Dernière mise à jour le 28/04/2026
EZETIMIBE KRKA 10 mg, comprimé
Indications thérapeutiques
EZETIMIBE KRKA est un médicament utilisé pour diminuer les taux élevés de cholestérol.
EZETIMIBE KRKA diminue les taux de cholestérol total, le « mauvais » cholestérol (LDL-cholestérol) et les substances grasses appelées triglycérides dans le sang. De plus, EZETIMIBE KRKA augmente le taux de « bon » cholestérol (HDL-cholestérol).
L’ézétimibe, la substance active d’EZETIMIBE KRKA, agit en réduisant le cholestérol absorbé par votre tube digestif.
EZETIMIBE KRKA complète l'effet hypocholestérolémiant des statines, une famille de médicaments qui réduit le cholestérol fabriqué par votre organisme.
Le cholestérol est une des nombreuses substances grasses trouvées dans le sang. Votre cholestérol total est composé principalement de LDL-cholestérol et de HDL-cholestérol.
Le LDL-cholestérol est souvent appelé « mauvais » cholestérol parce qu’il peut s’accumuler sur les parois de vos artères sous forme de plaque. La constitution de cette plaque peut entrainer un rétrécissement des artères. Ce rétrécissement peut ralentir ou bloquer le flux sanguin vers les organes vitaux comme le cœur et le cerveau. Ce blocage du flux sanguin peut entraîner une crise cardiaque ou un accident vasculaire cérébral.
Le HDL-cholestérol est souvent appelé « bon » cholestérol, parce qu’il empêche le « mauvais » cholestérol de s’accumuler dans les artères et protège contre les maladies cardiaques.
Les triglycérides sont une autre forme de graisse présente dans votre sang qui peut augmenter votre risque de maladie cardiaque.
EZETIMIBE KRKA est utilisé chez les patients chez qui les taux de cholestérol ne peuvent être contrôlés uniquement par un régime alimentaire pauvre en graisse. Vous devez continuer votre régime pauvre en graisse tout en prenant ce médicament.
EZETIMIBE KRKA est utilisé en complément de votre régime pauvre en graisse, si vous avez :
· un taux élevé de cholestérol dans votre sang (hypercholestérolémie primaire [familiale hétérozygote et non-familiale]) :
o en association à une statine, si votre taux de cholestérol n'est pas bien contrôlé par une statine seule,
o seul, lorsqu'un traitement par une statine est inapproprié ou mal toléré.
· une maladie héréditaire (hypercholestérolémie familiale homozygote) qui augmente votre taux de cholestérol sanguin. Une statine sera également prescrite et vous pourrez également recevoir un autre traitement.
· une maladie héréditaire (sitostérolémie homozygote également connue sous le nom de phytostérolémie) qui augmente le taux des stérols végétaux sanguins.
Si vous avez une maladie cardiaque, EZETIMIBE KRKA, pris avec des médicaments faisant baisser le cholestérol appelés « statines », réduit le risque de crise cardiaque, d'accident vasculaire cérébral, d’intervention chirurgicale pour augmenter le flux sanguin cardiaque ou d'hospitalisation pour des douleurs thoraciques.
EZETIMIBE KRKA ne vous aide pas à perdre du poids.
Présentations
> plaquette(s) OPA : polyamide orienté aluminium PVC-Aluminium de 30 comprimé(s)
Code CIP : 34009 301 252 1 2
Déclaration de commercialisation : 18/04/2018
Cette présentation est agréée aux collectivités
- Prix hors honoraire de dispensation : 11,75 €
- Honoraire de dispensation : 1,02 €
- Prix honoraire compris : 12,77 €
- Taux de remboursement :65%
> plaquette(s) OPA : polyamide orienté aluminium PVC-Aluminium de 90 comprimé(s)
Code CIP : 34009 301 252 3 6
Déclaration de commercialisation : 18/04/2018
Cette présentation est agréée aux collectivités
- Prix hors honoraire de dispensation : 33,23 €
- Honoraire de dispensation : 2,76 €
- Prix honoraire compris : 35,99 €
- Taux de remboursement :65%
Service médical rendu (SMR)
Amélioration du service médical rendu (ASMR)
Ce médicament étant un générique, l'ASMR n'a pas été évalué par la commission de la transparence (CT), il est possible de se référer à la /aux spécialité(s) de référence du groupe générique auquel appartient ce médicament (cliquez ici pour aller à la rubrique des groupes génériques)
ANSM - Mis à jour le : 17/01/2024
EZETIMIBE KRKA 10 mg, comprimé
2. COMPOSITION QUALITATIVE ET QUANTITATIVE
Chaque comprimé contient 10 mg d’ézétimibe.
Pour la liste complète des excipients, voir rubrique 6.1.
Les comprimés sont blancs à presque blancs, en forme de gélule, à bords biseautés. Dimensions du comprimé : 8 x 4 mm.
4.1. Indications thérapeutiques
EZETIMIBE KRKA en association avec une statine (inhibiteur de l'HMG-CoA réductase) est indiqué comme traitement adjuvant au régime chez les patients ayant une hypercholestérolémie primaire (familiale hétérozygote et non familiale) et qui ne sont pas contrôlés de façon appropriée par une statine seule.
EZETIMIBE KRKA en monothérapie est indiqué comme traitement adjuvant au régime chez les patients ayant une hypercholestérolémie primaire (familiale hétérozygote et non familiale) pour lesquels un traitement par statine est inapproprié ou est mal toléré.
Prévention des événements cardiovasculaires
EZETIMIBE KRKA est indiqué pour réduire le risque d’événements cardiovasculaires (voir la rubrique 5.1) chez les patients présentant une maladie coronaire avec un antécédent de syndrome coronarien aigu (SCA), en complément d’un traitement en cours par statine ou avec l’initiation concomitante d’une statine.
Hypercholestérolémie familiale homozygote (HFHo)
EZETIMIBE KRKA en association avec une statine est indiqué comme traitement adjuvant au régime chez les patients ayant une HFHo. Ces patients peuvent recevoir également des traitements adjuvants (exemple : aphérèse des LDL).
Sitostérolémie homozygote (phytostérolémie)
EZETIMIBE KRKA est indiqué comme traitement adjuvant au régime, chez les patients ayant une sitostérolémie familiale homozygote.
4.2. Posologie et mode d'administration
Pendant toute la durée du traitement par EZETIMIBE KRKA, le patient devra suivre un régime hypolipidémiant adapté.
La posologie recommandée est d'un comprimé d'EZETIMIBE KRKA 10 mg par jour. EZETIMIBE KRKA peut être administré à tout moment de la journée, pendant ou en dehors des repas.
Quand EZETIMIBE KRKA est prescrit en association à une statine, la posologie initiale usuelle, ou la posologie déjà établie de cette statine, doit être poursuivie. Dans ce cas, les recommandations d'adaptation posologique de la statine utilisée doivent être consultées.
Utilisation chez les patients présentant une maladie coronaire avec un antécédent de SCA
Pour obtenir une réduction supplémentaire des événements cardiovasculaires chez des patients présentant une maladie coronaire avec un antécédent de SCA, EZETIMIBE KRKA 10 mg peut être administré avec une statine ayant démontré un bénéfice cardiovasculaire.
Administration en association avec une résine échangeuse d'ions
L'administration d'EZETIMIBE KRKA se fera soit ≥ 2 heures avant ou ≥ 4 heures après l'administration d'une résine échangeuse d'ions.
Sujets âgés
Aucun ajustement posologique n'est nécessaire chez le sujet âgé (voir rubrique 5.2).
Patient présentant une insuffisance hépatique
Aucun ajustement posologique n'est nécessaire chez les patients ayant une insuffisance hépatique légère (score de Child Pugh 5 à 6). Le traitement par EZETIMIBE KRKA n'est pas recommandé chez les patients ayant une insuffisance hépatique modérée (score de Child Pugh 7 à 9) ou sévère (score de Child Pugh > 9) (voir rubriques 4.4 et 5.2).
Patient présentant une insuffisance rénale
Aucun ajustement posologique n'est nécessaire chez l'insuffisant rénal (voir rubrique 5.2).
Population pédiatrique
Le traitement doit être initié sous la surveillance d'un spécialiste.
Enfants et adolescents ≥ 6 ans : la sécurité d’emploi et l’efficacité d’ézétimibe chez l’enfant âgé de 6 à 17 ans n’ont pas été établies. Les données actuellement disponibles sont décrites dans les rubriques 4.4, 4.8, 5.1 et 5.2 mais aucune recommandation de posologie ne peut être émise.
Quand EZETIMIBE KRKA est co-administré avec une statine, se conformer aux indications de posologie de la statine chez les enfants.
Enfants < 6 ans : la sécurité d’emploi et l’efficacité d’ézétimibe chez l’enfant âgé de < 6 ans n’ont pas été établies. Aucune donnée n’est disponible.
Mode d’administration
La voie d’administration est la voie orale.
Hypersensibilité à la substance active ou à l'un des excipients mentionnés à la rubrique 6.1.
Lorsqu’EZETIMIBE KRKA est administré en association avec une statine, se référer au Résumé des Caractéristiques du Produit de la statine utilisée.
Le traitement par EZETIMIBE KRKA en association avec une statine est contre-indiqué pendant la grossesse ou l'allaitement.
EZETIMIBE KRKA en association avec une statine est contre-indiqué chez les patients présentant une affection hépatique évolutive ou des élévations persistantes et inexpliquées des transaminases sériques.
4.4. Mises en garde spéciales et précautions d'emploi
Enzymes hépatiques
Des études contrôlées de l'association d'EZETIMIBE KRKA avec une statine ont montré des élévations consécutives des transaminases (≥ 3 x la limite supérieure de la normale [LSN]). Dans ce cas, des tests fonctionnels hépatiques doivent être pratiqués au début du traitement et selon les recommandations relatives à la statine (voir rubrique 4.8).
Dans l’étude randomisée IMPROVE-IT (IMProved Reduction of Outcomes Vytorin Efficacy International Trial), 18 144 patients présentant une maladie coronaire avec un antécédent de SCA ont reçu soit 10 mg/40 mg d’ézétimibe/simvastatine par jour (n = 9 067), soit 40 mg de simvastatine par jour (n = 9 077). Au cours d'un suivi médian de 6,0 ans, l'incidence d’élévations consécutives des transaminases (≥ 3 x LSN) a été de 2,5 % pour ézétimibe/simvastatine et de 2,3 % pour simvastatine (voir rubrique 4.8).
Dans une étude clinique randomisée plus de 9 000 patients insuffisants rénaux chroniques ont reçu EZETIMIBE KRKA 10 mg en association avec la simvastatine 20 mg une fois par jour (n = 4 650) ou un placebo (n = 4 620) et ont été suivis pendant 4,9 années (médiane).
L'incidence d’élévations persistantes des transaminases sériques (> 3 x LSN) était de 0,7 % pour ézétimibe en association avec la simvastatine et 0,6 % dans le groupe placebo (voir rubrique 4.8).
Muscles squelettiques
Depuis la mise sur le marché ézétimibe, des cas d'atteinte musculaire et de rhabdomyolyse ont été rapportés. La plupart des patients qui ont présenté une rhabdomyolyse prenaient une statine de façon concomitante à l’ézétimibe. Cependant, des rhabdomyolyses ont été très rarement rapportées avec l’ézétimibe en monothérapie, et très rarement rapportées lorsque l’ézétimibe a été associé à d'autres médicaments connus pour être liés à un risque accru de rhabdomyolyse. Si une atteinte musculaire est suspectée, face à des symptômes musculaires, ou confirmée par un taux de CPK > 10 x LSN, EZETIMIBE KRKA, la statine ou tout autre de ces agents pris de façon concomitante doivent être arrêtés immédiatement. Tous les patients débutant un traitement par EZETIMIBE KRKA doivent être informés du risque d'atteinte musculaire, et doivent signaler rapidement toute sensibilité douloureuse, toute douleur ou faiblesse musculaires inexpliquées (voir rubrique 4.8).
Dans l’étude randomisée IMPROVE-IT, 18 144 patients présentant une maladie coronaire avec un antécédent de SCA ont reçu soit 10 mg/40 mg d’ézétimibe/simvastatine par jour (n = 9 067), soit 40 mg de simvastatine par jour (n = 9 077). Au cours d'un suivi médian de 6,0 ans, l'incidence des myopathies a été de 0,2 % pour ézétimibe/simvastatine et de 0,1 % pour simvastatine, les myopathies étant définies comme une faiblesse ou une douleur musculaire inexpliquée avec des CPK sériques ≥ 10 x LSN ou avec deux résultats consécutifs de CPK ≥ 5 et < 10 x LSN. L'incidence des rhabdomyolyses a été de 0,1 % avec ézétimibe/simvastatine et de 0,2 % avec simvastatine, la rhabdomyolyse étant définie comme une faiblesse ou une douleur musculaire inexpliquée avec des CPK sériques ≥ 10 x LSN et une atteinte rénale prouvée, ou avec des CPK ≥ 5 x LSN et < 10 x LSN à deux reprises consécutives avec une atteinte rénale prouvée ou avec des CPK ≥ 10 000 UI/L sans atteinte rénale prouvée (voir rubrique 4.8).
Dans une étude clinique randomisée, plus de 9 000 patients insuffisants rénaux chroniques ont reçu de l’ézétimibe 10 mg en association avec la simvastatine 20 mg une fois par jour (n = 4 650) ou un placebo (n = 4 620) et ont été suivis pendant 4,9 années (médiane). L'incidence des myopathies / rhabdomyolyses était de 0,2 % pour l’ézétimibe en association avec la simvastatine et 0,1 % dans le groupe placebo (voir rubrique 4.8).
Patients présentant une insuffisance hépatique
Les effets d'une exposition accrue à l'ézétimibe n'étant pas connus chez des patients présentant une insuffisance hépatique modérée ou sévère, EZETIMIBE KRKA n'est pas recommandé dans cette population (voir rubrique 5.2).
Population pédiatrique
L'efficacité et la sécurité d’emploi de l’ézétimibe chez les enfants âgés de 6 ans à 10 ans, ayant une hypercholestérolémie familiale hétérozygote ou non-familiale ont été évaluées dans un essai clinique contrôlé versus placebo pendant une durée de 12 semaines. Les effets de l'ézétimibe sur une période de plus de 12 semaines de traitement n'ont pas été étudiés dans cette tranche d'âge (voir rubriques 4.2, 4.8, 5.1 et 5.2).
L’ézétimibe n'a pas été étudié chez des patients âgés de moins de 6 ans (voir rubriques 4.2 et 4.8).
L'efficacité et la sécurité d'emploi de l’ézétimibe, en association avec la simvastatine, ont été évaluées dans un essai clinique contrôlé, réalisé chez des patients âgés de 10 à 17 ans, ayant une hypercholestérolémie familiale hétérozygote : des adolescents (présentant un stade II pubertaire ou plus selon l'échelle de Tanner) et des adolescentes (1 an au moins après l'apparition des premières règles).
Dans cette étude contrôlée, aucun effet n’a été décelé sur la croissance ou la maturité sexuelle des adolescents garçons ou filles, ni sur la longueur du cycle menstruel des filles. Cependant, les effets de l'ézétimibe sur la croissance ou la maturité sexuelle n'ont pas été étudiés au-delà de 33 semaines de traitement (voir rubriques 4.2 et 4.8).
La sécurité d'emploi et l'efficacité de l’ézétimibe administré en association avec des doses de plus de 40 mg de simvastatine par jour n'ont pas été étudiées chez des patients âgés de 10 à 17 ans.
La sécurité d'emploi et l'efficacité de l’ézétimibe administré en association avec la simvastatine n’ont pas été étudiées chez les enfants âgés de moins de 10 ans (voir rubriques 4.2 et 4.8).
Chez des patients de moins de 17 ans, l'efficacité à long terme du traitement par l’ézétimibe, afin de diminuer la morbi-mortalité à l'âge adulte, n'a pas été étudiée.
Fibrates
L'efficacité et la sécurité d'emploi de l’ézétimibe administré en association avec des fibrates n'ont pas été établies.
Si une lithiase biliaire est suspectée chez un patient traité par EZETIMIBE KRKA et fénofibrate, des investigations biliaires sont indiquées et le traitement doit être interrompu (voir rubriques 4.5 et 4.8).
Ciclosporine
Chez les patients traités par ciclosporine, l'initiation d'un traitement par EZETIMIBE KRKA se fera avec prudence. Les concentrations de ciclosporine doivent être surveillées chez les patients prenant EZETIMIBE KRKA associé à la ciclosporine (voir rubrique 4.5).
Anticoagulants
Si EZETIMIBE KRKA est associé à la warfarine, à un autre anticoagulant coumarinique, ou à la fluindione, le temps de prothrombine exprimé en INR doit être surveillé de façon appropriée (voir rubrique 4.5).
Ce médicament contient moins de 1 mmol (23 mg) de sodium par comprimé, c’est-à-dire qu’il est essentiellement « sans sodium ».
4.5. Interactions avec d'autres médicaments et autres formes d'interactions
Des études cliniques d'interaction ont montré que l'ézétimibe n'a pas d'effet sur la pharmacocinétique du dapsone, du dextrométhorphane, de la digoxine, des contraceptifs oraux (éthinylestradiol et lévonorgestrel), du glipizide, du tolbutamide, ou du midazolam quand il est administré en association à ces substances. La cimétidine, associée à l'ézétimibe, n'entraîne aucun effet sur la biodisponibilité de l'ézétimibe.
Anti-acides
L'administration simultanée d'anti-acides diminue le taux d'absorption d'ézétimibe mais n'a aucun effet sur la biodisponibilité de l'ézétimibe. Cette diminution du taux d'absorption d'ézétimibe n'est pas considérée comme cliniquement significative.
Cholestyramine
L'administration simultanée de cholestyramine diminue d'environ 55 % l'aire sous la courbe (ASC) moyenne de l'ézétimibe total (ézétimibe + glycuronide d'ézétimibe). La diminution supplémentaire du LDL-cholestérol observée liée à l'association EZETIMIBE KRKA et cholestyramine pourrait être réduite par cette interaction (voir rubrique 4.2).
Fibrates
Chez les patients traités par EZETIMIBE KRKA et fénofibrate, les médecins doivent être informés du risque potentiel de survenue de lithiase et d'affection de la vésicule biliaire (voir rubriques 4.4 et 4.8).
Si une lithiase biliaire est suspectée chez un patient traité par EZETIMIBE KRKA et fénofibrate, des investigations biliaires sont indiquées et le traitement doit être interrompu (voir rubrique 4.8).
L'administration simultanée de fénofibrate ou de gemfibrozil augmente modérément les concentrations totales d'ézétimibe, respectivement d'environ 1,5 et 1,7 fois. L'administration d'EZETIMIBE KRKA en association avec d'autres fibrates n'a pas été étudiée.
Les fibrates peuvent majorer l'excrétion du cholestérol dans la bile et entraîner une lithiase biliaire. Dans les études animales, l'ézétimibe augmente parfois la teneur en cholestérol de la bile vésiculaire mais pas chez toutes les espèces (voir rubrique 5.3). Un risque lithogène lié à la prise d'EZETIMIBE KRKA ne peut être écarté.
Statines
En association avec l'atorvastatine, la simvastatine, la pravastatine, la lovastatine, la fluvastatine ou la rosuvastatine, aucune interaction pharmacocinétique cliniquement significative n'a été observée.
Ciclosporine
Une étude réalisée chez 8 patients transplantés rénaux ayant une clairance de la créatinine > 50 mL/min, recevant une dose fixe de ciclosporine et une dose unique de 10 mg d’ézétimibe a montré une augmentation de l'ASC (aire sous la courbe) moyenne de l'ézétimibe total de 3,4 fois (2,3 à 7,9 fois) par rapport à des volontaires sains d'une autre étude (n = 17), traités par l'ézétimibe seul.
Dans une autre étude chez un patient transplanté rénal ayant une insuffisance rénale sévère et recevant de la ciclosporine ainsi que de nombreux médicaments, l'exposition totale à l'ézétimibe était 12 fois supérieure à celle des témoins. Dans une étude, en cross over, de deux périodes, chez 12 sujets sains, l'administration quotidienne de 20 mg d'ézétimibe pendant 8 jours, avec une seule dose de 100 mg de ciclosporine, au 7ème jour, comparée à l'administration d'une dose unique de 100 mg de ciclosporine (sans ézétimibe), a entraîné une augmentation moyenne de 15 % de l'ASC (aire sous la courbe) de la ciclosporine (avec une variation allant de - 10 % à + 51 %).
Aucune étude contrôlée sur les effets de l’administration de l’ézétimibe en association à la ciclosporine n’a été conduite chez les patients transplantés rénaux. La prudence est requise lors de l'initiation d'un traitement par EZETIMIBE KRKA chez les patients traités par la ciclosporine. Les concentrations de ciclosporine doivent être surveillées chez les patients prenant EZETIMIBE KRKA associé à la ciclosporine (voir rubrique 4.4).
Anticoagulants
Dans une étude chez 12 volontaires sains de sexe masculin, l'administration concomitante d'ézétimibe (10 mg une fois par jour) n'a pas eu d'effet significatif sur la biodisponibilité de la warfarine et sur le temps de prothrombine. Cependant, depuis la mise sur le marché, une augmentation du temps de prothrombine exprimé en INR a été rapportée chez des patients prenant de l’ézétimibe associé à la warfarine ou à la fluindione. Si EZETIMIBE KRKA est associé à la warfarine, à un autre anticoagulant coumarinique, ou à la fluindione, le temps de prothrombine exprimé en INR doit être surveillé de façon appropriée (voir rubrique 4.4).
Population pédiatrique
Les études d’interaction n’ont été réalisées que chez l’adulte.
4.6. Fertilité, grossesse et allaitement
En association avec une statine, EZETIMIBE KRKA est contre-indiqué pendant la grossesse et l'allaitement (voir rubrique 4.3). Se référer au Résumé des Caractéristiques du Produit de la statine utilisée.
Grossesse
EZETIMIBE KRKA ne sera administré à une femme enceinte qu'en cas de nécessité absolue. Aucune donnée clinique n'est disponible sur l'utilisation d’EZETIMIBE KRKA pendant la grossesse. Les études chez l'animal sur l'utilisation de l'ézétimibe en monothérapie n'ont pas mis en évidence d'effet délétère direct ou indirect sur la grossesse, le développement embryonnaire ou fœtal, la naissance ou le développement post-natal (voir rubrique 5.3).
Allaitement
EZETIMIBE KRKA ne doit pas être utilisé pendant l'allaitement. Des études réalisées chez le rat ont montré que l'ézétimibe est sécrété dans le lait maternel. Aucune donnée n'existe sur la sécrétion de l'ézétimibe dans le lait maternel chez la femme.
Fertilité
Il n’existe pas de données cliniques sur les effets de l’ézétimibe sur la fertilité chez l’être humain. L’ézétimibe n’a eu aucun effet sur la fertilité des rats mâles et femelles (voir rubrique 5.3).
4.7. Effets sur l'aptitude à conduire des véhicules et à utiliser des machines
Liste des effets indésirables (études cliniques et données rapportées depuis la mise sur le marché)
Dans des études cliniques d'une durée allant jusqu'à 112 semaines, ézétimibe 10 mg par jour a été administré seul chez 2 396 patients, en association avec une statine chez 11 308 patients ou avec le fénofibrate chez 185 patients. Les effets indésirables étaient habituellement légers et transitoires. L'incidence globale d'effets secondaires observés était identique entre l’ézétimibe et un placebo. De même, le taux d'arrêt du traitement dû aux événements indésirables était comparable entre l’ézétimibe et le placebo.
L’ézétimibe est administré seul ou en association avec une statine.
Les effets indésirables suivants ont été observés chez les patients traités par l’ézétimibe (N = 2 396), et avec une plus grande incidence qu'avec le placebo (N = 1 159) ou chez les patients traités par l’ézétimibe co-administré avec une statine (N = 11 308) et avec une plus grande incidence qu'avec une statine administrée seule (N = 9 361). Les effets indésirables rapportés depuis la commercialisation ont été observés avec l’ézétimibe administré seul ou avec une statine. Les effets indésirables observés au cours des études cliniques d’ézétimibe (en monothérapie ou co-administré avec une statine) ou qui ont été rapportés depuis la commercialisation d’ézétimibe administré seul ou avec une statine sont listés dans le tableau 1. Ces effets indésirables sont présentés par classe de systèmes d’organes et par fréquence.
Les fréquences sont définies comme : très fréquent (≥ 1/10) ; fréquent (≥ 1/100 à < 1/10) ; peu fréquent (≥ 1/1 000 à < 1/100) ; rare (≥ 1/10 000 à < 1/1 000) ; très rare (< 1/10 000) et indéterminée (ne peut être estimée sur la base des données disponibles).
Tableau 1
Effets Indésirables
|
Classes de systèmes d'organes Fréquence |
Effet indésirable |
|
Affection hématologique et du système lymphatique |
|
|
Indéterminée |
thrombocytopénie |
|
Affections du système immunitaire |
|
|
Indéterminée |
hypersensibilité ; incluant rash ; urticaire ; anaphylaxie et angio-œdème |
|
Troubles du métabolisme et de la nutrition |
|
|
Peu fréquent |
diminution de l'appétit |
|
Affections psychiatriques |
|
|
Indéterminée |
dépression |
|
Affections du système nerveux |
|
|
Fréquent |
maux de tête |
|
Peu fréquent |
paresthésie |
|
Indéterminée |
étourdissements |
|
Affections vasculaires |
|
|
Peu fréquent |
bouffées vasomotrices ; hypertension |
|
Affections respiratoires, thoraciques et médiastinales |
|
|
Peu fréquent |
toux |
|
Indéterminée |
dyspnée |
|
Affections gastro-intestinales |
|
|
Fréquent |
douleurs abdominales ; diarrhée ; flatulence |
|
Peu fréquent |
dyspepsie ; reflux gastro-œsophagien ; nausées : sécheresse buccale ; gastrite |
|
Indéterminée |
pancréatite ; constipation |
|
Affections hépatobiliaires |
|
|
Indéterminée |
hépatite ; lithiase biliaire ; cholécystite |
|
Affections de la peau et du tissu sous-cutané |
|
|
Peu fréquent |
prurit ; rash ; urticaire |
|
Indéterminée |
érythème multiforme |
|
Affections musculo-squelettiques et systémiques |
|
|
Fréquent |
myalgie |
|
Peu fréquent |
arthralgie ; crampes musculaires ; douleurs cervicales ; douleurs dorsales ; faiblesse musculaire ; douleur des extrémités |
|
Indéterminée |
myopathie/rhabdomyolyse (voir rubrique 4.4) |
|
Troubles généraux et anomalies au site d'administration |
|
|
Fréquent |
fatigue |
|
Peu fréquent |
douleur à la poitrine ; douleur ; asthénie ; œdème périphérique |
|
Investigations |
|
|
Fréquent |
augmentation des ALAT et/ou ASAT |
|
Peu fréquent |
augmentation des CPK ; augmentation des γ-GT ; anomalie du test fonctionnel hépatique |
L’ézétimibe en association avec le fénofibrate
Troubles gastro-intestinaux : douleurs abdominales (fréquent).
Dans une étude multicentrique, en double-aveugle, contrôlée, versus placebo, 625 patients présentant une hyperlipidémie mixte ont été traités jusqu'à 12 semaines et 576 jusqu'à un an. Dans cette étude, 172 patients ont été traités par l’ézétimibe associé au fénofibrate jusqu'à la fin des 12 semaines, et 230 patients ont été traités par l’ézétimibe associé au fénofibrate (incluant 109 patients ayant reçu l’ézétimibe en monothérapie pendant les 12 premières semaines) jusqu'à la fin de la période de 1 an. Cette étude n'avait pas pour but de comparer les groupes traités en termes d'effets peu fréquents.
Les taux d'incidence (IC 95 %) des augmentations cliniquement importantes des transaminases sériques (> 3 x LSN, consécutives) étaient de 4,5 % (1,9 ; 8,8) et 2,7 % (1,2 ; 5,4) respectivement pour les groupes fénofibrate en monothérapie et ézétimibe en association avec le fénofibrate, ajusté à l'exposition au traitement. Les taux d'incidence correspondant à la cholécystectomie étaient respectivement de 0,6 % (0,0 ; 3,1) et 1,7 % (0,6 ; 4,0) pour le fénofibrate en monothérapie et pour l’ézétimibe en association avec le fénofibrate (voir rubriques 4.4 et 4.5).
Population pédiatrique (6 à 17 ans)
Dans une étude menée chez des enfants (âgés de 6 à 10 ans) ayant une hypercholestérolémie familiale hétérozygote ou non-familiale (n = 138), des élévations des ALAT et/ou ASAT (≥ 3 x LSN, consécutives) ont été observées chez 1,1 % des patients (1 patient) dans le groupe ézétimibe, versus 0 % des patients dans le groupe placebo. Aucune élévation des CPK (≥ 10 x LSN) n’a été rapportée. Aucun cas de myopathie n'a été rapporté.
Dans une autre étude menée chez des adolescents (âgés de 10 à 17 ans) ayant une hypercholestérolémie familiale hétérozygote (n = 248), des élévations des ALAT et/ou ASAT (≥ 3 x LSN, consécutives) ont été observées chez 3 % des patients (4 patients) dans le groupe ézétimibe/simvastatine, versus 2 % des patients (2 patients) dans le groupe simvastatine en monothérapie. Pour l'élévation des CPK (≥ 10 x LSN) les données étaient respectivement de 2 % des patients (2 patients) et 0 %. Aucun cas de myopathie n'a été rapporté.
Ces essais n'avaient pas pour objet la comparaison des effets indésirables rares.
Patients présentant une maladie coronaire avec un antécédent de SCA
Dans l'étude IMPROVE-IT (voir rubrique 5.1), menée chez 18 144 patients traités soit par ézétimibe/simvastatine 10 mg/40 mg (n = 9 067 ; dont 6 % a reçu une posologie augmentée à 10 mg/80 mg d'ézétimibe/simvastatine) soit par 40 mg de simvastatine (n = 9 077 ; dont 27 % a reçu une posologie augmentée à 80 mg de simvastatine), les profils de sécurité ont été similaires pendant une période médiane de suivi de 6,0 ans. Les taux d'arrêt de traitement dus aux événements indésirables ont été de 10,6 % pour les patients traités par ézétimibe/simvastatine et de 10,1 % pour les patients traités par simvastatine.
L'incidence des myopathies a été de 0,2 % avec ézétimibe/simvastatine et de 0,1 % avec simvastatine ; les myopathies étant définies comme une faiblesse ou une douleur musculaire inexpliquée avec des CPK sériques ≥ 10 x LSN ou avec deux résultats consécutifs de CPK ≥ 5 x LSN et < 10 x LSN. L'incidence des rhabdomyolyses a été de 0,1 % avec ézétimibe/simvastatine et de 0,2 % avec simvastatine, les rhabdomyolyses étant définies comme une faiblesse ou une douleur musculaire inexpliquée avec des CPK sériques ≥ 10 x LSN et une atteinte rénale prouvée ou avec ≥ 5 x LSN et < 10 x LSN à deux reprises consécutives et une atteinte rénale prouvée ou avec des CPK ≥ 10 000 UI/L sans atteinte rénale prouvée. L'incidence d'élévations consécutives des transaminases (≥ 3 x LSN) a été de 2,5 % avec ézétimibe/simvastatine et de 2,3 % avec simvastatine (voir rubrique 4.4). Des effets indésirables liés à la vésicule biliaire ont été rapportés chez 3,1 % des patients traités par ézétimibe/simvastatine versus 3,5 % des patients traités par simvastatine. L'incidence des hospitalisations pour une cholécystectomie a été de 1,5 % dans les deux groupes de traitement. Des cancers (définis comme toute nouvelle tumeur maligne) ont été diagnostiqués au cours de l’étude chez 9,4 % des patients traités par ézétimibe/simvastatine versus 9,5 % des patients traités par simvastatine.
Patients insuffisants rénaux chroniques
Dans l'étude SHARP (Study of Heart and Renal Protection) (voir rubrique 5.1), réalisée chez plus de 9 000 patients traités, recevant de l’ézétimibe 10 mg en association avec la simvastatine 20 mg à dose fixe une fois par jour (n = 4 650) ou du placebo (n = 4 620), les profils de sécurité ont été comparables pendant une période de suivi de 4,9 années (médiane). Dans cet essai, seuls les effets indésirables graves et les arrêts de traitement dus à des effets indésirables ont été rapportés. Les taux d'arrêt de traitement en raison d'effets indésirables étaient comparables (10,4 % chez les patients traités par l’ézétimibe en association avec de la simvastatine, et 9,8 % chez ceux traités par placebo). L'incidence d'atteinte musculaire/rhabdomyolyse était de 0,2 % chez les patients traités par l’ézétimibe en association avec de la simvastatine et de 0,1 % chez ceux traités par placebo. L'incidence d’élévations persistantes des transaminases sériques (> 3 x LSN) était de 0,7 % chez les patients traités par l’ézétimibe en association avec de la simvastatine comparé à 0,6 % chez ceux traités par placebo (voir rubrique 4.4).
Dans cet essai, il n'y a pas eu d'augmentation significative des effets indésirables pré-spécifiés, tels que cancer (9,4 % pour l’ézétimibe en association avec de la simvastatine et 9,5 % pour le placebo), hépatite, cholécystectomie ou complication de calculs biliaires ou de pancréatite.
Valeurs biologiques
Une augmentation cliniquement significative des transaminases sériques (ALAT et/ou ASAT ≥ 3 x LSN, consécutives) a été observée de manière similaire entre l’ézétimibe (0,5 %) et le placebo (0,3 %) au cours des études cliniques contrôlées en monothérapie.
Dans les études cliniques de co-administration, l'incidence était de 1,3 % chez les patients traités par l’ézétimibe en association à une statine, et de 0,4 % pour les patients traités par une statine seule. Ces augmentations sont généralement asymptomatiques, non associées à une cholestase ; les valeurs reviennent à leur valeur initiale à l'arrêt du traitement ou lors de la poursuite du traitement (voir rubrique 4.4).
Dans les essais cliniques, l'incidence des CPK > 10 x LSN concernait 4 des 1 674 patients sous ézétimibe seul (soit 0,2 %) versus 1 des 786 patients sous placebo (soit 0,1 %) ; et 1 des 917 patients sous ézétimibe associé à une statine (soit 0,1 %) versus 4 des 929 patients sous statine seule (soit 0,4 %). Il n'y a pas eu d'augmentation d'atteinte musculaire ou de rhabdomyolyse associée à l’ézétimibe en comparaison avec le groupe contrôle (placebo ou statine seule) (voir rubrique 4.4).
Déclaration des effets indésirables suspectés
La déclaration des effets indésirables suspectés après autorisation du médicament est importante. Elle permet une surveillance continue du rapport bénéfice/risque du médicament. Les professionnels de santé déclarent tout effet indésirable suspecté via le système national de déclaration : Agence nationale de sécurité du médicament et des produits de santé (ANSM) et réseau des Centres Régionaux de Pharmacovigilance - Site internet : https://signalement.social-sante.gouv.fr/.
Dans des études cliniques, l'administration d'ézétimibe à la dose de 50 mg/jour chez 15 sujets sains sur une période allant jusqu'à 14 jours ou à la dose de 40 mg/jour chez 18 patients ayant une hypercholestérolémie primaire sur une période allant jusqu'à 56 jours, a été généralement bien tolérée. Chez l'animal, aucune toxicité n'a été observée après des doses orales uniques de 5 000 mg/kg d'ézétimibe chez le rat et la souris et de 3 000 mg/kg chez le chien.
Quelques cas de surdosage ont été rapportés avec l’ézétimibe. La plupart d'entre eux n'ont pas été associés à des effets indésirables. Il n'a pas été rapporté d'effets indésirables graves. En cas de surdosage, un traitement symptomatique voire des mesures complémentaires doivent être utilisées.
5. PROPRIETES PHARMACOLOGIQUES
5.1. Propriétés pharmacodynamiques
Mécanisme d’action
EZETIMIBE KRKA appartient à une nouvelle classe d'agents hypolipidémiants qui inhibent de façon sélective l'absorption intestinale du cholestérol et des phytostérols apparentés. EZETIMIBE KRKA est utilisé par voie orale. Son mécanisme d'action diffère de celui des autres classes d'agents hypocholestérolémiants (statines, résines échangeuses d'ions, dérivés de l'acide fibrique et stanols végétaux). La cible moléculaire de l'ézétimibe est le transporteur de stérols, Niemann-Pick C1-Like 1 (NPC1L1), qui est responsable de l'absorption intestinale du cholestérol et des phytostérols.
L'ézétimibe se localise au niveau de la bordure en brosse de l'intestin grêle et inhibe l'absorption du cholestérol, entraînant une diminution des apports au foie du cholestérol intestinal alors que les statines diminuent la synthèse hépatique du cholestérol. Ainsi, ces deux molécules administrées simultanément avec des mécanismes distincts entraînent une diminution complémentaire du cholestérol. Une étude clinique de 2 semaines réalisée chez 18 patients hypercholestérolémiques a montré que l’ézétimibe inhibe l'absorption intestinale du cholestérol de 54 % par rapport à un placebo.
Effets pharmacodynamiques
Plusieurs études précliniques visant à déterminer la sélectivité de l'ézétimibe sur l'inhibition de l'absorption du cholestérol ont été réalisées. L'ézétimibe inhibe l'absorption du [14C]-cholestérol mais n'a pas d'effet sur l'absorption des triglycérides, des acides gras, des acides biliaires, de la progestérone, de l'éthinylestradiol, ou des vitamines liposolubles A et D.
Des études épidémiologiques ont montré que la morbidité et la mortalité cardiovasculaires varient directement avec le taux de cholestérol total et LDL-cholestérol et est inversement proportionnel au taux de HDL-cholestérol.
L'administration d'ézétimibe avec une statine est efficace pour réduire le risque d'événements cardiovasculaires chez les patients présentant une maladie coronaire avec un antécédent de SCA.
Efficacité et sécurité clinique
Des études cliniques contrôlées ont montré que, chez des patients ayant une hypercholestérolémie, ézétimibe seul ou en association avec une statine, diminue significativement le cholestérol total (C-total), le LDL-cholestérol (lié aux lipoprotéines de basse densité), les apolipoprotéines B (Apo B), les triglycérides (TG) et augmente le HDL-cholestérol (lié aux lipoprotéines de haute densité).
Hypercholestérolémie primaire
Dans une étude en double aveugle, contrôlée contre placebo, de 8 semaines, 769 patients ayant une hypercholestérolémie déjà traités par une statine en monothérapie, mais n'atteignant pas l'objectif de LDL-cholestérol du NCEP (National Cholesterol Education Program) (soit 2,6 à 4,1 mmol/L [100 à 160 mg/dL]), selon les caractéristiques initiales, ont été randomisés pour recevoir soit 10 mg d'ézétimibe, soit un placebo, en association avec le traitement par la statine en cours.
Chez les patients traités par statine et n'ayant pas atteint l'objectif de LDL-C lors du bilan initial (~82 %), l'objectif du LDL-cholestérol a été atteint en fin d'étude par 72 % des patients randomisés et traités par ézétimibe contre 19 % des patients traités par placebo. Les diminutions correspondantes du LDL-cholestérol étaient significativement différentes entre ézétimibe et le placebo (25 % et 4 %, respectivement). De plus, ézétimibe, en association au traitement par la statine en cours a significativement diminué le taux de cholestérol total, l’Apo B et les triglycérides et augmenté le taux de HDL-cholestérol par rapport au placebo. L'association d'ézétimibe ou d'un placebo à un traitement par une statine a diminué la concentration moyenne de la protéine C-réactive de respectivement 10 % et 0 % par rapport à la valeur initiale.
Deux études randomisées en double aveugle contrôlées versus placebo d'une durée de 12 semaines réalisées chez 1 719 patients ayant une hypercholestérolémie primaire ont montré que 10 mg d'ézétimibe diminuait significativement le cholestérol total (13 %), le LDL-cholestérol (19 %), le taux d'Apo B (14 %) et les triglycérides (8 %) et augmentait le HDL-cholestérol (3 %), comparativement au placebo. De plus, ézétimibe n'a eu aucun effet sur les concentrations plasmatiques des vitamines liposolubles A, D et E, et sur le temps de prothrombine, et comme pour les autres hypolipidémiants, aucune modification de la production d'hormones stéroïdiennes cortico-surrénaliennes n'a été observée.
Dans une étude clinique contrôlée, multicentrique, en double aveugle (ENHANCE), 720 patients ayant une hypercholestérolémie familiale hétérozygote ont été randomisés pour recevoir 10 mg d'ézétimibe en association à 80 mg de simvastatine (n = 357) ou 80 mg de simvastatine (n = 363) pendant 2 ans. L'objectif principal de cette étude était d'évaluer l'effet de l'association ézétimibe/simvastatine sur l'épaisseur intima-média (EIM) de l'artère carotide par rapport à la simvastatine seule. L'impact de ce marqueur de substitution sur la morbi-mortalité cardiovasculaire n'a pas encore été démontré.
La mesure par échographie en mode B de la modification de l'EIM moyenne des 6 segments de l'artère carotide (critère principal), n'a pas mis en évidence de différence significative (p = 0,29) entre les 2 groupes de traitement. Avec l'association ézétimibe 10 mg/simvastatine 80 mg ou avec la simvastatine 80 mg seule, l'épaisseur intima-média a augmenté respectivement de 0,0111 mm et 0,0058 mm, pendant les 2 ans de la durée de l'étude (EIM initiale moyenne = 0,68 mm et 0,69 mm, respectivement).
Les taux de LDL-cholestérol, de cholestérol total, d'Apo B et de triglycérides étaient significativement plus diminués avec l'association ézétimibe 10 mg/simvastatine 80 mg qu'avec la simvastatine 80 mg. L'augmentation du pourcentage de l'HDL-cholestérol a été similaire dans les deux groupes de traitement. Les effets indésirables rapportés avec l'association ézétimibe 10 mg/simvastatine 80 mg étaient cohérents avec le profil connu de sécurité d'emploi.
Population pédiatrique
Dans une étude contrôlée, multicentrique, en double aveugle, 138 patients (59 garçons et 79 filles) âgés de 6 à 10 ans (âge moyen 8,3 ans) ayant une hypercholestérolémie familiale hétérozygote (HFHe) ou non-familiale, avec des taux de LDL-cholestérol à l’inclusion entre 3,74 et 9,92 mmol/L, ont été randomisés pour recevoir ézétimibe 10 mg ou un placebo pendant 12 semaines.
A la 12ème semaine, ézétimibe a significativement diminué le cholestérol total (-21 % vs 0 %), le LDL-cholestérol (-28 % vs -1 %), le taux d'Apo B (-22 % vs -1 %), et le non HDL-cholestérol (-26 % vs 0 %) par rapport au placebo. Les résultats étaient non significativement différents dans les deux groupes de traitement pour les triglycérides et le HDL-cholestérol (-6 % vs +8 % et +2 % vs +1 %, respectivement).
Dans une étude contrôlée, multicentrique, en double aveugle, 142 garçons (présentant un stade pubertaire de grade II et plus selon l'échelle de Tanner) et 106 filles (1 an au moins après l'apparition des premières règles) âgés de 10 à 17 ans (âge moyen de 14,2 ans) ayant une hypercholestérolémie familiale hétérozygote (HFHe) avec des taux initiaux de LDL-cholestérol de 4,1 à 10,4 mmol/L ont été randomisés pour recevoir de l’ézétimibe 10 mg en association avec de la simvastatine (10, 20 ou 40 mg) ou de la simvastatine seule (10, 20 ou 40 mg) pendant 6 semaines.
Ils ont reçu ensuite pendant les 27 semaines suivantes ézétimibe en association à 40 mg de simvastatine ou 40 mg de simvastatine seule. Cet essai s'est poursuivi en ouvert avec ézétimibe associé à de la simvastatine (10, 20 ou 40 mg) pendant 20 semaines.
Après 6 semaines de traitement, ézétimibe associé à la simvastatine (toutes doses confondues) a significativement diminué le cholestérol total (38 % vs 26 %), le LDL-cholestérol (49 % vs 34 %), le taux d'Apo B (39 % vs 27 %), et le non HDL-cholestérol (47 % vs 33 %) en comparaison à la simvastatine seule (toutes doses). Les résultats étaient similaires dans les deux groupes de traitement pour les triglycérides et le HDL-cholestérol (-17 % vs -12 % et + 7 % vs + 6 %, respectivement). Après 33 semaines, les résultats étaient cohérents avec ceux observés après 6 semaines. Plus de patients traités par l'association ézétimibe/simvastatine 40 mg (62 %) ont atteint l’objectif idéal du NCEP/AAP (National Cholesterol Education Program/American Academy of Paediatrics) (< 2,8 mmol/l [110 mg/dL] pour le LDL-cholestérol en comparaison à ceux recevant 40 mg de simvastatine (25 %). A la fin de la période de prolongation de cet essai, après 53 semaines, les effets sur les paramètres lipidiques étaient maintenus.
L'efficacité et la sécurité d'emploi d'ézétimibe en association à des doses de simvastatine supérieures à 40 mg par jour n'ont pas été étudiées chez des patients âgés de 10 à 17 ans. L'efficacité et la sécurité d'emploi d'ézétimibe en association avec de la simvastatine n’ont pas été étudiées chez les enfants âgés de moins de 10 ans.
L'efficacité à long terme du traitement par ézétimibe chez des patients de moins de 17 ans pour réduire la morbi-mortalité à l'âge adulte n'a pas été étudiée.
Prévention des événements cardiovasculaires
IMPROVE-IT (IMProved Reduction of Outcomes : Vytorin Efficacy International Trial) était une étude multicentrique, randomisée, en double-insu, menée versus comparateur actif chez 18 144 patients recrutés dans les 10 jours suivant une hospitalisation pour syndrome coronarien aigu (SCA ; infarctus du myocarde aigu [IDM] ou angor instable). A l’inclusion, les patients avec SCA avaient un taux de LDL-cholestérol soit ≤ 125 mg/dL (≤ 3,2 mmol/L) s’ils n’avaient pas de traitement hypolipidémiant, soit ≤ 100 mg/dL (≤ 2,6 mmol/L) s’ils prenaient un traitement hypolipidémiant. Tous les patients ont été randomisés selon un ratio 1 : 1 pour recevoir soit ézétimibe/simvastatine 10 mg/40 mg (n = 9 067) soit simvastatine 40 mg (n = 9 077) et ont été suivis pendant une période médiane de 6,0 ans.
Les patients avaient un âge moyen de 63,6 ans ; 76 % étaient des hommes, 84 % étaient d’origine caucasienne et 27 % étaient diabétiques. La valeur moyenne du LDL-cholestérol au moment de l’événement qualifiant l’entrée dans l’étude était de 80 mg/dL (2,1 mmol/L) pour ceux déjà sous traitement hypolipidémiant (n = 6 390) et de 101 mg/dL (2,6 mmol/L) pour ceux sans traitement hypolipidémiant antérieur (n = 11 594). Avant l’hospitalisation pour le SCA qualifiant l’inclusion dans l’étude, 34 % des patients étaient sous traitement par statine. Après un an, le LDL-cholestérol moyen des patients toujours sous traitement étaient de 53,2 mg/dL (1,4 mmol/L) dans le groupe ézétimibe/simvastatine et de 69,9 mg/dL (1,8 mmol/L) dans le groupe simvastatine en monothérapie. Les valeurs lipidiques ont été principalement obtenues chez les patients poursuivant le traitement de l'étude.
Le critère d'évaluation principal était un critère composite comprenant le décès d’origine cardiovasculaire, l’événement coronarien majeur (définis comme infarctus du myocarde non fatal, angor instable documenté nécessitant une hospitalisation, ou intervention de revascularisation coronaire intervenant au moins 30 jours après la randomisation des groupes de traitement) et l’AVC non fatal. L’étude a démontré que le traitement par ézétimibe ajouté à la simvastatine apporte un bénéfice supplémentaire en réduisant le critère composite principal comprenant le décès d’origine cardiovasculaire, l’événement coronarien majeur et l’AVC non fatal, comparé au traitement par simvastatine seule (réduction du risque relatif de 6,4 %, p = 0,016).
Le critère d’évaluation principal est survenu chez 2 572 des 9 067 patients (taux de survie à 7 ans selon Kaplan-Meyer [KM] de 32,72 %) dans le groupe ézétimibe/simvastatine et chez 2 742 des 9 077 patients (taux de survie à 7 ans selon KM de 34,67 %) dans le groupe simvastatine seule (voir Figure 1 et Tableau 2).
Ce bénéfice supplémentaire dans la réduction du risque d'événements cardiovasculaires devrait être similaire lorsque ézétimibe est co-administré avec les statines ayant prouvé une réduction du risque cardiovasculaire. La mortalité totale est restée inchangée dans ce groupe à haut risque (voir Tableau 2).
Un bénéfice global a été observé pour tous les AVC ; cependant il y a eu une petite augmentation non-significative d’AVC hémorragique dans le groupe ézétimibe/simvastatine en comparaison avec simvastatine seule (voir Tableau 2). Le risque d’AVC hémorragique lorsqu’ézétimibe est co-administré avec des statines plus puissantes n’a pas été évalué dans des études à long-terme.
L’effet du traitement de l’association ézétimibe/simvastatine était généralement cohérent sur l’ensemble des résultats recueillis dans de nombreux sous-groupes, incluant sexe, âge, origine ethnique, antécédent de diabète, taux de lipides à l’inclusion, traitement antérieur par statine, antécédent d’AVC, et hypertension.
Figure 1 : Effet d’ézétimibe/simvastatine sur le critère composite principal comprenant le décès d’origine cardiovasculaire, l’événement coronarien majeur, ou l’AVC non fatal.
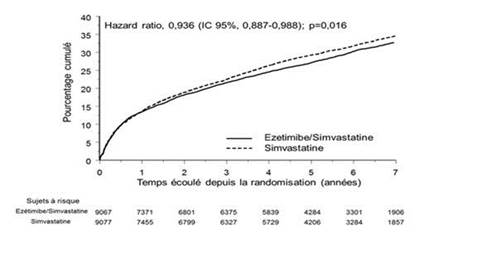
Tableau 2
Evénements cardiovasculaires majeurs par groupe de traitement chez tous les patients randomisés d’IMPROVE-IT
|
Résultat |
Ezétimibe/Simvastatine 10 mg/40 mga (N = 9 067) |
Simvastatine 40 mgb (N = 9 077) |
Hazard Ratio (IC 95 %) |
Valeur p |
||
|
n |
KM % c |
n |
KM % c |
|||
|
Critère d’efficacité composite principal |
||||||
|
(Décès d’origine cardiovasculaire, événements coronariens majeurs et AVC non fatal) |
2 572 |
32,72 % |
2 742 |
34,67 % |
0,936 (0,887 ; 0,988) |
0,016 |
|
Critère d’efficacité composite secondaire |
||||||
|
Décès d’origine coronarienne, infarctus du myocarde non fatal, revascularisation coronaire urgente après 30 jours |
1 322 |
17,52 % |
1 448 |
18,88 % |
0,912 (0,847 ; 0,983) |
0,016 |
|
Evénement coronarien majeur, AVC non fatal, décès (toutes causes) |
3 089 |
38,65 % |
3 246 |
40,25 % |
0,948 (0,903 ; 0,996) |
0,035 |
|
Décès d’origine cardiovasculaire, infarctus du myocarde non fatal, angor instable nécessitant une hospitalisation, toute revascularisation, AVC non fatal |
2 716 |
34,49% |
2 869 |
36,20 % |
0,945 (0,897 ; 0,996) |
0,035 |
|
Composants du critère d’efficacité composite principal et critères d’efficacité sélectionnés (premières apparitions d’un événement défini pouvant survenir à tout moment) |
||||||
|
Décès d’origine cardiovasculaire |
537 |
6,89 % |
538 |
6,84 % |
1,000 (0,887 ; 1,127) |
0,997 |
|
Evénement coronarien majeur : |
||||||
|
Infarctus du myocarde non fatal |
945 |
12,77 % |
1 083 |
14,41 % |
0,871 (0,798 ; 0,950) |
0,002 |
|
Angor instable nécessitant une hospitalisation |
156 |
2,06 % |
148 |
1,92 % |
1,059 (0,846 ; 1,326) |
0,618 |
|
Revascularisation coronaire après 30 jours |
1 690 |
21,84 % |
1 793 |
23,36 % |
0,947 (0,886 ; 1,012) |
0,107 |
|
AVC non fatal |
245 |
3,49 % |
305 |
4,24 % |
0,802 (0,678 ; 0,949) |
0,010 |
|
Tout infarctus du myocarde (fatal et non fatal) |
977 |
13,13 % |
1 118 |
14,82 % |
0,872 (0,800 : 0,950) |
0,002 |
|
Tout AVC (fatal et non fatal) |
296 |
4,16 % |
345 |
4,77 % |
0,857 (0,734 ; 1,001) |
0,052 |
|
AVC non hémorragiqued |
242 |
3,48 % |
305 |
4,23 % |
0,793 (0,670 ; 0,939) |
0,007 |
|
AVC hémorragique |
59 |
0,77 % |
43 |
0,59% |
1,377 (0,930 ; 2,040) |
0,110 |
|
Décès toute cause |
1 215 |
15,36 % |
1 231 |
15,28 % |
0,989 (0,914 ; 1,070) |
0,782 |
a 6 % ont reçu une augmentation de la dose à 10 mg/80 mg d’ézétimibe/simvastatine
b 27 % ont reçu une augmentation de la dose à 80 mg de simvastatine
c Taux de survie à 7 ans selon Kaplan-Meyer
d inclut un AVC ischémique ou un AVC de type indéterminé
Prévention des accidents vasculaires majeurs dans la maladie rénale chronique (IRC)
L'étude SHARP (Study of Heart and Renal Protection) était une étude multicentrique, randomisée, en double aveugle versus placebo menée chez 9 438 patients insuffisants rénaux chroniques, dont un tiers de dialysés à l'inclusion. Un total de 4 650 patients ont été traités par ézétimibe 10 mg en association avec de la simvastatine 20 mg et 4 620 ont reçu un placebo ; ils ont été suivis pendant une durée médiane de 4,9 ans. Les patients avaient un âge moyen de 62 ans, 63 % étaient des hommes, 72 % de type caucasien, 23 % étaient diabétiques, et pour ceux qui n'étaient pas dialysés, le débit moyen de filtration glomérulaire estimé (DFGe) était de 26,5 mL/min/1,73 m2. Il n'y avait pas de critère d'inclusion basé sur les lipides.
Le taux moyen initial de LDL-cholestérol était de 108 mg/dL. Après un an, en incluant les patients ne prenant plus de médicament de l'étude, le LDL-cholestérol était réduit de 26 % dans le groupe simvastatine 20 mg seule et de 38 % dans le groupe ézétimibe 10 mg associé à la simvastatine 20 mg, par rapport au placebo.
Le critère principal du protocole SHARP était une analyse en intention de traiter des événements vasculaires majeurs (définis comme infarctus du myocarde non fatal, décès d'origine cardiaque, accident vasculaire cérébral ou toute intervention de revascularisation), uniquement chez les patients randomisés dans le groupe ézétimibe en association avec simvastatine (n = 4 193) ou le groupe placebo (n = 4 191). Les analyses secondaires incluaient le même critère composite pour la population entière (à l'inclusion ou à un an) randomisée dans le groupe ézétimibe en association avec simvastatine (n = 4 650) ou le groupe placebo (n = 4 620), ainsi que les composants de ces critères.
L'analyse du critère principal montrait qu'ézétimibe en association avec simvastatine réduisait significativement le risque d'événements vasculaires majeurs (749 patients avec des événements dans le groupe placebo contre 639 dans le groupe ézétimibe en association avec simvastatine) avec une réduction du risque relatif de 16 % (p = 0,001).
Cependant, le schéma de cette étude n'a pas permis de déterminer la contribution spécifique de l'ézétimibe pour réduire significativement le risque d'accidents vasculaires majeurs chez les patients ayant une insuffisance rénale chronique.
Les composants individuels des événements vasculaires majeurs chez l’ensemble des patients randomisés sont présentés dans le Tableau 3. Ezétimibe en association avec simvastatine a significativement réduit le risque d'accident vasculaire cérébral et de revascularisation, avec des différences numériques non significatives en faveur d'ézétimibe en association avec simvastatine pour les infarctus du myocarde non fatals et les décès d'origine cardiaque.
Tableau 3
Evénements vasculaires majeurs, par groupe de traitement, chez tous les patients inclus dans SHARPa
|
Résultats |
Ezétimibe 10 mg en association avec simvastatine 20 mg (n = 4 650) |
Placebo (n = 4 620) |
Rapport risques (IC 95 %) |
Valeur p |
|
Evénements vasculaires majeurs |
701 (15,1 %) |
814 (17,6 %) |
0,85 (0,77-0,94) |
0,001 |
|
Infarctus du myocarde non fatal |
134 (2,9 %) |
159 (3,4 %) |
0,84 (0,66 -1,05) |
0,12 |
|
Mort cardiaque |
253 (5.4 %) |
272 (5.9 %) |
0.93 (0.78-1.10) |
0.38 |
|
Accident vasculaire cérébral |
171 (3,7 %) |
210 (4,5 %) |
0,81 (0,66 - 0,99) |
0,038 |
|
AVC non hémorragique |
131 (2,8 %) |
174 (3,8 %) |
0,75 (0,60 - 0,94) |
0,011 |
|
AVC hémorragique |
45 (1,0 %) |
37 (0,8 %) |
1,21 (0,78 - 1,86) |
0,40 |
|
Toute revascularisation |
284 (6,1 %) |
352 (7,6 %) |
0,79 (0,68 - 0,93) |
0,004 |
|
Evénements athérosclérotiques majeurs b |
526 (11,3 %) |
619 (13,4 %) |
0,83 (0,74 - 0,94) |
0,002 |
a Analyse en intention de traiter sur la totalité des patients randomisés au début de l'étude SHARP ou à 1 an, dans le groupe ézétimibe en association avec simvastatine ou le groupe placebo.
b Evénements athérosclérotiques majeurs tels que infarctus du myocarde non fatals, décès d'origine coronaire, accident vasculaire non hémorragique, toute revascularisation.
La réduction absolue du LDL-cholestérol atteinte avec ézétimibe en association avec simvastatine, était inférieure chez les patients ayant un taux de LDL-cholestérol plus bas (< 2,5 mmol/L) à l’inclusion et chez les patients dialysés à l'inclusion, par rapport aux autres patients, et les réductions de risque correspondant dans ces deux groupes étaient atténuées.
Hypercholestérolémie familiale homozygote (HFHo)
Une étude randomisée, en double aveugle, d'une durée de 12 semaines, réalisée chez 50 patients présentant une hypercholestérolémie familiale homozygote HFHo (diagnostic clinique et/ou génotypique) et traitée par atorvastatine ou simvastatine (40 mg), associée ou non à une aphérèse du LDL-cholestérol a montré que ézétimibe, associé à un traitement par atorvastatine (40 ou 80 mg) ou par simvastatine (40 ou 80 mg) diminuait significativement le LDL-cholestérol de 15 %, alors qu'une augmentation de dose de 40 à 80 mg était nécessaire chez les patients traités en monothérapie par simvastatine ou atorvastatine.
Sitostérolémie homozygote (phytostérolémie)
Une étude randomisée, en double aveugle, versus placebo, d'une durée de 8 semaines, réalisée chez 37 patients présentant une sitostérolémie homozygote recevant soit 10 mg d'ezetimibe (n = 30) soit un placebo (n = 7), associé ou non chez certains patients à d'autres traitements (tels que statines, résines) a montré que l’ezetimibe diminuait significativement les taux des deux principaux phytostérols, le sitostérol et le campestérol, de 21 % et 24 % respectivement par rapport à leur valeur initiale. Les effets du sitostérol sur la diminution de la morbidité et de la mortalité dans cette population ne sont pas connus.
Sténose aortique
L'étude SEAS (Simvastatine et ézétimibe dans le traitement de la Sténose Aortique) était une étude multicentrique, en double aveugle, versus placebo, d'une durée de 4,4 ans (médiane), réalisée chez 1 873 patients avec une sténose aortique asymptomatique documentée par mesure écho-doppler du pic de vélocité du flux aortique situé dans l'intervalle 2,5 - 4,0 m/s. Seuls ont été inclus dans l'étude les patients ne nécessitant pas un traitement par une statine pour réduire le risque cardiovasculaire lié à l'athérosclérose. Les patients ont été randomisés (ratio 1 : 1) pour recevoir un placebo ou l'ézétimibe 10 mg associé à la simvastatine 40 mg par jour.
Le critère principal était un composite d'événements cardiovasculaires majeurs associant le décès d'origine cardiovasculaire, le remplacement chirurgical de la valve aortique, l'insuffisance cardiaque congestive (ICC) consécutive à l'évolution de la sténose aortique, l'infarctus du myocarde non fatal, le pontage coronarien (CABG), l'intervention coronarienne percutanée (ICP), l'hospitalisation pour angor instable et l'AVC non hémorragique. Les critères secondaires principaux étaient des composites de sous-groupes du critère principal.
L'ézétimibe/simvastatine 10 mg/40 mg n'a pas réduit de façon significative le risque d'événements cardiovasculaires majeurs, en comparaison au placebo.
Les événements du critère principal ont été observés chez 333 patients (35,3 %) dans le groupe ézétimibe/simvastatine et chez 355 patients (38,2 %) dans le groupe placebo (risque relatif dans le groupe ézétimibe/simvastatine : 0,96 ; intervalle de confiance à 95 % : 0,83 à 1,12 ; p = 0,59). La valve aortique a été remplacée chez 267 patients (28,3 %) dans le groupe ézétimibe/simvastatine et chez 278 patients (29,9 %) dans le groupe placebo (risque relatif : 1,00 ; IC 95 % : 0,84 à 1,18 ; p = 0,97). Moins de patients ont eu des événements cardiovasculaires ischémiques dans le groupe ézétimibe/simvastatine (n = 148) que dans le groupe placebo (n = 187) (risque relatif : 0,78 ; IC 95 % : 0,63 à 0,97 ; p = 0,02), principalement en raison du nombre plus faible de patients ayant bénéficié d'un pontage coronarien.
L'apparition de cancer est plus fréquente dans le groupe ézétimibe/simvastatine (105 versus 70, p = 0,01). La signification clinique de cette observation est incertaine, d'autant que dans l'étude SHARP plus importante en termes de nombre de patients, le nombre de cancers (438 dans le groupe ézétimibe/simvastatine contre 439 pour le groupe placebo) n'était pas différent. De plus, dans l'étude IMPROVE-IT, le nombre total de patients présentant toute nouvelle apparition de tumeur maligne (853 dans le groupe ézétimibe/simvastatine versus 863 dans le groupe simvastatine) n’était pas significativement différent et, par conséquent, les résultats de l’étude SEAS n’ont pas été confirmés par ceux des études SHARP ou IMPROVE-IT.
5.2. Propriétés pharmacocinétiques
Après administration orale, l'ézétimibe est rapidement absorbé et subit une importante glycuroconjugaison conduisant à la formation d'un composé glycuronide phénolique pharmacologiquement actif (ézétimibe-glycuronide). Les concentrations plasmatiques maximales moyennes (Cmax) sont atteintes en 1 à 2 heures pour l'ézétimibe-glycuronide et en 4 à 12 heures pour l'ézétimibe. La biodisponibilité absolue de l'ézétimibe ne peut être déterminée car le produit est pratiquement insoluble dans les milieux aqueux adaptés aux injections.
L'administration concomitante d'aliments (repas riches en graisses ou repas sans graisses) n'a pas d'effet sur la biodisponibilité orale de l'ézétimibe administré sous forme de comprimés d'EZETIMIBE KRKA 10 mg. De ce fait, EZETIMIBE KRKA peut être administré pendant ou en dehors des repas.
Distribution
La liaison aux protéines plasmatiques humaines est de 99,7 % pour l'ézétimibe et de 88 à 92 % pour l'ézétimibe-glycuronide.
Biotransformation
L'ézétimibe est principalement métabolisé dans l'intestin grêle et le foie par glycuroconjugaison (réaction de phase II), suivie d'une excrétion biliaire. Un métabolisme oxydatif minimal (réaction de phase I) a été observé dans toutes les espèces évaluées.
L'ézétimibe et l'ézétimibe-glycuronide sont les principaux produits dérivés détectés dans le plasma, représentant respectivement environ 10 à 20 % et 80 à 90 % du produit total dans le plasma. L'ézétimibe et l'ézétimibe-glycuronide sont tous deux éliminés lentement du plasma ; il existe un recyclage entérohépatique significatif. La demi-vie de l'ézétimibe et de l'ézétimibe-glycuronide est d'environ 22 heures.
Élimination
Chez l'Homme, après administration orale de 14C-ézétimibe (20 mg), l'ézétimibe total représente environ 93 % de la radioactivité totale du plasma. Environ 78 % de la radioactivité est récupérée dans les selles et 11 % dans les urines sur une période de recueil de 10 jours. Après 48 heures, aucune radioactivité n'était détectable dans le plasma.
Populations particulières
Population pédiatrique
La pharmacocinétique de l’ézétimibe est similaire chez l'enfant (≥ 6 ans) et chez l'adulte. Aucune donnée pharmacocinétique chez l'enfant de moins de 6 ans n'est disponible. L'expérience clinique concernant les enfants et adolescents est limitée aux patients avec HFHo ou HFHe ou sitostérolémie.
Sujets âgés
Les concentrations plasmatiques d'ézétimibe total sont environ 2 fois plus élevées chez le sujet âgé (≥ 65 ans) que chez le sujet jeune (18 à 45 ans). La diminution du LDL-cholestérol et la sécurité d'emploi sont comparables chez les sujets âgés et les sujets jeunes traités par EZETIMIBE KRKA. Aucun ajustement posologique n'est nécessaire chez le sujet âgé.
Insuffisance hépatique
Après administration d'une dose unique de 10 mg d'ézétimibe, l'ASC moyenne de l'ézétimibe total augmente d'environ 1,7 fois chez les patients présentant une insuffisance hépatique légère (score de Child Pugh de 5 ou 6), par rapport aux sujets sains. Une étude de doses répétées (10 mg par jour) d'une durée de 14 jours réalisée chez des patients présentant une insuffisance hépatique modérée (score de Child Pugh de 7 à 9), montre que l'ASC moyenne de l'ézétimibe total augmente d'environ 4 fois au Jour 1 et au Jour 14, par rapport aux volontaires sains. Aucun ajustement posologique n'est nécessaire chez les patients présentant une insuffisance hépatique légère.
Chez les patients présentant une insuffisance hépatique modérée ou sévère (score de Child Pugh > 9), compte-tenu des effets inconnus d'une exposition accrue, EZETIMIBE KRKA n'est pas recommandé (voir rubrique 4.4).
Insuffisance rénale
Chez les patients présentant une insuffisance rénale sévère (n = 8, clairance de la créatinine ≤ 0.5 mL/s/1.73 m2 (30 mL/min/1,73 m2)), l'administration d'une dose unique de 10 mg d'ézétimibe entraîne une augmentation d'environ 1,5 fois de l'ASC moyenne de l'ézétimibe total par rapport aux volontaires sains (n = 9). Ce résultat n'étant pas considéré comme cliniquement significatif, aucun ajustement posologique n'est nécessaire chez ces patients.
Dans cette étude, chez un patient transplanté rénal recevant de nombreux médicaments dont la ciclosporine, l'exposition à l'ézétimibe total était 12 fois supérieure.
Sexe
Les concentrations plasmatiques de l'ézétimibe total sont légèrement plus élevées (approximativement 20 %) chez la femme que chez l'homme. La diminution de LDL-cholestérol et la sécurité d'emploi étant comparables chez l'homme et la femme traités par ézétimibe, aucun ajustement posologique n'est nécessaire en fonction du sexe.
5.3. Données de sécurité préclinique
Aucun organe cible n'a été identifié dans les études de toxicité chronique réalisées chez l'animal. Chez le chien traité pendant quatre semaines par ézétimibe (≥ 0,03 mg/kg/jour), la concentration de cholestérol dans la bile vésiculaire a été multipliée par un facteur de 2,5 à 3,5. Par contre, une étude d'un an réalisée chez le chien recevant des doses allant jusqu'à 300 mg/kg/jour n'a pas montré d'augmentation de l'incidence de lithiase biliaire ou d'autres effets hépatobiliaires. La signification de ces données chez l'homme n'est pas connue. Un risque lithogène chez des patients traités par l’ézétimibe ne peut être exclu.
En association avec une statine, les effets toxiques observés sont essentiellement ceux observés avec les statines. Certains effets sont cependant plus importants que ceux observés par un traitement par une statine seule ; ceci pouvant être attribué aux interactions pharmacocinétiques et pharmacodynamiques observées avec l'association. Aucune interaction de ce type n'a été observée dans les études cliniques. Chez le rat, des cas de myopathies ont été observés uniquement après exposition à des doses plusieurs fois supérieures à la dose thérapeutique chez l'homme (environ 20 fois le niveau de l'ASC des statines et 500 à 2 000 fois le niveau de l'ASC des métabolites actifs).
Une série de tests in vivo et in vitro n'a pas mis en évidence d'effet génotoxique quand l'ézétimibe est administré seul ou en association à une statine. Les tests de carcinogénicité à long terme de l'ézétimibe étaient négatifs.
L'ézétimibe n'a pas d'effet sur la fertilité du rat mâle ou femelle ni d'effet tératogène chez le rat et le lapin, que ce soit sur le développement pré-ou post-natal. L'ézétimibe franchit la barrière placentaire chez la rate et la lapine gravide recevant des doses multiples de 1 000 mg/kg/jour. L'administration concomitante d'ézétimibe et de statines n'a pas d'effet tératogène chez le rat. Chez la lapine gravide, quelques déformations squelettiques (fusion des vertèbres thoraciques et caudales, réduction du nombre des vertèbres caudales) ont été observées. L'administration concomitante d'ézétimibe et de lovastatine a entraîné des morts embryonnaires.
5 ans.
6.4. Précautions particulières de conservation
A conserver dans l’emballage d’origine à l’abri de l’humidité.
6.5. Nature et contenu de l'emballage extérieur
14, 28, 30, 50, 56, 60, 90, 98, 100 comprimés sous plaquettes (OPA/Aluminium/PVC//Aluminium).
14 x 1, 28 x 1, 30 x 1, 50 x 1, 56 x 1, 60 x 1, 90 x 1, 98 x 1, 100 x 1 comprimé sous plaquettes (OPA/Aluminium/PVC//Aluminium) prédécoupées unitaires.
Toutes les présentations peuvent ne pas être commercialisées.
6.6. Précautions particulières d’élimination et de manipulation
Pas d’exigences particulières.
7. TITULAIRE DE L’AUTORISATION DE MISE SUR LE MARCHE
MARJEKA CESTA 6
8501 NOVO MESTO
SLOVENIE
8. NUMERO(S) D’AUTORISATION DE MISE SUR LE MARCHE
· 34009 301 252 1 2 : 30 comprimés sous plaquettes (OPA/Aluminium/PVC/Aluminium)
· 34009 301 252 2 9 : 30x1 comprimés sous plaquettes prédécoupées unitaires (OPA/Aluminium/PVC/Aluminium)
· 34009 301 252 3 6 : 90 comprimés sous plaquettes (OPA/Aluminium/PVC/Aluminium)
· 34009 301 252 4 3 : 90x1 comprimés sous plaquettes prédécoupées unitaires (OPA/Aluminium/PVC/Aluminium)
9. DATE DE PREMIERE AUTORISATION/DE RENOUVELLEMENT DE L’AUTORISATION
[à compléter ultérieurement par le titulaire]
10. DATE DE MISE A JOUR DU TEXTE
[à compléter ultérieurement par le titulaire]
Sans objet.
12. INSTRUCTIONS POUR LA PREPARATION DES RADIOPHARMACEUTIQUES
Liste I.
ANSM - Mis à jour le : 17/01/2024
EZETIMIBE KRKA 10 mg, comprimé
Ezétimibe
Veuillez lire attentivement cette notice avant de prendre ce médicament car elle contient des informations importantes pour vous.
· Gardez cette notice. Vous pourriez avoir besoin de la relire.
· Si vous avez d’autres questions, interrogez votre médecin ou votre pharmacien.
· Ce médicament vous a été personnellement prescrit. Ne le donnez pas à d’autres personnes. Il pourrait leur être nocif, même si les signes de leur maladie sont identiques aux vôtres.
· Si vous ressentez un quelconque effet indésirable, parlez-en à votre médecin ou votre pharmacien. Ceci s’applique aussi à tout effet indésirable qui ne serait pas mentionné dans cette notice. Voir rubrique 4.
1. Qu'est-ce que EZETIMIBE KRKA 10 mg, comprimé et dans quels cas est-il utilisé ?
2. Quelles sont les informations à connaître avant de prendre EZETIMIBE KRKA 10 mg, comprimé ?
3. Comment prendre EZETIMIBE KRKA 10 mg, comprimé ?
4. Quels sont les effets indésirables éventuels ?
5. Comment conserver EZETIMIBE KRKA 10 mg, comprimé ?
6. Contenu de l’emballage et autres informations.
1. QU’EST-CE QUE EZETIMIBE KRKA 10 mg, comprimé ET DANS QUELS CAS EST-IL UTILISE ?
EZETIMIBE KRKA est un médicament utilisé pour diminuer les taux élevés de cholestérol.
EZETIMIBE KRKA diminue les taux de cholestérol total, le « mauvais » cholestérol (LDL-cholestérol) et les substances grasses appelées triglycérides dans le sang. De plus, EZETIMIBE KRKA augmente le taux de « bon » cholestérol (HDL-cholestérol).
L’ézétimibe, la substance active d’EZETIMIBE KRKA, agit en réduisant le cholestérol absorbé par votre tube digestif.
EZETIMIBE KRKA complète l'effet hypocholestérolémiant des statines, une famille de médicaments qui réduit le cholestérol fabriqué par votre organisme.
Le cholestérol est une des nombreuses substances grasses trouvées dans le sang. Votre cholestérol total est composé principalement de LDL-cholestérol et de HDL-cholestérol.
Le LDL-cholestérol est souvent appelé « mauvais » cholestérol parce qu’il peut s’accumuler sur les parois de vos artères sous forme de plaque. La constitution de cette plaque peut entrainer un rétrécissement des artères. Ce rétrécissement peut ralentir ou bloquer le flux sanguin vers les organes vitaux comme le cœur et le cerveau. Ce blocage du flux sanguin peut entraîner une crise cardiaque ou un accident vasculaire cérébral.
Le HDL-cholestérol est souvent appelé « bon » cholestérol, parce qu’il empêche le « mauvais » cholestérol de s’accumuler dans les artères et protège contre les maladies cardiaques.
Les triglycérides sont une autre forme de graisse présente dans votre sang qui peut augmenter votre risque de maladie cardiaque.
EZETIMIBE KRKA est utilisé chez les patients chez qui les taux de cholestérol ne peuvent être contrôlés uniquement par un régime alimentaire pauvre en graisse. Vous devez continuer votre régime pauvre en graisse tout en prenant ce médicament.
EZETIMIBE KRKA est utilisé en complément de votre régime pauvre en graisse, si vous avez :
· un taux élevé de cholestérol dans votre sang (hypercholestérolémie primaire [familiale hétérozygote et non-familiale]) :
o en association à une statine, si votre taux de cholestérol n'est pas bien contrôlé par une statine seule,
o seul, lorsqu'un traitement par une statine est inapproprié ou mal toléré.
· une maladie héréditaire (hypercholestérolémie familiale homozygote) qui augmente votre taux de cholestérol sanguin. Une statine sera également prescrite et vous pourrez également recevoir un autre traitement.
· une maladie héréditaire (sitostérolémie homozygote également connue sous le nom de phytostérolémie) qui augmente le taux des stérols végétaux sanguins.
Si vous avez une maladie cardiaque, EZETIMIBE KRKA, pris avec des médicaments faisant baisser le cholestérol appelés « statines », réduit le risque de crise cardiaque, d'accident vasculaire cérébral, d’intervention chirurgicale pour augmenter le flux sanguin cardiaque ou d'hospitalisation pour des douleurs thoraciques.
EZETIMIBE KRKA ne vous aide pas à perdre du poids.
2. QUELLES SONT LES INFORMATIONS A CONNAITRE AVANT DE PRENDRE EZETIMIBE KRKA 10 mg, comprimé ?
Ne prenez jamais EZETIMIBE KRKA 10 mg, comprimé
· si vous êtes allergique à la substance active ou à l’un des autres composants contenus dans ce médicament, mentionnés dans la rubrique 6.
Ne prenez jamais EZETIMIBE KRKA en association à une statine :
· Si vous présentez une affection hépatique,
· Si vous êtes enceinte ou allaitez.
Avertissements et précautions
Adressez-vous à votre médecin ou pharmacien avant de prendre EZETIMIBE KRKA 10 mg, comprimé.
· Informez votre médecin de tout problème de santé y compris les allergies.
· Votre médecin doit vous prescrire un examen sanguin avant de commencer le traitement par EZETIMIBE KRKA avec une statine. Ceci afin de vérifier le bon fonctionnement de votre foie.
· Votre médecin peut également prescrire un bilan hépatique en cours de traitement afin de vérifier le bon fonctionnement de votre foie lors de la prise d'EZETIMIBE KRKA avec une statine.
· Si vous présentez des problèmes hépatiques modérés ou sévères, EZETIMIBE KRKA 10 mg, comprimé n'est pas recommandé.
· L'efficacité et la sécurité d'emploi de l'association d’EZETIMIBE KRKA avec certains médicaments destinés à faire baisser le cholestérol, tels que les fibrates, n'ont pas été établies.
Enfants et adolescents
Ne donnez pas ce médicament à des enfants et adolescents (âgés de 6 à 17 ans), sauf s’il est prescrit par un spécialiste, car il existe peu de données sur sa sécurité d’emploi et son efficacité. Ne donnez pas ce médicament à des enfants de moins de 6 ans car il n'existe aucune information dans cette tranche d'âge.
Autres médicaments et EZETIMIBE KRKA 10 mg, comprimé
Informez votre médecin ou pharmacien si vous prenez, avez récemment pris ou pourriez prendre tout autre médicament. En particulier, il convient de signaler à votre médecin la prise de médicaments contenant l’une des substances actives suivantes :
· ciclosporine (souvent utilisée chez les patients transplantés)
· médicaments contenant une substance active qui évite la formation de caillots sanguins, tels que warfarine, phenprocoumone, acénocoumarol ou fluindione (anticoagulants)
· cholestyramine (utilisée également pour baisser le cholestérol), parce qu'elle modifie le mode d’action d'EZETIMIBE KRKA
· fibrates (utilisés également pour baisser le cholestérol).
EZETIMIBE KRKA 10 mg, comprimé avec des aliments et de l’alcool
Vous pouvez prendre EZETIMIBE KRKA pendant ou en dehors des repas.
Vous ne devez pas prendre EZETIMIBE KRKA avec une statine si vous êtes enceinte, si vous envisagez de l'être ou si vous pensez l'être. Si vous découvrez que vous êtes enceinte alors que vous êtes traitée par EZETIMIBE KRKA et une statine, arrêtez les deux médicaments immédiatement et informez votre médecin.
Il n'y a pas d'étude sur l'utilisation d'EZETIMIBE KRKA sans statine pendant la grossesse. Demandez l'avis de votre médecin avant de prendre EZETIMIBE KRKA si vous êtes enceinte.
Ne prenez pas EZETIMIBE KRKA et une statine si vous allaitez, car on ne sait pas si ce médicament passe dans le lait maternel.
Même sans statine, vous ne devez pas prendre EZETIMIBE KRKA si vous allaitez. Demandez conseil à votre médecin.
Demandez conseil à votre médecin avant de prendre tout médicament.
Conduite de véhicules et utilisation de machines
Aucun effet sur l'aptitude à conduire des véhicules ou à utiliser des machines n'est attendu avec EZETIMIBE KRKA. Il est à noter que des cas d'étourdissements ont cependant été observés chez les patients après la prise d’EZETIMIBE KRKA.
EZETIMIBE KRKA 10 mg, comprimé contient du sodium
Ce médicament contient moins de 1 mmol (23 mg) de sodium par comprimé, c’est-à-dire qu’il est essentiellement « sans sodium ».
3. COMMENT PRENDRE EZETIMIBE KRKA 10 mg, comprimé ?
Veillez à toujours prendre ce médicament en suivant exactement les indications de votre médecin. Continuez à prendre vos autres médicaments hypocholestérolémiants, sauf si votre médecin vous demande d'arrêter. Demander conseil auprès de votre médecin ou pharmacien si vous n'êtes pas sûr.
· Avant de débuter le traitement par EZETIMIBE KRKA, vous devez suivre un régime hypocholestérolémiant.
· Vous devez poursuivre ce régime pauvre en graisse pendant toute la durée du traitement par EZETIMIBE KRKA.
La dose recommandée est d’un comprimé d'EZETIMIBE KRKA à 10 mg, par voie orale, une fois par jour.
Prenez EZETIMIBE KRKA à n'importe quel moment de la journée, pendant ou en dehors des repas.
Si EZETIMIBE KRKA 10 mg, comprimé vous est prescrit en association à une statine, les deux médicaments peuvent être pris simultanément. Dans ce cas, lisez également le mode d'administration dans la notice du médicament utilisé.
Si EZETIMIBE KRKA 10 mg, comprimé vous est prescrit en association à un autre médicament utilisé pour baisser le cholestérol, contenant de la cholestyramine, ou tout autre médicament contenant un chélateur des acides biliaires, vous devez impérativement prendre EZETIMIBE KRKA au moins 2 heures avant ou 4 heures après avoir pris le chélateur des acides biliaires.
Si vous avez pris plus d’EZETIMIBE KRKA 10 mg, comprimé que vous n’auriez dû
Consultez immédiatement votre médecin ou votre pharmacien.
Si vous oubliez de prendre EZETIMIBE KRKA 10 mg, comprimé
Ne prenez pas de dose double pour compenser le comprimé que vous avez oublié de prendre, continuez à prendre la posologie usuelle le lendemain à l’heure habituelle.
Si vous arrêtez de prendre EZETIMIBE KRKA 10 mg, comprimé
Si vous arrêtez de prendre EZETIMIBE KRKA votre cholestérol pourrait augmenter à nouveau.
Si vous avez d’autres questions sur l’utilisation de ce médicament, demandez plus d’informations à votre médecin ou à votre pharmacien.
4. QUELS SONT LES EFFETS INDESIRABLES EVENTUELS ?
Contactez immédiatement votre médecin, si vous ressentez des douleurs musculaires inexpliquées, une sensibilité musculaire douloureuse ou une faiblesse musculaire. En effet, dans de rares cas, les problèmes musculaires dont les atteintes musculaires entraînant des lésions rénales peuvent être graves, et potentiellement menacer le pronostic vital.
Des réactions allergiques incluant gonflement du visage, des lèvres, de la langue, et/ou de la gorge qui peuvent provoquer des difficultés à respirer ou à avaler (qui demandent un traitement immédiat) ont été rapportées lors de l'utilisation habituelle.
Utilisé seul, les effets indésirables suivants ont été rapportés :
Fréquent (peut affecter jusqu’à 1 patient sur 10) : douleurs abdominales ; diarrhée ; flatulence ; sensation de fatigue.
Peu fréquent (peut affecter jusqu’à 1 patient sur 100) : augmentations de certains tests de laboratoire sanguins de la fonction hépatique (transaminases) ou musculaire (CPK) ; toux ; indigestion ; brûlures d'estomac ; nausée ; douleurs articulaires ; spasmes musculaires ; douleurs cervicales ; diminution d’appétit ; douleur ; douleur à la poitrine ; bouffées de chaleur ; hypertension.
De plus, en association avec une statine, les effets indésirables suivants ont été observés :
Fréquent (peut affecter jusqu’à 1 patient sur 10) : augmentations de certains tests sanguins de la fonction hépatique (transaminases) ; maux de tête ; douleurs musculaires ; sensibilité ou faiblesse.
Peu fréquent (peut affecter jusqu’à 1 patient sur 100) : sensation de fourmillements ; sécheresse buccale ; démangeaison ; rash ; urticaire ; douleur dorsale ; faiblesse musculaire ; douleurs des bras et des jambes ; fatigue ou faiblesse inhabituelles ; gonflement surtout des mains et des pieds.
En association avec le fénofibrate, l'effet indésirable fréquent suivant a été observé : douleurs abdominales.
De plus les effets indésirables suivants ont été rapportés depuis la mise sur le marché (fréquence inconnue) : étourdissements ; douleurs musculaires ; problèmes hépatiques ; réactions allergiques y compris rash et urticaire ; éruption de plaques rouges en relief, parfois en forme de cocarde (érythème multiforme) ; douleurs musculaires ; sensibilité ou faiblesse ; atteinte musculaire ; calculs biliaires ou inflammation de la vésicule biliaire (pouvant causer douleur abdominale, nausées, vomissements) ; inflammation du pancréas souvent avec douleur abdominale sévère ; constipation ; diminution des cellules sanguines, pouvant entrainer des bleus/saignements (thrombocytopénie) ; sensation de fourmillement ; dépression ; fatigue ou faiblesse inhabituelles ; essoufflement.
Déclaration des effets secondaires
Si vous ressentez un quelconque effet indésirable, parlez-en à votre médecin ou votre pharmacien. Ceci s’applique aussi à tout effet indésirable qui ne serait pas mentionné dans cette notice. Vous pouvez également déclarer les effets indésirables directement via le système national de déclaration : Agence nationale de sécurité du médicament et des produits de santé (ANSM) et réseau des Centres Régionaux de Pharmacovigilance - Site internet: https://signalement.social-sante.gouv.fr/
En signalant les effets indésirables, vous contribuez à fournir davantage d’informations sur la sécurité du médicament.
5. COMMENT CONSERVER EZETIMIBE KRKA 10 mg, comprimé ?
Tenir ce médicament hors de la vue et de la portée des enfants.
N’utilisez pas ce médicament après la date de péremption indiquée sur l’emballage et la plaquette après EXP. La date de péremption fait référence au dernier jour de ce mois.
A conserver dans l’emballage d’origine à l’abri de l’humidité.
Ne jetez aucun médicament au tout-à-l’égout ou avec les ordures ménagères. Demandez à votre pharmacien d’éliminer les médicaments que vous n’utilisez plus. Ces mesures contribueront à protéger l’environnement.
6. CONTENU DE L’EMBALLAGE ET AUTRES INFORMATIONS
Ce que contient EZETIMIBE KRKA 10 mg, comprimé
· La substance active est :
Ezétimibe............................................................................................................................... 10 mg
Pour un comprimé.
· Les autres composants sont :
Laurilsulfate de sodium, povidone K30, mannitol (E421), croscarmellose sodique (E468), cellulose microcristalline (E460), stéaryle (fumarate de) sodique. Voir rubrique 2 « EZETIMIBE KRKA 10 mg, comprimé contient du sodium ».
Qu’est-ce que EZETIMIBE KRKA 10 mg, comprimé et contenu de l’emballage extérieur
Les comprimés sont blancs à presque blancs, en forme de gélule, à bords biseautés. Dimensions du comprimé : 8 x 4 mm.
Boîte de 14, 28, 30, 50, 56, 60, 90, 98, 100 comprimés sous plaquette (OPA/Aluminium/PVC//Aluminium).
Boîte de 14 x 1, 28 x 1, 30 x 1, 50 x 1, 56 x 1, 60 x 1, 90 x 1, 98 x 1, 100 x 1 comprimé sous plaquettes (OPA/Aluminium/PVC//Aluminium) prédécoupées unitaires.
Toutes les présentations peuvent ne pas être commercialisées.
Titulaire de l’autorisation de mise sur le marché
MARJEKA CESTA 6
8501 NOVO MESTO
SLOVENIE
Exploitant de l’autorisation de mise sur le marché
12-14 RUE DE L’EGLISE
75015 PARIS
FRANCE
MARJEKA CESTA 6
8501 NOVO MESTO
SLOVENIE
OU
TAD PHARMA GMBH
HEINZ-LOHMANN STRASSE 5
27472 CUXHAVEN
ALLEMAGNE
Noms du médicament dans les Etats membres de l'Espace Economique Européen
Ce médicament est autorisé dans les Etats membres de l'Espace Economique Européen et au Royaume-Uni (Irlande du Nord) sous les noms suivants : Conformément à la réglementation en vigueur.
[à compléter ultérieurement par le titulaire]
La dernière date à laquelle cette notice a été révisée est :
[à compléter ultérieurement par le titulaire]
Des informations détaillées sur ce médicament sont disponibles sur le site Internet de l’ANSM (France).