Dernière mise à jour le 01/06/2026
SOMATULINE L.P. 60 mg, solution injectable à libération prolongée en seringue préremplie
Indications thérapeutiques
Classe pharmacothérapeutique - code ATC : H01C B03
Qu’est-ce que SOMATULINE L.P. 60 mg et dans quels cas est-il utilisé ?
SOMATULINE L.P. est une forme à libération prolongée de lanréotide. Le lanréotide (substance active) appartient à une famille de médicaments appelée « antihormones de croissance ». Il est similaire à une autre substance (hormone) appelée « somatostatine ».
Le lanréotide diminue le taux de certaines hormones dans le corps, telles que l’hormone de croissance (GH) et l’IGF-1. Il inhibe la libération de certaines hormones au niveau du tractus digestif ainsi que les sécrétions intestinales. Il permet aussi la diminution ou l’arrêt de la croissance de certaines tumeurs avancées de l’intestin et du pancréas (appelées tumeurs neuroendocrines).
SOMATULINE L.P. 60 mg, solution injectable à libération prolongée en seringue préremplie est utilisé :
· Dans le traitement à long terme de l’acromégalie (une maladie au cours de laquelle le corps produit trop d’hormone de croissance).
· Dans le traitement des symptômes liés à l’acromégalie, tels que sensation de fatigue, maux de tête, transpiration, douleurs articulaires et engourdissement des mains et des pieds.
· Dans le traitement des symptômes tels que bouffées de chaleur et diarrhée, qui peuvent survenir chez les patients souffrant de certaines tumeurs appelées tumeurs carcinoïdes.
· Dans le traitement et le contrôle de la croissance de certaines tumeurs avancées de l’intestin et du pancréas, appelées tumeurs neuroendocrines gastro-entéro-pancréatiques, lorsque ces tumeurs ne peuvent pas être retirées par la chirurgie.
Présentations
> 1 seringue préremplie polypropylène avec 1 aiguille fixe en acier inoxydable munie d'un système de sécurité automatique
Code CIP : 357 287-2 ou 34009 357 287 2 2
Déclaration de commercialisation : 15/01/2002
Cette présentation est agréée aux collectivités
- Prix hors honoraire de dispensation : 713,47 €
- Honoraire de dispensation : 1,02 €
- Prix honoraire compris : 714,49 €
- Taux de remboursement :100%
- Traitement de l'acromégalie lorsque la chirurgie et/ou la radiothérapie ne peuvent pas être envisagés.
- Traitement des symptômes cliniques des tumeurs carcinoïdes. ; JOURNAL OFFICIEL ; 08/08/13
Service médical rendu (SMR)
Les libellés affichés ci-dessous ne sont que des résumés ou extraits issus des avis rendus par la Commission de la Transparence. Seul l'avis complet de la Commission de la Transparence fait référence.
Cet avis est consultable à partir du lien `Avis du jj/mm/aaaa` ou encore sur demande auprès de la HAS (plus d'informations dans l'aide). Les avis et synthèses d'avis contiennent un paragraphe sur la place du médicament dans la stratégie thérapeutique.
| Valeur du SMR | Avis | Motif de l'évaluation | Résumé de l'avis |
|---|---|---|---|
| Important | Avis du 06/04/2016 | Extension d'indication | Le service médical rendu par SOMATULINE LP 120 mg est important dans le traitement des tumeurs neuroendocrines gastro?entéro-pancréatiques non résécables et non progressives, localement avancées ou métastatiques, de grade 1 ou de grade 2 avec un index KI 67 = 10 %, ayant pour origine l’intestin moyen, le pancréas, ou d’origine inconnue après exclusion d’un site primitif au niveau de l’intestin postérieur. |
| Insuffisant | Avis du 06/04/2016 | Extension d'indication | Le service médical rendu par SOMATULINE LP 120 mg est insuffisant pour justifier d’une prise en charge par la solidarité nationale lorsque ces tumeurs sont progressives. |
| Important | Avis du 16/12/2015 | Renouvellement d'inscription (CT) | Le service médical rendu par SOMATULINE LP 30 mg, 60 mg 90 mg et 120 mg reste important dans : • le traitement de l'acromégalie lorsque les taux circulants d'hormone de croissance (GH) et d'IGF-1 ne sont pas normalisés après chirurgie et/ou radiothérapie ou lorsque la chirurgie et/ou la radiothérapie ne peuvent pas être envisagés, • le traitement des symptômes cliniques des tumeurs carcinoïdes. Le service médical rendu par SOMATULINE LP 60 mg 90 mg et 120 mg reste important dans le traitement des symptômes cliniques au cours de l’acromégalie. Le service médical rendu par SOMATULINE LP 30 mg reste important dans le traitement des adénomes thyréotropes primitifs responsables d’une hyperthyroïdie : en préparation ou en complément de la chirurgie et/ou de la radiothérapie, ou lorsque ces thérapeutiques ne sont pas appropriées. |
Amélioration du service médical rendu (ASMR)
Les libellés affichés ci-dessous ne sont que des résumés ou extraits issus des avis rendus par la Commission de la Transparence. Seul l'avis complet de la Commission de la Transparence fait référence.
Cet avis est consultable à partir du lien `Avis du jj/mm/aaaa` ou encore sur demande auprés de la HAS (plus d'informations dans l'aide). Les avis et synthéses d'avis contiennent un paragraphe sur la place du médicament dans la stratégie thérapeutique.
| Valeur de l'ASMR | Avis | Motif de l'évaluation | Résumé de l'avis |
|---|---|---|---|
| V (Inexistant) | Avis du 06/04/2016 | Extension d'indication | Considérant le bénéfice démontré en termes de survie sans progression mais l’absence de gain en survie globale à un stade de la maladie où l’abstention thérapeutique peut être préconisée, la Commission considère que SOMATULINE LP 120 mg n’apporte pas d’amélioration sur service médical rendu (ASMR V) dans la stratégie de prise en charge des patients adultes atteints de TNE gastro-entéropancréatiques non résécables et non progressives, localement avancées ou métastatiques. |
| V (Inexistant) | Avis du 03/01/2007 | Modification des conditions d'inscription (CT) | Le nouveau schéma posologique de SOMATULINE LP 120 mg n'apporte pas d'amélioration du service médical rendu par rapport à la prise en charge actuelle par les autres hormones de croissance chez les patients acromégales. |
Autres informations
- Titulaire de l'autorisation : IPSEN PHARMA
- Conditions de prescription et de délivrance :
- liste I
- prescription initiale hospitalière annuelle
- Statut de l'autorisation : Valide
- Type de procédure : Procédure nationale
- Code CIS : 6 875 807 6
ANSM - Mis à jour le : 16/06/2025
SOMATULINE L.P. 60 mg, solution injectable à libération prolongée en seringue préremplie
2. COMPOSITION QUALITATIVE ET QUANTITATIVE
Lanréotide ............................................................................................................................. 60 mg
(sous forme d’acétate de lanréotide)
Pour une seringue préremplie.
La seringue préremplie contient une solution sursaturée d’acétate de lanréotide correspondant à 0,246 mg de lanréotide base/mg de solution, permettant l’injection d’une dose de 60 mg de lanréotide.
Pour la liste complète des excipients, voir rubrique 6.1.
Solution injectable à libération prolongée en seringue préremplie.
Préparation semi solide blanche à jaune pâle.
4.1. Indications thérapeutiques
Traitement des symptômes cliniques au cours de l’acromégalie.
Traitement des symptômes cliniques des tumeurs carcinoïdes.
Traitement des tumeurs neuroendocrines (TNE) gastro-entéro-pancréatiques non résécables de l’adulte, localement avancées ou métastatiques, de grade 1 ou de grade 2 avec un index Ki67 ≤ 10%, ayant pour origine l’intestin moyen, le pancréas, ou d’origine inconnue après exclusion d’un site primitif au niveau de l’intestin postérieur (voir rubrique 5.1).
4.2. Posologie et mode d'administration
SOMATULINE L.P., solution injectable à libération prolongée, existe sous 3 dosages différents : 60 mg, 90 mg et 120 mg de lanréotide.
Acromégalie
La posologie initiale recommandée varie de 60 à 120 mg tous les 28 jours.
Par la suite, la dose doit être individualisée en fonction de la réponse du patient (évaluée par une réduction des symptômes et/ou une réduction des taux de GH et/ou d'IGF-1).
Si la réponse attendue n'est pas obtenue, la dose peut être augmentée.
La dose peut être augmentée lorsque les concentrations de GH sont supérieures à 2,5 ng/mL.
Lorsque les concentrations de GH sont comprises entre 2,5 ng/mL et 1 ng/mL, la dose peut être maintenue si le taux d'IGF-1 ajusté à l'âge est normal.
Si un contrôle complet est obtenu (basé sur des taux de GH inférieurs à 1 ng/mL, des taux d'IGF-1 normalisés et/ou la disparition des symptômes), la dose peut être diminuée.
Chez les patients bien contrôlés par un analogue de la somatostatine, SOMATULINE L.P. 120 mg peut être administré tous les 42 ou 56 jours. Par exemple, les patients qui sont bien contrôlés par SOMATULINE L.P. 60 mg injecté tous les 28 jours, peuvent être traités par SOMATULINE L.P. 120 mg tous les 56 jours et les patients, qui sont bien contrôlés par SOMATULINE L.P. 90 mg injecté tous les 28 jours, peuvent être traités par SOMATULINE L.P. 120 mg tous les 42 jours.
Une surveillance à long terme des symptômes, des taux de GH et d'IGF-1 doit être effectuée selon les indications cliniques.
Symptômes cliniques des tumeurs carcinoïdes
La dose initiale recommandée est de 60 mg à 120 mg administrés tous les 28 jours.
Par la suite, la dose doit être individualisée en fonction du degré de soulagement des symptômes obtenu. La dose maximale recommandée est de 120 mg de SOMATULINE L.P. tous les 28 jours.
Chez les patients bien contrôlés par un analogue de la somatostatine, SOMATULINE L.P. 120 mg peut être administré tous les 42 ou 56 jours. Par exemple, les patients qui sont bien contrôlés par SOMATULINE L.P. 60 mg injecté tous les 28 jours, peuvent être traités par SOMATULINE L.P. 120 mg tous les 56 jours et les patients, qui sont bien contrôlés par SOMATULINE L.P. 90 mg injecté tous les 28 jours, peuvent être traités par SOMATULINE L.P. 120 mg tous les 42 jours.
Une surveillance étroite des symptômes doit être effectuée lorsque le traitement est modifié pour passer à l’intervalle posologique étendu.
Tumeurs neuroendocrines (TNE) gastro-entéro-pancréatiques non résécables de l’adulte, localement avancées ou métastatiques, de grade 1 ou de grade 2 avec un index Ki67 ≤ 10%, ayant pour origine l’intestin moyen, le pancréas, ou d’origine inconnue après exclusion d’un site primitif au niveau de l’intestin postérieur
La dose recommandée est de 1 injection de SOMATULINE L.P. 120 mg tous les 28 jours. Le traitement doit être poursuivi pendant la durée nécessaire pour assurer le contrôle tumoral.
Insuffisance rénale et/ou hépatique
Chez les patients présentant une insuffisance rénale ou hépatique, il n’est pas nécessaire d’ajuster la posologie (voir section 5.2).
Sujets âgés
Chez le sujet âgé, aucun ajustement posologique n’est nécessaire (voir section 5.2).
Population pédiatrique
La sécurité d’emploi et l’efficacité de SOMATULINE L.P. n’a pas été établie chez l’enfant et l’adolescent.
Mode d’administration
La solution doit être injectée par voie sous-cutanée profonde dans le quadrant supéro-externe de la fesse ou dans la région supéro-externe de la cuisse.
L’injection est pratiquée par un professionnel de santé. Toutefois, chez les patients recevant une dose stable de SOMATULINE L.P., le produit peut être administré soit par le patient, soit par une personne de son entourage après une formation appropriée par un professionnel de santé.
Dans le cas d’une auto-injection, celle-ci devra être réalisée dans la région supéro-externe de la cuisse.
La décision d’administration par le patient ou par une autre personne formée devra être prise par le professionnel de santé.
Quel que soit le site d’injection, la peau ne doit pas être pincée et l’aiguille doit être introduite rapidement sur toute sa longueur, perpendiculairement à la peau.
Les injections seront réalisées alternativement du côté droit et du côté gauche.
4.4. Mises en garde spéciales et précautions d'emploi
Lithiase biliaire et complications de la lithiase biliaire :
Le lanréotide peut diminuer la motilité de la vésicule biliaire et entraîner la formation de calculs. Un contrôle régulier des patients peut donc être nécessaire. Il est conseillé, lors de traitements prolongés, de pratiquer auparavant, et tous les 6 mois, une échographie de la vésicule biliaire (voir rubrique 4.8). Des cas de calculs biliaires entraînant des complications, notamment une cholécystite, une cholangite et une pancréatite, ayant nécessité une cholécystectomie ont été rapportés après la commercialisation chez des patients traités par du lanréotide. En cas de suspicion de complications de la lithiase biliaire, arrêter le lanréotide et traiter la lithiase biliaire de manière appropriée.
Hyperglycémie et hypoglycémie :
Des études de pharmacologie animale et humaine ont montré que le lanréotide, comme la somatostatine et ses analogues, inhibe la sécrétion d’insuline et de glucagon. Ainsi, une hypoglycémie ou une hyperglycémie peuvent survenir chez les patients traités par du lanréotide. La glycémie doit être surveillée lors de l’initiation du traitement par du lanréotide et lors de toute modification de posologie. Le traitement antidiabétique sera adapté en conséquence. Chez le diabétique insulino-traité, les doses d’insuline seront a priori réduites de 25 %, puis adaptées aux glycémies, qui devront être contrôlées attentivement chez ces patients dès l’instauration du traitement.
Hypothyroïdie :
Chez le patient acromégale traité par du lanréotide, on a pu observer une légère diminution de la fonction thyroïdienne s’accompagnant rarement d’une hypothyroïdie clinique. Un examen de la fonction thyroïdienne est recommandé selon la pertinence clinique.
Bradycardie :
Chez le patient sans pathologie cardiaque sous-jacente, le lanréotide peut entraîner une diminution du rythme cardiaque, sans atteindre nécessairement le seuil de bradycardie. En cas de problèmes cardiaques préexistants, une bradycardie sinusale peut survenir. Chez les patients souffrant de bradycardie, il convient d’être prudent lors de l’initiation du traitement par du lanréotide (voir section 4.5).
Fonction pancréatique :
Une insuffisance pancréatique exocrine a été observée chez certains patients traités par du lanréotide pour des tumeurs neuroendocrines gastro-entéro-pancréatiques. Les symptômes d’une insuffisance pancréatique exocrine peuvent inclure une stéatorrhée, des selles molles, des ballonnements abdominaux et une perte de poids. La détection et le traitement approprié de l’insuffisance pancréatique exocrine, conformément aux recommandations cliniques, doivent être envisagés chez les patients symptomatiques.
L’apparition d’une élévation franche et durable de la stéatorrhée justifie la prescription complémentaire d’extraits pancréatiques.
Surveillance des tumeurs hypophysaires :
Chez les patients acromégales, l’utilisation du lanréotide ne dispense pas de la surveillance du volume de la tumeur hypophysaire.
4.5. Interactions avec d'autres médicaments et autres formes d'interactions
Les interactions avec les médicaments fortement liés aux protéines plasmatiques sont peu probables compte tenu de la liaison modeste du lanréotide aux protéines plasmatiques.
Risque d’hypoglycémie ou d’hyperglycémie : diminution des besoins en traitement antidiabétique, par diminution ou augmentation de la sécrétion de glucagon endogène.
Pendant le traitement par du lanréotide, l’autosurveillance glycémique doit être renforcée et la posologie du traitement antidiabétique doit être adaptée si nécessaire.
L’administration concomitante de médicaments induisant une bradycardie (ex : bêta-bloquants) peut avoir un effet additif sur la légère diminution du rythme cardiaque associée au lanréotide. Un ajustement posologique de ces médicaments peut être nécessaire (voir rubrique 4.4).
Selon des données limitées de la littérature, les analogues de la somatostatine peuvent diminuer la clairance métabolique des produits métabolisés par le système enzymatique cytochrome P450, ce qui pourrait être lié à la suppression de l’hormone de croissance. Cet effet ne pouvant être exclu avec le lanréotide, les médicaments métabolisés principalement par le CYP3A4 et possédant un faible index thérapeutique (ex : quinidine) doivent être utilisés avec prudence.
4.6. Fertilité, grossesse et allaitement
Grossesse
Les études effectuées chez l’animal ont mis en évidence une toxicité sur la reproduction mais aucune preuve d’effets tératogènes (voir rubrique 5.3). Le risque potentiel pour l’Homme est inconnu.
Par mesure de précaution, il est préférable d’éviter l’utilisation de SOMATULINE L.P. pendant la grossesse.
Il n’y a pas de données disponibles sur l’excrétion du lanréotide dans le lait maternel.
Un risque pour les nouveau-nés/nourrissons ne peut être exclu.
SOMATULINE L.P. ne doit pas être utilisé pendant l’allaitement.
Fertilité
Une diminution de la fertilité a été observée chez la rate en raison de l’inhibition de la sécrétion de GH, à des doses supérieures à celles utilisées chez l’homme en thérapeutique.
4.7. Effets sur l'aptitude à conduire des véhicules et à utiliser des machines
SOMATULINE L.P. a une influence mineure ou modérée sur l’aptitude à conduire des véhicules et à utiliser des machines. Aucune étude n’a été réalisée sur les effets sur l’aptitude à conduire des véhicules et utiliser des machines. Cependant, des sensations vertigineuses ont été rapportées avec SOMATULINE L.P. (voir rubrique 4.8). En cas de survenue de tels effets, le patient doit s’abstenir de conduire ou d’utiliser des machines.
Les effets indésirables rapportés au cours des études cliniques lors du traitement de l’acromégalie et des tumeurs neuroendocrines gastro-entéro-pancréatiques par le lanréotide sont listés par classe-organe selon les fréquences suivantes : très fréquent (≥ 1/10) ; fréquent (≥ 1/100 à < 1/10) ; peu fréquent (≥ 1/1 000 à < 1/100) ; inconnu (ne peut pas être estimé sur la base des données disponibles).
Les effets indésirables les plus fréquents lors du traitement par du lanréotide sont les troubles gastro-intestinaux (le plus souvent diarrhée et douleurs abdominales, habituellement légères à modérées et transitoires), la lithiase biliaire (souvent asymptomatique) et les réactions au site d’injection (douleur, nodule et induration).
Le profil de tolérance observé est similaire dans les toutes les indications.
|
Système Classe Organe |
Très fréquent (≥ 1/10) |
Fréquent (≥ 1/100 à < 1/10) |
Peu fréquent (≥ 1/1 000 à < 1/100) |
Expérience post-commercialisation (fréquence inconnue) |
|
Affections du système immunitaire |
|
|
|
Réactions allergiques (incluant angio-œdème, anaphylaxie, hypersensibilité) |
|
Troubles du métabolisme et de la nutrition |
|
Hypoglycémie, appétit diminué**, hyperglycémie, diabète |
|
|
|
Affections psychiatriques |
|
|
Insomnie* |
|
|
Affections du système nerveux |
|
Sensations vertigineuses, céphalées, léthargie** |
|
|
|
Affections cardiaques |
|
Bradycardie sinusale* |
|
|
|
Affections vasculaires |
|
|
Bouffées de chaleur* |
|
|
Affections gastro-intestinales |
Diarrhée, selles molles*, douleurs abdominales |
Nausées, vomissements, constipation, flatulence, distension abdominale, inconfort abdominal*, dyspepsie, stéatorrhée** |
Décoloration des selles* |
Insuffisance pancréatique exocrine, pancréatite |
|
Affections hépatobiliaires |
Lithiase biliaire |
Dilatation biliaire* |
|
Cholécystite, cholangite |
|
Affections de la peau et du tissu sous-cutané |
|
Alopécie, hypotrichose* |
|
|
|
Affections musculo-squelettiques et du tissu conjonctif |
|
Douleurs musculo-squelettiques**, myalgie** |
|
|
|
Troubles généraux et anomalies au site d’administration |
|
Asthénie, fatigue, réactions au point d’injection (douleur, induration, nodule, prurit) |
|
Abcès au site d’injection |
|
Investigations |
|
Elévation des ALAT*, anomalies des ASAT et des ALAT*, élévation de la bilirubinémie*, élévation de la glycémie*, élévation de l’hémoglobine glycosylée*, perte de poids, enzymes pancréatiques diminuées** |
Elévation des ASAT*, élévation des phosphatases alcalines*, anomalies de la bilirubinémie*, diminution de la natrémie* |
|
* sur la base d’un ensemble d’études effectuées dans l’acromégalie
** sur la base d’un ensemble d’études effectuées dans les tumeurs neuroendocrines gastro-entéro-pancréatiques
Déclaration des effets indésirables suspectés
La déclaration des effets indésirables suspectés après autorisation du médicament est importante. Elle permet une surveillance continue du rapport bénéfice/risque du médicament. Les professionnels de santé déclarent tout effet indésirable suspecté via le système national de déclaration : Agence nationale de sécurité du médicament et des produits de santé (ANSM) et réseau des Centres Régionaux de Pharmacovigilance - Site internet : https://signalement.social-sante.gouv.fr.
En cas de surdosage, un traitement symptomatique est recommandé.
5. PROPRIETES PHARMACOLOGIQUES
5.1. Propriétés pharmacodynamiques
Mécanisme d’action
Le lanréotide est un octapeptide analogue de la somatostatine naturelle. Comme la somatostatine, le lanréotide est un inhibiteur de différentes fonctions endocrines, neuro-endocrines, exocrines et paracrines. Il présente une forte affinité pour les récepteurs à la somatostatine humaine (SSTR) 2 et 5, et une affinité faible sur les SSTR 1, 3 et 4. L'inhibition de l'hormone de croissance s’expliquerait principalement par cette activité au niveau des récepteurs SSTR 2 et 5. Le lanréotide est plus actif que la somatostatine naturelle et présente une durée d'action plus longue.
Sa sélectivité marquée vis-à-vis de la sécrétion de l'hormone de croissance par rapport à celle de l'insuline en fait un produit adapté au traitement de l'acromégalie.
Comme la somatostatine, le lanréotide a une action anti-sécrétoire exocrine générale. Il inhibe la sécrétion basale de motiline, de peptide inhibiteur gastrique et de polypeptide pancréatique, mais n'a pas d’action significative sur la sécrétion à jeun de sécrétine ou de gastrine. En outre, il diminue le taux plasmatique de chromogranine A et le taux urinaire de 5-HIAA (acide 5-hydroxyindolacétique) chez les patients présentant une TNE gastro-entéro-pancréatique associée à des taux élevés de ces marqueurs tumoraux. Le lanréotide inhibe l’augmentation du débit sanguin de l’artère mésentérique supérieure et de la veine porte en post-prandial.
Le lanréotide diminue significativement la sécrétion jéjunale d’eau, de sodium, de potassium et de chlorures, stimulée par la prostaglandine E1. Le lanréotide diminue les taux de prolactine chez les patients acromégales traités au long cours.
Chez les patients acromégales, le lanréotide peut provoquer une réduction du volume tumoral.
Au cours d’une étude non contrôlée en ouvert, SOMATULINE L.P. 120 mg a été administré tous les 28 jours pendant 48 semaines chez 90 patients acromégales présentant un macroadénome hypophysaire et non traités préalablement.
Les patients qui devaient subir une intervention chirurgicale ou une radiothérapie hypophysaire au cours de la période étaient exclus de l’étude.
À la semaine 48, 63% des patients ont montré une réduction cliniquement pertinente du volume tumoral ≥ 20% (critère principal d’efficacité), bien que cette réduction ne soit pas statistiquement significative (IC 95% : 52% -73%). Le pourcentage moyen de réduction du volume tumoral était de 26,8%. La concentration de GH était inférieure à 2,5 µg/L chez 77,8% des patients et la concentration d’IGF-1 était normalisée chez 50% des patients. Quarante-trois pour cent (43,5%) des patients ont présenté à la fois une concentration d’IGF-1 normalisée et une concentration de GH-inférieure à 2,5 µg/L.
La plupart des patients ont rapporté une amélioration des symptômes d'acromégalie tels que céphalées (38,7%), fatigue (56,5%), transpiration excessive (66,1%), arthralgies (59,7%) et gonflement des tissus mous (66,1%).
Une réduction précoce et durable du volume tumoral et des concentrations de GH et d'IGF-1 a été mise en évidence dès la douzième semaine de traitement et s’est maintenue pendant 48 semaines.
L'action inhibitrice du lanréotide sur la sécrétion exocrine intestinale, les hormones digestives et les mécanismes de prolifération cellulaire est particulièrement intéressante pour son application au traitement symptomatique des tumeurs endocrines digestives, notamment carcinoïdes.
Une étude multicentrique de phase III, randomisée, en double-aveugle, contrôlée contre placebo, d’une durée fixe de 96 semaines a été menée dans le but d’évaluer l’effet antiprolifératif du lanréotide chez des patients présentant une TNE gastro-entéro-pancréatique.
Les patients ont été randomisés selon un ratio 1:1 entre le groupe SOMATULINE L.P. 120 mg tous les 28 jours (n=101) et le groupe placebo (n=103). La randomisation était stratifiée en fonction des traitements reçus avant l’entrée dans l’étude et de la présence/absence de progression à l’état initial, évaluée selon les critères RECIST 1.0 (Response Evaluation Criteria in Solid Tumours) au cours d’une phase de sélection d’une durée de 3 à 6 mois.
Les patients présentaient une tumeur inopérable, métastatique et/ou localement avancée, confirmée au plan histologique comme étant bien ou moyennement différenciée, avec une localisation primitive au niveau du pancréas (44,6% des patients), de l’intestin moyen (35,8%), de l’intestin postérieur (6,9%) ou d’origine autre ou inconnue (12,7%).
69% des patients présentaient une TNE gastro-entéro-pancréatique de grade G1, définie soit par un index de prolifération Ki67 ≤ 2% (50,5% de la population totale), soit par un index mitotique < 2 mitoses /10 HPF (18,5% de la population totale). 30% des patients présentaient une TNE gastro-entéro-pancréatique se situant dans la fourchette basse du grade G2 (2% < Ki67 ≤ 10%). Le grade n’était pas connu chez 1% des patients. Les patients présentant une TNE gastro-entéro-pancréatique de grade G2 avec index de prolifération plus élevé (10% < Ki67 ≤ 20%) ou présentant un carcinome neuroendocrine de grade G3 (Ki67 > 20%) étaient exclus de l’étude.
Globalement, la charge tumorale hépatique était ≤ 10%, comprise entre 10 et 25% et > 25% chez respectivement 52,5% ,14,5% et 33% des patients.
Le critère principal était la survie sans progression (SSP) définie comme le délai entre soit la progression de la maladie selon les critères RECIST 1.0, soit le décès, au cours des 96 semaines suivant la première administration du traitement. L’analyse de la SSP était basée sur l’évaluation radiologique de la progression tumorale, effectuée de façon centralisée et indépendante.
Tableau 1: Résultats d’efficacité de l’étude de phase III
|
Médiane de survie sans progression (semaines) |
Hazard Ratio (IC 95%) |
Réduction du risque de progression ou de décès |
p |
|
|
Lanréotide LP (n=101 ) |
Placebo (n=103 ) |
|||
|
> 96 semaines |
72,00 semaines (IC 95% :48,57, 96,00) |
0,470 (0,304, 0,729) |
53%
|
0,0002 |
Figure 1 : Survie sans progression : courbes de Kaplan-Meier
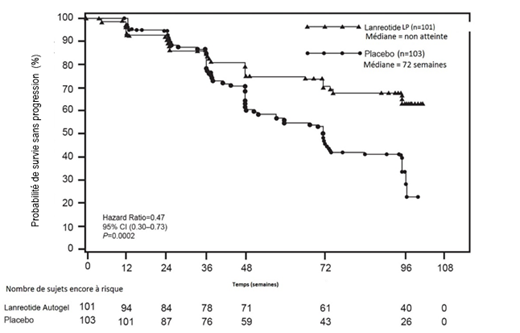
L’effet bénéfique du lanréotide sur la réduction du risque de progression ou de décès était cohérent, quel que soit le site primitif de la tumeur, la charge hépatique tumorale, la chimiothérapie antérieure, le Ki67 à l’état initial, le grade tumoral ou les autres caractéristiques pré-spécifiées (voir Figure 2).
Le bénéfice clinique du traitement par SOMATULINE L.P. a été observé chez les patients présentant une tumeur ayant pour origine le pancréas, l’intestin moyen ou une origine autre/inconnue, ainsi que dans la population totale de l’étude. Compte tenu du nombre limité de patients présentant une tumeur originaire de l’intestin postérieur (14/204), l’interprétation des résultats est difficile dans ce sous-groupe. Les données disponibles suggèrent une absence de bénéfice du lanréotide chez ces patients.
Figure 2 : Analyse des co-facteurs de la SSP par le modèle de Cox à risques proportionnels
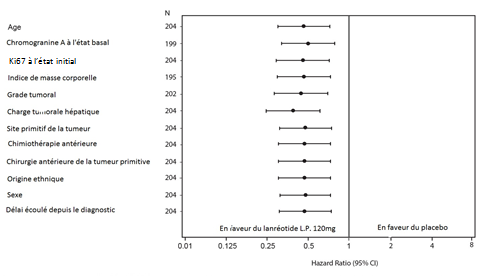
Note : Tous les Hazard ratios expriment le risque relatif pour SOMATULINE L.P. versus placebo. Les résultats des co-facteurs sont issus de modèles de Cox à risques proportionnels avec les facteurs traitement, progression à l’état initial, traitement antérieur à l’inclusion dans l’étude et le facteur identifié sur l’axe vertical.
Dans la phase d’extension en ouvert de l’étude, 45,6% des patients (47/103) sont passés du placebo au traitement par SOMATULINE L.P.
Population pédiatrique
L'Agence européenne des médicaments a accordé une dérogation à l’obligation de soumettre les résultats d’études avec SOMATULINE L.P. dans tous les sous-groupes de la population pédiatrique, pour l’acromégalie et le gigantisme d’origine pituitaire (voir les informations concernant l'usage pédiatrique en rubrique 4.2). L'Agence européenne des médicaments a inscrit les TNE gastro-entéro-pancréatiques (à l’exception des neuroblastomes, des neuroganglioblastomes et des phéochromocytomes) sur la liste des exemptions de classe.
5.2. Propriétés pharmacocinétiques
Au cours des études d’élimination, moins de 5% du lanréotide était excrété par voie urinaire et moins de 0,5% était retrouvé sous forme inchangée dans les fèces, indiquant une excrétion biliaire.
Chez le volontaire sain, après injection sous-cutanée profonde de SOMATULINE L.P. 60 mg, 90 mg et 120 mg, les concentrations sériques du lanréotide augmentent pour atteindre un pic de concentration (Cmax) de 4,25, 8,39 et 6,79 ng/mL. La Cmax est atteinte le jour de l’administration en 8, 12 et 7 heures (valeurs médianes). Les concentrations diminuent ensuite lentement en suivant une cinétique de premier ordre, avec une demi-vie terminale d’élimination de respectivement 23,3, 27,4 et 30,1 jours. Quatre semaines après l’administration, les concentrations sériques moyennes du lanréotide sont respectivement de 0,9, 1,11 et 1,69 ng/mL. La biodisponibilité absolue est de 73,4, 69,0 et 78,4%.
Chez le patient acromégale, après injection sous-cutanée profonde de SOMATULINE L.P. 60 mg, 90 mg et 120 mg, les concentrations sériques de lanréotide augmentent pour atteindre un pic de concentration (Cmax) de 1,6, 3,5 et 3,1 ng/mL. La Cmax est atteinte le jour de l’administration en 6, 6 et 24 heures. Les concentrations diminuent ensuite lentement en suivant une cinétique de premier ordre. Quatre semaines après l’administration, les concentrations sériques moyennes du lanréotide sont respectivement de 0,7, 1,0 et 1,4 ng/mL.
Les concentrations sériques du lanréotide à l’équilibre sont obtenues en moyenne après 4 injections pratiquées toutes les 4 semaines.
Après administration répétée toutes les 4 semaines de SOMATULINE L.P. 60 mg, 90 mg et 120 mg, la Cmax à l’équilibre est respectivement de 3,8, 5,7 et 7,7 ng/mL et la Cmin est respectivement de 1,8, 2,5 et 3,8 ng/mL. L’index de fluctuation pic-nadir est modéré, allant de 81 à 108%.
Après injection sous-cutanée profonde de SOMATULINE L.P. 60 mg, 90 mg et 120 mg chez le patient acromégale, un profil pharmacocinétique de libération linéaire a été observé.
Les concentrations sériques minimales du lanréotide obtenues après trois injections sous-cutanées profondes de SOMATULINE L.P. 60 mg, 90 mg ou 120 mg administrées tous les 28 jours sont identiques à celles obtenues à l’état d’équilibre chez des patients acromégales précédemment traités tous les 14, 10 ou 7 jours respectivement par SOMATULINE L.P. 30 mg, par voie intramusculaire.
Une libération initiale rapide a été observée, avec une Cmax moyenne de 7,49 ± 7,58 ng/mL, atteinte au cours du 1er jour suivant une injection unique, lors d’une analyse pharmacocinétique portant sur une population de 290 patients souffrant de TNE gastro-entéro-pancréatique recevant SOMATULINE L.P. 120 mg. Les concentrations à l’équilibre ont été atteintes après 5 injections de SOMATULINE L.P. 120 mg tous les 28 jours ; elles ont été maintenues jusqu’à la dernière mesure (jusqu’à 96 semaines suivant la 1ère injection). A l’équilibre, la Cmax moyenne était de 13,9 ± 7,44 ng/mL et la concentration sérique moyenne la plus basse était de 6,56 ± 1,99 ng/mL. La demi-vie terminale apparente moyenne était de 49,8 ± 28,0 jours.
Insuffisance hépatique/rénale :
En cas d’insuffisance rénale sévère, on observe une réduction de moitié environ de la clairance sérique totale du lanréotide, avec pour conséquence une augmentation de la demi-vie et l’ASC. En cas d’insuffisance hépatique modérée à sévère, on observe une diminution de la clairance de 30 %. Le volume de distribution et le temps de résidence moyen augmentent quel que soit le degré d’insuffisance hépatique.
Aucun effet sur la clairance du lanréotide n’a été observé au cours d’une analyse pharmacocinétique conduite sur une population de 165 patients atteints d’une insuffisance rénale légère et modérée (respectivement 106 et 59) et souffrant de TNE gastro-entéro-pancréatique traités par SOMATULINE L.P. Les patients atteints d’une insuffisance rénale sévère et souffrant de TNE gastro-entéro-pancréatique n’ont pas été étudiés. Aucun patient souffrant de TNE gastro-entéro-pancréatique présentant une insuffisance hépatique (selon le score de Child-Pugh) n’a été étudié.
Il n’est pas nécessaire de modifier la dose initiale en cas d’insuffisance hépatique ou rénale, dans la mesure où les concentrations sériques du lanréotide attendues chez ces patients restent dans la fourchette des concentrations bien tolérées chez le volontaire sain.
Sujets âgés
Une augmentation de la demi-vie et du temps de résidence moyen est observée chez le sujet âgé comparativement aux sujets sains jeunes.
Il n’est pas nécessaire de modifier la dose initiale chez le sujet âgé, dans la mesure où les concentrations sériques du lanréotide attendues chez ces patients restent dans la fourchette des concentrations bien tolérées chez le volontaire sain.
Aucun effet de l’âge sur la clairance et le volume de distribution du lanréotide n’a été observé, lors d’une analyse pharmacocinétique de population conduite chez 122 patients âgés de 65 à 85 ans souffrant de TNE gastro-entéro-pancréatique.
5.3. Données de sécurité préclinique
Au cours des études de cancérogenèse chez le rat et la souris, aucune modification néoplasique systémique n’a été observée à des doses supérieures à celles utilisées chez l’homme en thérapeutique. Une augmentation de l’incidence des tumeurs sous-cutanées a été observée au site d’injection, probablement en raison de la plus grande fréquence d’administration chez l’animal (quotidienne) que chez l’homme (administration mensuelle) et n’est donc pas pertinente en clinique.
Au cours des tests standard in vitro et in vivo, le lanréotide n’a pas montré de potentiel génotoxique.
Les études sur la reproduction menées chez les rates gravides ayant reçues des injections sous-cutanées de 30 mg/kg toutes les 2 semaines (cinq fois la dose chez l’Homme, selon la surface corporelle) ont montré une diminution de la survie embryo-fœtale. Les études chez les lapines gravides ayant reçues des injections sous-cutanées de 0,45 mg/kg/jour (deux fois l’exposition thérapeutique chez l’Homme à la dose maximale recommandée de 120 mg, selon les comparaisons de la surface corporelle relative) montrent une diminution de la survie des fœtus et une augmentation des anomalies fœtales du squelette et des tissus mous.
Eau pour préparations injectables, acide acétique glacial (pour ajustement du pH).
3 ans.
Après ouverture de l’emballage laminé protecteur, le produit doit être administré immédiatement.
6.4. Précautions particulières de conservation
A conserver au réfrigérateur (entre 2°C et 8°C).
A conserver dans l’emballage d’origine à l’abri de la lumière.
Après sortie du réfrigérateur, le produit qui est resté dans son étui de protection scellé peut être remis au réfrigérateur pour y être conservé en vue d’une utilisation ultérieure, à condition de ne pas avoir subi plus de trois excursions de température et d’avoir été conservé pendant moins de 72 heures au total à une température inférieure à 40°C.
6.5. Nature et contenu de l'emballage extérieur
SOMATULINE L.P. se présente sous forme d’une seringue préremplie (polypropylène) munie d’un système de sécurité automatique, d’un bouchon-piston (bromobutyle) et d’une aiguille (acier inoxydable) recouverte d’un capuchon en plastique.
La seringue préremplie prête à l'emploi est présentée dans un support en plastique et conditionnée dans un emballage laminé et une boite en carton.
Boite contenant 1 seringue préremplie avec 1 aiguille fixe.
6.6. Précautions particulières d’élimination et de manipulation
La solution injectable contenue dans la seringue préremplie de SOMATULINE L.P. est une solution prête à l'emploi.
A usage immédiat et unique après ouverture.
Il est important que le produit soit injecté conformément aux instructions données dans la notice d’information de l’utilisateur présente dans la boîte.
Ne pas utiliser si l’emballage laminé est endommagé ou ouvert.
Tout produit non utilisé ou déchet doit être éliminé conformément à la réglementation en vigueur.
7. TITULAIRE DE L’AUTORISATION DE MISE SUR LE MARCHE
70 RUE BALARD
75015 PARIS
8. NUMERO(S) D’AUTORISATION DE MISE SUR LE MARCHE
· 34009 357 287 2 2 : seringue préremplie ; boîte de 1.
9. DATE DE PREMIERE AUTORISATION/DE RENOUVELLEMENT DE L’AUTORISATION
[à compléter ultérieurement par le titulaire]
10. DATE DE MISE A JOUR DU TEXTE
[à compléter ultérieurement par le titulaire]
Sans objet.
12. INSTRUCTIONS POUR LA PREPARATION DES RADIOPHARMACEUTIQUES
Liste I
Médicament soumis à prescription initiale hospitalière annuelle.
ANSM - Mis à jour le : 16/06/2025
SOMATULINE L.P. 60 mg, solution injectable à libération prolongée en seringue préremplie
Lanréotide
· Gardez cette notice. Vous pourriez avoir besoin de la relire.
· Si vous avez d’autres questions, interrogez votre médecin ou votre pharmacien.
· Ce médicament vous a été personnellement prescrit. Ne le donnez pas à d’autres personnes. Il pourrait leur être nocif, même si les signes de leur maladie sont identiques aux vôtres.
· Si vous ressentez un quelconque effet indésirable, parlez-en à votre médecin ou votre pharmacien. Ceci s’applique aussi à tout effet indésirable qui ne serait pas mentionné dans cette notice. Voir rubrique 4.
1. Qu'est-ce que SOMATULINE L.P. 60 mg, solution injectable à libération prolongée en seringue préremplie et dans quels cas est-il utilisé ?
2. Quelles sont les informations à connaître avant d'utiliser SOMATULINE L.P. 60 mg, solution injectable à libération prolongée en seringue préremplie ?
3. Comment utiliser SOMATULINE L.P. 60 mg, solution injectable à libération prolongée en seringue préremplie ?
4. Quels sont les effets indésirables éventuels ?
5. Comment conserver SOMATULINE L.P. 60 mg, solution injectable à libération prolongée en seringue préremplie ?
6. Contenu de l’emballage et autres informations.
1. QU’EST-CE QUE SOMATULINE L.P. 60 mg, solution injectable à libération prolongée en seringue préremplie ET DANS QUELS CAS EST-IL UTILISE ?
Classe pharmacothérapeutique - code ATC : H01C B03
Qu’est-ce que SOMATULINE L.P. 60 mg et dans quels cas est-il utilisé ?
SOMATULINE L.P. est une forme à libération prolongée de lanréotide. Le lanréotide (substance active) appartient à une famille de médicaments appelée « antihormones de croissance ». Il est similaire à une autre substance (hormone) appelée « somatostatine ».
Le lanréotide diminue le taux de certaines hormones dans le corps, telles que l’hormone de croissance (GH) et l’IGF-1. Il inhibe la libération de certaines hormones au niveau du tractus digestif ainsi que les sécrétions intestinales. Il permet aussi la diminution ou l’arrêt de la croissance de certaines tumeurs avancées de l’intestin et du pancréas (appelées tumeurs neuroendocrines).
SOMATULINE L.P. 60 mg, solution injectable à libération prolongée en seringue préremplie est utilisé :
· Dans le traitement à long terme de l’acromégalie (une maladie au cours de laquelle le corps produit trop d’hormone de croissance).
· Dans le traitement des symptômes liés à l’acromégalie, tels que sensation de fatigue, maux de tête, transpiration, douleurs articulaires et engourdissement des mains et des pieds.
· Dans le traitement des symptômes tels que bouffées de chaleur et diarrhée, qui peuvent survenir chez les patients souffrant de certaines tumeurs appelées tumeurs carcinoïdes.
· Dans le traitement et le contrôle de la croissance de certaines tumeurs avancées de l’intestin et du pancréas, appelées tumeurs neuroendocrines gastro-entéro-pancréatiques, lorsque ces tumeurs ne peuvent pas être retirées par la chirurgie.
2. QUELLES SONT LES INFORMATIONS A CONNAITRE AVANT D’UTILISER SOMATULINE L.P. 60 mg, solution injectable à libération prolongée en seringue préremplie ?
N’utilisez jamais SOMATULINE L.P. 60 mg, solution injectable à libération prolongée en seringue préremplie :
· Si vous êtes allergique (hypersensible) au lanréotide, à la somatostatine ou aux médicaments de la même famille (analogues de la somatostatine) ou à l’un des autres composants contenus dans ce médicament, mentionnés dans la rubrique 6.
Avertissements et précautions
Adressez-vous à votre médecin ou pharmacien avant d’utiliser SOMATULINE L.P. 60 mg, solution injectable à libération prolongée en seringue préremplie :
· Si vous êtes diabétique. Le lanréotide peut agir sur le taux de sucre dans le sang. Dans ce cas, votre médecin pourra juger nécessaire de vérifier le taux de sucre dans votre sang et éventuellement de modifier votre traitement anti-diabétique pendant votre traitement par le lanréotide.
· Si vous avez des calculs biliaires. Le lanréotide peut favoriser la formation de calculs biliaires dans la vésicule biliaire. Une surveillance régulière de la vésicule biliaire peut être nécessaire dans ce cas. Votre médecin pourra décider d’arrêter le traitement par le lanréotide si des complications liées à des calculs biliaires surviennent.
· Si vous avez des problèmes de thyroïde. Le lanréotide peut diminuer légèrement le fonctionnement de votre thyroïde.
· Si vous avez des problèmes cardiaques. Une bradycardie sinusale (ralentissement du rythme du cœur) peut survenir au cours du traitement par le lanréotide. Si vous souffrez de bradycardie (ralentissement du rythme du cœur), il faudra être particulièrement prudent au début du traitement par le lanréotide.
Si vous êtes dans l’une des situations décrites ci-dessus, prévenez votre médecin ou votre pharmacien avant d’utiliser SOMATULINE L.P.
Adressez-vous à votre médecin ou pharmacien pendant le traitement :
· Si vous avez des selles grasses, des selles molles, des ballonnements abdominaux ou une perte de poids, car le lanréotide peut affecter la sécrétion des enzymes pancréatiques impliquées dans la digestion des aliments.
Enfants et adolescents
L’utilisation de SOMATULINE L.P. 60 mg n’est pas recommandée chez les enfants et les adolescents.
Autres médicaments et SOMATULINE L.P. 60 mg solution injectable à libération prolongée en seringue préremplie
Certains médicaments ont un effet sur l’action d’autres médicaments. Informez votre médecin ou pharmacien si vous prenez, avez récemment pris ou pourriez prendre tout autre médicament.
Il faut être particulièrement prudent si vous prenez en même temps :
· De la ciclosporine (un médicament qui diminue la réaction immunitaire, utilisé après une transplantation d’organe ou en cas de maladie auto-immune) ;
· Un traitement antidiabétique (par exemple insuline, glitazones, répaglinide ou sulfamides hypoglycémiants) ;
· De la bromocriptine (agoniste dopaminergique utilisé dans le traitement de certains types de tumeurs du cerveau et de la maladie de Parkinson ou pour prévenir la production de lait après accouchement) ;
· Un médicament induisant une bradycardie (médicament qui ralentit le rythme du cœur comme par exemple les bêta-bloquants).
Dans ce cas, votre médecin pourra décider de modifier la dose des médicaments associés.
SOMATULINE L.P. 60 mg, solution injectable à libération prolongée en seringue préremplie avec des aliments et boissons
Sans objet.
Si vous êtes enceinte ou si vous allaitez, si vous pensez être enceinte ou planifiez une grossesse, demandez conseil à votre médecin ou pharmacien avant de prendre ce médicament. SOMATULINE L.P. doit vous être administré qu'en cas de nécessité absolue.
Conduite de véhicules et utilisation de machines
Il est peu probable que votre capacité à conduire ou à utiliser des machines soit affectée par votre traitement. Toutefois, des effets indésirables comme des sensations de vertiges peuvent survenir avec SOMATULINE L.P. Si vous présentez de tels effets, il faudra être prudent en cas de conduite de véhicule ou d’utilisation de machines.
SOMATULINE L.P. 60 mg, solution injectable à libération prolongée en seringue préremplie contient
Sans objet.
3. COMMENT UTILISER SOMATULINE L.P. 60 mg, solution injectable à libération prolongée en seringue préremplie ?
Veillez à toujours utiliser ce médicament en suivant exactement les instructions de votre médecin. Vérifiez auprès de votre médecin ou pharmacien en cas de doute.
Posologie
Traitement de l’acromégalie
La dose recommandée est d’une injection tous les 28 jours.
Votre médecin pourra adapter la dose administrée en choisissant l’un des 3 dosages existants de SOMATULINE L.P. (60 mg, 90 mg ou 120 mg).
Si vous êtes bien contrôlé par votre traitement, votre médecin pourra réduire la fréquence des injections à 1 injection de SOMATULINE L.P. 120 mg tous les 42 ou 56 jours.
Votre médecin décidera pendant combien de temps vous devrez prendre ce traitement.
Traitement des symptômes (tels que bouffées de chaleur et diarrhée) liés aux tumeurs carcinoïdes
La dose recommandée est d’une injection tous les 28 jours.
Votre médecin pourra adapter la dose administrée en choisissant l’un des 3 dosages existants de SOMATULINE L.P. (60 mg, 90 mg ou 120 mg).
Si vous êtes bien contrôlé par votre traitement, votre médecin pourra réduire la fréquence des injections à 1 injection de SOMATULINE L.P. 120 mg tous les 42 ou 56 jours.
Votre médecin décidera pendant combien de temps vous devrez prendre ce traitement.
Traitement des tumeurs neuroendocrines digestives avancées de l’intestin et du pancréas, lorsqu’elles ne peuvent pas être retirées par la chirurgie
La dose recommandée est d’une injection de 120 mg tous les 28 jours.
Votre médecin décidera pendant combien de temps vous devrez recevoir SOMATULINE L.P. pour le contrôle de la tumeur.
Mode d’administration
SOMATULINE L.P. doit être administré par une injection sous-cutanée profonde.
L'injection doit être effectuée par un professionnel de la santé ou par une personne de votre entourage (membre de la famille ou ami) ou par vous-même, après avoir reçu une formation appropriée par un professionnel de la santé.
La décision d’administration par vous-même ou par une autre personne formée sera prise par votre médecin. Si vous avez un doute sur la façon d’effectuer cette injection, veuillez contacter votre médecin ou un professionnel de santé afin d’avoir des conseils ou une formation complémentaire.
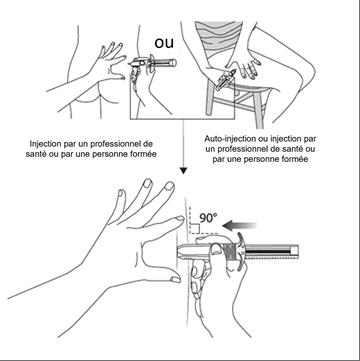
Si l’injection est pratiquée par un professionnel de santé ou par une personne de votre entourage formée, elle devra être effectuée dans le quadrant supéro-externe de la fesse ou dans le haut de la face externe de la cuisse (voir figures 5a et 5b ci-dessous).
Si vous réalisez vous-même l’injection (auto-injection), elle devra être effectuée dans le haut de la face externe de la cuisse (voir figure 5b ci-dessous).
Instructions pour l’administration du produit :
Attention : lisez attentivement les instructions ci-après avant de pratiquer l’injection. Il s’agit d’une injection sous-cutanée profonde qui nécessite une technique spécifique, différente de l’injection sous-cutanée normale.
Les instructions suivantes expliquent comment injecter SOMATULINE L.P.
SOMATULINE L.P. se présente sous forme d’une seringue préremplie à usage unique, prête à l’emploi, munie d’un système de sécurité automatique. L'aiguille se rétracte automatiquement après administration complète du produit afin d’éviter les blessures accidentelles par piqûre.
|
|
|
|
1. Sortez SOMATULINE L.P. du réfrigérateur 30 minutes avant l’administration. L’injection peut être douloureuse si le médicament est froid. Maintenez l’étui de protection plastifié scellé jusqu’au moment de l’injection.
|
|
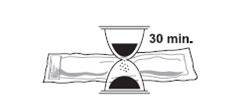
2. Attention : Avant l’ouverture, vérifiez que l’étui de protection est intact et que la date de péremption n’est pas dépassée.
N’utilisez pas la seringue préremplie :
· Si vous laissez tomber ou endommagez la seringue préremplie, ou si la seringue préremplie ou l’emballage vous semble endommagé ;
· Si la date de péremption est dépassée ; elle figure sur la boite extérieure et sur l’étui de protection.
Si vous rencontrez l’un des cas ci-dessus, contactez votre médecin ou votre pharmacien.
3. Lavez-vous les mains au savon.
4. Ouvrez l’étui de protection en suivant la ligne pointillée et sortez la seringue préremplie. Le contenu de la seringue préremplie se présente sous forme d’une phase semi-solide visqueuse ayant un aspect de gel, de couleur blanc à jaune pâle. La solution sursaturée peut également contenir des microbulles pouvant disparaître lors de l'injection. Ces différences sont normales et n’influent pas sur la qualité du produit.
|
|
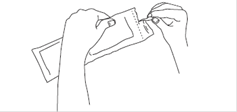
Après ouverture de l’étui de protection plastifié, le produit doit être administré immédiatement.
5. Déterminez le site d’injection :
· Si l’injection est pratiquée par un professionnel de santé ou par une personne de votre entourage qui a reçu une formation appropriée : injection dans le quadrant supéro-externe de la fesse (figure 5a) ou dans le haut de la face externe de la cuisse (figure 5b),
· Si vous pratiquez vous-même l’injection (auto-injection) : injection dans le haut de la face externe de la cuisse (figure 5b).
|
|
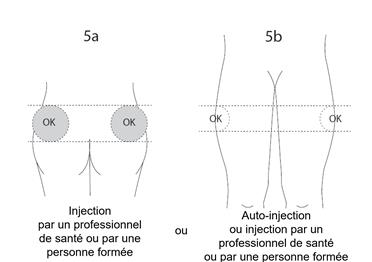
Alternez le site d’injection entre le côté droit et le côté gauche lors de chaque injection de SOMATULINE L.P. Évitez les zones présentant des grains de beauté, des cicatrices, une peau rougie ou bosselée.
6. Désinfectez le site d’injection.
7. Avant l’injection, retirez la seringue préremplie de son support. Jetez le support.
|
|
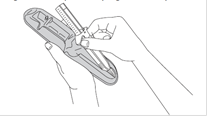
8. Otez le capuchon protecteur de l’aiguille en tirant et jetez-le.
|
|
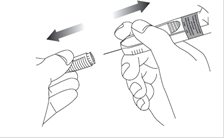
9. Tendez la peau autour de la zone d’injection entre le pouce et l’index de la main qui ne tient pas la seringue pour aplatir cette zone. Ne pas pincer la peau. Effectuez un geste ferme, comme avec une fléchette, pour insérer rapidement l'aiguille perpendiculairement à la peau (angle de 90 degrés), dans toute sa longueur.
Il est très important d’enfoncer l’aiguille complètement. Aucune partie de l’aiguille ne doit être visible lorsqu’elle est complètement enfoncée.
Ne pas aspirer (ne pas tirer le piston).
|
|
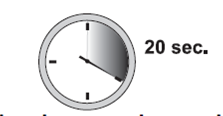
10. Relâchez le site d'injection qui était aplati avec la main. Poussez le piston avec une pression très ferme et constante. Le médicament est plus épais et plus difficile à pousser qu’attendu. En général, 20 secondes sont nécessaires. Injectez la dose complète et donnez une dernière pression pour vous assurer de ne pas pouvoir pousser davantage
|
|
Remarque : Maintenez une pression sur le piston avec votre pouce pour éviter de déclencher le système de sécurité automatique.
|
|
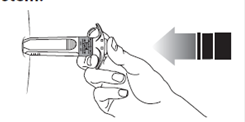
11. Retirez l’aiguille de la peau sans relâcher la pression sur le piston.
|
|
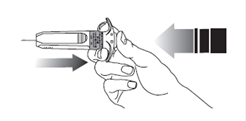
12. Relâchez la pression sur le piston. L’aiguille va se rétracter automatiquement dans un étui protecteur où elle sera bloquée définitivement.
|
|
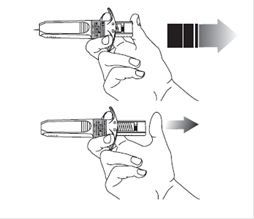
13. Appuyez légèrement sur le point d’injection avec un coton sec ou de la gaze stérile afin d’éviter le saignement. Ne pas frotter ni masser le point d’injection après administration.
14. Jetez la seringue usagée de manière appropriée. Votre médecin ou votre infirmière vous expliquera les précautions particulières d’élimination du matériel d’injection usagé. Ne pas jeter la seringue usagée dans votre poubelle ménagère.
Si vous avez utilisé plus de SOMATULINE L.P. 60 mg, solution injectable à libération prolongée en seringue préremplie que vous n’auriez dû
Si vous avez utilisé plus de SOMATULINE L.P. que vous n’auriez dû, consultez votre médecin.
Si vous avez reçu une dose trop importante de SOMATULINE L.P., vous pouvez présenter plus d’effets indésirables ou des effets indésirables plus sévères (voir rubrique 4 Quels sont les effets indésirables éventuels).
Si vous oubliez d’utiliser SOMATULINE L.P. 60 mg, solution injectable à libération prolongée en seringue préremplie
Dès que vous vous apercevez que vous avez oublié une injection, prévenez votre médecin qui vous conseillera sur le moment le mieux adapté pour effectuer votre prochaine injection.
Ne faites pas vous-même d’injection supplémentaire pour compenser l’injection que vous avez oublié de faire sans en discuter au préalable avec votre médecin.
Si vous arrêtez d’utiliser SOMATULINE L.P. 60 mg, solution injectable à libération prolongée en seringue préremplie
Une interruption de plus d’une injection ou un arrêt prématuré de votre traitement par SOMATULINE L.P. peut compromettre le succès de votre traitement. Demandez conseil à votre médecin avant d’arrêter votre traitement.
Si vous avez d’autres questions sur l’utilisation de ce médicament, demandez plus d’informations à votre médecin ou à votre pharmacien.
4. QUELS SONT LES EFFETS INDESIRABLES EVENTUELS ?
Prévenez immédiatement votre médecin si vous remarquez l’un des effets indésirables suivants :
· Si vous avez plus soif ou si vous vous sentez plus fatigué que d’habitude et si vous avez la bouche sèche. Ces signes peuvent indiquer que vous avez un taux élevé de sucre dans le sang ou que vous développez un diabète.
· Si vous avez une sensation de faim, des tremblements, une transpiration plus importante que d’habitude, ou si vous vous sentez confus. Ces signes peuvent indiquer que vous avez un taux bas de sucre dans le sang.
Ces effets indésirables sont fréquents ; ils peuvent concerner jusqu’à 1 personne sur 10.
Prévenez immédiatement votre médecin si vous remarquez que
· Votre visage devient rouge ou gonflé, ou si vous présentez des boutons ou une éruption cutanée.
· Vous sentez votre poitrine se serrer, vous êtes gêné pour respirer ou votre respiration est sifflante.
· Vous vous sentez faible, peut-être en relation avec une chute de la pression artérielle.
Ces signes peuvent être la conséquence d’une réaction allergique.
La fréquence de cet effet indésirable n’est pas connue car les données disponibles ne permettent pas de l’estimer.
Autres effets indésirables
Si vous remarquez l’un des effets indésirables mentionnés ci-dessous, veuillez en informer votre médecin ou votre pharmacien.
Les effets indésirables les plus fréquents sont des troubles gastro-intestinaux, des problèmes au niveau de la vésicule biliaire et des réactions au niveau du point d’injection.
Les effets indésirables qui peuvent survenir avec SOMATULINE L.P. sont listés ci-dessous par ordre de fréquence.
Très fréquents (pouvant survenir chez plus de 1 personne sur 10)
· Diarrhée, selles molles, douleurs abdominales.
· Calculs biliaires et autres problèmes de vésicule biliaire. Vous pouvez présenter des symptômes tels que douleur abdominale sévère et soudaine, fièvre élevée, jaunisse (peau et blanc des yeux devenant jaune), frissons, perte d’appétit, démangeaisons.
Fréquents (pouvant survenir jusque chez 1 personne sur 10)
· Perte de poids.
· Manque d’énergie.
· Battements cardiaques lents.
· Sensation de fatigue très importante.
· Diminution de l’appétit.
· Sensation de faiblesse généralisée.
· Excès de graisse dans les selles.
· Sensation de vertiges, maux de tête.
· Perte des cheveux ou diminution du développement des poils.
· Douleurs au niveau des muscles des ligaments, des tendons et des os.
· Réactions au niveau du point d’injection telles que douleur, induration ou démangeaisons.
· Anomalies des tests hépatiques et pancréatiques ; modification du taux de sucre dans le sang.
· Nausées, vomissements, constipation, flatulence, ballonnement ou inconfort gastrique, indigestion.
· Dilatation biliaire (élargissement des voies biliaires entre le foie et la vésicule biliaire et l’intestin). Vous pouvez ressentir des symptômes tels que douleurs de l’estomac, nausées, jaunisse et fièvre.
Peu fréquents (pouvant survenir jusque chez 1 personne sur 100)
· Bouffées de chaleur.
· Difficultés d’endormissement.
· Changement de couleur des selles.
· Modification des taux sanguins de phosphatases alcalines et de sodium.
Fréquence inconnue (la fréquence ne peut pas être estimée sur la base des données disponibles)
· Douleur soudaine et intense dans la partie basse de l’estomac. Ceci peut être un signe d’inflammation du pancréas (pancréatite).
· Rougeur, douleur, chaleur et gonflement au site d’injection qui peut sembler rempli de liquide lorsqu’il est pressé, et fièvre. Ceci peut être le signe d’un abcès.
· Douleur intense et soudaine en haut à droite de l’abdomen ou en son centre, pouvant irradier vers l’épaule ou le dos, une sensibilité de l’abdomen, des nausées, des vomissements et une forte fièvre. Ceci peut être le signe de calculs biliaires (cholécystite).
· Douleur dans la partie supérieure droite de votre ventre (abdomen), fièvre, frissons, jaunissement de la peau et des yeux (jaunisse), nausées, vomissements, selles couleur argile, urine foncée, et fatigue. Il peut s’agir des signes d'une inflammation du canal biliaire (cholangite).
· Diminution des enzymes pancréatiques. Comme le lanréotide peut affecter la libération des enzymes pancréatiques impliquées dans la digestion des aliments, vous pouvez présenter des symptômes tels que des selles grasses, des selles molles, des ballonnements abdominaux ou une perte de poids.
Comme le lanréotide peut modifier le taux de sucre dans le sang, votre médecin pourra juger nécessaire de vérifier le taux de sucre dans votre sang, surtout en début du traitement.
De même, comme ce type de médicament peut entraîner des problèmes au niveau de la vésicule biliaire, votre médecin pourra juger nécessaire de surveiller votre vésicule biliaire au début du traitement par SOMATULINE L.P., puis périodiquement lors de la poursuite du traitement.
Informez votre médecin ou votre pharmacien si vous remarquez l’un des effets secondaires ci-dessus.
Déclaration des effets secondaires
Si vous ressentez un quelconque effet indésirable, parlez-en à votre médecin ou votre pharmacien. Ceci s’applique aussi à tout effet indésirable qui ne serait pas mentionné dans cette notice. Vous pouvez également déclarer les effets indésirables directement via le système national de déclaration : Agence nationale de sécurité du médicament et des produits de santé (ANSM) et réseau des Centres Régionaux de Pharmacovigilance - Site internet : https://signalement.social-sante.gouv.fr.
En signalant les effets indésirables, vous contribuez à fournir davantage d’informations sur la sécurité du médicament.
5. COMMENT CONSERVER SOMATULINE L.P. 60 mg, solution injectable à libération prolongée en seringue préremplie ?
Tenir ce médicament hors de la vue et de la portée des enfants.
N’utilisez pas ce médicament après la date de péremption indiquée sur l’emballage après EXP. La date de péremption fait référence au dernier jour de ce mois.
A conserver au réfrigérateur (entre 2°C et 8°C), dans l’emballage d’origine à l’abri de la lumière.
Après sortie du réfrigérateur, le produit qui est resté dans son étui de protection scellé peut être remis au réfrigérateur pour y être conservé en vue d’une utilisation ultérieure, à condition de ne pas avoir subi plus de trois excursions de température et d’avoir été conservé pendant moins de 72 heures au total à une température inférieure à 40°C.
Chaque seringue préremplie est conditionnée individuellement.
Ne jetez aucun médicament au tout-à-l’égout ou avec les ordures ménagères. Demandez à votre pharmacien d’éliminer les médicaments que vous n’utilisez plus. Ces mesures contribueront à protéger l’environnement.
6. CONTENU DE L’EMBALLAGE ET AUTRES INFORMATIONS
· La substance active est :
Lanréotide....................................................................................................................... 60 mg
(sous forme d’acétate de lanréotide)
Pour une seringue préremplie.
· Les autres composants sont :
Eau pour préparations injectables, acide acétique glacial (pour ajustement du pH).
SOMATULINE L.P. est une solution injectable visqueuse à libération prolongée en seringue préremplie prête à l’emploi, comportant un système de sécurité automatique. Le produit est une solution blanche à jaune pâle, visqueuse et sursaturée.
Chaque seringue préremplie prête à l’emploi est emballée dans un étui de protection plastifié et dans une boîte en carton.
Boite contenant 1 seringue préremplie avec système de sécurité avec 1 aiguille fixe.
Titulaire de l’autorisation de mise sur le marché
70 RUE BALARD
75015 PARIS
Exploitant de l’autorisation de mise sur le marché
70 RUE BALARD
75015 PARIS
Parc d’Activités du Plateau de Signes
Chemin départemental n°402
83870 SIGNES
Noms du médicament dans les Etats membres de l'Espace Economique Européen
Sans objet.
La dernière date à laquelle cette notice a été révisée est :
[à compléter ultérieurement par le titulaire]
Des informations détaillées sur ce médicament sont disponibles sur le site Internet de l’ANSM (France).