seconde dose près de leur lit.
- Le parent ou soignant d’un adolescent ou d’un enfant de 7 ans et plus ne doivent pas laisser la deuxième dose près du lit de l’enfant ni dans un endroit pouvant être aisément atteint par l’enfant.
- Vous aurez peut-être besoin d’un réveil pour pouvoir prendre votre seconde dose au moins 2h30 mais pas plus de 4 heures après la prise de la première dose.
§ Puis:
· Otez le bouchon du premier godet doseur en appuyant dessus tout en tournant dans le sens contraire des aiguilles d’une montre.
· Buvez la première dose lorsque vous serez assis sur votre lit puis refermez le godet doseur et allongez-vous immédiatement. Pour les enfants qui dorment plus de 8 heures, mais moins de 12 heures, la première dose peut être donnée après que l’enfant ait dormi pendant 1 à 2 heures.
· Lorsque vous vous réveillerez 2h30 à 4 heures plus tard, enlevez le bouchon du deuxième godet doseur. Buvez la seconde dose entièrement, assis sur votre lit, puis refermez le godet doseur et allongez-vous de nouveau pour continuer à dormir.
Si vous avez l’impression que les effets de OXYBATE DE SODIUM REIG JOFRE sont trop forts ou trop faibles, veuillez en parler à votre médecin ou à votre pharmacien.
Si vous avez pris plus de OXYBATE DE SODIUM REIG JOFRE que vous n’auriez dû
Les symptômes d’un surdosage peuvent inclure : agitation, confusion, troubles des mouvements, atteinte respiratoire, vision trouble, transpiration abondante, mal de tête, vomissements, diminution de la conscience conduisant au coma et crises convulsives, soif excessive, crampes musculaires et faiblesse. Si vous avez pris plus de ce médicament que vous n’auriez dû, ou si vous en avez pris par accident, prévenez immédiatement les urgences médicales. Prenez le flacon étiqueté du médicament avec vous, même si celui-ci est vide.
Si vous oubliez de prendre OXYBATE DE SODIUM REIG JOFRE
Si vous n'êtes pas sûr(e) d’avoir pris OXYBATE DE SODIUM REIG JOFRE
En cas de doute sur l'administration d'une dose, ne pas administrer une nouvelle dose pour réduire le risque de surdosage.
Si vous arrêtez de prendre OXYBATE DE SODIUM REIG JOFRE
Vous devez poursuivre votre traitement par OXYBATE DE SODIUM REIG JOFRE aussi longtemps que votre médecin vous l’aura indiqué. En cas d’arrêt du traitement, vous pouvez constater une reprise de vos accès de cataplexie ainsi que la survenue d’insomnies, maux de tête, anxiété, vertiges, troubles du sommeil, somnolence, hallucinations, troubles de la pensée.
Si vous arrêtez de prendre ce médicament pendant plus de 14 jours consécutifs, vous devez consulter votre médecin, car vous devez le reprendre à une dose réduite.
Si vous avez d’autres questions sur l’utilisation de ce médicament, demandez plus d’informations à votre médecin ou à votre pharmacien.
4. QUELS SONT LES EFFETS INDESIRABLES EVENTUELS ?
Adultes - effets indésirables les plus fréquents observés dans les études cliniques (survenant chez 10 à 20 % des patients):
· sensations vertigineuses
· nausées
· maux de tête
Si vous présentez l’un de ces effets indésirables, informez immédiatement votre médecin.
Enfants et adolescents - effets indésirables les plus fréquents observés dans une étude clinique:
· pipi au lit (18,3 %)
· nausées (12,5 %)
· vomissements (8,7 %)
· perte de poids (8,7 %)
· perte d’appétit (6,7 %)
· maux de tête (5,8 %)
· sensations vertigineuses (5,8 %)
· pensées suicidaires (1 %)
· sensation de mal-être mental (perte de contact avec la réalité) (1 %)
Si vous présentez l’un de ces effets indésirables, informez immédiatement votre médecin.
Les effets indésirables chez les adultes et les enfants sont les mêmes. Si vous présentez l’un des effets indésirables ci-dessous, informez immédiatement votre médecin:
Très fréquents (pouvant survenir chez plus d’1 personne sur 10):
· nausées
· sensations vertigineuses
· maux de tête
Fréquents (pouvant survenir chez 1 à 10 personnes sur 100):
· troubles du sommeil, y compris insomnie, rêves anormaux, paralysie du sommeil, somnolence,
· cauchemars, somnambulisme, pipi au lit, somnolence diurne excessive, difficulté à s’endormir
· au milieu de la nuit
· sensation d’ébriété, tremblements, confusion/désorientation, vision floue, troubles de
· l’équilibre, chute, sensation de « tournis » (vertiges),
· sentir ses battements cardiaques, augmentation de la tension artérielle, essoufflement
· vomissements, maux d’estomac, diarrhée
· anorexie, perte d’appétit, perte de poids
· faiblesse, fatigue, sédation
· sudation
· dépression
· crampes musculaires, gonflement
· douleurs des articulations et du dos
· troubles de l’attention, troubles de la sensibilité particulièrement au toucher, sensation du
· toucher anormale, goût anormal
· anxiété, nervosité
· incontinence urinaire
· ronflement, congestion du nez
· éruption cutanée
· inflammation des sinus, inflammation du nez et de la gorge
Peu fréquents (pouvant survenir chez 1 à 10 personnes sur 1 000):
· psychose (un trouble mental qui peut impliquer hallucinations, élocution incohérente, ou
· comportement désorganisé et agité)
· paranoïa, pensées anormales, hallucinations, agitation, tentative de suicide
· difficultés à s’endormir, syndrome des jambes sans repos
· troubles de la mémoire
· myoclonies (contractions involontaires des muscles)
· passage involontaire de selles
· hypersensibilité
Fréquence indéterminée (ne peut être estimée sur la base des données disponibles):
· crise convulsive
· diminution de l’amplitude ou de la fréquence de la respiration, bref arrêt de la respiration
· pendant le sommeil
· urticaire
· pensées suicidaires, délire, pensées de commettre des actes violents (y compris faire du mal à
· autrui)
· irritabilité, agressivité
· humeur euphorique
· crise de panique
· manie / trouble bipolaire
· sécheresse de la bouche, déshydratation
· gonflement du visage (oedème de Quincke)
· bruxisme (grincer des dents et serrer les mâchoires)
· pollakiurie / miction impérieuse (besoin accru d’uriner)
· acouphènes (bruit dans les oreilles tels que des tintements ou des bourdonnements)
· trouble de l’alimentation lié au sommeil
· augmentation de l’appétit
· perte de conscience
· dyskinésie (par ex. mouvements anormaux, incontrôlés des membres)
· pellicules
· augmentation du désir sexuel
· nycturie (miction excessive la nuit)
· sensation d’étouffement
Si vous présentez l’un des effets indésirables listés ci-dessus, informez immédiatement votre médecin.
Déclaration des effets secondaires
Si vous ressentez un quelconque effet indésirable, parlez-en à votre médecin ou votre pharmacien ou à votre infirmier/ère. Ceci s’applique aussi à tout effet indésirable qui ne serait pas mentionné dans cette notice. Vous pouvez également déclarer les effets indésirables directement via le système national de déclaration : Agence nationale de sécurité du médicament et des produits de santé (ANSM) et réseau des Centres Régionaux de Pharmacovigilance - Site internet: www.signalement-sante.gouv.fr
En signalant les effets indésirables, vous contribuez à fournir davantage d’informations sur la sécurité du médicament.
5. COMMENT CONSERVER OXYBATE DE SODIUM REIG JOFRE 500 mg/mL, solution buvable ?
Tenir ce médicament hors de la vue et de la portée des enfants.
N’utilisez pas ce médicament après la date de péremption indiquée sur le flacon après EXP. La date de péremption fait référence au dernier jour de ce mois.
Après dilution dans le gobelet doseur, la préparation doit être utilisée dans les 24 heures.
Une fois le flacon de OXYBATE DE SODIUM REIG JOFRE ouvert, toute solution non utilisée dans les 90 jours après ouverture devra être jetée.
Ne jetez aucun médicament au tout-à-l’égout ou avec les ordures ménagères. Demandez à votre pharmacien d’éliminer les médicaments que vous n’utilisez plus. Ces mesures contribueront à protéger l’environnement.
6. CONTENU DE L’EMBALLAGE ET AUTRES INFORMATIONS
Ce que contient OXYBATE DE SODIUM REIG JOFRE 500 mg/mL, solution buvable
· La substance active est : oxybate de sodium.
Chaque mL contient 500 mg d’oxybate de sodium
· Les autres composants sont : eau purifiée, acide malique et hydroxyde de sodium.
Chaque boîte contient un flacon, un adaptateur pour flacon pressable (PIBA), une seringue-doseuse en plastique et deux godets doseurs avec deux bouchons sécurité enfant.
OXYBATE DE SODIUM REIG JOFRE 500 mg/mL, solution buvable est une solution claire et incolore.
Titulaire de l’autorisation de mise sur le marché
GRAN CAPITAN 10
08970 SANT JOAN DESPÍ (BARCELONA)
ESPAGNE
Exploitant de l’autorisation de mise sur le marché
LABORATOIRES FORTE PHARMA S.A.M.
41, AVENUE HECTOR OTTO
MC 98000 MONACO
GRAN CAPITAN 10
08970 SANT JOAN DESPÍ (BARCELONA)
ESPAGNE
Votre médecin a dû vous remettre du matériel d’information comprenant des explications sur la façon de prendre le médicament, des réponses aux questions fréquemment posées par les patients et une carte patient.
Noms du médicament dans les Etats membres de l'Espace Economique Européen
Ce médicament est autorisé dans les Etats membres de l'Espace Economique Européen sous les noms suivants : Conformément à la réglementation en vigueur.
[à compléter ultérieurement par le titulaire]
La dernière date à laquelle cette notice a été révisée est :
[à compléter ultérieurement par le titulaire]
Votre médecin a dû vous remettre du matériel d'information comprenant des explications sur la façon de prendre le médicament, des réponses aux questions fréquemment posées par les patients et une carte patient.
Pour toute information complémentaire concernant ce médicament, veuillez prendre contact avec le représentant local du titulaire de l'autorisation de mise sur le marché.
Des informations détaillées sur ce médicament sont disponibles sur le site Internet de l’ANSM (France).