Dernière mise à jour le 03/08/2026
ALPHAGAN 0,2 % (2mg/ml), collyre en solution
Indications thérapeutiques
Classe pharmacothérapeutique - code ATC : S01EA 05
Il peut être utilisé soit seul quand un collyre bêtabloquant est contre-indiqué, soit en association avec un autre collyre quand celui-ci n'a pas assez réduit la pression à l'intérieur de l'œil. Il est utilisé dans le traitement du glaucome à angle ouvert ou de l'hypertension oculaire.
Présentations
> flacon(s) compte-gouttes polyéthylène de 5 ml
Code CIP : 345 473-0 ou 34009 345 473 0 0
Déclaration de commercialisation : 01/09/1998
Cette présentation est agréée aux collectivités
- Prix hors honoraire de dispensation : 5,75 €
- Honoraire de dispensation : 1,02 €
- Prix honoraire compris : 6,77 €
- Taux de remboursement :65%
Service médical rendu (SMR)
Les libellés affichés ci-dessous ne sont que des résumés ou extraits issus des avis rendus par la Commission de la Transparence. Seul l'avis complet de la Commission de la Transparence fait référence.
Cet avis est consultable à partir du lien `Avis du jj/mm/aaaa` ou encore sur demande auprès de la HAS (plus d'informations dans l'aide). Les avis et synthèses d'avis contiennent un paragraphe sur la place du médicament dans la stratégie thérapeutique.
| Valeur du SMR | Avis | Motif de l'évaluation | Résumé de l'avis |
|---|---|---|---|
| Important | Avis du 20/04/2016 | Renouvellement d'inscription (CT) | Le service médical rendu par ALPHAGAN reste important dans les indications de l’AMM. |
Amélioration du service médical rendu (ASMR)
Pas d'ASMR disponible pour ce médicament (plus d'informations dans l\'aide)
ANSM - Mis à jour le : 19/03/2026
ALPHAGAN 0,2 % (2 mg/ml), collyre en solution
2. COMPOSITION QUALITATIVE ET QUANTITATIVE
Un ml de solution contient 2,0 mg de tartrate de brimonidine, équivalent à 1,3 mg de brimonidine.
Excipient(s) à effet notoire : chlorure de benzalkonium 0,05 mg/ml.
Pour la liste complète des excipients, voir rubrique 6.1.
Collyre en solution (collyre).
Solution limpide, jaune-vert à jaune-vert pâle.
4.1. Indications thérapeutiques
· En monothérapie chez les patients présentant une contre-indication aux bêtabloquants à usage local.
· En association à d'autres traitements diminuant la pression intraoculaire dans le cas où une monothérapie ne permet pas d'atteindre la PIO cible (voir rubrique 5.1).
4.2. Posologie et mode d'administration
Dose recommandée chez l'adulte (y compris le patient âgé)
La posologie recommandée est d'une goutte d'ALPHAGAN deux fois par jour dans l'œil ou les yeux atteint(s), les deux instillations devant être espacées d'environ 12 heures. L'utilisation du collyre chez les patients âgés ne requiert aucune adaptation posologique.
Utilisation en cas d'insuffisance rénale ou hépatique
ALPHAGAN n'a pas été étudié chez les patients ayant une insuffisance rénale ou hépatique (voir rubrique 4.4).
Population pédiatrique
Il n'a pas été conduit d'études cliniques chez les adolescents (12 à 17 ans).
ALPHAGAN n'est pas recommandé chez les enfants de moins de 12 ans et est contre indiqué chez les nouveau-nés et les nourrissons (moins de 2 ans) (voir rubriques 4.3, 4.4, 4.9). Il est connu que des réactions indésirables graves peuvent se produire chez les nouveau-nés. La sécurité et l’efficacité d’ALPHAGAN chez les enfants âgés de 2 à 12 ans n’ont pas été établies..
Mode d’administration
Comme c'est le cas avec tous les collyres, afin de réduire une possible absorption systémique, il est recommandé de comprimer le sac lacrymal au niveau du canthus interne (occlusion ponctuelle) pendant une minute. Ceci doit être fait immédiatement après l'instillation de chaque goutte. Cela pourrait entrainer une diminution des effets indésirables systémiques et une augmentation de l'activité locale. Pour éviter la contamination de l'œil ou du collyre, ne laissez pas l'embout du flacon entrer en contact avec une quelconque surface.
En cas d'utilisation de plusieurs produits ophtalmiques à usage local, les instillations des différents produits doivent être espacées de 5 à 15 minutes.
· Hypersensibilité à la substance active ou à l’un des excipients mentionnés à la rubrique 6.1.
· Nouveau-nés et nourrissons (moins de 2 ans) (voir rubrique 4.8).
· Patients recevant un traitement inhibiteur de la monoamine oxydase (IMAO) et patients sous antidépresseurs affectant la transmission noradrénergique (par exemple antidépresseurs tricycliques et miansérine).
4.4. Mises en garde spéciales et précautions d'emploi
Les enfants de deux ans ou plus, en particulier ceux âgés de 2 à 7 ans et/ou de poids inférieur ou égal à 20 kg, doivent être traités avec précaution et étroitement surveillés en raison de l’incidence élevée et de la sévérité de la somnolence (voir rubrique 4.8).
Troubles cardiaques
Une attention particulière est nécessaire chez les patients atteints d'une maladie cardio-vasculaire sévère ou instable et incontrôlée.
Troubles oculaires
Lors des essais cliniques, certains patients (12,7 %) ont présenté une réaction oculaire de type allergique avec ALPHAGAN (voir rubrique 4.8). En cas de réactions allergiques, le traitement avec ALPHAGAN doit être arrêté.
Des réactions d’hypersensibilité oculaires retardées ont été rapportées avec ALPHAGAN 0,2 %, certaines étaient associées à une augmentation de la pression intra-oculaire.
Troubles vasculaires
ALPHAGAN doit être utilisé avec précaution chez les patients souffrant de dépression, d'insuffisance cérébrale ou coronarienne, de phénomène de Raynaud, d'hypotension orthostatique ou de thromboangéite oblitérante.
Insuffisance rénale et hépatique
ALPHAGAN n’a pas été étudié chez les patients présentant une insuffisance hépatique ou rénale. Une attention particulière est nécessaire en cas de traitement de ces patients.
Chlorure de benzalkonium
Le chlorure de benzalkonium, agent conservateur présent dans ALPHAGAN, est susceptible d'entraîner une irritation oculaire, des symptômes du syndrome de l’œil sec et peut endommager le film lacrymal et la surface cornéenne. Les patients doivent retirer les lentilles de contact avant l’utilisation du collyre et attendre au moins 15 minutes avant de les remettre. Le chlorure de benzalkonium est susceptible de modifier la couleur des lentilles de contact souples. Les patients doivent éviter la mise en contact avec les lentilles de contact souples.
ALPHAGAN doit être utilisé avec précaution chez les patients souffrant du syndrome de l’œil sec et chez les patients dont l’état de la cornée pourrait être endommagé. Les patients doivent faire l’objet d’un suivi en cas d’utilisation prolongée.
4.5. Interactions avec d'autres médicaments et autres formes d'interactions
Bien qu’il n’y ait pas d'étude spécifique des interactions médicamenteuses avec ALPHAGAN, la possibilité d'un effet additif ou potentialisateur en cas de prise de dépresseurs du système nerveux central (alcool, barbituriques, opiacés, sédatifs ou anesthésiques) doit être prise en compte.
On ne dispose d'aucune donnée concernant le taux de catécholamines circulantes après l’instillation d'ALPHAGAN. En conséquence, il convient de faire preuve de vigilance avec les patients prenant des médicaments risquant d'affecter le métabolisme et la fixation des amines circulantes (ex : chlorpromazine, méthylphénidate, réserpine).
Après l’instillation d'ALPHAGAN, on a observé chez certains patients des diminutions de la tension artérielle non significatives du point de vue clinique. Il convient d’être vigilant en cas de prise concomitante d’ALPHAGAN et d'antihypertenseurs et/ou de glucosides cardiotoniques.
Une attention particulière est recommandée lors de l’instauration (ou de la modification de la posologie) d’un traitement systémique concomitant (quelle que soit la forme pharmaceutique) susceptible d'interagir avec des agonistes α-adrénergiques ou d’interférer avec leur activité, tels que les agonistes ou antagonistes des récepteurs adrénergiques (ex : isoprénaline, prazosine).
4.6. Fertilité, grossesse et allaitement
Grossesse
La sécurité d'utilisation chez la femme durant la grossesse n'a pas été établie. Lors des études chez l’animal, le tartrate de brimonidine n’a pas provoqué d’effet tératogène. Chez le lapin, le tartrate de brimonidine, à des concentrations plasmatiques supérieures à celles obtenues lors du traitement chez l’homme, a provoqué des pertes plus importantes lors de la période de préimplantation et une réduction de la croissance postnatale. ALPHAGAN ne doit être utilisé pendant la grossesse que lorsque les bénéfices potentiels pour la mère dépassent les risques potentiels encourus par le fœtus. Pour réduire l’absorption systémique, voir rubrique 4.2.
Il n’a pas été établi si la brimonidine est excrétée dans le lait maternel. Le produit est excrété dans le lait des rates allaitantes. ALPHAGAN ne doit pas être utilisé pendant l’allaitement.
4.7. Effets sur l'aptitude à conduire des véhicules et à utiliser des machines
Les effets indésirables les plus communément rapportés sont sécheresse buccale, hyperhémie oculaire, sensations de brûlure ou sensations de picotement, survenant chez 25,9 à 31,2 % des patients. Ils sont généralement transitoires et rarement assez sévères pour nécessiter l’arrêt du traitement.
Dans les études cliniques, des symptômes de réactions allergiques oculaires sont apparus chez 12,7 % des patients (entraînant l’arrêt du traitement chez 11,5 % des patients), leur apparition survenant entre 3 et 9 mois chez la majorité des patients.
Dans chaque catégorie, les effets indésirables sont présentés dans l’ordre décroissant de gravité. Pour classer la fréquence des effets indésirables les terminologies suivantes ont été utilisées :
Très fréquents (³1/10) ;
Fréquents (³1/100, <1/10) ;
Peu fréquents (³1/1 000, <1/100) ;
Rares (³1/10 000, <1/1 000) ;
Très rares (<1/10 000) ;
Fréquence indéterminée (ne peut être estimée sur la base des données disponibles).
Tableau 1 : Tabulation de la liste des effets indésirables
|
Classe de système d’organe |
Fréquence |
Effets indésirables |
|
Affections du système immunitaire
|
Peu fréquent |
Réactions allergiques systémiques |
|
Affections psychiatriques |
Peu fréquent |
Dépression |
|
|
Très rare |
Insomnie |
|
Affections du système nerveux |
Très fréquents |
Maux de tête |
|
|
Fréquents |
Vertiges Goût anormal |
|
Affections oculaires |
Très fréquents |
Hyperhémie oculaire Brûlures et picotements des yeux Vision floue Sensation de corps étrangers Follicules conjonctivaux Prurit Réactions oculaires allergiques (incluant blépharite allergique, blépharoconjonctivite allergique, conjonctivite allergique, réaction allergique et conjonctivite folliculaire). |
|
|
Fréquents |
Hyperhémie de la paupière Œdème de la paupière Blépharite Œdème conjonctival Sécrétions conjonctivales Douleur oculaire Larmoiement Photophobie Erythème de la paupière Erosion et coloration cornéenne Sécheresse oculaire Blanchiment conjonctival Vision anormale Conjonctivite Irritation oculaire Papilles conjonctivales |
|
|
Très rares |
Iritis Myosis |
|
Affections cardiaques |
Peu fréquents |
Palpitations/arythmies (incluant bradycardie et tachycardie) |
|
Affections vasculaires
|
Très rares |
Hypertension Hypotension |
|
Affections respiratoires, thoraciques et médiastinales |
Fréquents |
Symptômes des voies respiratoires supérieures |
|
|
Peu fréquent |
Sécheresse nasale |
|
|
Rare |
Dyspnée |
|
Affections gastro-intestinales |
Très fréquent |
Sécheresse buccale |
|
|
Fréquent |
Symptômes gastro-intestinaux |
|
Troubles généraux et anomalies au site d'administration |
Très fréquent |
Fatigue/somnolence |
|
|
Fréquent |
Asthénie |
.
Les effets indésirables suivants ont été identifiés après la commercialisation d’ALPHAGAN. Sachant qu’ils ont été rapportés sur la base des notifications spontanées dans une population de taille inconnue, une estimation de leur fréquence n’a pu être effectuée.
Tableau 2 : Effets indésirables identifiés lors de la surveillance post-commercialisation
|
Classe de système d’organe |
Effets indésirables |
|
|
Affections oculaires |
Iritis Iridocyclite (uvéite antérieure) Myosis Conjonctivite Prurit de la paupière |
|
|
Affections de la peau et du tissus sous-cutané |
Hypersensibilité Réaction cutanée incluant érythème, œdème facial, prurit, rash et vasodilatation |
|
|
Affections cardiaque |
Palpitations/arythmies (incluant bradycardie ou tachycardie) |
|
|
Affections psychiatrique |
Dépression |
|
|
Affections vasculaire |
Hypotension Syncope |
|
.
Population pédiatrique
Dans des cas où la brimonidine a été utilisée dans le cadre du traitement médical d’un glaucome congénital, des symptômes de surdosage à la brimonidine tels que perte de conscience, léthargie, somnolence, hypotension, hypotonie, bradycardie, hypothermie, cyanose, pâleur, dépression respiratoire et apnée ont été rapportés chez des nouveau-nés et nourrissons qui avaient reçu de la brimonidine (voir rubrique 4.3).
Dans une étude de phase III d’une durée de 3 mois, chez des enfants âgés de 2 à 7 ans, présentant un glaucome insuffisamment contrôlé sous bêta-bloquants, une prévalence importante (55%) de somnolence a été rapportée avec ALPHAGAN utilisé en association. Chez 8% des enfants, celle-ci était sévère et a conduit à un arrêt du traitement dans 13% des cas. L’incidence de la somnolence a diminué avec l’augmentation de l’âge, en étant la plus faible dans le groupe de sujets âgés de 7 ans (25%) mais elle était davantage influencée par le poids, apparaissant plus fréquemment chez les enfants d’un poids inférieur ou égal à 20 kg (63%) que chez ceux d’un poids supérieur à 20 kg (25%) (voir rubrique 4.4).
Déclaration des effets indésirables suspectés
La déclaration des effets indésirables suspectés après autorisation du médicament est importante. Elle permet une surveillance continue du rapport bénéfice/risque du médicament. Les professionnels de santé déclarent tout effet indésirable suspecté via le système national de déclaration : Agence nationale de sécurité du médicament et des produits de santé (ANSM) et réseau des Centres Régionaux de Pharmacovigilance - Site internet : https://signalement.social-sante.gouv.fr/.
Surdosage ophtalmique (adultes)
Dans les cas reçus, les événements rapportés ont été généralement listés parmi les effets indésirables.
Surdosage systémique résultant d'une ingestion accidentelle (adultes)
Il existe très peu d’information concernant l’ingestion accidentelle de brimonidine chez les adultes. Le seul effet indésirable rapporté à ce jour est l’hypotension. Cet épisode hypotensif a été suivi d’une hypertension de rebond.
Le traitement d’un surdosage par voie orale repose sur des mesures symptomatiques et de support. Les voies respiratoires doivent être maintenues dégagées.
Les cas rapportés de surdosage oral avec d'autres alpha-2-stimulants ont été associés à des symptômes tels que : hypotension, asthénie, vomissements, léthargie, sédation, bradycardie, arythmies, myosis, apnée, hypotonie, hypothermie, dépression respiratoire et convulsions.
Population pédiatrique
Des notifications d’effets indésirables graves consécutifs à l’ingestion accidentelle d’ALPHAGAN par des enfants ont été publiées ou rapportées. Les sujets ont présenté des symptômes de dépression du système nerveux central (SNC), typiquement coma temporaire ou faible niveau de conscience, léthargie, somnolence, hypotonie, bradycardie, hypothermie, pâleur, dépression respiratoire et apnée, ayant nécessité une admission en soins intensifs avec intubation si nécessaire. Un rétablissement complet a été rapporté chez tous les sujets, en général dans les 6 à 24 heures.
5. PROPRIETES PHARMACOLOGIQUES
5.1. Propriétés pharmacodynamiques
Classe pharmacothérapeutique : Sympathomimétiques antiglaucomateux, code ATC : S01EA 05
La brimonidine est un agoniste des récepteurs alpha-2 adrénergiques 1000 fois plus sélectif pour les récepteurs alpha-2 adrénergiques que pour les récepteurs alpha-1 adrénergiques.
Du fait de cette sélectivité, on observe aucune mydriase ni vasoconstriction des microvaisseaux, associée aux xénogreffes de rétine humaine.
Une administration locale de tartrate de brimonidine diminue la pression intra-oculaire (PIO) chez l'homme et n'affecte que de façon minime les paramètres cardio-vasculaires ou pulmonaires.
Des données restreintes sur des patients atteints d’asthme bronchique n’ont pas mis en évidence d’effet néfaste.
ALPHAGAN a un délai d'action rapide, l'effet hypotenseur oculaire maximum étant observé deux heures après l'administration. Dans 2 études d’un an, les diminutions moyennes de la PIO obtenues avec ALPHAGAN étaient approximativement comprises entre 4 et 6 mmHg.
Des études fluorophotométriques menées chez l’animal et chez l’homme suggèrent que le tartrate de brimonidine possède un double mécanisme d'action. On pense qu'ALPHAGAN diminue la PIO en réduisant la formation d'humeur aqueuse et en augmentant l'écoulement de l'humeur aqueuse par la voie uvéosclérale.
Des études cliniques montrent qu’ALPHAGAN est efficace en association aux bêtabloquants à usage local. Des études plus courtes suggèrent également qu’ALPHAGAN a un effet additionnel cliniquement significatif en association au travoprost (6 semaines) et au latanoprost (3 mois).
5.2. Propriétés pharmacocinétiques
Après une administration oculaire d'une solution à 0,2 % deux fois par jour pendant 10 jours, les concentrations plasmatiques étaient faibles (Cmax moyenne égale à 0,06 ng/ml). On a observé une légère accumulation dans le sang après de multiples instillations (2 fois par jour pendant 10 jours). L'aire sous la courbe « concentration plasmatique/temps » sur 12 heures à l’état d’équilibre (ASC0-12h) était égale à 0,31 ng.h/ml, par rapport à 0,23 ng.h/ml après la première dose. La demi-vie apparente moyenne dans la circulation générale était d'environ 3 heures chez l'homme après administration locale.
Chez l'homme, le taux de fixation aux protéines plasmatiques de la brimonidine après une administration locale est d'environ 29%.
La brimonidine se lie de façon réversible à la mélanine dans les tissus oculaires, in vitro et in vivo. Après 2 semaines d’instillations oculaires, les concentrations de brimonidine dans l’iris, le corps ciliaire et la choroïde-rétine, étaient 3 à 17 fois supérieures à celles observées après instillation unique. Il n’y a pas d’accumulation en l’absence de mélanine.
L’importance de la liaison avec la mélanine chez l’homme est mal connue. Cependant, aucun effet oculaire néfaste n’a été trouvé lors de l’examen biomicroscopique des yeux de patients traités avec ALPHAGAN pendant des périodes allant jusqu’à un an ; de même, aucune toxicité oculaire significative n’a été trouvée durant une étude de sécurité oculaire d’un an chez le singe recevant environ quatre fois la dose recommandée de tartrate de brimonidine.
Après administration orale chez l’homme, la brimonidine est bien absorbée et rapidement éliminée. La plus grande partie de la dose (environ 75 %) a été excrétée dans l’urine sous forme de métabolites en 5 jours ; moins de 5 % du médicament non métabolisé a été détecté dans l’urine sur la base d'une analyse semi-quantitative par chromatographie sur couche mince. Des études in vitro, utilisant du foie animal et humain, montrent que le métabolisme fait largement intervenir l’aldéhyde oxydase et le cytochrome P450. En conséquence, l’élimination systémique semble être due en priorité à un métabolisme hépatique.
Profil cinétique :
Après une administration locale unique à des concentrations 0,08 %, 0,2 % et 0,5 %, aucun écart significatif de la linéarité de la cinétique d’absorption (mesurée par les Cmax plasmatiques et les ASC) n’est observé.
Caractéristiques chez les patients âgés
Chez les patients âgés (sujets de 65 ans ou plus), la Cmax, l'aire sous la courbe (ASC) et la demi-vie apparente de la brimonidine sont similaires, après une instillation unique, à celles observées chez les adultes jeunes, ce qui indique que l'âge n'influe pas sur l'absorption et l'élimination systémiques du produit.
D’après les données d’une étude clinique de 3 mois qui incluait des patients âgés, l’exposition systémique à la brimonidine a été très faible.
5.3. Données de sécurité préclinique
· 2 ans pour les flacons de 2,5 ml
· 3 ans pour les flacons de 5 ml et 10 ml
Après ouverture :
· 28 jours
6.4. Précautions particulières de conservation
A conserver à une température ne dépassant pas 25°C.
6.5. Nature et contenu de l'emballage extérieur
Flacon en polyéthylène de basse densité blanc équipé d'un compte-gouttes de 35µl. Le système de fermeture est, soit un bouchon traditionnel, en polystyrène, soit un bouchon d'observance (C-Cap).
Flacon de 2,5 ml, 5 ml et 10 ml, par boîte de 1,3 ou 6 unités.
Toutes les présentations peuvent ne pas être commercialisées.
6.6. Précautions particulières d’élimination et de manipulation
Pas d’exigences particulières.
7. TITULAIRE DE L’AUTORISATION DE MISE SUR LE MARCHE
5/13 BOULEVARD DE LA REPUBLIQUE
92100 BOULOGNE-BILLANCOURT
8. NUMERO(S) D’AUTORISATION DE MISE SUR LE MARCHE
· 34009 345 472 4 9 : 2,5 ml, en flacon compte-gouttes (PE)
· 34009 345 473 0 0 : 5 ml, en flacon compte-gouttes (PE)
· 34009 345 474 7 8 : 10 ml, en flacon compte-gouttes (PE)
9. DATE DE PREMIERE AUTORISATION/DE RENOUVELLEMENT DE L’AUTORISATION
[à compléter ultérieurement par le titulaire]
Date de première autorisation : {JJ mois AAAA}
10. DATE DE MISE A JOUR DU TEXTE
[À compléter ultérieurement par le titulaire]
{JJ mois AAAA}
Sans objet.
12. INSTRUCTIONS POUR LA PREPARATION DES RADIOPHARMACEUTIQUES
Liste I
ANSM - Mis à jour le : 19/03/2026
ALPHAGAN 0,2 % (2 mg/ml), collyre en solution
Tartrate de brimonidine
· Gardez cette notice. Vous pourriez avoir besoin de la relire.
· Si vous avez d’autres questions, interrogez votre médecin ou votre pharmacien.
· Ce médicament vous a été personnellement prescrit. Ne le donnez pas à d’autres personnes. Il pourrait leur être nocif, même si les signes de leur maladie sont identiques aux vôtres.
· Si vous ressentez un quelconque effet indésirable, parlez-en à votre médecin ou votre pharmacien. Ceci s’applique aussi à tout effet indésirable qui ne serait pas mentionné dans cette notice. Voir rubrique 4.
1. Qu'est-ce que ALPHAGAN 0,2 % (2 mg/ml), collyre en solution et dans quels cas est-il utilisé ?
2. Quelles sont les informations à connaître avant d'utiliser ALPHAGAN 0,2 % (2 mg/ml), collyre en solution ?
3. Comment utiliser ALPHAGAN 0,2 % (2 mg/ml), collyre en solution ?
4. Quels sont les effets indésirables éventuels ?
5. Comment conserver ALPHAGAN 0,2 % (2 mg/ml), collyre en solution ?
6. Contenu de l’emballage et autres informations.
1. QU’EST-CE QUE ALPHAGAN 0,2 % (2 mg/ml), collyre en solution ET DANS QUELS CAS EST-IL UTILISE ?
Classe pharmacothérapeutique - code ATC : S01EA 05
Il peut être utilisé soit seul quand un collyre bêtabloquant est contre-indiqué, soit en association avec un autre collyre quand celui-ci n'a pas assez réduit la pression à l'intérieur de l'œil. Il est utilisé dans le traitement du glaucome à angle ouvert ou de l'hypertension oculaire.
2. QUELLES SONT LES INFORMATIONS A CONNAITRE AVANT D’UTILISER ALPHAGAN 0,2 % (2 mg/ml), collyre en solution ?
· si vous êtes allergique au tartrate de brimonidine ou à l’un des autres composants contenus dans ce médicament, mentionnés dans la rubrique 6.
· si vous prenez des inhibiteurs de la monoamine oxydase (IMAO), ou certains autres antidépresseurs. Vous devez informer votre médecin si vous prenez un médicament antidépresseur.
· si vous allaitez.
· chez les nouveau-nés et les nourrissons (de la naissance jusqu'à 2ans).
Avertissements et précautions
Adressez-vous à votre médecin avant d’utiliser ALPHAGAN.
· Si vous souffrez ou avez souffert de dépression, d'une réduction de vos capacités mentales, d'une réduction de la circulation sanguine au niveau du cerveau, de problèmes cardiaques, de troubles circulatoires des membres, ou de troubles de la tension artérielle.
· Si vous avez ou avez eu des maladies du foie ou des reins.
Enfants et adolescents
L’utilisation d’ALPHAGAN n'est pas recommandée chez les enfants âgés de 2 à 12 ans.
ALPHAGAN ne doit généralement pas être utilisé chez les adolescents âgés de 12 à 17 ans car aucune étude clinique n’a été menée dans ce groupe d’âge.
Autres médicaments et ALPHAGAN
Informez votre médecin ou pharmacien si vous utilisez, avez récemment utilisé ou pourriez utiliser tout autre médicament.
Indiquez à votre médecin si vous prenez l'un des médicaments suivants :
· Antidouleurs, sédatifs, opiacés, barbituriques ou si vous consommez régulièrement de l'alcool.
· Anesthésiques.
· Traitement pour le cœur ou pour réduire la tension artérielle.
· Médicament qui affecte le métabolisme tel que la chlorpromazine, le méthylphénidate et la réserpine.
· Médicament qui agit sur les mêmes récepteurs qu'ALPHAGAN par exemple : isoprénaline et prazosine.
· Inhibiteurs de la monoamine oxydase (IMAO) et d'autres antidépresseurs.
· Tout médicament, même s'il ne s'agit pas d'un médicament destiné à l'œil.
· Ou si la posologie d'un de vos médicaments actuels est changée.
Ceci peut affecter votre traitement avec ALPHAGAN
ALPHAGAN avec des aliments, boissons et de l’alcool
Sans objet.
Grossesse, allaitement et fertilité
Si vous êtes enceinte ou que vous allaitez, si vous pensez être enceinte ou planifiez une grossesse, demandez conseil à votre médecin ou pharmacien avant de prendre ce médicament.
N’utilisez pas ALPHAGAN si vous êtes enceinte sauf si votre médecin le juge nécessaire.
N’utilisez pas ALPHAGAN si vous allaitez.
Conduite de véhicules et utilisation de machines
ALPHAGAN est susceptible de provoquer une vision trouble ou anormale. Cet effet semblerait plus important la nuit ou en cas de réduction de la luminosité.
ALPHAGAN peut aussi provoquer une fatigue ou une somnolence chez certains patients.
Si vous présentez un de ces symptômes, ne conduisez pas et n'utilisez pas de machines jusqu'à ce que les symptômes aient disparu.
ALPHAGAN contient du chlorure de benzalkonium
ALPHAGAN contient 0,25 mg de chlorure de benzalkonium pour 5 ml de collyre équivalant à 0,05 mg/ml.
Le chlorure de benzalkonium peut être absorbé par les lentilles de contact souples et changer leur couleur. Retirez les lentilles de contact avant application et attendre au moins 15 minutes avant de les remettre.
Le chlorure de benzalkonium peut également provoquer une irritation des yeux, surtout si vous souffrez du syndrome de l’œil sec ou des troubles de la cornée (couche transparente à l’avant de l’œil). En cas de sensation anormale dans l’œil, de picotements ou de douleur dans les yeux après avoir utilisé ce médicament, prévenez votre médecin.
3. COMMENT UTILISER ALPHAGAN 0,2 % (2 mg/ml), collyre en solution ?
Adultes
La dose recommandée est d'une goutte d'ALPHAGAN deux fois par jour dans l'œil ou les yeux atteint(s), les deux instillations devant être espacées d'environ 12 heures. Ne modifiez pas la posologie et n’arrêtez pas d’utiliser ALPHAGAN avant d’en parler à votre médecin.
Utilisation chez les enfants et les adolescents
Enfants de moins de 12 ans
ALPHAGAN ne doit pas être utilisé chez les nourrissons de moins de 2 ans.
ALPHAGAN n'est pas recommandé chez les enfants (de 2 à 12 ans).
Mode d'administration
ALPHAGAN s'utilise comme un collyre. Lavez-vous toujours les mains avant d'appliquer le collyre. Votre prescription vous indique combien de gouttes vous devez utiliser. Si vous utilisez ALPHAGAN avec un autre collyre, attendez 5 à 15 minutes avant d'appliquer le second collyre.
Appliquez le collyre de la façon suivante :

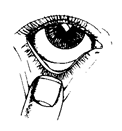

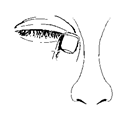
1. Inclinez la tête en arrière et regardez en l'air.
2. Tirez doucement la paupière inférieure jusqu'à ce qu'il y ait une petite poche.
3. Retournez le flacon à l'envers et appuyez dessus pour en détacher une goutte dans l'œil.
4. En gardant l'œil fermé, appuyez votre doigt contre le coin interne de votre œil fermé (du côté de l'œil proche du nez) et maintenir pendant une minute.
Si une goutte tombe à côté de votre œil, répétez la procédure.
Pour éviter toute contamination, ne laissez pas la pointe du flacon toucher votre œil ou quoi que ce soit d'autre. Remettez le bouchon et refermez le flacon aussitôt après utilisation.
Si vous avez utilisé plus d’ALPHAGAN que vous n’auriez dû
Adultes
Chez les patients adultes ayant utilisé plus de gouttes que ce qui leur avait été prescrit, les effets indésirables rapportés étaient ceux déjà connus pour ALPHAGAN.
Les adultes ayant accidentellement avalé ALPHAGAN ont présenté une baisse de la pression sanguine, suivie chez certains par une augmentation de la pression sanguine.
Enfants
Des effets indésirables graves ont été rapportés chez les enfants ayant accidentellement avalé ALPHAGAN. Les signes observés comprennent une somnolence, une mollesse, une baisse de la température corporelle, une pâleur et des difficultés respiratoires. Si l'un de ces signes se manifeste, consultez immédiatement votre médecin.
Adultes et enfants
En cas d'ingestion accidentelle d'ALPHAGAN, ou si vous avez utilisé plus d'ALPHAGAN que nécessaire, contactez immédiatement votre médecin.
Si vous oubliez d’utiliser ALPHAGAN
Si vous arrêtez d’utiliser ALPHAGAN
ALPHAGAN doit être utilisé chaque jour pour agir correctement.
N'interrompez pas votre traitement par ALPHAGAN sans l'avis de votre médecin.
Si vous avez d’autres questions sur l’utilisation de ce médicament, demandez plus d’informations à votre médecin ou à votre pharmacien.
4. QUELS SONT LES EFFETS INDESIRABLES EVENTUELS ?
Les effets indésirables au niveau de l'œil pouvant être observés avec ALPHAGAN sont les suivants :
Effets affectant les yeux
Très fréquents (pouvant affecter plus d’1 patient sur 10)
· Irritation de l’œil (œil rouge, brûlures, picotements, sensation de corps étranger dans l’œil, démangeaisons, follicules ou points blancs sur la membrane transparente qui couvre la surface de l’œil).
· Vision trouble.
· Réaction allergique dans l’œil.
Fréquents (pouvant affecter jusqu’à 1 patient sur 10)
· Irritation locale (inflammation, rougeurs et inconfort dans l’œil, gonflement de la paupière, gonflement de la membrane transparente qui couvre la surface de l’œil, œil collé, douleur et larmoiement).
· Sensibilité à la lumière.
· Erosion de la surface de l’œil et coloration.
· Sécheresse oculaire.
· Blanchiment de la membrane transparente qui couvre la surface de l’œil.
· Vision anormale.
· Inflammation de la membrane transparente qui couvre la surface de l’œil.
· De petites bosses sur la face arrière des paupières dues à une inflammation.
Très rares (pouvant affecter jusqu’à 1 patient sur 10 000)
· Inflammation dans l’œil.
· Rétrécissement de la pupille.
Fréquence indéterminée (ne pouvant être estimée sur la base des données disponibles)
· Démangeaisons des paupières.
Effets affectant le corps entier
Très fréquents (pouvant affecter plus d’1 patient sur 10)
· Maux de tête.
· Sécheresse de la bouche.
· Fatigue/somnolence.
Fréquents (pouvant affecter jusqu’à 1 patient sur 10)
· Vertiges.
· Symptômes de rhume.
· Symptômes au niveau de l’estomac ou de la digestion.
· Altération du goût.
· Faiblesse générale.
Peu fréquents (pouvant affecter jusqu’à 1 patient sur 100)
· Dépression.
· Palpitations ou troubles du rythme du cœur.
· Sécheresse du nez.
· Réactions allergiques générales.
Rares (pouvant affecter jusqu’à 1 patient sur 1 000)
· Essoufflement.
Très rares (pouvant affecter jusqu’à 1 patient sur 10 000)
· Insomnie.
· Evanouissement.
· Augmentation de la tension artérielle.
· Diminution de la tension artérielle.
Fréquence indéterminée (ne pouvant être estimée sur la base des données disponibles)
· Réactions cutanées incluant rougeur, gonflement du visage, démangeaisons, rash et dilatation des vaisseaux sanguins.
Déclaration des effets secondaires
Si vous ressentez un quelconque effet indésirable, parlez-en à votre médecin ou votre pharmacien. Ceci s’applique aussi à tout effet indésirable qui ne serait pas mentionné dans cette notice. Vous pouvez également déclarer les effets indésirables directement via le système national de déclaration : Agence nationale de sécurité du médicament et des produits de santé (ANSM) et réseau des Centres Régionaux de Pharmacovigilance - Site internet : https://signalement.social-sante.gouv.fr/.
En signalant les effets indésirables, vous contribuez à fournir davantage d’informations sur la sécurité du médicament.
5. COMMENT CONSERVER ALPHAGAN 0,2 % (2 mg/ml), collyre en solution ?
Tenir ce médicament hors de la vue et de la portée des enfants.
A conserver à une température ne dépassant pas 25°C.
N’utilisez pas ce médicament si vous remarquez que le système d'ouverture est endommagé avant la première utilisation.
N’utilisez pas ce médicament après la date de péremption indiquée sur l’étiquette du flacon et le carton après « EXP ». La date de péremption fait référence au dernier jour de ce mois.
Jetez le flacon 28 jours après ouverture même s'il reste de la solution.
Ne jetez aucun médicament au tout-à-l’égout ou avec les ordures ménagères. Demandez à votre pharmacien d’éliminer les médicaments que vous n’utilisez plus. Ces mesures contribueront à protéger l’environnement.
6. CONTENU DE L’EMBALLAGE ET AUTRES INFORMATIONS
· La substance active est le tartrate de brimonidine. Un ml de solution contient 2,0 mg de tartrate de brimonidine équivalent à 1,3 mg de brimonidine.
· Les autres composants sont : chlorure de benzalkonium comme conservateur, alcool polyvinylique , chlorure de sodium, citrate de sodium dihydraté, acide citrique monohydraté, acide chlorhydrique ou hydroxyde de sodium (pour ajuster le pH), eau purifiée.
Qu’est-ce que ALPHAGAN et contenu de l’emballage extérieur
ALPHAGAN est un collyre en solution limpide, jaune-vert à légèrement jaune-vert, dans un flacon en plastique.
Chaque flacon contient 2,5 ml, 5 ml ou 10 ml de solution.
ALPHAGAN se présente en boîte de 1, 3 ou 6 flacons.
Toutes les présentations peuvent ne pas être commercialisées.
Titulaire de l’autorisation de mise sur le marché
5/13 BOULEVARD DE LA REPUBLIQUE
92100 BOULOGNE-BILLANCOURT
Exploitant de l’autorisation de mise sur le marché
ABBVIE
5/13 BOULEVARD DE LA REPUBLIQUE
92100 BOULOGNE-BILLANCOURT
ALLERGAN PHARMACEUTICALS IRELAND
WESPORT
CO. MAYO
IRLANDE
Noms du médicament dans les Etats membres de l'Espace Economique Européen
Ce médicament est autorisé dans les Etats membres de l'Espace Economique Européen sous les noms suivants : Conformément à la réglementation en vigueur.
[À compléter ultérieurement par le titulaire]
La dernière date à laquelle cette notice a été révisée est :
[À compléter ultérieurement par le titulaire]
Des informations détaillées sur ce médicament sont disponibles sur le site Internet de l’ANSM (France).
Pour écouter cette notice ou pour en demander une copie en