Dernière mise à jour le 01/06/2026
LEPTOPROL 5 mg, implant en seringue pré-remplie
Indications thérapeutiques
La substance active de LEPTOPROL (acétate de leuproréline) appartient au groupe des inhibiteurs de certaines hormones sexuelles.
LEPTOPROL agit sur l’hypophyse en la stimulant brièvement puis en réduisant la production des hormones qui contrôlent la sécrétion des hormones sexuelles dans les testicules.
Cela signifie que les concentrations des hormones sexuelles sont ensuite fortement réduites et, lorsque l’administration est poursuivie, elles restent à ce niveau. Après l’arrêt de LEPTOPROL, les concentrations des hormones hypophysaires et sexuelles reviennent à des valeurs normales.
LEPTOPROL est indiqué dans le traitement symptomatique des tumeurs hormonosensibles avancées de la prostate (cancer de la prostate).
LEPTOPROL est aussi utilisé dans le traitement du cancer de la prostate localement avancé et localisé hormonosensible, en association avec ou après la radiothérapie externe.
Présentations
> 1 seringue(s) préremplie(s) polycarbonate de 1 implant(s) avec aiguille(s)
Code CIP : 34009 300 200 0 5
Déclaration de commercialisation : 14/03/2018
Cette présentation est agréée aux collectivités
- Prix hors honoraire de dispensation : 198,65 €
- Honoraire de dispensation : 1,02 €
- Prix honoraire compris : 199,67 €
- Taux de remboursement :100%
- Cancer de la prostate (en traitement palliatif) ; JOURNAL OFFICIEL ; 13/03/18
Service médical rendu (SMR)
Les libellés affichés ci-dessous ne sont que des résumés ou extraits issus des avis rendus par la Commission de la Transparence. Seul l'avis complet de la Commission de la Transparence fait référence.
Cet avis est consultable à partir du lien `Avis du jj/mm/aaaa` ou encore sur demande auprès de la HAS (plus d'informations dans l'aide). Les avis et synthèses d'avis contiennent un paragraphe sur la place du médicament dans la stratégie thérapeutique.
| Valeur du SMR | Avis | Motif de l'évaluation | Résumé de l'avis |
|---|---|---|---|
| Important | Avis du 05/02/2020 | Extension d'indication | Le service médical rendu par LEPTOPROL est important dans l’indication de l’AMM. |
| Important | Avis du 17/05/2017 | Inscription (CT) | Le service médical rendu par LEPTOPROL est important dans l’indication de l’AMM. |
Amélioration du service médical rendu (ASMR)
Les libellés affichés ci-dessous ne sont que des résumés ou extraits issus des avis rendus par la Commission de la Transparence. Seul l'avis complet de la Commission de la Transparence fait référence.
Cet avis est consultable à partir du lien `Avis du jj/mm/aaaa` ou encore sur demande auprés de la HAS (plus d'informations dans l'aide). Les avis et synthéses d'avis contiennent un paragraphe sur la place du médicament dans la stratégie thérapeutique.
| Valeur de l'ASMR | Avis | Motif de l'évaluation | Résumé de l'avis |
|---|---|---|---|
| V (Inexistant) | Avis du 05/02/2020 | Extension d'indication | LEPTOPROL n’apporte pas d’amélioration du service médical rendu (ASMR V) dans la prise charge du cancer de la prostate localement avancé hormonosensible, en association pendant ou après la radiothérapie externe et du cancer de la prostate localisé hormonosensible chez les patients à risque intermédiaire ou à haut risque, en association à la radiothérapie externe. |
| V (Inexistant) | Avis du 17/05/2017 | Inscription (CT) | Cette spécialité est un médicament hybride qui n’apporte pas d’amélioration du service médical rendu (ASMR V) par rapport à ENANTONE LP 11,25 mg, microsphères et solution pour usage parentéral (SC ou IM) à libération prolongée. |
ANSM - Mis à jour le : 09/04/2026
LEPTOPROL 5 mg, implant en seringue pré-remplie
2. COMPOSITION QUALITATIVE ET QUANTITATIVE
Chaque implant contient 5 mg de leuproréline (sous forme d’acétate de leuproréline).
Pour la liste complète des excipients, voir rubrique 6.1.
Implant en seringue pré-remplie.
Bâtonnet biodégradable blanc-jaunâtre de forme cylindrique (longueur 10 mm) dans une seringue pré-remplie.
4.1. Indications thérapeutiques
· Traitement à visée palliative chez les patients atteints de cancer de la prostate avancé hormonosensible.
· Traitement du cancer de la prostate localement avancé hormonosensible, en association pendant ou après la radiothérapie externe.
· Traitement du cancer de la prostate localisé hormonosensible chez les patients à risque intermédiaire ou à haut risque, en association à la radiothérapie externe.
4.2. Posologie et mode d'administration
Posologie
L’indication du traitement doit être établie et le suivi du traitement à long terme doit être réalisé par des médecins expérimentés dans le traitement des cancers.
La dose recommandée est une dose unique de 5 mg de Leptoprol une fois tous les 3 mois. Si, dans des cas exceptionnels, l’administration de l’implant devait être reportée dans la limite de 4 semaines, l’effet thérapeutique sera maintenu chez la majorité des patients (voir rubrique 5.2.).
Populations particulières
Aucun ajustement posologique n’est nécessaire pour les patients atteints d’insuffisance rénale ou hépatique ou chez les personnes âgées.
Population pédiatrique
Leptoprol est contre-indiqué chez les enfants et les adolescents (voir rubrique 4.3).
LEPTOPROL peut être utilisé comme traitement néoadjuvant et adjuvant à la radiothérapie externe dans le cancer de la prostate localement avancé hormonosensible ainsi que dans le cancer de la prostate localisé hormonosensible chez les patients à risque intermédiaire ou à haut risque.
Mode d’administration
LEPTOPROL doit être préparé et administré uniquement par un professionnel de santé ayant pris connaissance des instructions relatives aux étapes d’administration du produit.
Un implant est injecté par voie sous-cutanée dans la paroi abdominale antérieure. Avant l’injection, un anesthésique local peut être administré.
Il est recommandé d’initier une thérapie anti-androgénique adjuvante environ 5 jours avant de débuter LEPTOPROL (voir rubrique 4.4.).
Instructions d’utilisation
Veuillez lire attentivement ces instructions, l’applicateur fourni avec ce médicament pouvant être différent de ceux que vous avez déjà utilisés.
|
1. Désinfecter le point d’injection sur la paroi abdominale antérieure en-dessous du nombril.
|
|
||
|
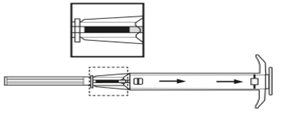
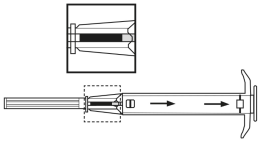
2. Retirer l’applicateur de son sachet stérile et vérifier que l’implant est visible dans le réservoir (voir encadré). Pour vérifier, visualiser l’applicateur contre une lumière ou le secouer légèrement.
|
|
||
|
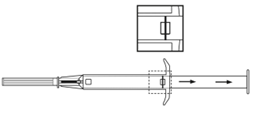
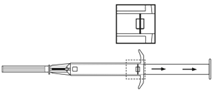
3. Tirer le piston de l’applicateur complètement vers l’arrière jusqu’à ce que vous puissiez voir une ligne complète dans la 2ème fenêtre. Remarque : Le piston ne peut être poussé vers l’avant pour injecter l’implant que s’il a déjà été préalablement complètement tiré vers l’arrière ! |
|
||
|
4. Retirer le capuchon protecteur de l’aiguille. |
|||
|
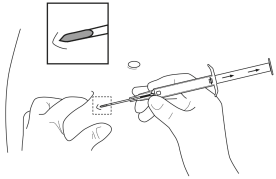
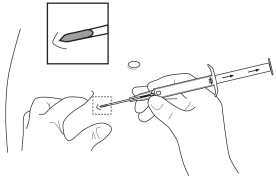
5. Tenir le corps principal de l’applicateur dans une main. Avec l’autre main, pincer la peau de la paroi abdominale antérieure du patient, en-dessous du nombril (voir l’illustration). Avec l’ouverture de l’aiguille dirigée vers le haut, insérer l’ensemble de l’aiguille, en formant un angle léger, presque parallèle à la peau dans le tissu sous-cutané. |
|
||
|
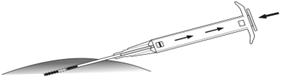
6. Tirer doucement l’applicateur sur environ 1 cm vers l’arrière (Cela forme le canal de ponction de l’implant). |
|||
|
7. Injecter l’implant dans le canal de ponction en poussant complètement le piston vers l’avant jusqu’à ce qu’il se mette en place et que vous entendiez un cliquetis. |
|
||
|
8. Retirer l’aiguille. Pour s’assurer que l’implant a été injecté correctement, vérifier que la pointe bleu clair du piston est visible à la pointe de l’aiguille. |
|

Les taux sériques de PSA et de testostérone totale doivent être déterminés au début et après 3 mois d’utilisation de LEPTOPROL. Le cancer de la prostate est sensible aux androgènes lorsque les concentrations de testostérone sont au niveau des valeurs obtenues par castration (≤ 0,5 ng/mL) après 3 mois et que le taux de PSA a diminué. Une baisse marquée précoce du taux de PSA (environ 80 % du taux de référence) peut être considérée comme un bon indicateur pronostique de la réponse à long terme à la privation androgénique. Un traitement hormono-ablatif (par exemple LEPTOPROL) est alors indiqué.
Lorsque les taux de PSA restent inchangés ou ont augmenté chez les patients dépourvus de testostérone, le cancer de la prostate est insensible aux androgènes. Dans de tels cas, la poursuite du traitement hormono-ablatif n’est pas adaptée.
Cependant, si le patient a montré une réponse clinique (par exemple une amélioration de la douleur et des symptômes de la dysurie, une réduction de la taille de la prostate), le résultat doit être considéré comme un faux négatif. Dans ces rares cas, l’administration de LEPTOPROL doit être poursuivie pendant encore 3 mois et le taux de PSA doit être revérifié. De plus, les symptômes cliniques du patient devront être surveillés de très près.
Le traitement du cancer avancé de la prostate hormono-dépendant par LEPTOPROL est généralement un traitement à long terme.
Au cours d’essais cliniques, il a été montré que, pour le traitement du cancer de la prostate localement avancé hormonosensible, une durée de traitement par privation androgénique de 3 ans chez les patients recevant une radiothérapie devait être privilégiée par rapport à une durée de 6 mois (voir rubrique 5.1). Les recommandations cliniques préconisent une durée de 2 à 3 ans de traitement par privation androgénique chez les patients (T3-T4) recevant une radiothérapie.
Dans le cancer de la prostate localisé hormonosensible chez les patients à risque intermédiaire, un traitement par privation androgénique avec des agonistes de la LHRH est recommandé pendant 4 à 6 mois en association à la radiothérapie, tandis que chez les patients à haut risque, la durée de traitement recommandée en association à la radiothérapie est de 2 à 3 ans.
Hypersensibilité à la substance active ou à l’un des excipients mentionnés à la rubrique 6.1, ou à d’autres analogues de la LHRH.
Indépendance hormonale confirmée du cancer.
LEPTOPROL est contre-indiqué chez les femmes et chez les enfants.
4.4. Mises en garde spéciales et précautions d'emploi
Les patients hypertendus doivent être surveillés de près.
Il existe un risque accru de cas de dépression (qui peut être sévère) chez les patients traités par des agonistes de la LHRH comme LEPTOPROL. Les patients doivent être informés de ce risque et traités comme il convient si des symptômes apparaissent.
Des réactions allergiques et anaphylactiques ont été observées. Elles comprennent des réactions locales au point d’injection et des symptômes systémiques.
Des cas post-commercialisation de convulsions ont été observés chez les patients traités par l’acétate de leuproréline avec ou sans antécédents d’épilepsie, de convulsions ou de facteurs de prédisposition.
Après une castration chirurgicale, LEPTOPROL n’entraîne pas de réduction supplémentaire de la concentration de testostérone.
En raison de l’augmentation à court terme de la concentration sérique de testostérone au début du traitement, qui peut temporairement intensifier certains symptômes de la maladie, les patients présentant un risque de complications neurologiques, de métastase spinale et d’obstruction de l’appareil urinaire doivent être surveillés en permanence au cours des premières semaines de traitement, en hospitalisation dans la mesure du possible.
L’administration supplémentaire d’un anti-androgène approprié doit être envisagée pour la phase initiale du traitement, afin d’atténuer les séquelles possibles du pic initial de testostérone et l’aggravation des symptômes cliniques.
L’efficacité du traitement doit être contrôlée régulièrement (mais en particulier s’il existe des signes de progression malgré un traitement approprié) au moyen d’examens cliniques (toucher rectal de la prostate, échographie, scintigraphie osseuse, tomodensitométrie) et en contrôlant les taux des phosphatases et/ou de l’antigène prostatique spécifique (PSA) et la concentration sérique de testostérone.
Un hypogonadisme se produisant suite à un traitement à long terme par des analogues de la LHRH et/ou une orchidectomie peuvent entraîner une ostéoporose avec un risque accru de fracture, le développement de l’ostéoporose étant plus marqué suite à une orchidectomie, avec une augmentation des taux de cortisol, que suite à l’administration des analogues de la LHRH. Chez les patients présentant un risque élevé, l’administration supplémentaire d’un bisphosphonate peut prévenir la déminéralisation osseuse.
Une thérapie de privation androgénique peut prolonger l’intervalle QT.
Avant l’initiation de traitement par LEPTOPROL chez les patients présentant des antécédents ou des facteurs de risque de prolongation de l’intervalle QT et chez les patients recevant concomitamment des médicaments pouvant prolonger l’intervalle QT (voir rubrique 4.5), le médecin devra évaluer le rapport bénéfice/ risque incluant la possibilité de torsade de pointes.
Modifications métaboliques
Une hyperglycémie et un risque accru d’apparition d’un diabète ont été rapportés chez des hommes recevant des agonistes de la GnRH. L’hyperglycémie est susceptible d’être liée à l’apparition d’un diabète sucré ou à la dégradation du contrôle glycémique chez les patients diabétiques. La glycémie et/ou le taux d’hémoglobine glycosylée (HbA1c) doivent être surveillés périodiquement chez les patients recevant un agoniste de la GnRH, et ces derniers doivent être pris en charge en suivant les pratiques en vigueur en ce qui concerne le traitement de l’hyperglycémie ou du diabète. Les modifications métaboliques liées aux agonistes de la GnRH peuvent également inclure la stéatose hépatique.
Hypertension intracrânienne idiopathique
Des cas d’hypertension intracrânienne idiopathique (méningite séreuse) ont été rapportés chez des patients recevant de la leuproréline. Les patients doivent être avertis de la possibilité de signes et symptômes d’hypertension intracrânienne idiopathique, notamment des céphalées sévères ou récurrentes, de troubles visuels et d’acouphènes. En présence d’une hypertension intracrânienne idiopathique, l’interruption du traitement par leuproréline doit être envisagée.
Réactions indésirables cutanées sévères
Des réactions indésirables cutanées sévères (SCAR), dont le syndrome de Stevens-Johnson (SSJ), et la nécrolyse épidermique toxique (NET, ou syndrome de Lyell), qui peuvent engager le pronostic vital ou être fatals, ont été rapportés en association avec le traitement par leuproréline. Au moment de la prescription, les patients doivent être informés des signes et symptômes, et surveillés étroitement en cas de réactions cutanées graves. En cas d’apparition de signes et symptômes évocateurs de ces réactions, le traitement par leuproréline doit être arrêté immédiatement et un autre traitement doit être envisagé (le cas échéant).
4.5. Interactions avec d'autres médicaments et autres formes d'interactions
Aucune étude d’interaction médicamenteuse n’a été conduite.
Une thérapie de privation androgénique pouvant prolonger l’intervalle QT, l’utilisation concomitante de LEPTOPROL et de médicaments connus pour prolonger l’intervalle QT ou de médicaments susceptibles d’entrainer des torsades de pointes, tels que les antiarythmiques de classe IA (ex. quinidine, disopyramide) ou de classe III (ex amiodarone, sotalol, dofétilide, ibutilide), la méthadone, la moxifloxacine, les antipsychotiques etc, doit être évaluée avec attention (voir rubrique 4.4).
4.6. Fertilité, grossesse et allaitement
LEPTOPROL est destiné uniquement à une utilisation chez les patients de sexe masculin.
4.7. Effets sur l'aptitude à conduire des véhicules et à utiliser des machines
LEPTOPROL peut altérer la réactivité, même lorsqu’il est utilisé conformément à l’usage auquel il est destiné, ce qui peut avoir une influence importante sur l’aptitude à conduire des véhicules et à utiliser des machines. Cela est dû à la fatigue survenant chez quelques patients, en particulier en début de traitement, qui peut également être causée par la tumeur sous-jacente.
Ce phénomène est accentué lorsque que LEPTOPROL est associé à une consommation d’alcool.
Au début du traitement, il est habituellement observé une augmentation de courte durée de la concentration sérique de testostérone, qui peut temporairement aggraver certains symptômes de la maladie (douleur osseuse ou augmentation de la douleur osseuse, obstruction des voies urinaires et ses conséquences, compression de la moelle épinière, faiblesse musculaire dans les jambes, œdème lymphatique). Cette augmentation des symptômes régresse normalement spontanément sans avoir à interrompre LEPTOPROL.
Des effets indésirables peuvent survenir en raison de la privation des hormones sexuelles.
Tableau récapitulatif des effets indésirables :
Les effets secondaires sont énumérés et classés par système organique et d’après la convention de fréquence MedDRA :
Très fréquents : ³ 1/10.
Fréquents : ³ 1/100, < 1/10.
Peu fréquents : ³ 1/1 000, < 1/100.
Rares : ³ 1/10 000, < 1/1 000.
Très rares : < 1/10 000.
Fréquence indéterminée : la fréquence ne peut pas être estimée à partir des données disponibles.
|
Très fréquents |
Fréquents |
Peu fréquents |
Rares |
Très rares |
Indéterminée |
|
|
Affections du système immunitaire |
Réactions allergiques générales (fièvre, démangeaisons, éosinophilie, éruption cutanée) |
Réactions anaphy-lactiques |
||||
|
Troubles du métabolisme et de la nutrition |
Diminution de l’appétit, augmentation de l’appétit |
Changements du statut métabolique des diabétiques (augmentation ou diminution de la glycémie) |
||||
|
Affections psychiatriques |
Sautes d’humeur, dépression, troubles du sommeil |
|||||
|
Affections du système nerveux |
Maux de tête, paresthésies |
Vertige, dysgueusie transitoire |
Comme pour les autres médicaments de cette classe thérapeutique, de très rares cas d’apoplexie hypophysaire ont été rapportés suite à l’administration initiale de leuproréline chez les patients présentant un adénome hypophysaire. |
Convulsions, hypertension intracrânienne idiopathique (méningite séreuse) (voir rubrique 4.4) |
||
|
Affections cardiaques |
|
|
|
|
|
Prolongation de l’intervalle QT (voir rubriques 4.4 et 4.5) |
|
Affections vasculaires |
Bouffées de chaleur |
Variation de la pression artérielle (hypertension ou hypotension), thrombose |
||||
|
Affections respiratoires, thoraciques et médiastinales |
Embolie pulmonaire |
Pneumopathie interstitielle |
||||
|
Affections gastro-intestinales |
Nausées/ vomissements |
Diarrhée |
||||
|
Affections de la peau et du tissu sous-cutané |
Peau ou muqueuses sèches, transpiration nocturne |
Alopécie |
Syndrome de Stevens-Johnson (SSJ) /nécrolyse épidermique toxique (NET ou syndrome de Lyell) (voir rubrique 4.4), éruption cutanée toxique, érythème polymorphe |
|||
|
Affections musculo-squelettiques et systémiques |
Douleurs osseuses |
Douleurs articulaires et/ou au dos, myasthénie |
Déminéralisation osseuse (voir section 4.4) |
|||
|
Affections du rein et des voies urinaires |
Nycturie, dysurie, pollakiurie |
Rétention urinaire |
||||
|
Affections des organes de reproduction et du sein |
Réduction ou perte de la libido et de la puissance sexuelle, réduction de la taille des testicules |
Gynécomastie |
Douleurs testiculaires |
|||
|
Troubles généraux et anomalies au site d’administration |
Augmentation de la transpiration ; réactions au point d’injection, par exemple érythème, douleur, œdème, prurit qui disparaissent généralement même lorsque le traitement est poursuivi |
Fatigue, œdème périphérique |
||||
|
Investigations |
Prise de poids |
Perte de poids, augmentations de la LDH, des transaminases (ALAT, ASAT), des gamma-GT et de la phosphatase alcaline, qui peuvent également être une manifestation de la maladie sous-jacente |
Après la commercialisation, des cas de pneumonie interstitielle ont été rapportés, principalement au Japon.
Remarques particulières
La réponse au traitement par LEPTOPROL peut être suivie en mesurant les concentrations sériques de testostérone, de phosphatase acide et de PSA (antigène prostatique spécifique). Les taux de testostérone augmentent initialement lorsque le traitement est débuté mais ils diminuent en 2 semaines. Après 2 à 4 semaines, les concentrations de testostérone atteintes sont comparables à celles observées après une orchidectomie bilatérale, puis ils restent constants tout au long du traitement.
Une augmentation transitoire des taux de phosphatase acide peut se produire dans la phase initiale du traitement. Des taux normaux ou des taux proches des valeurs normales sont habituellement atteints après quelques semaines.
De rares cas d’abcès survenu au site d’injection ont été rapportés. Dans un seul cas d’abcès, une absorption insuffisante de la leuproréline à partir de la forme implant été observée. Les taux de testostérone doivent alors être contrôlés dans de telles situations.
Déclaration des effets indésirables suspectés
La déclaration des effets indésirables suspectés après autorisation du médicament est importante. Elle permet une surveillance continue du rapport bénéfice/risque du médicament. Les professionnels de santé déclarent tout effet indésirable suspecté via le système national de déclaration : Agence nationale de sécurité du médicament et des produits de santé (ANSM) et réseau des Centres Régionaux de Pharmacovigilance – Site internet : https://signalement.social-sante.gouv.fr/.
A ce jour, aucun symptôme d’intoxication n’a été observé.
Même lorsque des doses allant jusqu’à 20 mg d’acétate de leuproréline par jour ont été administrées pendant 2 ans, comme c’était le cas dans les premières études cliniques, aucun autre ou nouvel effet indésirable n’a été observé en dehors de ceux survenant après l’administration quotidienne de 1 mg ou de l’administration de 11,25 mg d’acétate de leuproréline tous les 3 mois.
5. PROPRIETES PHARMACOLOGIQUES
5.1. Propriétés pharmacodynamiques
Classe pharmacothérapeutique : Hormones et agents apparentés, analogues de l’hormone de libération de la gonadotropine, code ATC : L02AE02.
L’acétate de leuproréline, la substance active du LEPTOPROL, est un analogue synthétique de la LHRH, une hormone hypothalamique qui est produite naturellement et qui contrôle la libération des hormones gonadotropes, la LH (hormone lutéinisante) et la FSH (hormone folliculo-stimulante) par le lobe antérieur de l’hypophyse. Ces hormones stimulent à leur tour la synthèse des stéroïdes gonadiques.
Contrairement à la LHRH physiologique qui est libérée de manière pulsatile à partir de l’hypothalamus, l’acétate de leuproréline – aussi appelé l’agoniste de la LHRH - bloque les récepteurs de la LHRH au niveau de l’hypophyse de façon continue au cours du traitement à long terme, et après la stimulation initiale à court terme, il les régule négativement. Par conséquent, il y a une suppression hypophysaire réversible de la libération des gonadotropines associée à une diminution des concentrations de testostérone.
La concentration de testostérone est réduite, ce qui influence la croissance du tissu prostatique cancéreux, qui est normalement stimulée par la dihydrotestostérone, produite suite à la réduction de la testostérone dans les cellules de la prostate.
L’administration continue d’acétate de leuproréline entraîne une diminution du nombre et/ou de la sensibilité (appelée régulation négative) des récepteurs au niveau de l’hypophyse et par conséquent, une diminution des concentrations de LH, de FSH et de DHT. Au cours du processus, le taux de testostérone est réduit au niveau des valeurs obtenues par castration.
Un effet anti-androgénique et une inhibition de la croissance des carcinomes de la prostate ont également été démontrés dans des modèles animaux.
Selon les études précliniques et cliniques, le traitement mensuel par l’acétate de leuproréline inhibe la libération de la gonadotropine après la stimulation initiale.
Chez l’homme, l’administration sous-cutanée d’acétate de leuproréline provoque une augmentation initiale des taux de LH (hormone lutéinisante) et de FSH (hormone folliculo-stimulante), caractérisée par une augmentation transitoire des concentrations de testostérone et de dihydrotestostérone.
Etant donné qu’une aggravation symptomatique de la maladie de courte durée a été observée au cours des 3 premières semaines dans des cas isolés, l’administration adjuvante d’anti-androgènes doit être envisagée chez les patients atteints de cancer de la prostate.
En revanche, le traitement à long terme par l’acétate de leuproréline entraîne une diminution des concentrations de LH et de FSH chez tous les patients ; les concentrations d’androgènes chez les hommes atteignent des valeurs similaires à celles obtenues suite à une orchidectomie bilatérale. Ces variations apparaissent généralement 2 à 3 semaines après le début du traitement et sont maintenues pendant toute la durée du traitement. C’est pourquoi la sensibilité hormonale des carcinomes de la prostate et le bénéfice thérapeutique possible de l’orchidectomie peuvent également être étudiés avec l’acétate de leuproréline. Si nécessaire, l’orchidectomie peut être remplacée par l’administration mensuelle d’acétate de leuproréline.
Jusqu’à présent, il a été possible de maintenir des taux de testostérone similaires à ceux obtenus suite à une castration après une administration continue d’acétate de leuproréline sur 5 ans.
Efficacité clinique
Dans une étude de phase III multicentrique et randomisée portant sur l'acétate de leuproréline, 263 patients atteints d'un cancer de la prostate localement avancé aux stades T3-T4 ou pT3, N0, M0 ont été évalués.
133 patients ont reçu un traitement par privation androgénique en association à la radiothérapie et 130 patients ont reçu un traitement par privation androgénique avec de l'acétate de leuproréline seul, pendant 3 ans.
Selon les critères ASTRO (et Phoenix), la survie sans progression à 5 ans était de 60,9 % (64,7 %) dans le groupe traité par l’association, versus 8,5 % (15,4 %) dans le groupe recevant l'hormonothérapie seule (p = 0, 0001 ; [p = 0,0005]).
Selon les critères ASTRO, le risque de progression était 3,8 fois plus élevé dans le groupe recevant un traitement hormonal seul (IC à 95% [2,17 ; 6,49]).
La survie médiane sans progression clinique ou biochimique selon les critères ASTRO était de 641 jours (IC à 95% [626 ; 812]) dans le groupe recevant un traitement hormonal seul par rapport à 2 ,804 jours (IC à 95% [2,090 ; - ] ; p < 0, 0001) dans le groupe traité par l’association.
Aucune différence statistiquement significative n’a été observée concernant la progression locorégionale (HR 3,6 [IC à 95% {1,9 ; 6,8} ; p < 0,0001]), la progression métastatique (p < 0,018) et la survie sans métastases (p = 0,018) dans le groupe de traité par l’association versus le groupe recevant un traitement par privation androgénique seul.
Dans cet essai clinique, l'association d'un traitement par privation androgénique associé à une radiothérapie pendant 3 ans pourrait clairement démontrer une supériorité par rapport au traitement par privation androgénique seul.
Les essais cliniques suivants montrent une supériorité du traitement en association avec des analogues de la LHRH par rapport à la radiothérapie seule chez des patients atteints d'un cancer de la prostate localement avancé.
Dans l’essai clinique randomisé RTOG 85-31, 977 patients atteints d’un cancer de la prostate localement avancé de stade T1-T3, avec métastases des ganglions lymphatiques, extension extra prostatiques ou infiltration des cellules cancéreuses dans les vésicules séminales ont été inclus.
488 patients ont reçu une association d’une radiothérapie avec un traitement à long terme de privation androgénique avec de la goséréline, tandis que 489 patients ont reçu une radiothérapie seule.
Les résultats montrent clairement une supériorité du traitement en association par rapport à la radiothérapie seule.
La survie sans progression à 10 ans était de 37 % versus 23 % (p < 0,001), la survie sans progression avec une valeur de PSA < 1,5 ng/mL était de 31 % versus 9 %, la récidive locale était de 23 % versus 38 % (p < 0,0001) et la progression avec des métastases de 24 % versus 39 % (p < 0,0001).
La survie globale était de 49 % versus 39 % (p = 0,002) et la mortalité spécifique due à la maladie était de 16 % versus 22 % (p = 0,0052).
La supériorité de l’association de la radiothérapie et du traitement par privation androgénique avec des analogues de la LHRH par rapport à la radiothérapie seule chez des patients atteints d'un cancer de la prostate localisé à risque profil de risque intermédiaire été démontrée lors de l'essai clinique suivant.
L’essai clinique de phase III randomisé RTOG 94-08 a été conduit chez des patients atteints d’un cancer localisé de la prostate au stade T1b, T1c, T2a ou T2b et dont le PSA était inférieur ou égal à 10 ng/mL.
Le sous-groupe de patients présentant un profil de risque intermédiaire défini par un score de Gleason de 6 associé à un PSA compris entre > 10 ng/mL et 20 ng/mL ou au stade clinique T2b incluait 524 patients dans le groupe traité par privation androgénique de courte durée sur 4 mois (2 mois avant et 2 mois en association avec la radiothérapie) et 544 patients dans le groupe traité par radiothérapie seule.
Dans ce sous-groupe de patients à profil de risque intermédiaire, le groupe ayant reçu en association la radiothérapie et le traitement par privation androgénique avec de l'acétate de goséréline ou de l'acétate de leuproréline présentait un bénéfice supérieur au groupe recevant une radiothérapie seule.
La survie globale à 10 ans était de 61 % versus 54 % (hazard ratio : 1,23, IC à 95 % [1,02 - 1,49 ; p = 0,03]). La mortalité spécifique due à la maladie était de 3 % versus 10 % (hazard ratio de 2,49, IC à 95 % [1,50 - 4,11 ; p = 0,004]) et la progression biochimique de 28 % versus 45 % (hazard ratio de 1,79, IC à 95 % [1,45 - 2,21; p < 0,001]).
L'utilisation chez les patients atteints d'un cancer de la prostate localisé à haut risque repose sur des essais cliniques publiés portant sur la radiothérapie en association avec des agonistes de la LHRH, incluant l'acétate de leuproréline.
Les données cliniques publiées dans le cadre de cinq essais cliniques montrent clairement l’intérêt de l’association de la radiothérapie avec des agonistes de la LHRH (EORTC 22863, RTOG 85-31, RTOG 92-02, RTOG 8610 et D’Amico et al., JAMA 2004). Une différenciation claire de la population étudiée entre les indications cancer de la prostate localement avancé et cancer de la prostate localisé à haut risque n’était pas possible.
Des données cliniques ont montré que la radiothérapie suivie d'un traitement par privation androgénique de 3 ans devrait être privilégiée à la radiothérapie suivie d'un traitement par privation androgénique de 6 mois.
Une durée de traitement par privation androgénique de 2 à 3 ans chez les patients du stade T3 au stade T4 est préconisée par les recommandations cliniques.
Chez les patients atteints d'un cancer de la prostate métastatique résistant à la castration, le bénéfice de l’ajout aux agonistes de la LHRH comme l'acétate de leuproréline, des médicaments tels que des inhibiteurs de la synthèse d'androgènes (par exemple, l’acétate d’abiratérone), des antiandrogènes (par exemple l'enzalutamide), des taxanes (par exemple le docétaxel ou le paclitaxel) ou des agents de radiothérapie (par exemple le radium 223) a été démontré.
5.2. Propriétés pharmacocinétiques
La substance active, acétate de leuproréline, est libérée de façon continue à partir du polymère de l’acide polylactique sur une période allant jusqu’à 182 jours (26 semaines) suite à l’injection de l’implant biodégradable de LEPTOPROL. Le polymère est absorbé de la même façon que le matériau de suture chirurgicale.
Dans les 2 heures suivant l’administration sous-cutanée d’une dose unique de LEPTOPROL, des pics sériques de leuproréline de 5216 pg/mL (5,2 ng/mL) ont été mesurés.
Pendant un traitement par LEPTOPROL de 3 mois, l’ASC était de 32,4 ng/mL*j.
Des taux détectables sont présents dans le sérum jusqu’à 182 jours (26 semaines) après l’administration.
Le volume de distribution de la leuproréline est de 36 L chez les hommes ; la clairance totale est de 139,6 mL/min.
Chez les patients dont la fonction rénale ou hépatique est altérée, les taux de leuproréline se situaient dans la fourchette des valeurs observées chez les patients ayant une fonction rénale ou hépatique normale. Chez certains patients atteints d’insuffisance rénale chronique, des taux sériques de leuproréline plus élevés étaient mesurés. Cependant, cette observation ne semble pas être pertinente d’un point de vue clinique.
5.3. Données de sécurité préclinique
Les études précliniques sur LEPTOPROL ont montré des effets sur les organes reproducteurs, ce qui était attendu compte tenu des propriétés pharmacologiques connues de la leuproréline.
Cancérogénicité
Chez le rat, une augmentation dose-dépendante dans les adénomes hypophysaires a été observée suite à l’injection sous-cutanée de doses comprises entre 0,6 et 4 mg/kg/jour sur jusqu’à 12 et 24 mois. Un tel effet n’a pas été observé chez la souris traitée pendant 24 mois.
Mutagénicité
Les études réalisées in vitro et in vivo sur l’acétate de leuproréline pour la détection de mutations génétiques et chromosomiques n’ont pas révélé de potentiel mutagène.
Toxicité pour la reproduction
Dans les études de toxicité pour la reproduction réalisées chez le lapin, une augmentation de la mortalité fœtale et une réduction du poids du fœtus ont été observées. Les effets sur la mortalité fœtale sont les conséquences attendues des effets pharmacodynamiques de cette substance.
Tolérance locale
Des études non cliniques réalisées chez le chien et le lapin ont révélé une bonne tolérance locale de LEPTOPROL.
Acide polylactique.
Sans objet.
4 ans.
6.4. Précautions particulières de conservation
A conserver à une température ne dépassant pas 30°C.
6.5. Nature et contenu de l'emballage extérieur
Seringue pré-remplie en plastique de polycarbonate avec un piston en copolymère d’acrylonitrile-butadiène-styrène et une aiguille scellée dans un sachet en film composite de polyéthylène téréphtalate/aluminium.
Format des boîtes :
1 x 1 implant avec 5 mg de leuproréline (sous forme d’acétate de leuproréline).
2 x 1 implant avec 5 mg de leuproréline (sous forme d’acétate de leuproréline).
3 x 1 implant avec 5 mg de leuproréline (sous forme d’acétate de leuproréline).
5 x 1 implant avec 5 mg de leuproréline (sous forme d’acétate de leuproréline).
Toutes les présentations peuvent ne pas être commercialisées.
6.6. Précautions particulières d’élimination et de manipulation
Pas d’exigences particulières pour l’élimination.
Tout médicament non utilisé ou déchet doit être éliminé conformément à la réglementation en vigueur.
7. TITULAIRE DE L’AUTORISATION DE MISE SUR LE MARCHE
SANDOZ
9 PLACE MARIE-JEANNE BASSOT
92300 LEVALLOIS-PERRET
8. NUMERO(S) D’AUTORISATION DE MISE SUR LE MARCHE
· 34009 300 200 0 5 : 1 seringue(s) préremplie(s) polycarbonate de 1 implant avec aiguille(s)
9. DATE DE PREMIERE AUTORISATION/DE RENOUVELLEMENT DE L’AUTORISATION
[à compléter ultérieurement par le titulaire]
10. DATE DE MISE A JOUR DU TEXTE
[à compléter ultérieurement par le titulaire]
Sans objet.
12. INSTRUCTIONS POUR LA PREPARATION DES RADIOPHARMACEUTIQUES
Sans objet.
Médicament soumis à prescription médicale.
Liste I.
ANSM - Mis à jour le : 09/04/2026
LEPTOPROL 5 mg, implant en seringue pré-remplie
Leuproréline
· Gardez cette notice. Vous pourriez avoir besoin de la relire.
· Si vous avez d’autres questions, interrogez votre médecin ou votre pharmacien ou votre infirmier/ère.
· Ce médicament vous a été personnellement prescrit. Ne le donnez pas à d’autres personnes. Il pourrait leur être nocif, même si les signes de leur maladie sont identiques aux vôtres.
· Si vous ressentez un quelconque effet indésirable, parlez-en à votre médecin ou votre pharmacien ou votre infirmier/ère. Ceci s’applique aussi à tout effet indésirable qui ne serait pas mentionné dans cette notice. Voir rubrique 4.
1. Qu'est-ce que LEPTOPROL 5 mg, implant en seringue pré-remplie et dans quels cas est-il utilisé ?
2. Quelles sont les informations à connaître avant d'utiliser LEPTOPROL 5 mg, implant en seringue pré-remplie ?
3. Comment utiliser LEPTOPROL 5 mg, implant en seringue pré-remplie ?
4. Quels sont les effets indésirables éventuels ?
5. Comment conserver LEPTOPROL 5 mg, implant en seringue pré-remplie ?
6. Contenu de l’emballage et autres informations.
1. QU’EST-CE QUE LEPTOPROL 5 mg, implant en seringue pré-remplie ET DANS QUELS CAS EST-IL UTILISE ?
La substance active de LEPTOPROL (acétate de leuproréline) appartient au groupe des inhibiteurs de certaines hormones sexuelles.
LEPTOPROL agit sur l’hypophyse en la stimulant brièvement puis en réduisant la production des hormones qui contrôlent la sécrétion des hormones sexuelles dans les testicules.
Cela signifie que les concentrations des hormones sexuelles sont ensuite fortement réduites et, lorsque l’administration est poursuivie, elles restent à ce niveau. Après l’arrêt de LEPTOPROL, les concentrations des hormones hypophysaires et sexuelles reviennent à des valeurs normales.
LEPTOPROL est indiqué dans le traitement symptomatique des tumeurs hormonosensibles avancées de la prostate (cancer de la prostate).
LEPTOPROL est aussi utilisé dans le traitement du cancer de la prostate localement avancé et localisé hormonosensible, en association avec ou après la radiothérapie externe.
2. QUELLES SONT LES INFORMATIONS A CONNAITRE AVANT D’UTILISER LEPTOPROL 5 mg, implant en seringue pré-remplie ?
N’utilisez jamais LEPTOPROL 5 mg, implant en seringue pré-remplie :
· si vous êtes allergique à la leuproréline ou à l’un des autres composants contenus dans ce médicament (mentionnés dans la rubrique 6),
· si vous êtes allergique à des substances similaires à la leuproréline, telles que la goséréline ou la buséréline,
· si votre cancer n’est pas sensible aux hormones,
· si vous êtes une femme ou un enfant.
Avertissements et précautions
Adressez-vous à votre médecin ou infirmier(ère) avant d’utiliser LEPTOPROL 5 mg, implant en seringue pré-remplie :
· S’il est établi que vous avez une pression artérielle élevée : dans ce cas, votre médecin vous surveillera avec attention.
· Si vos deux testicules ont été enlevés par une intervention chirurgicale : dans ce cas, LEPTOPROL ne produit pas de nouvelles baisses de la concentration sanguine de l’hormone sexuelle masculine.
· Si, avant de débuter le traitement, vous avez déjà des symptômes du système nerveux (pression sur la moelle épinière, métastases sur la colonne vertébrale) ou ressentez un inconfort au moment d’uriner en raison d’un déplacement de l’appareil urinaire, vous devez en informer votre médecin sans délai : il/elle vous surveillera de très près dans les premières semaines, si possible à l’hôpital.
· Si les symptômes de la maladie réapparaissent (tels que douleur, difficulté à uriner ou faiblesse dans les jambes lors de l’utilisation prolongée de LEPTOPROL) : dans ce cas, votre médecin vérifiera régulièrement l’efficacité du traitement en effectuant des examens cliniques (toucher rectal de la prostate, examens d’imagerie médicale) et en vérifiant les paramètres sanguins (les phosphatases et l’antigène prostatique spécifique (PSA) et l’hormone sexuelle masculine (la testostérone)).
· S’il existe un risque que vous développiez une ostéoporose, votre médecin vous prescrira un médicament supplémentaire si cela sera nécessaire, pour prévenir la perte osseuse.
· Si vous êtes diabétique : dans ce cas, votre médecin vous surveillera de très près.
· Si vous présentez une stéatose hépatique (une maladie dans laquelle un excès de graisse s’accumule dans le foie).
Si vous souffrez de maux de tête intenses ou récurrents, de problèmes de vue et de sifflements ou bourdonnements dans les oreilles, contacter votre médecin immédiatement.
Des cas de dépression chez les patients prenant LEPTOPROL, pouvant être grave, ont été rapportés. Si vous prenez LEPTOPROL et que vous développez une humeur dépressive, veuillez en informer votre médecin.
Prévenez votre médecin si vous souffrez de troubles cardiovasculaires, notamment des troubles du rythme cardiaque (arythmie), ou si vous êtes traité par des médicaments indiqués pour ces troubles. Le risque de troubles du rythme cardiaque pourrait être augmenté par la prise de LEPTOPROL.
Des éruptions cutanées sévères, dont le syndrome de Stevens-Johnson (SSJ) et la nécrolyse épidermique toxique (NET ou syndrome de Lyell) ont été rapportées en association avec la leuproréline. Arrêtez d’utiliser la leuproréline et consultez immédiatement un médecin si vous remarquez l’un des symptômes liés à ces réactions cutanées graves, décrits à la rubrique 4.
Autres médicaments et LEPTOPROL 5 mg, implant en seringue pré-remplie
Informez votre médecin ou pharmacien si vous utilisez, avez récemment utilisé ou pourriez utiliser tout autre médicament.
LEPTOPROL peut interagir avec des médicaments utilisés pour traiter les troubles du rythme cardiaque (ex. quinidine, procaïnamide, amiodarone et sotalol) ou peuvent augmenter le risque de troubles du rythme cardiaque quand ils sont utilisés avec d’autres médicaments (ex. méthadone (utilisée comme antalgique et dans le sevrage de la dépendance aux drogues), la moxifloxacine (antibiotique), les antipsychotiques utilisés dans le traitement des maladies mentales graves.
Enfants et adolescents
LEPTOPROL est uniquement destiné aux patients adultes.
LEPTOPROL est uniquement destiné aux patients de sexe masculin.
Conduite de véhicules et utilisation de machines
Ce médicament ainsi que la tumeur peuvent causer une fatigue. Cela est plus susceptible de se produire en consommant de l’alcool.
Par conséquent, ne conduisez pas ou n’utilisez pas de machines sans la permission de votre médecin si tel est votre cas.
3. COMMENT UTILISER LEPTOPROL 5 mg, implant en seringue pré-remplie ?
En utilisant LEPTOPROL
· Le point d’injection sera nettoyé.
· Un anesthésique local peut alors être administré pour soulager la douleur ressentie lors de l’injection de l’implant.
· LEPTOPROL sera administré par injection sous la peau (sous-cutanée) dans la région abdominale.
· LEPTOPROL doit être uniquement administré par votre médecin ou un(e) infirmier(e). Ce sont eux qui seront chargés de préparer le produit.
Posologie
La dose recommandée est de 1 implant de LEPTOPROL contenant 5 mg de leuproréline tous les 3 mois.
· Veillez à suivre les conseils de votre médecin en ce qui concerne la fréquence d’administration de LEPTOPROL et à respecter l’intervalle entre chaque injection.
· LEPTOPROL vous sera injecté tous les 3 mois. Si l’injection suivante est reportée dans des cas exceptionnels dans la limite de 4 semaines, l’effet thérapeutique n’est généralement pas altéré.
· Le contenu d’une seringue pré-remplie est injecté.
· La seringue contient un implant correspondant à une dose de 5 mg de leuproréline.
Analyses de sang
Votre médecin vous demandera de réaliser régulièrement des analyses de sang pour vérifier si le médicament est efficace.
Après 3 mois de traitement, votre médecin peut généralement indiquer si votre cancer de la prostate peut être traité par LEPTOPROL. Il/elle doit pour cela vérifier les taux d’antigène prostatique spécifique (PSA) et de testostérone.
Durée du traitement
La durée du traitement sera décidée par votre médecin traitant. Le traitement doit être poursuivi, même si les symptômes liés au cancer ont diminué ou si le cancer s’est amélioré.
Le cancer de la prostate peut être traité avec ce médicament pendant plusieurs années. Par conséquent, s’il est efficace et que vous pouvez le tolérer, vous pouvez l’utiliser en continu. Votre médecin réalisera des examens à des intervalles réguliers pour évaluer le traitement, en particulier si les symptômes suivants réapparaissent :
· douleur,
· difficulté à uriner,
· faiblesse dans les jambes.
Si vous avez utilisé plus de LEPTOPROL 5 mg, implant en seringue pré-remplie que vous n’auriez dû
Il est peu probable que votre médecin ou votre infirmier/ère vous administre trop de médicament. Si une plus grande quantité est administrée de façon accidentelle, votre médecin vous surveillera et, si nécessaire, vous donnera un traitement approprié.
Si vous oubliez d’utiliser LEPTOPROL 5 mg, implant en seringue pré-remplie
Si vous pensez que la dose trimestrielle de LEPTOPROL a été oubliée, parlez-en à votre médecin.
Si vous arrêtez d’utiliser LEPTOPROL 5 mg, implant en seringue pré-remplie
Si le traitement est arrêté sans l’approbation de votre médecin, les symptômes associés à votre maladie peuvent s’aggraver.
Le traitement ne doit donc pas être arrêté prématurément sans la permission de votre médecin.
Si vous avez d’autres questions sur l’utilisation de ce médicament, demandez plus d’informations à votre médecin ou à votre infirmier/ère.
4. QUELS SONT LES EFFETS INDESIRABLES EVENTUELS ?
Contactez votre médecin immédiatement ou rendez-vous dans l’hôpital le plus proche si vous ressentez les effets secondaires suivants :
· Réactions allergiques (réactions anaphylactiques). Les symptômes peuvent comprendre l’apparition soudaine de :
o sensation de chaleur, éruption cutanée, démangeaisons ou urticaire sur la peau et/ou les muqueuses,
o gonflement du visage, des lèvres ou de la langue ou d’autres parties du corps,
o essoufflement, respiration sifflante ou difficulté à respirer,
o chute de la pression artérielle, accélération du rythme cardiaque, convulsions, et dans les cas les plus sévères, insuffisance du système cardiovasculaire mettant en jeu le pronostic vital.
· Gonflement et douleur dans une partie du corps dus à un caillot de sang dans une veine.
· Difficulté à respirer, douleur thoracique, évanouissements, rythme cardiaque rapide, peau bleuâtre et décoloration dus à un caillot de sang dans les poumons.
Ces effets indésirables sont rares (ils peuvent affecter 1 personne sur 1000).
Une augmentation initiale à court terme du taux d’hormone sexuelle masculine (la testostérone) dans le sang est habituellement observée. Par conséquent, les symptômes liés à la maladie suivants peuvent être temporairement aggravés :
· Survenue ou augmentation des douleurs osseuses.
· Difficulté à uriner en raison d’un déplacement de l’appareil urinaire.
· Pression sur la moelle épinière.
· Faiblesse musculaire dans les jambes.
· Gonflement dû à un liquide se trouvant dans les tissus qui ne peut pas s’écouler (œdème lymphatique).
Cette augmentation des symptômes régresse généralement sans avoir à interrompre LEPTOPROL.
Lorsque le traitement est débuté, l’administration d’un antagoniste de l’hormone sexuelle masculine (un anti-androgène) doit être envisagée pour réduire les conséquences possibles de l’augmentation initiale de l’hormone sexuelle masculine.
Au cours du traitement, le taux d’hormone sexuelle masculine chute à un niveau très bas. Par conséquent, chez certains patients les effets secondaires suivants apparaissent :
Très fréquents : peuvent affecter plus de 1 personne sur 10
· bouffées de chaleur,
· augmentation de la transpiration,
· douleur osseuse,
· réduction ou perte de la libido et de la puissance sexuelle,
· réduction de la taille des testicules,
· prise de poids,
· réactions cutanées locales, telles que rougeur ou induration, douleur, gonflement et démangeaisons au point d’injection qui disparaissent normalement même lorsque le traitement est poursuivi ; dans des cas isolés, un abcès est apparu.
Fréquents : peuvent affecter jusqu’à 1 personne sur 10
· développement des seins chez l’homme,
· diminution de l’appétit,
· augmentation de l’appétit,
· dépression, changements d’humeur,
· troubles du sommeil,
· maux de tête,
· sensations anormales, telles que des sensations de picotements et/ou d’engourdissement,
· nausées/vomissements,
· douleurs articulaires ou dorsales,
· faiblesse musculaire,
· augmentation du besoin d’uriner la nuit,
· besoin fréquent et excessif d’uriner pendant la journée,
· difficulté à uriner et douleur en urinant,
· fatigue,
· gonflement des chevilles, des pieds ou des doigts (œdème périphérique),
· perte de poids,
· augmentation des taux sanguins des enzymes hépatiques (ALAT, ASAT, gamma-GT) et d’autres enzymes (LDH, phosphatases alcalines).
Peu fréquents : peuvent affecter jusqu’à 1 personne sur 100
· réactions allergiques générales telles que fièvre, démangeaisons, augmentation des cellules éosinophiles dans le sang, éruption cutanée,
· diarrhée,
· peau ou muqueuses sèches,
· douleur testiculaire,
· incapacité à vider spontanément sa vessie pleine,
· augmentation de la transpiration la nuit.
Rares : peuvent affecter jusqu’à 1 personne sur 1000
· augmentation ou réduction de la glycémie,
· étourdissements,
· altération du goût,
· augmentation ou réduction de la pression artérielle,
· perte de cheveux.
Très rares : peuvent affecter jusqu’à 1 personne sur 10000
Comme avec les autres médicaments de cette classe de substances : infarctus pituitaire après la première administration chez les patients atteints de tumeur hypophysaire.
Fréquence indéterminée (la fréquence ne peut pas être estimée à partir des données disponibles)
· maladie pulmonaire non infectieuse (pneumonie) (rapportée principalement au Japon).
· dans des cas isolés, un abcès est apparu au point d’injection,
· changement de l’ECG (allongement de l’intervalle QT),
· inflammation des poumons, pneumopathies,
· convulsions,
· hypertension intracrânienne idiopathique (augmentation de la pression intracrânienne dans la zone du cerveau caractérisée par des maux de tête, une vision double et autres symptômes visuels et des sifflements ou bourdonnements dans une ou les deux oreilles),
· plaques rougeâtres non surélevées, en forme de cible ou circulaires sur le tronc, souvent avec des cloques centrales, une desquamation de la peau, des aphtes de la bouche, de la gorge, du nez, des organes génitaux et des yeux. Ces éruptions cutanées graves peuvent être précédées de fièvre et de symptômes pseudo-grippaux (Syndrome de Stevens-Johnson/Nécrolyse épidermique toxique),
· rougeur cutanée et éruption cutanée accompagnée de démangeaisons (Éruption cutanée toxique),
· une éruption cutanée qui provoque l’apparition de boutons ou plaques rouges sur la peau, pouvant ressembler à une cible, avec un centre rouge entouré de cercles rouges plus pâles (Érythème polymorphe).
Informations particulières :
L’effet du traitement par LEPTOPROL peut être contrôlé en mesurant les concentrations sanguines de l’hormone sexuelle masculine (la testostérone) et en réalisant d’autres analyses de sang (phosphatase acide, PSA = antigène prostatique spécifique). Le taux de testostérone augmente d’abord en début de traitement puis chute sur une période de 2 semaines. Après 2 à 4 semaines, les concentrations de testostérone atteintes sont similaires à celles observées suite à l’ablation chirurgicale des deux testicules, puis elles restent constantes pendant toute la durée du traitement.
Une augmentation temporaire des taux sanguins de phosphatase acide peut se produire dans la phase initiale du traitement. Des taux normaux ou quasi-normaux sont atteints après quelques semaines.
Une diminution du taux d’hormone sexuelle masculine, comme c’est le cas après l’ablation des testicules ou sous traitement avec des médicaments qui inhibent les hormones sexuelles (comme LEPTOPROL), peut entraîner une réduction de la densité osseuse avec une augmentation du risque de fractures osseuses (voir : Mises en garde et précautions). La réduction de la densité osseuse après l’ablation des testicules est cependant plus marquée qu’après l’administration de LEPTOPROL. Votre médecin envisagera l’administration d’un médicament supplémentaire afin de réguler le métabolisme du calcium (appelé un bisphosphonate).
Déclaration des effets secondaires
Si vous ressentez un quelconque effet indésirable, parlez-en à votre médecin ou infirmier/ère. Ceci s’applique aussi à tout effet indésirable qui ne serait pas mentionné dans cette notice. Vous pouvez également déclarer les effets indésirables directement via le système national de déclaration : Agence nationale de sécurité du médicament et des produits de santé (ANSM) et réseau des Centres Régionaux de Pharmacovigilance – Site internet : https://signalement.social-sante.gouv.fr/
En signalant les effets indésirables, vous contribuez à fournir davantage d’informations sur la sécurité du médicament.
5. COMMENT CONSERVER LEPTOPROL 5 mg, implant en seringue pré-remplie ?
Tenir ce médicament hors de la vue et de la portée des enfants.
Ne pas stocker au-dessus de 30°C.
N’utilisez pas ce médicament après la date de péremption indiquée sur l’emballage extérieur ainsi que sur le sachet et l’étiquette de la seringue après EXP. La date de péremption fait référence au dernier jour de ce mois.
Ne jetez aucun médicament au tout-à-l’égout ou avec les ordures ménagères. Demandez à votre pharmacien d’éliminer les médicaments que vous n’utilisez plus. Ces mesures contribueront à protéger l’environnement.
6. CONTENU DE L’EMBALLAGE ET AUTRES INFORMATIONS
Ce que contient LEPTOPROL 5 mg, implant en seringue pré-remplie
· La substance active est la leuproréline (sous forme d’acétate de leuproréline).
Un implant contient 5 mg de leuproréline (sous forme d’acétate de leuproréline).
· L’autre composant est l’acide polylactique.
Qu’est-ce que LEPTOPROL 5 mg, implant en seringue pré-remplie et contenu de l’emballage extérieur
Seringue pré-remplie en plastique de polycarbonate avec un piston en copolymère d’acrylonitrile-butadiène-styrène et une aiguille scellée dans un sachet en film composite de polyéthylène/téréphtalate/aluminium.
L’emballage contient :
1 x 1 implant avec 5 mg de leuproréline (sous forme d’acétate de leuproréline).
2 x 1 implant avec 5 mg de leuproréline (sous forme d’acétate de leuproréline).
3 x 1 implant avec 5 mg de leuproréline (sous forme d’acétate de leuproréline).
5 x 1 implant avec 5 mg de leuproréline (sous forme d’acétate de leuproréline).
Toutes les présentations peuvent ne pas être commercialisées.
Titulaire de l’autorisation de mise sur le marché
9 PLACE MARIE-JEANNE BASSOT
92300 LEVALLOIS-PERRET
Exploitant de l’autorisation de mise sur le marché
Laboratoires bouchara-recordati s.a.s
TOUR HEKLA
52 avenue du general de gaulle
92800 puteaux
OTTO-SCHOTT-STRASSE 15,
07745 JENA
ALLEMAGNE
ou
SANDOZ GMBH
BIOCHEMIESTRASSE 10
6250 KUNDL
AuTRICHE
ou
EVER PHARMA JENA GMBH
BRÜSSELER STRASSE 18
07747 JENA
ALLEMAGNE
ou
LEK PHARMACEUTICALS D.D.
VEROVSKOVA ULICA 57
1526 LJUBLJANA
SLOVENIE
Noms du médicament dans les Etats membres de l'Espace Economique Européen
Ce médicament est autorisé dans les Etats membres de l'Espace Economique Européen sous les noms suivants : Conformément à la réglementation en vigueur.
[À compléter ultérieurement par le titulaire]
La dernière date à laquelle cette notice a été révisée est :
[à compléter ultérieurement par le titulaire]
Des informations détaillées sur ce médicament sont disponibles sur le site Internet de l’ANSM (France).
Les informations suivantes sont destinées exclusivement aux professionnels de santé :
Veuillez lire attentivement ces instructions, l’applicateur fourni avec ce médicament pouvant être différent de ceux que vous avez déjà utilisés.
Conseils d’utilisation
|
1. Désinfecter le point d’injection sur la paroi abdominale antérieure en-dessous du nombril.
|
|
||
|
2. Retirer l’applicateur de son sachet stérile et vérifier que l’implant est visible dans le réservoir (voir encadré). Pour vérifier, visualiser l’applicateur contre une lumière ou le secouer légèrement.
|
|
||
|
3. Tirer le piston de l’applicateur complètement vers l’arrière jusqu’à ce que vous puissiez voir une ligne complète dans la 2ème fenêtre. Remarque : Le piston ne peut être poussé vers l’avant pour injecter l’implant que s’il a déjà été préalablement complètement tiré vers l’arrière ! |
|
||
|
4. Retirer le capuchon protecteur de l’aiguille. |
|||
|
5. Tenir le corps principal de l’applicateur dans une main. Avec l’autre main, pincer la peau de la paroi abdominale antérieure du patient, en-dessous du nombril. Voir l’illustration. Avec l’ouverture de l’aiguille dirigée vers le haut, insérer l’ensemble de l’aiguille. Faire cela en formant un angle léger, presque parallèle à la peau dans le tissu sous-cutané. |
|
||
|
6. Tirer doucement l’applicateur sur environ 1 cm vers l’arrière (Cela forme le canal de ponction de l’implant). |
|
||
|
7. Injecter l’implant dans le canal de ponction en poussant complètement le piston vers l’avant jusqu’à ce qu’il se mette en place et que vous entendiez un cliquetis. |
|
||
|
8. Retirer l’aiguille. Pour s’assurer que l’implant a été injecté correctement, vérifier que la pointe bleu clair du piston est visible à la pointe de l’aiguille. |
|

Pour plus d’informations sur le dosage, veuillez-vous référer à la rubrique 3 : « Comment utiliser LEPTOPROL ».