Dernière mise à jour le 03/08/2026
TERIPARATIDE TEVA 20 microgrammes/80 microlitres, solution injectable en stylo prérempli
Indications thérapeutiques
TERIPARATIDE TEVA contient la substance active tériparatide qui est utilisée pour renforcer les os et réduire le risque de fractures en stimulant la formation osseuse.
TERIPARATIDE TEVA est utilisé pour traiter l'ostéoporose chez les adultes. L'ostéoporose est une maladie qui rend vos os fins et fragiles. Cette maladie touche particulièrement les femmes après la ménopause, mais elle peut également toucher les hommes. L’ostéoporose est également fréquente chez les patients recevant des corticoïdes.
Présentations
> 1 cartouche en verre siliconé(e) de 2,4 mL (28 doses) dans stylo prérempli
Code CIP : 34009 302 827 1 7
Déclaration de commercialisation : 25/06/2024
Cette présentation est agréée aux collectivités
- Prix hors honoraire de dispensation : 136,53 €
- Honoraire de dispensation : 1,02 €
- Prix honoraire compris : 137,55 €
- Taux de remboursement :65 %
Service médical rendu (SMR)
Amélioration du service médical rendu (ASMR)
Ce médicament étant un générique, l'ASMR n'a pas été évalué par la commission de la transparence (CT), il est possible de se référer à la /aux spécialité(s) de référence du groupe générique auquel appartient ce médicament (cliquez ici pour aller à la rubrique des groupes génériques)
ANSM - Mis à jour le : 22/05/2025
TERIPARATIDE TEVA 20 microgrammes/80 microlitres, solution injectable en stylo prérempli
2. COMPOSITION QUALITATIVE ET QUANTITATIVE
Chaque dose de 80 microlitres contient 20 microgrammes de tériparatide* (sous forme d’acétate de tériparatide).
Un stylo prérempli de 2,4 mL contient 600 microgrammes de tériparatide (correspondant à 250 microgrammes par mL).
* Le tériparatide (1-34) est identique à la séquence des 34 acides aminés de l’extrémité amino-terminale de la parathormone humaine endogène.
Pour la liste complète des excipients, voir rubrique 6.1.
Solution injectable en stylo prérempli.
Solution incolore, limpide.
pH : 3,5-4,5
Osmolalité : 260-320 mOsmol/kg
4.1. Indications thérapeutiques
TERIPARATIDE TEVA est indiqué chez les adultes.
Traitement de l'ostéoporose chez les femmes ménopausées et les hommes présentant un risque accru de fracture (voir rubrique 5.1). Chez les femmes ménopausées, une réduction significative de l'incidence des fractures vertébrales et périphériques, mais pas des fractures de la hanche, a été démontrée.
Traitement de l’ostéoporose associée à une corticothérapie au long cours par voie générale chez les femmes et les hommes présentant un risque accru de fracture (voir rubrique 5.1).
4.2. Posologie et mode d'administration
La dose recommandée de TERIPARATIDE TEVA est de 20 microgrammes administrés une fois par jour.
La durée totale maximale de traitement avec TERIPARATIDE TEVA doit être de 24 mois (voir rubrique 4.4). Ce cycle de 24 mois par tériparatide ne doit pas être renouvelé au cours de la vie d’un patient.
Les patients doivent être supplémentés en calcium et vitamine D si leurs apports alimentaires sont insuffisants.
Après l'arrêt du traitement par tériparatide, les patients peuvent poursuivre d'autres traitements contre l'ostéoporose.
Populations particulières
Patients avec une insuffisance rénale
Le tériparatide ne doit pas être utilisé chez les patients présentant une insuffisance rénale sévère (voir rubrique 4.3). Chez les patients présentant une insuffisance rénale modérée, le tériparatide doit être utilisé avec prudence. Aucune précaution particulière n’est requise chez les patients ayant une insuffisance rénale légère.
Patients avec une insuffisance hépatique
Aucune donnée n’est disponible chez les patients ayant une altération de la fonction hépatique (voir rubrique 5.3). Par conséquent, le tériparatide doit être utilisé avec prudence.
Population pédiatrique et adultes jeunes dont les épiphyses ne sont pas soudées
La sécurité et l’efficacité du tériparatide chez les enfants et adolescents âgés de moins de 18 ans n’ont pas été établies. Le tériparatide ne doit pas être utilisé chez les patients pédiatriques de moins de 18 ans ou chez les adultes jeunes dont les épiphyses ne sont pas soudées.
Patients âgés
Aucune adaptation posologique liée à l’âge n’est nécessaire (voir rubrique 5.2).
Mode d’administration
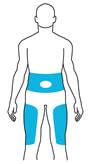
Le tériparatide doit être administré une fois par jour par injection sous-cutanée dans la cuisse ou l’abdomen.
Les patients doivent être formés pour appliquer les bonnes techniques d’injection (voir rubrique 6.6). Un manuel d’utilisation est également disponible pour apprendre aux patients à utiliser correctement le stylo.
· Hypersensibilité à la substance active ou à l'un des excipients mentionnés à la rubrique 6.1.
· Grossesse et allaitement (voir rubriques 4.4 et 4.6)
· Hypercalcémie pré-existante
· Insuffisance rénale sévère
· Maladies métaboliques osseuses (dont l’hyperparathyroïdie et la maladie osseuse de Paget) autres que l’ostéoporose primitive ou l’ostéoporose corticosonique.
· Elévations inexpliquées des phosphatases alcalines
· Antécédent de radiothérapie du squelette par méthode conventionnelle ou par implant
· Chez les patients atteints de tumeurs osseuses malignes ou de métastases osseuses, le traitement par tériparatide est contre-indiqué.
4.4. Mises en garde spéciales et précautions d'emploi
Calcémie et calciurie
Chez les patients normocalcémiques, des augmentations légères et transitoires de la calcémie ont été observées après l'injection de tériparatide. Après chaque dose de tériparatide, la calcémie atteint un maximum en 4 à 6 heures et revient aux valeurs initiales en 16 à 24 heures. Par conséquent, si des prélèvements sanguins sont réalisés pour le dosage de la calcémie, ceux-ci doivent être effectués au moins 16 heures après la dernière injection de TERIPARATIDE TEVA. Il n'est pas nécessaire de surveiller la calcémie pendant le traitement.
Le tériparatide peut induire de légères augmentations de l’excrétion urinaire du calcium mais, dans les études cliniques, l'incidence de l'hypercalciurie n’était pas différente de celle observée chez les patients recevant le placebo.
Lithiase urinaire
Le tériparatide n'a pas été étudié chez les patients ayant une lithiase urinaire évolutive. TERIPARATIDE TEVA doit être utilisé avec prudence chez les patients présentant une lithiase urinaire évolutive ou récente, en raison du risque d'aggravation de cette pathologie.
Hypotension orthostatique
Dans des études cliniques de courte durée avec le tériparatide, des épisodes isolés d'hypotension orthostatique transitoire ont été observés. En règle générale, ce type d'événement débutait dans les 4 heures suivant l'administration et disparaissait spontanément en quelques minutes à quelques heures. Lorsque l'hypotension orthostatique transitoire s'est produite, elle est survenue au cours des premières doses, a été soulagée en plaçant les sujets en position allongée et n'a pas empêché la poursuite du traitement.
Insuffisance rénale
La prudence s’impose chez les patients avec une insuffisance rénale modérée.
Population de jeunes adultes
Les données d’utilisation du tériparatide sont limitées dans la population de jeunes adultes, y compris les femmes non ménopausées (voir rubrique 5.1). Dans cette population, le traitement ne doit être initié que lorsque le bénéfice l’emporte clairement sur les risques.
Les femmes en âge de procréer doivent utiliser des méthodes de contraception efficaces lors de l’utilisation de TERIPARATIDE TEVA. En cas de survenue d’une grossesse, le traitement par TERIPARATIDE TEVA doit être interrompu.
Durée de traitement
Des études chez le rat ont montré une augmentation de l’incidence des ostéosarcomes après administration prolongée de tériparatide (voir rubrique 5.3). Dans l’attente de données cliniques complémentaires, la durée de traitement recommandée de 24 mois ne doit pas être dépassée.
Informations importantes concernant certains des composants de TERIPARATIDE TEVA :
Ce médicament contient moins de 1 mmol (23 mg) de sodium par dose, c’est-à-dire qu’il est essentiellement « sans sodium ».
4.5. Interactions avec d'autres médicaments et autres formes d'interactions
Le tériparatide a fait l'objet d'études d'interactions pharmacodynamiques avec l'hydrochlorothiazide. Aucune interaction cliniquement significative n'a été observée.
La co-administration de raloxifène ou d’un traitement hormonal substitutif avec le tériparatide n'a pas modifié les effets du tériparatide sur la calcémie ou la calciurie, ni les effets indésirables cliniques.
4.6. Fertilité, grossesse et allaitement
Femmes en âge de procréer/Contraception chez les femmes
Les femmes en âge de procréer doivent utiliser des méthodes de contraception efficaces lors de l’utilisation de TERIPARATIDE TEVA. En cas de survenue d’une grossesse, le traitement par TERIPARATIDE TEVA doit être interrompu.
Grossesse
TERIPARATIDE TEVA est contre-indiqué pendant la grossesse (voir rubrique 4.3).
Allaitement
TERIPARATIDE TEVA est contre-indiqué pendant l’allaitement. Le passage du tériparatide dans le lait maternel n’est pas connu.
Fertilité
Des études chez le lapin ont révélé une toxicité pour la reproduction (voir rubrique 5.3). L’effet du tériparatide sur le développement du fœtus humain n’a pas été étudié. Le risque potentiel chez l’être humain est inconnu.
4.7. Effets sur l'aptitude à conduire des véhicules et à utiliser des machines
Résumé du profil de sécurité
Les effets indésirables les plus fréquemment rapportés chez les patients traités par tériparatide sont : nausées, douleurs dans les membres, céphalées et sensations vertigineuses.
Tableau listant les effets indésirables
Parmi les patients inclus dans les essais cliniques menés avec le tériparatide, il a été rapporté au moins 1 événement indésirable chez 82,8 % des patients sous tériparatide et 84,5 % des patients sous placebo.
Les effets indésirables liés à l’utilisation du tériparatide survenus lors des essais cliniques portant sur l’ostéoporose et lors de l’expérience post-commercialisation sont résumés dans le tableau ci-dessous. La convention suivante a été utilisée pour la classification des effets indésirables : très fréquent (≥ 1/10), fréquent (≥ 1/100, < 1/10), peu fréquent (≥ 1/1 000, < 1/100), rare (≥ 1/10 000, < 1/1 000), très rare (< 1/10 000).
|
Affections hématologiques et du système lymphatique Fréquent : anémie |
|
Affections du système immunitaire Rare : anaphylaxie |
|
Troubles du métabolisme et de la nutrition Fréquent : hypercholestérolémie Peu fréquent : hypercalcémie supérieure à 2,76 mmol/L, hyperuricémie Rare : hypercalcémie supérieure à 3,25 mmol/L |
|
Affections psychiatriques Fréquent : dépression |
|
Affections du système nerveux Fréquent : sensations vertigineuses, céphalées, sciatique, syncope |
|
Affections de l’oreille et du labyrinthe Fréquent : vertiges |
|
Affections cardiaques Fréquent : palpitations Peu fréquent : tachycardie |
|
Affections vasculaires Fréquent : hypotension |
|
Affections respiratoires, thoraciques et médiastinales Fréquent : dyspnée Peu fréquent : emphysème |
|
Affections gastro-intestinales Fréquent : nausées, vomissements, hernie hiatale, maladie de reflux gastro-œsophagien Peu fréquent : hémorroïdes |
|
Affections de la peau et du tissu sous-cutané Fréquent : hypersudation |
|
Affections musculo-squelettiques et systémiques Très fréquent : douleurs dans les membres Fréquent : crampes musculaires Peu fréquent : myalgies, arthralgies, crampes/douleurs dorsales* |
|
Affections du rein et des voies urinaires Peu fréquent : incontinence urinaire, polyurie, impériosité mictionnelle, lithiase rénale Rare : insuffisance rénale/dysfonction rénale |
|
Troubles généraux et anomalies au site d’administration Fréquent : fatigue, douleur thoracique, asthénie, manifestations légères et transitoires au site d’injection incluant douleur, gonflement, érythème, hématomes localisés, prurit et saignement mineur au site d’injection Peu fréquent : érythème au site d’injection, réaction au site d’injection Rare : réactions allergiques possibles peu après l’injection : dyspnée aiguë, œdème bucco-facial, urticaire généralisée, douleurs thoraciques, œdèmes (principalement périphériques). |
|
Investigations Peu fréquent : prise de poids, souffle cardiaque, augmentation des phosphatases alcalines |
* Des cas graves de crampes ou de douleurs dorsales ont été rapportés dans les minutes suivant l’injection.
Description de certains effets indésirables
Dans les essais cliniques, les effets suivants ont été rapportés avec une différence de fréquence ≥ 1 % par rapport au placebo : vertiges, nausées, douleurs dans les membres, sensations vertigineuses, dépression, dyspnée.
Le tériparatide augmente les concentrations sériques d’acide urique. Dans les essais cliniques, 2,8 % des patientes traitées par tériparatide présentaient une uricémie au-dessus de la limite supérieure de la normale, par rapport à 0,7 % des patientes sous placebo. Cependant, cette hyperuricémie n’a pas entraîné d’augmentation des crises de goutte, d’arthralgies ou de lithiase urinaire.
Dans un vaste essai clinique, des anticorps dirigés contre tériparatide ont été détectés chez 2,8 % des femmes recevant le tériparatide. Généralement, les anticorps ont été détectés pour la première fois après 12 mois de traitement et leur taux a diminué après l’arrêt du traitement. Il n’a pas été mis en évidence de réactions d’hypersensibilité, de réactions allergiques, d’effets sur la calcémie ou sur les variations de densité minérale osseuse (DMO).
Déclaration des effets indésirables suspectés
La déclaration des effets indésirables suspectés après autorisation du médicament est importante. Elle permet une surveillance continue du rapport bénéfice/risque du médicament. Les professionnels de santé déclarent tout effet indésirable suspecté via le système national de déclaration : Agence nationale de sécurité du médicament et des produits de santé (ANSM) et réseau des Centres Régionaux de Pharmacovigilance - Site internet : https://signalement.social-sante.gouv.fr/.
Signes et symptômes
Le tériparatide a été administré en doses uniques allant jusqu'à 100 microgrammes et en doses répétées allant jusqu’à 60 microgrammes/jour pendant 6 semaines.
Les effets pouvant être attendus lors d’un surdosage sont une hypercalcémie différée dans le temps et un risque d’hypotension orthostatique. Peuvent aussi survenir : nausées, vomissements, sensations vertigineuses et céphalées.
Cas de surdosage basés sur les notifications spontanées après commercialisation
Dans les notifications spontanées après commercialisation, des cas d’erreur d’administration ont été observés, où la quantité totale de tériparatide contenue dans le stylo (jusqu’à 800 microgrammes) a été injectée en une seule fois. Des effets indésirables transitoires tels que des nausées, une faiblesse/somnolence et une hypotension ont été rapportés. Dans certains cas, le surdosage n’a entraîné aucun effet indésirable. Aucun décès lié à un surdosage n’a été rapporté.
Conduite à tenir en cas de surdosage
Il n'existe pas d'antidote spécifique pour le tériparatide. En cas de suspicion d’un surdosage, il convient d'interrompre temporairement le traitement par TERIPARATIDE TEVA, de surveiller la calcémie et de mettre en œuvre une prise en charge appropriée, telle qu’une réhydratation.
5. PROPRIETES PHARMACOLOGIQUES
5.1. Propriétés pharmacodynamiques
Mécanisme d’action
La parathormone (PTH) endogène de 84 acides aminés, est le principal régulateur du métabolisme phosphocalcique au niveau osseux et rénal. Le tériparatide représente le fragment actif (1-34) de la parathormone humaine endogène. Les actions physiologiques de la PTH incluent la stimulation de la formation osseuse par des effets directs sur les cellules de la formation osseuse (ostéoblastes), augmentant indirectement l'absorption intestinale du calcium, la réabsorption tubulaire du calcium et l'excrétion rénale du phosphate.
Effets pharmacodynamiques
Le tériparatide est un agent de la formation osseuse, destiné au traitement de l’ostéoporose. Les effets du tériparatide sur le squelette dépendent du mode d'exposition systémique. L'administration du tériparatide une fois par jour accroît l'apposition d’os nouvellement formé à la surface de l’os trabéculaire et cortical, en stimulant préférentiellement l'activité ostéoblastique par rapport à l'activité ostéoclastique.
Efficacité clinique
Facteurs de risque
L’identification des femmes et des hommes à risque accru de fractures ostéoporotiques qui pourraient bénéficier d’un traitement doit être faite sur la base des facteurs de risque indépendants, par exemple, une densité minérale osseuse (DMO) basse, l’âge, des antécédents de fractures, des antécédents familiaux de fracture de hanche, un remodelage osseux élevé ou un indice de masse corporelle bas.
Les femmes non ménopausées atteintes d’ostéoporose cortisonique doivent être considérées à haut risque de fracture en présence d’antécédent de fracture ou de facteurs de risque multiples les exposant à un risque élevé de fracture (comme une faible densité osseuse [par exemple, T score ≤ −2], un traitement prolongé à fortes doses de glucocorticoïdes [par exemple, ≥ 7,5 mg/jour pour une durée d’au moins 6 mois], une forte activité de la maladie sous-jacente, de faibles taux de stéroïdes sexuels).
Ostéoporose post-ménopausique
L'étude pivot a inclus 1 637 femmes ménopausées (âge moyen de 69,5 ans). A l’inclusion, quatre-vingt-dix pour cent des patientes avaient une ou plusieurs fractures vertébrales et, en moyenne, une DMO vertébrale de 0,82 g/cm2 (équivalente à un T score à -2,6). Toutes les patientes ont reçu 1 000 mg de calcium par jour et au moins 400 UI de vitamine D par jour. Pour une période de traitement par tériparatide allant jusqu’à 24 mois (médiane : 19 mois), les résultats démontrent une réduction fracturaire statistiquement significative (tableau 1). Pour prévenir la survenue d’une ou plusieurs nouvelles fractures vertébrales, 11 femmes ont dû être traitées pendant une période médiane de 19 mois.
Tableau 1
|
Incidence des fractures chez les femmes ménopausées : |
|||
|
|
Placebo (N = 544) (%) |
Tériparatide (N = 541) (%) |
Risque relatif (IC à 95 %) vs placebo |
|
Nouvelle fracture vertébrale (≥ 1)a |
14,3 |
5,0b |
0,35 (0,22 - 0,55) |
|
Fractures vertébrales multiples (≥ 2)a |
4,9 |
1,1b |
0,23 (0,09 - 0,60) |
|
Fractures périphériques par fragilité osseusec |
5,5 |
2,6d |
0,47 (0,25 - 0,87) |
|
Principales fractures périphériques par fragilité osseusec (hanche, radius, humérus, côtes et bassin) |
3,9 |
1,5d |
0,38 (0,17 - 0,86) |
Abréviations : N = nombre de patientes randomisées affectées à chaque groupe de traitement ; IC = intervalle de confiance.
a L’incidence des fractures vertébrales a été évaluée chez 448 patientes sous placebo et 444 patientes sous tériparatide présentant des radiographies de la colonne vertébrale à l’inclusion et au cours du suivi.
b p ≤ 0,001 par rapport au placebo.
c Une réduction significative de l’incidence des fractures de hanche n’a pas été démontrée.
d p ≤ 0,025 par rapport au placebo.
Après 19 mois de traitement (durée médiane), la densité minérale osseuse (DMO) a augmenté au niveau du rachis lombaire et de la hanche totale respectivement de 9 % et 4 % par rapport au placebo (p < 0,001).
Prise en charge post-traitement : suite à l’arrêt du traitement par tériparatide, 1 262 femmes ménopausées de l'étude pivot ont participé à une étude de suivi post-traitement. L’objectif principal de cette étude était de recueillir des données de sécurité sur le tériparatide. Durant cette période d’observation, d’autres traitements contre l’ostéoporose étaient autorisés et une évaluation complémentaire des fractures vertébrales a été réalisée.
Au cours d’une période d’une durée médiane de 18 mois après l’arrêt du tériparatide, une réduction de 41 % du nombre de patientes avec au moins une nouvelle fracture vertébrale (p = 0,004) par rapport au placebo a été observée.
Dans une étude en ouvert, 503 femmes ménopausées atteintes d’ostéoporose sévère et ayant eu une fracture par fragilité osseuse au cours des 3 années précédentes (83 % avaient reçu un traitement ostéoporotique antérieur) ont été traitées par tériparatide pendant une période allant jusqu’à 24 mois. A 24 mois, l’augmentation moyenne de la DMO par rapport à l’inclusion, au niveau du rachis lombaire, de la hanche totale et du col fémoral était respectivement de 10,5 %, 2,6 % et 3,9 %. L’augmentation moyenne de la DMO entre 18 et 24 mois était respectivement de 1,4 %, 1,2 % et 1,6 % au niveau du rachis lombaire, de la hanche totale et du col fémoral.
Une étude de phase 4 d’une durée de 24 mois, randomisée, en double aveugle et contrôlée avec un comparateur a inclus 1 360 femmes ménopausées avec un diagnostic d’ostéoporose. 680 sujets ont été randomisés dans le bras tériparatide et 680 sujets ont été randomisés dans le bras risédronate par voie orale à 35 mg/semaine. A l’inclusion, les femmes avaient une moyenne d’âge de 72,1 ans et avaient une médiane de 2 fractures vertébrales prévalentes ; 57,9 % des patientes ont reçu un traitement antérieur à base de bisphosphonates et 18,8 % ont pris un traitement concomitant de glucocorticoïdes pendant l’étude. 1 013 (74,5 %) patientes ont atteint le suivi des 24 mois. La dose cumulative moyenne (médiane) de glucocorticoïdes était de 474,3 (66,2) mg dans le bras tériparatide et de 898,0 (100,0) mg dans le bras risédronate. L’apport moyen (médian) en vitamine D était de 1 433 UI/jour (1 400 UI/jour) dans le bras tériparatide et de 1 191 UI/jour (900 UI/jour) dans le bras risédronate. Pour les sujets qui avaient des radiographies de la colonne vertébrale à l’inclusion et pendant le suivi, l’incidence de nouvelles fractures vertébrales était de 28/516 (5,4 %) chez les patientes traitées par tériparatide et de 64/533 (12,0 %) chez les patientes traitées par risédronate ; risque relatif (IC à 95 %) = 0,44 (0,29-0,68), p < 0,0001.
L’incidence cumulative de l’ensemble des fractures cliniques (fractures cliniques vertébrales et périphériques) était de 4,8 % chez les patientes traitées par tériparatide et de 9,8 % chez les patientes traitées par risédronate, rapport de risque (IC à 95 %) = 0,48 (0,32-0,74), p = 0,0009.
Ostéoporose masculine
437 patients (âge moyen 58,7 ans) atteints d’ostéoporose hypogonadique (définie par un taux matinal faible de testostérone libre ou une FSH ou LH élevée) ou idiopathique ont été inclus dans un essai clinique mené chez les hommes. A l’inclusion, les T scores moyens de la densité minérale osseuse au niveau du rachis et du col fémoral étaient respectivement de -2,2 et -2,1. A l’inclusion, 35 % des patients avaient une fracture vertébrale et 59 % avaient une fracture périphérique.
Tous les patients ont reçu 1 000 mg de calcium par jour et au moins 400 UI de vitamine D par jour. La DMO mesurée au niveau du rachis lombaire a significativement augmenté après 3 mois. Après 12 mois, la DMO au niveau du rachis lombaire et de la hanche totale a augmenté respectivement de 5 % et de 1 % par rapport au placebo. Cependant, aucun effet significatif sur les taux de fracture n’a été démontré.
Ostéoporose cortisonique
L’efficacité du tériparatide chez les hommes et les femmes (N = 428) recevant une corticothérapie au long cours par voie générale (équivalant à 5 mg ou plus de prednisone pendant au moins 3 mois) a été démontrée au cours de la phase principale de 18 mois d’une étude randomisée d’une durée de 36 mois, effectuée en double-aveugle et contrôlée par un comparateur (alendronate 10 mg/jour). A l’inclusion, 28 % des patients avaient une ou plusieurs fractures vertébrales radiographiques. Tous les patients ont reçu 1 000 mg de calcium par jour et 800 UI de vitamine D par jour.
Cette étude a inclus des femmes ménopausées (N = 277), des femmes non ménopausées (N = 67), et des hommes (N = 83). A l’inclusion, l’âge moyen des femmes ménopausées était de 61 ans, leur T score moyen de DMO lombaire était de -2,7, la dose médiane de traitement était de 7,5 mg/jour équivalent prednisone, et 34 % d’entre elles avaient eu une ou plusieurs fractures vertébrales radiographiques ; l’âge moyen des femmes non ménopausées était de 37 ans, leur T score moyen de DMO lombaire était de -2,5, la dose médiane de traitement était de 10 mg/jour équivalent prednisone, et 9 % d’entre elles avaient une ou plusieurs fractures vertébrales radiographiques ; enfin, l’âge moyen des hommes était de 57 ans, leur T score moyen de DMO lombaire était de -2,2, la dose médiane de traitement était de 10 mg/jour équivalent prednisone, et 24 % d’entre eux avaient une ou plusieurs fractures vertébrales radiographiques.
Soixante-neuf pour cent des patients ont terminé la phase principale de 18 mois. Au terme de ces 18 mois, le tériparatide avait significativement augmenté la DMO au niveau du rachis lombaire (7,2 %) par rapport à l’alendronate (3,4 %) (p < 0,001). Le tériparatide a entraîné une augmentation de la DMO au niveau de la hanche totale (3,6 %) par rapport à l’alendronate (2,2 %) (p < 0,01) et de la DMO au niveau du col fémoral (3,7 %) par rapport à l’alendronate (2,1 %) (p < 0,05). Chez les patients traités avec tériparatide, la DMO au niveau du rachis lombaire, de la hanche totale et du col fémoral a augmenté, respectivement, de 1,7 %, 0,9 % et 0,4 % entre 18 et 24 mois.
A 36 mois, l’analyse des radiographies du rachis portant sur 169 patients sous alendronate et 173 patients sous tériparatide a révélé que 13 patients du groupe alendronate (7,7 %) avaient eu une nouvelle fracture vertébrale, contre 3 patients du groupe tériparatide (1,7 %) (p = 0,01).De plus, 15 patients sur 214 du groupe alendronate (7,0 %) avaient eu une fracture périphérique, contre 16 patients sur 214 du groupe tériparatide (7,5 %) (p = 0,84).
Chez les femmes non ménopausées, l’augmentation de la DMO entre l’inclusion et la fin de 18 mois d’étude était significativement plus importante dans le groupe tériparatide que dans le groupe alendronate au niveau du rachis lombaire (4,2 % versus -1,9 % ; p < 0,001) et de la hanche totale (3,8 % versus 0,9 % ; p = 0,005). Cependant, aucun effet significatif sur le taux de fracture n’a été démontré.
5.2. Propriétés pharmacocinétiques
Le volume de distribution est d’environ 1,7 L/kg. La demi-vie du tériparatide est d'environ 1 heure après administration sous-cutanée, ce qui traduit le temps nécessaire à l’absorption à partir du site d’injection.
Biotransformation
Aucune étude de métabolisme ni d'élimination n'a été conduite avec le tériparatide, toutefois, le métabolisme périphérique de la parathormone semble être principalement hépatique et rénal.
Elimination
Le tériparatide est éliminé par clairance hépatique et extra-hépatique (environ 62 L/h chez les femmes et 94 L/h chez les hommes).
Personnes âgées
Aucune différence de pharmacocinétique du tériparatide n’a été détectée en fonction de l’âge des patients (de 31 à 85 ans). Aucune adaptation posologique liée à l’âge n’est nécessaire.
5.3. Données de sécurité préclinique
Des rats traités pendant presque toute leur durée de vie par des injections quotidiennes ont présenté une augmentation excessive dose-dépendante de la formation osseuse et une augmentation de l’incidence des cas d'ostéosarcome, dues vraisemblablement à un mécanisme épigénétique. Le tériparatide n'a pas augmenté l'incidence des autres types de tumeurs malignes chez le rat. En raison des différences de physiologie osseuse entre les rats et les êtres humains, la pertinence clinique de ces observations est probablement mineure. Aucune tumeur osseuse n'a été observée chez des guenons ovariectomisées traitées pendant 18 mois ou pendant la période de suivi de 3 ans après l’arrêt du traitement. De plus, aucun cas d’ostéosarcome n'a été observé lors des essais cliniques ou au cours de l’étude de suivi post-traitement.
Les études chez l'animal ont montré qu'une réduction importante du débit sanguin hépatique diminue l'exposition de la PTH au principal système de clivage (cellules de Kupffer) et, par conséquent, la clairance de la PTH (1-84).
2 ans
La stabilité chimique, physique et microbiologique du produit en cours d’utilisation a été démontrée pendant 28 jours entre 2 °C et 8 °C. Une fois ouvert, le produit peut être conservé pendant 28 jours entre 2 °C et 8 °C. En cours d’utilisation, toute autre condition de durée et de conservation relève de la responsabilité de l’utilisateur.
6.4. Précautions particulières de conservation
Ne pas conserver le dispositif d’injection avec l’aiguille fixée dessus.
Produit non ouvert
Le produit non ouvert peut être retiré du réfrigérateur et conservé à une température ne dépassant pas 25 °C pendant une seule période de 5 jours maximum, après quoi il doit être remis au réfrigérateur (2 °C– 8 °C). Eliminer le produit non ouvert s’il est conservé à une température supérieure à 8 °C pendant plus de 5 jours.
6.5. Nature et contenu de l'emballage extérieur
2,4 mL de solution en cartouche (verre de type I siliconé), avec un piston (halobutyl de caoutchouc) et un joint (laminé de polyisoprène/bromobutyl de caoutchouc)/aluminium assemblés en stylo jetable.
TERIPARATIDE TEVA est disponible en boîtes de 1 ou 3 stylos ou en conditionnement multiple contenant 3 stylos (3 boîtes de 1 stylo). Chaque stylo contient 28 doses de 20 microgrammes (par 80 microlitres).
Toutes les présentations peuvent ne pas être commercialisées.
6.6. Précautions particulières d’élimination et de manipulation
TERIPARATIDE TEVA est fourni dans un stylo prérempli. Chaque stylo doit être utilisé par un seul patient.
Dans chaque boîte de TERIPARATIDE TEVA, un manuel d’utilisation décrivant en détail le mode d’emploi du stylo est fourni.
Une nouvelle aiguille stérile doit être utilisée à chaque injection. Aucune aiguille n’est fournie avec le produit. Le dispositif peut être utilisé avec des aiguilles pour stylo injecteur d’insuline.
Après chaque injection, le stylo TERIPARATIDE TEVA doit être remis au réfrigérateur.
TERIPARATIDE TEVA ne doit pas être utilisé si la solution est trouble, colorée ou si elle contient des particules.
Veuillez consulter également le manuel d’utilisation pour savoir comment utiliser le stylo.
Tout médicament non utilisé ou déchet doit être éliminé conformément à la réglementation en vigueur.
7. TITULAIRE DE L’AUTORISATION DE MISE SUR LE MARCHE
SWENSWEG 5
2031GA HAARLEM
PAYS-BAS
8. NUMERO(S) D’AUTORISATION DE MISE SUR LE MARCHE
· 34009 302 827 1 7 : 2,4 mL (28 doses) en stylo prérempli. Boîte de 1.
9. DATE DE PREMIERE AUTORISATION/DE RENOUVELLEMENT DE L’AUTORISATION
[à compléter ultérieurement par le titulaire]
10. DATE DE MISE A JOUR DU TEXTE
[à compléter ultérieurement par le titulaire]
Sans objet.
12. INSTRUCTIONS POUR LA PREPARATION DES RADIOPHARMACEUTIQUES
Liste I
ANSM - Mis à jour le : 22/05/2025
TERIPARATIDE TEVA 20 microgrammes/80 microlitres, solution injectable en stylo prérempli
Tériparatide
· Gardez cette notice. Vous pourriez avoir besoin de la relire.
· Si vous avez d’autres questions, interrogez votre médecin ou votre pharmacien.
· Ce médicament vous a été personnellement prescrit. Ne le donnez pas à d’autres personnes. Il pourrait leur être nocif, même si les signes de leur maladie sont identiques aux vôtres.
· Si vous ressentez un quelconque effet indésirable, parlez-en à votre médecin ou votre pharmacien. Ceci s’applique aussi à tout effet indésirable qui ne serait pas mentionné dans cette notice. Voir rubrique 4.
1. Qu'est-ce que TERIPARATIDE TEVA 20 microgrammes/80 microlitres, solution injectable en stylo prérempli et dans quels cas est-il utilisé ?
2. Quelles sont les informations à connaître avant d'utiliser TERIPARATIDE TEVA 20 microgrammes/ 80 microlitres, solution injectable en stylo prérempli ?
3. Comment utiliser TERIPARATIDE TEVA 20 microgrammes/80 microlitres, solution injectable en stylo prérempli ?
4. Quels sont les effets indésirables éventuels ?
5. Comment conserver TERIPARATIDE TEVA 20 microgrammes/80 microlitres, solution injectable en stylo prérempli ?
6. Contenu de l’emballage et autres informations.
1. QU’EST-CE QUE TERIPARATIDE TEVA 20 microgrammes/80 microlitres, solution injectable en stylo prérempli ET DANS QUELS CAS EST-IL UTILISE ?
TERIPARATIDE TEVA contient la substance active tériparatide qui est utilisée pour renforcer les os et réduire le risque de fractures en stimulant la formation osseuse.
TERIPARATIDE TEVA est utilisé pour traiter l'ostéoporose chez les adultes. L'ostéoporose est une maladie qui rend vos os fins et fragiles. Cette maladie touche particulièrement les femmes après la ménopause, mais elle peut également toucher les hommes. L’ostéoporose est également fréquente chez les patients recevant des corticoïdes.
2. QUELLES SONT LES INFORMATIONS A CONNAITRE AVANT D’UTILISER TERIPARATIDE TEVA 20 microgrammes/80 microlitres, solution injectable en stylo prérempli ?
· si vous êtes allergique au tériparatide ou à l’un des autres composants contenus dans ce médicament, mentionnés dans la rubrique 6.
· si vous avez des taux de calcium élevés (hypercalcémie préexistante).
· si vous souffrez de graves problèmes rénaux.
· si, dans votre cas, le diagnostic de cancer des os ou de tout autre cancer étendu (métastasé) aux os a été établi.
· si vous avez certaines maladies osseuses. Si vous avez une maladie osseuse, parlez-en à votre médecin.
· si vous présentez des taux élevés et inexpliqués de phosphatase alcaline dans le sang, ce qui signifie que vous pourriez être atteint(e) de la maladie osseuse de Paget (maladie caractérisée par des modifications osseuses anormales). En cas de doute, parlez-en à votre médecin.
· si vous avez subi un traitement par radiothérapie au niveau des os.
· si vous êtes enceinte ou si vous allaitez.
Avertissements et précautions
TERIPARATIDE TEVA peut entraîner une augmentation de la quantité de calcium dans votre sang ou dans vos urines.
Adressez-vous à votre médecin ou pharmacien avant ou pendant l’utilisation de TERIPARATIDE TEVA :
· si vous avez continuellement des nausées, des vomissements, une constipation, une baisse d’énergie ou une faiblesse musculaire. Cela pourrait être des signes d’excès de calcium dans le sang.
· si vous avez ou avez eu des calculs rénaux.
· si vous souffrez de problèmes rénaux (insuffisance rénale modérée).
Quelques patients ont eu des sensations vertigineuses ou des palpitations après les premières doses. Pour les premières doses, faites l’injection de TERIPARATIDE TEVA à un endroit où vous pourrez vous asseoir ou vous allonger en cas de sensations vertigineuses.
La durée de traitement recommandée de 24 mois ne doit pas être dépassée.
TERIPARATIDE TEVA ne doit pas être utilisé chez les adultes en période de croissance.
Enfants et adolescents
TERIPARATIDE TEVA ne doit pas être utilisé chez les enfants et les adolescents (de moins de 18 ans).
Autres médicaments et TERIPARATIDE TEVA 20 microgrammes/80 microlitres, solution injectable en stylo prérempli
Informez votre médecin ou pharmacien si vous prenez, avez récemment pris ou pourriez prendre tout autre médicament, car il peut parfois se produire des interactions médicamenteuses (ex : digoxine/ digitaline, un médicament utilisé pour traiter les maladies cardiaques).
TERIPARATIDE TEVA 20 microgrammes/80 microlitres, solution injectable en stylo prérempli avec des aliments et boissons
Sans objet.
Grossesse, allaitement et fertilité
N’utilisez pas TERIPARATIDE TEVA si vous êtes enceinte ou si vous allaitez. Si vous êtes une femme susceptible d’avoir des enfants, vous devez utiliser des méthodes de contraception efficaces lors de l’utilisation de TERIPARATIDE TEVA. Si vous êtes enceinte, le traitement par TERIPARATIDE TEVA doit être interrompu. Demandez conseil à votre médecin ou à votre pharmacien avant de prendre tout médicament.
Conduite de véhicules et utilisation de machines
Certains patients peuvent ressentir une sensation vertigineuse après l’injection de TERIPARATIDE TEVA. Si vous ressentez cette sensation vertigineuse, vous ne devez pas conduire de véhicules ni utiliser de machines jusqu’à ce que vous vous sentiez mieux.
TERIPARATIDE TEVA 20 microgrammes/80 microlitres, solution injectable en stylo prérempli contient du sodium
Ce médicament contient moins de 1 mmol (23 mg) de sodium par dose, c’est-à-dire qu’il est essentiellement « sans sodium ».
3. COMMENT UTILISER TERIPARATIDE TEVA 20 microgrammes/80 microlitres, solution injectable en stylo prérempli ?
La dose recommandée est de 20 microgrammes administrés une fois par jour par injection sous la peau (injection sous-cutanée) au niveau de la cuisse ou de l’abdomen. Pour ne pas oublier d’utiliser votre médicament, effectuez l’injection à peu près à la même heure chaque jour.
Effectuez une injection de TERIPARATIDE TEVA chaque jour pendant toute la durée de traitement prescrite par votre médecin. La durée totale du traitement par TERIPARATIDE TEVA ne doit pas dépasser 24 mois. Vous ne devez pas recevoir plus de 24 mois de traitement au cours de votre vie.
TERIPARATIDE TEVA peut être injecté aux heures des repas.
Lisez le manuel d’utilisation inclus dans la boîte pour connaître le mode d’emploi du stylo TERIPARATIDE TEVA.
Les aiguilles pour injection ne sont pas fournies avec le stylo. Les aiguilles pour stylo des Laboratoires Becton-Dickinson de calibre 29 à 31 (diamètre : 0,25-0,33 mm) et de longueur 12,7 ; 8 ou 5 mm peuvent être utilisées.
Vous devez effectuer votre injection de TERIPARATIDE TEVA peu de temps après avoir sorti le stylo du réfrigérateur, comme indiqué dans le manuel d'utilisation. Replacez le stylo au réfrigérateur immédiatement après l’avoir utilisé.
Utilisez une nouvelle aiguille pour injection à chaque injection et jetez-la après chaque utilisation. Ne conservez jamais le stylo avec l'aiguille fixée dessus. Ne partagez jamais votre stylo TERIPARATIDE TEVA avec d’autres personnes.
Votre médecin pourra vous conseiller de prendre TERIPARATIDE TEVA avec du calcium et de la vitamine D. Votre médecin vous indiquera la quantité à prendre chaque jour.
TERIPARATIDE TEVA peut être administré au cours ou en dehors des repas.
Si vous avez utilisé plus de TERIPARATIDE TEVA 20 microgrammes/80 microlitres, solution injectable en stylo prérempli que vous n’auriez dû
Si, par erreur, vous avez utilisé plus de TERIPARATIDE TEVA que vous n’auriez dû, contactez votre médecin ou votre pharmacien.
Les effets pouvant être liés à un surdosage incluent des nausées, des vomissements, des sensations vertigineuses et des maux de tête.
Si vous arrêtez d’utiliser TERIPARATIDE TEVA 20 microgrammes/80 microlitres, solution injectable en stylo prérempli
Si vous envisagez d’arrêter le traitement par TERIPARATIDE TEVA, parlez-en avec votre médecin. Votre médecin vous conseillera et décidera de la durée du traitement par TERIPARATIDE TEVA.
Si vous avez d’autres questions sur l’utilisation de ce médicament, demandez plus d’informations à votre médecin ou à votre pharmacien.
4. QUELS SONT LES EFFETS INDESIRABLES EVENTUELS ?
Les effets indésirables les plus fréquents sont : douleurs dans les membres (très fréquent, pouvant affecter plus de 1 personne sur 10), nausées, maux de tête et sensations vertigineuses (fréquent). Si vous ressentez des étourdissements (sensation de vertige) après votre injection, vous devez vous asseoir ou vous allonger jusqu’à ce que vous vous sentiez mieux. Si vous ne vous sentez pas mieux, vous devez contacter un médecin avant de poursuivre le traitement. Des cas de malaise, associés à l’utilisation du tériparatide, ont été rapportés.
Si vous ressentez quelques désagréments au niveau du site d’injection, tels que rougeur cutanée, douleur, gonflement, démangeaisons, hématome ou saignement sans gravité (fréquent), cela devrait disparaître en quelques jours ou semaines. Dans le cas contraire, prévenez votre médecin dès que possible.
Certains patients ont présenté des réactions allergiques peu après l’injection, à type d’essoufflement, de gonflement du visage, d’éruption cutanée et de douleur dans la poitrine (rare). Dans de rares cas, des réactions allergiques graves et pouvant potentiellement mettre en jeu le pronostic vital, incluant l’anaphylaxie, peuvent se produire. Si vous présentez l'un de ces symptômes, ARRETEZ d’utiliser TERIPARATIDE TEVA et contactez IMMEDIATEMENT votre médecin.
Autres effets indésirables :
Fréquent : pouvant affecter jusqu’à 1 personne sur 10
· augmentation des taux de cholestérol sanguin ;
· dépression ;
· douleur névralgique dans les jambes ;
· sensation de faiblesse ;
· battements cardiaques irréguliers ;
· essoufflement ;
· augmentation de la transpiration ;
· crampes musculaires ;
· perte d’énergie ;
· fatigue ;
· douleur thoracique ;
· tension artérielle basse ;
· brûlures d’estomac (sensation de douleur ou de brûlure juste en-dessous du sternum) ;
· mal au cœur (vomissements) ;
· hernie au niveau du tube qui transporte la nourriture à l’estomac ;
· taux d’hémoglobine ou de globules rouges faible (anémie).
Peu fréquent : pouvant affecter jusqu’à 1 personne sur 100
· accélération du rythme cardiaque ;
· bruit anormal du cœur ;
· accélération de la respiration ;
· hémorroïdes ;
· pertes d’urine accidentelles ou fuites urinaires ;
· besoin accru d’uriner ;
· prise de poids ;
· calculs rénaux ;
· douleurs musculaires et articulaires. Certains patients ont présenté de sévères crampes ou douleurs dans le dos, ayant nécessité une hospitalisation ;
· augmentation du taux de calcium dans le sang ;
· augmentation du taux d’acide urique dans le sang ;
· augmentation du taux d’une enzyme appelée phosphatase alcaline.
Rare : pouvant affecter jusqu’à 1 personne sur 1 000
· diminution de la fonction rénale, dont l’insuffisance rénale ;
· gonflement, principalement au niveau des mains, des pieds et des jambes.
Déclaration des effets secondaires
Si vous ressentez un quelconque effet indésirable, parlez-en à votre médecin ou votre pharmacien. Ceci s’applique aussi à tout effet indésirable qui ne serait pas mentionné dans cette notice. Vous pouvez également déclarer les effets indésirables directement via le système national de déclaration : Agence nationale de sécurité du médicament et des produits de santé (ANSM) et réseau des Centres Régionaux de Pharmacovigilance - Site internet : https://signalement.social-sante.gouv.fr/
En signalant les effets indésirables, vous contribuez à fournir davantage d’informations sur la sécurité du médicament.
5. COMMENT CONSERVER TERIPARATIDE TEVA 20 microgrammes/80 microlitres, solution injectable en stylo prérempli ?
Tenir ce médicament hors de la vue et de la portée des enfants.
N’utilisez pas ce médicament après la date de péremption indiquée sur la boîte et le stylo après EXP. La date de péremption fait référence au dernier jour de ce mois.
TERIPARATIDE TEVA doit être conservé au réfrigérateur (entre 2 °C et 8 °C). Vous pouvez utiliser TERIPARATIDE TEVA dans un délai de 28 jours maximum après la première injection, tant que le stylo est conservé au réfrigérateur (entre 2 °C et 8 °C).
Ne pas congeler TERIPARATIDE TEVA. Eviter de placer les stylos à proximité du compartiment à glace du réfrigérateur pour éviter toute congélation. Ne pas utiliser TERIPARATIDE TEVA s’il est ou a été congelé.
Chaque stylo doit être éliminé selon la réglementation en vigueur au bout de 28 jours, même s’il n’est pas complètement vide.
TERIPARATIDE TEVA contient une solution limpide et incolore. N’utilisez pas TERIPARATIDE TEVA s'il présente des particules solides ou si la solution est trouble ou colorée.
Produit non ouvert
Le produit non ouvert peut être retiré du réfrigérateur et conservé à une température ne dépassant pas 25 °C pendant une seule période de 5 jours maximum, après quoi il doit être remis au réfrigérateur (2 °C– 8 °C). Eliminer le produit non ouvert s’il est conservé à une température supérieure à 8 °C pendant plus de 5 jours.
Ne jetez aucun médicament au tout-à-l’égout ou avec les ordures ménagères. Demandez à votre pharmacien d’éliminer les médicaments que vous n’utilisez plus. Ces mesures contribueront à protéger l’environnement.
6. CONTENU DE L’EMBALLAGE ET AUTRES INFORMATIONS
· La substance active est le tériparatide (sous forme d’acétate de tériparatide).
Chaque dose de 80 microlitres contient 20 microgrammes de tériparatide (sous forme d’acétate de tériparatide).
Un stylo prérempli de 2,4 mL contient 600 microgrammes de tériparatide (correspondant à 250 microgrammes par mL).
· Les autres composants sont :
Acide acétique glacial, acétate de sodium trihydraté, mannitol, métacrésol et eau pour préparations injectables. De plus, une solution d’acide chlorhydrique et/ou d’hydroxyde de sodium a pu être ajoutée pour ajuster le pH.
TERIPARATIDE TEVA est une solution incolore et limpide. Il est présenté en cartouche contenue dans un stylo prérempli jetable. Chaque stylo contient 2,4 mL de solution, soit 28 doses.
Les stylos sont disponibles en boîtes contenant 1 ou 3 stylos ou en conditionnement multiple contenant 3 stylos (3 boîtes de 1 stylo).
Toutes les présentations peuvent ne pas être commercialisées.
Titulaire de l’autorisation de mise sur le marché
SWENSWEG 5
2031GA HAARLEM
PAYS-BAS
Exploitant de l’autorisation de mise sur le marché
100-110, esplanade du Général de Gaulle
92931 Paris La Défense Cedex
PRILAZ BARUNA FILIPOVICA 25
ZAGREB, 10000
CROATIE
Noms du médicament dans les Etats membres de l'Espace Economique Européen
Ce médicament est autorisé dans les Etats membres de l'Espace Economique Européen sous les noms suivants : Conformément à la réglementation en vigueur.
[À compléter ultérieurement par le titulaire]
La dernière date à laquelle cette notice a été révisée est :
[à compléter ultérieurement par le titulaire]
{mois AAAA}.
Des informations détaillées sur ce médicament sont disponibles sur le site Internet de l’ANSM (France).
Stylo TERIPARATIDE TEVA
Tériparatide injectable en stylo prérempli
Important : veuillez commencer par lire la notice du médicament incluse dans votre boîte de TERIPARATIDE TEVA.
Avant d’utiliser votre nouveau stylo TERIPARATIDE TEVA, veuillez lire l’intégralité du recto et du verso de ce manuel d’utilisation. Suivez attentivement les instructions lorsque vous utilisez le stylo TERIPARATIDE TEVA.
Le stylo TERIPARATIDE TEVA contient 28 jours de traitement. Eliminez le stylo TERIPARATIDE TEVA au bout de 28 jours, même s’il n’est pas complètement vide. N’effectuez pas plus d’une injection de TERIPARATIDE TEVA le même jour.
Ne transférez pas TERIPARATIDE TEVA dans une seringue.
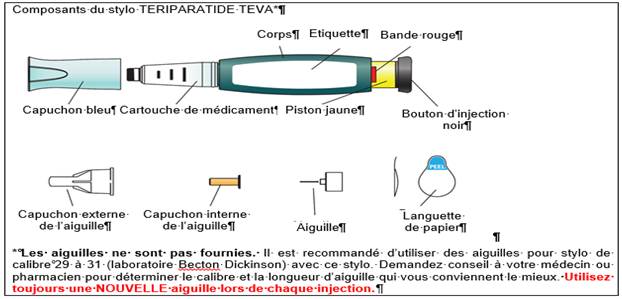
Lavez-vous les mains avant chaque injection. Préparez le site d’injection suivant les recommandations de votre médecin ou pharmacien.
1. Retirez le capuchon bleu

Vérifiez l’étiquette du stylo TERIPARATIDE TEVA afin de vous assurer que vous utilisez le médicament qui vous a été prescrit et que la date de péremption n’est pas dépassée.
N’utilisez pas le stylo TERIPARATIDE TEVA si le stylo semble endommagé, si le médicament dans la cartouche n’est pas limpide et incolore et s’il contient des particules.
2. Fixez une nouvelle aiguille
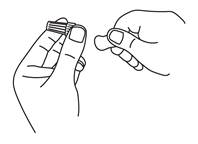
Retirez la languette de papier.
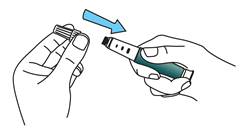
Fixez l’aiguille de façon bien droite sur la cartouche de médicament.
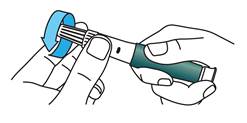
Capuchon externe de l’aiguille
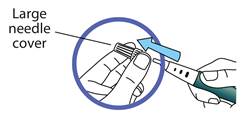
Retirez le capuchon externe de l’aiguille et conservez-le.
3. Sélectionnez la dose
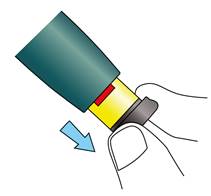
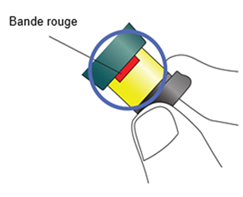
Tirez le bouton d’injection noir jusqu’à la butée et jusqu’à l’apparition de la bande rouge.
Si vous ne pouvez pas tirer le bouton d’injection noir, reportez-vous au Problème E de la rubrique Dépannage, en fin de page.
Capuchon interne de l’aiguille
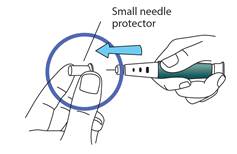
Retirez le capuchon interne de l’aiguille et jetez-le.
4. Injectez la dose
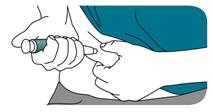
Pincez doucement un pli de peau de la cuisse ou de l’abdomen et insérez l’aiguille de façon bien droite dans la peau.
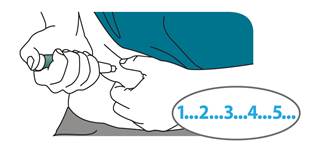
Appuyez sur le bouton d’injection noir jusqu’à la butée. Maintenez-le enfoncé et comptez lentement jusqu’à 5.
Vous devez compter jusqu’à 5 pour vous assurer que vous recevez la bonne dose.
Retirez ensuite l’aiguille de la peau.
5. Confirmez la dose
IMPORTANT
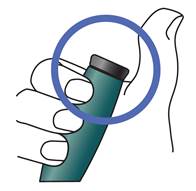
Après avoir effectué l’injection :
Une fois que l’aiguille est retirée de la peau, retirez votre pouce du bouton d’injection noir.
Assurez-vous que le bouton d’injection noir est complètement enfoncé. L’injection a été correctement effectuée si vous ne voyez plus le piston jaune.
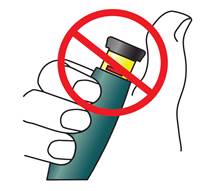
Vous NE devez PAS du tout voir le piston jaune. Si c’est le cas et que vous avez déjà effectué l’injection, n’effectuez pas une deuxième injection au cours de la même journée. Par contre, vous DEVEZ réinitialiser le stylo TERIPARATIDE TEVA (reportez-vous au Problème A de la rubrique Dépannage, en fin de page).
6. Retirez l’aiguille
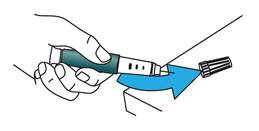
Remettez le capuchon externe de l’aiguille sur l’aiguille. N’essayez pas de remettre le capuchon de l’aiguille avec vos mains.
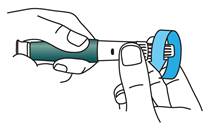
Dévissez l’aiguille recouverte jusqu’au bout en tournant le capuchon externe de l’aiguille de 3 à 5 tours dans le sens inverse des aiguilles d’une montre.
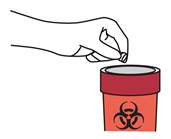
Retirez l’aiguille et jetez-la conformément aux instructions de votre médecin ou de votre pharmacien.
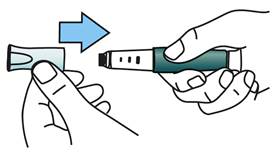
Remettez en place le capuchon bleu sur le stylo. Placez votre stylo TERIPARATIDE TEVA au réfrigérateur immédiatement après utilisation.
Pour plus d’informations ou si vous avez des questions, passez à la fin de cette page.
Tériparatide injectable en stylo prérempli

Dépannage
|
Problème |
Solution |
|
A. Le piston jaune est toujours visible après que j’ai appuyé sur le bouton d’injection noir. Comment dois-je réinitialiser mon stylo TERIPARATIDE TEVA ?
|
Vous pouvez éviter ce problème en utilisant toujours une NOUVELLE aiguille lors de chaque injection, en vous assurant que l’aiguille est correctement fixée et en appuyant sur le bouton d’injection noir jusqu’au bout. Pour réinitialiser le stylo TERIPARATIDE TEVA, suivez les étapes ci-après. 1) Si vous avez déjà effectué votre injection, N’effectuez PAS une deuxième injection au cours de la même journée. 2) Retirez l’aiguille. 3) Répétez l’étape 2 « Fixez une nouvelle aiguille » et l’étape 3 « Sélectionnez la dose » des instructions. 4) Dirigez l’aiguille vers le bas dans un récipient vide. Appuyez sur le bouton d’injection noir jusqu’à la butée. Maintenez le bouton enfoncé. Vous pourriez voir un petit jet ou une goutte de liquide. Lorsque vous avez terminé, le bouton d’injection noir doit être complètement enfoncé. Passez à l’étape 6 « Retirez l’aiguille ». 5) Si vous voyez toujours le piston jaune, contactez votre médecin ou votre pharmacien. Des réinitialisations répétées de votre stylo TERIPARATIDE TEVA pourraient entraîner une perte de doses quotidiennes. |
|
B. Comment savoir si mon stylo TERIPARATIDE TEVA fonctionne ? |
Lorsque le bouton d’injection noir est complètement enfoncé, cela indique que la dose complète du médicament a été injectée par le stylo TERIPARATIDE TEVA. Utilisez une nouvelle aiguille à chaque injection pour être sûr que votre stylo TERIPARATIDE TEVA fonctionne correctement. |
|
C. Je vois une bulle d’air dans mon stylo TERIPARATIDE TEVA. |
Une petite bulle d’air n’aura aucune incidence sur votre dose et sera sans danger. Vous pouvez continuer l’administration de votre dose comme d’habitude. |
|
D. Je n’arrive pas à retirer l’aiguille |
1) Remettez le capuchon externe de l’aiguille sur l’aiguille. 2) Utilisez le capuchon externe de l’aiguille pour dévisser l’aiguille. 3) Dévissez l’aiguille jusqu’au bout en tournant le capuchon externe de l’aiguille de 3 à 5 tours dans le sens inverse des aiguilles d’une montre. 4) Si vous n’arrivez toujours pas à retirer l’aiguille, demandez à quelqu’un de vous aider. |
|
E. Que dois-je faire si j’ai du mal à tirer le bouton d’injection noir ? |
Demandez conseil à votre médecin. Vous devrez peut-être utiliser un nouveau stylo TERIPARATIDE TEVA. Lorsqu’il n’est pas possible de tirer le bouton d’injection noir, cela indique qu’il n’y a plus assez de produit dans votre stylo TERIPARATIDE TEVA pour une nouvelle dose, même si vous pouvez encore voir du produit dans la cartouche. |
Nettoyage et conservation
Nettoyage de votre stylo TERIPARATIDE TEVA
· Nettoyez l’extérieur du stylo TERIPARATIDE TEVA avec un linge humide.
· Ne mettez pas votre stylo TERIPARATIDE TEVA dans l’eau ou n’utilisez pas de produit liquide pour le laver ou le nettoyer.
Conservation de votre stylo TERIPARATIDE TEVA
· Placez votre stylo TERIPARATIDE TEVA au réfrigérateur immédiatement après chaque utilisation. Lisez et suivez les instructions de la rubrique « Comment conserver TERIPARATIDE TEVA 20 microgrammes/80 microlitres, solution injectable en stylo prérempli ? » de la Notice d’information de l’utilisateur.
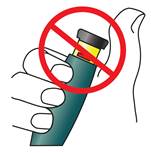
· Ne conservez pas le stylo TERIPARATIDE TEVA avec l’aiguille fixée dessus. Cela peut provoquer la formation de bulles d’air dans la cartouche de médicament.
· Conservez le stylo TERIPARATIDE TEVA avec le capuchon bleu fixé dessus.
· Ne conservez jamais le stylo TERIPARATIDE TEVA au congélateur. Si le stylo TERIPARATIDE TEVA a été congelé, jetez le stylo et utilisez un nouveau stylo TERIPARATIDE TEVA.
· Si le stylo TERIPARATIDE TEVA est resté en dehors du réfrigérateur, ne jetez pas le stylo. Replacez le stylo au réfrigérateur et contactez votre médecin ou votre pharmacien.
Autres remarques importantes
· Le stylo TERIPARATIDE TEVA contient 28 jours de traitement.
· Ne transférez pas le médicament dans une seringue car vous pourriez ainsi ne pas vous injecter la bonne dose de médicament.
· Ecrivez la date de votre première injection sur un calendrier.
· Lisez et suivez les instructions du manuel d’utilisation pour savoir comment utiliser correctement votre stylo TERIPARATIDE TEVA.
· Vérifiez l’étiquette du stylo TERIPARATIDE TEVA afin de vous assurer que vous utilisez le médicament qui vous a été prescrit et que la date de péremption n’est pas dépassée.
· N’utilisez pas le stylo TERIPARATIDE TEVA s’il semble endommagé. Examinez la solution de tériparatide dans la cartouche. Si le médicament n’est pas limpide et incolore ou s’il contient des particules, ne l’utilisez pas. Contactez votre médecin ou votre pharmacien si vous constatez l’un de ces problèmes.
· Utilisez une nouvelle aiguille à chaque injection.
· Pendant l’injection, vous pouvez entendre un ou plusieurs clics, cela correspond au fonctionnement normal du stylo.
· L’utilisation du stylo TERIPARATIDE TEVA par des personnes aveugles ou malvoyantes n’est pas recommandée sans l’aide d’une personne formée à la bonne utilisation du stylo.
· Conservez votre stylo TERIPARATIDE TEVA et les aiguilles hors de la portée des enfants.
Elimination des aiguilles et du stylo TERIPARATIDE TEVA
· Avant de jeter le stylo TERIPARATIDE TEVA, assurez-vous d’avoir retiré l’aiguille du stylo.
· Jetez votre stylo TERIPARATIDE TEVA et les aiguilles usagées conformément aux instructions de votre médecin ou de votre pharmacien.
Jetez le stylo TERIPARATIDE TEVA 28 jours après la première utilisation.
Date de la 1re utilisation _/_/_
Eliminer après le _/_/_
Coordonnées
Fabriqué pour : TEVA B.V.
La dernière date à laquelle ce manuel d’utilisation a été révisé est {MM/AAAA}