Dernière mise à jour le 28/04/2026
GAMMAGARD 50 mg/ml, poudre et solvant pour solution pour perfusion
Indications thérapeutiques
ANTISERUMS ET IMMUNOGLOBULINES.
GAMMAGARD appartient à la classe des médicaments dénommés immunoglobulines. Ces médicaments contiennent des anticorps humains, également présents dans votre sang. Les anticorps aident votre corps à combattre les infections. Les médicaments comme GAMMAGARD sont utilisés chez les patients ne possédant pas suffisamment d’anticorps dans leur sang et sujets à de fréquentes infections. Ils peuvent aussi être utilisés chez les patients ayant besoin d’anticorps supplémentaires lors du traitement de certains troubles inflammatoires (maladies auto-immunes).
GAMMAGARD est une immunoglobuline humaine normale indiquée chez les patients avec déficits en immunoglobuline A (IgA) et anticorps anti-IgA :
Le traitement de patients ne possédant pas suffisamment d’anticorps (traitement substitutif). Ces patients sont répartis en cinq groupes :
· Patients présentant un défaut congénital de production d’anticorps (déficits immunitaires primitifs, DIP) ;
· Patients présentant un cancer du sang (leucémie lymphoïde chronique) entraînant un déficit de production d’anticorps et des infections récurrentes en cas d’échec du traitement préventif par antibiotiques ;
· Patients présentant un cancer de la moelle osseuse (myélome multiple) et un déficit de production d’anticorps accompagnée d’infections récurrentes, qui ne répondent pas à un vaccin contre certaines bactéries (pneumocoques) ;
· Patients ayant un déficit immunitaire suite à une infection par le VIH depuis leur naissance et présentant des infections fréquentes ;
· Patients présentant une faible production d’anticorps après une greffe de cellules de moelle osseuse provenant d’un donneur.
Le traitement des patients souffrant de certains troubles inflammatoires (traitements immunomodulateurs). Ces patients sont répartis en trois groupes :
· Patients n’ayant pas assez de plaquettes dans le sang (purpura thrombopénique immunitaire primaire/idiopathique) et présentant des risques élevés d’hémorragie ou devant subir une opération chirurgicale dans un futur proche ;
· Patients atteints d’une maladie associée à une inflammation de plusieurs nerfs du corps (syndrome de Guillain Barré) ;
· Patients présentant une maladie provoquant une inflammation de plusieurs organes du corps (maladie de Kawasaki).
Présentations
> 1 flacon(s) en verre de 5 g - 1 flacon(s) en verre de 100 ml avec matériel de perfusion avec dispositif de transfert
Code CIP : 560 314-0 ou 34009 560 314 0 5
Déclaration de commercialisation : 01/10/1999
Cette présentation est agréée aux collectivités
Inscription sur la liste de rétrocession au titre de son AMM, selon les conditions précisées au Journal Officiel. Prix de cession publié au Journal Officiel.
> 1 flacon(s) en verre de 10 g - 1 flacon(s) en verre de 200 ml avec matériel de perfusion avec dispositif de transfert
Code CIP : 560 315-7 ou 34009 560 315 7 3
Déclaration de commercialisation : 01/10/1999
Cette présentation est agréée aux collectivités
Inscription sur la liste de rétrocession au titre de son AMM, selon les conditions précisées au Journal Officiel. Prix de cession publié au Journal Officiel.
Service médical rendu (SMR)
Pas de SMR disponible pour ce médicament ( plus d'informations dans l'aide )
Amélioration du service médical rendu (ASMR)
Pas d'ASMR disponible pour ce médicament (plus d'informations dans l'aide )
Autres informations
- Titulaire de l'autorisation : BAXALTA INNOVATIONS GMBH
- Conditions de prescription et de délivrance :
- liste I
- prescription hospitalière
- prescription par un médecin exerçant dans un établissement de transfusion sanguine
- Statut de l'autorisation : Valide
- Type de procédure : Procédure nationale
- Code CIS : 6 839 906 8
ANSM - Mis à jour le : 04/07/2025
GAMMAGARD 50 mg/ml, poudre et solvant pour solution pour perfusion.
2. COMPOSITION QUALITATIVE ET QUANTITATIVE
Immunoglobuline humaine normale (IgIV)* 50 mg**
pour 1 ml de solution reconstituée
* la poudre a une teneur maximale en IgA inférieure à 44 µg/g de protéines (soit 2,2 µg/ml).
** correspondant à une quantité totale de protéines, dont au moins 90% sont des Immunoglobulines G (IgG).
· Un flacon de 10 ml contient 0,5 g d’immunoglobuline humaine normale.
· Un flacon de 50 ml contient 2,5 g d’immunoglobuline humaine normale.
· Un flacon de 100 ml contient 5 g d’immunoglobuline humaine normale.
· Un flacon de 200 ml contient 10 g d’immunoglobuline humaine normale.
Distribution des sous-classes d’IgG :
· IgG1 > 56,9 %
· IgG2 > 16,0 %
· IgG3 > 3,3 %
· IgG4 > 0,3 %
Fabriqué à partir du plasma humain issu de dons.
Excipients à effet notoire : sodium et glucose.
Pour la liste complète des excipients, voir rubrique 6.1.
Poudre et solvant pour solution pour perfusion intraveineuse.
GAMMAGARD se présente sous la forme d'une poudre lyophilisée, blanche ou très légèrement jaune, pratiquement exempte de particule visible.
4.1. Indications thérapeutiques
Traitement de substitution chez les adultes, et chez les enfants et les adolescents (0-18 ans) :
· Déficits immunitaires primitifs (DIP) avec anomalies de la production d’anticorps (voir rubrique 4.4),
· Hypogammaglobulinémie et infections bactériennes récurrentes chez les patients atteints de leucémie lymphoïde chronique, après échec de l’antibioprophylaxie,
· Hypogammaglobulinémie et infections bactériennes récurrentes chez les patients atteints de myélome multiple en phase de plateau (stabilisé) n’ayant pas répondu à la vaccination anti-pneumococcique,
· Hypogammaglobulinémie chez les patients ayant bénéficié d’une allogreffe de cellules souches hématopoïétiques (allo-SCH),
· SIDA par infection VIH congénitale avec infections bactériennes récurrentes.
Traitement immunomodulateur chez les adultes, et chez les enfants et les adolescents (de 0 à 18 ans) :
· Thrombopénie immunitaire primaire (purpura thrombopénique idiopathique (PTI)) en cas de risque hémorragique important ou avant un acte chirurgical pour corriger le taux de plaquettes,
· Syndrome de Guillain et Barré,
· Maladie de Kawasaki.
4.2. Posologie et mode d'administration
Posologie
La dose et la posologie dépendent des indications.
Dans les traitements substitutifs, la dose peut être individualisée pour chaque patient en fonction de la réponse pharmacocinétique et clinique. Les posologies suivantes sont données à titre indicatif.
Traitement de substitution en cas de déficit immunitaire primitif :
La posologie doit permettre d’atteindre un taux d’IgG résiduel (mesuré avant la perfusion suivante) d'au moins 5 à 6 g/l. 3 à 6 mois sont nécessaires après le début du traitement pour atteindre l’équilibre. La dose de charge recommandée est de 0,4 à 0,8 g/kg de poids corporel, suivie d’au moins 0,2 g/kg de poids corporel toutes les 3 à 4 semaines.
La dose nécessaire pour atteindre un taux résiduel de 5 à 6 g/l est de l'ordre de 0,2 à 0,8 g/kg de poids corporel/mois.
L’intervalle entre les doses lorsque l’état d’équilibre est atteint varie entre 3 et 4 semaines.
Les taux résiduels doivent être mesurés et évalués en prenant en compte la fréquence des infections. Pour réduire la fréquence des infections, il peut être nécessaire d’augmenter les doses et d’atteindre des taux résiduels plus élevés.
Hypogammaglobulinémie et infections bactériennes récurrentes chez les patients atteints de leucémie lymphoïde chronique après échec de l’antibio-prophylaxie; hypogammaglobulinémie et infections bactériennes récidivantes chez les patients atteints de myélome multiple en phase de plateau (ou stabilisé) n’ayant pas répondu à la vaccination anti-pneumococcique ; infections bactériennes récurrentes ayant un SIDA par infection congénitale à VIH.
La dose recommandée est de 0,2 à 0,4 g/kg de poids corporel toutes les 3 à 4 semaines.
Hypogammaglobulinémie chez les patients ayant bénéficié d’une allogreffe de cellules souches hématopoïétiques.
La posologie est ajustée pour chaque patient dans le traitement des infections et la prévention de la maladie du greffon contre l’hôte.
La dose recommandée est de 0,2 à 0,4 g/kg de poids corporel toutes les 3 à 4 semaines. Les taux résiduels doivent être maintenus au-dessus de 5 g/l.
Thrombopénie immunitaire primaire.
Il existe deux schémas posologiques possibles :
· 0,8 à 1 g/kg de poids corporel administré le jour 1 ; cette dose peut être répétée une fois dans les 3 jours suivants,
· 0,4 g/kg de poids corporel administré chaque jour pendant deux à cinq jours.
Le traitement peut être répété en cas de rechute.
Syndrome de Guillain-Barré.
0,4 g/kg de poids corporel/jour pendant 5 jours.
Maladie de Kawasaki.
1,6 à 2,0 g/kg de poids corporel doivent être administrés en plusieurs doses réparties sur 2 à 5 jours ou 2 g/kg de poids corporel en dose unique.
Les patients doivent recevoir un traitement concomitant avec de l’acide acétylsalicylique.
Les doses recommandées sont résumées ci-dessous :
|
Indication |
Posologie |
Rythme des injections |
|
Traitement substitutif des déficits immunitaires primitifs |
· dose de charge : 0,4 à 0,8 g/kg de poids corporel |
|
|
|
· dose d'entretien : 0,2 à 0,8 g/kg de poids corporel |
toutes les 3 à 4 semaines pour obtenir un taux résiduel d'IgG d'au moins 5 à 6 g/L |
|
Traitement substitutif des déficits immunitaires secondaires |
0,2 à 0,4 g/kg de poids corporel |
toutes les 3 à 4 semaines pour obtenir un taux résiduel d’IgG d’au moins 5 à 6 g/L |
|
SIDA par Infection VIH congénitale |
0,2 à 0,4 g/kg de poids corporel |
toutes les 3 à 4 semaines |
|
Hypogammaglobulinémie (<4 g/L) chez les patients après une allogreffe de cellules souches hématopoïétiques |
0,2 à 0,4 g/kg de poids corporel |
toutes les 3 à 4 semaines afin d’obtenir un taux résiduel d’IgG supérieur à 5 g/l |
|
· le traitement des infections et la prévention de la maladie du greffon contre l’hôte |
|
toutes les semaines à partir de J-7 jusqu’à 3 mois après la greffe |
|
· le déficit persistant de production d’anticorps |
|
tous les mois jusqu’à ce que les taux d’anticorps soient revenus à la normale |
|
Traitement immunomodulateur |
|
|
|
Thrombopénie immunitaire primaire (Purpura thrombopénique idiopathique) |
0,8 à 1g/kg de poids corporel
ou
0,4 g/kg de poids corporel/j |
à J1, éventuellement répété une fois dans les 3 jours suivants
pendant 2 à 5 jours |
|
Syndrome de Guillain et Barré |
0,4 g/kg de poids corporel/j |
pendant 5 jours |
|
Maladie de Kawasaki |
1,6 à 2,0 g/kg de poids corporel
ou
2 g/kg de poids corporel |
en plusieurs doses réparties sur 2 à 5 j en association avec de l’acide acétylsalicylique
en une dose unique en association avec de l’acide acétylsalicylique |
Population pédiatrique
La posologie chez l’enfant et l’adolescent (de 0 à 18 ans) est identique à celle des adultes, la dose pour chaque indication étant calculée en fonction du poids corporel et ajustée selon les résultats cliniques des pathologies susmentionnées.
Mode d’administration
Cette immunoglobuline intraveineuse (IgIV) se présente sous la forme d'une poudre à reconstituer extemporanément avec de l'eau pour préparations injectables, selon les modalités décrites dans le paragraphe « 6.6. Instructions pour l'utilisation et la manipulation ».
Ne pas utiliser de solutions présentant un aspect non homogène ou contenant un dépôt.
Administration par voie intraveineuse.
GAMMAGARD doit être perfusé par voie intraveineuse à un débit initial de 0,5 ml/kg de poids corporel/heure. En général, il est recommandé que les patients débutant un traitement avec GAMMAGARD ou passant d’une spécialité pharmaceutique d’IgIV à GAMMAGARD, commencent au débit le plus faible et augmentent ensuite jusqu’au débit maximal, s’ils ont toléré plusieurs perfusions à des débits de perfusion intermédiaires (voir également rubrique 4.4.).
Si le débit de perfusion est bien toléré, il peut être augmenté progressivement jusqu’à un maximum de 4 mL/kg de poids corporel/heure. Si aucun effet indésirable ne survient, le débit peut être augmenté progressivement jusqu'à un maximum de 8 ml/kg de poids corporel/heure.
Ce médicament est contre-indiqué dans les situations suivantes :
· Hypersensibilité ou réaction anaphylactique connue à la substance active ou à l’un des excipients (voir rubrique 4.4),
· Hypersensibilité aux immunoglobulines humaines.
4.4. Mises en garde spéciales et précautions d'emploi
GAMMAGARD 50 mg/ml contient 21,7 mg de glucose par dose. Ceci devra être pris en compte en cas de diabète latent (possible apparition d’une glycosurie transitoire), de diabète, ou chez les patients avec un régime pauvre en sucre. En cas d’insuffisance rénale aiguë, voir ci-dessous.
Certains effets indésirables graves peuvent être associés au débit d'administration. Le débit recommandé au paragraphe « 4.2. Posologie et mode d'administration », doit être scrupuleusement respecté. Les patients doivent rester sous surveillance pendant toute la durée de la perfusion, afin de détecter d'éventuels symptômes.
Certains d’effets indésirables peuvent survenir plus fréquemment :
· en cas de débit de perfusion élevé,
· chez les patients hypo- ou agammaglobulinémiques avec ou sans déficit en IgA,
· chez les patients recevant la première administration d’une immunoglobuline humaine normale et particulièrement ceux présentant une immunodéficience, ou dans de rares cas, lors d’un changement d’immunoglobuline humaine normale ou si la dernière perfusion remonte à un certain temps.
Les complications potentielles peuvent être souvent évitées en s’assurant que :
· les patients ne sont pas sensibles à l’immunoglobuline humaine normale en démarrant la perfusion lentement (0,5 ml/kg de poids corporel/heure),
· les patients sont suivis pendant toute la durée de la perfusion afin de détecter d’éventuels symptômes. En particulier, s’ils reçoivent une immunoglobuline humaine normale pour la première fois, lors d’un changement de spécialité d’IgIV ou lorsque la dernière perfusion a été faite longtemps auparavant, ces patients doivent être suivis pendant toute la durée de la première administration et pendant l'heure qui suit la fin de cette perfusion, afin de détecter d'éventuels effets indésirables. Tous les autres patients devront être surveillés pendant au moins 20 minutes après la perfusion,
· la quantité de glucose (quantité maximale de 0,43 g/g d’IgG) est prise en compte en cas de diabète latent (possible apparition d’une glycosurie transitoire), de diabète, ou chez les patients avec un régime pauvre en sucre.
Chez tous les patients, l'administration d'IgIV impose :
· une hydratation appropriée avant le début de la perfusion d'IgIV,
· une surveillance de la diurèse,
· une surveillance de la créatininémie,
· d'éviter l’administration concomitante de diurétiques de l'anse.
En cas d’effets indésirables, le débit d’administration doit être réduit ou la perfusion arrêtée. Le traitement requis dépend de la nature et de la sévérité des effets indésirables.
En cas de choc, le traitement médical symptomatique relatif à l’état de choc doit être instauré.
Hypersensibilité
Les vraies réactions allergiques à ce médicament sont rares. Elles peuvent survenir dans de très rares cas de déficit en IgA avec anticorps anti-IgA.
Rarement, une immunoglobuline humaine normale peut entraîner une réaction anaphylactique avec une chute brutale de la tension artérielle même chez des patients qui ont présenté une bonne tolérance à une administration précédente d'immunoglobuline humaine normale.
Les patients, ayant des anticorps anti-IgA ou présentant un déficit en IgA dans le cadre d’un déficit immunitaire primitif sous-jacent pour lequel un traitement par IgIV est indiqué, peuvent présenter un risque plus élevé de réaction anaphylactique. L’anaphylaxie a été rapportée lors de l’utilisation de GAMMAGARD même si le médicament contient des taux faibles d’IgA (voir rubrique 4.8). L’administration de GAMMAGARD doit être réalisée avec la plus grande prudence chez les patients qui ont déjà présentés une réaction d’hypersensibilité grave et le lieu où s’effectue l’administration doit disposer de soins appropriés de support pour traiter les réactions mettant en jeu le pronostic vital.
Précautions supplémentaires
Thromboembolisme
Cliniquement, l’existence d’un lien est reconnue entre un traitement par IgIV (incluant GAMMAGARD) et des réactions thromboemboliques, comme par exemple l’infarctus du myocarde, l’atteinte vasculaire cérébrale (y compris l'accident vasculaire cérébral), l’embolie pulmonaire et la thrombose veineuse profonde. Ces évènements sont probablement liés à une élévation relative de la viscosité sanguine due à un apport important en immunoglobuline chez les patients à risque. Toutes les précautions doivent être prises lors de la prescription et de la perfusion d’IgIV chez les patients obèses, chez les patients présentant des facteurs de risques thrombotiques préexistants (tels que des antécédents d’athérosclérose, des facteurs de risques cardiovasculaires multiples, un âge avancé, un débit cardiaque altéré, une hyperviscosité connue ou suspectée, par exemple, déshydratation ou para protéines, des troubles d'hypercoagulabilité, des périodes prolongées d'immobilisation, l'obésité, l’utilisation d’œstrogènes, le diabète, des troubles thrombophiliques acquis ou hérités, un antécédent de maladie vasculaire, un cathéter vasculaire à demeure, un antécédent d'événement thrombotique ou thromboembolique).
Chez les patients présentant un risque d’effets indésirables thromboemboliques, GAMMAGARD doit être administré à un débit de perfusion minimal et à une posologie adaptée.
Assurer une hydratation appropriée aux patients avant et après administration. Surveiller les signes et symptômes de thrombose et évaluer la viscosité sanguine chez les patients à risque d’hyperviscosité.
Complications rénales
Des réactions rénales indésirables sévères ont été rapportées chez des patients recevant un traitement par IgIV, notamment une insuffisance rénale aiguë, une nécrose tubulaire aiguë, une néphropathie tubulaire proximale et une néphrose osmotique. Dans la plupart des cas, des facteurs de risque ont été identifiés, tels qu’une insuffisance rénale préexistante, un diabète sucré, une hypovolémie, un surpoids, la prise concomitante de médicaments néphrotoxiques, un âge supérieur à 65 ans, une septicémie ou une paraprotéinémie.
En cas d’atteinte rénale, une interruption de l’immunoglobuline doit être envisagée.
Bien que des cas d'atteinte rénale et d'insuffisance rénale aigüe aient été associés à l'utilisation de nombreuses spécialités pharmaceutiques d'IgIV contenant des excipients tels que le saccharose, le glucose et le maltose, celles contenant du saccharose (GAMMAGARD ne contient pas de saccharose) comme stabilisant sont les plus représentées. Chez les patients à risque, l'utilisation d’IgIV ne contenant pas ces excipients doit être envisagée. GAMMAGARD contient du glucose (voir la liste des excipients).
Chez les patients à risque, l'utilisation d’IgIV ne contenant pas de saccharose doit être envisagée. GAMMAGARD ne contient pas de saccharose ni de maltose.
Chez les patients présentant un risque d’insuffisance rénale aiguë, les IgIV doivent être administrées à un débit de perfusion minimal et à une posologie adaptée.
Syndrome de détresse respiratoire aigu post-transfusionnel
Des cas d’œdème pulmonaire non cardiogénique (TRALI) ont été rapportés chez les patients traités par des IgIV.
Syndrome de méningite aseptique
Des cas de syndrome de méningite aseptique ont été rapportés comme pouvant survenir lors d’un traitement par IgIV (incluant GAMMAGARD). L'arrêt du traitement par IgIV peut conduire à une régression du syndrome de méningite aseptique en plusieurs jours. Le syndrome apparaît généralement entre plusieurs heures et deux jours après le traitement par IgIV.
· Les résultats d’analyse du liquide céphalo-rachidien sont souvent positifs avec une pléocytose pouvant aller jusqu'à plusieurs milliers de cellules par mm³, essentiellement de type granulocytaire, et une hausse du taux de protéines jusqu'à plusieurs centaines de mg/dl,
· Des incidences plus élevées de syndrome de méningite aseptique ont été constatées chez les femmes.
Anémie hémolytique
Les immunoglobulines intraveineuses peuvent contenir des anticorps à des groupes sanguins susceptibles d'agir comme des hémolysines et d'induire le recouvrement in vivo des globules rouges par des immunoglobulines, ce qui entraîne une réaction antiglobuline directe positive (Test de Coombs) et, dans de rares cas, une hémolyse. Une anémie hémolytique peut se développer à la suite d'un traitement par IgIV en raison d’une augmentation de la séquestration des globules rouges. Des signes cliniques ou des symptômes d’hémolyse doivent être surveillés chez les patients recevant des IgIV (voir rubrique 4.8).
Déficit en IgA sélectif
Les IgIV ne sont pas indiquées chez les patients présentant un déficit en IgA sélectif sans autre anomalie immunitaire. Ces patients devront être traités seulement si leur déficit en IgA est associé à un déficit immunitaire pour lequel l’IgIV est clairement indiquée.
Hyperprotéinémie
Une hyperprotéinémie et une élévation de la viscosité sanguine peuvent survenir chez les patients recevant un traitement par IgIV.
Teneur en sodium
La teneur en sodium contenue dans une dose maximale journalière peut sensiblement s’ajouter à la teneur journalière en sodium alimentaire recommandée chez les patients suivant un régime hyposodé. Chez ces patients, la teneur en sodium du produit doit être calculée et prise en compte lors de la détermination de l’apport alimentaire en sodium. GAMMAGARD 50 mg/ml contient environ 3,4 mg/ml de sodium. Un patient de 70 kg recevant 1 g/kg (1,4 litres) recevra 4,7 g de sodium.
Agents transmissibles
GAMMAGARD est fabriqué à partir de plasma humain. Les mesures habituelles de prévention du risque de transmission d’agents infectieux par les médicaments préparés à partir de sang ou de plasma humain comprennent la sélection des donneurs, la recherche des marqueurs spécifiques d’infection sur chaque don et sur les mélanges de plasma ainsi que la mise en œuvre dans le procédé de fabrication d’étapes efficaces pour l’inactivation/élimination virale. Cependant, lorsque des médicaments préparés à partir de sang ou de plasma humain sont administrés, le risque de transmission d’agents infectieux ne peut pas être totalement exclu. Ceci s’applique également aux virus inconnus ou émergents ou autres types d’agents infectieux, comme l’agent de la maladie de Creutzfeldt-Jakob (MCJ).
Les mesures prises sont considérées comme efficaces vis-à-vis des virus enveloppés tels que le VIH, le VHB et le VHC, et vis-à-vis des virus non enveloppés VHA et parvovirus B19.
L'expérience clinique ne rapporte pas de transmission du virus de l’hépatite A ni du parvovirus B19 par les immunoglobulines, les anticorps présents contribuant probablement à la sécurité du produit.
Interférence avec des tests sérologiques
Après perfusion d'immunoglobuline, l'augmentation transitoire de la concentration de divers anticorps transférés de façon passive dans le sang du patient peut être responsable de résultats faussement positifs lors de tests sérologiques, par exemple pour l’Hépatite A, l’Hépatite B, la rougeole et la varicelle.
La transmission passive d'anticorps anti-érythrocytaires tels que les anticorps anti-A, anti-B et anti-D peut interférer avec certains tests sérologiques comme la recherche des anticorps anti-globules rouges, par exemple le test des antiglobulines (TDA, test de Coombs direct).
GAMMAGARD contient des anticorps des groupes sanguins qui peuvent agir comme des hémolysines et induire in vivo l’agglutination des globules rouges avec l'immunoglobuline. Cela peut entraîner un test de Coombs direct positif. GAMMAGARD peut également être responsable de résultats faussement positif lors des dosages sérologiques des β-D-glucanes au cours du diagnostic des infections fongiques et ce, durant plusieurs semaines après l’injection du produit.
Une anémie hémolytique retardée peut se développer à la suite du traitement par GAMMAGARD à cause d’une augmentation de la séquestration des globules rouges ; une hémolyse aiguë, consécutive à une hémolyse intravasculaire, a été rapportée.
Les facteurs de risque suivants peuvent être liés au développement d'une hémolyse : des doses élevées (administration unique ou répartie sur plusieurs jours) et le groupe sanguin non-O.
Un état inflammatoire sous-jacent chez un patient peut augmenter le risque d'hémolyse mais son rôle est incertain.
GAMMAGARD contient du sodium
Ce médicament contient 668 mg de sodium par flacon (de 10 g), ce qui équivaut à 34 % de l’apport alimentaire quotidien maximal recommandé par l’OMS de 2 g de sodium par adulte.
GAMMAGARD contient du glucose
GAMMAGARD contient approximativement 20 mg/ml de glucose.
4.5. Interactions avec d'autres médicaments et autres formes d'interactions
Vaccins constitués de virus vivants atténués
L'administration d'IgIV peut entraver l'efficacité des vaccins constitués de virus vivants atténués tels que les vaccins contre la rougeole, la rubéole, les oreillons et la varicelle. Après perfusion d'IgIV, attendre au minimum 6 semaines (de préférence 3 mois) avant d'administrer ce type de vaccins. Pour le vaccin contre la rougeole, cette altération de la réponse peut persister plus d’un an après la perfusion d’IgIV. Par conséquent, si un patient est vacciné contre la rougeole, un contrôle des anticorps protecteurs devrait être fait.
Si le patient a reçu des vaccins constitués de virus vivants atténués (rougeole, rubéole, oreillons, varicelle) au cours des 2 semaines précédant la perfusion, un contrôle des anticorps protecteurs post-vaccinaux peut être nécessaire en vue d'un éventuel rappel.
Population pédiatrique
Il n’existe aucune étude d’interaction avec GAMMAGARD dans la population pédiatrique.
4.6. Fertilité, grossesse et allaitement
Il n’y a pas de données pertinentes sur l’utilisation de GAMMAGARD chez les femmes enceintes ou qui allaitent.
Il est avéré que les médicaments à base d'IgIV administrés à la mère traversent la barrière placentaire, particulièrement lors du troisième trimestre.
Le médecin doit évaluer les risques potentiels et prescrire GAMMAGARD seulement si cela est clairement nécessaire.
Les effets de GAMMAGARD sur la fertilité n’ont pas été établis.
4.7. Effets sur l'aptitude à conduire des véhicules et à utiliser des machines
Résumé du profil de sécurité
Les effets secondaires liés à l'administration d'IgIV sont plus fréquents chez les malades atteints de déficits immunitaires primitifs.
Avec les immunoglobulines humaines normales pour administration intraveineuse, des réactions de type frissons, céphalées, vertiges, fièvre, vomissements, manifestations allergiques, nausées, arthralgies, chute de la tension artérielle et lombalgies modérées peuvent survenir occasionnellement.
Rarement, les immunoglobulines humaines normales peuvent entraîner une chute brutale de la tension artérielle et dans des cas isolés, un choc anaphylactique même lorsque le patient n'a pas présenté d’hypersensibilité lors d’une administration antérieure.
Des cas réversibles de méningites aseptiques, des cas isolés réversibles d’anémies hémolytiques/hémolyses et de rares cas de réactions cutanées transitoires (y compris lupus érythémateux cutané -fréquence indéterminée- et autres réactions cutanées eczématiformes régressives) ont été observés avec les immunoglobulines humaines normales, surtout chez les patients des groupes A, B et AB.
Rarement, une anémie hémolytique nécessitant une transfusion peut apparaître après une forte dose d’IgIV (voir aussi section 4.4).
Une élévation de la créatininémie et/ou une insuffisance rénale aiguë ont été observés.
Très rarement : réactions thromboemboliques telles que, infarctus du myocarde, accident vasculaire cérébral, embolie pulmonaire et thrombose veineuse profonde ont été observées.
Un possible lien entre l’administration d’IgIV et l’éventuelle survenue d’événements thromboemboliques a été confirmé par la clinique.
Les effets indésirables ont été compilés à partir d’une étude clinique pivot de GAMMAGARD et d’une étude de phase 4 évaluant la tolérance à court et moyen terme de GAMMAGARD. Les effets indésirables rapportés dans les deux études et après la mise sur le marché sont résumés et présentés par système – organe selon la classification MedDRA et suivant leur fréquence dans le tableau ci-dessous.
Tableau récapitulatif des effets indésirables
Le tableau récapitulatif présenté ci-dessous fait référence à la classification des systèmes d’organes MedDRA (Classe de Système Organe et terme préférentiel).
La fréquence est définie selon la règle suivante : très fréquent (≥1/10) ; fréquent (≥1/100 à <1/10) ; peu fréquent (≥1/1 000 à <1/100) ; rare (≥1/10 000 à <1/1 000) ; très rare (<1/10 000) ; indéterminé (ne peut être estimé sur la base des données disponibles).
Au sein de chaque groupe de fréquence, les effets indésirables sont présentés suivant un ordre décroissant de gravité.
|
Fréquence des effets indésirables |
||
|
Classification des systèmes d’organes MedDRA |
Terme préférentiel MedDRA (Version 17.0) |
Fréquence des effets indésirables* |
|
Infections et infestations |
Grippe |
Peu fréquent |
|
Méningite aseptique |
Indéterminé |
|
|
Affections hématologiques et du système lymphatique |
Hémolyse, Anémie, Thrombocytopénie, Lymphadénopathie |
Indéterminé |
|
Affections du système immunitaire |
Choc anaphylactique, Réaction anaphylactique ou anaphylactoïde, Hypersensibilité |
Indéterminé |
|
Affections psychiatriques |
Anxiété, Agitation |
Peu fréquent |
|
Nervosité |
Indéterminé |
|
|
Affections du système nerveux |
Céphalées |
Fréquent |
|
Léthargie |
Peu fréquent |
|
|
Accident vasculaire cérébral, Accident ischémique transitoire, Convulsions, Migraine, Sensation vertigineuses, Paresthésie, Syncope, Tremblements |
Indéterminé |
|
|
Affections oculaires |
Vision trouble |
Peu fréquent |
|
Thrombose veineuse rétinienne, Troubles visuels, Douleur oculaire, Photophobie |
Indéterminé |
|
|
Affections cardiaques |
Palpitations |
Peu fréquent |
|
Infarctus du myocarde, Cyanose, Tachycardie |
Indéterminé |
|
|
Affections vasculaires |
Flush |
Fréquent |
|
Fluctuations de la tension artérielle |
Peu fréquent |
|
|
Thrombose artérielle, Thrombose de la veine cave, Thrombose veineuse profonde, Thrombophlébite, Hypotension, Hypertension, Pâleur |
Indéterminé |
|
|
Affections respiratoires, thoraciques et médiastinales |
Dyspnée, Épistaxis |
Peu fréquent |
|
Embolie pulmonaire, Œdème pulmonaire, Hypoxie, Bronchospasme, Respiration sifflante, Hyperventilation, Sensation de gorge serrée, Toux |
Indéterminé |
|
|
Affections gastro-intestinales |
Nausée, Vomissement |
Fréquent |
|
Diarrhée, Douleur abdominale, Gêne abdominale, Stomatite |
Peu fréquent |
|
|
Douleur abdominale, Dyspepsie |
Indéterminé |
|
|
Affections hépato-biliaires |
Atteinte hépatique (non infectieuse) |
Indéterminé |
|
Affections de la peau et du tissu sous-cutané |
Urticaire, Prurit, sueur froide, Hyperhidrose |
Peu fréquent |
|
Angiœdème, Dermatite, Érythème, Rash, Eruption eczématiforme |
Indéterminé |
|
|
Affections des musculo-squelettiques et systémiques |
Dorsalgies, Crampes musculaires, Douleur aux extrémités |
Peu fréquent |
|
Arthralgie, Myalgie |
Indéterminé |
|
|
Affections rénales et urinaires |
Insuffisance rénale |
Indéterminé |
|
Troubles généraux et anomalies au site d’administration |
Fatigue, Frissons, Fièvre |
Fréquent |
|
Douleur thoracique, Malaise, Douleur, gêne thoracique, Sensation de malaise, Sensation de froid ou de chaleur, Symptômes pseudo-grippaux, Érythème / douleur au site de perfusion, Extravasation au site de perfusion |
Peu fréquent |
|
|
Réactions au site de perfusion, Asthénie, Œdème |
Indéterminé |
|
|
Investigations |
Augmentation de la tension artérielle |
Peu fréquent |
|
Test de Coombs direct positif |
Indéterminé |
|
|
Affections nutritionnelles et du métabolisme |
Baisse de l’appétit |
Peu fréquent |
* Basée sur le pourcentage par perfusions.
Pour la sécurité relative aux agents transmissibles, voir rubrique 4.4.
Déclaration des effets indésirables suspectés
La déclaration des effets indésirables suspectés après autorisation du médicament est importante. Elle permet une surveillance continue du rapport bénéfice/risque du médicament. Les professionnels de santé déclarent tout effet indésirable suspecté via le système national de déclaration : Agence nationale de sécurité du médicament et des produits de santé (ANSM) et réseau des Centres Régionaux de Pharmacovigilance - Site internet : https://signalement.social-sante.gouv.fr/.
5. PROPRIETES PHARMACOLOGIQUES
5.1. Propriétés pharmacodynamiques
GAMMAGARD contient principalement des immunoglobulines G (IgG) entières à large spectre d’activité anticorps vis à vis de divers agents infectieux et de toxines.
GAMMAGARD contient des anticorps de la classe des anti-IgG présents dans la population normale. En général, ce produit est préparé à partir de pools de plasma provenant d'au moins 1000 dons. La répartition des sous classes d’IgG est proportionnelle à celui du plasma humain natif. Des administrations appropriées de GAMMAGARD sont susceptibles de ramener à une valeur normale des taux anormalement bas d’immunoglobuline G.
Le mécanisme d’action pour les indications autres que le traitement substitutif n’est pas totalement élucidé. Il comprend toutefois des effets immunomodulateurs.
Le taux d'IgG n'est pas inférieur à 92 % et est en moyenne de 94,7 %
Titre des anticorps : Anti-viraux
|
anti-CMV |
≥ |
472 U-PEI/g de protéines |
|
anti-HBs |
≥ |
1,9 UI/g de protéines |
|
anti-hépatite A |
≥ |
530 UI/g de protéines |
|
anti-rougeole |
≥ |
283 UI/g de protéines |
|
anti-zona-varicelle |
|
Indéterminé |
Le respect des fonctions biologiques des immunoglobulines a été validé par un test de la fonction Fc.
5.2. Propriétés pharmacocinétiques
La demi-vie de GAMMAGARD est de 37.7 ± 15 jours. Cette demi-vie peut être variable d’un patient à un autre, particulièrement en cas de déficit immunitaire primitif.
Les IgG et les complexes d’IgG sont dégradés dans les cellules du système réticulo-endothélial.
5.3. Données de sécurité préclinique
Les immunoglobulines sont des composants naturels du corps humain.
La tolérance de GAMMAGARD a été démontrée dans plusieurs études pré-cliniques. Les données non cliniques issues des études conventionnelles de pharmacologie de sécurité et de génotoxicité n’ont pas révélé de risque particulier pour l’homme.
Les résultats cliniques n’ayant apporté aucune preuve d’un potentiel carcinogène des immunoglobulines, aucune étude expérimentale sur des espèces variées n’a été menée.
Poudre : sodium, glucose, albumine humaine, glycine et macrogol 3350.
Solvant : eau pour préparations injectables.
La stabilité physico-chimique de la solution reconstituée a été démontrée pendant 2 heures à une température ne dépassant pas 25°C. D’un point de vue microbiologique, une utilisation immédiate est recommandée. La durée et les conditions de conservation lors de l’utilisation sont de la responsabilité de l’utilisateur et ne devraient pas dépasser 24 heures entre 2 et 8°C, dès lors que la reconstitution a été réalisée dans des conditions d’asepsie contrôlées et validées.
6.4. Précautions particulières de conservation
À conserver à une température ne dépassant pas 25°C.
Ne pas congeler, le flacon de solvant pourrait se casser.
À conserver à l'abri de la lumière dans l’emballage extérieur.
Ne pas utiliser après la date d’expiration.
Tenir hors de la portée et de la vue des enfants.
Pour les conditions de conservation du médicament reconstitué, voir rubrique 6.3.
6.5. Nature et contenu de l'emballage extérieur
5 g de poudre en flacon (verre de type I) + 100 ml de solvant en flacon (verre de type I) muni de bouchon (bromobutyle) avec un système de transfert, un nécessaire de perfusion et un suspensoir en plastique - boîte de 1.
10 g de poudre en flacon (verre de type I) + 200 ml de solvant en flacon (verre de type I) muni de bouchon (bromobutyle) avec un système de transfert, un nécessaire de perfusion et un suspensoir en plastique - boîte de 1.
Toutes les présentations peuvent ne pas être commercialisées.
6.6. Précautions particulières d’élimination et de manipulation
Lorsque la reconstitution est réalisée de manière aseptique en dehors d’une hotte à flux laminaire stérile, l’administration doit être initiée dès que possible, mais pas plus de 2 heures après reconstitution. Lorsque la reconstitution est réalisée de manière aseptique sous une hotte à flux laminaire stérile, le produit reconstitué peut être conservé jusqu’à 24 heures, au réfrigérateur (2-8°C). Si ces conditions ne sont pas respectées, la stérilité du produit reconstitué ne peut pas être maintenue. Les flacons partiellement utilisés doivent être jetés.
Une dissolution totale doit être obtenue au bout de 30 minutes. Le produit doit être amené à température ambiante ou à température du corps avant utilisation.
Avant reconstitution :
La poudre doit être blanche ou avoir un aspect très légèrement jaune pratiquement exempt de particules visibles. Le produit reconstitué doit faire l’objet d’une inspection visuelle avant administration (particules, coloration). La solution doit être limpide ou légèrement opalescente, incolore ou jaune pâle.
Reconstitution de la solution :
Amener les deux flacons (de poudre et de solvant) à température ambiante. Maintenir cette température pendant la reconstitution et l'administration.
|
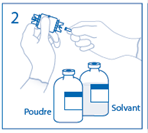
1. Retirer la capsule protectrice du flacon de solvant et du flacon de poudre et désinfecter la surface de chaque bouchon à l'aide d'une solution antiseptique. |
|
|
|
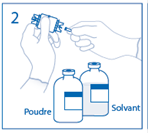
2. Retirer le capuchon du perforateur de l’une des extrémités du dispositif de transfert. Ne pas toucher le perforateur dénudé. |
|
|
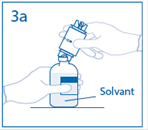
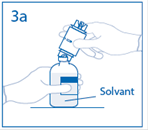
3a. Placer le flacon de solvant sur une surface plane. Enfoncer le perforateur dénudé du dispositif de transfert au centre du bouchon du flacon de solvant. Attention: si le perforateur n’est pas inséré au centre du bouchon, celui-ci pourrait se détacher. |
|
|
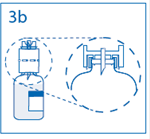
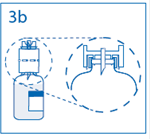
3b. Exercer une pression ferme sur le dispositif de transfert pour s’assurer que le col du flacon s’encastre totalement dans celui-ci. Retirer le capuchon du perforateur de l’autre extrémité du dispositif de transfert, tout en maintenant le dispositif en place. Ne pas toucher le perforateur dénudé. |
|
|
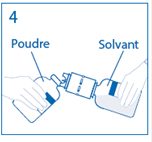
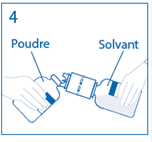
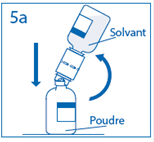
4. Maintenir le flacon de solvant fixé au dispositif de transfert incliné tout contre le flacon de poudre afin d’éviter toute perte de solvant.
Remarque : ne pas retourner complètement le flacon de solvant, car cela pourrait entraîner une perte de solvant. |
|
|
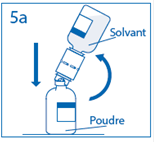
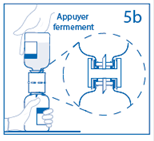
5a. Percer le centre du bouchon du flacon de poudre en retournant simultanément le flacon de solvant afin d’éviter toute perte de solvant. Attention : si le perforateur n’est pas inséré au centre du bouchon, celui-ci pourrait se détacher et de l’air pourrait rentrer dans le flacon. |
|
|
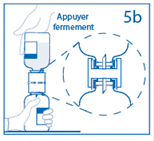
5b. Exercer une pression ferme sur le flacon de solvant pour s’assurer que le col du flacon de poudre s’encastre totalement dans le dispositif de transfert. |
|
|
6. Lorsque le solvant est transféré dans le flacon de poudre, retirer le dispositif de transfert fixé au flacon vide de solvant. Agiter modérément le flacon de poudre par un mouvement de rotation doux jusqu’à dissolution complète de la poudre en moins de 30 minutes. Attention : ne pas secouer le flacon afin d’éviter la formation de mousse. Eliminer le dispositif de transfert réservé à un usage unique. |
Le produit reconstitué doit faire l’objet d’une inspection visuelle avant administration, afin de s'assurer qu'il ne contient pas de particules. La solution reconstituée doit être limpide ou légèrement opalescente, incolore ou jaune pâle.
Ne pas utiliser de solutions présentant un aspect non homogène ou contenant un dépôt.
Administration :
· Le produit doit être administré immédiatement par voie intraveineuse en une seule fois après reconstitution.
· Retirer l'embout protecteur du perforateur du nécessaire de perfusion et enfoncer ce perforateur dans le bouchon du flacon de produit reconstitué.
· Fermer hermétiquement la tubulure de perfusion à l'aide du clamp à roulette.
· Presser légèrement la chambre compte-gouttes pour y faire couler la solution; remplir la chambre compte-gouttes au 1/3 de son volume environ.
· Ouvrir le clamp à roulette avec précaution et amorcer la tubulure en prenant soin d'éliminer toute bulle d'air dans la tubulure.
· Connecter le nécessaire de perfusion au cathéter de façon aseptique et en évitant tout passage d'air.
Tout produit non utilisé ou déchet doit être éliminé conformément à la réglementation en vigueur.
7. TITULAIRE DE L’AUTORISATION DE MISE SUR LE MARCHE
INDUSTRIESTRASSE 67
A-1221 VIENNE
AUTRICHE
8. NUMERO(S) D’AUTORISATION DE MISE SUR LE MARCHE
· 34009 560 315 7 3 : 10 g de poudre en flacon (verre de type I) + 200 ml de solvant en flacon (verre de type I) muni de bouchon (bromobutyle) avec un système de transfert, un nécessaire de perfusion et un suspensoir en plastique - boîte de 1.
9. DATE DE PREMIERE AUTORISATION/DE RENOUVELLEMENT DE L’AUTORISATION
[à compléter ultérieurement par le titulaire]
10. DATE DE MISE A JOUR DU TEXTE
[à compléter ultérieurement par le titulaire]
<{JJ mois AAAA}>
Sans objet.
12. INSTRUCTIONS POUR LA PREPARATION DES RADIOPHARMACEUTIQUES
Liste I.
Médicament soumis à prescription hospitalière. La prescription par un médecin exerçant dans un établissement de transfusion sanguine autorisé à dispenser des médicaments aux malades qui y sont traités est également autorisée.
ANSM - Mis à jour le : 04/07/2025
GAMMAGARD 50 mg/ml, poudre et solvant pour solution pour perfusion
Immunoglobuline humaine normale
· Gardez cette notice. Vous pourriez avoir besoin de la relire.
· Si vous avez d’autres questions, interrogez votre médecin ou votre pharmacien.
· Ce médicament vous a été personnellement prescrit. Ne le donnez pas à d’autres personnes. Il pourrait leur être nocif, même si les signes de leur maladie sont identiques aux vôtres.
· Si vous ressentez un quelconque effet indésirable, parlez-en à votre médecin ou à votre pharmacien. Ceci s’applique aussi à tout effet indésirable qui ne serait pas mentionné dans cette notice. Voir rubrique 4.
1. Qu'est-ce que GAMMAGARD 50 mg/ml, poudre et solvant pour solution pour perfusion et dans quels cas est-il utilisé ?
2. Quelles sont les informations à connaître avant d'utiliser GAMMAGARD 50 mg/ml, poudre et solvant pour solution pour perfusion?
3. Comment utiliser GAMMAGARD 50 mg/ml, poudre et solvant pour solution pour perfusion ?
4. Quels sont les effets indésirables éventuels ?
5. Comment conserver GAMMAGARD 50 mg/ml, poudre et solvant pour solution pour perfusion ?
6. Contenu de l’emballage et autres informations.
1. QU’EST-CE QUE GAMMAGARD 50 mg/ml, poudre et solvant pour solution pour perfusion ET DANS QUELS CAS EST-IL UTILISE ?
ANTISERUMS ET IMMUNOGLOBULINES.
GAMMAGARD appartient à la classe des médicaments dénommés immunoglobulines. Ces médicaments contiennent des anticorps humains, également présents dans votre sang. Les anticorps aident votre corps à combattre les infections. Les médicaments comme GAMMAGARD sont utilisés chez les patients ne possédant pas suffisamment d’anticorps dans leur sang et sujets à de fréquentes infections. Ils peuvent aussi être utilisés chez les patients ayant besoin d’anticorps supplémentaires lors du traitement de certains troubles inflammatoires (maladies auto-immunes).
GAMMAGARD est une immunoglobuline humaine normale indiquée chez les patients avec déficits en immunoglobuline A (IgA) et anticorps anti-IgA :
Le traitement de patients ne possédant pas suffisamment d’anticorps (traitement substitutif). Ces patients sont répartis en cinq groupes :
· Patients présentant un défaut congénital de production d’anticorps (déficits immunitaires primitifs, DIP) ;
· Patients présentant un cancer du sang (leucémie lymphoïde chronique) entraînant un déficit de production d’anticorps et des infections récurrentes en cas d’échec du traitement préventif par antibiotiques ;
· Patients présentant un cancer de la moelle osseuse (myélome multiple) et un déficit de production d’anticorps accompagnée d’infections récurrentes, qui ne répondent pas à un vaccin contre certaines bactéries (pneumocoques) ;
· Patients ayant un déficit immunitaire suite à une infection par le VIH depuis leur naissance et présentant des infections fréquentes ;
· Patients présentant une faible production d’anticorps après une greffe de cellules de moelle osseuse provenant d’un donneur.
Le traitement des patients souffrant de certains troubles inflammatoires (traitements immunomodulateurs). Ces patients sont répartis en trois groupes :
· Patients n’ayant pas assez de plaquettes dans le sang (purpura thrombopénique immunitaire primaire/idiopathique) et présentant des risques élevés d’hémorragie ou devant subir une opération chirurgicale dans un futur proche ;
· Patients atteints d’une maladie associée à une inflammation de plusieurs nerfs du corps (syndrome de Guillain Barré) ;
· Patients présentant une maladie provoquant une inflammation de plusieurs organes du corps (maladie de Kawasaki).
2. QUELLES SONT LES INFORMATIONS A CONNAITRE AVANT D’UTILISER GAMMAGARD 50 mg/ml, poudre et solvant pour solution pour perfusion?
N’utilisez jamais GAMMAGARD 50 mg/ml, poudre et solvant pour solution pour perfusion :
· Si vous êtes allergique (hypersensible) à l'immunoglobuline humaine normale ou à l’un des autres composants contenus dans ce médicament, mentionnés dans la rubrique 6.
Faites attention avec GAMMAGARD 50 mg/ml, poudre et solvant pour solution pour perfusion :
Une surveillance est requise au cours de la perfusion
Vous serez attentivement surveillé lors de la période de perfusion de GAMMAGARD pour éviter tout risque de réaction. Le médecin adaptera le débit de la perfusion de GAMMAGARD en fonction de votre cas.
· Si GAMMAGARD est administré à un débit élevé,
· Si vous possédez une faible quantité d’anticorps dans votre sang (hypo- ou agammaglobulinémie),
· Si vous n’avez jamais reçu ce médicament auparavant ou,
· Si la dernière perfusion a été effectuée il y a longtemps (depuis plusieurs semaines par exemple).
Dans ces cas, vous devez faire l’objet d’une surveillance attentive lors de votre perfusion et durant 1 heure après la fin de cette dernière.
Si GAMMAGARD vous a déjà été administré auparavant et que vous avez reçu le dernier traitement récemment, alors vous ferez l’objet d’une surveillance pendant au moins 20 minutes après votre perfusion.
Un ralentissement ou un arrêt de la perfusion est requis
Dans de rares cas, votre corps peut réagir à des médicaments contenant des anticorps. Cela peut arriver particulièrement si vous souffrez d’un déficit en immunoglobuline A. Dans ces rares cas, vous pourriez voir apparaître des réactions allergiques, comme une chute brutale de la tension artérielle ou un choc, même si vous avez déjà été traité avec des médicaments contenant des anticorps dans le passé.
Si vous présentez l’un des effets indésirables suivants, vous devez en informer votre médecin ou votre infirmière immédiatement :
· Respiration sifflante soudaine, difficulté à respirer ou oppression dans la poitrine,
· Mal de tête,
· Fièvre,
· Gonflement des paupières, des lèvres, du visage ou des vaisseaux sanguins,
· Boutons gonflés (type urticaire) sur la peau ou boutons rouges avec démangeaisons,
· Démangeaisons de tout le corps.
Selon la décision du médecin, le débit de perfusion peut être diminué voire totalement arrêté.
Groupes spécifiques de patients
Votre médecin devra vous surveiller tout particulièrement si vous souffrez d’une surcharge pondérale, si vous êtes âgé, diabétique ou si vous êtes immobilisé, si vous utilisez des œstrogènes ou si vous avez une sonde vasculaire à demeure.
Il vous surveillera particulièrement :
· Si vous présentez une augmentation de la pression du sang,
· Si vous présentez un faible volume sanguin (hypovolémie),
· Si vous avez une viscosité du sang augmentée ou des problèmes circulatoires (maladies vasculaires). Dans ces conditions, les immunoglobulines peuvent accroître dans de très rares cas le risque d'attaque cardiaque (infarctus), d’attaque cérébrale (accident vasculaire cérébral), d’occlusion des vaisseaux du poumon (embolie pulmonaire) ou d’occlusion des vaisseaux du corps (thrombose veineuse profonde).
Informez votre médecin si vous êtes diabétique. GAMMAGARD ne contient pas de saccharose ou de maltose. GAMMAGARD 50 mg/ml contient 20 mg de glucose par dose, ce qui peut modifier votre taux de sucre dans le sang.
Votre médecin devra également prendre des précautions particulières :
· Si vous avez ou avez eu des problèmes rénaux auparavant, ou,
· Si vous prenez des médicaments qui risquent d’affecter vos reins (médicaments néphrotoxiques), car il existe un risque très faible d’insuffisance rénale aiguë. Veuillez informer votre médecin si vous avez des affections rénales,
· Le taux de protéines du sang peut augmenter et entrainer une diminution de la fluidité du sang (augmentation de la viscosité). Votre médecin vous hydratera avant administration. Il surveillera votre état de santé et fera des analyses sanguines.
Informations sur les substances de base de GAMMAGARD
GAMMAGARD est préparé à partir de plasma humain (le liquide composant le sang). Lorsque des médicaments sont préparés à partir de sang ou de plasma humain, des mesures de prévention contre la transmission d’agents infectieux aux patients sont mises en place. Celles-ci comprennent une sélection soigneuse des donneurs de sang et de plasma de façon à exclure les donneurs risquant d’être porteurs d’infections, et un contrôle de chaque don et mélanges de plasma pour la recherche de virus/d’infections. Les fabricants de ces médicaments mettent également en œuvre dans leur procédé de fabrication des étapes capables d’éliminer ou d’inactiver les virus. Cependant, lorsque des médicaments préparés à partir de sang ou de plasma humain sont administrés, le risque de transmission de maladies infectieuses ne peut pas être totalement exclu. Ceci s’applique également aux virus inconnus ou émergents ou autres types d’agents infectieux.
Les mesures prises pour la fabrication de GAMMAGARD sont considérées comme efficaces pour lutter contre le risque d’infection par les virus enveloppés tels que le virus de l’immunodéficience humaine (VIH), le virus de l’hépatite B, le virus de l’hépatite C, et les virus non enveloppés de l’hépatite A et le parvovirus B19.
GAMMAGARD contient aussi des anticorps qui peuvent protéger contre une infection par le virus de l’hépatite A et le parvovirus B19.
Enfants
Sans objet.
Autres médicaments et GAMMAGARD 50 mg/ml, poudre et solvant pour solution pour perfusion
Veuillez avertir votre médecin si vous avez reçu un vaccin au cours des six dernières semaines.
La perfusion d’immunoglobulines telles que GAMMAGARD peut atténuer l’effet de certains vaccins à virus vivants, comme le vaccin contre la rougeole, la rubéole, les oreillons et la varicelle. Par conséquent, après avoir reçu des immunoglobulines, il est conseillé d’attendre un délai de 3 mois avant de pouvoir recevoir un vaccin vivant atténué. Pour le vaccin contre la rougeole, vous pourriez avoir à attendre 1 an après une perfusion d’immunoglobulines.
Si le patient a reçu des vaccins constitués de virus vivants atténués (rougeole, rubéole, oreillons, varicelle), au cours des deux semaines précédant la perfusion, un contrôle des anticorps protecteurs post-vaccinaux peut être nécessaire en vue d'un éventuel rappel.
Effet sur les tests sanguins
GAMMAGARD contient une grande variété d'anticorps différents. Certains peuvent avoir des effets sur les analyses de sang. Si vous devez subir une analyse de sang, veuillez-en informer la personne chargée de prélever votre sang ou votre médecin que vous avez reçu GAMMAGARD.
Si vous prenez ou avez pris récemment un autre médicament, y compris un médicament obtenu sans ordonnance, parlez-en à votre médecin ou à votre pharmacien.
Informez votre médecin si vous êtes enceinte ou si vous allaitez. Votre médecin vous dira si GAMMAGARD peut être utilisé durant votre grossesse ou en période d’allaitement.
Il n’existe pas d’études cliniques avec GAMMAGARD chez la femme enceinte ou allaitant. Cependant, les traitements par anticorps sont utilisés chez la femme enceinte ou allaitant. L’expérience acquise suggère qu'aucun effet nocif n'est attendu au cours de la grossesse ou pour le bébé.
Si vous allaitez et que vous recevez GAMMAGARD, les anticorps de ce produit se retrouveront dans votre lait. Ainsi votre bébé pourra être protégé contre certaines infections.
Demandez conseil à votre médecin ou à votre pharmacien avant de prendre tout médicament.
Conduite de véhicules et utilisation de machines
Les patients peuvent présenter des réactions (par exemple des étourdissements ou des nausées) pendant le traitement par GAMMAGARD, ce qui risque d’affecter la capacité à conduire ou à utiliser des machines. Le cas échéant, vous devez attendre que les réactions disparaissent.
GAMMAGARD 50 mg/ml, poudre et solvant pour solution pour perfusion contient du sodium et du glucose.
Informez votre médecin si vous êtes diabétique. GAMMAGARD contient 0,4 g de glucose maximum. Ceci est à prendre en compte chez les patients diabétiques.
GAMMAGARD contient 3,4 mg/ml de sodium. Cela équivaut à 34 % de l’apport alimentaire quotidien maximal recommandé de sodium pour un adulte.
3. COMMENT UTILISER GAMMAGARD 50 mg/ml, poudre et solvant pour solution pour perfusion ?
Au début de la perfusion, vous recevrez GAMMAGARD à un débit faible. Selon votre état, votre médecin pourra augmenter progressivement le débit de la perfusion.
Utilisation chez les enfants
Les mêmes indications, doses et fréquences de perfusion s’appliquent aux adultes et aux enfants (de 0 à 18 ans).
Si vous avez utilisé plus de GAMMAGARD 50 mg/ml, poudre et solvant pour solution pour perfusion que vous n'auriez dû :
Consultez immédiatement votre médecin ou votre pharmacien.
Votre sang peut devenir trop épais (hyperviscosité). Le sang a alors du mal à circuler dans les vaisseaux de votre corps. Cela peut entrainer une oxygénation plus faible des organes vitaux, tels que le cerveau, les poumons, etc. Cela peut se produire particulièrement si vous êtes un patient à risque, comme par exemple un patient âgé ou un patient ayant un problème au cœur ou aux reins. Informez votre médecin si vous présentez des problèmes de santé connus.
Si vous oubliez d’utiliser GAMMAGARD 50 mg/ml, poudre et solvant pour solution pour perfusion :
Ne prenez pas de dose double pour compenser la dose simple que vous avez oublié de prendre.
Si vous arrêtez d'utiliser GAMMAGARD 50 mg/ml, poudre et solvant pour solution pour perfusion :
Si vous avez d'autres questions sur l'utilisation de ce médicament, demandez plus d'informations à votre médecin ou à votre pharmacien.
4. QUELS SONT LES EFFETS INDESIRABLES EVENTUELS ?
Cependant, des effets indésirables peuvent être réduits en diminuant le débit de perfusion.
Les effets indésirables suivants peuvent généralement survenir après le traitement par des immunoglobulines (médicaments comme GAMMAGARD) :
· Les effets indésirables fréquents ou peu fréquents (touchent moins de 1 patient sur 10 mais plus de 1 patient sur 1 000) sont frissons, mal de tête, fièvre, vomissements, réactions allergiques, nausées, douleur articulaire, tension artérielle faible et douleur modérée dans le bas du dos.
· Les effets indésirables rares (touchent moins de 1 patient sur 1 000) sont :
o Chute brutale de la tension artérielle,
o Symptômes ressemblant à de l’eczéma (réactions cutanées transitoires).
· Les effets indésirables très rares (touchent moins de 1 patient sur 10 000 ou dont la fréquence ne peut pas être estimée à partir des données disponibles) sont :
o Cas isolés de réactions allergiques (choc anaphylactique), même si vous n’en avez pas eu lors de précédentes perfusions,
o Cas d’inflammation passagère des méninges (méningite aseptique réversible)
o Cas isolés de réduction passagère du nombre de globules rouges (anémie hémolytique réversible/hémolyse)
o Augmentation transitoire des paramètres de la fonction hépatique, une augmentation de la teneur en créatinine sanguine et insuffisance rénale
o Formation de caillot sanguin dans les veines (réactions thromboemboliques), qui peut provoquer une attaque cardiaque (infarctus), une attaque cérébrale (accident vasculaire cérébral), une occlusion des vaisseaux du poumon (embolie pulmonaire) et une occlusion des vaisseaux du corps (thrombose veineuse profonde).
Les effets indésirables listés ci-dessous ont été rapportés par quelques patients lors d’essais cliniques ou après commercialisation :
· Effets indésirables très fréquents (observés chez plus de 1 patient sur 10) : aucun
· Effets indésirables fréquents (observés chez moins de 1 patient sur 10) : mal de tête, rougeurs, nausées, vomissements, fatigue, frissons, fièvre
· Effets indésirables peu fréquents (observés chez moins de 1 patient sur 100) :
o grippe,
o anxiété, agitation,
o somnolence anormale,
o vision trouble,
o palpitations,
o respiration coupée,
o saignement de nez,
o diarrhées, douleur abdominale, inconfort au niveau de l’estomac, inflammation de l’estomac,
o démangeaisons, urticaire, sudation excessive, sueur froide,
o mal de dos, crampe musculaire, douleur aux extrémités,
o malaise, douleur sensation de malaise, douleur dans la poitrine, gêne thoracique, sensation de froid ou de chaud, maladie ressemblant à la grippe, rougeur au site de perfusion, douleur au site de perfusion,
o tension artérielle élevée,
o baisse de l’appétit.
· Fréquence indéterminée (ne peut être estimée sur la base des données disponibles) :
o Inflammation des membranes du cerveau non provoquées par une infection bactérienne,
o destruction des globules rouges, diminution du nombre des globules rouges, diminution des plaquettes, ganglions lymphatiques gonflés,
o réaction allergique de gravité variable comprenant choc allergique, sensation cutanée anormale,
o agitation,
o vertiges, fourmillements, engourdissement, tremblements involontaires, convulsions, attaque cérébrale (accident vasculaire cérébral), migraine, perte de conscience,
o gêne par la lumière (photophobie), trouble visuel, douleur à l’œil, occlusion du vaisseau sanguin central de l’œil,
o attaque cardiaque, coloration bleue de la peau, augmentation du rythme cardiaque, réduction du rythme cardiaque,
o tension artérielle élevée, pâleur, faible tension artérielle, inflammation des veines, occlusion des vaisseaux sanguins,
o toux, sensation de gorge serrée, saturation en oxygène diminuée dans le sang, respiration accélérée, respiration sifflante, spasme des voies aériennes, occlusion des vaisseaux sanguins dans les poumons, liquide dans les poumons,
o digestion perturbée,
o inflammation du foie (non infectieuse),
o rougeur de la peau, éruptions cutanées de type eczéma, inflammation de la peau, gonflement (type urticaire) sous la peau,
o douleur articulaire et musculaire,
o insuffisance rénale,
o faiblesse générale, œdème, réactions au niveau du site de perfusion,
o résultat positif au test de Coombs.
Déclaration des effets secondaires
Si vous ressentez un quelconque effet indésirable, parlez-en à votre médecin ou à votre pharmacien ou à votre infirmier/ère. Ceci s’applique aussi à tout effet indésirable qui ne serait pas mentionné dans cette notice. Vous pouvez également déclarer les effets indésirables directement via le système national de déclaration : Agence nationale de sécurité du médicament et des produits de santé (ANSM) et réseau des Centres Régionaux de Pharmacovigilance (Site internet : https://signalement.social-sante.gouv.fr/).
En signalant les effets indésirables, vous contribuez à fournir davantage d’informations sur la sécurité du médicament.
5. COMMENT CONSERVER GAMMAGARD 50 mg/ml, poudre et solvant pour solution pour perfusion ?
Tenir ce médicament hors de la vue et de la portée des enfants.
N’utilisez pas ce médicament après la date de péremption indiquée sur la boîte après « Péremption ». La date de péremption fait référence au dernier jour de ce mois.
A conserver à une température ne dépassant pas + 25°C.
Conservez dans l'emballage extérieur à l'abri de la lumière.
Ne pas congeler.
N’utilisez pas ce médicament si vous remarquez des particules visibles ou une décoloration.
Après reconstitution, la solution doit être utilisée immédiatement.
La stabilité physico-chimique de la solution reconstituée a été démontrée pendant 2 heures à une température ne dépassant pas 25°C. D’un point de vue microbiologique, une utilisation immédiate est recommandée. La durée et les conditions de conservation lors de l’utilisation sont de la responsabilité de l’utilisateur et ne devraient pas dépasser 24 heures entre 2 et 8°C, dès lors que la reconstitution a été réalisée dans des conditions d’asepsie contrôlées et validées.
Ne jetez aucun médicament au tout-à-l’égout ou avec les ordures ménagères. Demandez à votre pharmacien d’éliminer les médicaments que vous n’utilisez plus. Ces mesures contribueront à protéger l’environnement.
6. CONTENU DE L’EMBALLAGE ET AUTRES INFORMATIONS
Ce que contient GAMMAGARD 50 mg/ml, poudre et solvant pour solution pour perfusion
· La substance active est :
Immunoglobuline humaine normale*........................................................................................ 50 mg
Pour 1 ml de solution reconstituée.
* La poudre a une teneur maximale en IgA inférieure à 44 μg/g de protéines (soit 2,2 μg/ml).
Un flacon de 100 ml contient 5 g d'immunoglobuline humaine normale.
Un flacon de 200 ml contient 10 g d'immunoglobuline humaine normale.
· Les autres composants sont :
Pour la poudre: le sodium, le glucose, l'albumine humaine, la glycine, le macrogol 3350.
Pour le solvant : l'eau pour préparations injectables.
Titulaire de l’autorisation de mise sur le marché
INDUSTRIESTRASSE 67
A-1221 VIENNE
AUTRICHE
Exploitant de l’autorisation de mise sur le marché
16 PLACE DE L’IRIS
92400 COURBEVOIE
BAXALTA BELGIUM MANUFACTURING SA
BOULEVARD RENÉ BRANQUART 80
B 7860 - LESSINES
BELGIQUE
Noms du médicament dans les Etats membres de l'Espace Economique Européen
Sans objet.
La dernière date à laquelle cette notice a été révisée est :
Des informations détaillées sur ce médicament sont disponibles sur le site Internet de l’ANSM (France).
Il existe aussi des liens vers d’autres sites concernant les maladies rares et leur traitement.
Les informations suivantes sont destinées exclusivement aux professionnels de santé :
Précautions particulières de conservation :
Lorsque la reconstitution est réalisée de manière aseptique en dehors d’une hotte à flux laminaire stérile, l’administration doit être initiée dès que possible, mais pas plus de 2 heures après reconstitution. Lorsque la reconstitution est réalisée de manière aseptique sous une hotte à flux laminaire stérile, le produit reconstitué peut être conservé jusqu’à 24 heures, sous réfrigération constante (2-8°C). Les flacons partiellement utilisés doivent être jetés.
Reconstitution de la solution :
Amener les deux flacons (de poudre et de solvant) à température ambiante. Maintenir cette température pendant la reconstitution et l'administration.
|
1. Retirer la capsule protectrice du flacon de solvant et du flacon de poudre et désinfecter la surface de chaque bouchon à l'aide d'une solution antiseptique. |
|
|
|
2. Retirer le capuchon du perforateur de l’une des extrémités du dispositif de transfert. Ne pas toucher le perforateur dénudé. |
|
|
3a. Placer le flacon de solvant sur une surface plane. Enfoncer le perforateur dénudé du dispositif de transfert au centre du bouchon du flacon de solvant. Attention : si le perforateur n’est pas inséré au centre du bouchon, celui-ci pourrait se détacher. |
|
|
3b. Exercer une pression ferme sur le dispositif de transfert pour s’assurer que le col du flacon s’encastre totalement dans celui-ci. Retirer le capuchon du perforateur de l’autre extrémité du dispositif de transfert, tout en maintenant le dispositif en place. Ne pas toucher le perforateur dénudé. |
|
|
4. Maintenir le flacon de solvant fixé au dispositif de transfert incliné tout contre le flacon de poudre afin d’éviter toute perte de solvant.
Remarque : ne pas retourner complètement le flacon de solvant, car cela pourrait entraîner une perte de solvant. |
|
|
5a. Percer le centre du bouchon du flacon de poudre en retournant simultanément le flacon de solvant afin d’éviter toute perte de solvant. Attention : si le perforateur n’est pas inséré au centre du bouchon, celui-ci pourrait se détacher et de l’air pourrait rentrer dans le flacon. |
|
|
5b. Exercer une pression ferme sur le flacon de solvant pour s’assurer que le col du flacon de poudre s’encastre totalement dans le dispositif de transfert. |
|
|
6. Lorsque le solvant est transféré dans le flacon de poudre, retirer le dispositif de transfert fixé au flacon vide de solvant. Agiter modérément le flacon de poudre par un mouvement de rotation doux jusqu’à dissolution complète de la poudre en moins de 30 minutes. Attention : ne pas secouer le flacon afin d’éviter la formation de mousse. Eliminer le dispositif de transfert réservé à un usage unique. |
Le produit reconstitué doit faire l’objet d’une inspection visuelle avant administration, afin de s'assurer qu'il ne contient pas de particules. La solution reconstituée doit être limpide ou légèrement opalescente, incolore ou jaune pâle.
Ne pas utiliser de solutions présentant un aspect non homogène ou contenant un dépôt.
Administration :
· Le produit doit être administré immédiatement par voie intraveineuse en une seule fois après reconstitution.
· Retirer l'embout protecteur du perforateur du nécessaire de perfusion et enfoncer ce perforateur dans le bouchon du flacon de produit reconstitué.
· Fermer hermétiquement la tubulure de perfusion à l'aide du clamp à roulette.
· Presser légèrement la chambre compte-gouttes pour y faire couler la solution; remplir la chambre compte-gouttes au 1/3 de son volume environ.
· Ouvrir le clamp à roulette avec précaution et amorcer la tubulure en prenant soin d'éliminer toute bulle d'air dans la tubulure.
· Connecter le nécessaire de perfusion au cathéter de façon aseptique et en évitant tout passage d'air.
GAMMAGARD doit être perfusé par voie intraveineuse à un débit initial de 0,5 ml/kg de poids corporel/heure. En général, il est recommandé que les patients débutant un traitement avec GAMMAGARD ou passant d’une spécialité pharmaceutique d’IgIV à une autre, commencent à un débit faible et augmentent ensuite jusqu’au débit maximal, s’ils ont toléré plusieurs perfusions à des débits de perfusion intermédiaires.
Si le débit de perfusion est bien toléré, il peut être augmenté progressivement jusqu’à un maximum de 4 ml/kg de poids corporel/heure. Si aucun effet indésirable ne survient, le débit peut être augmenté progressivement jusqu'à un maximum de 8 ml/kg de poids corporel/heure.
Tout produit non utilisé ou déchet doit être éliminé conformément à la réglementation en vigueur.
Modalités de manipulation et d’élimination
· Une dissolution totale doit être obtenue au bout de 30 minutes.
· Le produit doit être amené à température ambiante ou à température du corps avant utilisation.
· Le produit reconstitué doit être une solution claire à légèrement opalescente et incolore à jaune pâle. Ne pas utiliser de solutions troubles ou présentant un dépôt. Les produits reconstitués doivent faire l’objet d’une inspection visuelle avant administration (particules, coloration).
· Tout produit non utilisé ou déchet doit être éliminé conformément à la réglementation en vigueur.
· Jeter le dispositif de transfert après usage unique.
Précautions particulières
Tous les effets indésirables liés à la perfusion doivent être traités en diminuant le débit de perfusion ou en arrêtant la perfusion.
Il est recommandé qu’à chaque administration de GAMMAGARD le nom du produit et le numéro de lot soit consigné.
Incompatibilités
GAMMAGARD ne doit pas être mélangé avec d’autres médicaments.
Il est recommandé d’administrer séparément GAMMAGARD et d’autres médicaments que le patient pourrait recevoir.
Recommandations posologiques
|
Indication |
Posologie |
Rythme des injections |
|
Traitement substitutif des déficits immunitaires primitifs |
· dose de charge : 0,4 à 0,8 g/kg de poids corporel |
|
|
|
· dose d'entretien : 0,2 à 0,8 g/kg de poids corporel |
toutes les 3 à 4 semaines pour obtenir un taux résiduel d'IgG d'au moins 5 à 6 g/L |
|
Traitement substitutif des déficits immunitaires secondaires |
0,2 à 0,4 g/kg de poids corporel |
toutes les 3 à 4 semaines pour obtenir un taux résiduel d’IgG d’au moins 5 à 6 g/L |
|
SIDA par Infection VIH congénitale |
0,2 à 0,4 g/kg de poids corporel |
toutes les 3 à 4 semaines |
|
Hypogammaglobulinémie (<4 g/L) chez les patients après une allogreffe de cellules souches hématopoïétiques |
0,2 à 0,4 g/kg de poids corporel |
toutes les 3 à 4 semaines afin d’obtenir un taux résiduel d’IgG supérieur à 5 g/l |
|
· le traitement des infections et la prévention de la maladie du greffon contre l’hôte |
|
toutes les semaines à partir de J-7 jusqu’à 3 mois après la greffe |
|
· le déficit persistant de production d’anticorps |
|
tous les mois jusqu’à ce que les taux d’anticorps soient revenus à la normale |
|
Traitement immunomodulateur |
|
|
|
Thrombopénie immunitaire primaire (Purpura thrombopénique idiopathique) |
0,8 à 1g/kg de poids corporel
ou
0,4 g/kg de poids corporel/j |
à J1, éventuellement répété une fois dans les 3 jours suivants
pendant 2 à 5 jours |
|
Syndrome de Guillain et Barré |
0,4 g/kg de poids corporel/j |
pendant 5 jours |
|
Maladie de Kawasaki |
1,6 à 2,0 g/kg de poids corporel
ou
2 g/kg de poids corporel |
en plusieurs doses réparties sur 2 à 5 j en association avec de l’acide acétylsalicylique
en une dose unique en association avec de l’acide acétylsalicylique |