Dernière mise à jour le 01/06/2026
BOSUTINIB EG 400 mg, comprimé pelliculé
Indications thérapeutiques
Classe pharmacothérapeutique : Antinéoplasiques, inhibiteurs de protéine kinase - code ATC : L01EA04
BOSUTINIB EG contient la substance active bosutinib. Il est utilisé dans le traitement de patients adultes atteints d'un type de leucémie appelée leucémie myéloïde chronique (LMC) à chromosome Philadelphie positif (Ph+) nouvellement diagnostiquée ou pour lesquels les traitements précédents destinés à traiter la LMC n’ont pas fonctionné ou ne sont pas adaptés. La LMC Ph+ est un cancer du sang qui conduit l'organisme à produire un trop grand nombre de globules blancs d'un certain type, appelés granulocytes.
Si vous avez des questions sur le mécanisme d'action de BOSUTINIB EG et sur les raisons pour lesquelles ce médicament vous a été prescrit, parlez-en à votre médecin.
Présentations
> plaquette(s) aluminium PVC-Aluminium OPA : polyamide orienté de 28 comprimé(s)
Code CIP : 34009 302 879 0 3
Déclaration de commercialisation : 03/10/2024
Cette présentation est agréée aux collectivités
- Prix hors honoraire de dispensation : 902,14 €
- Honoraire de dispensation : 1,02 €
- Prix honoraire compris : 903,16 €
- Taux de remboursement :100 %
Service médical rendu (SMR)
Amélioration du service médical rendu (ASMR)
Ce médicament étant un générique, l'ASMR n'a pas été évalué par la commission de la transparence (CT), il est possible de se référer à la /aux spécialité(s) de référence du groupe générique auquel appartient ce médicament (cliquez ici pour aller à la rubrique des groupes génériques)
Autres informations
- Titulaire de l'autorisation : EG LABO - Laboratoires EuroGenerics
- Conditions de prescription et de délivrance :
- liste I
- médicament nécessitant une surveillance particulière pendant le traitement
- prescription initiale hospitalière semestrielle
- prescription initiale réservée à certains spécialistes
- renouvellement de la prescription réservé aux médecins compétents en CANCEROLOGIE
- renouvellement de la prescription réservé aux spécialistes en HEMATOLOGIE
- renouvellement de la prescription réservé aux spécialistes en ONCOLOGIE MEDICALE
- Statut de l'autorisation : Valide
- Type de procédure : Procédure décentralisée
- Code CIS : 6 838 871 6
ANSM - Mis à jour le : 05/03/2026
BOSUTINIB EG 400 mg, comprimé pelliculé
2. COMPOSITION QUALITATIVE ET QUANTITATIVE
Bosutinib............................................................................................................................. 400 mg
Pour un comprimé pelliculé.
Pour la liste complète des excipients, voir rubrique 6.1.
Comprimé pelliculé, ovale, orange (largeur : 9 mm ; longueur : 17 mm), biconvexe, comportant la mention « C19 » gravée en creux sur une face.
4.1. Indications thérapeutiques
BOSUTINIB EG est indiqué dans le traitement des patients adultes atteints de :
· leucémie myéloïde chronique à chromosome Philadelphie positif (LMC Ph+) en phase chronique (PC) nouvellement diagnostiquée ;
· LMC Ph+ en PC, en phase accélérée (PA) et en crise blastique (CB) précédemment traités par un ou plusieurs inhibiteurs de la tyrosine kinase (ITK) et pour lesquels l'imatinib, le nilotinib et le dasatinib ne sont pas considérés comme des options thérapeutiques appropriées.
4.2. Posologie et mode d'administration
La thérapie doit être instaurée par un médecin expérimenté dans le diagnostic et le traitement des patients atteints de LMC.
Posologie
LMC Ph+ en PC nouvellement diagnostiquée
La dose recommandée est de 400 mg de bosutinib une fois par jour.
LMC Ph+ en PC, PA ou CB avec résistance ou intolérance au traitement précédent
La dose recommandée est de 500 mg de bosutinib une fois par jour.
Au cours des essais cliniques menés sur les deux indications, le traitement par bosutinib a été poursuivi jusqu’à progression de la maladie ou intolérance du traitement.
Ajustements posologiques
Au cours de l’étude clinique de phase I/II menée chez des patients atteints de LMC présentant une résistance ou une intolérance au traitement précédent, des augmentations de la dose de 500 mg à 600 mg une fois par jour (au moment des repas) ont été autorisées chez les patients n’ayant pas présenté de réponse hématologique complète (RHC) dans les 8 semaines ou de réponse cytogénétique complète (RCyC) dans les 12 semaines et n’ayant pas présenté d’événements indésirables de grade 3 ou supérieur susceptibles d’être liés au produit expérimental. Au cours de l’étude clinique de phase III menée chez des patients atteints de LMC en PC nouvellement diagnostiquée traités par 400 mg de bosutinib, des augmentations de dose par paliers de 100 mg jusqu’à 600 mg maximum une fois par jour (au moment des repas) ont été autorisées si le patient ne présentait pas un taux du transcrit breakpoint cluster region-Abelson (BCR-ABL) ≤ 10 % au Mois 3, s’il ne présentait pas d’effet indésirable de grade 3 ou 4 au moment de l’augmentation, et si toutes les toxicités non hématologiques de grade 2 avaient été résolues et étaient revenues au moins au grade 1.
Au cours de l’étude clinique de phase IV menée chez des patients atteints de LMC Ph+ précédemment traités par un ou plusieurs ITK, des augmentations de dose de 500 mg à 600 mg une fois par jour (au moment des repas) ont été autorisées chez les patients présentant une réponse insatisfaisante ou des signes de progression de la maladie en l’absence d’événements indésirables de grade 3 ou 4 ou de grade 2 persistants.
Au cours de l’étude de phase I/II menée chez des patients atteints de LMC présentant une résistance ou une intolérance au traitement précédent et ayant débuté un traitement ≤ 500 mg, 93 (93/558 ; 16,7 %) patients ont eu des augmentations de dose jusqu’à 600 mg par jour.
Au cours de l’étude de phase III menée chez des patients atteints de LMC en PC nouvellement diagnostiquée ayant débuté le traitement par bosutinib 400 mg, 58 patients (21,6 %) au total ont eu des augmentations de dose journalière jusqu’à 500 mg. De plus, 10,4 % des patients du groupe traité par bosutinib ont eu des augmentations de dose journalière jusqu’à 600 mg.
Au cours de l’étude de phase IV menée chez des patients atteints de LMC Ph+ précédemment traités par 1 ou plusieurs ITK et ayant débuté le traitement par bosutinib 500 mg par jour, 1 patient (0,6 %) a eu une augmentation de la dose journalière jusqu’à 600 mg.
Les doses supérieures à 600 mg/jour n’ont pas fait l’objet d’études et ne doivent pas être administrées.
Ajustements posologiques consécutifs à des effets indésirables
Effets indésirables non hématologiques
En cas de développement d'une toxicité non hématologique modérée ou sévère cliniquement significative, le traitement par bosutinib doit être interrompu et peut être repris à une dose réduite d’un palier de 100 mg une fois par jour après résolution de la toxicité. Si cliniquement indiqué, une nouvelle augmentation peut être envisagée à la dose reçue avant la réduction de la dose en une prise une fois par jour (voir rubrique 4.4). Des doses inférieures à 300 mg/jour ont été utilisées chez certains patients. Toutefois, l’efficacité n’a pas été établie.
Elévation des transaminases hépatiques : en cas d'élévation des transaminases hépatiques > 5 × la limite supérieure de la normale (LSN), le traitement par bosutinib doit être interrompu jusqu'à un retour à un taux ≤ 2,5 × LSN, et pourra être repris à une dose de 400 mg une fois par jour. Si le taux est toujours trop élevé après 4 semaines, l'arrêt du traitement par bosutinib doit être envisagé. Si l'élévation des transaminases ≥ 3 × LSN s'accompagne d'une élévation de la bilirubine > 2 × LSN et un taux de phosphatase alcaline < 2 × LSN, le traitement par bosutinib doit être arrêté (voir rubrique 4.4).
Diarrhée : en cas de diarrhée de grade 3– 4 selon les critères de terminologie communs pour les événements indésirables (CTCAE) du NCI, le traitement par bosutinib doit être interrompu et peut être repris à une dose de 400 mg une fois par jour après un retour au grade ≤ 1 (voir rubrique 4.4).
Effets indésirables hématologiques
Des réductions de la dose sont recommandées en cas de neutropénie et de thrombopénie sévère ou persistante, comme indiqué dans le tableau 1 :
Tableau 1 Ajustements posologiques en cas de neutropénie et de thrombopénie
|
PNNa < 1,0 × 109/L et/ou Plaquettes < 50 × 109/L |
Interrompre le traitement par bosutinib jusqu'à l'obtention d’une numération avec des PNN ≥ 1,0 × 109/L et un taux de plaquettes ≥ 50 × 109/L. Reprendre le traitement par bosutinib à la même dose si récupération dans les 2 semaines. Si la numération reste basse pendant une durée > 2 semaines, réduire la dose de 100 mg et reprendre le traitement après la résolution de l’événement. En cas de rechute de la cytopénie, réduire la dose d’un palier supplémentaire de 100 mg et reprendre le traitement après la résolution de l’événement. Des doses inférieures à 300 mg/jour ont été utilisées. Toutefois, l’efficacité n’a pas été établie. |
a PNN = polynucléaires neutrophiles
Populations particulières
Patients âgés (≥ 65 ans)
Aucune recommandation posologique spécifique n'est nécessaire chez les personnes âgées. Etant donné que les informations disponibles chez les personnes âgées sont limitées, la prudence est de mise chez ces patients.
Insuffisance rénale
Les patients dont la créatinine sérique est > 1,5 × LSN n’ont pas été inclus dans les études portant sur la LMC. Une augmentation de l'exposition (aire sous la courbe [ASC]) chez les patients présentant une insuffisance rénale modérée ou sévère a été observée au cours des études.
LMC Ph+ en PC nouvellement diagnostiquée
Chez les patients présentant une insuffisance rénale modérée (clairance de la créatinine [ClCr] comprise entre 30 et 50 mL/min, selon la formule de Cockcroft-Gault), la dose recommandée de bosutinib est de 300 mg par jour au moment des repas (voir rubriques 4.4 et 5.2).
Chez les patients présentant une insuffisance rénale sévère (ClCr < 30 mL/min, selon la formule de Cockcroft-Gault), la dose recommandée de bosutinib est de 200 mg par jour au moment des repas (voir rubriques 4.4 et 5.2).
Une augmentation de la dose à 400 mg une fois par jour (au moment des repas) chez les patients présentant une insuffisance rénale modérée ou à 300 mg une fois par jour chez les patients présentant une insuffisance rénale sévère peut être envisagée sous réserve qu’ils n’aient pas présenté d’effets indésirables sévères ou modérés persistants, et qu’ils ne soient pas en réponse hématologique, cytogénétique ou moléculaire adéquate.
LMC Ph+ en PC, PA ou CB avec résistance ou intolérance au traitement précédent
Chez les patients présentant une insuffisance rénale modérée (ClCr comprise entre 30 et 50 mL/min, selon la formule de Cockcroft-Gault), la dose recommandée de bosutinib est de 400 mg par jour (voir rubriques 4.4 et 5.2).
Chez les patients présentant une insuffisance rénale sévère (ClCr < 30 mL/min, selon la formule de Cockcroft-Gault), la dose recommandée de bosutinib est de 300 mg par jour (voir rubriques 4.4 et 5.2).
Une augmentation de la dose à 500 mg une fois par jour chez les patients présentant une insuffisance rénale modérée et à 400 mg une fois par jour chez les patients présentant une insuffisance rénale sévère peut être envisagée sous réserve qu’ils n’aient pas présenté d’effets indésirables sévères ou modérés persistants, et qu’ils ne soient pas en réponse hématologique, cytogénétique ou moléculaire adéquate.
Affections cardiaques
Les patients présentant des affections cardiaques non contrôlées ou significatives (par exemple, infarctus du myocarde récent, insuffisance cardiaque congestive ou angor instable) n’ont pas été inclus dans les études cliniques. La prudence est de mise chez les patients présentant des affections cardiaques pertinentes (voir rubrique 4.4).
Affections gastro-intestinales cliniquement significatives récentes ou en cours
Les patients présentant des affections gastro-intestinales cliniquement significatives récentes ou en cours (par exemple, diarrhée et/ou vomissements sévères) n’ont pas été inclus dans les études cliniques. La prudence est de mise chez les patients présentant des affections gastro-intestinales cliniquement significatives récentes ou en cours (voir rubrique 4.4).
Population pédiatrique
La sécurité et l’efficacité du bosutinib chez les enfants et les adolescents âgés de moins de 18 ans n’ont pas été établies. Aucune donnée n’est disponible.
Mode d’administration
BOSUTINIB EG doit être pris par voie orale une fois par jour, au moment des repas (voir rubrique 5.2). En cas d'omission d'une dose depuis plus de 12 heures, le patient ne doit pas recevoir de dose supplémentaire. Le patient doit prendre la dose prescrite habituelle le lendemain.
Hypersensibilité à la substance active ou à l’un des excipients mentionnés à la rubrique 6.1.
Insuffisance hépatique (voir rubriques 5.1 et 5.2).
4.4. Mises en garde spéciales et précautions d'emploi
Anomalies de la fonction hépatique
Le traitement par bosutinib est associé à une élévation des transaminases sériques (alanine aminotransférase [ALT], aspartate aminotransférase [AST]).
Des élévations des transaminases ont généralement été observées en début de traitement (parmi tous les patients ayant présenté une élévation des transaminases, quel qu’en soit le grade, > 80 % ont connu leur premier événement au cours des 3 premiers mois). Les patients sous bosutinib doivent subir des tests de la fonction hépatique avant l’instauration du traitement et tous les mois au cours des 3 premiers mois du traitement ou quand cela est cliniquement indiqué.
Les patients présentant des élévations des transaminases doivent être pris en charge en interrompant temporairement le traitement par bosutinib (en envisageant une réduction de la dose après un retour au grade 1 ou à la valeur initiale) et/ou en arrêtant définitivement le bosutinib. Des élévations des transaminases, notamment dans le cadre d’élévations concomitantes de la bilirubine, peuvent être le signe précoce d’une lésion du foie induite par le médicament et ces patients doivent être pris en charge en conséquence (voir rubriques 4.2 et 4.8).
Diarrhée et vomissements
Le traitement par bosutinib est associé à une diarrhée et des vomissements ; par conséquent, les patients souffrant de troubles gastro-intestinaux cliniquement significatifs récents ou en cours doivent utiliser ce médicament avec prudence et uniquement si une évaluation minutieuse du rapport bénéfice/risque a été menée, dans la mesure où les patients respectifs n’ont pas été inclus dans les études cliniques. Les patients souffrant de diarrhée et de vomissements doivent être pris en charge avec un traitement standard, comprenant un antidiarrhéique ou un antiémétique et/ou une réhydratation. La diarrhée et les vomissements peuvent également être pris en charge en interrompant temporairement, en réduisant la dose et/ou en arrêtant définitivement le traitement par bosutinib (voir rubriques 4.2 et 4.8). La dompéridone, utilisée comme antiémétique, peut entraîner un allongement de l’intervalle QT (QTc) et induire des arythmies de type « torsades de pointes ». Par conséquent, l’utilisation concomitante de dompéridone doit être évitée. Elle ne doit être utilisée que si les autres médicaments ne sont pas efficaces. Dans ce cas, une évaluation du rapport bénéfice/risque individuelle doit impérativement être effectuée et les patients doivent être surveillés quant à la survenue d’un allongement de l’intervalle QTc.
Myélosuppression
Le traitement par bosutinib est associé à une myélosuppression, définie comme une anémie, une neutropénie et une thrombopénie. Une numération de la formule sanguine complète doit être réalisée une fois par semaine pendant le premier mois du traitement et tous les mois par la suite, ou quand cela est cliniquement indiqué. La myélosuppression doit/peut être prise en charge en interrompant temporairement, en réduisant la dose et/ou en arrêtant définitivement le traitement par bosutinib (voir rubriques 4.2 et 4.8).
Rétention liquidienne
Le traitement par bosutinib peut être associé à une rétention liquidienne avec épanchement péricardique, épanchement pleural, œdème pulmonaire et/ou œdème périphérique. Les patients doivent être surveillés et pris en charge à l'aide d'un traitement standard. La rétention liquidienne peut également être prise en charge en interrompant temporairement, en réduisant la dose et/ou en arrêtant définitivement le traitement par bosutinib (voir rubriques 4.2 et 4.8).
Lipase sérique
Une élévation de la lipase sérique a été observée. La prudence est recommandée chez les patients présentant des antécédents de pancréatite. Si les élévations de la lipase sont accompagnées de symptômes abdominaux, le traitement par bosutinib doit être interrompu et des mesures appropriées pour établir le diagnostic et exclure une pancréatite doivent être envisagées (voir rubrique 4.2).
Infections
Le bosutinib peut prédisposer les patients à des infections causées par des bactéries, des champignons, des virus ou des protozoaires.
Potentiel pro-arythmique
Un allongement de l'intervalle QTc (mesuré automatiquement) sans arythmie a été observé. Le bosutinib doit être administré avec prudence chez les patients présentant des antécédents ou une prédisposition à l’allongement de l'intervalle QTc, souffrant d'une maladie cardiaque non contrôlée ou significative y compris un infarctus du myocarde récent, une insuffisance cardiaque congestive, un angor instable ou une bradycardie cliniquement significative, ou traités avec des médicaments ayant pour effet d’allonger le QTc (par exemple, médicaments anti-arythmiques et autres substances susceptibles d’allonger le QTc [rubrique 4.5]). La présence d'une hypokaliémie et d'une hypomagnésémie peut exacerber cet effet.
Il est conseillé de contrôler l'effet exercé sur le QTc et un électrocardiogramme (ECG) à l’inclusion est recommandé avant d’instaurer le traitement par bosutinib et quand cela est cliniquement indiqué. L'hypokaliémie ou l’hypomagnésémie doit être corrigée avant l'administration de bosutinib et doit être surveillée régulièrement au cours du traitement.
Insuffisance rénale
Le traitement par bosutinib peut entraîner un déclin cliniquement significatif de la fonction rénale chez les patients atteints de LMC. Un déclin dans le temps du débit de filtration glomérulaire estimé (DFGe) a été observé dans des études cliniques chez les patients traités par bosutinib. Chez les patients atteints de LMC en PC nouvellement diagnostiquée, traités à la dose de 400 mg, le déclin médian du DFGe par rapport à l’inclusion était de 11,1 mL/min/1,73 m2 à 1 an et de 14,1 mL/min/1,73 m2 à 5 ans pour les patients sous traitement. Les patients atteints de LMC naïfs de traitement et traités à la dose de 500 mg ont présenté un déclin médian du DFGe de 9,2 mL/min/1,73 m2 à 1 an, de 12,0 mL/min/1,73 m2 à 5 ans et de 16,6 mL/min/1,73 m2 à 10 ans pour les patients sous traitement. Chez les patients atteints de LMC en PC ou en phase avancée précédemment traités et traités par bosutinib à la dose de 500 mg, le déclin médian du DFGe était de 7,6 mL/min/1,73 m2 à 1 an, de 12,3 mL/min/1,73 m2 à 5 ans et de 15,9 mL/min/1,73 m2 à 10 ans pour les patients sous traitement. Chez les patients atteints de LMC Ph+ précédemment traités par au moins un ITK et traités par bosutinib à la dose de 500 mg, la diminution médiane du DFGe par rapport à l’inclusion était de 9,2 mL/min/1,73 m2 à 1 an et de 14,5 mL/min/1,73 m2 à 4 ans pour les patients sous traitement.
Il est important d’évaluer la fonction rénale avant l’instauration du traitement et de la surveiller étroitement pendant le traitement par bosutinib, en particulier chez les patients présentant une altération préexistante de la fonction rénale ou des facteurs de risque de dysfonctionnement rénal, y compris en cas d’utilisation concomitante de médicaments potentiellement néphrotoxiques tels que les diurétiques, les inhibiteurs de l'enzyme de conversion de l'angiotensine (IEC), les antagonistes des récepteurs de l’angiotensine et les anti-inflammatoires non stéroïdiens (AINS).
Au cours d’une étude sur l’insuffisance rénale, les expositions au bosutinib ont augmenté chez les patients présentant une altération de la fonction rénale modérée ou sévère. Une diminution de la dose est recommandée chez les patients présentant une insuffisance rénale modérée ou sévère (voir rubriques 4.2 et 5.2).
Les patients dont la créatinine sérique était > 1,5 × LSN n’ont pas été inclus dans les études sur la LMC. Sur la base d'une analyse pharmacocinétique de population, une augmentation de l'exposition (ASC) au bosutinib a été observée, à l’instauration du traitement, chez les patients présentant une insuffisance rénale modérée ou sévère, au cours des études cliniques (voir rubriques 4.2 et 5.2).
Les données cliniques sont très limitées (n = 3) chez les patients atteints de LMC présentant une insuffisance rénale modérée et ayant reçu une dose de bosutinib augmentée à 600 mg.
Population asiatique
D’après les analyses pharmacocinétiques de population, les Asiatiques ont présenté une clairance inférieure, ce qui a entraîné une exposition accrue. Par conséquent, ces patients doivent faire l’objet d’une surveillance étroite afin de détecter tout effet indésirable, notamment en cas d’augmentation de la dose.
Réactions cutanées sévères
Le bosutinib peut provoquer des réactions cutanées sévères telles qu’un syndrome de Stevens-Johnson ou une nécrolyse épidermique toxique. Le bosutinib doit être arrêté définitivement chez les patients présentant une réaction cutanée sévère au cours du traitement.
Syndrome de lyse tumorale
En raison de l’apparition possible d’un syndrome de lyse tumorale (SLT), il est recommandé de corriger toute déshydratation cliniquement significative et de traiter l’hyperuricémie avant l’instauration du traitement par bosutinib (voir rubrique 4.8).
Réactivation de l'hépatite B
Des cas de réactivation du virus de l’hépatite B (VHB) ont été observés chez des patients porteurs chroniques du virus et traités par des ITK BCR-ABL. Certains de ces cas ont évolué vers une insuffisance hépatique aiguë ou une hépatite fulminante nécessitant une transplantation hépatique ou dont l’issue a été fatale.
Les patients doivent faire l’objet d’un dépistage d’une infection par le VHB avant l’instauration d’un traitement par bosutinib. Des experts en maladies hépatiques et en traitement du VHB doivent être consultés avant l'instauration du traitement chez les patients présentant une sérologie VHB positive (y compris ceux présentant une maladie active) et chez les patients dont le test de dépistage de l'infection par le VHB est positif au cours du traitement. Les porteurs du VHB qui doivent être traités par bosutinib doivent faire l’objet d’une surveillance étroite en vue de détecter tout signe ou symptôme d'une infection active par le VHB pendant toute la durée du traitement et pendant plusieurs mois après la fin du traitement (voir rubrique 4.8).
Photosensibilité
Une exposition directe au soleil ou au rayonnement ultraviolet (UV) doit être évitée ou réduite en raison du risque de photosensibilité associé au traitement par bosutinib. Les patients doivent être informés des mesures à prendre telles que le port de vêtements de protection et l’utilisation d'une crème solaire avec un indice de protection (SPF) élevé.
Inhibiteurs du cytochrome P450 (CYP)3A
L’utilisation concomitante de bosutinib et d’inhibiteurs puissants ou modérés du CYP3A doit être évitée, car elle entraîne une élévation de la concentration plasmatique en bosutinib (voir rubrique 4.5).
Dans la mesure du possible, il est recommandé de s’orienter vers un autre traitement concomitant sans potentiel d'inhibition du CYP3A (ou avec un faible potentiel).
Si un inhibiteur du CYP3A puissant ou modéré doit être administré pendant le traitement par bosutinib une interruption du traitement par bosutinib ou une réduction de la dose de bosutinib doit être envisagée.
Inducteurs du CYP3A
L’utilisation concomitante de bosutinib et d’inducteurs puissants ou modérés du CYP3A doit être évitée, car elle entraîne une baisse de la concentration plasmatique en bosutinib (voir rubrique 4.5).
Effet des aliments
Les produits à base de pamplemousse, y compris le jus de pamplemousse, et les autres aliments exerçant un effet inhibiteur du CYP3A doivent être évités (voir rubrique 4.5).
Sodium alimentaire
Ce médicament contient moins de 1 mmol (23 mg) de sodium par comprimé de 100 mg, 400 mg ou 500 mg. Les patients suivant un régime hyposodé doivent être informés que ce produit est essentiellement « sans sodium ».
4.5. Interactions avec d'autres médicaments et autres formes d'interactions
Effets des autres médicaments sur le bosutinib
Inhibiteurs du CYP3A
L’utilisation concomitante de bosutinib et d’inhibiteurs puissants du CYP3A (y compris, sans s'y limiter, itraconazole, kétoconazole, posaconazole, voriconazole, clarithromycine, télithromycine, néfazodone, mibéfradil, indinavir, lopinavir/ritonavir, nelfinavir, ritonavir, saquinavir, bocéprévir, télaprévir, produits à base de pamplemousse y compris le jus de pamplemousse) ou d’inhibiteurs modérés du CYP3A (y compris, sans s'y limiter, fluconazole, ciprofloxacine, érythromycine, diltiazem, vérapamil, amprénavir, atazanavir, darunavir/ritonavir, fosamprénavir, aprépitant, crizotinib, imatinib) doit être évitée, car elle entraîne une élévation de la concentration plasmatique du bosutinib.
La prudence est de mise en cas d’utilisation concomitante d'inhibiteurs faibles du CYP3A et de bosutinib.
Dans la mesure du possible, il est recommandé de s’orienter vers un autre traitement concomitant sans (ou avec un faible) potentiel d'inhibition de l'enzyme CYP3A.
Si un inhibiteur du CYP3A puissant ou modéré doit être administré pendant le traitement par bosutinib, une interruption du traitement par bosutinib ou une réduction de la dose de bosutinib doit être envisagée.
Au cours d’une étude portant sur 24 sujets sains recevant 5 doses quotidiennes de 400 mg de kétoconazole (un inhibiteur puissant du CYP3A) de façon concomitante avec une dose unique de 100 mg de bosutinib à jeun, le kétoconazole a multiplié par 5,2 la Cmax du bosutinib et par 8,6 l'ASC du bosutinib dans le plasma, par rapport à l'administration du bosutinib en monothérapie.
Au cours d’une étude portant sur 20 sujets sains, recevant une dose unique de 125 mg d'aprépitant (un inhibiteur modéré du CYP3A) en association avec une dose unique de 500 mg de bosutinib après un repas, l'aprépitant a multiplié par 1,5 la Cmax du bosutinib et par 2,0 l'ASC du bosutinib dans le plasma, par rapport à l'administration du bosutinib en monothérapie.
Inducteurs du CYP3A
L’utilisation concomitante de bosutinib et d’inducteurs puissants du CYP3A (y compris, sans s'y limiter, carbamazépine, phénytoïne, rifampicine, millepertuis) ou d’inducteurs modérés du CYP3A (y compris, sans s'y limiter, bosentan, éfavirenz, étravirine, modafinil, nafcilline) doit être évitée, car elle entraîne une baisse de la concentration plasmatique du bosutinib.
Etant donné l’importante diminution de l'exposition au bosutinib observée en cas de co-administration de bosutinib et de rifampicine, il est peu probable que l'augmentation de la dose de bosutinib lors de l'administration concomitante d'inducteurs puissants ou modérés du CYP3A compense suffisamment la perte d'exposition.
La prudence est de mise en cas d’utilisation concomitante d'inducteurs faibles du CYP3A avec le bosutinib.
Suite à l’administration concomitante d'une dose unique de bosutinib et de 6 doses quotidiennes de 600 mg de rifampicine chez 24 sujets sains prises après un repas, l'exposition au bosutinib (Cmax et ASC dans le plasma) a diminué de 14 % et 6 %, respectivement, par rapport aux valeurs enregistrées lors de l'administration de 500 mg de bosutinib en monothérapie.
Inhibiteurs de la pompe à protons (IPP)
La prudence est de mise lors de l'administration concomitante de bosutinib et d’IPP. Des antiacides d'action rapide doivent être envisagés en remplacement des IPP et les heures d'administration du bosutinib et des antiacides doivent être différentes dans la mesure du possible (par exemple, prise de bosutinib le matin et des antiacides le soir). Le bosutinib présente une hydrosolubilité dépendante du pH in vitro. Lorsqu'une dose unique de bosutinib (400 mg) par voie orale était co-administrée avec des doses multiples de lansoprazole (60 mg) par voie orale chez 24 sujets sains à jeun, la Cmax et l'ASC du bosutinib ont chuté à 54 % et 74 %, respectivement, par rapport aux valeurs obtenues lorsque le bosutinib (400 mg) était administré en monothérapie.
Effets du bosutinib sur les autres médicaments
Au cours d’une étude portant sur 27 sujets sains, recevant une dose unique de 500 mg de bosutinib en association avec une dose unique de 150 mg de mésilate de dabigatran étexilate (un substrat de la glycoprotéine P [P-gp]) après un repas, le bosutinib n'a pas augmenté la Cmax ni l'ASC du dabigatran dans le plasma, par rapport à l'administration de mésilate de dabigatran étexilate en monothérapie. Les résultats de l'étude indiquent que le bosutinib ne présente pas d'effets inhibiteurs de la P-gp cliniquement significatifs.
Une étude in vitro indique que les interactions médicamenteuses sont peu probables aux doses thérapeutiques en raison de l'induction par le bosutinib du métabolisme de médicaments substrats du CYP1A2, du CYP2B6, du CYP2C9, du CYP2C19 et du CYP3A4.
Des études in vitro indiquent que les interactions médicamenteuses cliniques sont peu probables aux doses thérapeutiques en raison de l'inhibition par le bosutinib du métabolisme de médicaments substrats du CYP1A2, du CYP2A6, du CYP2C8, du CYP2C9, du CYP2C19, du CYP2D6 ou du CYP3A4/5.
Des études in vitro indiquent que le bosutinib présente un faible potentiel d’inhibition de la protéine de résistance au cancer du sein (BCRP, systémique), du polypeptide de transport des anions organiques (OATP)1B1, OATP1B3, du transporteur des anions organiques (OAT)1, OAT3, du transporteur des cations organiques (OCT)2 à des concentrations cliniquement pertinentes, mais il est susceptible d’inhiber la BCRP dans le tractus gastro-intestinal et l’OCT1.
Anti-arythmiques et autres substances susceptibles d’allonger l'intervalle QT
Le bosutinib doit être utilisé avec prudence chez les patients présentant, ou susceptibles de développer, un allongement de l'intervalle QT, notamment les patients sous anti-arythmiques tels que l'amiodarone, le disopyramide, le procaïnamide, la quinidine et le sotalol, ou d'autres médicaments pouvant entraîner un allongement de l'intervalle QT tels que la chloroquine, l'halofantrine, la clarithromycine, la dompéridone, l'halopéridol, la méthadone et la moxifloxacine (voir rubrique 4.4).
4.6. Fertilité, grossesse et allaitement
Femmes en capacité de procréer/Contraception
Les femmes en capacité de procréer doivent être informées de la nécessité d'utiliser une méthode de contraception efficace pendant le traitement par bosutinib et pendant au moins 1 mois après l'administration de la dernière dose afin d'éviter toute grossesse pendant le traitement par bosutinib. En outre, la patiente doit être avertie que des vomissements ou diarrhées sont susceptibles de réduire l'efficacité des contraceptifs oraux en empêchant leur absorption complète.
Grossesse
Les données sur l'utilisation du bosutinib chez la femme enceinte sont limitées. Les études effectuées chez l'animal ont mis en évidence une toxicité sur la reproduction (voir rubrique 5.3). Le bosutinib n'est pas recommandé pendant la grossesse ou chez les femmes en capacité de procréer n'utilisant pas de contraception. Si le bosutinib est administré pendant la grossesse ou si la patiente débute une grossesse pendant le traitement par bosutinib, elle doit être informée des risques potentiels pour le fœtus.
Allaitement
On ne sait pas si le bosutinib et ses métabolites sont excrétés dans le lait maternel. Une étude avec le bosutinib radiomarqué [au 14C] chez des rats a démontré l'excrétion d'une radioactivité dérivée du bosutinib dans le lait maternel (voir rubrique 5.3). Un risque pour les nourrissons allaités ne peut être exclu. L'allaitement doit être interrompu au cours du traitement par bosutinib.
Fertilité
Sur base de résultats non cliniques, il est possible que bosutinib nuise à la fonction de reproduction et à la fertilité chez l’être humain (voir rubrique 5.3). Il est conseillé aux hommes traités par bosutinib de se renseigner sur la conservation des spermatozoïdes avant le traitement en raison d'une possible diminution de la fertilité due au traitement par bosutinib.
4.7. Effets sur l'aptitude à conduire des véhicules et à utiliser des machines
Le bosutinib n’a aucun effet ou un effet négligeable sur l’aptitude à conduire des véhicules et à utiliser des machines. Cependant, si un patient sous bosutinib présente des sensations vertigineuses, de la fatigue, des troubles visuels ou d'autres effets indésirables susceptibles d'avoir un impact sur la capacité à conduire ou à utiliser des machines en toute sécurité, le patient doit renoncer à ces activités pendant toute la durée de manifestation des effets indésirables.
Résumé du profil de sécurité
Au total, 1 372 patients atteints de leucémie ont reçu au moins 1 dose unique de bosutinib en monothérapie. La durée médiane du traitement était de 26,30 mois (intervalle : 0,03 à 170,49 mois). Ces patients étaient soit atteints d’une LMC en PC nouvellement diagnostiquée, soit résistants ou intolérants aux traitements précédents d’une LMC en phase chronique ou accélérée, ou en crise blastique, ou encore atteints d’une leucémie aiguë lymphoblastique (LAL) Ph+. Parmi ces patients, 268 (dose initiale de 400 mg) et 248 (dose initiale de 500 mg) étaient issus de 2 études de phase III menées sur des patients atteints d’une LMC n’ayant reçu aucun traitement antérieur, 60 (dose initiale de 400 mg) étaient issus d’une étude de phase II menée sur des patients atteints d’une LMC n’ayant reçu aucun traitement antérieur, 570 et 63 (phase II : dose initiale de 500 mg) étaient issus de 2 études de phase I/II menées sur des patients atteints de leucémies Ph+ précédemment traitées, et 163 (dose initiale de 500 mg) étaient issus d’une étude de phase IV menée sur des patients atteints d’une LMC précédemment traitée.
La durée médiane du traitement était de 55,1 mois (intervalle : 0,2 à 60,05 mois), 61,6 mois (intervalle : 0,03 à 145,86 mois), 15,3 mois (intervalle : 0,3 à 21,8 mois), 11,1 mois (intervalle : 0,03 à 170,49 mois), 30,2 mois (intervalle : 0,2 à 85,6 mois) et 37,8 mois (intervalle : 0,16 à 50,0 mois), respectivement. Les analyses de sécurité ont inclus des données provenant d’une étude d’extension terminée.
Au moins 1 effet indésirable, tous grades de toxicité confondus, a été rapporté chez 1 349 (98,3 %) patients. Les effets indésirables les plus fréquemment rapportés chez ≥ 20 % des patients étaient la diarrhée (80,4 %), les nausées (41,5 %), les douleurs abdominales (35,6 %), la thrombopénie (34,4 %), les vomissements (33,7 %), le rash (32,8 %), l’ALT augmentée (28,0 %), l’anémie (27,2 %), la fièvre (23,4 %), l’AST augmentée (22,5 %), la fatigue (32,0 %) et les céphalées (20,3 %). Au moins 1 effet indésirable de grade 3 ou 4 a été rapporté chez 943 (68,7 %) patients. Les effets indésirables de grade 3 ou 4 rapportés chez ≥ 5 % des patients étaient la thrombopénie (19,7 %), l’ALT augmentée (14,6 %), la neutropénie (10,6 %), la diarrhée (10,6 %), l’anémie (10,3 %), la lipase augmentée (10,1 %), l’AST augmentée (6,7 %) et le rash (5,0 %).
Tableau listant les effets indésirables
Les effets indésirables suivants ont été rapportés au cours des essais cliniques portant sur le bosutinib (tableau 2). Ils représentent une évaluation des données concernant les effets indésirables survenus chez 1 372 patients atteints soit de LMC en PC nouvellement diagnostiquée, de LMC en phase chronique ou accélérée ou en crise blastique résistante ou intolérante à des traitements antérieurs soit de LAL Ph+ ayant reçu au moins 1 dose unique de bosutinib en monothérapie. Ces effets indésirables sont présentés par classe de systèmes d'organes et par fréquence. Les catégories de fréquence sont définies comme suit : très fréquent (≥ 1/10), fréquent (≥ 1/100, < 1/10), peu fréquent (≥ 1/1 000, < 1/100), rare (≥ 1/10 000, < 1/1 000), très rare (< 1/10 000), fréquence indéterminée (ne peut être estimée sur la base des données disponibles). Au sein de chaque groupe de fréquence, les effets indésirables sont présentés par ordre décroissant de gravité.
Tableau 2 Effets indésirables du bosutinib
|
Infections et infestations |
|
|
Très fréquent |
Infection des voies respiratoires (incluant infection des voies respiratoires inférieures, infection virale des voies respiratoires, infection des voies respiratoires supérieures, infection virale des voies respiratoires supérieures), rhinopharyngite |
|
Fréquent |
Pneumonie (incluant pneumonie atypique, pneumonie bactérienne, pneumonie fongique, pneumonie nécrosante, pneumonie streptococcique, grippe (incluant la grippe H1N1), bronchite |
|
Tumeurs bénignes, malignes et non précisées (incl. kystes et polypes) |
|
|
Peu fréquent |
Syndrome de lyse tumorale** |
|
Très fréquent |
Thrombopénie (incluant diminution du taux de plaquettes), neutropénie (incluant diminution du taux de neutrophiles), anémie (incluant diminution du taux d’hémoglobine, diminution du taux de globules rouges) |
|
Fréquent |
Leucopénie (incluant diminution du taux de globules blancs) |
|
Peu fréquent |
Neutropénie fébrile, granulocytopénie |
|
Affections du système immunitaire |
|
|
Fréquent |
Hypersensibilité médicamenteuse |
|
Peu fréquent |
Choc anaphylactique |
|
Troubles du métabolisme et de la nutrition |
|
|
Très fréquent |
Appétit diminué |
|
Fréquent |
Déshydratation, hyperkaliémie (incluant augmentation du potassium sanguin), hypophosphatémie (incluant diminution du phosphore sanguin) |
|
Affections du système nerveux |
|
|
Très fréquent |
Sensations vertigineuses, céphalées |
|
Fréquent |
Dysgueusie |
|
Affections de l'oreille et du labyrinthe |
|
|
Fréquent |
Acouphènes |
|
Affections cardiaques |
|
|
Fréquent |
Epanchement péricardique |
|
Peu fréquent |
Péricardite |
|
Affections vasculaires |
|
|
Fréquent |
Hypertension (incluant augmentation de la pression artérielle, augmentation de la pression artérielle systolique, hypertension essentielle, crise d'hypertension) |
|
Affections respiratoires, thoraciques et médiastinales |
|
|
Très fréquent |
Epanchement pleural, dyspnée, toux |
|
Fréquent |
Hypertension pulmonaire (incluant hypertension artérielle pulmonaire, augmentation de la pression artérielle pulmonaire), insuffisance respiratoire |
|
Peu fréquent |
Œdème pulmonaire aigu (incluant œdème pulmonaire) |
|
Fréquence indéterminée |
Pneumopathie interstitielle diffuse |
|
Affections gastro-intestinales |
|
|
Très fréquent |
Diarrhée, vomissements, nausées, douleurs abdominales (incluant gêne abdominale, douleurs abdominales basse et haute, abdomen sensible, douleurs gastro-intestinales) |
|
Fréquent |
Hémorragie gastro-intestinale (incluant hémorragie anale, hémorragie gastrique, hémorragie intestinale, hémorragie gastro-intestinale basse, hémorragie rectale, hémorragie gastro-intestinale haute), pancréatite (incluant pancréatite aiguë), gastrite |
|
Affections hépatobiliaires |
|
|
Fréquent |
Hépatotoxicité (incluant hépatite, hépatite toxique, troubles hépatiques), fonction hépatique anormale (incluant augmentation des enzymes hépatiques, test de la fonction hépatique anormal, test de la fonction hépatique augmenté, transaminases augmentées) |
|
Peu fréquent |
Lésion du foie (incluant lésion du foie d’origine médicamenteuse, lésion hépatocellulaire) |
|
Affections de la peau et du tissu sous-cutané |
|
|
Très fréquent |
Rash (incluant rash maculaire, rash maculopapulaire, rash papuleux, rash pruritique), prurit |
|
Fréquent |
Réaction de photosensibilité (incluant lucite polymorphe), urticaire, acné |
|
Peu fréquent |
Erythème polymorphe, rash avec exfoliation, éruption d’origine médicamenteuse, vascularite cutanée |
|
Fréquence indéterminée |
Syndrome de Stevens-Johnson**, nécrolyse épidermique toxique** |
|
Affections musculosquelettiques et du tissu conjonctif |
|
|
Très fréquent |
Arthralgie, dorsalgie |
|
Fréquent |
Myalgie |
|
Affections du rein et des voies urinaires |
|
|
Fréquent |
Insuffisance rénale aiguë, insuffisance rénale, atteinte de la fonction rénale |
|
Troubles généraux et anomalies au site d'administration |
|
|
Très fréquent |
Œdème (incluant œdème des paupières, œdème de la face, œdème généralisé, œdème localisé, œdème périphérique, œdème périorbitaire, gonflement périorbitaire, gonflement périphérique, gonflement, gonflement des paupières), fièvre, fatigue (incluant asthénie, malaise) |
|
Fréquent |
Douleur thoracique (incluant gêne thoracique), douleur |
|
Investigations |
|
|
Très fréquent |
Lipase augmentée (incluant hyperlipasémie), alanine aminotransférase augmentée (incluant alanine aminotransférase anormale), aspartate aminotransférase augmentée, créatinine sanguine augmentée |
|
Fréquent |
Intervalle QT allongé à l’électrocardiogramme (incluant syndrome du QT long), amylase augmentée (incluant hyperamylasémie), créatine phosphokinase sanguine augmentée, gamma-glutamyltransférase augmentée, bilirubine sanguine augmentée (incluant hyperbilirubinémie, bilirubine conjuguée augmentée, bilirubine non conjuguée augmentée) |
** Effet indésirable identifié après la commercialisation.
Description de certains effets indésirables
Les descriptions figurant ci-dessous sont basées sur la population de sécurité de 1 372 patients ayant reçu au moins 1 dose de bosutinib et ayant été traités soit pour une LMC en PC nouvellement diagnostiquée, soit pour une LMC, qu’elle soit en PC, PA ou CB, ou une LAL Ph+ présentant une résistance ou une intolérance au traitement précédent.
Affections hématologiques et du système lymphatique
Sur les 372 (27,1 %) patients ayant rapporté des effets indésirables d’anémie, 6 ont arrêté le traitement par bosutinib en raison de l’anémie. Une toxicité maximale de grade 1 a été rapportée chez 95 (25,5 %) patients, de grade 2 chez 135 (36,3 %) patients, de grade 3 chez 113 patients (30,4 %) et de grade 4 chez 29 (7,8 %) patients. Parmi ces patients, le délai médian d'apparition du premier événement était de 29 jours (intervalle : 1 à 3 999 jours) et la durée médiane par événement était de 22 jours (intervalle : 1 à 3 682 jours).
Sur les 209 (15,2 %) patients ayant rapporté une neutropénie comme effet indésirable, 19 ont arrêté le traitement par bosutinib en raison de la neutropénie. Une toxicité maximale de grade 1 a été rapportée chez 19 (9,1 %) patients, de grade 2 chez 45 (21,5 %) patients, de grade 3 chez 95 (45,5 %) patients et de grade 4 chez 50 (23,9 %) patients. Parmi ces patients, le délai médian d'apparition du premier événement était de 56 jours (intervalle : 1 à 1 769 jours) et la durée médiane par événement était de 15 jours (intervalle : 1 à 913 jours).
Sur les 472 (34,4 %) patients ayant rapporté une thrombopénie comme effet indésirable, 42 sujets ont arrêté le bosutinib en raison de la thrombopénie. Une toxicité maximale de grade 1 a été rapportée chez 114 (24,2 %) patients, de grade 2 chez 88 (18,6 %) patients, de grade 3 chez 172 (36,4 %) patients et de grade 4 chez 98 (20,8 %) patients. Parmi ces patients, le délai médian d’apparition du premier événement était de 28 jours (intervalle : 1 à 1 688 jours) et la durée médiane par événement était de 15 jours (intervalle : 1 à 3 921 jours).
Affections hépatobiliaires
Parmi les patients ayant rapporté des élévations de l'ALT ou de l'AST (tous grades confondus) comme effets indésirables, le délai médian d’apparition observé était de 29 jours, avec un intervalle d’apparition de 1 à 3 995 jours pour l’ALT et l’AST. La durée médiane d'un événement était de 17 jours (intervalle : 1 à 1 148 jours), et de 15 jours (intervalle : 1 à 803 jours) pour l’ALT et l'AST, respectivement.
Deux cas compatibles avec une toxicité hépatique d’origine médicamenteuse (définis comme des élévations concomitantes des ALT ou des AST ≥ 3 × LSN et de la bilirubine totale > 2 × LSN avec une phosphatase alcaline < 2 × LSN) sans autres causes ont été rapportés chez 2/1 711 (0,1 %) sujets ayant reçu du bosutinib.
Réactivation de l'hépatite B
Des cas de réactivation de l’hépatite B ont été rapportés chez des patients traités par ITK BCR-ABL. Certains de ces cas ont évolué vers une insuffisance hépatique aiguë ou une hépatite fulminante nécessitant une transplantation hépatique ou dont l’issue a été fatale (voir rubrique 4.4).
Affections gastro-intestinales
Sur les 1 103 (80,4 %) patients ayant présenté une diarrhée, 14 patients ont arrêté le traitement par bosutinib en raison de cet événement. Des médicaments concomitants ont été administrés pour traiter la diarrhée chez 756 (68,5 %) patients. Une toxicité maximale de grade 1 est survenue chez 575 (52,1 %) patients, une toxicité maximale de grade 2 chez 383 (34,7 %) patients, une toxicité maximale de grade 3 chez 144 (13,1 %) patients ; 1 seul patient (0,1 %) a présenté un événement de grade 4. Parmi les patients ayant présenté une diarrhée, le délai médian d'apparition du premier événement était de 2 jours (intervalle : 1 à 2 702 jours) et la durée médiane de la diarrhée (tous grades confondus) de 2 jours (intervalle : 1 à 4 247 jours).
Parmi les 1 103 patients ayant présenté une diarrhée, 218 patients (19,8 %) ont été pris en charge par une interruption de leur traitement et 208 (95,4 %) d'entre eux ont repris ensuite le traitement par bosutinib. Parmi les patients ayant repris le traitement, 201 (96,6 %) n'ont pas constaté d'autre événement ni arrêté le traitement par bosutinib en raison d'un nouvel événement de diarrhée.
Affections cardiaques
Sept patients (0,5 %) ont présenté un allongement du QTcF (supérieur à 500 ms). Onze patients (0,8 %) ont présenté un allongement du QTcF > 60 ms par rapport à l’inclusion. Les patients atteints d'une maladie cardiovasculaire non contrôlée ou significative, y compris un allongement du QTc, à l'inclusion, n’ont pas été inclus dans les études cliniques (voir rubriques 5.1 et 5.3).
Déclaration des effets indésirables suspectés
La déclaration des effets indésirables suspectés après autorisation du médicament est importante. Elle permet une surveillance continue du rapport bénéfice/risque du médicament. Les professionnels de santé déclarent tout effet indésirable suspecté via le système national de déclaration : Agence nationale de sécurité du médicament et des produits de santé (ANSM) et réseau des Centres Régionaux de Pharmacovigilance - Site internet : https://signalement.social-sante.gouv.fr/.
L'expérience concernant un surdosage de bosutinib dans les études cliniques s'est limitée à des cas isolés. Les patients qui prennent un surdosage de bosutinib doivent faire l’objet d’une surveillance et recevoir un traitement de soutien adapté.
5. PROPRIETES PHARMACOLOGIQUES
5.1. Propriétés pharmacodynamiques
Classe pharmacothérapeutique : Antinéoplasiques, inhibiteurs de protéine kinase, code ATC : L01EA04.
Mécanisme d’action
Le bosutinib appartient à une classe pharmacologique de médicaments appelée inhibiteurs des protéines kinases. Le bosutinib inhibe la protéine kinase anormale BCR-ABL à l'origine de la LMC. Selon les études de modélisation, le bosutinib se lie au domaine kinase de BCR-ABL. Le bosutinib est également un inhibiteur des kinases de la famille Src, dont Src, Lyn et Hck. Le bosutinib inhibe de manière minimale le récepteur du facteur de croissance dérivé des plaquettes (PDGF) et le c-Kit.
Dans les études in vitro, le bosutinib inhibe la prolifération et la survie des lignées cellulaires établies de la LMC, des lignées cellulaires de la LAL Ph+ et des cellules primitives de LMC dérivées du patient. Le bosutinib a inhibé 16 des 18 formes de BCR-ABL résistantes à l'imatinib exprimées dans des lignées cellulaires myéloïdes murines. Le traitement par bosutinib a réduit la taille des tumeurs de la LMC se développant chez des souris nude et a inhibé la croissance des tumeurs myéloïdes murines exprimant des formes de BCR-ABL résistantes à l'imatinib. Le bosutinib inhibe également le récepteur des tyrosine kinases c-Fms, les récepteurs EphA et B, les kinases des familles Trk, Axl et Tec, certains membres de la famille ErbB, la tyrosine kinase non réceptrice Csk, les sérine/thréonine kinases de la famille Ste20, et 2 protéines kinases dépendantes de la calmoduline.
Effets pharmacodynamiques
L'effet de l'administration du bosutinib 500 mg sur le QTc a été évalué dans le cadre d'une étude randomisée, à dose unique, en double aveugle (en ce qui concerne le bosutinib), avec permutation, contrôlée contre placebo et avec le moxifloxacine en ouvert chez des sujets sains.
Les résultats de cette étude indiquent que le bosutinib n’allonge pas le QTc chez les sujets sains à la dose journalière de 500 mg prise au moment des repas et dans des conditions entraînant des concentrations plasmatiques supra-thérapeutiques. Après l'administration d'une dose orale unique de 500 mg de bosutinib (dose thérapeutique) et de 500 mg de bosutinib avec 400 mg de kétoconazole (pour atteindre des concentrations supra-thérapeutiques de bosutinib) chez des sujets sains, la limite supérieure de l'intervalle de confiance (IC) unilatéral à 95 % concernant la variation moyenne du QTc était inférieure à 10 ms à tous les temps d’évaluation après la dose, et aucun événement indésirable évoquant un allongement du QTc n'a été observé.
Au cours d’une étude portant sur des sujets insuffisants hépatiques, une fréquence accrue de l’allongement du QTc > 450 ms, accompagnée d'une baisse de la fonction hépatique, a été observée. Au cours de l'étude clinique de phase I/II portant sur des patients atteints de leucémies Ph+ précédemment traitées par 500 mg de bosutinib, un allongement du QTcF > 60 ms par rapport à l’inclusion a été observé chez 9 (1,6 %) des 570 patients. Au cours de l’étude clinique de phase III portant sur des patients atteints de LMC en PC nouvellement diagnostiquée, traités par 400 mg de bosutinib, aucun allongement du QTcF > 60 ms (N = 268) par rapport à l’inclusion n’a été observé. Au cours de l'étude clinique de phase III portant sur des patients atteints de LMC Ph+ en PC nouvellement diagnostiquée, traités par 500 mg de bosutinib, un allongement du QTcF > 60 ms par rapport à l’inclusion a été observé chez 2 (0,8 %) des 248 patients sous bosutinib. Au cours de l’étude clinique de phase IV menée chez des patients atteints de LMC Ph+ précédemment traitée par 1 ou plusieurs ITK et traités par 500 mg de bosutinib (N = 163), aucun patient n’a présenté un allongement du QTcF > 60 ms par rapport à l’inclusion. Un effet proarythmique du bosutinib ne peut être exclu.
Efficacité et sécurité clinique
Etude clinique sur la LMC en PC non précédemment traitée
Etude évaluant le bosutinib 400 mg
Un essai de supériorité, de phase III, multicentrique, en ouvert, à 2 bras a été mené pour évaluer l’efficacité et la sécurité du bosutinib 400 mg une fois par jour en monothérapie, par rapport à l’imatinib 400 mg une fois par jour en monothérapie chez les patients adultes atteints de LMC Ph+ en PC nouvellement diagnostiquée. L’essai a randomisé 536 patients (268 dans chaque groupe de traitement) atteints de LMC Ph+ ou Ph- en PC nouvellement diagnostiquée (population en intention de traiter [ITT]) incluant 487 patients atteints d’une LMC Ph+ présentant des transcrits b2a2 et/ou b3a2 et un nombre de copies BCR– ABL identifiées à l’inclusion > 0 (population en intention de traiter modifiée [ITTm]).
La proportion de réponse moléculaire majeure (RMM) à 12 mois (48 semaines) dans le groupe traité par bosutinib par rapport à celle du groupe traité par imatinib dans la population ITTm constituait le critère d’efficacité principal. La RMM a été définie comme étant un rapport BCR– ABL/ABL ≤ 0,1 % selon l’échelle internationale (correspondant à une réduction ≥ 3 log par rapport aux valeurs initiales standard) avec un minimum de 3 000 transcrits ABL évalués par le laboratoire central.
Les principaux critères d’évaluation secondaires comprenaient la réponse cytogénétique complète (RCyC) cumulée sur 12 mois, la durée de la RCyC, la durée de la RMM, la survie sans événement (SSE) et la survie globale (SG). La RCyC cumulée sur 12 mois a été définie comme l’absence de métaphases Ph+ dans l’analyse des bandes chromosomiques de ≥ 20 métaphases provenant de l’aspiration de moelle osseuse ou de la RMM en l’absence d’une évaluation cytogénétique adéquate. Les valeurs de p pour les critères d’évaluation autres que la RMM à 12 mois et la RCyC cumulée sur 12 mois n’ont pas été ajustées pour les comparaisons multiples.
Les caractéristiques à l’inclusion pour la population ITTm étaient bien équilibrées entre les 2 groupes de traitement en fonction de l’âge (l’âge médian était de 52 ans pour le groupe bosutinib et de 53 ans pour le groupe imatinib avec 19,5 % et 17,4 % de patients âgés de 65 ans et plus, respectivement), du sexe (42,3 % et 44,0 % de femmes, respectivement), de l’origine ethnique (78,0 % et 77,6 % de Caucasiens, 12,2 % et 12,4 % d’Asiatiques, 4,1 % et 4,1 % de Noirs ou d’Afroaméricains, et 5,7 % et 5,4 % de la catégorie « Autre », respectivement, ainsi qu’une personne d’origine ethnique « inconnue » dans le groupe imatinib), et du score de risque de Sokal (risque faible : 35,0 % et 39,4 %, risque intermédiaire : 43,5 % et 38,2 %, risque élevé : 21,5 % et 22,4 %, respectivement).
Après une période de suivi de 60 mois dans la population ITTm, 60,2 % des patients traités par bosutinib (N = 246) et 59,8 % des patients traités par imatinib (N = 239) recevaient toujours un traitement de première intention.
Après une période de suivi de 60 mois dans la population ITTm, les arrêts de traitement dus à la progression de la maladie vers une LMC en PA ou CB pour les patients traités par bosutinib ont été de 0,8 % contre 1,7 % pour les patients traités par imatinib.
Six (2,4 %) patients sous bosutinib et 7 (2,9 %) patients sous imatinib ont développé une progression de la maladie vers une LMC en PA ou une LMC en CB. Des arrêts de traitement dus à une réponse sous-optimale ou à un échec du traitement, tels qu'évalués par l’investigateur, sont survenus chez 5,3 % des patients du groupe traité par bosutinib contre 15,5 % des patients du groupe traité par imatinib. Douze (4,9 %) patients sous bosutinib et 14 (5,8 %) patients sous imatinib sont décédés au cours de l’étude. Aucune progression supplémentaire n’est survenue dans la population ITT, 2 décès supplémentaires sont survenus dans le bras bosutinib dans la population ITT.
Les résultats d’efficacité de la RMM et de la RCyC sont récapitulés dans le tableau 3.
Tableau 3 Résumé des taux de RMM aux Mois 12 et 18 et des taux de la RCyC cumulée sur 12 mois, par groupe de traitement dans la population ITTm
|
Réponse |
Bosutinib (N = 246) |
Imatinib (N = 241) |
Odds ratio (IC à 95 %)a |
|
Réponse moléculaire majeure RMM au Mois 12, n (%) (IC à 95 %) |
116 (47,2)b (40,9 ; 53,4) |
89 (36,9) (30,8 ; 43,0) |
1,55 (1,07 ; 2,23) |
|
Valeur de p unilatérale |
0,0100b |
||
|
RMM au Mois 18, n (%) (IC à 95 %) |
140 (56,9) (50,7 ; 63,1) |
115 (47,7) (41,4 ; 54,0) |
1,45 (1,02 ; 2,07) |
|
Valeur de p unilatérale |
0,0208c |
||
|
Réponse cytogénétique complète RCyC cumulée sur 12 mois, n (%) (IC à 95 %) |
190 (77,2)b (72,0 ; 82,5) |
160 (66,4) (60,4 ; 72,4) |
1,74 (1,16 ; 2,61) |
|
Valeur de p unilatérale |
0,0037b |
||
Remarque : la RMM a été définie comme étant un rapport BCR– ABL/ABL ≤ 0,1 % selon l’échelle internationale (correspondant à une réduction ≥ 3 log par rapport aux valeurs initiales standard) avec un minimum de 3 000 transcrits ABL évalués par le laboratoire central. La réponse cytogénétique complète a été définie comme l’absence de métaphases Ph+ dans l’analyse des bandes chromosomiques de ≥ 20 métaphases provenant de l’aspiration de moelle osseuse ou de la RMM en l’absence d’une évaluation cytogénétique adéquate.
Abréviations : BCR– AB = breakpoint cluster region-Abelson ; IC = intervalle de confiance ; CMH = Cochran– Mantel– Haenszel ; RCyC = réponse cytogénétique complète ; ITTm = intention de traiter modifiée ; RMM = réponse moléculaire majeure ; N/n = nombre de patients ; Ph+ = chromosome Philadelphie positif.
a Ajusté en fonction de la zone géographique et du score de Sokal au moment de la randomisation.
b Comparaison statistiquement significative au niveau du seuil de significativité prédéfini ; d’après le test de CMH stratifié en fonction de la zone géographique et du score de Sokal au moment de la randomisation.
c D’après le test de CMH stratifié en fonction de la zone géographique et du score de Sokal au moment de la randomisation.
Au Mois 12, le taux de RM4 (défini comme BCR– ABL ≤ 0,01 % [correspondant à une réduction ≥ 4 log par rapport aux valeurs initiales standard] avec un minimum de 9 800 transcrits ABL) était plus élevé dans le groupe traité par bosutinib que dans le groupe traité par imatinib dans la population ITTm (20,7 % [IC à 95 % : 15,7 % ; 25,8 %] versus 12,0 % [IC à 95 % : 7,9 % ; 16,1 %], respectivement, odds ratio (OR) de 1,88 [IC à 95 % : 1,15 ; 3,08], valeur de p unilatérale = 0,0052).
Aux Mois 3, 6 et 9, la proportion de patients présentant une RMM était plus élevée dans le groupe traité par bosutinib que dans le groupe traité par imatinib (tableau 4).
Tableau 4 Comparaison des taux de RMM aux Mois 3, 6 et 9 de traitement dans la population ITTm
|
Délai |
Nombre (%) de sujets présentant une RMM |
Odds ratio (IC à 95 %)a |
|
|
Bosutinib (N = 246) |
Imatinib (N = 241) |
||
|
Mois 3 (IC à 95 %) |
10 (4,1) (1,6 ; 6,5) |
4 (1,7) (0,0 ; 3,3) |
2,48 (0,77 ; 7,98) |
|
Valeur de p unilatéraleb |
0,0578 |
||
|
Mois 6 (IC à 95 %) |
86 (35,0) (29,0 ; 40,9) |
44 (18,3) (13,4 ; 23,1) |
2,42 (1,59 ; 3,69) |
|
Valeur de p unilatéraleb |
< 0,0001 |
||
|
Mois 9 (IC à 95 %) |
104 (42,3) (36,1 ; 48,4) |
71 (29,5) (23,7 ; 35,2) |
1,78 (1,22 ; 2,60) |
|
Valeur de p unilatéraleb |
0,0015 |
||
Remarque : les pourcentages étaient basés sur le nombre de patients dans chaque groupe de traitement. La RMM a été définie comme étant un rapport BCR– ABL/ABL ≤ 0,1 % selon l’échelle internationale (correspondant à une réduction ≥ 3 log par rapport aux valeurs initiales standard) avec un minimum de 3 000 transcrits ABL évalués par le laboratoire central.
Abréviations : BCR– ABL = breakpoint cluster region-Abelson ; IC = intervalle de confiance ; CMH = Cochran– Mantel– Haenszel ; ITTm = intention de traiter modifiée ; RMM = réponse moléculaire majeure ; N = nombre de patients.
a Ajusté en fonction de la zone géographique et du score de Sokal au moment de la randomisation.
b D’après le test de CMH stratifié en fonction de la zone géographique et du score de Sokal au moment de la randomisation.
Cumulée sur 60 mois, dans la population ITTm, la proportion de patients présentant une RMM, une RM4 et une RM4,5 était plus élevée dans le groupe bosutinib que dans le groupe imatinib (tableau 5). Les taux de RMM cumulée sur 60 mois dans les sous-groupes de risque de Sokal sont récapitulés dans le tableau 6.
Tableau 5 Résumé de la réponse moléculaire cumulée sur 60 mois dans la population ITTm
|
Réponse |
Bosutinib (N = 246) |
Imatinib (N = 241) |
Odds ratio (IC à 95 %)a |
|
Réponse moléculaire cumulée sur 60 mois, n (%) (IC à 95 %) |
|
|
|
|
RMM |
182 (74,0) (68,5 ; 79,5) |
158 (65,6) (59,6 ; 71,6)
|
1,52 (1,02 ; 2,25) |
|
RM4 |
145 (58,9) (52,8 ; 65,1) |
120 (49,8) (43,5 ; 56,1)
|
1,46 (1,02 ; 2,09) |
|
RM4,5 |
119 (48,4) (42,1 ; 54,6) |
93 (38,6) (32,4 ; 44,7) |
1,50 (1,05 ; 2,16) |
Remarque : les RMM/RM4/RM4,5 ont été définies comme étant un rapport BCR– ABL/ABL ≤ 0,1/0,01/0,0032 % selon l’échelle internationale (correspondant à une réduction ≥ 3/4/4,5 log par rapport aux valeurs initiales standard) avec un minimum de 3 000/9 800/30 990 transcrits ABL évalués par le laboratoire central.
Abréviations : BCR– ABL = breakpoint cluster region-Abelson ; IC = intervalle de confiance ; ITTm = intention de traiter modifiée ; RMM = réponse moléculaire majeure ; RM = réponse moléculaire ; N/n = nombre de patients.
a Ajusté en fonction de la zone géographique et du score de Sokal au moment de la randomisation.
Tableau 6 Résumé de la RMM cumulée sur 60 mois par score de risque de Sokal dans la population ITTm
|
Réponse |
Bosutinib |
Imatinib |
Odds ratio (IC à 95 %) |
|
Risque de Sokal faible RMM, n (%) (IC à 95 %) |
N = 86 67 (77,9) (69,1 ; 86,7) |
N = 95 68 (71,6) (62,5 ; 80,6) |
1,40 (0,71 ; 2,76) |
|
Risque de Sokal intermédiaire RMM, n (%) (IC à 95 %) |
N = 107 79 (73,8) (65,5 ; 82,2) |
N = 92 62 (67,4) (57,8 ; 77,0) |
1,37 (0,74 ; 2,52) |
|
Risque de Sokal élevé RMM, n (%) (IC à 95 %) |
N = 53 36 (67,9) (55,4 ; 80,5) |
N = 54 28 (51,9) (38,5 ; 65,2) |
1,97 (0,90 ; 4,32) |
Remarque : les pourcentages étaient basés sur le nombre de patients dans chaque groupe de traitement. La RMM a été définie comme étant un rapport BCR– ABL/ABL ≤ 0,1 % selon l’échelle internationale (correspondant à une réduction ≥ 3 log par rapport aux valeurs initiales standard) avec un minimum de 3 000 transcrits ABL évalués par le laboratoire central.
Abréviations : BCR– ABL = breakpoint cluster region-Abelson ; IC = intervalle de confiance ; ITTm = intention de traiter modifiée ; RMM = réponse moléculaire majeure ; N/n = nombre de patients.
L’incidence cumulée de la RCyC ajustée en fonction du risque concurrent d’arrêt du traitement sans RCyC était plus élevée dans le groupe traité par bosutinib que dans le groupe traité par imatinib dans la population ITTm (83,3 % [IC à 95 % : 78,1 % ; 87,4 %] versus 76,8 % [IC à 95 % : 70,9 % ; 81,6 %] au Mois 60 ; rapport de risque [RR] à partir d’un modèle de sous-répartition proportionnelle stratifié : 1,35 [IC à 95 % : 1,11 ; 1,64]). Le délai médian d’obtention de la RCyC (répondeurs uniquement) était de 24,0 semaines (intervalle : 11,4 à 120,7) dans le groupe bosutinib versus 24,3 semaines (intervalle : 11,4 à 96,6) dans le groupe imatinib.
Le délai médian d’obtention de la RMM, de la RM4 et de la RM4,5 (répondeurs uniquement) était de 36,1 semaines (intervalle : 11,9 à 241,9), 83,7 semaines (intervalle : 12,4 à 244,3) et 108,0 semaines (intervalle : 24,1 à 242,1), respectivement, pour le groupe bosutinib versus 47,7 semaines (intervalle : 12,1 à 216,1), 84,4 semaines (intervalle : 23,6 à 241,9), et 120,4 semaines (intervalle : 24,6 à 240,7), respectivement, pour le groupe traité par imatinib dans la population ITTm.
L’incidence cumulée de la RMM, de la RM4 et de la RM4,5 ajustée en fonction du risque concurrent d’arrêt du traitement sans l’événement était plus élevée dans le groupe traité par bosutinib que dans le groupe traité par imatinib, comme illustré aux figures 1 à 3.
Figure 1 Incidence cumulée de la RMM (population ITTm)
|
|
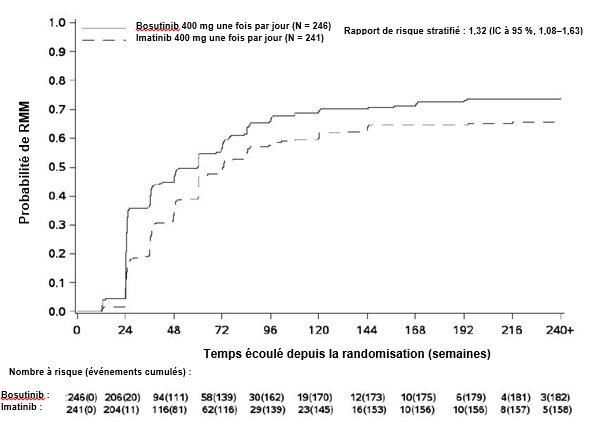
Figure 2 Incidence cumulée de la RM4 (population ITTm)
|
|
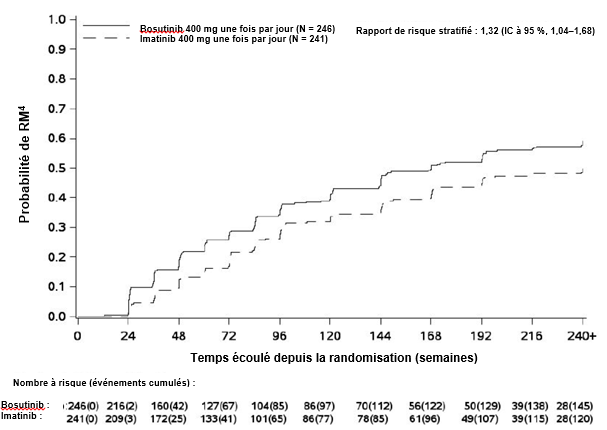
Figure 3 Incidence cumulée de la RM4,5 (population ITTm)
|
|

Dans la population ITTm, parmi les patients ayant obtenu une RCyC, l’estimation de Kaplan-Meier du maintien d’une réponse à 4 ans était de 97,4 % (IC à 95 % : 93,9 % ; 98,9 %) et de 93,7 % (IC à 95 % : 88,9 % ; 96,5 %) dans les groupes bosutinib et imatinib (RR : 0,39 [IC à 95 % : 0,14 ; 1,13]), respectivement. Parmi les patients ayant obtenu une RMM, l’estimation de Kaplan-Meier du maintien d’une réponse à 4 ans était de 92,2 % (IC à 95 % : 86,8 % ; 95,4 %) et de 92,0 % (IC à 95 % : 85,9 % ; 95,5 %) dans les groupes bosutinib et imatinib (RR : 1,09 [IC à 95 % : 0,49 ; 2,44]), respectivement.
Cumulé sur 60 mois, dans la population ITTm, 43,9 % (IC à 95 % : 37,7 % ; 50,1 %) et 38,6 % (IC à 95 % : 32,4 % ; 44,7 %) des patients traités par bosutinib et imatinib (OR : 1,24 [IC à 95 % : 0,87 ; 1,78]), respectivement, avaient une RM4 stable définie par les critères suivants : sous traitement pendant au moins 3 ans avec au moins une RM4 à toutes les évaluations pendant une période de 1 an.
L’incidence cumulée des événements de SSE sous traitement au Mois 60 dans la population ITTm était de 6,9 % (IC à 95 % : 4,2 % ; 10,5 %) dans le bras bosutinib et de 10,4 % (IC à 95 % : 6,9 % ; 14,6 %) dans le bras imatinib (RR : 0,64 ; IC à 95 % : 0,35 ; 1,17).
Les estimations de Kaplan– Meier de SG au Mois 60 pour les patients sous bosutinib et imatinib dans la population ITTm étaient de 94,9 % (IC à 95 % : 91,1 % ; 97,0 %) et 94,0 % (IC à 95 % : 90,1 % ; 96,4 %), respectivement (RR : 0,80 ; IC à 95 % : 0,37 ; 1,73).
Dans une analyse rétrospective, parmi les patients évaluables de la population ITT, un nombre supérieur de patients du bras bosutinib 200/248 (80,6 %) ont obtenu une réponse moléculaire précoce (transcrits BCR-ABL ≤ 10 % à 3 mois) par rapport aux patients du bras imatinib 153/253 (60,5 %), OR : 2,72 (IC à 95 % : 1,82 ; 4,08). La RMM et la SSE au Mois 60 chez les patients sous bosutinib avec et sans réponse moléculaire précoce sont récapitulées dans le tableau 7.
Tableau 7 Résultats au Mois 60 chez les patients sous bosutinib avec un BCR-ABL ≤ 10 % vs > 10 % au Mois 3 dans la population ITT
|
Bosutinib (N = 248) |
Patients avec un BCR-ABL ≤ 10 % à 3 mois (N = 200) |
Patients avec un BCR-ABL > 10 % à 3 mois (N = 48) |
Rapport de risque (IC à 95 %)a |
|
Incidence cumulée de la RMM, % (IC à 95 %) |
84,0 (78,1 ; 88,4) |
56,5 (41,1 ; 69,4) |
2,67 (1,90 ; 3,75) |
|
Incidence cumulée des événements de SSE, % (IC à 95 %) |
5,5 (2,9 ; 9,3) |
12,5 (5,1 ; 23,4) |
0,40 (0,14 ; 1,17) |
Abréviations : BCR– ABL = breakpoint cluster region-Abelson ; IC = intervalle de confiance ; ITT = intention de traiter ; RMM = réponse moléculaire majeure ; SSE = survie sans événement ; N = nombre de patients présentant des copies ABL ≥ 3 000 au Mois 3.
a Ajusté en fonction de la zone géographique et du score de Sokal au moment de la randomisation.
Un nombre inférieur de patients du bras bosutinib [6 (2,4 %) pour le bosutinib et 12 (5,0 %) pour l’imatinib] présentaient des nouvelles mutations à 60 mois dans la population ITTm.
Etude clinique de phase I/II chez des patients atteints de LMC résistante ou intolérante à l'imatinib en PC, PA et en CB
Un essai de phase I/II multicentrique, en ouvert, à bras unique a été mené afin d'évaluer l'efficacité et la sécurité du bosutinib 500 mg une fois par jour chez des patients atteints de LMC résistante ou intolérante à l'imatinib avec des cohortes distinctes pour les malades en phase chronique, accélérée et en crise blastique précédemment traités avec 1 ITK (imatinib) ou plusieurs ITK (imatinib suivi du dasatinib et/ou du nilotinib).
Au cours de cet essai, 570 patients ont été traités par bosutinib, dont des patients atteints de LMC en PC précédemment traités avec 1 seul ITK (imatinib), des patients atteints de LMC-PC précédemment traités avec de l'imatinib et au moins 1 ITK supplémentaire (dasatinib et/ou nilotinib), des patients atteints de LMC en phase accélérée ou en crise blastique précédemment traités avec au moins 1 ITK (imatinib) et des patients atteints de LAL Ph+ précédemment traités avec au moins 1 ITK (imatinib).
Le critère d'efficacité principal de l'étude était le taux de réponse cytogénétique majeure (RCyM) à la Semaine 24 chez les patients atteints de LMC en PC résistants à l'imatinib précédemment traités avec 1 seul ITK (imatinib). Les autres critères d’efficacité comprenaient les taux cumulés de réponse cytogénétique et moléculaire, le délai d’obtention et la durée des réponses cytogénétiques et moléculaires, la réponse en fonction des mutations identifiées à l’inclusion, le taux de transformation en PA/CB, la survie sans progression et la SG pour toutes les cohortes.
Les patients qui recevaient encore du bosutinib à la fin de l’étude de phase I/II et qui bénéficiaient du traitement par bosutinib selon le jugement de l’investigateur, ainsi que les patients qui avaient déjà arrêté le bosutinib dans le cadre de l’étude de phase I/II et qui faisaient l’objet d’un suivi de survie à long terme ou qui avaient terminé l’étude de phase I/II, étaient éligibles à l’étude d’extension. Chaque patient est resté dans l’étude d’extension, soit sous traitement par bosutinib, soit en suivi de survie à long terme, jusqu’à ce que le dernier patient atteigne 10 ans de suivi, calculé à partir de la date de sa première dose de bosutinib administrée dans l’étude de phase I/II.
Les critères d’efficacité de l’étude d’extension comprenaient la durée des réponses cytogénétiques et moléculaires, le taux de transformation en PA/CB, la survie sans progression et la SG.
Les analyses d’efficacité ont inclus les données finales de cette étude d’extension achevée.
Patients atteints de LMC en PC
Les résultats d'efficacité chez les patients atteints de LMC Ph+ en PC précédemment traités avec de l'imatinib et au moins 1 ITK supplémentaire (suivi minimum de 120 mois, durée médiane de traitement de 9 mois [intervalle : 0,23 à 164,28 mois] et 20,2 % et 7,6 % toujours sous traitement à 60 et 120 mois, respectivement) et les résultats de patients atteints de LMC Ph+ en PC précédemment traités avec de l’imatinib uniquement (suivi minimum de 120 mois, durée médiane du traitement de 26 mois [intervalle : 0,16 à 170,49 mois] et 40,5 % et 19,4 % toujours sous traitement à 60 et 120 mois, respectivement) sont présentés dans le tableau 8.
Patients atteints de LMC en PA et CB
Les résultats d'efficacité chez les patients atteints de LMC Ph+ en PA (suivi minimum de 120 mois, durée médiane du traitement de 10 mois [intervalle : 0,10 à 156,15 mois] et 12,7 % et 7,6 % toujours sous traitement à 60 et 120 mois, respectivement) et en CB (suivi minimum de 120 mois, durée médiane du traitement de 2,8 mois [intervalle : 0,03 à 71,38 mois] et 3,1 % et 0 % toujours sous traitement à 60 et 120 mois respectivement) sont présentés dans le tableau 8.
Tableau 8 Résultats d'efficacité chez des patients précédemment traités atteints de LMC* en phase chronique et avancée
|
|
LMC Ph+ en PC avec traitement antérieur par imatinib uniquement |
LMC Ph+ en PC avec traitement antérieur par imatinib et dasatinib ou nilotinib |
Phase accélérée avec traitement antérieur par imatinib au moins |
Crise blastique avec traitement antérieur par imatinib au moins |
|
Réponse cytogénétique cumuléea RCyM, % (IC à 95 %) RCyC, % (IC à 95 %) |
N = 262 59,9 (53,7 ; 65,9) 49,6 (43,4 ; 55,8) |
N = 112 42,0 (32,7 ; 51,7) 32,1 (23,6 ; 41,6) |
N = 72 40,3 (28,9 ; 52,5) 30,6 (20,2 ; 42,5) |
N = 54 37,0 (24,3 ; 51,3) 27,8 (16,5 ; 41,6) |
|
Réponse moléculaire cumuléea RMM, % (IC à 95 %) RM4, % (IC à 95 %) |
N = 197 42,1 (35,1 ; 49,4) 37,1 (30,3 ; 44,2) |
N = 107 17,8 (11,0 ; 26,3) 15,0 (8,8 ; 23,1) |
N = 54 16,7 (7,9 ; 29,3) 13,0 (5,4 ; 24,9) |
N = 48 10,4 (3,5 ; 22,7) 10,4 (3,5 ; 22,7) |
|
Délai d’obtention de la RCyM chez les répondeurs uniquementb, médiane (intervalle), semaines |
12,3 (4,0 ; 346,0) |
12,3 (3,9 ; 550,6) |
12,0 (3,9 ; 144,7) |
8,2 (3,9 ; 25,1) |
|
Durée de la RCyMb K-M à 5 ans, % (IC à 95 %) K-M à 10 ans, % (IC à 95 %) Médiane, semaines (IC à 95 %) |
N = 157 70,7 (63,1 ; 78,3) 65,3 (56,6 ; 74,0) N/Att |
N = 47 66,6 (51,5 ; 81,7) 55,3 (36,3 ; 74,4) N/Att |
N = 29 40,8 (20,9 ; 60,7) 40,8 (20,9 ; 60,7) 84,0 (24,0 ; N/E) |
N = 20 21,2 (0,1 ; 42,3) N/E 29,1 (11,9 ; 38,3) |
|
Délai d’obtention de la RCyC pour les répondeurs uniquementb, médiane (intervalle), semaines |
24,0 (7,7 ; 240,6) |
24,0 (11,6 ; 216,0) |
23,8 (4,1 ; 120,0) |
8,4 (3,9 ; 25,1) |
|
Durée de la RCyCb K-M à 5 ans, % (IC à 95 %) K-M à 10 ans, % (IC à 95 %) Médiane, semaines (IC à 95 %) |
N = 130 69,7 (61,3 ; 78,2) 63,4 (54,0 ; 72,8) N/Att |
N = 36 54,4 (36,7 ; 72,1) 40,8 (22,0 ; 59,6) 252,0 (24,0 ; N/E) |
N = 22 40,0 (18,5 ; 61,5) 40,0 (18,5 ; 61,5) 72,0 (36,1 ; N/E) |
N = 15 24,9 (0,9 ; 48,9) N/E 20,0 (9,1 ; 29,6) |
|
Délai d’obtention de la RMM pour les répondeurs uniquementb, médiane (intervalle), semaines |
35,6 (3,1 ; 367,1) |
12,4 (4,0 ; 171,7) |
36,1 (12,1 ; 144,1) |
4,7 (3,9 ; 168,9) |
|
Durée de la RMMb K-M à 5 ans, % (IC à 95 %) K-M à 10 ans, % (IC à 95 %) Médiane, semaines (IC à 95 %) |
N = 83 74,1 (64,2 ; 83,9) 63,4 (50,2 ; 76,6) N/Att |
N = 19 70,0 (47,5 ; 92,5) 70,0 (47,5 ; 92,5) N/Att |
N = 9 66,7 (35,9 ; 97,5) 66,7 (35,9 ; 97,5) N/Att |
N = 5 60,0 (17,1 ; 100,0) N/E N/Att |
|
Délai d’obtention de la RM4 pour les répondeurs uniquementb, médiane (intervalle), semaines |
28,0 (3,1 ; 583,1) |
23,8 (4,0 ; 240,1) |
24,1 (22,9 ; 96,0) |
4,7 (3,9 ; 284,9) |
|
Durée de la RM4b,e K-M à 5 ans, % (IC à 95 %) K-M à 10 ans, % (IC à 95 %) Médiane, semaines (IC à 95 %) |
N = 73 74,7 (64,2 ; 85,2) 60,8 (46,1 ; 75,4) N/Att |
N/A |
N/A |
N/A |
|
Transformation en PA/CBc en cours de traitement, n |
N = 284 15 |
N = 119 5 |
N = 79 3 |
N/A |
|
Survie sans progressionc IncCum à 5 ans, % (IC à 95 %)d IncCum à 10 ans, % (IC à 95 %)d |
N = 284 19,7 (15,6 ; 24,9) 23,9 (19,5 ; 29,5) |
N = 119 24,4 (17,8 ; 33,4) 26,9 (20,0 ; 36,2) |
N = 79 41,8 (32,2 ; 54,2) 41,8 (32,2 ; 54,2) |
N = 64 67,2 (56,6 ; 79,7) N/E |
|
Survie globalec K-M à 5 ans, % (IC à 95 %) K-M à 10 ans, % (IC à 95 %) Médiane, mois (IC à 95 %) |
N = 284 83,5 (78,7 ; 88,3) 71,5 (64,4 ; 78,7) N/Att |
N = 119 74,1 (64,8 ; 83,4) 60,4 (47,2 ; 73,7) N/Att |
N = 79 58,5 (46,9 ; 70,2) 50,7 (36,5 ; 65,0) N/Att |
N = 64 22,5 (7,1 ; 37,9) 22,5 (7,1 ; 37,9) 10,9 (8,7 ; 19,7) |
Date de consultation des données : 2 oct 2015 pour l’étude de phase I/II, 2 sept 2020 pour l’étude d’extension.
Critères de réponse cytogénétique : la RCyM incluait les réponses complètes [0 % de métaphases Ph+ de la moelle osseuse ou < 1 % de cellules positives après hybridation in situ en fluorescence (FISH) ou partielles (1 %– 35 %). Les réponses cytogénétiques étaient basées sur le pourcentage de métaphases Ph+ sur ≥ 20 cellules métaphasiques dans chaque prélèvement de moelle osseuse.
L'analyse FISH (≥ 200 cellules) a pu être utilisée pour les évaluations cytogénétiques post-inclusion en l'absence de ≥ 20 métaphases. Dans l’étude d’extension, la RCyC a été imputée à partir de la RMM si une évaluation cytogénétique valide n’était pas disponible à une date spécifique.
Critères de réponse moléculaire : au cours de l’étude de phase I/II, les RMM/RM4 ont été définies comme étant ≤ 0,1/0,01 % de transcrits BCR– ABL/ABL selon l’évaluation du laboratoire central (pas sur l'échelle internationale). Au cours de l’étude d’extension, les répondeurs avaient une RMM/RM4 indiquée sur le rapport de cas, telle qu’évaluée par un laboratoire local.
Abréviations : PA = phase accélérée ; CB = crise blastique ; Ph+ = chromosome Philadelphie positif ; PC = phase chronique ; LMC = leucémie myéloïde chronique ; K-M = Kaplan– Meier ; N/n = nombre de patients ; N/A = non applicable ; N/Att = non atteint lors du suivi minimum ; N/E = non estimable ; IC = intervalle de confiance ; RCyM = réponse cytogénétique majeure ; RCyC = réponse cytogénétique complète ; IncCum = incidence cumulée ; RMM = réponse moléculaire majeure ; BCR ABL = breakpoint cluster region-Abelson.
a Inclut les patients (N) présentant une évaluation valide à l'inclusion pour l’évaluation cytogénétique et les patients ne provenant pas de Chine, d’Afrique du Sud, d’Inde ou de Russie pour l’évaluation moléculaire car les échantillons ne pouvaient pas être exportés pour l’évaluation moléculaire dans ces pays. Les analyses permettent d'inclure les patients présentant une réponse à l'inclusion et la maintenant par la suite. Durée minimum de suivi (délai entre la première dose du dernier patient et la date de consultation des données) de 120 mois.
b Inclut les patients (N) qui ont atteint ou maintenu une réponse.
c Inclut les patients (N) qui ont reçu au moins 1 dose de bosutinib.
d Analyse de l’incidence cumulée ajustée pour le risque concurrent d’arrêt du traitement sans l’événement.
e Non analysé pour les groupes à effectif réduit.
La survie globale dans les cohortes PC, PA et CB est représentée graphiquement dans la figure 4.
Figure 4 Estimation de Kaplan-Meier de la survie globale (SG) dans les cohortes PC2L, PC3L, PA et CB
|
|
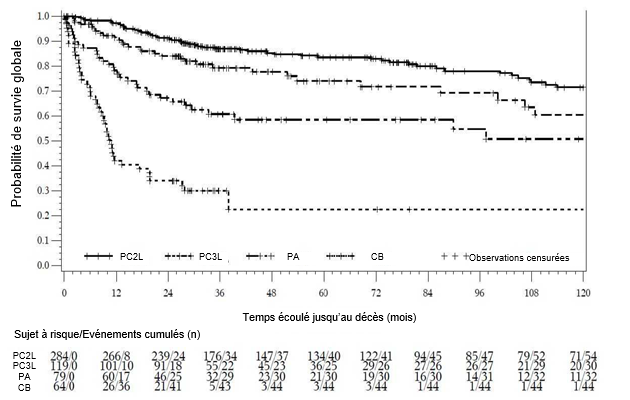
![]() Tableau 9 Réponse en fonction des mutations BCR-ABL identifiées à l’inclusion chez les patients évaluables atteints de LMC-PC : avant traitement par imatinib et dasatinib et/ou nilotinib (troisième ligne)
Tableau 9 Réponse en fonction des mutations BCR-ABL identifiées à l’inclusion chez les patients évaluables atteints de LMC-PC : avant traitement par imatinib et dasatinib et/ou nilotinib (troisième ligne)
|
Mutation BCR-ABL à l’inclusion |
Fréquence à l’inclusion (%)a |
RcyM atteinte ou maintenue Rép/Evalb (%) N = 112 |
|
Mutation évaluée |
98 (100,0) |
36/92 (39,1) |
|
Pas de mutation |
59 (60,2) |
23/55 (41,8) |
|
Au moins 1 mutation |
39 (39,8) |
13/37 (35,1) |
|
Mutations résistantes au dasatinib |
10 (10,2) |
1/9 (11,1) |
|
E255K/V |
2 (2,0) |
0/2 |
|
F317L |
8 (8,2) |
1/7 (14,3) |
|
Mutations résistantes au nilotinibc |
13 (13,3) |
8/13 (61,5) |
|
Y253H |
6 (6,1) |
5/6 (83,3) |
|
E255K/V |
2 (2,0) |
0/2 |
|
F359C/I/V |
7 (7,1) |
5/7 (71,4) |
Date de consultation des données : 2 oct 2015 pour la phase I/II, 2 sept 2020 pour l’étude d’extension.
Remarque : les mutations à l’inclusion ont été identifiées avant l’administration de la première dose de médicament à l’étude.
Abréviations : BCR– ABL = breakpoint cluster region-Abelson ; PC = phase chronique ; LMC = leucémie myéloïde chronique ; RCyM = réponse cytogénétique majeure ; N/n = nombre de patients ; Rép = répondeurs ; Eval = évaluable.
a Le pourcentage est basé sur l’évaluation du nombre de patients porteurs de mutations à l’inclusion.
b la population évaluable comprend les patients atteints d’une évaluation valide de la maladie à inclusion.
c 2 patients présentaient plus de 1 mutation dans cette catégorie.
Un patient porteur d’une mutation E255V précédemment traitée par nilotinib a atteint une RHC comme meilleure réponse.
Les essais in vitro indiquaient une activité limitée du bosutinib contre les mutations T315I ou V299L.
Aucune activité clinique n’est donc attendue chez les patients porteurs de ces mutations.
Etude clinique de phase IV chez des patients atteints de LMC Ph+ et ayant déjà reçu au moins 1 ITK
Une étude de phase IV, à bras unique, en ouvert, non randomisée, multicentrique a été menée pour évaluer l’efficacité et la sécurité du bosutinib à la dose de 500 mg une fois par jour chez des patients atteints de LMC résistants ou intolérants aux ITK, avec des cohortes séparées distinctes par phase de la maladie (PC, PA ou CB) et par ligne de traitement précédente (1 ITK ou plus).
163 patients ont été traités par bosutinib dans cet essai, dont 46 patients atteints de LMC Ph+ en PC ayant été traités précédemment par 1 ITK (imatinib, dasatinib ou nilotinib), 61 patients atteints de LMC Ph+ en PC ayant été traités précédemment par 2 ITK (imatinib et/ou dasatinib et/ou nilotinib), 49 patients atteints de LMC Ph+ en PC ayant été traités précédemment par 3 ITK (imatinib et dasatinib et nilotinib), 4 patients atteints de LMC Ph+ en PA ayant été traités précédemment par au moins 1 ITK (2 patients traités précédemment par 2 ITK et 2 patients traités précédemment par 3 ITK) et 3 patients atteints de LMC Ph- traités précédemment par au moins 1 ITK.
Le critère d’efficacité principal était la RCyM confirmée cumulée sur 1 an (Semaine 52) chez les patients atteints de LMC Ph+ en PC précédemment traités par 1 ou 2 ITK et chez les patients atteints de LMC Ph+ en PC traités précédemment par 3 ITK. Chez les patients atteints de LMC Ph+ en PA et CB ayant reçu précédemment un traitement par ITK, le critère d'efficacité principal était la réponse hématologique globale (RHG) confirmée cumulée sur 1 an (Semaine 52). Les autres critères d’efficacité chez les patients atteints de LMC Ph+ en PC comprenaient le taux de réponse cytogénétique et moléculaire cumulée, la durée des réponses cytogénétiques et moléculaires, le taux de réponse en fonction des mutations identifiées à l’inclusion, le taux de transformation en PA/CB, la SSP et la SG.
Les critères d’évaluation supplémentaires dans la cohorte Ph+ PA/CB comprenaient les taux de réponses cytogénétiques et moléculaires cumulées, la SSP et la SG.
Patients atteints de LMC en PC
Le critère d’évaluation principal du taux de RCyM confirmées cumulées (IC à 95 %) sur 1 an (52 semaines) était de 76,5 % (66,9 ; 84,5) chez les patients traités précédemment par 1 ou 2 ITK et de 62,2 % (46,5 ; 76,2) chez les patients traités précédemment par 3 ITK.
Le tableau 10 présente d’autres résultats d’efficacité à la clôture de l’étude, après un suivi minimal de 3 ans, chez les patients atteints de LMC Ph+ traités précédemment par 1 (durée médiane du traitement de 47,5 mois [intervalle : 0,9 à 50,1 mois] et 60,9 % toujours sous traitement), 2 (durée médiane du traitement de 41,9 mois [intervalle : 0,4 à 48,9 mois] et 45,9 % toujours sous traitement) et 3 (durée médiane du traitement de 20,0 mois [intervalle : 0,2 à 48,9 mois] et 38,8 % toujours sous traitement) ITK.
Tableau 10 Résultats d’efficacité chez des patients précédemment traités atteints de LMC Ph+ en phase chronique
|
|
LMC Ph+ en PC traitée précédemment par 1 ITK |
LMC Ph+ en PC traitée précédemment par 2 ITK |
LMC Ph+ en PC traitée précédemment par 3 ITK |
Cohorte totale LMC Ph+ en PC |
|
RCyMa confirmée cumulée sur 1 an, % (IC à 95 %) |
N = 43 83,7 (69,3 ; 93,2) |
N = 55 70,9 (57,1 ; 82,4) |
N = 45 62,2 (46,5 ; 76,2) |
N = 143 72,0 (63,9 ; 79,2) |
|
Réponse cytogénétique cumuléea,b
RCyM, % (IC à 95 %)
RCyC, % (IC à 95 %) |
N = 46
88,4 (74,9 ; 96,1)
86,0 (72,1 ; 94,7) |
N = 55
85,5 (73,3 ; 93,5)
83,6 (71,2 ; 92,2) |
N = 45
77,8 (62,9 ; 88,8)
73,3 (58,1 ; 85,4) |
N = 143
83,9 (76,9 ; 89,5)
81,1 (73,7 ; 87,2) |
|
Réponse moléculaire cumuléea,b
RMM, % (IC à 95 %)
RM4, % (IC à 95 %)
RM4,5, % (IC à 95 %) |
N = 46
82,6 (68,6 ; 92,2)
73,9 (58,9 ; 85,7)
58,7 (43,2 ; 73,0) |
N = 55
76,4 (63 ; 0,86,8)
63,6 (49,6 ; 76,2)
50,9 (37,1 ; 64,6) |
N = 48
56,3 (41,2 ; 70,5)
41,7 (27,6 ; 56,8)
35,4 (22,2 ; 50,5) |
N = 149
71,8 (63,9 ; 78,9)
59,7 (51,4 ; 67,7)
48,3 (40,1 ; 56,6)
|
|
Délai d’obtention de la réponse cytogénétique pour les répondeurs uniquementb, médiane (intervalle), mois
RCyM RcyC |
3,0 (1,0 ; 11,8) 3,0 (1,0 ; 17,6) |
2,9 (0,3 ; 6,4) 2,9 (0,3 ; 6,4) |
3,0 (1,8 ; 8,8) 3,0 (1,8 ; 8,8) |
3,0 (0,3 ; 11,8) 3,0 (0,3 ; 17,6) |
|
Durée d’obtention de la réponse cytogénétiqueb
RCyM, K-M à 3 ans, % (IC à 95 %)
RCyC, K-M à 3 ans, % (IC à 95 %) |
96,6 (77,9 ; 99,5)
94,4 (77,2 ; 99,5) |
94,4 (79,2 ; 98,6)
94,4 (79,2 ; 98,6) |
96,9 (79,8 ; 99,6)
100,0 (100,0 ; 100,0) |
95,6 (88,7 ; 98,4)
96,5 (89,5 ; 98,9) |
|
Délai d’obtention de la réponse moléculaire pour les répondeurs uniquement, médiane (intervalle), mois
RMM
RM4
RM4,5 |
3,0 (2,8 ; 23,3)
6,0 (2,8 ; 47,4)
9,2 (2,8 ; 47,6)
|
3,0 (1,0 ; 35,9)
3,1 (1,0 ; 36,1)
6,0 (2,8 ; 36,2) |
3,1 (1,8 ; 9,3)
3,2 (1,8 ; 47,9)
5,8 (1,8 ; 18,0) |
3,0 (1,0 ; 35,9)
5,5 (1,0 ; 47,9)
6,0 (1,8 ; 47,6) |
|
Durée de la réponse moléculaireb
RMM, K-M à 3 ans, % (IC à 95 %)
RM4, K-M à 3 ans, % (IC à 95 %) |
90,7 (73,9 ; 96,9)
89,5 (70,9 ; 96,5) |
81,5 (63,2 ; 91,3)
68,7 (48,0 ; 82,5) |
90,2 (65,9 ; 97,5)
85,2 (51,9 ; 96,2) |
87,2 (78,0 ; 92,7)
80,7 (69,4 ; 88,1) |
|
Date de consultation des données : 23 nov 2020. Abréviations : Ph+ = chromosome de Philadelphie positif ; PC = phase chronique ; LMC = leucémie myéloïde chronique ; K-M = Kaplan-Meier ; N = nombre de patients ; IC = intervalle de confiance ; RCyM = réponse cytogénétique majeure ; RCyC = réponse cytogénétique complète ; RMM = réponse moléculaire majeure ; RM4 = réduction de transcrits BCR– ABL/ABL ≥ 4 log par rapport aux valeurs initiales standard ; RM4,5 = réduction de transcrits BCR– ABL/ABL ≥ 4,5 log par rapport aux valeurs initiales standard. Définition de la RCyM confirmée cumulée : la réponse est confirmée par 2 évaluations consécutives espacées d’au moins 28 jours. Pour être considéré comme un répondeur, le patient doit avoir maintenu la réponse présente à l’inclusion pendant au moins 52 semaines ou la réponse doit s’être améliorée par rapport à celle présente à l’inclusion. Les patients ayant une réponse cytogénétique partielle (RCyP) à l’inclusion doivent atteindre une RCyC sous traitement pour être considérés comme répondeurs cytogénétiques. Les patients présentant au moins une RMM et une réponse moléculaire plus profonde que la réponse présente à l’inclusion sont considérés comme des RCyC confirmés. Définition de la réponse cytogénétique cumulée : la réponse cytogénétique majeure comprenait les réponses cytogénétiques complètes (0 % de métaphases Ph+ dans la moelle osseuse ou < 1 % de cellules positives par hybridation in situ fluorescente [FISH]) ou partielles (1 %– 35 %). Les réponses cytogénétiques étaient basées sur le pourcentage de métaphases Ph+ parmi ≥ 20 cellules en métaphase dans chaque échantillon de moelle osseuse. Une analyse FISH (≥ 200 cellules) pouvait être utilisée pour évaluer la RCyC si ≥ 20 métaphases n’étaient pas évaluables. Les patients sans évaluation valide de la moelle osseuse ou de FISH et présentant au moins une RMM étaient comptés comme RCyC.
Définition de la réponse moléculaire cumulée : RMM, RM4 et RM4,5 ont été définies comme étant respectivement un rapport BCR-ABL/ABL ≤ 0,1 %, ≤ 0,01 % et ≤ 0,0032 selon l’échelle internationale (correspondant à une réduction ≥ 3, ≥ 4 et ≥ 4,5 log par rapport aux valeurs initiales standard) avec un minimum de 10 000, 10 000 et 32 000 transcrits ABL évalués par le laboratoire central, respectivement. a Inclut les patients (N) avec une évaluation à l’inclusion valide. Durée minimale de suivi (temps écoulé entre la première dose administrée au patient et la date de consultation des données) de 36 mois. b Inclut les patients (N) qui ont obtenu ou maintenu une réponse. |
||||
L’incidence cumulée de RMM, RM4 et RM4,5 ajustée pour le risque concurrent d’arrêt du traitement sans l’événement est présentée dans la figure 5.
Figure 5 Incidence cumulée de la réponse moléculaire (population évaluable en PC)
|
|
Les réponses moléculaires obtenues par ligne de traitement sont présentées dans le tableau 11.
Tableau 11 Réponses moléculaires obtenues
|
|
LMC Ph+ en PC traitée précédemment par 1 ITK |
LMC Ph+ en PC traitée précédemment par 2 ITK |
LMC Ph+ en PC traitée précédemment par 3 ITK |
Cohorte totale LMC Ph+ en PC |
|
Patients sans RMM à l’inclusiona
RMM, % (IC à 95 %) |
N = 25
76,0 (54,9 ; 90,6) |
N = 28
64,3 (44,1 ; 81,4) |
N = 26
38,5 (20,2 ; 59,4) |
N = 79
59,5 (47,9 ; 70,4) |
|
Patients sans RM4 à l’inclusiona
RM4, % (IC à 95 %) |
N = 37
70,3 (53,0 ; 84,1) |
N = 38
55,3 (38,3 ; 71,4) |
N = 37
32,4 (18,0 ; 49,8) |
N = 112
52,7 (43,0 ; 62,2) |
|
Patients sans RM4,5 à l’inclusiona
RM4,5, % (IC à 95 %) |
N = 42
54,8 (38,7 ; 70,2) |
N = 46
43,5 (28,9 ; 58,9) |
N = 43
30,2 (17,2 ; 46,1) |
N = 131
42,7 (34,1 ; 51,7) |
|
Patients avec RMM à l’inclusiona
RM plus profonde, % (IC à 95 %) |
N = 21
85,7 (63,7 ; 97,0) |
N = 27
66,7 (46,0 ; 83,5) |
N = 22
63,6 (40,7 ; 82,8) |
N = 70
71,4 (59,4 ; 81,6) |
|
Date de consultation des données : 23 nov 2020. Abréviations : Ph+ = chromosome de Philadelphie positif ; PC = phase chronique ; LMC = leucémie myéloïde chronique ; N = nombre de patients ; IC = intervalle de confiance ; RMM = réponse moléculaire majeure ; RM = réponse moléculaire ; RM4 = réduction des transcrits BCR– ABL/ABL ≥ 4 log par rapport aux valeurs initiales standard ; RM4,5 = réduction des transcrits BCR– ABL/ABL ≥ 4,5 log par rapport aux valeurs initiales standard. a Inclut les patients (N) avec une évaluation à l’inclusion valide. Pour être considérés comme répondeurs, les patients doivent avoir obtenu une réponse améliorée par rapport à la réponse présente à l’inclusion. Les RMM, RM4 et RM4,5 ont été définies comme étant respectivement un rapport BCR-ABL/ABL ≤ 0,1 %, ≤ 0,01 % et ≤ 0,0032 % selon l’échelle internationale (correspondant à une réduction ≥ 3, ≥ 4 et ≥ 4,5 log par rapport aux valeurs initiales standard) avec un minimum de 10 000, 10 000 et 32 000 transcrits ABL évalués par le laboratoire central, respectivement |
||||
Chez les patients PC, il n’y a pas eu de progression en cours de traitement vers une LMC en PA ou CB.
Patients atteints de LMC en PA
Chez les patients atteints de LMC Ph+ en PA, la durée médiane du traitement était de 22,1 mois (intervalle : 1,6 à 50,1 mois), le taux de RHC confirmée cumulé sur 1 an (52 semaines) était de 75,0 % (IC à 95 % : 19,4 à 99,4), tout comme le taux cumulé de RCyC, les 3 patients ont maintenu leur RCyC sous traitement.
Réponse en fonction des mutations BCR-ABL présentes à l’inclusion
Dix patients de la cohorte PC présentaient des mutations à l’inclusion (A365V, E453K, E255K, E255V, Q252H, L298V [n = 1 chacune], Y253F et G250E [n = 2 chacune]). Un patient de la cohorte PC présentait une mutation F359I identifiée au Jour 8 de l’étude. Un patient de la cohorte PA présentait 2 mutations (F311L et L387F) à l’inclusion. Dans la cohorte PC, parmi les patients présentant des mutations, des réponses moléculaires ont été observées chez 4/11 (36,4 %) patients, 1 patient présentant une mutation E255V a atteint une RMM et 3 patients présentant respectivement F359I, Y253F et A365V ont obtenu une RM4,5. Le patient présentant des mutations dans la cohorte PA n’a pas obtenu de réponse.
Population pédiatrique
L’Agence européenne des médicaments a différé l’obligation de soumettre les résultats d’études réalisées avec le médicament de référence contenant du bosutinib dans un ou plusieurs sous-groupes de la population pédiatrique atteinte de LMC (voir rubrique 4.2 pour les informations concernant l’usage pédiatrique).
5.2. Propriétés pharmacocinétiques
Après l'administration d'une dose unique de bosutinib (500 mg) au moment du repas à des sujets sains, la biodisponibilité absolue était de 34 %. L’absorption était relativement lente, le temps médian nécessaire pour atteindre la concentration maximale (tmax) étant de plus de 6 heures. Le bosutinib présente des augmentations proportionnelles de l'ASC et de la Cmax aux doses de 200 à 600 mg. L'absorption d'aliments a multiplié la Cmax de bosutinib par 1,8 et son ASC par 1,7 par rapport à une prise à jeun. Chez les patients atteints de LMC à l’état d’équilibre, la Cmax (moyenne géométrique, coefficient de variation [CV] en %) était de 145 (14) ng/mL et l’ASCéé (moyenne géométrique, CV en %) était de 2 700 (16) ngh/mL après l’administration quotidienne de 400 mg de bosutinib au moment du repas. Après l’administration quotidienne de 500 mg de bosutinib au moment du repas, la Cmax était de 200 (6) ngh/mL et l'ASCéé de 3 640 (12) ngh/mL. La solubilité du bosutinib est dépendante du pH et l’absorption est réduite lorsque le pH gastrique augmente (voir rubrique 4.5).
Distribution
Après l’administration d’une dose intraveineuse unique de 120 mg de bosutinib à des sujets sains, le bosutinib possédait un volume de distribution moyen (% de coefficient de variation [CV]) de 2 331 (32) L, ce qui suggère que le bosutinib est largement distribué dans les tissus extravasculaires.
Le bosutinib était fortement lié aux protéines plasmatiques humaines in vitro (94 %) et ex vivo chez les sujets sains (96 %), indépendamment de la concentration.
Biotransformation
Des études in vitro et in vivo ont indiqué que le bosutinib (composé d'origine) était principalement soumis au métabolisme hépatique chez l’être humain. Après l'administration d'une ou plusieurs doses de bosutinib (400 ou 500 mg) à des êtres humains, les principaux métabolites en circulation semblaient être du bosutinib oxychloré (M2) et N-déméthylé (M5), le N-oxyde (M6) de bosutinib étant un métabolite mineur en circulation. L'exposition systémique au métabolite N-déméthylé correspondait à 25 % du composé d'origine, contre 19 % pour le métabolite oxychloré. Les 3 métabolites présentaient une activité ≤ 5 % de celle de bosutinib au cours d'un test de prolifération des fibroblastes transformés par Src indépendamment de l'ancrage. Le bosutinib et le bosutinib N-déméthylé étaient les principaux composants médicamenteux présents dans les selles. Des études in vitro sur des microsomes hépatiques humains ont indiqué que le principal isoenzyme cytochrome P450 intervenant dans le métabolisme du bosutinib était le CYP3A4 et des études sur les interactions médicamenteuses ont indiqué que le kétoconazole et la rifampicine avaient produit des effets sur la pharmacocinétique du bosutinib (voir rubrique 4.5). Aucun métabolisme du bosutinib n'a été observé avec les CYP 1A2, 2A6, 2B6, 2C8, 2C9, 2C19, 2D6, 2E1 ou 3A5.
Elimination
Chez des sujets sains ayant reçu une dose intraveineuse unique de 120 mg de bosutinib, la demi-vie d'élimination terminale moyenne (% CV) était de 35,5 (24) heures et la clairance moyenne (% CV) de 61,9 (26) L/h. Dans une étude de bilan de masse avec bosutinib par voie orale, 94,6 % de la dose totale a été retrouvée en moyenne en 9 jours ; la principale voie d'excrétion était les fèces (91,3 %), 3,29 % de la dose étant retrouvée dans les urines. Soixante-quinze pour cent de la dose a été retrouvée dans les 96 heures. L'excrétion de bosutinib inchangé dans les urines était faible (avec environ 1 % de la dose), tant chez les sujets sains que chez ceux souffrant de tumeurs solides malignes au stade avancé.
Populations particulières
Insuffisance hépatique
Une dose de 200 mg de bosutinib administrée au moment des repas, a été évaluée dans une cohorte de 18 sujets présentant une atteinte hépatique (Child-Pugh de classes A, B et C) et chez 9 sujets sains correspondants. La Cmax du bosutinib dans le plasma a été multipliée par 2,4, 2 et 1,5, respectivement, dans les classes A, B et C de Child-Pugh, l'ASC du bosutinib dans le plasma étant multipliée par 2,3, 2 et 1,9, respectivement. La t½ du bosutinib a augmenté chez les patients présentant une atteinte hépatique par rapport aux sujets sains.
Insuffisance rénale
Au cours d’une étude sur l’insuffisance rénale, une dose unique de 200 mg de bosutinib a été administrée au moment du repas à 26 sujets présentant une insuffisance rénale légère, modérée ou sévère et à 8 volontaires sains appariés. L’insuffisance rénale reposait sur les valeurs de ClCr (calculée avec la formule Cockcroft-Gault) suivantes : < 30 mL/min (insuffisance rénale sévère), 30 ≤ ClCr ≤ 50 mL/min (insuffisance rénale modérée) ou 50 < ClCr ≤ 80 mL/min (insuffisance rénale légère). Les sujets présentant une insuffisance rénale modérée ou sévère ont présenté une augmentation de l’ASC de 35 % et de 60 %, respectivement, par rapport aux volontaires sains. La Cmax après exposition maximale a augmenté de 28 % et 34 % dans les groupes insuffisance rénale modérée et insuffisance rénale sévère, respectivement. L’exposition au bosutinib n’a pas augmenté chez les sujets présentant une insuffisance rénale légère. La demi-vie d’élimination du bosutinib chez les sujets présentant une insuffisance rénale était similaire à celle observée chez les sujets sains.
Les ajustements posologiques en cas d’insuffisance rénale ont été effectués en fonction des résultats de cette étude, et des propriétés pharmacocinétiques linéaires connues du bosutinib dans l’intervalle de dose compris entre 200 et 600 mg.
Age, sexe et origine ethnique
Aucune étude formelle n'a été menée pour évaluer l'influence de ces facteurs démographiques. Les analyses pharmacocinétiques de population chez des patients atteints de leucémie Ph+ ou d'une tumeur solide maligne et chez des sujets sains indiquent une absence d'impact cliniquement significatif de l'âge, du sexe ou du poids corporel. Les analyses pharmacocinétiques de population ont révélé que les Asiatiques avaient une clairance inférieure de 18 % correspondant à une augmentation d’environ 25 % de l’exposition au bosutinib (ASC).
Population pédiatrique
Le bosutinib n'a pas encore été étudié chez les enfants et les adolescents âgés de moins de 18 ans.
5.3. Données de sécurité préclinique
Le bosutinib a été évalué dans le cadre d'études de pharmacologie de sécurité, toxicologie en administration répétée, génotoxicité, toxicité pour les fonctions de reproduction et phototoxicité.
Pharmacologie de sécurité
Le bosutinib n'exerçait aucun effet sur les fonctions respiratoires. Dans une étude du système nerveux central (SNC), les rats traités par bosutinib présentaient une contraction des pupilles et une démarche anormale. Aucune relation dose-effet pour la taille des pupilles n'a été établie, mais celle-ci a été observée pour la démarche anormale et correspondait à une exposition équivalente à environ 11 fois l’exposition chez l’être humain avec la dose clinique de 400 mg et 8 fois l’exposition chez l’être humain avec la dose clinique de 500 mg (sur la base de la Cmax de la fraction non liée dans chaque espèce). L'activité du bosutinib in vitro lors de tests hERG suggérait un risque d’allongement de la repolarisation ventriculaire (QTc). Dans une étude portant sur le bosutinib administré par voie orale à des chiens, le bosutinib n'a pas entraîné de modification de la pression artérielle, d'arythmies auriculaires ou ventriculaires anormales ni d’allongement de l'intervalle PR, QRS ou QTc à l'ECG à des expositions allant jusqu’à 3 fois l’exposition chez l’être humain avec la dose clinique de 400 mg et 2 fois l’exposition chez l’être humain avec la dose clinique de 500 mg (sur la base de la Cmax de la fraction non liée dans chaque espèce). Une accélération de la fréquence cardiaque tardive a été observée. Au cours d’une étude portant sur l'administration par voie intraveineuse à des chiens, des accélérations transitoires de la fréquence cardiaque, des chutes de la pression artérielle et un allongement minimal du QTc (< 10 ms) ont été observées à des expositions d’environ 6 à 20 fois l’exposition chez l’être humain avec la dose clinique de 400 mg et de 4 à 15 fois l’exposition chez l’être humain avec la dose clinique de 500 mg (sur la base de la Cmax de la fraction non liée dans chaque espèce). Aucune conclusion n'a pu être tirée concernant le rapport entre les effets observés et le traitement médicamenteux.
Toxicologie en administration répétée
Des études de toxicologie en administration répétée chez des rats (pendant 6 mois maximum) et des chiens (pendant 9 mois maximum) ont révélé que le système gastro-intestinal était le principal organe cible pour la toxicité du bosutinib. Les signes cliniques de toxicité incluaient une modification des selles et étaient associés à une perte de l'appétit et une perte de poids pouvant entraîner un décès ou une euthanasie programmée.
Sur le plan histopathologique, une dilatation luminale, une hyperplasie des cellules caliciformes, une hémorragie, une érosion et un œdème des voies intestinales, ainsi qu'une érythrocytose sinusale et une hémorragie des ganglions mésentériques ont été observés. Le foie a également été identifié comme un organe cible chez les rats. Des toxicités ont été caractérisées par une augmentation du poids du foie en corrélation avec une hypertrophie hépatocellulaire survenue en l’absence d’enzymes hépatiques élevées ou des signes microscopiques de cytotoxicité hépatocellulaire. La pertinence de ces observations reste inconnue pour l’être humain. La comparaison de l'exposition entre les espèces indique que les expositions n'ayant pas entraîné d'événements indésirables au cours des études de toxicologie de 6 et 9 mois chez le rat et le chien, respectivement, étaient semblables à l’exposition chez l’être humain avec la dose clinique de 400 mg ou 500 mg (sur la base de l’ASC de la fraction non liée dans chaque espèce).
Génotoxicité
Les études de génotoxicité dans des systèmes in vitro bactériens et des systèmes in vitro et in vivo mammaliens, avec et sans activation métabolique, n'ont révélé aucun signe de potentiel mutagène du bosutinib.
Toxicité pour les fonctions de reproduction et de développement
Dans une étude de fertilité menée chez les rats, la fertilité des mâles a légèrement diminué. Chez les femelles, une augmentation des résorptions embryonnaires et une diminution du nombre d'implantations et d'embryons viables ont été observées. La dose ne présentant aucun effet indésirable pour la reproduction chez les mâles (30 mg/kg/jour) et les femelles (3 mg/kg/jour) a entraîné des expositions correspondant à 0,6 fois et 0,3 fois, respectivement, l’exposition chez l’être humain avec la dose clinique de 400 mg, et correspondant à 0,5 fois et 0,2 fois, respectivement, l'exposition chez l’être humain avec la dose clinique de 500 mg (sur la base de l'ASC de la fraction non liée dans chaque espèce). Un effet sur la fertilité masculine ne peut être exclu (voir rubrique 4.6).
L'exposition fœtale à la radioactivité dérivée du bosutinib pendant la gestation a été démontrée par une étude du passage transplacentaire chez des rates Sprague Dawley gravides. Au cours d'une étude portant sur le développement prénatal et postnatal chez le rat, une réduction du nombre de petits nés à ≥ 30 mg/kg/jour et une incidence accrue de perte totale de la portée ainsi qu’une croissance réduite de la progéniture après la naissance à 70 mg/kg/jour ont été observées. La dose à laquelle aucun effet indésirable sur le développement n'a été observé (10 mg/kg/jour) a entraîné des expositions correspondant à 1,3 fois et 1,0 fois l'exposition chez l’être humain avec la dose clinique de 400 mg et 500 mg, respectivement (sur la base de l'ASC de la fraction non liée dans chaque espèce). Une étude de la toxicité pour les fonctions de développement chez le lapin, à une dose toxique administrée à la mère, a relevé des anomalies fœtales (fusion des sternèbres, ainsi que diverses modifications viscérales chez 2 fœtus) et une légère baisse du poids fœtal. L'exposition à la dose maximale testée chez le lapin (10 mg/kg/jour) n'entraînant pas d'effets indésirables pour le fœtus correspondait à 0,9 et 0,7 fois l'exposition chez l’être humain avec la dose clinique de 400 mg ou 500 mg, respectivement (sur la base de l'ASC de la fraction non liée dans chaque espèce).
Après l'administration d'une dose unique (10 mg/kg) par voie orale de bosutinib radiomarqué [au 14C] à des rates Sprague Dawley allaitantes, la radioactivité a été rapidement excrétée dans le lait maternel dès 0,5 h après l'administration. La concentration de radioactivité dans le lait était jusqu'à 8 fois supérieure à la concentration plasmatique, ce qui a permis l'apparition de concentrations mesurables de radioactivité dans le plasma des petits allaités.
Carcinogénicité
Le bosutinib ne s'est pas révélé carcinogène au cours des études de carcinogénicité de 2 ans chez le rat et de 6 mois chez la souris rasH2.
Phototoxicité
Le bosutinib a démontré une capacité à absorber les rayons UV-B et UV-A et se distribue dans la peau et le canal uvéal de rats pigmentés. Néanmoins, le bosutinib n'a pas révélé de risque de phototoxicité pour la peau ou les yeux des rats pigmentés exposés au bosutinib en présence de rayons UV à des expositions jusqu’à 3 et 2 fois l’exposition chez l’être humain avec la dose clinique de 400 mg ou 500 mg, respectivement (sur la base de la Cmax de la fraction non liée dans chaque espèce).
Noyau du comprimé
Cellulose microcristalline (E460), croscarmellose sodique (E468), silice colloïdale anhydre, stéarate de magnésium.
Pelliculage
Alcool polyvinylique (E1203), macrogols, talc (E553b), dioxyde de titane (E171), oxyde de fer jaune (E172), oxyde de fer rouge (E172).
3 ans.
6.4. Précautions particulières de conservation
Ce médicament ne nécessite pas de précautions particulières de conservation.
6.5. Nature et contenu de l'emballage extérieur
Les comprimés sont conditionnés sous plaquettes en (Aluminium/PVC/Aluminium/OPA) ou sous plaquettes prédécoupées unitaires en (Aluminium/PVC/Aluminium/OPA).
Chaque boîte contient 28 ou 112 comprimés
Chaque boîte contient 28 × 1 ou 112 × 1 comprimés
Toutes les présentations peuvent ne pas être commercialisées.
6.6. Précautions particulières d’élimination et de manipulation
Tout médicament non utilisé ou déchet doit être éliminé conformément à la réglementation en vigueur.
7. TITULAIRE DE L’AUTORISATION DE MISE SUR LE MARCHE
EG LABO - LABORATOIRES EUROGENERICS
CENTRAL PARK
9-15 RUE MAURICE MALLET
92130 ISSY-LES-MOULINEAUX
8. NUMERO(S) D’AUTORISATION DE MISE SUR LE MARCHE
· 34009 302 879 0 3 : 28 comprimés sous plaquettes (Aluminium/PVC/Aluminium/OPA).
9. DATE DE PREMIERE AUTORISATION/DE RENOUVELLEMENT DE L’AUTORISATION
[à compléter ultérieurement par le titulaire]
Date de première autorisation:{JJ mois AAAA}
10. DATE DE MISE A JOUR DU TEXTE
[à compléter ultérieurement par le titulaire]
{JJ mois AAAA}
Sans objet.
12. INSTRUCTIONS POUR LA PREPARATION DES RADIOPHARMACEUTIQUES
Liste I
Médicament soumis à une prescription initiale hospitalière semestrielle.
Prescription initiale et renouvellement réservés aux spécialistes en oncologie ou en hématologie, ou aux médecins compétents en cancérologie.
Médicament nécessitant une surveillance particulière pendant le traitement.
ANSM - Mis à jour le : 05/03/2026
BOSUTINIB EG 400 mg, comprimé pelliculé
Bosutinib
· Gardez cette notice. Vous pourriez avoir besoin de la relire.
· Si vous avez d’autres questions, interrogez votre médecin ou votre pharmacien.
· Ce médicament vous a été personnellement prescrit. Ne le donnez pas à d’autres personnes. Il pourrait leur être nocif, même si les signes de leur maladie sont identiques aux vôtres.
· Si vous ressentez un quelconque effet indésirable, parlez-en à votre médecin. Ceci s’applique aussi à tout effet indésirable qui ne serait pas mentionné dans cette notice. Voir rubrique 4.
1. Qu'est-ce que BOSUTINIB EG 400 mg, comprimé pelliculé et dans quels cas est-il utilisé ?
2. Quelles sont les informations à connaître avant de prendre BOSUTINIB EG 400 mg, comprimé pelliculé ?
3. Comment prendre BOSUTINIB EG 400 mg, comprimé pelliculé ?
4. Quels sont les effets indésirables éventuels ?
5. Comment conserver BOSUTINIB EG 400 mg, comprimé pelliculé ?
6. Contenu de l’emballage et autres informations.
1. QU’EST-CE QUE BOSUTINIB EG 400 mg, comprimé pelliculé ET DANS QUELS CAS EST-IL UTILISE ?
Classe pharmacothérapeutique : Antinéoplasiques, inhibiteurs de protéine kinase - code ATC : L01EA04
BOSUTINIB EG contient la substance active bosutinib. Il est utilisé dans le traitement de patients adultes atteints d'un type de leucémie appelée leucémie myéloïde chronique (LMC) à chromosome Philadelphie positif (Ph+) nouvellement diagnostiquée ou pour lesquels les traitements précédents destinés à traiter la LMC n’ont pas fonctionné ou ne sont pas adaptés. La LMC Ph+ est un cancer du sang qui conduit l'organisme à produire un trop grand nombre de globules blancs d'un certain type, appelés granulocytes.
Si vous avez des questions sur le mécanisme d'action de BOSUTINIB EG et sur les raisons pour lesquelles ce médicament vous a été prescrit, parlez-en à votre médecin.
2. QUELLES SONT LES INFORMATIONS A CONNAITRE AVANT DE PRENDRE BOSUTINIB EG 400 mg, comprimé pelliculé ?
Ne prenez jamais BOSUTINIB EG 400 mg, comprimé pelliculé
· si vous êtes allergique au bosutinib ou à l’un des autres composants contenus dans ce médicament, mentionnés dans la rubrique 6 ;
· si votre médecin vous a informé que votre foie était endommagé et ne fonctionnait pas normalement.
Avertissements et précautions
Adressez-vous à votre médecin, pharmacien ou infirmier/ère avant de prendre BOSUTINIB EG
· si vous souffrez, ou avez souffert dans le passé, de problèmes de foie. Informez votre médecin si vous avez des antécédents de problèmes de foie, notamment d’hépatite (infection ou inflammation du foie) de n'importe quel type, ou si vous avez déjà présenté des signes et symptômes de problèmes hépatiques suivants : démangeaisons, coloration jaune de la peau ou des yeux, urines foncées et douleur ou gêne dans la région supérieure droite de l'estomac. Votre médecin doit pratiquer des analyses de sang pour vérifier votre fonction hépatique avant le début du traitement par BOSUTINIB EG, pendant les 3 premiers mois de ce traitement, et dès lors qu’elles sont cliniquement indiquées ;
· si vous souffrez de diarrhée et de vomissements. Informez votre médecin en cas d'apparition de l'un des signes et symptômes suivants : augmentation du nombre de selles produites quotidiennement au-dessus de la normale, augmentation des épisodes de vomissements, présence de sang dans les vomissures, les selles ou les urines, ou selles noires (goudronneuses). Vous devez demander à votre médecin si la prise de votre traitement contre les vomissements est susceptible d’augmenter le risque d’arythmie cardiaque. En particulier, vous devez vous adresser à votre médecin si vous souhaitez prendre un médicament contenant de la dompéridone pour le traitement des nausées et/ou des vomissements. Le traitement des nausées ou vomissements avec ce type de médicaments associé avec BOSUTINIB EG peut augmenter le risque d’arythmies cardiaques dangereuses ;
· si vous souffrez de problèmes de saignement. Informez votre médecin en cas d'apparition de l'un des signes et symptômes suivants : saignement anormal ou présence de bleus en l'absence de choc ;
· si vous souffrez d'une infection. Informez votre médecin en cas d'apparition de l'un des signes et symptômes suivants : fièvre, problèmes urinaires tels qu'une brûlure lorsque vous urinez, apparition d'une toux ou de maux de gorge ;
· si vous souffrez de rétention liquidienne. Informez votre médecin en cas d'apparition de l'un des signes et symptômes de rétention liquidienne suivants pendant le traitement par BOSUTINIB EG : gonflement des chevilles, des pieds ou des jambes, difficultés respiratoires, douleurs dans la poitrine ou toux (il peut s’agir de signes de rétention liquidienne dans les poumons ou le thorax) ;
· si vous souffrez de problèmes cardiaques. Informez votre médecin si vous souffrez d’un problème cardiaque, tel que des arythmies ou la présence d'un signal électrique anormal appelé « allongement de l'intervalle QT ». Cela est toujours important, mais plus particulièrement si vous présentez des diarrhées fréquentes ou prolongées, comme décrit ci-dessus. Si vous vous évanouissez (perte de conscience) ou si vous avez des battements de cœur irréguliers pendant le traitement par BOSUTINIB EG, informez-en immédiatement votre médecin, car cela peut être le signe d'un trouble cardiaque grave ;
· si l'on vous a informé que vous aviez des problèmes rénaux. Informez votre médecin si vous urinez plus fréquemment et en plus grande quantité avec des urines de couleur pâle ou si vous urinez moins fréquemment et en plus faible quantité avec des urines de couleur foncée. Signalez-lui également si vous perdez du poids ou si vous présentez un gonflement des pieds, des chevilles, des jambes, des mains ou du visage ;
· si vous avez déjà eu ou avez actuellement une hépatite B. En effet, BOSUTINIB EG pourrait réactiver votre hépatite B, ce qui peut être fatal dans certains cas. Les patients seront étroitement surveillés par leur médecin afin de détecter tout signe d’infection avant le début du traitement ;
· si vous souffrez ou avez souffert de problèmes de pancréas. Informez votre médecin en cas d'apparition de douleur ou de gêne abdominale ;
· si vous présentez l’un de ces symptômes : éruptions cutanées graves. Informez votre médecin si vous développez l’un des signes ou symptômes suivants : éruption cutanée rouge ou violacée, douloureuse, qui s’étend et s’accompagne de cloques et/ou autres lésions commençant à apparaître sur les muqueuses (par exemple, la bouche et les lèvres) ;
· si vous remarquez l’un de ces symptômes : douleur au flanc, sang dans les urines ou diminution de la quantité d’urine éliminée. Si votre maladie est très sévère, il est possible que votre organisme ne puisse pas éliminer tous les déchets provenant des cellules cancéreuses mourantes. Ce phénomène s’appelle « syndrome de lyse tumorale » et peut provoquer une insuffisance rénale et des troubles cardiaques au cours des 48 heures suivant l’administration de la première dose de BOSUTINIB EG. Votre médecin en sera informé et pourra s’assurer que vous êtes suffisamment hydraté(e). Il pourra également vous prescrire d’autres traitements préventifs.
Protection au soleil/ aux UV
Vous pouvez devenir plus sensible au soleil ou aux rayons UV au cours du traitement par bosutinib. Il est important de couvrir les zones de la peau exposées au soleil et d’utiliser une crème solaire avec un indice de protection (SPF) élevé.
Enfants et adolescents
BOSUTINIB EG n’est pas recommandé chez les enfants de moins de 18 ans. Ce médicament n'a pas été étudié chez les enfants et les adolescents.
Autres médicaments et BOSUTINIB EG 400 mg, comprimé pelliculé
Informez votre médecin ou pharmacien si vous prenez, avez récemment pris ou pourriez prendre tout autre médicament, y compris des médicaments obtenus sans ordonnance, des vitamines et des produits de phytothérapie.
Certains médicaments peuvent affecter la concentration de BOSUTINIB EG dans votre organisme. Vous devez informer votre médecin si vous prenez des médicaments contenant des substances actives telles que les suivantes :
Les substances actives suivantes peuvent augmenter le risque d'effets indésirables liés à BOSUTINIB EG :
· kétoconazole, itraconazole, voriconazole, posaconazole et fluconazole, utilisés pour traiter les infections fongiques ;
· clarithromycine, télithromycine, érythromycine et ciprofloxacine, utilisées pour traiter les infections bactériennes ;
· néfazodone, utilisée pour traiter la dépression ;
· mibéfradil, diltiazem et vérapamil, utilisés pour abaisser la pression artérielle chez les personnes présentant une pression artérielle élevée ;
· ritonavir, lopinavir/ritonavir, indinavir, nelfinavir, saquinavir, atazanavir, amprénavir, fosamprénavir et darunavir, utilisés pour traiter le virus de l'immunodéficience humaine (VIH)/SIDA ;
· bocéprévir et télaprévir, utilisés pour traiter l'hépatite C ;
· aprépitant, utilisé pour la prévention et le contrôle des nausées (envie de vomir) et des vomissements ;
· imatinib, utilisé pour traiter un type de leucémie ;
· crizotinib, utilisé pour traiter un type de cancer du poumon appelé cancer bronchopulmonaire « non à petites cellules ».
Les substances actives suivantes peuvent réduire l'efficacité de BOSUTINIB EG :
· rifampicine, utilisée pour traiter la tuberculose ;
· phénytoïne et carbamazépine, utilisées pour traiter l'épilepsie ;
· bosentan, utilisé pour abaisser la pression artérielle élevée dans les poumons (hypertension artérielle pulmonaire) ;
· nafcilline, un antibiotique utilisé pour traiter les infections bactériennes ;
· millepertuis (préparation de phytothérapie disponible sans ordonnance), utilisé pour traiter la dépression ;
· éfavirenz et étravirine, utilisés pour traiter le VIH/SIDA ;
· modafinil, utilisé pour traiter certains types de troubles du sommeil.
Ces médicaments doivent être évités pendant le traitement par BOSUTINIB EG. Si vous prenez l'un de ces médicaments, informez votre médecin. Votre médecin pourra modifier la dose de ces médicaments ou de BOSUTINIB EG, ou vous prescrire un médicament différent.
Les substances actives suivantes peuvent affecter le rythme cardiaque :
· amiodarone, disopyramide, procaïnamide, quinidine et sotalol, utilisés pour traiter les affections cardiaques ;
· chloroquine, halofantrine, utilisées pour traiter le paludisme ;
· clarithromycine et moxifloxacine, des antibiotiques utilisés pour traiter les infections bactériennes ;
· halopéridol, utilisé pour traiter les troubles psychotiques tels que la schizophrénie ;
· dompéridone, utilisée pour traiter les nausées et vomissements ou pour stimuler la production de lait maternel ;
· méthadone, utilisée pour traiter la douleur.
Ces médicaments doivent être pris avec prudence pendant le traitement par BOSUTINIB EG. Si vous prenez l'un de ces médicaments, informez-en votre médecin.
Les médicaments mentionnés ici ne sont peut-être pas les seuls susceptibles d'interagir avec BOSUTINIB EG.
BOSUTINIB EG 400 mg, comprimé pelliculé avec des aliments et boissons
Ne prenez pas BOSUTINIB EG avec du pamplemousse ou du jus de pamplemousse, car cela peut augmenter le risque d'effets indésirables.
Grossesse, allaitement et fertilité
BOSUTINIB EG ne doit pas être utilisé pendant la grossesse, sauf en cas d’absolue nécessité, car BOSUTINIB EG pourrait nuire à l’enfant à naître. Demandez conseil à votre médecin avant de prendre BOSUTINIB EG si vous êtes enceinte ou susceptible de le devenir.
Les femmes prenant BOSUTINIB EG doivent être avisées d’utiliser une méthode de contraception efficace pendant le traitement et pendant au moins 1 mois après l’administration de la dernière dose. Les vomissements ou la diarrhée peuvent réduire l'efficacité des contraceptifs oraux.
Il existe un risque que le traitement par BOSUTINIB EG entraîne une diminution de la fertilité et vous pouvez, si vous le souhaitez, demander des conseils sur la conservation du sperme avant le début du traitement.
Informez votre médecin si vous allaitez. N'allaitez pas pendant le traitement par BOSUTINIB EG car cela peut être nocif pour votre bébé.
Conduite de véhicules et utilisation de machines
Si vous avez des sensations vertigineuses, une vision trouble ou si vous ressentez une fatigue inhabituelle, ne conduisez pas de véhicules et n'utilisez pas de machines jusqu'à la disparition de ces effets indésirables.
BOSUTINIB EG 400 mg, comprimé pelliculé contient du sodium
Ce médicament contient moins de 1 mmol (23 mg) de sodium par comprimé, c’est-à-dire qu’il est essentiellement « sans sodium ».
3. COMMENT PRENDRE BOSUTINIB EG 400 mg, comprimé pelliculé ?
BOSUTINIB EG ne peut vous être prescrit que par un médecin expérimenté dans le traitement de la leucémie.
Posologie et mode d’administration
La dose recommandée est de 400 mg une fois par jour pour les patients présentant une LMC nouvellement diagnostiquée. La dose recommandée est de 500 mg une fois par jour pour les patients chez lesquels les traitements précédents destinés à traiter la LMC n’ont pas fonctionné ou ne sont pas adaptés. Si vous souffrez de problèmes rénaux modérés ou sévères, votre médecin diminuera votre dose de 100 mg par jour si vos problèmes rénaux sont modérés, et de 100 mg supplémentaires une fois par jour si vos problèmes rénaux sont sévères. Le médecin pourra ajuster la dose à l'aide de comprimés de 100 mg en fonction de votre état de santé, de votre réponse au traitement et/ou des effets indésirables que vous pourriez ressentir. Prenez le(s) comprimé(s) une fois par jour au cours d’un repas. Avalez-le(s) comprimé(s) entier(s) avec de l’eau.
Si vous avez pris plus de BOSUTINIB EG 400 mg, comprimé pelliculé que vous n’auriez dû
Si vous prenez accidentellement un trop grand nombre de comprimés de BOSUTINIB EG ou une dose plus élevée que celle prescrite, demandez immédiatement conseil à un médecin. Si possible, montrez-lui la boîte ou cette notice. Vous aurez peut-être besoin de soins médicaux.
Si vous oubliez de prendre BOSUTINIB EG 400 mg, comprimé pelliculé
Ne prenez pas de dose double pour compenser le comprimé que vous avez oublié de prendre.
Si vous arrêtez de prendre BOSUTINIB EG 400 mg, comprimé pelliculé
N'arrêtez pas de prendre BOSUTINIB EG sauf si votre médecin vous le demande. Si vous n'êtes pas en capacité de prendre le médicament comme votre médecin l’a prescrit ou si vous pensez ne plus en avoir besoin, contactez immédiatement votre médecin.
Si vous avez d’autres questions sur l’utilisation de ce médicament, demandez plus d’informations à votre médecin ou à votre pharmacien.
4. QUELS SONT LES EFFETS INDESIRABLES EVENTUELS ?
Vous devez immédiatement contacter votre médecin en cas d'apparition de l'un de ces effets indésirables graves (voir également rubrique 2 « Quelles sont les informations à connaître avant de prendre BOSUTINIB EG 400 mg, comprimé pelliculé ? ») :
Troubles sanguins. Informez immédiatement votre médecin si vous présentez l'un des symptômes suivants : saignement, fièvre ou apparition fréquente de bleus (vous souffrez peut-être de problèmes sanguins ou du système lymphatique).
Troubles du foie. Informez immédiatement votre médecin si vous présentez l'un des symptômes suivants : démangeaisons, coloration jaune de la peau ou des yeux, urines foncées et douleur ou gêne dans la région supérieure droite de l'estomac ou fièvre.
Troubles gastro-intestinaux. Informez votre médecin si vous constatez l'apparition d'une douleur d'estomac, de brûlures d'estomac, de diarrhée, de constipation, de nausées et de vomissements.
Troubles cardiaques. Informez votre médecin si vous souffrez d'un problème cardiaque, tel qu'un signal électrique anormal appelé « allongement de l'intervalle QT », ou si vous vous évanouissez (perte de conscience) ou si les battements de votre cœur sont irréguliers pendant le traitement par BOSUTINIB EG.
Réactivation de l'hépatite B. Réapparition (réactivation) de l’hépatite B si vous avez déjà eu une hépatite B dans le passé (infection hépatique).
Réactions cutanées sévères. Informez immédiatement votre médecin si vous présentez l’un de ces symptômes : éruption cutanée rouge ou violacée, douloureuse, qui s’étend et s’accompagne de cloques et/ou d’autres lésions commençant à apparaître sur les muqueuses (par exemple, la bouche et les lèvres).
Les effets indésirables de BOSUTINIB EG peuvent inclure :
Effets indésirables très fréquents (pouvant affecter plus de 1 personne sur 10) :
· diminution du nombre de plaquettes, de globules rouges et/ou de neutrophiles (type de globules blancs) ;
· diarrhée, vomissements, maux d'estomac, nausées ;
· fièvre, gonflement des mains, des pieds ou du visage, fatigue, faiblesse ;
· infection des voies respiratoires ;
· rhinopharyngite ;
· modifications des résultats des analyses sanguines indiquant que BOSUTINIB EG affecte votre foie et/ou votre pancréas, vos reins ;
· diminution de l’appétit ;
· douleurs au niveau des articulations, mal de dos ;
· maux de tête ;
· éruption cutanée, pouvant s’accompagner de démangeaisons et/ou être généralisée ;
· toux ;
· essoufflement ;
· sensation de déséquilibre (sensations vertigineuses) ;
· présence de liquide dans les poumons (épanchement pleural) ;
· démangeaisons.
Effets indésirables fréquents (pouvant affecter jusqu’à 1 personne sur 10) :
· faible nombre de globules blancs (leucopénie) ;
· inflammation de l'estomac (gastrite), saignement de l’estomac ou de l’intestin ;
· douleurs dans la poitrine, douleurs ;
· atteinte toxique du foie, fonction hépatique anormale y compris troubles hépatiques ;
· infection des poumons (pneumonie), grippe, bronchite ;
· trouble du rythme cardiaque prédisposant aux évanouissements, aux sensations vertigineuses et aux palpitations ;
· augmentation de la pression artérielle ;
· taux élevé de potassium dans le sang, faible taux de phosphore dans le sang, perte excessive de liquides corporels (déshydratation) ;
· douleurs musculaires ;
· altération du goût (dysgueusie) ;
· insuffisance rénale aiguë, insuffisance rénale, altération de la fonction rénale ;
· présence de liquide autour du cœur (épanchement péricardique) ;
· bourdonnement dans les oreilles (acouphènes) ;
· urticaire, acné ;
· réaction de photosensibilité (sensibilité aux rayons UV du soleil et d’autres sources lumineuses) ;
· réaction allergique ;
· pression sanguine anormalement élevée dans les artères des poumons (hypertension pulmonaire) ;
· inflammation aiguë du pancréas (pancréatite aiguë) ;
· insuffisance respiratoire
Effets indésirables peu fréquents (pouvant affecter jusqu’à 1 personne sur 100) :
· fièvre associée à une baisse du nombre de globules blancs (neutropénie fébrile) ;
· atteinte du foie ;
· réaction allergique pouvant engager le pronostic vital (choc anaphylactique) ;
· accumulation anormale de liquide dans les poumons (œdème pulmonaire aigu) ;
· éruption cutanée ;
· inflammation de la membrane recouvrant le cœur (péricardite) ;
· baisse marquée du nombre de granulocytes (un type de globules blancs) ;
· trouble cutané sévère (érythème polymorphe) ;
· nausées, essoufflement, battements cardiaques irréguliers, crampes musculaires, convulsions, troubles urinaires et fatigue associés à des résultats anormaux d’analyses biologiques (taux élevés de potassium, d’acide urique et de phosphore et faibles taux de calcium dans le sang) pouvant entraîner des modifications de la fonction rénale et une insuffisance rénale aiguë (syndrome de lyse tumorale [SLT]) ;
· inflammation des vaisseaux sanguins de la peau pouvant entraîner une éruption cutanée ou des ecchymoses (vascularite cutanée).
Fréquence indéterminée (ne peut être estimée sur la base des données disponibles) :
· trouble cutané sévère (syndrome de Stevens-Johnson, nécrolyse épidermique toxique) dû à une réaction allergique, éruption cutanée avec exfoliation (peau écailleuse, desquamation) ;
· pneumopathie interstitielle diffuse (troubles provoquant des cicatrices dans les poumons) : les signes incluent toux, difficultés pour respirer, respiration douloureuse.
Déclaration des effets secondaires
Si vous ressentez un quelconque effet indésirable, parlez-en à votre médecin, à votre pharmacien ou à votre infirmier/ère. Ceci s’applique aussi à tout effet indésirable qui ne serait pas mentionné dans cette notice. Vous pouvez également déclarer les effets indésirables directement via le système national de déclaration : Agence nationale de sécurité du médicament et des produits de santé (ANSM) et réseau des Centres Régionaux de Pharmacovigilance - Site internet : https://signalement.social-sante.gouv.fr/
En signalant les effets indésirables, vous contribuez à fournir davantage d’informations sur la sécurité du médicament.
5. COMMENT CONSERVER BOSUTINIB EG 400 mg, comprimé pelliculé ?
Tenir ce médicament hors de la vue et de la portée des enfants.
N’utilisez pas ce médicament après la date de péremption indiquée sur la plaquette et sur l’emballage après « EXP ». La date de péremption fait référence au dernier jour de ce mois.
Ce médicament ne nécessite pas de précautions particulières de conservation.
N’utilisez pas ce médicament si vous remarquez que l’emballage est endommagé ou semble abimé.
Ne jetez aucun médicament au tout-à-l’égout ou avec les ordures ménagères. Demandez à votre pharmacien d’éliminer les médicaments que vous n’utilisez plus. Ces mesures contribueront à protéger l’environnement.
6. CONTENU DE L’EMBALLAGE ET AUTRES INFORMATIONS
Ce que contient BOSUTINIB EG 400 mg, comprimé pelliculé
· La substance active est :
Bosutinib....................................................................................................................... 400 mg
Pour un comprimé pelliculé.
· Les autres composants sont : cellulose microcristalline (E460), croscarmellose sodique (E468), silice colloïdale anhydre, stéarate de magnésium.
Le pelliculage contient : alcool polyvinylique (E1203), macrogols, talc (E553b), dioxyde de titane (E171), oxyde de fer jaune (E172), oxyde de fer rouge (E172).
Qu’est-ce que BOSUTINIB EG 400 mg, comprimé pelliculé et contenu de l’emballage extérieur
BOSUTINIB EG 400 mg, comprimé pelliculé se présente sous forme de comprimés pelliculés, ovales, orange, biconvexes, comportant la mention « C19 » gravée en creux sur une face.
BOSUTINIB EG 400 mg est disponible sous plaquettes contenant 28 ou 112 comprimés pelliculés.
BOSUTINIB EG 400 mg est disponible sous plaquettes prédécoupées unitaires contenant 28 × 1 ou 112 × 1 comprimés pelliculés.
Toutes les présentations peuvent ne pas être commercialisées.
Titulaire de l’autorisation de mise sur le marché
EG LABO - LABORATOIRES EUROGENERICS
CENTRAL PARK
9-15 RUE MAURICE MALLET
92130 ISSY-LES-MOULINEAUX
Exploitant de l’autorisation de mise sur le marché
EG LABO - LABORATOIRES EUROGENERICS
CENTRAL PARK
9-15 RUE MAURICE MALLET
92130 ISSY-LES-MOULINEAUX
REYKJAVIKURVEGUR 78
IS 220 HAFNARFJORDUR
ISLANDE
Ou
STADA ARZNEIMITTEL AG
STADASTRASSE 2 - 18
DORTWEIL, BAD VILBEL
61118 HASSIA
ALLEMAGNE
Ou
CLONMEL HEALTHCARE LTD.
WATERFORD ROAD
GURTNAFLEUR, CLONMEL,
E91 D768, CO. TIPPERARY
IRLANDE
Noms du médicament dans les Etats membres de l'Espace Economique Européen
Ce médicament est autorisé dans les Etats membres de l'Espace Economique Européen sous les noms suivants : Conformément à la réglementation en vigueur.
[À compléter ultérieurement par le titulaire]
La dernière date à laquelle cette notice a été révisée est :
[à compléter ultérieurement par le titulaire]
{mois AAAA}.
Des informations détaillées sur ce médicament sont disponibles sur le site Internet de l’ANSM (France).