Dernière mise à jour le 29/06/2026
CEFPODOXIME VIATRIS ENFANTS ET NOURRISSONS 40 mg/5 mL, poudre pour suspension buvable
Indications thérapeutiques
Classe pharmacothérapeutique : antibactériens à usage systémique, code ATC : J01DD13.
(Céphalosporines de 3ème génération)
Ce médicament est un antibiotique antibactérien de la famille des bêta-lactamines.
Ce médicament est indiqué chez l'enfant dans le traitement de certaines infections bactériennes à germes sensibles.
Présentations
> 1 flacon(s) en verre brun avec fermeture de sécurité enfant de 50 ml avec seringue(s) pour administration orale polyéthylène polypropylène
Code CIP : 387 363-9 ou 34009 387 363 9 7
Déclaration de commercialisation : 27/10/2008
Cette présentation est agréée aux collectivités
- Prix hors honoraire de dispensation : 3,34 €
- Honoraire de dispensation : 1,02 €
- Prix honoraire compris : 4,36 €
- Taux de remboursement :65%
> 1 flacon(s) en verre brun avec fermeture de sécurité enfant de 100 ml avec seringue(s) pour administration orale polyéthylène polypropylène
Code CIP : 387 364-5 ou 34009 387 364 5 8
Déclaration de commercialisation : 27/10/2008
Cette présentation est agréée aux collectivités
- Prix hors honoraire de dispensation : 5,57 €
- Honoraire de dispensation : 1,02 €
- Prix honoraire compris : 6,59 €
- Taux de remboursement :65%
Documents de bon usage du médicament
- Choix et durée de l'antibiothérapie : Otite moyenne aiguë purulente de l’enfant
Auteur : Haute autorité de santé
Type : Fiche mémo
Date de mise à jour :Mai 2025
- Choix et durée de l'antibiothérapie : Rhinopharyngite aiguë enfant
Auteur : Haute autorité de santé
Type : Fiche mémo
Date de mise à jour :Mai 2025
- Choix et durée de l'antibiothérapie : Rhinopharyngite aiguë l’adulte
Auteur : Haute autorité de santé
Type : Fiche mémo
Date de mise à jour :Mai 2025
- Choix et durée de l'antibiothérapie : Otite moyenne aiguë purulente de l’adulte
Auteur : Haute autorité de santé
Type : Fiche mémo
Date de mise à jour :Mai 2025
- Choix et durée de l'antibiothérapie : Sinusites de l'enfant
Auteur : Haute autorité de santé
Type : Fiche mémo
Date de mise à jour :Mai 2025
- Choix et durée de l'antibiothérapie : Sinusite de l'adulte
Auteur : Haute autorité de santé
Type : Fiche mémo
Date de mise à jour :Mai 2025
- Choix et durées d'antibiothérapies : Angine aiguë de l’adulte
Auteur : Haute autorité de santé
Type : Fiche mémo
Date de mise à jour :Mai 2025
- Choix et durées d'antibiothérapies : Angine aiguë de l’enfant
Auteur : Haute autorité de santé
Type : Fiche mémo
Date de mise à jour :Mai 2025
Service médical rendu (SMR)
Amélioration du service médical rendu (ASMR)
Ce médicament étant un générique, l'ASMR n'a pas été évalué par la commission de la transparence (CT), il est possible de se référer à la /aux spécialité(s) de référence du groupe générique auquel appartient ce médicament (cliquez ici pour aller à la rubrique des groupes génériques)
ANSM - Mis à jour le : 09/07/2025
CEFPODOXIME VIATRIS ENFANTS ET NOURRISSONS 40 mg/5 mL, poudre pour suspension buvable
2. COMPOSITION QUALITATIVE ET QUANTITATIVE
Cefpodoxime......................................................................................................................... 40 mg
Sous forme de cefpodoxime proxétil
Pour 5 mL de suspension reconstituée.
Excipients à effet notoire : saccharose, aspartam (E951), benzoate de sodium.
Pour la liste complète des excipients, voir rubrique 6.1.
4.1. Indications thérapeutiques
Elles sont limitées chez l'enfant au traitement des infections dues aux germes sensibles au cefpodoxime et notamment :
· otites moyennes aiguës ;
· angines documentées à streptocoque A bêta-hémolytique ;
· sinusites ;
· infections respiratoires basses.
Il convient de tenir compte des recommandations officielles concernant l'utilisation appropriée des antibactériens.
4.2. Posologie et mode d'administration
La présentation en suspension buvable est préconisée chez l'enfant. Pour certains cas, le recours à une présentation sous forme de comprimé est envisageable dans les indications et posologies pédiatriques validées si l'enfant peut avaler le comprimé.
Enfant normo-rénal
La posologie moyenne est de 8 mg/kg/jour, sans dépasser la dose adulte (200 mg/jour dans l’angine et 400 mg/jour dans les autres indications), répartis en 2 prises à 12 heures d'intervalle.
|
La dose par prise est indiquée, en fonction du poids de l’enfant sur la seringue doseuse, pour une administration orale graduée en kg de poids corporel (graduations de 0 à 25 kg). La dose par prise se lit donc directement. Ainsi, le point indiqué correspond à la dose pour une prise. La dose à administrer pour une prise est donc obtenue en tirant le piston jusqu’à la graduation correspondant au poids de l’enfant. Deux prises par jour sont nécessaires. Par exemple, la graduation n°12 correspond à la dose à administrer par prise pour un enfant de 12 kg, et ce, deux fois par jour. |
La durée de traitement des angines est de 5 jours.
Au-delà de 25 kg (200 mg/j), le comprimé à 100 mg peut être utilisé.
Insuffisant rénal
Si la clairance de la créatinine est > 40 mL/min/1,73 m2, se référer à la posologie du sujet normo-rénal.
Si la clairance de la créatinine est < 40 mL/min/1,73 m2, voir le tableau ci-dessous :
|
Clairance de la créatinine (mL/min/1.73 m2) |
Doses |
|
10 - 39 |
une dose unitaire toutes les 24 heures |
|
< 10 |
une dose unitaire toutes les 48 heures |
En cas d'hémodialyse, une dose unitaire sera administrée après chaque séance de dialyse.
Insuffisant hépatique
Il n'est pas nécessaire de modifier la posologie.
Mode d'administration
Voie orale.
La suspension buvable est à administrer au cours d’un repas.
Avant la mise en suspension, la capsule contenant le déshydratant, située à l'intérieur du flacon, doit être retirée et jetée.
La poudre contenue dans le flacon est mise en suspension par addition d'eau au moment de l'emploi jusqu'au trait de jauge.
Le flacon doit être agité afin d'homogénéiser la suspension ainsi obtenue.
· Hypersensibilité à la substance active ou à l’un des excipients mentionnés à la rubrique 6.1.
· En cas d'allergie connue aux antibiotiques du groupe des céphalosporines.
4.4. Mises en garde spéciales et précautions d'emploi
· La survenue de toute manifestation allergique impose l'arrêt du traitement.
· La prescription de céphalosporines nécessite un interrogatoire préalable, l'allergie aux pénicillines étant croisée avec celle aux céphalosporines dans 5 à 10 % des cas :
o l'utilisation des céphalosporines doit être extrêmement prudente chez les patients pénicillinosensibles : une surveillance médicale stricte est nécessaire dès la première administration,
o l'emploi des céphalosporines est à proscrire formellement chez les sujets ayant des antécédents d'allergie de type immédiat aux céphalosporines. En cas de doute, la présence du médecin auprès du patient est indispensable à la première administration, afin de traiter l'accident anaphylactique possible.
· Les réactions d'hypersensibilité (anaphylaxie) observées avec ces deux types de bêta-lactamines peuvent être graves et parfois fatales.
· La survenue d'un épisode diarrhéique peut être symptomatique, de façon exceptionnelle, d'une colite pseudo-membraneuse dont le diagnostic repose sur la coloscopie.
Cet accident, rare avec les céphalosporines, impose l'arrêt immédiat du traitement et la mise en route d'une antibiothérapie spécifique appropriée (vancomycine). Dans ce cas, l'administration de produits favorisant la stase fécale doit absolument être évitée.
· Affections hématologiques
Comme pour les autres antibiotiques de la classe des bêta-lactamines, une neutropénie et plus rarement une agranulocytose peuvent se développer pendant le traitement avec cefpodoxime, notamment si le traitement est de longue durée. Dans ce cas, une surveillance hématologique doit être envisagée.
· Eruptions bulleuses
Comme pour les autres céphalosporines, des cas d’éruptions bulleuses (érythème polymorphe, syndrome de Stevens-Johnson, syndrome de Lyell) ont été rapportés. Si une atteinte de la peau et/ou des muqueuses survient, les patients doivent immédiatement contacter leur médecin, et ce, avant de continuer le traitement.
· Surinfection
Comme pour les autres antibiotiques, l’administration de cefpodoxime, notamment si le traitement est de longue durée, peut entraîner une croissance excessive des micro-organismes non sensibles. Des évaluations régulières de l’état du patient sont essentielles. Si une surinfection survient pendant le traitement, des mesures appropriées doivent être prises.
· Encéphalopathie
Les bêta-lactamines y compris le cefpodoxime prédisposent le patient au risque d’encéphalopathie (qui peut inclure des convulsions, une confusion, des troubles de la conscience ou des mouvements anormaux) et, particulièrement, en cas de surdosage ou d’insuffisance rénale.
· Réactions indésirables cutanées sévères (SCARs)
Des réactions indésirables cutanées sévères (SCARs) de fréquence indéterminée, telles que le syndrome de Stevens-Johnson (SJS), la nécrolyse épidermique toxique (NET), la réaction médicamenteuse avec éosinophilie et symptômes systémiques (DRESS), et la pustulose exanthématique aiguë généralisée (PEAG), pouvant menacer le pronostic vital ou être fatales, ont été rapportées en association avec un traitement par cefpodoxime.
Les patients doivent être informés des signes et symptômes et faire l’objet d’une surveillance étroite en cas de réactions cutanées.
En cas d’apparition de signes ou symptômes évocateurs de ces réactions, le traitement par cefpodoxime doit être immédiatement interrompu et un traitement alternatif doit être envisagé.
Si le patient développe une réaction grave telle que SJS, NET, DRESS ou PEAG associé à l’utilisation de cefpodoxime, le traitement par cefpodoxime ne doit à aucun moment être repris chez ce patient.
Précautions d'emploi
· Chez les patients allergiques à d'autres bêta-lactamines, il faut tenir compte de la possibilité d'allergie croisée.
· En cas d'insuffisance rénale sévère, il peut être nécessaire d'adapter la dose quotidienne en fonction de la clairance de la créatinine (voir rubriques 4.2 et 5.2 paragraphe « Sujets à risque »).
· Ce médicament ne doit pas être utilisé chez l'enfant de moins de 15 jours, en l'absence d'études précises.
· Comme avec d'autres antibiotiques à large spectre, l'utilisation prolongée de cefpodoxime proxétil peut entraîner une sélection des germes non sensibles, ce qui peut nécessiter l'interruption du traitement.
· Interactions avec les examens de laboratoire
o Une positivation du test de Coombs a été décrite au cours du traitement par les céphalosporines.
o Il peut se produire une réaction faussement positive lors de la recherche de glucose dans les urines avec des substances réductrices, mais non lorsqu'on utilise des méthodes à la glucose oxydase.
Excipients
· Ce médicament contient du saccharose. Les patients présentant une intolérance au fructose, un syndrome de malabsorption du glucose et du galactose ou un déficit en sucrase/isomaltase (maladies héréditaires rares) ne doivent pas prendre ce médicament.
· Ce médicament contient du sodium. Ce médicament contient moins de 1 mmol (23 mg) de sodium par dose de 5 mL, c.-à-d. qu’il est essentiellement « sans sodium ».
· Ce médicament contient 25 mg d’aspartam pour 5 mL de suspension reconstituée équivalent à 5 mg/mL. L’aspartam contient une source de phénylalanine. Peut être dangereux pour les personnes atteintes de phénylcétonurie (PCU), une maladie génétique rare caractérisée par l’accumulation de phénylalanine ne pouvant être éliminée correctement. Il n’existe aucune donnée clinique ou non clinique concernant l’utilisation de l’aspartam chez les enfants âgés de moins de 12 semaines.
· Ce médicament contient 10 mg de sel de benzoate pour 5 mL de suspension reconstituée équivalent à 2 mg/mL. L’augmentation de la bilirubinémie suite à son déplacement grâce à l’albumine peut accroître le risque d’ictère néonatal pouvant se transformer en ictère nucléaire (dépôts de bilirubine non conjuguée dans le tissu cérébral).
4.5. Interactions avec d'autres médicaments et autres formes d'interactions
Augmentation du pH gastrique : les anti-H2 (ranitidine) et anti-acides (hydroxyde d'aluminium, bicarbonate de sodium) conduisent à une diminution de la biodisponibilité.
En revanche, une diminution du pH gastrique (pentagastrine) provoque une augmentation de la biodisponibilité.
Les conséquences cliniques restent à établir.
Problèmes particuliers du déséquilibre de l'INR
De nombreux cas d'augmentation de l'activité des anticoagulants oraux ont été rapportés chez des patients recevant des antibiotiques. Le contexte infectieux ou inflammatoire marqué, l'âge et l'état général du patient apparaissent comme des facteurs de risque. Dans ces circonstances, il apparaît difficile de faire la part entre la pathologie infectieuse et son traitement dans la survenue du déséquilibre de l'INR. Cependant, certaines classes d'antibiotiques sont davantage impliquées : il s'agit notamment des fluoroquinolones, des macrolides, des cyclines, du cotrimoxazole et de certaines céphalosporines.
4.6. Fertilité, grossesse et allaitement
4.7. Effets sur l'aptitude à conduire des véhicules et à utiliser des machines
En cas de survenue d’effets indésirables tels que des vertiges ou une encéphalopathie (qui peut inclure des convulsions, une confusion, des troubles de la conscience ou des mouvements anormaux) (voir rubriques 4.4, 4.8, 4.9), le patient ne doit pas conduire des véhicules ou utiliser des machines.
Les fréquences ont été définies en utilisant les critères suivants : très fréquent (≥ 10 %) ; fréquent (≥ 1 % et < 10 %) ; peu fréquent (≥ 0,1 % et < 1 %) ; rare (≥ 0,01 % et < 0,1 %) ; très rare (< 0,01 %), fréquence indéterminée (ne peut être estimée sur la base des données disponibles).
· Affections hématologiques et du système lymphatique
Peu fréquent : neutropénie.
Rare : thrombocytose, leucopénie.
Fréquence indéterminée : agranulocytose, éosinophilie, thrombocytopénie, anémie hémolytique.
· Affections de l’oreille et du labyrinthe
Fréquent : acouphènes.
· Affections gastro-intestinales
Très fréquent : douleurs abdominales, diarrhée.
Fréquent : nausées, vomissements.
Peu fréquent : entérocolite.
Fréquence indéterminée : hématochézie, colite pseudomembraneuse, colite à Clostridioides difficile.
· Troubles généraux et anomalies au site d'administration
Fréquence indéterminée : malaise, asthénie.
· Affections hépatobiliaires
Fréquent : élévation des ASAT (aspartate aminotransférase), élévation des ALAT (alanine aminotransférase), élévation des phosphatases alcalines.
Fréquence indéterminée : élévation de la bilirubine sanguine, atteinte hépatique, atteinte hépatique cholestatique.
· Affections du système immunitaire
Peu fréquent : réactions anaphylactiques, bronchospasme.
Fréquence indéterminée : choc anaphylactique, œdème de Quincke.
· Infections et infestations
Fréquence indéterminée : surinfection.
· Affections du système nerveux
Très fréquent : céphalées.
Fréquent : vertiges.
Fréquence indéterminée : paresthésie, convulsions.
Les bêta-lactamines y compris le cefpodoxime prédisposent le patient au risque d’encéphalopathie (qui peut inclure des convulsions, une confusion, des troubles de la conscience ou des mouvements anormaux) et, particulièrement, en cas de surdosage ou d’atteinte de la fonction rénale.
· Affections de la peau et du tissu sous-cutané :
Fréquent : rash, prurit, urticaire.
Fréquence indéterminée : purpura, dermatite bulleuse, érythème polymorphe, syndrome de Stevens-Johnson, nécrolyse épidermique toxique (syndrome de Lyell), pustulose exanthématique aiguë généralisée (PEAG), réaction médicamenteuse avec éosinophilie et symptômes systémiques (DRESS)..
· Affections du rein et des voies urinaires :
Rare : faible augmentation de l’urée sanguin et de la créatininémie.
Fréquence indéterminée : des atteintes de la fonction rénale ont été rapportées avec des antibiotiques appartenant à la même classe thérapeutique que le cefpodoxime, notamment lorsqu’ils sont associés à des aminoglycosides et/ou des diurétiques puissants.
Déclaration des effets indésirables suspectés
La déclaration des effets indésirables suspectés après autorisation du médicament est importante. Elle permet une surveillance continue du rapport bénéfice/risque du médicament. Les professionnels de santé déclarent tout effet indésirable suspecté via le système national de déclaration : Agence nationale de sécurité du médicament et des produits de santé (ANSM) et réseau des Centres Régionaux de Pharmacovigilance - Site internet : https://signalement.social-sante.gouv.fr/.
5. PROPRIETES PHARMACOLOGIQUES
5.1. Propriétés pharmacodynamiques
Classe pharmacothérapeutique : antibactériens à usage systémique, code ATC : J01DD13.
(Céphalosporines de 3ème génération)
Le cefpodoxime proxétil est un antibiotique semi-synthétique de la famille des bêta-lactamines, du groupe des céphalosporines orales de troisième génération, prodrogue du cefpodoxime.
Après administration par voie orale, le cefpodoxime proxétil est absorbé en milieu intestinal et rapidement hydrolysé par des estérases non spécifiques en cefpodoxime, antibiotique bactéricide.
Mécanisme d’action
Le mécanisme d'action du cefpodoxime repose sur l'inhibition de la synthèse des parois bactériennes.
Le cefpodoxime est stable vis à vis de nombreuses bêta-lactamases.
Seuils des tests de sensibilité
Les critères d’interprétation des concentrations minimales inhibitrices (CMI) pour les tests de sensibilité ont été établis par le Comité européen des antibiogrammes (EUCAST) pour la Cefpodoxime et sont énumérés ci-dessous : https://www.ema.europa.eu/documents/other/minimum-inhibitory-concentration-mic-breakpoints_en.xlsx
Prévalence
La prévalence de la résistance acquise peut varier en fonction de la géographie et du temps pour certaines espèces. Il est donc utile de disposer d'informations sur la prévalence de la résistance locale, surtout pour le traitement d'infections sévères. Ces données ne peuvent apporter qu'une orientation sur les probabilités de la sensibilité d'une souche bactérienne à cet antibiotique.
Lorsque la variabilité de la prévalence de la résistance en France est connue pour une espèce bactérienne, elle est indiquée dans le tableau ci-dessous :
|
Catégories |
Fréquence de résistance acquise en France (> 10 %) (valeurs extrêmes) |
|
ESPÈCES SENSIBLES |
|
|
Aérobies à Gram positif |
|
|
Corynebacterium diphtheriae |
|
|
Streptococcus |
|
|
Streptococcus pneumoniae |
20 - 60 % |
|
Aérobies à Gram négatif |
|
|
Branhamella catarrhalis |
|
|
Citrobacter koseri |
|
|
Escherichia coli |
|
|
Haemophilus influenzae |
|
|
Klebsiella |
0 - 30 % |
|
Neisseria gonorrhoeae |
|
|
Pasteurella |
|
|
Proteus mirabilis |
|
|
Proteus vulgaris |
29 - 38 % |
|
Providencia |
|
|
Anaérobies |
|
|
Fusobacterium |
10 - 20 % |
|
Prevotella |
30 - 70 % |
|
Propionibacterium acnes |
|
|
ESPÈCES MODÉRÉMENT SENSIBLES |
|
|
(in vitro de sensibilité intermédiaire) |
|
|
Aérobies à Gram positif |
|
|
Staphylococcus méti-S |
|
|
ESPÈCES RÉSISTANTES Aérobies à Gram positif Entérocoques Listeria monocytogenes Staphylococcus méti-R * Aérobies à Gram négatif Acinetobacter Citrobacter freundii Enterobacter Morganella morganii Pseudomonas Serratia Anaérobies Bacteroides fragilis ClostridioidesPeptostreptococcus |
|
* La fréquence de résistance à la méticilline est environ de 30 à 50 % de l'ensemble des staphylocoques et se rencontre surtout en milieu hospitalier.
5.2. Propriétés pharmacocinétiques
Absorption
L'absorption du cefpodoxime proxétil administré par voie orale au sujet à jeun, sous forme d'un comprimé correspondant à 100 mg de cefpodoxime, est de 40 à 50 %.
Celle-ci est augmentée par la prise d'aliments de sorte qu'il est recommandé que le produit soit administré au cours des repas.
Distribution
· Concentrations plasmatiques :
o Après administration par voie orale d'une dose unique de 100 mg, les concentrations plasmatiques maximales de cefpodoxime (Cmax) sont de 1 mg/L à 1,2 mg/L. Après administration d'une dose de 200 mg, les concentrations plasmatiques maximales sont de 2,2 à 2,5 mg/L. Dans les deux cas (100 ou 200 mg), elles sont atteintes (Tmax) en 2 à 3 heures.
o Les concentrations résiduelles à 12 heures sont respectivement de 0,08 mg/L et de 0,18 mg/L après administration de 100 mg et de 200 mg.
o Après administration pendant 14,5 jours de 100 à 200 mg, 2 fois par jour, les paramètres pharmacocinétiques plasmatiques du cefpodoxime ne sont pas modifiés, traduisant l'absence d'accumulation du principe actif.
· Le volume de distribution du cefpodoxime est de 30-35 L chez le sujet sain jeune (=0,43 L/kg).
· Fixation aux protéines plasmatiques :
o Le taux de fixation du cefpodoxime est de l'ordre de 40 % et se fait principalement sur l'albumine. Cette fixation est de type non saturable.
· Diffusion humorale et tissulaire :
o Le cefpodoxime a une bonne diffusion dans le parenchyme pulmonaire, la muqueuse bronchique, le liquide pleural, les amygdales et le liquide interstitiel.
o 4 à 7 heures après une prise unique de 100 mg, les concentrations amygdaliennes sont de 0,24 à 0,1 µg/g (20 à 25 % des concentrations plasmatiques).
o Après une prise unique de 200 mg de cefpodoxime, les concentrations dans le liquide interstitiel sont de 1,5 à 2,0 mg/L (80 % des concentrations plasmatiques).
o 3 à 12 heures après une prise unique de 200 mg de cefpodoxime, les concentrations dans le poumon sont de 0,6 à 0,2 µg/g ; dans la plèvre, elles sont de 0,6 à 0,8 mg/L.
o Dans la muqueuse bronchique, entre 1 et 4 heures après administration de 200 mg, les concentrations de cefpodoxime sont aux alentours de 1 µg/g (40 à 45 % des concentrations plasmatiques).
o Les concentrations mesurées sont supérieures aux CMI des micro-organismes sensibles.
Biotransformation
· Après absorption, le métabolite principal est le cefpodoxime, résultant de l'hydrolyse du cefpodoxime proxétil.
· Le cefpodoxime est très peu métabolisé.
Elimination
· Après absorption du cefpodoxime proxétil, 80 % du cefpodoxime libéré sont éliminés sous forme inchangé dans les urines.
· La demi-vie d'élimination du cefpodoxime est en moyenne de 2,4 heures.
Sujets à risque
· Les paramètres pharmacocinétiques du cefpodoxime sont très légèrement modifiés chez le sujet âgé à fonction rénale normale. Toutefois, la faible augmentation des concentrations sériques maximales et de la demi-vie d'élimination, ne justifie aucune réduction de posologie dans ce type de population, sauf chez les sujets dont la clairance rénale est inférieure à 40 mL/min.
· En cas d'insuffisance rénale, correspondant à une clairance de la créatinine inférieure à 40 mL/min, l'augmentation de la demi-vie d'élimination plasmatique et des concentrations plasmatiques maximales conduit à réduire la dose de moitié en l'administrant en une seule prise quotidienne.
· En cas d'insuffisance hépatique, les faibles modifications cinétiques observées ne justifient pas une adaptation spécifique de la posologie.
CHEZ L'ENFANT
Après administration par voie orale d'une dose unique de 5 mg/kg (200 mg maximum) exprimé en cefpodoxime, à des sujets âgés de 4 à 12 ans, les concentrations plasmatiques maximales de cefpodoxime (Cmax) sont en moyenne de 2,6 mg/L. Elles sont atteintes en 2 à 4 heures.
Chez les patients de moins de 2 ans lors d'une administration répétée de 5 mg/kg toutes les 12 heures, les concentrations plasmatiques moyennes, 2 heures après administration, sont comprises entre 2,7 mg/L (1 mois - 6 mois) et 2,0 mg/L (7 mois -2 ans).
Chez les patients âgés de 1 mois à 12 ans après administration répétée de 5 mg/kg toutes les 12 heures, les concentrations plasmatiques résiduelles de cefpodoxime (C12h) à l'état d'équilibre sont comprises entre 0,2 à 0,3 mg/L (1 mois - 2 ans) et 0,1 mg/L (2 ans - 12 ans).
5.3. Données de sécurité préclinique
Silice colloïdale hydratée, talc, trioléate de sorbitan, benzoate de sodium, chlorure de sodium, acide citrique anhydre, aspartam, galactomannane du guar, arôme citron, arôme orange, oxyde de fer jaune, saccharose.
Après reconstitution : 14 jours
6.4. Précautions particulières de conservation
A conserver à une température ne dépassant pas 25°C.
Après reconstitution : à conserver au réfrigérateur (entre 2°C et 8°C).
6.5. Nature et contenu de l'emballage extérieur
Toutes les présentations peuvent ne pas être commercialisées.
6.6. Précautions particulières d’élimination et de manipulation
Pas d'exigences particulières pour l’élimination.
Tout médicament non utilisé ou déchet doit être éliminé conformément à la réglementation en vigueur.
7. TITULAIRE DE L’AUTORISATION DE MISE SUR LE MARCHE
1 RUE DE TURIN
69007 LYON
8. NUMERO(S) D’AUTORISATION DE MISE SUR LE MARCHE
· 34009 387 364 5 8 : flacon de 100 mL (verre brun) avec fermeture de sécurité enfant et seringue (PE/PP) pour administration orale graduée en kg. Boîte de 1.
9. DATE DE PREMIERE AUTORISATION/DE RENOUVELLEMENT DE L’AUTORISATION
[à compléter ultérieurement par le titulaire]
10. DATE DE MISE A JOUR DU TEXTE
[à compléter ultérieurement par le titulaire]
Sans objet.
12. INSTRUCTIONS POUR LA PREPARATION DES RADIOPHARMACEUTIQUES
Liste I.
ANSM - Mis à jour le : 09/07/2025
CEFPODOXIME VIATRIS ENFANTS ET NOURRISSONS 40 mg/5 mL, poudre pour suspension buvable
Cefpodoxime proxétil
· Gardez cette notice. Vous pourriez avoir besoin de la relire.
· Si vous avez d’autres questions, interrogez votre médecin, votre pharmacien ou votre infirmier/ère.
· Ce médicament vous a été personnellement prescrit. Ne le donnez pas à d’autres personnes. Il pourrait leur être nocif, même si les signes de leur maladie sont identiques aux vôtres.
· Si vous ressentez un quelconque effet indésirable, parlez-en à votre médecin, votre pharmacien ou votre infirmier/ère. Ceci s’applique aussi à tout effet indésirable qui ne serait pas mentionné dans cette notice. Voir rubrique 4.
2. Quelles sont les informations à connaître avant d'utiliser CEFPODOXIME VIATRIS ENFANTS ET NOURRISSONS 40 mg/5 mL, poudre pour suspension buvable ?
3. Comment utiliser CEFPODOXIME VIATRIS ENFANTS ET NOURRISSONS 40 mg/5 mL, poudre pour suspension buvable ?
4. Quels sont les effets indésirables éventuels ?
5. Comment conserver CEFPODOXIME VIATRIS ENFANTS ET NOURRISSONS 40 mg/5 mL, poudre pour suspension buvable ?
6. Contenu de l’emballage et autres informations.
1. QU’EST-CE QUE CEFPODOXIME VIATRIS ENFANTS ET NOURRISSONS 40 mg/5 mL, poudre pour suspension buvable ET DANS QUELS CAS EST-IL UTILISE ?
Classe pharmacothérapeutique : antibactériens à usage systémique, code ATC : J01DD13.
(Céphalosporines de 3ème génération)
Ce médicament est un antibiotique antibactérien de la famille des bêta-lactamines.
Ce médicament est indiqué chez l'enfant dans le traitement de certaines infections bactériennes à germes sensibles.
2. QUELLES SONT LES INFORMATIONS A CONNAITRE AVANT D’UTILISER CEFPODOXIME VIATRIS ENFANTS ET NOURRISSONS 40 mg/5 mL, poudre pour suspension buvable ?
N’utilisez jamais CEFPODOXIME VIATRIS ENFANTS ET NOURRISSONS 40 mg/5 mL, poudre pour suspension buvable :
· en cas d'allergie connue aux antibiotiques du groupe des céphalosporines.
Avertissements et précautions
Adressez-vous à votre médecin, pharmacien ou votre infirmier/ère avant d’utiliser CEFPODOXIME VIATRIS ENFANTS ET NOURRISSONS 40 mg/5 mL, poudre pour suspension buvable.
· Il existe une possibilité d'allergie (5 à 10 % des cas) chez les sujets allergiques aux pénicillines.
· Signalez à votre médecin toute allergie ou manifestations allergiques survenues lors de traitement par les antibiotiques du groupe des pénicillines.
· La survenue de diarrhée au cours d'un traitement antibiotique ne doit pas être traitée sans avis médical.
· En raison de la nécessité d'adapter le traitement, il est important de prévenir le médecin de toute maladie rénale.
· Chez le nourrisson de moins de 15 jours, il est recommandé de ne pas utiliser ce produit.
· Ce médicament peut provoquer une fausse réaction positive de certains examens de laboratoire (recherche de glucose dans les urines, test de Coombs).
· Comme pour l'ensemble des médicaments appartenant à cette classe d’antibiotiques (les bêta-lactamines), l'administration de ce médicament peut entraîner un risque d'encéphalopathie pouvant se traduire par des convulsions, une confusion, des troubles de la conscience, ou encore des mouvements anormaux, particulièrement en cas de surdosage ou en cas d’altération du fonctionnement du rein. Si de tels troubles apparaissent, consultez immédiatement votre médecin ou votre pharmacien (voir rubriques 3 et 4).
· Des réactions cutanées graves, dont le syndrome de Stevens-Johnson, la nécrolyse épidermique toxique, la réaction médicamenteuse avec éosinophilie et symptômes systémiques (DRESS), et la pustulose exanthématique généralisée aiguë (PEAG) ont été rapportées en association avec un traitement par cefpodoxime. Arrêtez la prise de cefpodoxime et consultez immédiatement un médecin si vous remarquez l’un des symptômes associés aux réactions cutanées graves décrites à la rubrique 4.
Enfants
Sans objet.
Autres médicaments et CEFPODOXIME VIATRIS ENFANTS ET NOURRISSONS 40 mg/5 mL, poudre pour suspension buvable
Prévenez en particulier votre médecin si vous prenez :
· des médicaments qui augmentent le pH gastrique (ranitidine, hydroxyde d’aluminium, bicarbonate de sodium) car cela conduit à une diminution de l’absorption de Cefpodoxime Viatris enfants et nourrisson 40 mg/5 mL, poudre pour suspension buvable ;
· des médicaments qui diminuent le pH gastrique (pentagastrine) car cela conduit à une augmentation de l’absorption de Cefpodoxime Viatris enfants et nourrisson 40 mg/5 mL, poudre pour suspension buvable ;
· des médicaments utilisés pour fluidifier le sang (anticoagulant) : leur activité peut être augmentée avec certains antibiotiques.
CEFPODOXIME VIATRIS ENFANTS ET NOURRISSONS 40 mg/5 mL, poudre pour suspension buvable avec des aliments et boissons
Sans objet.
Sans objet.
Conduite de véhicules et utilisation de machines
CEFPODOXIME VIATRIS ENFANTS ET NOURRISSONS 40 mg/5 mL, poudre pour suspension buvable contient du saccharose, de l’aspartam (E951), du benzoate de sodium et du sodium
Si votre médecin vous a informé(e) d'une intolérance à certains sucres, contactez-le avant de prendre ce médicament.
Ce médicament contient 25 mg d’aspartam pour 5 mL de suspension reconstituée équivalent à 5 mg/mL.
L’aspartam contient une source de phénylalanine. Peut être dangereux pour les personnes atteintes de phénylcétonurie (PCU), une maladie génétique rare caractérisée par l’accumulation de phénylalanine ne pouvant être éliminée correctement.
Ce médicament contient 10 mg de sel de benzoate pour 5 mL de suspension reconstituée équivalent à 2 mg/mL. Le sel de benzoate peut accroître le risque ictère (jaunissement de la peau et des yeux) chez les nouveau-nés (jusqu’à 4 semaines).
3. COMMENT UTILISER CEFPODOXIME VIATRIS ENFANTS ET NOURRISSONS 40 mg/5 mL, poudre pour suspension buvable ?
Veillez à toujours utiliser ce médicament en suivant exactement les indications de votre médecin ou pharmacien. Vérifiez auprès de votre médecin ou pharmacien en cas de doute.
Au-delà de 25 kg (200 mg/j), le comprimé à 100 mg peut être utilisé.
La posologie chez l’enfant varie selon l'âge et le poids.
A titre indicatif, la posologie usuelle chez l'enfant est de 8 mg par kg et par jour, sans dépasser la dose adulte (c’est à dire 200 mg/jour en cas d’angine et 400 mg/jour dans les autres indications), à prendre en 2 prises, à 12 heures d'intervalle.
|
La dose par prise est indiquée sur la seringue graduée pour administration orale, par les graduations correspondant au poids de l'enfant en kilogrammes (kg) (graduations de 0 à 25 kg). La dose par prise se lit donc directement. Ainsi, le point indiqué correspond à la dose pour une prise. La dose à administrer pour une prise est donc obtenue en tirant le piston jusqu’à la graduation correspondant au poids de l’enfant. Deux prises par jour sont nécessaires. Par exemple, la graduation n°12 correspond à la dose à administrer par prise pour un enfant de 12 kg, et ce, deux fois par jour. |
Si votre enfant a une maladie des reins, votre médecin adaptera la dose à votre situation.
Mode et voie d'administration
Ce médicament s’administre par voie orale avec la seringue graduée en kilogrammes qui correspondent au poids de l’enfant (les graduations se lisent directement sur la seringue).
Il doit être pris pendant les repas.
A lire attentivement avant ouverture du flacon.
Flacon (verre brun) avec bouchon contenant une capsule déshydratante
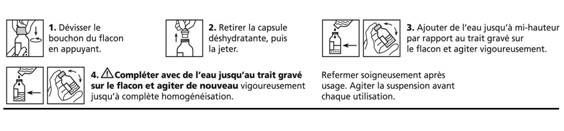
· Dévisser le bouchon du flacon en appuyant.
· Retirer la capsule déshydratante, puis la jeter.
· Ajouter de l'eau jusqu'à mi-hauteur par rapport au trait gravé sur le flacon. Agiter vigoureusement.
· Compléter avec de l'eau jusqu'au trait gravé sur le flacon et agiter de nouveau vigoureusement jusqu'à complète homogénéisation.
· Refermer soigneusement après usage. Rincer puis sécher la seringue pour administration. Agiter la suspension avant chaque utilisation.
Utiliser la seringue graduée pour l'administration chez l'enfant.
Attention, celle-ci ne doit pas être réutilisée pour un autre médicament, la graduation de la seringue pour administration orale étant spécifique à ce produit.
Fréquence d'administration
Deux prises par jour, au cours des repas.
Durée du traitement
Pour être efficace, cet antibiotique doit être utilisé régulièrement aux doses prescrites, et aussi longtemps que votre médecin vous l'aura conseillé.
La disparition de la fièvre, ou de tout autre symptôme, ne signifie pas que vous êtes complètement guéri(e).
L'éventuelle impression de fatigue n'est pas due au traitement antibiotique, mais à l'infection elle-même. Le fait de réduire ou de suspendre votre traitement serait sans effet sur cette impression et retarderait votre guérison.
Cas particulier : la durée du traitement de certaines angines est de 5 jours.
Si vous avez utilisé plus de CEFPODOXIME VIATRIS ENFANTS ET NOURRISSONS 40 mg/5 mL, poudre pour suspension buvable que vous n’auriez dû
Comme pour l'ensemble des médicaments appartenant à cette classe d’antibiotiques (les bêta-lactamines), l'administration de ce médicament, en particulier en cas de surdosage, peut entraîner un risque d'encéphalopathie pouvant se traduire par des convulsions, une confusion, des troubles de la conscience, ou encore des mouvements anormaux. Si de tels troubles apparaissent, consultez immédiatement votre médecin ou votre pharmacien (voir rubriques 2 et 4).
Si vous oubliez d’utiliser CEFPODOXIME VIATRIS ENFANTS ET NOURRISSONS 40 mg/5 mL, poudre pour suspension buvable
Si vous arrêtez d’utiliser CEFPODOXIME VIATRIS ENFANTS ET NOURRISSONS 40 mg/5 mL, poudre pour suspension buvable
Sans objet.
Si vous avez d’autres questions sur l’utilisation de ce médicament, demandez plus d’informations à votre médecin, à votre pharmacien ou à votre infirmier/ère.
4. QUELS SONT LES EFFETS INDESIRABLES EVENTUELS ?
Arrêtez de prendre cefpodoxime et consultez immédiatement un médecin si vous présentez l’un des symptômes suivants :
· Éruption cutanée étendue, température corporelle élevée et gonflement des ganglions lymphatiques (syndrome DRESS ou syndrome d’hypersensibilité médicamenteuse).
· Éruption cutanée étendue rouge et squameuse s’accompagnant de bosses sous la peau et de cloques accompagnées de fièvre. Les symptômes apparaissent habituellement en début de traitement (pustulose exanthématique aiguë généralisée).
Très fréquents (peuvent toucher plus d’1 patient sur 10)
· Manifestations digestives : douleurs au ventre, diarrhée.
· Maux de tête.
Fréquents (peuvent toucher jusqu’à 1 patient sur 10)
· Bourdonnements dans les oreilles (acouphènes).
· Manifestations digestives : nausées, vomissements.
· Augmentation des enzymes du foie (transaminases et phosphatases alcalines).
· Sensations de vertiges.
· Manifestations cutanées : éruptions cutanées, démangeaisons, urticaire.
Peu fréquents (peuvent toucher jusqu’à 1 patient sur 100)
· Diminution du taux de certains globules blancs (neutropénie).
· Inflammation de l'intestin (entérocolite).
· Réactions allergiques pouvant être sévères (réactions anaphylactiques), difficultés soudaines à respirer (bronchospasme).
Rares (peuvent toucher 1 à 10 patients sur 10 000)
· Faible augmentation de l’urée et de la créatinine dans le sang.
Fréquence indéterminée (ne peut pas être évaluée à partir des données disponibles)
· Manifestations hématologiques : diminution du taux de plaquettes, diminution du taux de globules rouges (anémie), chute importante du taux de certains globules blancs (agranulocytose), augmentation du taux de certains globules blancs (éosinophilie).
· Saignement anal, maladie du gros intestin caractérisée par l'expulsion de fausses membranes ou de glaires, diarrhée et douleurs au ventre (colite pseudomembraneuse), diarrhée due à une bactérie Clostridioides difficile.
· Malaise, fatigue.
· Augmentation de certains éléments du sang (bilirubine), atteinte du foie.
· Manifestations allergiques : manifestation allergique pouvant conduire à un arrêt circulatoire (choc allergique), brusque gonflement du visage et du cou d’origine allergique (œdème de Quincke).
· Aggravation de l’infection (surinfection).
· Sensation de picotements dans les mains et les pieds (paresthésies).
· Manifestations cutanées : éruption de taches pourpres ne s'effaçant pas à la vitropression (purpura), éruption bulleuse localisée (dermatite bulleuse), maladie de la peau sous forme de rougeur (érythème polymorphe), décollement de la peau pouvant rapidement s'étendre de façon très grave à tout le corps (syndrome de Stevens-Johnson et syndrome de Lyell).
· Atteinte de la fonction rénale (observée avec des antibiotiques appartenant à la même classe thérapeutique que le cefpodoxime, notamment quand ils étaient associés à d’autres médicaments (aminoglycosides et/ou des diurétiques puissants)).
· Troubles neurologiques graves appelés encéphalopathies à type de convulsions, de confusion, de troubles de la conscience, ou encore de mouvements anormaux en particulier en cas de surdosage ou d’altération du fonctionnement du rein (voir rubriques 2 et 3).
Déclaration des effets secondaires
Si vous ressentez un quelconque effet indésirable, parlez-en à votre médecin, votre pharmacien ou à votre infirmier/ère. Ceci s’applique aussi à tout effet indésirable qui ne serait pas mentionné dans cette notice. Vous pouvez également déclarer les effets indésirables directement via le système national de déclaration : Agence nationale de sécurité du médicament et des produits de santé (ANSM) et réseau des Centres Régionaux de Pharmacovigilance - Site internet : https://signalement.social-sante.gouv.fr/.
En signalant les effets indésirables, vous contribuez à fournir davantage d’informations sur la sécurité du médicament.
5. COMMENT CONSERVER CEFPODOXIME VIATRIS ENFANTS ET NOURRISSONS 40 mg/5 mL, poudre pour suspension buvable ?
Tenir ce médicament hors de la vue et de la portée des enfants.
N’utilisez pas ce médicament après la date de péremption indiquée sur le flacon après EXP. La date de péremption fait référence au dernier jour de ce mois.
A conserver à une température ne dépassant pas 25°C.
Après reconstitution : la suspension se conserve au maximum 14 jours au réfrigérateur (entre 2°C et 8°C).
Ne jetez aucun médicament au tout-à-l’égout ou avec les ordures ménagères. Demandez à votre pharmacien d’éliminer les médicaments que vous n’utilisez plus. Ces mesures contribueront à protéger l’environnement.
6. CONTENU DE L’EMBALLAGE ET AUTRES INFORMATIONS
cefpodoxime................................................................................................................... 40 mg
sous forme de cefpodoxime proxétil
pour 5 mL de suspension reconstituée.
· Les autres composants sont :
silice colloïdale hydratée, talc, trioléate de sorbitan, benzoate de sodium, chlorure de sodium, acide citrique anhydre, aspartam (E951), galactomannane du guar, arôme citron, arôme orange, oxyde de fer jaune, saccharose.
Ce médicament se présente sous forme de poudre pour suspension buvable.
Flacon de 50 mL ou 100 mL avec fermeture de sécurité enfant et seringue pour administration orale graduée.
Titulaire de l’autorisation de mise sur le marché
VIATRIS SANTE
1 RUE DE TURIN
69007 LYON
Exploitant de l’autorisation de mise sur le marché
1 RUE DE TURIN
69007 LYON
BIOCHEMIESTRASSE 10
6250 KUNDL
AUTRICHE
Noms du médicament dans les Etats membres de l'Espace Economique Européen
La dernière date à laquelle cette notice a été révisée est :
[à compléter ultérieurement par le titulaire]
Des informations détaillées sur ce médicament sont disponibles sur le site Internet de l’ANSM (France).
CONSEILS / EDUCATION SANITAIRE
QUE SAVOIR SUR LES ANTIBIOTIQUES ?
Les antibiotiques sont efficaces pour combattre les infections dues aux bactéries. Ils ne sont pas efficaces contre les infections dues aux virus.
Aussi, votre médecin a choisi de vous prescrire cet antibiotique parce qu’il convient précisément à votre cas et à votre maladie actuelle.
Les bactéries ont la capacité de survivre ou de se reproduire malgré l’action d’un antibiotique. Ce phénomène est appelé résistance : il rend certains traitements antibiotiques inactifs.
La résistance s’accroît par l’usage abusif ou inapproprié des antibiotiques.
Vous risquez de favoriser l’apparition de bactéries résistantes et donc de retarder votre guérison ou même de rendre inactif ce médicament, si vous ne respectez pas :
· la dose à prendre ;
· les moments de prise ;
· et la durée de traitement.
En conséquence, pour préserver l’efficacité de ce médicament
1- N’utilisez un antibiotique que lorsque votre médecin vous l’a prescrit.
2- Respectez strictement votre ordonnance.
3- Ne réutilisez pas un antibiotique sans prescription médicale même si vous pensez combattre une maladie apparemment semblable.
4- Ne donnez jamais votre antibiotique à une autre personne, il n’est peut-être pas adapté à sa maladie.
5- Une fois votre traitement terminé, rapportez à votre pharmacien toutes les boîtes entamées pour une destruction correcte et appropriée de ce médicament.