Dernière mise à jour le 29/06/2026
RYLIGENCY 20 mg/mL, solution injectable en seringue préremplie
Indications thérapeutiques
Classe pharmacothérapeutique : Antiarythmiques, classe Ib, code ATC : C01B B01.
RYLIGENCY 20 mg/mL, solution injectable en seringue préremplie est un médicament utilisé chez l’adulte pour contrôler une anomalie sévère des battements cardiaques (arythmie ventriculaire). Ce médicament est administré uniquement si votre médecin juge que votre affection met votre vie en danger.
Présentations
> 10 seringues préremplies graduées en polypropylène de 5 mL
Code CIP : 34009 301 606 8 8
Déclaration de commercialisation : 18/02/2021
Cette présentation est agréée aux collectivités
Service médical rendu (SMR)
Les libellés affichés ci-dessous ne sont que des résumés ou extraits issus des avis rendus par la Commission de la Transparence. Seul l'avis complet de la Commission de la Transparence fait référence.
Cet avis est consultable à partir du lien `Avis du jj/mm/aaaa` ou encore sur demande auprès de la HAS (plus d'informations dans l'aide). Les avis et synthèses d'avis contiennent un paragraphe sur la place du médicament dans la stratégie thérapeutique.
| Valeur du SMR | Avis | Motif de l'évaluation | Résumé de l'avis |
|---|---|---|---|
| Important | Avis du 12/06/2019 | Inscription (CT) | Le service médical rendu par RYLIGENCY est important dans l’indication de l’AMM. |
Amélioration du service médical rendu (ASMR)
Les libellés affichés ci-dessous ne sont que des résumés ou extraits issus des avis rendus par la Commission de la Transparence. Seul l'avis complet de la Commission de la Transparence fait référence.
Cet avis est consultable à partir du lien `Avis du jj/mm/aaaa` ou encore sur demande auprés de la HAS (plus d'informations dans l'aide). Les avis et synthéses d'avis contiennent un paragraphe sur la place du médicament dans la stratégie thérapeutique.
| Valeur de l'ASMR | Avis | Motif de l'évaluation | Résumé de l'avis |
|---|---|---|---|
| V (Inexistant) | Avis du 12/06/2019 | Inscription (CT) | Cette spécialité n’apporte pas d’amélioration du service médical rendu (ASMR V) par rapport aux autres spécialités injectables à base de lidocaïne déjà inscrites. |
Autres informations
- Titulaire de l'autorisation : Laboratoire AGUETTANT
- Conditions de prescription et de délivrance :
- liste II
- réservé à l'usage professionnel
- Statut de l'autorisation : Valide
- Type de procédure : Procédure décentralisée
- Code CIS : 6 856 313 3
ANSM - Mis à jour le : 20/10/2023
RYLIGENCY 20 mg/mL, solution injectable en seringue préremplie
2. COMPOSITION QUALITATIVE ET QUANTITATIVE
Chaque millilitre de solution injectable contient 20 mg de chlorhydrate de lidocaïne.
Chaque seringue préremplie de 5 mL contient 100 mg de chlorhydrate de lidocaïne.
Excipient à effet notoire :
Ce médicament contient du sodium
Chaque millilitre de solution injectable contient 2,0 mg de sodium, équivalent à 0,09 mmol.
Chaque seringue préremplie de 5 mL contient 10 mg de sodium, équivalent à 0,4 mmol.
Pour la liste complète des excipients, voir rubrique 6.1.
Solution injectable en seringue préremplie.
Solution limpide et incolore.
pH : 5,0 à 6,5
Osmolalité : 270 - 330 mOsm/Kg
4.1. Indications thérapeutiques
4.2. Posologie et mode d'administration
Adultes
La dose doit être ajustée en fonction des besoins individuels et de l’effet thérapeutique recherché.
Bolus :
La dose de charge habituelle est de 50 à 100 mg en bolus, soit 1 à 1,5 mg/kg de poids corporel de RYLIGENCY en injection intraveineuse directe, ce qui correspond approximativement à 2,5 - 5 mL ou 0,05 – 0,075 mL/kg de poids corporel.
La vitesse d’injection ne doit pas dépasser 25 à 50 mg/min, ce qui correspond approximativement à 1,25 – 2,5 mL/min.
Si l’effet thérapeutique de la première dose est insuffisant durant les 5 à 10 premières minutes, il est possible de renouveler l’injection une ou deux fois jusqu’à atteindre une dose maximale de 200 – 300 mg en 1 heure.
La dose maximale journalière en bolus est de 300 mg.
Patients atteints d’insuffisance rénale
Les patients doivent être surveillés car l’insuffisance rénale peut être responsable d’effets toxiques en raison de l’accumulation de métabolites actifs (voir rubriques 4.4 et 5.2). RYLIGENCY doit être utilisé avec précautions.
Patients atteints d’insuffisance hépatique ou d’insuffisance cardiaque
La dose doit être réduite de moitié au maximum chez les patients atteints d’insuffisance cardiaque ou hépatique (voir rubrique 4.4).
Personnes âgées
Chez les personnes âgées, les doses sont calculées individuellement en fonction de l’âge et du poids corporel du patient. Les doses peuvent nécessiter une adaptation car le débit cardiaque et le débit sanguin hépatique diminuent avec l’âge, impliquant une diminution de la clairance de la lidocaïne (voir rubrique 5.2).
Population pédiatrique
La tolérance et l’efficacité dans la population pédiatrique n’ont pas été établies. La seringue préremplie n’est pas adaptée à une administration à la population pédiatrique ; la graduation ne permet pas une mesure exacte.
Mode d’administration
RYLIGENCY doit être administré en injection intraveineuse lente. La seringue préremplie ne convient pas à une utilisation avec un pousse-seringue.
· Troubles de la conduction sévères (tels que blocs AV de type II-III) sans insertion d’un pacemaker.
4.4. Mises en garde spéciales et précautions d'emploi
Un surdosage peut entraîner des réactions toxiques systémiques.
L’administration de lidocaïne doit s’accompagner d’une surveillance continue de l’ECG, de la pression artérielle, de l’état de conscience et de la respiration. Une surveillance cardiologique minutieuse est requise en particulier en cas d’ajustement posologique du médicament anti-arythmique. Un équipement cardiologique d’urgence doit être disponible. Si un ou plusieurs paramètres indiquent une aggravation de la fonction cardiaque, il faut envisager une révision du traitement qui peut comprendre, si nécessaire, un arrêt du traitement par lidocaïne.
RYLIGENCY doit être utilisé avec précautions dans les cas suivants :
· épilepsie : les patients atteints de troubles épileptiques cérébraux doivent faire l’objet d’une surveillance très étroite afin de déceler toute manifestation symptomatique de troubles du système nerveux central. Même de faibles doses de lidocaïne peuvent entraîner une augmentation de la tendance aux convulsions ;
· hypovolémie ;
· insuffisance rénale ou hépatique ;
· myasthénie grave ;
· bloc de conduction cardiaque ;
· insuffisance cardiaque ou défaillance cardiaque ;
· dépression respiratoire ;
· choc sévère.
L’équipement et les médicaments nécessaires à la surveillance et à une réanimation en urgence doivent être disponibles.
En cas d’acidose, la liaison de la lidocaïne aux protéines plasmatiques est réduite et par conséquent, la concentration de lidocaïne libre est augmentée. L’effet de la lidocaïne peut donc être majoré en cas d’acidose.
Une hypokaliémie, une hypoxie et des troubles de l’équilibre acido-basique doivent être corrigés avant d’utiliser la lidocaïne chez des patients nécessitant de fortes doses d’antiarythmiques.
Remarque : Chez les patients sous anesthésie, les troubles du système nerveux central peuvent passer inaperçus et des effets indésirables cardiaques peuvent apparaître brutalement sans autres symptômes d’alerte antérieurs.
Ce médicament contient moins de 1 mmol de sodium (23 mg) par seringue, c’est-à-dire qu’il est essentiellement « sans sodium ».
4.5. Interactions avec d'autres médicaments et autres formes d'interactions
Interactions pharmacodynamiques
Antiarythmiques de classe I
L’administration simultanée de lidocaïne et d’autres antiarythmiques de classe I doit être évitée en raison du risque d’apparition d’effets indésirables cardiaques graves.
Autres antiarythmiques
Si la lidocaïne est associée à d'autres antiarythmiques tels que les bêta-bloquants ou les inhibiteurs calciques, l'effet inhibiteur sur la conduction auriculo-ventriculaire et intraventriculaire et sur la contractilité peut être renforcé.
Association à d’autres anesthésiques locaux
L’association de différents anesthésiques locaux peut entraîner des effets additifs sur le système cardiovasculaire et le système nerveux central.
Relaxants musculaires
L'effet des relaxants musculaires (par ex. le suxaméthonium) est prolongé par la lidocaïne.
Sédatifs, hypnotiques
La lidocaïne doit être administrée avec précaution aux patients recevant des sédatifs qui affectent également la fonction du SNC et qui peuvent donc altérer la toxicité de la lidocaïne. Il peut se produire un effet additif entre l'effet anesthésique local et les sédatifs ou hypnotiques.
Anesthésiques volatils
Si de la lidocaïne et des anesthésiques volatils sont administrés simultanément, les effets dépressifs des deux peuvent être intensifiés.
Médicaments pouvant abaisser le seuil convulsif
Comme la lidocaïne elle-même peut réduire le seuil convulsif, son administration concomitante avec d'autres médicaments abaissant le seuil convulsif (par ex. le tramadol ou le bupropion) peut augmenter le risque de convulsions.
Médicaments pouvant élever le seuil convulsif
L'administration simultanée de diazépam élève le seuil convulsif de la lidocaïne. Ceci doit être pris en compte lorsqu'on surveille les patients pour déceler d’éventuels signes de toxicité de la lidocaïne.
Vasoconstricteurs :
L'effet anesthésique local est prolongé par une association avec un vasoconstricteur, par exemple l'adrénaline. Si la lidocaïne est administrée en tant qu’agent antiarythmique, l'administration d'autres médicaments contenant de l'adrénaline ou de la noradrénaline peut entraîner une potentialisation des effets indésirables cardiaques.
Interactions pharmacocinétiques
La lidocaïne est principalement métabolisée par les isoenzymes CYP 3A4 et CYP 1A2 du cytochrome P 450 (voir la rubrique 5.2). Une administration concomitante avec des substances actives qui sont des substrats, des inhibiteurs ou des inducteurs d'enzymes hépatiques, des isoenzymes CYP3A4 et CYP1A2, peut avoir une influence sur la pharmacocinétique de la lidocaïne et donc aussi sur son effet.
Inhibiteurs du CYP 3A4 et/ou de CYP 1A2
Une administration concomitante de lidocaïne et d'inhibiteurs du CYP3A4 et/ou du CYP1A2 peut entraîner une élévation des concentrations plasmatiques de lidocaïne. Il a été signalé une augmentation des concentrations plasmatiques avec, par exemple :
· Amiodarone (inhibiteur de CYP3A4) : L'amiodarone diminue la puissance du métabolisme hépatique de la lidocaïne, ce qui entraîne un risque d'augmentation du taux de lidocaïne et, par conséquent, une augmentation de la toxicité neurologique et cardiovasculaire. Un suivi clinique, un ECG, et enfin un contrôle de la concentration plasmatique de lidocaïne doivent être réalisés. Au besoin, la posologie de la lidocaïne doit être ajustée pendant et après le traitement par l'amiodarone.
· Cimétidine (inhibiteur de CYP3A4 et de CYP1A2) : Cimétidine utilisée à des doses égales ou supérieures à 800 mg/jour : augmentation de la concentration plasmatique de lidocaïne avec augmentation subséquente de la toxicité neurologique et cardiovasculaire. Un suivi clinique, un ECG, et enfin un contrôle de la concentration plasmatique de lidocaïne doivent être réalisés. Au besoin, la posologie de la lidocaïne doit être ajustée pendant et après le traitement par la cimétidine.
· Fluvoxamine (inhibiteur de CYP3A4 et de CYP1A2) : Hausse de la concentration plasmatique de lidocaïne, augmentant ainsi le risque de toxicité neurologique et cardiovasculaire. Un suivi clinique, un ECG, et enfin un contrôle de la concentration plasmatique de lidocaïne doivent être réalisés. Au besoin, la posologie de la lidocaïne doit être ajustée pendant et après l'association.
· Bêta-bloquants (excepté esmolol) : Lidocaïne intraveineuse : hausse de la concentration plasmatique de lidocaïne, avec augmentation subséquente de la toxicité neurologique et cardiovasculaire. Un suivi clinique, un ECG, et enfin un contrôle de la concentration plasmatique de lidocaïne doivent être réalisés. Au besoin, la posologie de la lidocaïne doit être adaptée pendant et après le traitement par les bêtabloquants.
· Autres inhibiteurs connus de CYP3A4 : Inhibiteurs de protéase (par ex. ritonavir), antibiotiques de la famille des macrolides (par ex. érythromycine), antifongiques (par ex. kétoconazole, itraconazole).
· Autres inhibiteurs connus de CYP1A2 : ciprofloxacine
Inducteurs de CYP 3A4 et/ou de CYP 1A2
Les substances actives induisant le CYP3A4 et/ou le CYP 1A2 telles que les barbituriques (principalement le phénobarbital), la carbamazépine, la phénytoïne ou la primidone, accélèrent la clairance plasmatique de la lidocaïne et réduisent ainsi l'efficacité de la lidocaïne.
Autres interactions pharmacocinétiques
Les médicaments qui modifient le métabolisme, le débit sanguin hépatique, le débit cardiaque ou la distribution périphérique de la lidocaïne peuvent modifier les concentrations plasmatiques de lidocaïne.
Médicaments provoquant l’hypokaliémie
Les effets électrophysiologiques de la lidocaïne dépendent fortement de la concentration extracellulaire en potassium et peuvent être presque complètement bloqués par l'hypokaliémie. Une utilisation concomitante de médicaments pouvant provoquer une hypokaliémie grave (par ex. acétazolamide, diurétiques de l'anse et thiazides) doit donc être évitée ou utilisée sous une surveillance étroite de la concentration sérique en potassium.
4.6. Fertilité, grossesse et allaitement
Grossesse
Il n'existe pas de données appropriées sur l'utilisation de la lidocaïne chez les femmes enceintes.
La lidocaïne traverse la barrière placentaire (voir la rubrique 5.2). On peut supposer qu'un grand nombre de femmes enceintes et de femmes en âge de procréer ont reçu de la lidocaïne. Aucune perturbation spécifique du processus de reproduction n'a été signalée à ce jour, par exemple, aucune incidence accrue de malformations ou d'effets directs ou indirects sur le fœtus. Toutefois, les risques chez l’être humain ne sont pas encore complètement étudiés.
Les études réalisées chez l’animal ont démontré une toxicité pour la reproduction (voir la rubrique 5.3).
Allaitement
La lidocaïne est excrétée en petites quantités dans le lait maternel humain. Il est peu probable qu'elle ait un effet délétère sur le nourrisson lorsqu'elle est utilisée aux doses recommandées. L'allaitement peut donc être poursuivi pendant le traitement par la lidocaïne.
Fertilité
On ne dispose d'aucune donnée humaine sur l'effet de la lidocaïne sur la fertilité.
4.7. Effets sur l'aptitude à conduire des véhicules et à utiliser des machines
Résumé du profil de sécurité d’emploi
La fréquence et la gravité des effets indésirables de la lidocaïne dépendent de la dose, du mode d’administration et de la sensibilité individuelle du patient.
Des effets indésirables systémiques peuvent être attendus à des concentrations plasmatiques de lidocaïne supérieures à 5-10 mg/L. Ces effets se manifestent par des symptômes au niveau du SNC et des symptômes cardiovasculaires.
Liste sous forme de tableau des effets indésirables
Les effets indésirables énumérés dans la présente rubrique appartiennent aux catégories de fréquence suivantes : Très fréquent (≥ 1/10) ; fréquent (≥ 1/100 à < 1/10) ; peu fréquent (≥ 1/1 000 à< 1/100) ; rare (≥ 1/10 000 à < 1/1 000) ; très rare ( < 1/10 000) ; indéterminées (ne peuvent être estimées sur la base des données disponibles).
Le tableau ci-dessous énumère les effets indésirables associés à l’utilisation de la lidocaïne comme antiarythmique.
|
Classes de systèmes d’organes |
Très fréquent |
Fréquent |
Peu fréquent |
Rare |
Très rare |
Fréquence indéterminée |
|
Affections du sang et du système lymphatique |
|
|
|
|
|
Méthémoglo-binémie |
|
Affections du système immunitaire |
|
|
|
Réactions anaphylactiques se manifestant par une urticaire, un œdème, un bronchospasme, une détresse respiratoire et des symptômes circulatoires allant jusqu’au choc anaphylactique. |
|
|
|
Affections psychiatriques |
Dysphorie. |
Confusion, agitation, irritabilité, euphorie, hallucinations et dépression. |
|
|
|
|
|
Affections du système nerveux |
|
Somnolence, étourdissements, états de vertige, dysarthrie, acouphènes, tremblements, fourmillements et paresthésies (peau), vision floue |
|
Spasmes musculaires, allant jusqu’à des convulsions généralisées, altération du niveau de conscience allant jusqu’au coma |
|
|
|
Affections cardiaques |
|
|
|
Bradycardie, bloc auriculo-ventriculaire allant jusqu’à l’arrêt cardiaque |
Tachycardie ventriculaire
|
|
|
Affections vasculaires |
|
|
|
Hypotension |
|
|
|
Affections respiratoires, thoraciques et médiastinales |
|
|
|
Dépression respiratoire ou même arrêt |
|
|
|
Affections gastro-intestinales |
Nausées, vomissements, dysphagie. |
|
|
|
|
|
Autres populations spécifiques
Chez les personnes âgées, l’incidence des effets indésirables peut être augmentée.
Déclaration des effets indésirables suspectés
La déclaration des effets indésirables suspectés après autorisation du médicament est importante. Elle permet une surveillance continue du rapport bénéfice/risque du médicament. Les professionnels de santé déclarent tout effet indésirable suspecté via le système national de déclaration : Agence nationale de sécurité du médicament et des produits de santé (ANSM) et réseau des Centres Régionaux de Pharmacovigilance - Site internet : https://signalement.social-sante.gouv.fr.
La concentration plasmatique létale pour les humains est comprise entre 6 et 33 mg de lidocaïne par litre.
Le surdosage entraîne des signes de toxicité aiguë qui peuvent conduire à des effets indésirables très graves. Les effets toxiques de la lidocaïne dépendent de la concentration plasmatique ; les effets toxiques sont d’autant plus fréquents et graves que la concentration plasmatique est élevée et l’élévation rapide. Ces effets toxiques touchent le système nerveux central et le système cardiovasculaire.
Symptômes
De faibles surdoses toxiques de lidocaïne entraînent une stimulation du SNC. Un surdosage important, produisant des concentrations plasmatiques toxiques élevées, provoque une dépression des fonctions centrales.
La toxicité sur le système nerveux central se manifeste de façon progressive par des signes et des symptômes de sévérité croissante.
Les premiers symptômes observés sont les suivants : étourdissements, vertiges, agitation, hallucination, euphorie, appréhension, bâillements, logorrhée, céphalées, nausées, vomissements, paresthésies labiales, engourdissement de la langue, acouphènes et dysarthrie, déficience auditive et visuelle.
D’autres symptômes subjectifs du système nerveux central comprennent : désorientation, sensation occasionnelle de somnolence. Des cas de tachycardie, d’hypertension et de bouffées vasomotrices ont été également rapportées.
Ces signes d’alarme suivants nécessitent une surveillance attentive : contractions musculaires, tremblements, frissons et convulsions généralisées.
En cas d’administration d’une dose très élevée : dépression généralisée du système nerveux central, dépression respiratoire, coma et arrêt respiratoire.
Une toxicité cardiovasculaire peut être observée dans les cas graves : troubles du rythme cardiaque tels qu'une extrasystole ventriculaire, fibrillation ventriculaire, pouls impalpable, pâleur, bradycardie majeure, troubles de la conduction auriculo-ventriculaire, diminution de la contractilité cardiaque, hypotension et arrêt cardiaque.
Traitement
S’il apparaît des signes de toxicité aiguë pendant l’administration de RYLIGENCY, arrêter immédiatement l’administration du produit. Puisque la lidocaïne agit également comme anesthésique local, il convient d’administrer une perfusion intraveineuse afin d’éviter une hypoxie et une acidose qui potentialiseraient la toxicité systémique due à l’anesthésie locale (LAST - Local Anesthetic Systemic Toxicity en anglais) et accéléreraient l’évolution vers un collapsus cardiovasculaire et des convulsions.
Un traitement s’imposera en cas de convulsions et de dépression du SNC afin de maintenir l’oxygénation, arrêter les convulsions et soutenir la circulation. Une ventilation avec oxygène pure au masque doit être immédiatement instaurée pour obtenir l’arrêt des convulsions: c’est quelque fois suffisant pour obtenir l’arrêt des convulsions. Il est également nécessaire de s’assurer que les voies aériennes sont libres.
Commencer immédiatement une réanimation cardio-respiratoire, si nécessaire.
Si les convulsions persistent, administrer rapidement par voie intraveineuse un anticonvulsivant comme du thiopental ou une benzodiazépine (diazépam ou midazolam). Il convient d’envisager l’utilisation d’une émulsion lipidique intraveineuse. De la succinylcholine intraveineuse sera utilisée pour assurer une myorelaxation à condition que le médecin soit en mesure de réaliser une intubation endotrachéale et de prendre en charge un patient présentant une paralysie complète.
Après l’arrêt des convulsions et la réalisation d’une ventilation pulmonaire appropriée, aucun autre traitement n’est habituellement nécessaire. Néanmoins, en cas d’hypotension, il convient d’envisager un traitement approprié par vasopresseur intraveineux. Une bradycardie due à une hypertonie vagale doit être traitée par atropine intraveineuse.
Les patients présentant des signes manifestes de LAST devront être surveillés pendant au moins 12 heures, car la dépression cardiovasculaire peut persister ou réapparaître après le traitement.
Les analeptiques à action centrale sont contre-indiqués.
Il n’existe pas d’antidote spécifique.
La lidocaïne ne peut pas être éliminée par hémodialyse.
5. PROPRIETES PHARMACOLOGIQUES
5.1. Propriétés pharmacodynamiques
Classe pharmacothérapeutique : Antiarythmiques, classe Ib, code ATC : C01B B01.
La lidocaïne est un antiarythmique de classe Ib d’après la classifisaction de Vaughan Williams.
Mécanisme d’action et électrophysiologie
Des études évaluant les effets des concentrations thérapeutiques de lidocaïne sur les propriétés électrophysiologiques des fibres de Purkinje de mammifères ont montré que la lidocaïne atténuait la dépolarisation diastolique de phase 4, réduisait l’automatisme et entraînait une diminution (mais parfois aucune modification) de l’excitabilité et de la réactivité membranaire.
La durée du potentiel d’action (APD) et la période réfractaire effective (ERP) des fibres de Purkinje sont réduites alors que le rapport ERP/APD est augmenté. La durée du potentiel d’action et la période réfractaire effective du myocarde ventriculaire sont également diminuées. La période réfractaire effective du nœud AV peut augmenter, diminuer ou rester inchangée et la période réfractaire effective atriale est inchangée. La lidocaïne élève le seuil de fibrillation ventriculaire.
Aucune interaction significative n’a été décrite entre la lidocaïne et le système nerveux autonome et par conséquent, la lidocaïne a un effet minime voire nul sur le tonus du système nerveux autonome. Des études électrophysiologiques cliniques réalisées avec la lidocaïne ont montré une absence de modification du temps de récupération du nœud sinusal et du temps de conduction sino-auriculaire. Le temps de conduction du nœud AV est inchangé ou raccourci et le temps de conduction His-Purkinje est inchangé. Normalement, la lidocaïne n’entraîne pas de modifications au niveau de l’ECG, mais l’intervalle QT peut parfois être légèrement raccourci.
A doses thérapeutiques, la lidocaïne a des effets hémodynamiques minimes chez les sujets normaux et chez les sujets atteints de cardiopathie. Il a été montré que la lidocaïne n’entraînait aucune diminution, ou alors une diminution minime de la contractilité ventriculaire, du débit cardiaque, de la pression artérielle ou du débit cardiaque.
Efficacité et tolérance clinique
Les seuils d’excitation et de fibrillation du myocarde sont augmentés.
La lidocaïne supprime les foyers ectopiques de stimulation cardiaque et les potentiels d’action résultant des potentiels retardés et des tachy-arythmies dues à un circuit de réentrée.
Les canaux sodiques se lient plus rapidement à la lidocaïne lorsque la membrane est dépolarisée. L’effet anti-arythmique de la lidocaïne est donc particulièrement marqué en cas d’augmentation de la fréquence d’excitation.
L’effet de la lidocaïne est majoré si le potentiel de repos est moins négatif, par ex. en cas d’hyperkaliémie et/ou d’ischémie myocardique. Dans les situations d’hyperpolarisation, dues par ex. à une hypokaliémie, l’effet de la lidocaïne est diminué.
Il a été montré que la lidocaïne éliminait les arythmies ventriculaires dues à un phénomène de ré-entrée à la phase myocardique tardive en déprimant et en bloquant la voie de conduction de ré-entrée.
Les concentrations plasmatiques thérapeutiques doivent être comprises entre 1,5 et 5 microgrammes/mL. Au-delà de 5 microgrammes/mL, il faut s’attendre à des effets toxiques sur le SNC et le système cardiovasculaire.
5.2. Propriétés pharmacocinétiques
Après administration intraveineuse, la biodisponibilité est de 100 %.
Après administration intraveineuse, l’apparition de l’effet thérapeutique de la lidocaïne est rapide. Des concentrations plasmatiques thérapeutiques (1,5 – 5 microgrammes/mL) sont atteintes en 1 à 2 min. L’effet d’une injection en bolus dure 10 à 20 min ; afin de maintenir l’effet thérapeutique de la lidocaïne, son administration doit être continuée et se fera alors par perfusion intraveineuse.
Après une perfusion continue et lorsqu’aucune dose de charge n’est administrée, la concentration plasmatique atteint un état d’équilibre au plus tôt en 5 heures (entre 5 et 10 heures) après le début de la perfusion. Néanmoins, des concentrations thérapeutiques ont déjà été atteintes après 30 à 60 min.
Distribution
La lidocaïne suit une cinétique d’élimination biphasique. Après administration intraveineuse, la substance active est d’abord rapidement distribuée du compartiment central aux tissus et organes intensément perfusés (phase de distribution alpha). Cette phase est suivie d’une redistribution dans les muscles squelettiques et le tissu adipeux. La demi-vie pendant la phase de distribution alpha est de 4 à 8 minutes environ. Il est prévu que la distribution dans les tissus périphériques se produise dans les 15 minutes.
Le taux de liaison aux protéines plasmatiques est d'environ 60 à 80 % chez les adultes. Elle dépend de la concentration de substance active et de la concentration d'alpha-1-glycoprotéine acide (AGP). L'AGP est une protéine de phase aiguë qui se lie à la lidocaïne libre et peut être augmentée, par exemple après un traumatisme, une intervention chirurgicale ou des brûlures, selon l'état pathophysiologique du patient. Au contraire, il a été démontré que les concentrations d'AGP sont faibles chez les nouveau-nés et les patients souffrant d'une insuffisance hépatique, ce qui entraîne une réduction marquée de la liaison de la lidocaïne aux protéines plasmatiques.
Le volume de distribution à l'état d’équilibre est de 91 litres. Le volume de distribution peut être modifié chez les patients souffrant d'autres pathologies, comme par exemple une insuffisance cardiaque, une insuffisance hépatique ou une insuffisance rénale.
Biotransformation
La lidocaïne est rapidement métabolisée dans le foie par des mono-oxygénases principalement par N-désalkylation oxydative, hydroxylation au niveau du cycle aromatique et hydrolyse de la liaison amide. Les dérivés hydroxylés subissent une conjugaison.
Au total, environ 90 % de la lidocaïne sont métabolisés en 4-hydroxy-2,6-xylidine, en 4-hydroxy-2,6-xylidine-glucuronide et, dans une moindre mesure, en métabolites actifs monoéthyl glycine xylidide (MEGX) et glycine xylidide (GX).
Ces derniers métabolites peuvent s’accumuler pendant les perfusions de plus longue durée ou en cas d’insuffisance rénale sévère en raison de leur plus longue demi-vie par rapport à la lidocaïne. En cas d’affections hépatiques, le métabolisme peut être réduit à 10-50 % de la normale.
Les résultats obtenus avec des microsomes hépatiques humains et des isoformes recombinantes de CYP humain ont montré que les enzymes CYP1A2 et CYP3A4 sont les principales isoformes de CYP impliquées dans la N-déséthylation de la lidocaïne.
Élimination
Moins de 10 % de la lidocaïne sont excrétés sous forme inchangée dans les urines, la fraction restante est éliminée sous forme de métabolites.
La demi-vie d’élimination est de 1,5 à 2 heures chez l’adulte et approximativement de 3 heures chez le nouveau-né. La demi-vie d’élimination peut être augmentée en cas d’insuffisance cardiaque sévère (jusqu’à 4 – 12 heures) ou d’affection hépatique chronique (jusqu’à 4,5 – 6 heures).
Les demi-vies des métabolites actifs monoéthyl glycine xylidide (MEGX) et glycine xylidide (GX) sont respectivement de 2 - 6 heures et de 10 heures. Comme leur demi-vie plasmatique est plus longue que celle de la lidocaïne, une accumulation des métabolites, en particulier de GX, est possible au cours d’une perfusion prolongée.
De plus, la vitesse d’élimination dépend du pH ; elle peut être augmentée par acidification des urines. La clairance plasmatique est d’environ 0,95 mL/min.
Le débit sanguin hépatique semble limiter le métabolisme de la lidocaïne.
Populations particulières
Patients atteints d’insuffisance rénale
La demi-vie plasmatique de la lidocaïne semblait ne pas être modifiée sauf lorsqu’il existait une certaine accumulation de GX au cours d’une perfusion de 12 heures ou plus. Cette accumulation semblait être associée à l’administration à long terme du médicament. Néanmoins, chez des patients présentant une insuffisance rénale sévère, la clairance de la lidocaïne était approximativement diminuée de moitié et la demi-vie de la lidocaïne était environ deux fois plus élevée que chez des sujets sains.
Patients atteints d’insuffisance hépatique
La demi-vie plasmatique de la lidocaïne et de ses métabolites peut être allongée et il faut s’attendre à des effets significatifs sur la pharmacocinétique et à devoir ajuster la posologie chez les patients présentant une altération de la perfusion hépatique, par ex. après un infarctus aigu du myocarde, en cas d’insuffisance cardiaque, d’affection hépatique ou d’insuffisance cardiaque congestive.
Patients âgés
La demi-vie d’élimination et le volume of distribution peuvent être augmentés chez les patients âgés en raison d’une diminution du débit cardiaque et/ou du débit sanguin hépatique.
Femmes enceintes ou qui allaitent
La lidocaïne traverse la barrière placentaire par simple diffusion et atteint le fœtus en quelques minutes après l’administration.
Le fœtus est capable de métaboliser la lidocaïne. La concentration atteinte dans le sang fœtal est approximativement égale à 60 % de la concentration dans le sang maternel. En raison d’une plus faible fixation aux protéines plasmatiques dans le sang fœtal, la concentration de lidocaïne libre pharmacologiquement active est égale à 1,4 fois la concentration maternelle.
La lidocaïne est excrétée dans le lait maternel uniquement en faibles quantités.
5.3. Données de sécurité préclinique
La lidocaïne ne présentait aucun potentiel génotoxique dans les études de génotoxicité in vitro et in vivo. En revanche, la 2,6-xylidine, un métabolite de la lidocaïne, a présenté des signes d’activité génotoxique.
Des études de cancérogénèse n’ont pas été réalisées avec la lidocaïne. Il a été montré que la 2,6- xylidine avait un potentiel cancérogène dans des études de toxicologie clinique évaluant une exposition chronique. La portée clinique de ces résultats est inconnue.
Dans les études de toxicité sur les fonctions de reproduction, des effets embryotoxiques et foetotoxiques ont été décelés à des doses de 25 mg/kg s.c. chez le lapin. Chez le rat, à des doses inférieures aux valeurs toxiques maternelles, la lidocaïne n’a aucun effet sur le développement postnatal de la progéniture. Une altération de la fertilité des rats mâles et femelles n’a pas été observée avec la lidocaïne.
La lidocaïne traverse la barrière placentaire par simple diffusion (voir rubrique 5.2).
Hydroxyde de sodium (pour ajustement du pH),
Acide chlorhydrique concentré (pour ajustement du pH),
Eau pour préparations injectables.
Après ouverture, le médicament doit être utilisé immédiatement.
6.4. Précautions particulières de conservation
Conserver la seringue préremplie dans son blister non ouvert jusqu’à l’utilisation.
Ne pas congeler.
6.5. Nature et contenu de l'emballage extérieur
Seringue préremplie de 5 mL en polypropylène, conditionnée individuellement sous blister. La seringue préremplie est graduée (par graduations de 0,5 mL) de 0 à 5 mL à l’aide d’une étiquette auto-adhésive.
Boîte en carton de 1 et 10 seringues préremplies.
Toutes les présentations peuvent ne pas être commercialisées.
6.6. Précautions particulières d’élimination et de manipulation
Instruction d’utilisation
La seringue préremplie doit être préparée soigneusement de la façon suivante
La seringue préremplie est à usage unique, c’est-à-dire destinée à un seul patient. Jeter la seringue préremplie après utilisation. NE PAS RÉUTILISER.
Le contenu du blister non ouvert et non endommagé est stérile, et par conséquent il ne doit être ouvert qu’au moment de l’utilisation.
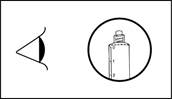
Le produit doit être inspecté visuellement avant l’administration pour déceler la présence de particules et d’une coloration anormale. Seule une solution limpide et incolore dépourvue de particules ou de précipité doit être utilisée.
Le médicament ne doit pas être utilisé si le sceau d'inviolabilité de la seringue est brisé.
La surface externe de la seringue préremplie est stérile jusqu’à l’ouverture du blister.
Lorsqu'il est manipulé en utilisant une technique aseptique, ce médicament peut être placé sur un champ stérile.
1) Retirez la seringue préremplie du blister stérile.
|
|
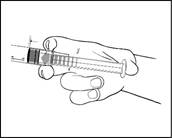
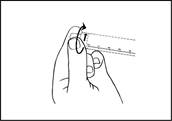
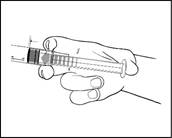
2) Presser la tige du piston pour libérer le joint. Le procédé de stérilisation a pu entrainer une adhérence du joint sur le corps de la seringue préremplie |
|
|
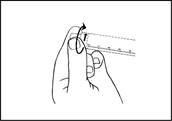
3) Dévisser l'embout protecteur pour rompre l’embout de scellage. Ne pas toucher l’embout de connexion Luer afin d'éviter toute contamination |
|
|
4) Vérifier que l’embout de scellage de la seringue préremplie a bien été totalement retiré. Dans le cas contraire, replacer le protège embout et dévisser à nouveau |
|
|
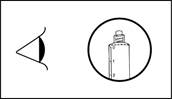
5) Purger l’air de la seringue en poussant légèrement le piston. |
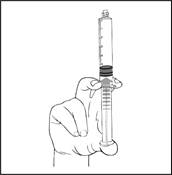
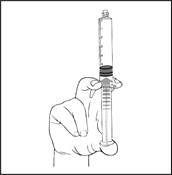
6) Raccordez la seringue préremplie à l’abord intraveineux. Poussez lentement le piston pour injecter le volume requis.
Tout médicament non utilisé ou déchet doit être éliminé conformément à la réglementation en vigueur.
7. TITULAIRE DE L’AUTORISATION DE MISE SUR LE MARCHE
1, RUE ALEXANDER FLEMING
69007 LYON
8. NUMERO(S) D’AUTORISATION DE MISE SUR LE MARCHE
· 34009 301 606 8 8 : Solution injectable en seringue préremplie de 5 mL (polypropylène), boîte de 10.
9. DATE DE PREMIERE AUTORISATION/DE RENOUVELLEMENT DE L’AUTORISATION
[à compléter ultérieurement par le titulaire]
10. DATE DE MISE A JOUR DU TEXTE
[à compléter ultérieurement par le titulaire]
Sans objet.
12. INSTRUCTIONS POUR LA PREPARATION DES RADIOPHARMACEUTIQUES
Liste II.
Médicament réservé à l'usage professionnel selon l'article R.5121-80 du code de la santé publique.
ANSM - Mis à jour le : 20/10/2023
RYLIGENCY 20 mg/mL, solution injectable en seringue préremplie
Chlorhydrate de lidocaïne
Veuillez lire attentivement cette notice avant que ce médicament ne vous soit administré car elle contient des informations importantes pour vous.
· Gardez cette notice. Vous pourriez avoir besoin de la relire.
· Si vous avez d’autres questions, interrogez votre médecin, votre pharmacien ou votre infirmier/ère.
· Si vous ressentez un quelconque effet indésirable, parlez-en à votre médecin, votre pharmacien ou votre infirmier/ère. Ceci s’applique aussi à tout effet indésirable qui ne serait pas mentionné dans cette notice. Voir rubrique 4.
1. Qu'est-ce que RYLIGENCY 20 mg/mL, solution injectable en seringue préremplie et dans quels cas est-il utilisé ?
2. Quelles sont les informations à connaître avant que RYLIGENCY 20 mg/mL, solution injectable en seringue préremplie ne vous soit administré ?
3. Comment RYLIGENCY 20 mg/mL, solution injectable en seringue préremplie est-il administré ?
4. Quels sont les effets indésirables éventuels ?
5. Comment conserver RYLIGENCY 20 mg/mL, solution injectable en seringue préremplie ?
6. Contenu de l’emballage et autres informations.
1. QU’EST-CE QUE RYLIGENCY 20 mg/mL, solution injectable en seringue préremplie ET DANS QUELS CAS EST-IL UTILISE ?
Classe pharmacothérapeutique : Antiarythmiques, classe Ib, code ATC : C01B B01.
RYLIGENCY 20 mg/mL, solution injectable en seringue préremplie est un médicament utilisé chez l’adulte pour contrôler une anomalie sévère des battements cardiaques (arythmie ventriculaire). Ce médicament est administré uniquement si votre médecin juge que votre affection met votre vie en danger.
2. QUELLES SONT LES INFORMATIONS A CONNAITRE AVANT D’UTILISER RYLIGENCY 20 mg/mL, solution injectable en seringue préremplie ?
N’utilisez jamais RYLIGENCY 20 mg/mL, solution injectable en seringue préremplie :
· si vous êtes allergique à la lidocaïne, aux anesthésiques locaux de type amide ou à l’un des autres composants contenus dans ce médicament mentionnés dans la rubrique 6.
· si vous avez des troubles sévères de la conduction cardiaque (par ex. blocs cardiaques) non corrigés par un pacemaker.
Avertissements et précautions
Adressez-vous à votre médecin, pharmacien ou votre infirmier/ère avant d’utiliser RYLIGENCY 20 mg/mL, solution injectable en seringue préremplie :
· si vous souffrez d’épilepsie. Votre médecin vous placera sous surveillance étroite afin de déceler toute manifestation de symptômes.
· si vous souffrez d’un état résultant d’une diminution de volume sanguin (hypovolémie).
· si vous avez une maladie des reins ou du foie.
· si vous avez une maladie entraînant une faiblesse musculaire (myasthénie).
· si vous avez des troubles cardiaques, y compris des troubles de la conduction, une insuffisance cardiaque, un ralentissement des battements cardiaques.
· si vous avez une dépression respiratoire (difficultés respiratoires avec respiration lente et superficielle).
· si vous êtes en état de choc.
Votre médecin vous administrera ce médicament pour traiter des problèmes cardiaques uniquement en prenant des précautions particulières si vous avez une importante acidité sanguine (acidose).
Avant de vous administrer de grandes quantités de ce médicament, votre médecin corrigera toute insuffisance de concentration de potassium dans votre sang, déficit en oxygène et troubles de votre équilibre acido-basique.
Remarque :
Si vous êtes sous anesthésie, votre médecin surveillera très attentivement votre état car des effets indésirables affectant votre système nerveux et votre cœur peuvent passer inaperçus et survenir sans symptômes d’alerte antérieurs.
Autres médicaments et RYLIGENCY 20 mg/mL, solution injectable en seringue préremplie
Informez votre médecin ou pharmacien si vous utilisez, avez récemment utilisé ou pourriez utiliser tout autre médicament. RYLIGENCY 20 mg/mL, solution injectable en seringue préremplie peut modifier l’action d’autres médicaments ou son action peut être modifiée par ces derniers.
Il est particulièrement important d’informer votre médecin si vous prenez un des médicaments suivants :
· médicaments utilisés pour traiter l’hypertension artérielle comme les diurétiques ;
· médicaments comme les bêta-bloquants (par ex. métoprolol, propranolol) ou les antagonistes calciques (par ex. amiodarone) utilisés pour traiter les troubles cardiaques, y compris une irrégularité des battements cardiaques ;
· médicaments qui diminuent le diamètre des vaisseaux sanguins (vasoconstricteurs, par ex. adrénaline, noradrénaline) ;
· médicaments utilisés pour relâcher les muscles pendant une anesthésie générale (par ex. suxaméthonium) ;
· somnifères et médicaments qui réduisent le niveau de conscience (sédatif) ;
· médicaments qui augmentent le risque de développer des spasmes et des crises convulsives (par ex. tramadol, bupropion) ;
· médicaments qui diminuent le risque de développer des spasmes et des crises convulsives (par ex. diazépam) ;
· cimétidine, un médicament utilisé pour traiter les brûlures d'estomac ;
· médicaments antiviraux (par ex.médicaments utilisés pour traiter une affection due au VIH), antibiotiques de la famille des macrolides (par ex. érythromycine) ou antifongiques (par ex. kétoconazole, itraconazole) ;
· ciprofloxacines (antibiotiques) ;
· médicaments utilisés pour traiter l’épilepsie (phénobarbital, phénytoïne, carbamazépine ou primidone) ;
· fluvoxamine, un médicament utilisé dans le traitement des maladies mentales ;
· médicaments utilisés pour réduire la pression à l’intérieur de l’œil (par ex. acétazolamide) ;
· autres anesthésiques, y compris les anesthésiques locaux.
Si vous êtes enceinte ou que vous allaitez, si vous pensez être enceinte ou planifiez une grossesse, demandez conseil à votre médecin ou pharmacien avant de prendre ce médicament. Votre médecin décidera alors si ce médicament doit vous être administré.
Grossesse
Votre médecin ne vous administrera ce médicament pendant que vous êtes enceinte que s’il juge cela nécessaire. La dose doit être aussi faible que possible.
Allaitement
La lidocaïne passe en faibles quantités dans le lait maternel. Il est peu probable que la lidocaïne utilisée aux doses recommandées affecte l’enfant qui est allaité. L’allaitement peut donc être poursuivi pendant l’utilisation de la lidocaïne.
Conduite de véhicules et utilisation de machines
RYLIGENCY 20 mg/mL, solution injectable en seringue préremplie peut affecter votre aptitude à conduire des véhicules et à utiliser des machines. Demandez à votre médecin quand vous pourrez de nouveau conduire des véhicules et utiliser des machines en toute sécurité.
RYLIGENCY 20 mg/mL, solution injectable en seringue préremplie contient du sodium
Ce médicament contient moins de 1 mmol de sodium (23 mg) par seringue, c’est-à-dire qu’il est essentiellement « sans sodium ».
3. COMMENT UTILISER RYLIGENCY 20 mg/mL, solution injectable en seringue préremplie ?
Si vous avez pris plus de RYLIGENCY 20 mg/mL, solution injectable en seringue préremplie que vous n’auriez dû
Puisque ce médicament est administré par un professionnel de santé qualifié, il est peu probable que vous receviez une trop grande quantité de RYLIGENCY.
L’apparition ou non de symptômes d’un surdosage dépend de la concentration de médicament dans votre sang. Les symptômes que vous pourriez présenter en cas de surdosage sont d’autant plus fréquents et plus sévères qu’il y a plus de lidocaïne dans votre sang et que l’administration est rapide.
Un surdosage léger affecte principalement le système nerveux central. En cas de survenue d’effets indésirables, ceux-ci disparaissent dans la plupart des cas après l’arrêt de l’administration de la lidocaïne.
Néanmoins, si vous pensez que vous avez reçu trop de médicament ou que vous commencez à ressentir des étourdissements ou une sensation de vertige, un engourdissement de la langue, un bourdonnement d’oreille, des vomissements ou des frissons, vous devez le signaler immédiatement à la personne qui effectue l’injection. Votre médecin saura comment prendre en charge ces symptômes et vous administrera le traitement nécessaire.
Si vous avez d’autres questions sur l’utilisation de ce médicament, demandez plus d’informations à votre médecin, à votre pharmacien ou à votre infirmier/ère.
4. QUELS SONT LES EFFETS INDESIRABLES EVENTUELS ?
Certains effets indésirables peuvent être graves. Consultez immédiatement un médecin si vous avez une réaction allergique entraînant :
· un gonflement des mains, des pieds, du visage, des lèvres, de la bouche, de la langue ou de la gorge
· des difficultés respiratoires
· une éruption cutanée accompagnée de démangeaisons
· une fièvre
· une chute de la tension artérielle et un état de choc
Ces effets indésirables sont rares (ils peuvent affecter jusqu’à 1 personne sur 1 000).
Les autres effets indésirables peuvent inclure :
Effets indésirables très fréquents (pouvant affecter plus d’1 personne sur 10)
· insatisfaction, susceptibilité ou état de détresse et de malaise (dysphorie)
· nausées, vomissements
· difficultés à avaler
Effets indésirables fréquents (pouvant affecter jusqu’à 1 personne sur 10)
· confusion, agitation, irritabilité, euphorie, hallucinations, dépression
· insomnie, étourdissements, sensation que tout tourne autour de vous, troubles de l’élocution, acouphènes, vision floue
· sensation de chatouillement, fourmillement, brulûre, picotement ou d’engourdissement de la peau (paresthésies)
Effets indésirables rares (pouvant affecter jusqu’à 1 personne sur 1 000)
· spasmes musculaires pouvant aller jusqu’à des crises convulsives généralisées et épileptiques
· rythme cardiaque lent, bloc cardiaque pouvant aller jusqu’à l’arrêt cardiaque
· faible tension artérielle
· respiration lente ou arrêt respiratoire
Effets indésirables très rares (pouvant affecter jusqu’à 1 personne sur 10 000)
· rythme cardiaque rapide
Effets indésirables fréquence indéterminée (ne peut être estimée sur la base des données disponibles)
· une coloration bleuâtre de la peau, des maux te tête, un essoufflement et une fatigue dus à des quantités anormales de méthémoglobine (une forme d’hémoglobine qui a une capacité réduite à fixer l’oxygène) dans le sang (méthémoglobinémie)
Déclaration des effets secondaires
Si vous ressentez un quelconque effet indésirable, parlez-en à votre médecin, votre pharmacien ou à votre infirmier/ère. Ceci s’applique aussi à tout effet indésirable qui ne serait pas mentionné dans cette notice. Vous pouvez également déclarer les effets indésirables directement via le système national de déclaration : Agence nationale de sécurité du médicament et des produits de santé (ANSM) et réseau des Centres Régionaux de Pharmacovigilance - Site internet : https://signalement.social-sante.gouv.fr
5. COMMENT CONSERVER RYLIGENCY 20 mg/mL, solution injectable en seringue préremplie ?
Tenir ce médicament hors de la vue et de la portée des enfants.
N’utilisez pas ce médicament après la date de péremption indiquée sur l’étiquette de la seringue, le blister et l’étui. La date de péremption fait référence au dernier jour de ce mois.
Conserver la seringue préremplie dans son blister non ouvert jusqu'à l'utilisation. Ne pas congeler.
N’utilisez pas ce médicament s’il y a des signes visibles de détérioration.
Tout médicament non utilisé ou déchet matériel doit être éliminé conformément à la réglementation locale en vigueur.
6. CONTENU DE L’EMBALLAGE ET AUTRES INFORMATIONS
Ce que contient RYLIGENCY 20 mg/mL, solution injectable en seringue préremplie
· La substance active est : chlorhydrate de lidocaïne
Chaque millilitre de solution injectable contient 20 mg de chlorhydrate de lidocaïne.
Chaque seringue préremplie de 5 mL contient 100 mg de chlorhydrate de lidocaïne.
· Les autres composants sont : chlorure de sodium, hydroxyde de sodium (pour l’ajustement du pH), acide chlorhydrique concentré (pour l’ajustement du pH), eau pour préparations injectables.
RYLIGENCY 20 mg/mL, solution injectable en seringue préremplie est une solution injectable limpide et incolore. RYLIGENCY se présente sous forme de seringue préremplie de 5 mL en polypropylène, conditionnée individuellement dans un blister transparent.
Boîte en carton de 1 ou 10 seringues préremplies.
Toutes les présentations peuvent ne pas être commercialisées.
Titulaire de l’autorisation de mise sur le marché
1, RUE ALEXANDER FLEMING
69007 LYON
FRANCE
Exploitant de l’autorisation de mise sur le marché
LABORATOIRE AGUETTANT
1, RUE ALEXANDER FLEMING
69007 LYON
FRANCE
1, RUE ALEXANDER FLEMING
69007 LYON
FRANCE
Noms du médicament dans les Etats membres de l'Espace Economique Européen
Ce médicament est autorisé dans les Etats membres de l'Espace Economique Européen sous les noms suivants : Conformément à la réglementation en vigueur.
[à compléter ultérieurement par le titulaire]
La dernière date à laquelle cette notice a été révisée est :
[à compléter ultérieurement par le titulaire]
Des informations détaillées sur ce médicament sont disponibles sur le site Internet de l’ANSM (France).
Les informations suivantes sont destinées exclusivement aux professionnels de la santé :
La seringue préremplie doit être préparée soigneusement de la façon suivante
La seringue préremplie est à usage unique, c’est-à-dire destinée à un seul patient. Jeter la seringue préremplie après utilisation. NE PAS RÉUTILISER.
Le contenu du blister non ouvert et non endommagé est stérile, et par conséquent il ne doit être ouvert qu’au moment de l’utilisation.
Le produit doit être inspecté visuellement avant l’administration pour déceler la présence de particules et d’une coloration anormale. Seule une solution limpide et incolore dépourvue de particules ou de précipité doit être utilisée.
Le médicament ne doit pas être utilisé si le sceau d'inviolabilité de la seringue est brisé.
La surface externe de la seringue préremplie est stérile jusqu’à l’ouverture du blister.
Lorsqu'il est manipulé en utilisant une technique aseptique, ce médicament peut être placé sur un champ stérile.
1) Retirez la seringue préremplie du blister stérile.
|
|
2) Presser la tige du piston pour libérer le joint. Le procédé de stérilisation a pu entrainer une adhérence du joint sur le corps de la seringue préremplie. |
|
|
3) Dévisser l'embout protecteur pour rompre l’embout de scellage. Ne pas toucher l’embout de connexion Luer afin d'éviter toute contamination. |
|
|
4) Vérifier que l’embout de scellage de la seringue préremplie a bien été totalement retiré. Dans le cas contraire, replacer le protège embout et dévisser à nouveau. |
|
|
5) Purger l’air de la seringue en poussant légèrement le piston. |
6) Raccordez la seringue préremplie à l’abord intraveineux. Poussez lentement le piston pour injecter le volume requis.