Dernière mise à jour le 01/06/2026
PREGABALINE BGR 300 mg, gélule
Ce médicament n'est ou ne sera bientôt plus disponible sur le marché.
Si vous prenez actuellement ce médicament, il vous est recommandé d'en parler avec votre médecin
ou avec votre pharmacien qui pourra vous orienter vers un autre traitement.
Indications thérapeutiques
Classe pharmacothérapeutique : antiépileptiques, autres antiépileptiques – code ATC : N03AX16.
PREGABALINE BGR appartient à une classe de médicaments utilisés pour traiter l'épilepsie, les douleurs neuropathiques et le Trouble Anxieux Généralisé (TAG) chez l’adulte.
Douleurs neuropathiques périphériques et centrales
PREGABALINE BGR est utilisé pour traiter les douleurs persistantes causées par des lésions des nerfs. Différentes pathologies comme le diabète ou le zona peuvent induire des douleurs neuropathiques périphériques. Les manifestations douloureuses peuvent être décrites comme étant des sensations de chaleur, de brûlure, de douleur lancinante, d’élancement, de coup de poignard, de douleur fulgurante, de crampe, d’endolorissement, de picotements, d’engourdissement, de pincements et de coups d’aiguille. Les douleurs neuropathiques périphériques et centrales peuvent aussi être associées à des changements de l’humeur, des troubles du sommeil, de la fatigue, et peuvent avoir un impact sur le fonctionnement physique et social, et sur la qualité de vie en général.
Épilepsie
PREGABALINE BGR est utilisé pour traiter un type particulier d’épilepsie (crises épileptiques partielles avec ou sans généralisation secondaire) chez l’adulte. Votre médecin vous prescrira PREGABALINE BGR pour aider à traiter votre épilepsie lorsque votre traitement actuel ne permet pas de contrôler complètement vos crises. Vous devez prendre PREGABALINE BGR en association à votre traitement actuel. PREGABALINE BGR ne doit pas être utilisé seul, mais doit toujours être utilisé en association à un autre traitement antiépileptique.
Trouble Anxieux Généralisé
PREGABALINE BGR est utilisé pour traiter le Trouble Anxieux Généralisé (TAG).
Les symptômes du TAG comportent une anxiété excessive prolongée et une inquiétude difficiles à contrôler. Le TAG peut également induire une agitation ou une sensation d'excitation ou d'énervement, une sensation d'être facilement fatigué(e), des difficultés à se concentrer ou des trous de mémoire, une irritabilité, une tension musculaire ou des troubles du sommeil. Ceci est différent du stress et des tensions de la vie quotidienne.
Service médical rendu (SMR)
Amélioration du service médical rendu (ASMR)
Ce médicament étant un générique, l'ASMR n'a pas été évalué par la commission de la transparence (CT), il est possible de se référer à la /aux spécialité(s) de référence du groupe générique auquel appartient ce médicament (cliquez ici pour aller à la rubrique des groupes génériques)
ANSM - Mis à jour le : 16/07/2024
PREGABALINE BGR 300 mg, gélule
2. COMPOSITION QUALITATIVE ET QUANTITATIVE
Chaque gélule contient 300 mg de prégabaline.
Pour la liste complète des excipients, voir rubrique 6.1.
Gélule présentant un corps blanc et une coiffe orange, portant l’inscription « 300 » sur le corps à l’encre noire.
4.1. Indications thérapeutiques
PREGABALINE BGR est indiqué dans le traitement des douleurs neuropathiques périphériques et centrales chez l’adulte.
Épilepsie
PREGABALINE BGR est indiqué chez l'adulte en association dans le traitement des crises épileptiques partielles avec ou sans généralisation secondaire.
Trouble anxieux généralisé
PREGABALINE BGR est indiqué dans le traitement du Trouble Anxieux Généralisé (TAG) chez l’adulte.
4.2. Posologie et mode d'administration
La posologie varie de 150 à 600 mg par jour, en deux ou en trois prises.
Douleurs neuropathiques
Le traitement par prégabaline peut être instauré à la dose de 150 mg par jour administrée en deux ou en trois prises. En fonction de la réponse et de la tolérance du patient, la dose peut être augmentée à 300 mg par jour après un intervalle de 3 à 7 jours, et peut si nécessaire être augmentée à la dose maximale de 600 mg par jour après un intervalle supplémentaire de 7 jours.
Épilepsie
Le traitement par prégabaline peut être instauré à la dose de 150 mg par jour administrée en deux ou en trois prises. En fonction de la réponse et de la tolérance du patient, la dose peut être augmentée à 300 mg par jour après 1 semaine. La dose maximale de 600 mg par jour peut être atteinte après un délai supplémentaire d'une semaine.
Trouble anxieux généralisé
La posologie varie de 150 à 600 mg par jour, en deux ou en trois prises. La nécessité de poursuivre le traitement doit être réévaluée régulièrement.
Le traitement par prégabaline peut être instauré à la dose de 150 mg par jour. En fonction de la réponse et de la tolérance du patient, la dose peut être augmentée à 300 mg par jour après 1 semaine.
Après un délai supplémentaire d'une semaine, la dose peut être augmentée à 450 mg par jour. La dose maximale de 600 mg par jour peut être atteinte après un délai supplémentaire d'une semaine.
Interruption du traitement par la prégabaline
Conformément aux pratiques cliniques actuelles, si le traitement par la prégabaline doit être interrompu, il est recommandé de le faire progressivement sur une période minimale d’1 semaine quelle que soit l'indication (voir rubriques 4.4. et 4.8).
Insuffisance rénale
La prégabaline est éliminée de la circulation générale principalement par voie rénale sous forme inchangée. La clairance de la prégabaline étant directement proportionnelle à la clairance de la créatinine (voir rubrique 5.2), chez les patients présentant une insuffisance rénale, une réduction de la dose devra être établie individuellement en tenant compte de la clairance de la créatinine (CLcr), comme indiqué dans le Tableau 1, calculée selon la formule suivante :
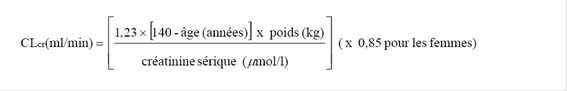
La prégabaline est éliminée efficacement du plasma par hémodialyse (50 % du médicament en 4 heures). Pour les patients hémodialysés, la dose journalière de prégabaline doit être adaptée en tenant compte de la fonction rénale. En plus de la dose journalière, une dose supplémentaire doit être administrée immédiatement après chaque hémodialyse de 4 heures (voir Tableau 1).
Tableau 1. Adaptation de la dose de prégabaline selon la fonction rénale
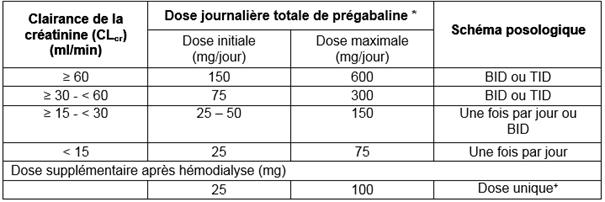
TID = trois doses séparées
BID = deux doses séparées
* La dose journalière totale (mg/jour) doit être divisée par le nombre de prises indiqué pour obtenir le nombre de mg par prise.
+ La dose supplémentaire est une dose complémentaire administrée en une seule prise.
Insuffisance hépatique
Aucun ajustement de la dose n'est nécessaire chez les patients insuffisants hépatiques (voir rubrique 5.2).
Population pédiatrique
La sécurité d’emploi et l’efficacité de la prégabaline chez les enfants de moins de 12 ans et chez les adolescents (12-17 ans) n’ont pas été établies. Les données actuellement disponibles sont décrites aux rubriques 4.8, 5.1 et 5.2, mais aucune recommandation sur la posologie ne peut être donnée.
Sujet âgé
En raison d’une diminution de la fonction rénale, une réduction de la dose de prégabaline peut être nécessaire chez les patients âgés (voir rubrique 5.2).
Mode d’administration
PREGABALINE BGR peut être pris au moment ou en dehors des repas.
PREGABALINE BGR est administré uniquement par voie orale.
Hypersensibilité à la substance active ou à l’un des excipients mentionnés à la rubrique 6.1.
4.4. Mises en garde spéciales et précautions d'emploi
Conformément aux pratiques cliniques actuelles, une adaptation du traitement hypoglycémiant peut être nécessaire chez certains patients diabétiques ayant présenté une augmentation de poids sous prégabaline.
Réactions d’hypersensibilité
Des notifications de réactions d’hypersensibilité, y compris des cas d’œdème de Quincke, ont été rapportées après commercialisation. La survenue de symptômes d’œdème de Quincke tels qu’un gonflement du visage, un gonflement péri-oral ou des voies aériennes supérieures, impose l’arrêt immédiat de la prégabaline.
Réactions indésirables cutanées graves
De rares cas de réactions indésirables cutanées graves, dont le syndrome de Stevens-Johnson (SSJ) et la nécrolyse épidermique toxique (NET), pouvant menacer le pronostic vital ou être fatales, ont été signalés dans le cadre d’un traitement par prégabaline. Au moment de la prescription, les patients doivent être informés des signes et symptômes et doivent faire l’objet d’une surveillance étroite pour les réactions cutanées. Si des signes et symptômes évocateurs de ces réactions apparaissent, la prégabaline doit être arrêtée immédiatement et un traitement de substitution doit être envisagé (le cas échéant).
Etourdissements, somnolence, perte de connaissance, confusion et altération de la fonction mentale
Le traitement par prégabaline a été associé à des étourdissements et de la somnolence, qui pourraient augmenter la survenue de blessures accidentelles (chutes) dans la population âgée. Après la mise sur le marché, les notifications suivantes ont été rapportées : perte de connaissance, confusion et altération de la fonction mentale. Il doit donc être conseillé aux patients d’être prudents jusqu’à ce qu’ils soient habitués aux effets potentiels du médicament.
Effets sur la vision
Dans les essais cliniques contrôlés, une proportion plus importante de patients traités par la prégabaline que de patients sous placebo a signalé une vision trouble qui a disparu dans la majorité des cas malgré la poursuite du traitement. Dans les études cliniques comportant des examens ophtalmologiques, l’incidence de la baisse de l’acuité visuelle et des modifications du champ visuel était supérieure chez les patients du groupe prégabaline par rapport au groupe placebo ; l’incidence des anomalies du fond d’œil était plus élevée sous placebo (voir rubrique 5.1).
Au cours de l’expérience post-commercialisation, ont également été rapportés des effets indésirables visuels qui incluaient une perte de la vue, une vision trouble ou d’autres modifications de l’acuité visuelle, la plupart desquels étant à caractère transitoire. L’arrêt de la prégabaline peut entraîner la disparition de cette symptomatologie visuelle ou son amélioration.
Insuffisance rénale
Des cas d'insuffisance rénale ont été rapportés et une interruption du traitement a montré une réversibilité de cet effet indésirable dans certains cas.
Suppression des médicaments antiépileptiques concomitants
Il n'existe pas de données suffisantes permettant une suppression des médicaments antiépileptiques concomitants dans le but d'instaurer une monothérapie, lorsque le contrôle des crises est atteint avec la prégabaline en association.
Insuffisance cardiaque congestive
Des notifications d'insuffisance cardiaque congestive ont été rapportées après commercialisation chez certains patients traités par la prégabaline.
Ces effets sont observés essentiellement pendant le traitement par la prégabaline pour une indication de douleurs neuropathiques chez les patients âgés dont la fonction cardiovasculaire est altérée. La prégabaline doit être utilisée avec prudence chez ces patients. Cet effet indésirable peut disparaître à l’arrêt de la prégabaline.
Traitement des douleurs neuropathiques centrales dues à une lésion de la moelle épinière
Dans le traitement des douleurs neuropathiques centrales dues à une lésion de la moelle épinière, l’incidence des effets indésirables en général, des effets indésirables touchant le système nerveux central et de la somnolence en particulier, a été accrue. Ceci peut être attribué à un effet additif dû à des médicaments concomitants (par exemple les antispastiques) nécessaires pour ce type d’affection. Ceci doit être pris en compte lors de la prescription de la prégabaline pour cette affection.
Des cas de dépression respiratoire sévère ont été rapportés en lien avec l’utilisation de la prégabaline. Les patients dont la fonction respiratoire est altérée ou atteints d’une affection respiratoire ou d’une maladie neurologique, d’insuffisance rénale, ou utilisant en association des médicaments dépresseurs du système nerveux central ainsi que les personnes âgées peuvent être plus à risque de présenter cet effet indésirable grave. Une adaptation de la posologie peut être nécessaire pour ces patients (voir rubrique 4.2).
Idées et comportement suicidaires
Des idées et un comportement suicidaires ont été rapportés chez des patients traités avec des agents antiépileptiques dans plusieurs indications. Une méta-analyse d’essais randomisés contrôlés contre placebo de médicaments antiépileptiques a également montré un risque légèrement accru d’idées et de comportement suicidaires. Le mécanisme de ce risque n’est pas connu. Des cas d’idées et de comportement suicidaires ont été observés chez des patients traités par prégabaline après commercialisation (voir rubrique 4.8). Une étude épidémiologique où chaque patient est son propre témoin (comparant les périodes de traitement et les périodes sans traitement chez un même individu) a mis en évidence un risque augmenté de décès par suicide et d’apparition de comportement suicidaire chez les patients traités par prégabaline.
Il convient de conseiller aux patients (et aux aidants) de consulter un médecin en cas d’apparition de signes de comportement suicidaire ou d’idées suicidaires. Les patients doivent être surveillés pour détecter d’éventuels signes d’idées et de comportement suicidaires, et un traitement adapté doit être envisagé. L’arrêt du traitement par prégabaline doit être envisagé en cas d’idées et de comportement suicidaires.
Ralentissement du transit du tractus gastro-intestinal inférieur
Des notifications d’effets indésirables liés à un ralentissement du transit du tractus gastro-intestinal inférieur (par exemple obstruction intestinale, iléus paralytique, constipation) ont été rapportées après commercialisation lorsque la prégabaline était administrée en association avec des médicaments pouvant entraîner une constipation tels que les analgésiques opioïdes. Lorsque la prégabaline est utilisée en association à des opioïdes, des mesures de prévention de la constipation doivent être envisagées (en particulier chez les femmes et les personnes âgées).
Utilisation concomitante avec des opioïdes
La prudence est requise lors de la prescription concomitante de prégabaline avec des opioïdes en raison du risque de dépression du système nerveux central (SNC) (voir rubrique 4.5). Au cours d’une étude cas-témoins menée auprès d’utilisateurs d’opioïdes, les patients qui prenaient de la prégabaline conjointement avec un opioïde présentaient un risque accru de décès lié aux opioïdes par rapport à ceux qui prenaient uniquement un opioïde (odds ratio ajusté [ORa], 1,68 [IC à 95 %, 1,19 à 2,36]). Ce risque accru a été observé à des doses faibles de prégabaline (≤ 300 mg, ORa 1,52 [IC 95 %, 1,04 - 2,22]), et avec une tendance à l’augmentation du risque à des doses plus élevées de prégabaline (> 300 mg, ORa 2,51 [95 % IC 1,24 – 5,06]).
Mésusage, abus médicamenteux ou dépendance
Les patients traités par prégabaline doivent être surveillés afin de détecter la survenue de symptômes de mésusage, d’abus ou de dépendance à la prégabaline, tels que le développement d’une tolérance, une augmentation de dose et un comportement de recherche de médicament.
Après l’arrêt d’un traitement à court ou à long terme par la prégabaline, des symptômes de sevrage ont été observés. Les symptômes suivants ont été rapportés : insomnie, céphalées, nausées, anxiété, diarrhée, syndrome grippal, nervosité, dépression, idées suicidaires, douleurs, convulsions, hyperhidrose et étourdissements. L’apparition de symptômes de sevrage après l’arrêt de la prégabaline peut indiquer une dépendance au médicament (voir rubrique 4.8). Le patient doit en être informé au début du traitement. Si la prégabaline doit être arrêtée, il est recommandé de le faire progressivement sur une période minimale de 1 semaine, indépendamment de l’indication (voir rubrique 4.2).
Les convulsions, notamment les états de mal épileptiques et les crises tonico-cloniques généralisées, peuvent apparaître pendant ou peu après l’arrêt du traitement par la prégabaline.
Concernant l’arrêt d’un traitement à long terme par la prégabaline, des données suggèrent que l’incidence et la sévérité des symptômes de sevrage peuvent être dose-dépendantes.
Encéphalopathie
Des cas d'encéphalopathie ont été rapportés, principalement chez les patients présentant des antécédents qui peuvent favoriser l’apparition d’une encéphalopathie.
Femmes en âge de procréer/Contraception
L’utilisation de PREGABALINE BGR au cours du premier trimestre de la grossesse peut entraîner des malformations congénitales majeures chez l’enfant à naître. La prégabaline ne doit pas être utilisée pendant la grossesse, sauf si le bénéfice pour la mère l’emporte clairement sur les risques potentiels pour le fœtus. Les femmes en âge de procréer doivent utiliser une méthode de contraception efficace pendant le traitement (voir rubrique 4.6).
4.5. Interactions avec d'autres médicaments et autres formes d'interactions
Etant donné que la prégabaline est essentiellement éliminée sous forme inchangée dans les urines, qu'elle n'est que très faiblement métabolisée chez l'homme (moins de 2 % de la dose sont retrouvés dans les urines sous forme de métabolites), qu'elle n'inhibe pas le métabolisme des médicaments in vitro et qu’elle ne se lie pas aux protéines plasmatiques, celle-ci est peu susceptible d’induire ou de subir des interactions pharmacocinétiques.
Etudes in vivo et analyse pharmacocinétique de population
Aucune interaction pharmacocinétique cliniquement significative n’a été observée dans les études in vivo entre la prégabaline et la phénytoïne, la carbamazépine, l’acide valproïque, la lamotrigine, la gabapentine, le lorazépam, l’oxycodone ou l’éthanol. Les analyses pharmacocinétiques de population ont montré que les antidiabétiques oraux, les diurétiques, l’insuline, le phénobarbital, la tiagabine et le topiramate, n’avaient pas d’effet cliniquement significatif sur la clairance de la prégabaline.
Contraceptifs oraux, noréthistérone et/ou éthinylestradiol
L'administration concomitante de prégabaline avec les contraceptifs oraux tels que la noréthistérone et/ou l'éthinylestradiol n'influence pas les paramètres pharmacocinétiques à l'état d'équilibre de l'une ou l'autre de ces substances.
Médicaments affectant le système nerveux central
La prégabaline peut potentialiser les effets de l'éthanol et du lorazépam.
Des notifications d'insuffisance respiratoire, de coma et de décès ont été rapportées après commercialisation chez des patients sous prégabaline et opioïdes et/ou autres médicaments dépresseurs du système nerveux central (SNC). L'effet de la prégabaline semble s'additionner à celui de l'oxycodone sur l'altération de la fonction cognitive et motrice globale.
Interactions et sujet âgé
Aucune étude pharmacodynamique spécifique d’interaction n’a été conduite chez les sujets âgés volontaires. Les études d’interaction n’ont été réalisées que chez l’adulte.
4.6. Fertilité, grossesse et allaitement
Femmes en âge de procréer / Contraception
Les femmes en âge de procréer doivent utiliser une méthode de contraception efficace pendant le traitement (voir rubrique 4.4).
Grossesse
Les études effectuées chez l’animal ont mis en évidence une toxicité sur la reproduction (voir rubrique 5.3).
Il a été démontré que la prégabaline traversait le placenta chez le rat (voir rubrique 5.2). La prégabaline pourrait traverser le placenta humain.
Malformations congénitales majeures
Les données d’une étude observationnelle réalisée dans les pays nordiques portant sur plus de 2 700 grossesses exposées à la prégabaline au cours du premier trimestre ont révélé une prévalence plus élevée de malformations congénitales majeures (MCM) dans la population pédiatrique (vivante ou mort-née) exposée à la prégabaline par rapport à la population non exposée (5,9 % contre 4,1 %).
Le risque de MCM dans la population pédiatrique exposée à la prégabaline au cours du premier trimestre était légèrement plus élevé que dans la population non exposée (rapport de prévalence ajusté et intervalle de confiance à 95 % : 1,14 [0,96– 1,35]), et que dans la population exposée à la lamotrigine (1,29 [1,01– 1,65]) ou à la duloxétine (1,39 [1,07– 1,82]).
Les analyses sur les malformations spécifiques ont révélé des risques plus élevés pour les malformations du système nerveux, de l’œil, du visage (fentes orofaciales), les malformations urinaires et les malformations génitales, mais les effectifs étaient faibles et les estimations imprécises.
PREGABALINE BGR ne doit pas être utilisé au cours de la grossesse à moins d'une nécessité absolue (si les bénéfices pour la mère l'emportent clairement sur les risques potentiels pour le fœtus).
Allaitement
La prégabaline est excrétée dans le lait maternel (voir rubrique 5.2). L’effet de la prégabaline sur les nouveau-nés/nourrissons n’est pas connu. La décision soit d’interrompre l’allaitement soit d’interrompre le traitement avec la prégabaline doit être prise en tenant compte du bénéfice de l’allaitement pour l’enfant au regard du bénéfice du traitement pour la femme.
Fertilité
Aucune donnée clinique n’est disponible concernant les effets de la prégabaline sur la fertilité chez la femme.
Lors d’un essai clinique évaluant l’effet de la prégabaline sur la motilité des spermatozoïdes, les sujets hommes sains ont été exposés à une dose de 600 mg/jour. Aucun effet sur la motilité des spermatozoïdes n’a été observé après 3 mois de traitement.
Une étude de fertilité chez des rats femelles a montré des effets délétères sur la reproduction. Des études de fertilité chez des rats mâles ont montré des effets délétères sur la reproduction et le développement. La pertinence clinique de ces données n’est pas connue (voir rubrique 5.3).
4.7. Effets sur l'aptitude à conduire des véhicules et à utiliser des machines
Le programme d’évaluation clinique de la prégabaline a été mené chez plus de 8 900 patients exposés à la prégabaline, plus de 5 600 d’entre eux l'ayant été dans le cadre d'essais en double aveugle contrôlés contre placebo. Les effets indésirables le plus fréquemment rapportés ont été les étourdissements et la somnolence. Ces effets indésirables étaient généralement d'intensité légère à modérée. Dans toutes les études contrôlées, les interruptions de traitement liées aux effets indésirables ont été de 12 % pour les patients recevant la prégabaline et de 5 % pour ceux recevant le placebo. Les effets indésirables les plus fréquents ayant entraîné l’arrêt du traitement par la prégabaline ont été les étourdissements et la somnolence.
Le tableau 2 ci-dessous énumère, par type et par fréquence, tous les effets indésirables survenus à une incidence supérieure à celle du placebo et chez plus d'un patient (très fréquent (≥ 1/10), fréquent (≥ 1/100 à <1/10), peu fréquent (≥ 1/1 000 à < 1/100), rare (≥ 1/10 000 à < 1/1 000), très rare (< 1/10 000), fréquence indéterminée (ne peut être estimée sur la base des données disponibles)).
Au sein de chaque groupe de fréquence, les effets indésirables sont présentés par ordre de gravité décroissante.
Les effets indésirables cités peuvent aussi être associés à la maladie sous-jacente et/ou aux médicaments concomitants.
Dans le traitement des douleurs neuropathiques centrales dues à une lésion de la moelle épinière, l’incidence des effets indésirables en général, des effets indésirables touchant le SNC et de la somnolence en particulier, a été accrue (voir rubrique 4.4).
Les effets supplémentaires rapportés après commercialisation figurent dans la liste ci-dessous en italique.
Tableau 2. Effets indésirables de la prégabaline
|
Classe de systèmes d’organes |
Effets indésirables |
|
Infections et infestations |
|
|
Fréquent |
Nasopharyngite |
|
Affections hématologiques et du système lymphatique |
|
|
Peu fréquent |
Neutropénie |
|
Affections du système immunitaire |
|
|
Peu fréquent
Rare |
Hypersensibilité
Œdème de Quincke, réaction allergique |
|
Troubles du métabolisme et de la nutrition |
|
|
Fréquent
Peu fréquent |
Augmentation de l’appétit
Anorexie, hypoglycémie |
|
Affections psychiatriques |
|
|
Fréquent
Peu fréquent
Rare
Fréquence indéterminée |
Humeur euphorique, confusion, irritabilité, désorientation, insomnie, diminution de la libido
Hallucinations, crises de panique, nervosité, agitation, dépression, humeur dépressive, exaltation, agression, humeur changeante, dépersonnalisation, manque du mot, rêves anormaux, augmentation de la libido, anorgasmie, apathie
Désinhibition, comportement suicidaire, idées suicidaires
Dépendance au médicament |
|
Affections du système nerveux |
|
|
Très fréquent
Fréquent
Peu fréquent
Rare |
Etourdissements, somnolence, céphalées
Ataxie, troubles de la coordination, tremblements, dysarthrie, amnésie, troubles de la mémoire, troubles de l’attention, paresthésies, hypoesthésie, sédation, troubles de l’équilibre, léthargie
Syncope, stupeur, myoclonie, perte de connaissance, hyperactivité psychomotrice, dyskinésie, vertiges de position, tremblement intentionnel, nystagmus, trouble cognitif, altération de la fonction mentale, trouble du langage, hyporéflexie, hyperesthésie, sensation de brûlure, agueusie, malaise
Convulsions, parosmie, hypokinésie, dysgraphie, syndrome parkinsonien |
|
Affections oculaires |
|
|
Fréquent Peu fréquent
Rare |
Vision trouble, diplopie Perte de la vision périphérique, troubles visuels, gonflement des yeux, anomalies du champ visuel, diminution de l’acuité visuelle, douleur oculaire, fatigue visuelle, photopsie, sécheresse oculaire, larmoiement, irritation des yeux
Perte de la vue, kératite, oscillopsie, altération de la vision stéréoscopique, mydriase, strabisme, halo visuel |
|
Affections de l’oreille et du labyrinthe |
|
|
Fréquent Peu fréquent |
Vertiges Hyperacousie |
|
Affections cardiaques |
|
|
Peu fréquent
Rare |
Tachycardie, bloc auriculo-ventriculaire du premier degré, bradycardie sinusale, insuffisance cardiaque congestive
Allongement de l’intervalle QT, tachycardie sinusale, arythmie sinusale |
|
Affections vasculaires |
|
|
Peu fréquent |
Hypotension, hypertension, bouffées de chaleur, bouffées vasomotrices, sensation de froid aux extrémités |
|
Affections respiratoires, thoraciques et médiastinales |
|
|
Peu fréquent
Rare
Fréquence indéterminée |
Dyspnée, épistaxis, toux, congestion nasale, rhinite, ronflement, sécheresse nasale
Œdème pulmonaire, sensation de constriction du pharynx
Dépression respiratoire |
|
Affections gastro-intestinales |
|
|
Fréquent
Peu fréquent
Rare |
Vomissements, nausées, constipation, diarrhée, flatulences, distension abdominale, bouche sèche
Reflux gastro-œsophagien, sialorrhée, hypoesthésie orale
Ascite, pancréatite, gonflement de la langue, dysphagie |
|
Affections hépatobiliaires |
|
|
Peu fréquent
Rare
Très rare |
Augmentation des enzymes hépatiques*
Ictère
Insuffisance hépatique, hépatite |
|
Affections de la peau et du tissu sous-cutané |
|
|
Peu fréquent
Rare |
Eruption papuleuse, urticaire, hyperhidrose, prurit
Nécrolyse épidermique toxique, syndrome de Stevens-Johnson, sueurs froides |
|
Affections musculo-squelettiques et systémiques |
|
|
Fréquent
Peu fréquent
Rare |
Crampes musculaires, arthralgie, dorsalgie, douleur des membres, spasmes cervicaux
Gonflements articulaires, myalgie, contractions musculaires, douleurs cervicales, rigidité musculaire
Rhabdomyolyse |
|
Affections du rein et des voies urinaires |
|
|
Peu fréquent
Rare |
Incontinence urinaire, dysurie
Insuffisance rénale, oligurie, rétention urinaire |
|
Affections des organes de reproduction et du sein |
|
|
Fréquent
Peu fréquent
Rare |
Troubles de l’érection
Dysfonction sexuelle, retard de l’éjaculation, dysménorrhée, douleur mammaire
Aménorrhée, écoulement mammaire, hypertrophie mammaire, gynécomastie |
|
Troubles généraux et anomalies au site d’administration |
|
|
Fréquent
Peu fréquent |
Œdème périphérique, œdème, troubles de la marche, chutes, sensation d’ébriété, sensations anormales, fatigue
Œdème généralisé, œdème de la face, oppression thoracique, douleur, fièvre, soif, frissons, asthénie |
|
Investigations |
|
|
Fréquent
Peu fréquent
Rare |
Prise de poids
Augmentation de la créatine phosphokinase sanguine, augmentation de la glycémie, diminution de la numération des plaquettes, augmentation de la créatininémie, diminution de la kaliémie, perte de poids
Diminution de la numération des globules blancs |
* augmentation de l’alanine aminotransférase (ALAT), augmentation de l’aspartate aminotransférase (ASAT)
Population pédiatrique
Le profil de sécurité d’emploi de la prégabaline observé dans cinq études pédiatriques chez des patients présentant des crises épileptiques partielles avec ou sans généralisation secondaire (étude d’efficacité et de sécurité d’emploi pendant 12 semaines chez des patients âgés de 4 à 16 ans , n = 295 ; étude d’efficacité et de sécurité d’emploi pendant 14 jours chez des patients âgés de 1 mois à moins de 4 ans, n = 175 ; étude de pharmacocinétique et de tolérance, n = 65 ; et deux études de suivi de la sécurité d’emploi en ouvert pendant 1 an, n = 54 et n = 431) était similaire à celui observé dans les études menées chez les patients adultes épileptiques. Les évènements indésirables les plus fréquemment observés au cours de l’étude de 12 semaines avec le traitement par prégabaline ont été : somnolence, fièvre, infection des voies aériennes supérieures, augmentation de l’appétit, prise de poids et nasopharyngite. Les évènements indésirables les plus fréquemment observés au cours de l’étude de 14 jours avec le traitement par prégabaline ont été : somnolence, infection des voies aériennes supérieures et fièvre (voir rubriques 4.2, 5.1 et 5.2).
Déclaration des effets indésirables suspectés
La déclaration des effets indésirables suspectés après autorisation du médicament est importante. Elle permet une surveillance continue du rapport bénéfice/risque du médicament. Les professionnels de santé déclarent tout effet indésirable suspecté via le système national de déclaration : Agence nationale de sécurité du médicament et des produits de santé (ANSM) et réseau des Centres Régionaux de Pharmacovigilance - Site internet : https://signalement.social-sante.gouv.fr/.
Des cas de coma ont été rapportés dans de rares occasions.
Le traitement d'un surdosage avec la prégabaline est symptomatique et une hémodialyse peut être réalisée si nécessaire (voir rubrique 4.2 Tableau 1).
5. PROPRIETES PHARMACOLOGIQUES
5.1. Propriétés pharmacodynamiques
Classe pharmacothérapeutique : Antiépileptiques, autres antiépileptiques, code ATC : N03AX16.
La substance active, prégabaline, est un analogue [(S)-3-(aminométhyl)-5-acide méthylhexanoïque] de l’acide gamma-aminobutyrique.
Mécanisme d’action
La prégabaline se lie à une sous-unité auxiliaire (protéine α2-δ) des canaux calciques voltage- dépendants dans le système nerveux central.
Efficacité et sécurité cliniques
Douleurs neuropathiques
L'efficacité de la prégabaline a été démontrée dans des essais sur la neuropathie diabétique, la névralgie post-zostérienne et la lésion de la moelle épinière. L’efficacité n’a pas été étudiée dans d’autres modèles de douleur neuropathique.
La prégabaline a été étudiée au cours de 10 essais cliniques contrôlés à raison de 2 prises par jour (BID) pendant 13 semaines au maximum et de 3 prises par jour (TID) pendant 8 semaines au maximum. Dans l’ensemble, les profils de sécurité et d’efficacité ont été similaires pour les schémas posologiques BID et TID.
Dans des essais cliniques allant jusqu’à 12 semaines sur les douleurs neuropathiques périphériques et centrales, une diminution de la douleur a été observée dès la première semaine et s’est maintenue tout au long de la période de traitement.
Dans les essais cliniques contrôlés portant sur les douleurs neuropathiques périphériques, 35 % des patients traités par la prégabaline et 18 % des patients sous placebo ont présenté une amélioration de 50 % du score de douleur. Pour les patients n'ayant pas présenté de somnolence, cette amélioration a été observée chez 33 % des patients traités par la prégabaline et chez 18 % des patients sous placebo. Pour les patients ayant présenté une somnolence, les taux de réponse étaient de 48 % sous prégabaline et de 16 % sous placebo.
Dans l’essai clinique contrôlé portant sur les douleurs neuropathiques centrales, 22 % des patients traités par la prégabaline et 7 % des patients sous placebo ont présenté une amélioration de 50 % du
score de douleur.
Epilepsie
Traitement en association
La prégabaline a été étudiée dans le cadre de 3 essais cliniques contrôlés d’une durée de 12 semaines à la posologie BID ou TID. Dans l’ensemble, les profils de sécurité et d’efficacité ont été similaires pour les schémas posologiques BID et TID.
Une diminution de la fréquence des crises a été observée dès la première semaine.
Population pédiatrique
L’efficacité et la sécurité d’emploi de la prégabaline n’ont pas été établies dans le traitement en association de l’épilepsie chez les patients pédiatriques de moins de 12 ans et chez les adolescents. Les événements indésirables observés lors d’une étude pharmacocinétique et de tolérance qui incluait des patients âgés de 3 mois à 16 ans (n = 65) présentant des crises épileptiques partielles étaient similaires à ceux observés chez l’adulte. Les résultats d’une étude menée versus placebo pendant 12 semaines auprès de 295 patients pédiatriques âgés de 4 à 16 ans et d’une étude menée versus placebo pendant 14 jours auprès de 175 patients pédiatriques âgés de 1 mois à moins de 4 ans portant sur l’évaluation de l’efficacité et de la sécurité d’emploi de la prégabaline comme traitement adjuvant des crises épileptiques partielles et de deux études de sécurité d’emploi en ouvert pendant 1 an menées auprès de 54 et 431 patients pédiatriques épileptiques respectivement, âgés de 3 mois à 16 ans montrent que les événements indésirables de fièvre et d’infections des voies aériennes supérieures étaient observés plus fréquemment que dans les études chez les patients adultes épileptiques (voir rubriques 4.2, 4.8 et 5.2).
Dans le cadre d’une étude contrôlée contre placebo pendant 12 semaines, des sujets pédiatriques (âgés de 4 à 16 ans) se sont vu attribuer la prégabaline à la posologie de 2,5 mg/kg/jour (150 mg/jour au maximum), la prégabaline à la posologie de 10 mg/kg/jour (600 mg/jour au maximum), ou le placebo. Le pourcentage de sujets ayant présenté une réduction de survenue de crises épileptiques partielles d’au moins 50 % par rapport à l’inclusion était de 40,6 % des sujets traités par la prégabaline à la posologie de 10 mg/kg/jour (p = 0,0068 versus placebo), 29,1 % des sujets traités par prégabaline à la posologie de 2,5 mg/kg/jour (p = 0,2600 versus placebo) et 22,6 % de ceux recevant le placebo.
La prégabaline 14 mg/kg/jour a réduit significativement la fréquence transformée logarithmiquement des crises épileptiques partielles par rapport au placebo (p = 0,0223) ; la prégabaline 7 mg/kg/jour n’a pas montré d’amélioration par rapport au placebo.
Monothérapie (patients nouvellement diagnostiqués)
La prégabaline a été étudiée lors d’un essai clinique contrôlé d’une durée de 56 semaines à la posologie BID. La prégabaline n’a pas démontré sa non-infériorité par rapport à la lamotrigine basée sur le critère d’absence de crise pendant 6 mois. La prégabaline et la lamotrigine avaient des profils de sécurité similaires et étaient bien tolérées.
Trouble Anxieux Généralisé
La prégabaline a été étudiée au cours de 6 essais contrôlés d’une durée de 4 à 6 semaines, d’une étude de 8 semaines chez des sujets âgés, et d’une étude de prévention des rechutes à long terme comportant une phase de prévention en double aveugle d'une durée de 6 mois.
Un soulagement des symptômes du TAG, évalué par l’échelle d’anxiété de Hamilton (HAM-A) a été observé dès la première semaine.
Dans les essais cliniques contrôlés (d’une durée de 4 à 8 semaines), 52 % des patients traités par la prégabaline et 38 % des patients recevant un placebo ont présenté une amélioration d’au moins 50 % du score total HAM-A entre le début et la fin de l’étude.
Dans les essais cliniques contrôlés, une proportion plus importante de patients traités par la prégabaline que de patients sous placebo a signalé une vision trouble qui a disparu dans la majorité des cas malgré la poursuite du traitement. Des examens ophtalmologiques (y compris mesure de l’acuité visuelle, champ visuel standard et examen du fond d’œil avec dilatation) ont été réalisés chez plus de 3 600 patients dans le cadre des essais cliniques contrôlés. Chez ces patients, 6,5 % de ceux traités par la prégabaline et 4,8 % de ceux traités par le placebo ont présenté une baisse d’acuité visuelle. Des modifications du champ visuel ont été mises en évidence chez 12,4 % des patients sous prégabaline et 11,7 % des patients recevant le placebo. Des anomalies du fond d’œil ont été observées dans 1,7 % des cas au sein du groupe prégabaline et 2,1 % dans le groupe placebo.
5.2. Propriétés pharmacocinétiques
Absorption
La prégabaline est rapidement absorbée lorsqu'elle est administrée à jeun, les pics plasmatiques apparaissant dans l’heure suivant l'administration d'une dose unique ou de doses multiples. La biodisponibilité orale de la prégabaline est estimée comme étant ≥ 90 % et est indépendante de la dose.
Après administration répétée du produit, l'état d'équilibre est atteint dans un délai de 24 à 48 heures. Le taux d'absorption de la prégabaline diminue lorsque le médicament est administré avec des aliments, entraînant une diminution de la Cmax d'environ 25-30 % et un retard du tmax d'environ 2,5 heures. Toutefois, l'administration de la prégabaline au cours du repas n'entraîne pas d'effet cliniquement significatif sur son taux d'absorption.
Distribution
Les études précliniques ont montré que la prégabaline traverse la barrière hémato-encéphalique chez les souris, les rats et les singes. Il a également été démontré que la prégabaline traverse le placenta chez les rates et est présente dans le lait des rates allaitantes. Chez l'homme, le volume de distribution apparent de la prégabaline après administration orale est d'environ 0,56 l/kg. La prégabaline ne se lie pas aux protéines plasmatiques.
Biotransformation
La prégabaline est très faiblement métabolisée chez l'homme. Après administration d'une dose de prégabaline radio-marquée, environ 98 % de la radioactivité retrouvés dans l'urine étaient de la prégabaline sous forme inchangée.
Le dérivé N-méthylé de la prégabaline, le principal métabolite de la prégabaline retrouvé dans l'urine, représentait 0,9 % de la dose. Dans les études précliniques, aucune racémisation de l’énantiomère S de la prégabaline en énantiomère R n'a été mise en évidence.
Élimination
La prégabaline est éliminée de la circulation générale principalement par voie rénale sous forme inchangée.
La demi-vie d'élimination de la prégabaline est d'environ 6,3 heures. La clairance plasmatique et la clairance rénale de la prégabaline sont directement proportionnelles à la clairance de la créatinine (voir rubrique 5.2 Insuffisance rénale).
L'adaptation de la dose chez les patients ayant une fonction rénale diminuée ou traités par hémodialyse est nécessaire (voir rubrique 4.2 Tableau 1).
Linéarité/non-linéarité
La prégabaline présente une pharmacocinétique linéaire aux doses journalières recommandées. La variabilité pharmacocinétique inter-individuelle observée avec la prégabaline est faible (< 20 %). La pharmacocinétique de la prégabaline administrée à dose multiple est extrapolable à partir de celle obtenue lorsqu’elle est administrée à dose unique. Il n’est donc pas nécessaire d’effectuer des contrôles de routine des concentrations plasmatiques de prégabaline.
Sexe
Les essais cliniques montrent que les concentrations plasmatiques de prégabaline ne sont pas cliniquement différentes entre les hommes et les femmes.
Insuffisance rénale
La clairance de la prégabaline est directement proportionnelle à la clairance de la créatinine. De plus, la prégabaline est éliminée du plasma par hémodialyse (après une hémodialyse de 4 heures, les concentrations plasmatiques de la prégabaline sont réduites d’environ 50 %). Etant donné que l’élimination rénale est la voie d’élimination principale, une réduction posologique chez les insuffisants rénaux et un complément de dose après hémodialyse s’avèrent nécessaires (voir rubrique 4.2 Tableau 1).
Insuffisance hépatique
Aucune étude pharmacocinétique spécifique n’a été menée chez les insuffisants hépatiques. Etant donné que la prégabaline ne subit pas de métabolisme important et qu’elle est essentiellement excrétée sous forme inchangée dans l’urine, une insuffisance hépatique ne devrait pas modifier significativement les concentrations plasmatiques de prégabaline.
Population pédiatrique
La pharmacocinétique de la prégabaline a été évaluée chez des patients pédiatriques épileptiques (tranches d’âge : de 1 à 23 mois, de 2 à 6 ans, de 7 à 11 ans et de 12 à 16 ans) à des niveaux de dose de 2,5, 5, 10 et 15 mg/kg/jour dans une étude pharmacocinétique et de tolérance.
Après administration orale de prégabaline chez des patients pédiatriques à jeun, le temps nécessaire pour atteindre le pic plasmatique était en général similaire dans toutes les tranches d’âge. Ce pic était atteint entre 0,5 et 2 heures après administration de la dose.
Les paramètres de Cmax et d’ASC de la prégabaline augmentaient de manière linéaire par rapport à l’augmentation de la dose dans chaque tranche d’âge. L’ASC était inférieure de 30 % chez les patients pédiatriques pesant moins de 30 kg en raison d’une plus forte clairance ajustée sur le poids corporel, de 43 %, chez ces patients par comparaison aux patients dont le poids était ≥ 30 kg.
La demi-vie terminale de la prégabaline était en moyenne de 3 à 4 heures environ chez les patients pédiatriques jusqu'à l’âge de 6 ans et de 4 à 6 heures à partir de l’âge de 7 ans.
L’analyse pharmacocinétique de population a montré que la clairance de la créatinine était une covariable significative de la clairance orale de la prégabaline, que le poids corporel était une covariable significative du volume de distribution oral apparent de la prégabaline et que ces corrélations étaient similaires chez les patients pédiatriques et adultes.
La pharmacocinétique de la prégabaline n’a pas été étudiée chez les patients de moins de 3 mois (voir rubriques 4.2, 4.8 et 5.1).
Sujets âgés
La clairance de la prégabaline tend à diminuer avec l'âge. Cette diminution de la clairance orale de la prégabaline correspond à la diminution de la clairance de la créatinine liée à l'âge. Une réduction de la dose de prégabaline peut s'avérer nécessaire chez les patients qui présentent une fonction rénale diminuée en rapport avec l'âge (voir rubrique 4.2 Tableau 1).
Mères allaitantes
La pharmacocinétique a été évaluée chez 10 femmes allaitantes recevant 150 mg de prégabaline toutes les 12 heures (300 mg par jour), et cela au moins 12 semaines après l’accouchement. L’allaitement n’a eu que peu ou pas d’influence sur la pharmacocinétique de la prégabaline. A l’état d’équilibre, la prégabaline a été excrétée dans le lait maternel à des concentrations moyennes égales à environ 76 % des concentrations plasmatiques maternelles. La quantité ingérée par le nourrisson via le lait maternel (en supposant une consommation de lait moyenne de 150 ml/kg/j) d’une mère recevant 300 mg/j ou la dose maximale de 600 mg/j a été estimée respectivement à 0,31 ou 0,62 mg/kg/j. Ces quantités correspondent à environ 7 % de la dose maternelle quotidienne totale rapportée au poids (mg/kg).
5.3. Données de sécurité préclinique
La prégabaline ne s'est pas révélée tératogène chez la souris, le rat et le lapin. Une toxicité fœtale chez le rat et le lapin est uniquement apparue lors d'expositions largement supérieures à l'exposition chez l'homme. Dans les études de toxicité pré- et postnatales, la prégabaline a induit une toxicité de la descendance chez le rat lors d’expositions > 2 fois la dose maximale recommandée chez l'homme.
Les effets indésirables observés sur la fertilité chez les rats mâles et femelles n’ont été observés qu’à des doses nettement supérieures aux doses thérapeutiques. Les effets indésirables observés sur l’appareil reproducteur mâle et sur les spermatozoïdes ont été réversibles et n’ont été observés qu’à des doses nettement supérieures aux doses thérapeutiques ou étaient associés à un processus dégénératif spontané de l’organe reproducteur mâle chez le rat. Ces effets sont donc considérés comme ayant peu ou pas de pertinence clinique.
La prégabaline n’est pas génotoxique comme le montrent les résultats d’une batterie de tests in vitro et in vivo.
Des études de carcinogénicité de deux ans ont été menées avec la prégabaline chez le rat et la souris.
Aucune tumeur n'a été observée chez le rat lors d'expositions atteignant jusqu'à 24 fois l'exposition moyenne chez l'homme correspondant à la dose clinique maximale recommandée de 600 mg/jour.
Chez la souris, aucune augmentation de l'incidence de tumeurs n'a été observée à des expositions similaires à l'exposition moyenne chez l'homme, mais une augmentation de l'incidence des hémangiosarcomes a été observée à des expositions supérieures. Le mécanisme non génotoxique de la formation de tumeurs induite par la prégabaline chez la souris implique des modifications plaquettaires et une prolifération associée de cellules endothéliales. Ces modifications plaquettaires n’ont pas été retrouvées chez le rat ou chez l'homme, sur la base des résultats cliniques à court ou à long terme. Il n'y a aucune preuve suggérant qu'il existe un tel risque chez l'homme.
Chez le rat jeune, les données de toxicité n’étaient pas qualitativement différentes de celles observées chez le rat adulte. Les rats jeunes sont cependant plus sensibles. Aux doses thérapeutiques, des signes cliniques évidents d’hyperactivité du SNC et de bruxisme ainsi que des modifications de la croissance (suppression transitoire de la prise de poids) ont été observés. Des effets sur le cycle œstral ont été observés à des doses correspondant à 5 fois l’exposition thérapeutique chez l’homme.
Une diminution de la réponse acoustique a été observée chez les rats jeunes 1 à 2 semaines après exposition à des doses 2 fois supérieures à la dose thérapeutique humaine. Neuf semaines après exposition, cet effet n’était plus observé.
Gélule : amidon de maïs prégélatinisé, mannitol, talc.
Enveloppe de la gélule : gélatine, dioxyde de titane (E171), oxyde de fer rouge (E172).
Encre d’impression : gomme laque, oxyde de fer noir (E172), propylèneglycol (E1520), hydroxyde d’ammonium (E527).
6.4. Précautions particulières de conservation
Ce médicament ne nécessite pas de précautions particulières de conservation.
6.5. Nature et contenu de l'emballage extérieur
14, 56, 100 et 112 gélules sous plaquettes (PVC/Aluminium).
60 gélules en flacon (PEHD).
Toutes les présentations peuvent ne pas être commercialisées.
6.6. Précautions particulières d’élimination et de manipulation
Pas d'exigences particulières pour l’élimination.
7. TITULAIRE DE L’AUTORISATION DE MISE SUR LE MARCHE
15 BOULEVARD CHARLES DE GAULLE
92700 COLOMBES
8. NUMERO(S) D’AUTORISATION DE MISE SUR LE MARCHE
· 34009 300 604 0 7 : Flacon (PEHD) de 60 gélules.
· 34009 300 603 8 4 : 14 gélules sous plaquettes (PVC/Aluminium).
· 34009 300 603 9 1 : 56 gélules sous plaquettes (PVC/Aluminium).
· 34009 550 219 7 1 : 100 gélules sous plaquettes (PVC/Aluminium).
· 34009 550 219 8 8 : 112 gélules sous plaquettes (PVC/Aluminium).
9. DATE DE PREMIERE AUTORISATION/DE RENOUVELLEMENT DE L’AUTORISATION
[à compléter ultérieurement par le titulaire]
10. DATE DE MISE A JOUR DU TEXTE
[à compléter ultérieurement par le titulaire]
Sans objet.
12. INSTRUCTIONS POUR LA PREPARATION DES RADIOPHARMACEUTIQUES
Liste I.
Prescription limitée à 6 mois sur ordonnance sécurisée.
Ce médicament n'est ou ne sera bientôt plus disponible sur le marché.
Si vous prenez actuellement ce médicament, il vous est recommandé d'en parler avec votre médecin
ou avec votre pharmacien qui pourra vous orienter vers un autre traitement.
Informations importantes
Les informations importantes disponibles pour ce médicament sont les suivantes :
- Antiépileptiques pendant la grossesse entre 2013 et 2021 : une étude du GIS EPI-PHARE met en évidence une baisse importante de l'exposition au valproate et dérivés, mais moindre pour le topiramate et la carbamazépine
- Antiépileptiques et grossesse : mieux connaître les risques pour l'enfant à naître
- Le risque de malformation chez les enfants exposés pendant la grossesse à la prégabaline est confirmé
ANSM - Mis à jour le : 16/07/2024
PREGABALINE BGR 300 mg, gélule
Prégabaline
Veuillez lire attentivement cette notice avant de prendre ce médicament car elle contient des informations importantes pour vous.
· Gardez cette notice. Vous pourriez avoir besoin de la relire.
· Si vous avez d’autres questions, interrogez votre médecin, votre pharmacien ou votre infirmier/ère.
· Ce médicament vous a été personnellement prescrit. Ne le donnez pas à d’autres personnes. Il pourrait leur être nocif, même si les signes de leur maladie sont identiques aux vôtres.
· Si vous ressentez un quelconque effet indésirable, parlez-en à votre médecin, votre pharmacien ou votre infirmier/ère. Ceci s’applique aussi à tout effet indésirable qui ne serait pas mentionné dans cette notice. Voir rubrique 4.
1. Qu’est-ce que PREGABALINE BGR 300 mg, gélule et dans quels cas est-il utilisé ?
2. Quelles sont les informations à connaître avant de prendre PREGABALINE BGR 300 mg, gélule ?
3. Comment prendre PREGABALINE BGR 300 mg, gélule ?
4. Quels sont les effets indésirables éventuels ?
5. Comment conserver PREGABALINE BGR 300 mg, gélule ?
6. Contenu de l’emballage et autres informations.
1. QU’EST-CE QUE PREGABALINE BGR 300 mg, gélule ET DANS QUELS CAS EST-IL UTILISE ?
Classe pharmacothérapeutique : antiépileptiques, autres antiépileptiques – code ATC : N03AX16.
PREGABALINE BGR appartient à une classe de médicaments utilisés pour traiter l'épilepsie, les douleurs neuropathiques et le Trouble Anxieux Généralisé (TAG) chez l’adulte.
Douleurs neuropathiques périphériques et centrales
PREGABALINE BGR est utilisé pour traiter les douleurs persistantes causées par des lésions des nerfs. Différentes pathologies comme le diabète ou le zona peuvent induire des douleurs neuropathiques périphériques. Les manifestations douloureuses peuvent être décrites comme étant des sensations de chaleur, de brûlure, de douleur lancinante, d’élancement, de coup de poignard, de douleur fulgurante, de crampe, d’endolorissement, de picotements, d’engourdissement, de pincements et de coups d’aiguille. Les douleurs neuropathiques périphériques et centrales peuvent aussi être associées à des changements de l’humeur, des troubles du sommeil, de la fatigue, et peuvent avoir un impact sur le fonctionnement physique et social, et sur la qualité de vie en général.
Épilepsie
PREGABALINE BGR est utilisé pour traiter un type particulier d’épilepsie (crises épileptiques partielles avec ou sans généralisation secondaire) chez l’adulte. Votre médecin vous prescrira PREGABALINE BGR pour aider à traiter votre épilepsie lorsque votre traitement actuel ne permet pas de contrôler complètement vos crises. Vous devez prendre PREGABALINE BGR en association à votre traitement actuel. PREGABALINE BGR ne doit pas être utilisé seul, mais doit toujours être utilisé en association à un autre traitement antiépileptique.
Trouble Anxieux Généralisé
PREGABALINE BGR est utilisé pour traiter le Trouble Anxieux Généralisé (TAG).
Les symptômes du TAG comportent une anxiété excessive prolongée et une inquiétude difficiles à contrôler. Le TAG peut également induire une agitation ou une sensation d'excitation ou d'énervement, une sensation d'être facilement fatigué(e), des difficultés à se concentrer ou des trous de mémoire, une irritabilité, une tension musculaire ou des troubles du sommeil. Ceci est différent du stress et des tensions de la vie quotidienne.
2. QUELLES SONT LES INFORMATIONS A CONNAITRE AVANT DE PRENDRE PREGABALINE BGR 300 mg, gélule ?
Ne prenez jamais PREGABALINE BGR 300 mg, gélule
· Si vous êtes allergique à la prégabaline ou à l’un des autres composants contenus dans ce médicament, mentionnés dans la rubrique 6.
Avertissements et précautions
Adressez-vous à votre médecin ou pharmacien avant de prendre PREGABALINE BGR.
· Quelques patients prenant de la prégabaline ont fait état de symptômes évocateurs d’une réaction allergique. Il s’agissait d’un gonflement du visage, des lèvres, de la langue et de la gorge ainsi que d’un rash cutané diffus. Contactez immédiatement votre médecin si vous présentez l’une de ces réactions.
· La prégabaline a été associée à des étourdissements et de la somnolence, pouvant augmenter la survenue de blessures accidentelles (chutes) chez les patients âgés. Vous devez par conséquent être prudent(e) jusqu’à ce que vous soyez habitué(e) aux éventuels effets que le médicament pourrait produire.
· La prégabaline peut faire apparaître une vision trouble ou une perte de la vue ou d’autres modifications de la vue, la plupart desquelles étant transitoires. Informez immédiatement votre médecin si vous observez une quelconque modification de votre vision.
· Une adaptation des médicaments utilisés en cas de diabète peut être nécessaire chez certains patients diabétiques qui ont pris du poids lors d’un traitement sous prégabaline.
· Certains effets indésirables peuvent être plus fréquents, notamment la somnolence, car les patients présentant une lésion de la moelle épinière peuvent recevoir d'autres médicaments, destinés à traiter par exemple la douleur ou la spasticité, qui ont des effets indésirables similaires à ceux de la prégabaline et dont la sévérité peut être augmentée lorsque ces traitements sont pris en même temps.
· Des cas d'insuffisance cardiaque ont été rapportés chez certains patients prenant de la prégabaline ; ces patients étaient pour la plupart âgés et présentaient des problèmes cardiovasculaires. Avant de prendre ce médicament, vous devez informer votre médecin si vous avez déjà eu des maladies cardiaques dans le passé.
· Des cas d'insuffisance rénale ont été rapportés chez certains patients prenant de la prégabaline. Si au cours de votre traitement par PREGABALINE BGR, vous observez une diminution de votre miction, vous devez informer votre médecin car l’arrêt de votre traitement peut améliorer ces troubles.
· Des problèmes gastro-intestinaux peuvent apparaître (par exemple constipation, transit intestinal bloqué ou paralysé) lorsque PREGABALINE BGR est utilisé avec d’autres médicaments pouvant entraîner une constipation (tels que certaines classes de médicaments contre la douleur). Informez votre médecin si vous souffrez de constipation, en particulier si vous êtes sujet(te) à ce problème.
· Avant de prendre ce médicament, informez votre médecin si vous avez déjà abusé ou été dépendant(e) de l’alcool, de médicaments sur ordonnance ou de drogues illégales ; cela pourrait signifier que vous présentez un plus grand risque de devenir dépendant de PREGABALINE BGR.
· Des cas de convulsions ont été rapportés lors de la prise de prégabaline ou peu après l’arrêt du traitement. En cas de convulsions, contactez votre médecin immédiatement.
· Des cas de modification de la fonction cérébrale (encéphalopathie) ont été rapportés chez certains patients prenant de la prégabaline et présentant des facteurs favorisants. Prévenez votre médecin en cas d’antécédents médicaux graves y compris les maladies du foie ou des reins.
· Des cas de difficultés respiratoires ont été rapportés. Si vous souffrez d’une maladie neurologique ou respiratoire, d’insuffisance rénale, ou que vous avez plus de 65 ans, votre médecin pourra vous prescrire des doses adaptées. Contactez votre médecin si vous avez des difficultés à respirer ou si votre respiration vous semble superficielle.
Certaines personnes peuvent devenir dépendantes de PREGABALINE BGR (besoin de continuer à prendre le médicament). Elles peuvent présenter des effets de sevrage lorsqu’elles arrêtent de prendre PREGABALINE BGR (voir rubrique 3, « Comment prendre PREGABALINE BGR ? » et « Si vous arrêtez de prendre PREGABALINE BGR »). Si vous craignez de devenir dépendant(e) de PREGABALINE BGR, il est important que vous consultiez votre médecin.
Si vous remarquez l’un des signes suivants pendant votre traitement par PREGABALINE BGR, il peut s’agir d’un signe indiquant que vous êtes devenu dépendant(e) :
· Vous devez prendre le médicament plus longtemps que ce qui vous a été conseillé par votre médecin.
· Vous ressentez le besoin de prendre une dose plus importante que celle qui vous a été recommandée.
· Vous utilisez le médicament pour des raisons autres que celles pour lesquelles il vous a été prescrit.
· Vous avez tenté à plusieurs reprises, sans succès, d’arrêter ou de contrôler l’utilisation de ce médicament.
· Lorsque vous arrêtez de prendre le médicament, vous vous sentez mal, et vous vous sentez mieux lorsque vous le reprenez.
Si vous remarquez l’un de ces symptômes, parlez-en à votre médecin pour discuter de la meilleure solution de traitement pour vous, notamment du moment opportun pour arrêter le traitement et de la façon de le faire en toute sécurité.
Enfants et adolescents
La sécurité d’emploi et l’efficacité chez les enfants et les adolescents (moins de 18 ans) n’ont pas été établies. La prégabaline ne doit donc pas être utilisée dans cette population.
Autres médicaments et PREGABALINE BGR 300 mg, gélule
Informez votre médecin ou pharmacien si vous prenez, avez récemment pris ou pourriez prendre tout autre médicament.
PREGABALINE BGR peut interagir avec d’autres médicaments, c’est-à-dire qu’il peut avoir une influence sur d’autres médicaments et inversement. Pris avec certains médicaments qui ont des effets sédatifs (y compris les opioïdes), PREGABALINE BGR peut potentialiser ces effets et pourrait entraîner une insuffisance respiratoire, le coma et le décès. L’intensité des étourdissements, de la somnolence et de la diminution de la concentration peut être augmentée si PREGABALINE BGR est pris en même temps que des médicaments contenant :
· de l’oxycodone (utilisé pour traiter la douleur) ;
· du lorazépam (utilisé pour traiter l’anxiété) ;
· de l’alcool.
PREGABALINE BGR peut être pris en même temps que les contraceptifs oraux.
PREGABALINE BGR 300 mg, gélule avec des aliments, boissons et de l’alcool
Les gélules de PREGABALINE BGR peuvent être prises au moment ou en dehors des repas.
La prise simultanée de PREGABALINE BGR et d'alcool n’est pas recommandée.
Grossesse et allaitement
Si vous êtes enceinte ou que vous allaitez, si vous pensez être enceinte ou planifiez une grossesse, demandez conseil à votre médecin ou pharmacien avant de prendre ce médicament.
PREGABALINE BGR ne doit pas être pris pendant la grossesse ou l’allaitement, sauf avis contraire de votre médecin.
Les femmes en âge de procréer doivent utiliser une méthode contraceptive efficace.
Conduite de véhicules et utilisation de machines
PREGABALINE BGR peut entraîner des étourdissements, de la somnolence et une diminution de la concentration.
Vous ne devez ni conduire, ni utiliser des machines complexes, ni vous engager dans des activités potentiellement dangereuses jusqu’à ce qu’il soit déterminé si ce médicament affecte votre aptitude à exercer de telles activités.
PREGABALINE BGR 300 mg, gélule contient
Sans objet.
3. COMMENT PRENDRE PREGABALINE BGR 300 mg, gélule ?
Vérifiez auprès de votre médecin ou pharmacien en cas de doute. Ne prenez pas une dose plus importante que celle qui vous a été prescrite.
Votre médecin déterminera la posologie qui convient dans votre cas.
PREGABALINE BGR est destiné à la voie orale uniquement.
Douleurs neuropathiques périphériques et centrales, épilepsie ou Trouble Anxieux Généralisé :
· Prenez le nombre de gélules prescrit par votre médecin.
· La dose habituelle, qui a été adaptée à vous et à votre état, est comprise entre 150 mg et 600 mg par jour.
· Votre médecin vous dira si vous devez prendre PREGABALINE BGR deux fois ou trois fois par jour. En cas de deux prises par jour, prenez PREGABALINE BGR une fois le matin et une fois le soir, environ aux mêmes heures chaque jour. En cas de trois prises par jour, prenez PREGABALINE BGR une fois le matin, une fois le midi et une fois le soir, environ aux mêmes heures chaque jour.
Si vous avez l’impression que l’effet de PREGABALINE BGR est trop fort ou trop faible, consultez votre médecin ou votre pharmacien.
Si vous êtes un patient âgé (de plus de 65 ans), vous devez prendre PREGABALINE BGR normalement sauf en cas de maladie des reins.
Votre médecin peut vous prescrire un horaire de prise et/ou une dose différents en cas de maladie des reins.
Avalez la gélule entière avec de l’eau.
Continuez à prendre PREGABALINE BGR jusqu'à ce que votre médecin vous dise d’arrêter.
Si vous avez pris plus de PREGABALINE BGR 300 mg, gélule que vous n’auriez dû :
Vous devez immédiatement contacter votre médecin ou vous rendre au service des urgences de l’hôpital le plus proche. Prenez votre boîte ou votre flacon de gélules de PREGABALINE BGR avec vous. Vous pouvez ressentir somnolence, confusion, agitation ou nervosité si vous avez pris plus de PREGABALINE BGR que vous n’auriez dû. Des convulsions et des pertes de conscience (coma) ont également été rapportées.
Si vous oubliez de prendre PREGABALINE BGR 300 mg, gélule :
Il est important de prendre vos gélules de PREGABALINE BGR de façon régulière aux mêmes heures chaque jour. Si vous avez oublié de prendre une dose, prenez-la dès que vous vous en rendez compte, à moins que ce ne soit le moment de prendre la dose suivante. Dans ce cas, prenez simplement la dose suivante comme convenu. Ne prenez pas de dose double pour compenser la dose que vous avez oublié de prendre.
Si vous arrêtez de prendre PREGABALINE BGR 300 mg, gélule :
N’arrêtez pas brusquement de prendre PREGABALINE BGR. Si vous souhaitez arrêter de prendre PREGABALINE BGR, parlez-en d’abord à votre médecin. Il/Elle vous dira comment procéder. Si votre traitement est arrêté, l’arrêt doit s’effectuer de façon progressive pendant 1 semaine minimum.
Après l'arrêt d'un traitement à court ou à long terme par PREGABALINE BGR, vous devez savoir que vous pouvez ressentir certains effets indésirables, appelés effets de sevrage. Ces effets comprennent des troubles du sommeil, des maux de tête, des nausées, une sensation d’anxiété, de la diarrhée, des symptômes pseudo-grippaux, des convulsions, de la nervosité, de la dépression, des idées d’auto-agression ou de suicide, de la douleur, de la transpiration et des étourdissements.
Ces effets peuvent apparaître plus fréquemment ou de façon plus sévère lorsque PREGABALINE BGR a été administré pendant une période prolongée. Si vous présentez des effets de sevrage, vous devez contacter votre médecin.
Si vous avez d’autres questions sur l’utilisation de ce médicament, demandez plus d’informations à votre médecin, à votre pharmacien ou à votre infirmier/ère.
4. QUELS SONT LES EFFETS INDESIRABLES EVENTUELS ?
Très fréquents : susceptibles d’affecter plus de 1 personne sur 10
· étourdissements, somnolence, maux de tête
Fréquents : susceptibles d’affecter jusqu’à 1 personne sur 10
· augmentation de l’appétit
· sensation d’euphorie, état confusionnel, désorientation, diminution de la libido, irritabilité
· troubles de l’attention, maladresse, troubles de la mémoire, perte de mémoire, tremblements, difficulté à parler, sensation de picotement, engourdissement, sédation, léthargie, insomnie, fatigue, sensations anormales
· vision floue, vision double
· vertiges, troubles de l’équilibre, chutes
· sécheresse de la bouche, constipation, vomissements, flatulences, diarrhée, nausées, ballonnement
· troubles de l’érection
· gonflement du corps y compris des extrémités
· sensation d’ivresse, troubles de la marche
· prise de poids
· crampes musculaires, douleurs articulaires, douleurs dorsales, douleurs dans les membres
· mal de gorge
Peu fréquents : susceptibles d’affecter jusqu’à 1 personne sur 100
· perte d’appétit, perte de poids, taux faible de sucre dans le sang, taux de sucre élevé dans le sang
· modification de la personnalité, nervosité, dépression, agitation, humeur changeante, difficulté à trouver les mots, hallucinations, rêves anormaux, crises de panique, apathie, agression, exaltation, altération de la fonction mentale, difficulté à se concentrer, augmentation de la libido, problèmes de fonctionnement sexuel incluant l'incapacité de parvenir à un orgasme, éjaculation retardée
· trouble de la vue, mouvements oculaires anormaux, troubles de la vision y compris rétrécissement du champ visuel, éclairs de lumière, mouvements saccadés, diminution des réflexes, hyperactivité, vertiges en position debout, peau sensible, perte du goût, sensation de brûlure, tremblements lors des mouvements, diminution de la vigilance, perte de connaissance, syncope, sensibilité au bruit augmentée, sensation de malaise
· yeux secs, yeux gonflés, douleurs oculaires, faiblesse oculaire, yeux larmoyants, irritation des yeux
· troubles du rythme du cœur, accélération du rythme cardiaque, diminution de la pression sanguine, augmentation de la pression sanguine, modifications des battements du cœur, insuffisance cardiaque
· rougeur de la face, bouffées de chaleur
· difficulté à respirer, sécheresse du nez, congestion nasale
· augmentation de la salive, brûlures d’estomac, engourdissement autour de la bouche
· transpiration, rash cutané, frissons, fièvre
· contractions musculaires, gonflements articulaires, rigidité musculaire, douleurs y compris douleurs musculaires, douleurs de la nuque
· douleur dans les seins
· miction difficile ou douloureuse, incontinence
· sensation de faiblesse, sensation de soif, oppression dans la poitrine
· modifications des résultats des tests sanguins et du foie (augmentation de la créatinine phosphokinase du sang, augmentation de l’alanine aminotransférase, augmentation de l’aspartate aminotransférase, diminution du nombre des plaquettes, neutropénie, augmentation de la créatinine dans le sang, diminution du potassium dans le sang)
· hypersensibilité, gonflement du visage, démangeaisons, urticaire, écoulement nasal, saignement nasal, toux, ronflements
· douleurs menstruelles
· froideur des mains et des pieds
Rares : susceptibles d’affecter jusqu’à 1 personne sur 1000
· troubles de l’odorat, vision oscillante, altération de la perception de la profondeur, éclat visuel, perte de la vue
· dilatation des pupilles, strabisme
· sueurs froides, contraction de la gorge, gonflement de la langue
· inflammation du pancréas
· difficultés à avaler
· mouvement lent ou réduit du corps
· difficultés à écrire correctement
· accumulation de liquide dans l’abdomen
· liquide dans les poumons
· convulsions
· modifications de l’enregistrement des paramètres électriques (ECG) du cœur correspondant à des troubles du rythme du cœur
· lésion musculaire
· écoulement mammaire, croissance anormale des seins, augmentation de la taille des seins chez l’homme
· interruption des règles
· insuffisance rénale, diminution du volume urinaire, rétention urinaire
· diminution du nombre de globules blancs
· comportement anormal, comportement suicidaire, idées suicidaires
· réactions allergiques (pouvant comprendre des difficultés à respirer, une inflammation des yeux (kératite), et une réaction cutanée grave qui se manifeste par des taches rougeâtres non surélevées, en forme de cibles ou de cercles, sur le tronc, souvent accompagnées de cloques centrales, d’une desquamation de la peau, d’ulcères de la bouche, de la gorge, du nez, des organes génitaux et des yeux. Ces éruptions cutanées graves peuvent être précédées par de la fièvre et des symptômes de type grippal (syndrome de Stevens-Johnson, nécrolyse épidermique toxique)
· jaunisse (jaunissement de la peau et des yeux)
· syndrome parkinsonien, c’est-à-dire des symptômes ressemblant à ceux de la maladie de Parkinson ; tels que tremblements, bradykinésie (diminution de la capacité à bouger) et rigidité (raideur musculaire)
Très rares : susceptibles d’affecter jusqu’à 1 personne sur 10 000
· insuffisance hépatique
· hépatite (inflammation du foie)
Fréquence indéterminée : ne peut être estimée sur la base des données disponibles
· devenir dépendant(e) de PREGABALINE BGR (« dépendance au médicament »).
Après l’arrêt d’un traitement à court ou à long terme par PREGABALINE BGR, vous devez savoir que vous pouvez présenter certains effets indésirables, appelés effets de sevrage (voir « Si vous arrêtez de prendre PREGABALINE BGR »).
En cas de gonflement du visage ou de la langue ou si votre peau devient rouge et commence à former des ampoules ou à peler, vous devez immédiatement demander un avis médical.
Certains effets indésirables peuvent être plus fréquents, notamment la somnolence, car les patients présentant une lésion de la moelle épinière peuvent recevoir d'autres médicaments, destinés à traiter par exemple la douleur ou la spasticité, qui ont des effets indésirables similaires à ceux de la prégabaline et dont la sévérité peut être augmentée lorsque ces traitements sont pris en même temps.
Les effets indésirables suivants ont été rapportés après commercialisation : difficultés à respirer, respiration superficielle.
Déclaration des effets secondaires
Si vous ressentez un quelconque effet indésirable, parlez-en à votre médecin, votre pharmacien ou à votre infirmier/ère. Ceci s’applique aussi à tout effet indésirable qui ne serait pas mentionné dans cette notice. Vous pouvez également déclarer les effets indésirables directement via le système national de déclaration : Agence Nationale de Sécurité du Médicament et des produits de santé (ANSM) et réseau des Centres Régionaux de Pharmacovigilance - Site internet : https://signalement.social-sante.gouv.fr/
En signalant les effets indésirables, vous contribuez à fournir davantage d’informations sur la sécurité du médicament.
5. COMMENT CONSERVER PREGABALINE BGR 300 mg, gélule ?
Tenir ce médicament hors de la vue et de la portée des enfants.
N’utilisez pas ce médicament après la date de péremption indiquée sur l’emballage. La date de péremption fait référence au dernier jour de ce mois.
Ce médicament ne nécessite pas de précautions particulières de conservation.
Ne jetez aucun médicament au tout-à-l’égout ou avec les ordures ménagères. Demandez à votre pharmacien d’éliminer les médicaments que vous n’utilisez plus. Ces mesures contribueront à protéger l’environnement.
6. CONTENU DE L’EMBALLAGE ET AUTRES INFORMATIONS
Ce que contient PREGABALINE BGR 300 mg, gélule
Pour une gélule.
· Les autres composants sont :
Amidon de maïs prégélatinisé, mannitol, talc, gélatine, dioxyde de titane (E171), oxyde de fer rouge (E172), encre d’impression (gomme laque, oxyde de fer noir (E172), propylèneglycol (E1520), hydroxyde d’ammonium (E527)).
Qu’est-ce que PREGABALINE BGR 300 mg, gélule et contenu de l’emballage extérieur
Boîte de 14, 56, 100 et 112 gélules.
Flacon de 60 gélules.
Toutes les présentations peuvent ne pas être commercialisées.
Titulaire de l’autorisation de mise sur le marché
15 BOULEVARD CHARLES DE GAULLE
92700 COLOMBES
Exploitant de l’autorisation de mise sur le marché
15 BOULEVARD CHARLES DE GAULLE
92700 COLOMBES
INDUSTRIAL PARK SAPES
RODOPI PREFECTURE, BLOCK N°5
69300 RODOPI
GRÈCE
OU
PHARMATHEN S.A.
DERVENAKION 6
PALLINI
15351 ATTIKIS
GRÈCE
Noms du médicament dans les Etats membres de l'Espace Economique Européen
Sans objet.
La dernière date à laquelle cette notice a été révisée est :
[à compléter ultérieurement par le titulaire]
Des informations détaillées sur ce médicament sont disponibles sur le site internet de l’ANSM (France).