Dernière mise à jour le 29/06/2026
TRIAFEMI, comprimé
Indications thérapeutiques
Ce médicament est un contraceptif et un antiacnéique.
Il empêche l’ovulation. Il est préconisé dans le but d’éviter une grossesse, chez la femme ayant une acné légère à modérée.
Ce contraceptif contient deux types d’hormones féminines : un estrogène et un progestatif.
Ces hormones agissent en empêchant la libération de l’ovule (ovulation) de vos ovaires ce qui vous empêche d’être enceinte. Par ailleurs, TRIAFEMI rend plus épaisse la glaire (mucus) du col de l’utérus ce qui rend plus difficile l’entrée des spermatozoïdes dans l’utérus. L’effet contraceptif commence à la prise du premier comprimé.
Composition en substances actives
-
Comprimé blanc ( Composition pour un comprimé )
- > éthinylestradiol 0,035 mg
- > norgestimate 0,18 mg
-
Comprimé bleu ciel ( Composition pour un comprimé )
- > éthinylestradiol 0,035 mg
- > norgestimate 0,215 mg
-
Comprimé bleu foncé ( Composition pour un comprimé )
- > éthinylestradiol 0,035 mg
- > norgestimate 0,25 mg
Présentations
> 3 plaquette(s) thermoformée(s) PVC PVDC Aluminium de 21 comprimés (7 comprimés blancs, 7 comprimés bleu ciel et 7 comprimés bleu foncé)
Code CIP : 358 996-7 ou 34009 358 996 7 5
Déclaration de commercialisation : 09/04/2003
Cette présentation n'est pas agréée aux collectivités
Documents de bon usage du médicament
- Contraception chez la femme en post-partum
Auteur : Haute autorité de santé
Type : Fiche mémo
Date de mise à jour :Septembre 2019
- Contraception d’urgence
Auteur : Haute autorité de santé
Type : Fiche mémo
Date de mise à jour :Septembre 2019
- Contraception chez l’homme
Auteur : Haute autorité de santé
Type : Fiche mémo
Date de mise à jour :Septembre 2019
- Stérilisation à visée contraceptive chez l’homme et chez la femme
Auteur : Haute autorité de santé
Type : Fiche mémo
Date de mise à jour :Septembre 2019
- Contraception chez la femme adulte et de l'adolescente en âge de procréer (hors post-partum et post-IVG)
Auteur : Haute autorité de santé
Type : Fiche mémo
Date de mise à jour :Septembre 2019
- Contraception chez la femme à risque cardio-vasculaire
Auteur : Haute autorité de santé
Type : Fiche mémo
Date de mise à jour :Septembre 2019
- Contraception : prescriptions et conseils aux femmes
Auteur : Haute autorité de santé
Type : Fiche mémo
Date de mise à jour :Septembre 2019
- Contraception hormonale orale : dispensation en officine
Auteur : Haute autorité de santé
Type : Fiche mémo
Date de mise à jour :Septembre 2019
- Contraception d’urgence : dispensation en officine
Auteur : Haute autorité de santé
Type : Fiche mémo
Date de mise à jour :Septembre 2019
- Contraception estroprogestative transdermique ou vaginale : dispensation en officine
Auteur : Haute autorité de santé
Type : Fiche mémo
Date de mise à jour :Septembre 2019
- Contraception chez la femme après une interruption volontaire de grossesse (IVG)
Auteur : Haute autorité de santé
Type : Fiche mémo
Date de mise à jour :Septembre 2019
- Méthodes contraceptives : Focus sur les méthodes les plus efficaces disponibles
Auteur : Haute autorité de santé
Type : Evaluation des technologies de santé
Date de mise à jour :Novembre 2017
- Contraceptifs oraux estroprogestatifs : préférez les «pilules» de 1re ou 2e génération
Auteur : Haute autorité de santé
Type : Fiche Bon Usage du Médicament
Date de mise à jour :Janvier 2013
Service médical rendu (SMR)
Amélioration du service médical rendu (ASMR)
Pas d'ASMR disponible pour ce médicament (plus d'informations dans l\'aide)
ANSM - Mis à jour le : 19/02/2026
TRIAFEMI, comprimé
2. COMPOSITION QUALITATIVE ET QUANTITATIVE
Norgestimate ................................................................................................................... 0,180 mg
Ethinylestradiol ................................................................................................................ 0,035 mg
Pour un comprimé blanc.
Norgestimate ................................................................................................................... 0,215 mg
Ethinylestradiol ................................................................................................................ 0,035 mg
Pour un comprimé bleu ciel.
Norgestimate ................................................................................................................... 0,250 mg
Ethinylestradiol ................................................................................................................ 0,035 mg
Pour un comprimé bleu foncé.
Excipient à effet notoire : lactose anhydre
Pour la liste complète des excipients, voir rubrique 6.1.
7 comprimés blancs, 7 comprimés bleu ciel, 7 comprimés bleu foncé.
4.1. Indications thérapeutiques
Contraception orale chez la femme ayant une acné légère à modérée; ce traitement contraceptif ne dispense pas d'un traitement spécifique de l'acné si celui-ci est nécessaire.
La décision de prescrire TRIAFEMI doit être prise en tenant compte des facteurs de risque de la patiente, notamment ses facteurs de risque de thrombo-embolie veineuse (TEV), ainsi que du risque de TEV associé à TRIAFEMI en comparaison aux autres CHC (Contraceptifs Hormonaux Combinés) (voir rubriques 4.3 et 4.4).
4.2. Posologie et mode d'administration
Voie d’administration : voie orale.
Posologie
Comment prendre TRIAFEMI
Prendre régulièrement et sans oubli 1 comprimé par jour au même moment de la journée (celui où vous risquez le moins d’oublier), pendant 21 jours consécutifs avec un arrêt de 7 jours entre deux plaquettes.
Le 1er jour d’utilisation de votre plaquette, collez l’étiquette* du jour de la semaine correspondant et prenez le comprimé numéro 1. Les jours suivants prenez les comprimés environ à la même heure de la journée, dans l’ordre chronologique jusqu’au 21ème jour.
Ensuite, vous ne prendrez plus de comprimés pendant 7 jours. Une hémorragie de privation doit apparaître pendant cet intervalle, généralement le 2ème ou le 3ème jour après la prise du dernier comprimé.
Commencez la plaquette suivante dès le 8ème jour, même si l’hémorragie de privation persiste. Vous devez toujours commencer la plaquette suivante le même jour de la semaine.
*Les étiquettes sont disponibles dans votre boite.
Mode d’administration
Comment débuter TRIAFEMI
· Début du traitement de l'acné et de la contraception orale (Pas de prise antérieure d’une contraception hormonale (au cours du mois précédent) :
La prise des comprimés doit commencer le jour 1 du cycle naturel de la femme (c'est-à-dire le premier jour de ses règles).
On peut commencer entre les jours 2-5, mais dans ce cas une méthode barrière supplémentaire est recommandée pendant les 7 premiers jours du premier cycle.
· En relais d’une contraception hormonale combinée (contraceptif oral combiné (COC), anneau vaginal ou dispositif transdermique) :
La femme doit commencer TRIAFEMI de préférence le jour après le dernier comprimé actif (le dernier comprimé contenant les substances actives) de son COC précédent, mais au plus tard le jour suivant l’intervalle sans comprimés habituel ou l’intervalle des comprimés placebo de son COC précédent.
Dans le cas où un anneau vaginal ou un dispositif transdermique a été utilisé, la femme devra commencer à prendre TRIAFEMI de préférence le jour du retrait, mais au plus tard le jour où la pose suivante est prévue.
· En relais d’une contraception par un progestatif seul (contraceptif oral, forme injectable ou implant contenant un progestatif seul) ou d’un système intra-utérin (SIU) contenant un progestatif :
La femme peut changer n’importe quel jour de la pilule progestative seule (d'un implant ou d’un SIU le jour de son retrait, d'un produit injectable quand l'injection suivante est prévue), mais doit dans tous ces cas être conseillée d’utiliser une méthode barrière supplémentaire pendant les 7 premiers jours de prise de comprimés.
· Après un avortement au cours du premier trimestre :
La femme peut commencer immédiatement. Ce faisant, elle n’a pas besoin de prendre de mesures contraceptives supplémentaires.
· Après un accouchement ou un avortement au cours du deuxième trimestre :
Les femmes doivent être informées de commencer au jour 21 à 28 après l'accouchement ou l'avortement du deuxième trimestre. En commençant plus tard, la femme devra être informée d'utiliser une méthode barrière supplémentaire pendant les 7 premiers jours. Cependant, si des rapports sexuels ont déjà eu lieu, une grossesse devra être exclue avant de commencer le COC ou la femme doit attendre ses premières règles.
En cas d'allaitement, voir rubrique 4.6.
Conduite en cas d'oubli de comprimés
Si l’utilisatrice à moins de 12 heures de retard dans la prise d’un comprimé, la protection contraceptive n’est pas réduite. La femme doit prendre le comprimé dès qu'elle s’en souvient et prendre les comprimés suivants à l’heure habituelle.
Si elle a plus de 12 heures de retard dans la prise d’un comprimé, la protection contraceptive peut être réduite.
La conduite à tenir pour les comprimés oubliés peut être guidée par les deux règles de base suivantes :
1. La prise des comprimés ne doit jamais être interrompue pendant plus de 7 jours.
2. 7 jours de prise ininterrompue de comprimés sont requis pour atteindre une inhibition adéquate de l'axe hypothalamo-hypophyso-ovarien.
En conséquence, les conseils suivants peuvent être donnés dans la pratique quotidienne :
· Semaine 1 (Jour 1 à 7)
La femme doit prendre le dernier comprimé oublié dès qu'elle s’en souvient, même si cela signifie de prendre deux comprimés en même temps. Elle continue ensuite à prendre les comprimés à son heure habituelle. En plus, une méthode barrière comme un préservatif doit être utilisée pendant les 7 jours suivants. Si des rapports sexuels ont eu lieu pendant les 7 jours précédents, la possibilité d’une grossesse doit être considérée. Plus il y a de comprimés oubliés et plus ils sont proches de la phase de l’intervalle sans comprimés, plus le risque de grossesse est élevé.
· Semaine 2 (Jour 8 à 14)
La femme doit prendre le dernier comprimé oublié dès qu'elle s’en souvient, même si cela signifie de prendre deux comprimés en même temps. Elle continue ensuite à prendre les comprimés à son heure habituelle. Pour autant que la femme a pris ses comprimés correctement au cours des 7 jours précédant le premier comprimé oublié, il n'est pas nécessaire d’utiliser de précautions contraceptives supplémentaires. Toutefois, si elle a oublié plus d’un comprimé, il sera conseillé à la femme d’utiliser des précautions supplémentaires pendant 7 jours.
· Semaine 3 (Jour 15 à 21)
Le risque de diminution de la fiabilité est imminent en raison de la proximité des 7 jours d’intervalle sans comprimés. Toutefois, en ajustant le schéma de prise des comprimés, une diminution de la protection contraceptive peut encore être prévenue. En suivant l’une des deux options suivantes, il n’est ainsi pas nécessaire de prendre des précautions contraceptives supplémentaires, à condition que pendant les 7 jours précédant le premier comprimé oublié la femme a pris tous les comprimés correctement. Si ce n’est pas le cas, elle doit suivre la première de ces deux options et prendre aussi des précautions supplémentaires pendant les 7 jours suivants.
1. La femme doit prendre le dernier comprimé oublié dès qu'elle s’en souvient, même si cela signifie de prendre deux comprimés en même temps. Elle continue ensuite à prendre les comprimés à son heure habituelle. La plaquette suivante doit être commencée dès que la plaquette en cours est terminée, c’est-à-dire aucun intervalle ne doit être laissé entre les plaquettes. Il est peu probable que la femme ait une hémorragie de privation jusqu’à la fin des comprimés actifs de la seconde plaquette, mais elle peut présenter des spottings ou hémorragies de rupture pendant les jours de prise des comprimés.
2. La femme devra être informée d'arrêter la prise des comprimés de la plaquette en cours. Elle devra alors avoir un intervalle sans comprimés de 7 jours, incluant les jours où elle a oublié les comprimés, et ensuite continuer avec la plaquette suivante.
Si la femme a oublié des comprimés et n’a donc pas d’hémorragie de privation lors du premier intervalle sans comprimés, la possibilité d’une grossesse doit être considérée.
Comment changer le jour de début des règles ou reporter une hémorragie de privation
Pour retarder une hémorragie de privation, la femme doit continuer avec une autre plaquette de TRIAFEMI sans intervalle sans comprimés. La prolongation peut être poursuivie aussi longtemps que souhaité jusqu’à la fin de la seconde plaquette. Durant cette prolongation la femme peut avoir une hémorragie de rupture ou des spottings. La prise habituelle de TRIAFEMI est ensuite réinstaurée après l’habituel intervalle sans comprimés de 7 jours.
Si la femme veut changer le jour de début de son hémorragie de privation à un autre jour de la semaine, on peut lui conseiller de réduire le prochain intervalle sans comprimés d’autant de jours qu'elle le souhaite. Plus l'intervalle est court, plus le risque est élevé qu’elle n’ait pas d'hémorragie de privation mais une hémorragie de rupture ou des spottings pendant la plaquette suivante (tout comme pour retarder l’hémorragie de privation).
Conseils en cas de troubles gastro-intestinaux
En cas de vomissements ou diarrhée sévère, l’absorption des substances actives peut ne pas être complète et des mesures contraceptives supplémentaires sont à prendre.
Si des vomissements ou diarrhée sévère surviennent dans les 3-4 heures après la prise d’un comprimé actif, un nouveau (remplacement) comprimé doit être pris dès que possible. Le nouveau comprimé doit être pris dans les 12 heures suivant le moment de prise habituelle si possible. Si plus de 12 heures se sont écoulées, le conseil concernant l’oubli de comprimés comme donné en rubrique 4.2 « Conduite en cas d'oubli de comprimés » est applicable. Si la femme ne souhaite pas modifier son calendrier habituel de prise de comprimés, elle doit prendre le(s) comprimé(s) supplémentaire(s) d’une autre plaquette.
Personnes âgées
L’utilisation de ce produit n’est pas indiquée chez les femmes post-ménopausées.
Population pédiatrique
TRIAFEMI est contre-indiqué chez les filles qui n’ont pas atteint l’âge de la puberté – avant la ménarche.
En cas de survenue pour la première fois de l'une de ces pathologies lors de la prise d'une contraception orale, interrompre immédiatement le traitement.
· Présence ou risque de thrombo-embolie veineuse (TEV) :
o Thrombo-embolie veineuse – présence de TEV (patient traité par des anticoagulants) ou antécédents de TEV (p. ex. thrombose veineuse profonde [TVP] ou embolie pulmonaire [EP]);
o Prédisposition connue, héréditaire ou acquise, à la thrombo-embolie veineuse, telle qu’une résistance à la protéine C activée (PCa) (y compris une mutation du facteur V de Leiden), un déficit en antithrombine III, un déficit en protéine C, un déficit en protéine S ;
o Intervention chirurgicale majeure avec immobilisation prolongée (voir rubrique 4.4) ;
o Risque élevé de thrombo-embolie veineuse dû à la présence de multiples facteurs de risque (voir rubrique 4.4).
· Présence ou risque de thrombo-embolie artérielle (TEA) :
o Thrombo-embolie artérielle – présence ou antécédents de thrombo-embolie artérielle (p. ex. infarctus du myocarde [IM]) ou de prodromes (p. ex. angine de poitrine) ;
o Affection cérébrovasculaire – présence ou antécédents d’accident vasculaire cérébral (AVC) ou de prodromes (p. ex. accident ischémique transitoire [AIT]) ;
o Prédisposition connue, héréditaire ou acquise, à la thrombo-embolie artérielle, telle qu’une hyperhomocystéinémie ou la présence d’anticorps anti-phospholipides (anticorps anti-cardiolipine, anticoagulant lupique) ;
o Antécédents de migraine avec signes neurologiques focaux ;
o Risque élevé de thrombo-embolie artérielle dû à la présence de multiples facteurs de risque (voir rubrique 4.4) ou d’un facteur de risque sévère tel que :
§ diabète avec symptômes vasculaires,
§ hypertension artérielle sévère,
§ dyslipoprotéinémie sévère.
· Présence ou antécédents d’affection hépatique sévère tant que les valeurs de la fonction hépatique ne sont pas revenues à la normale.
· Présence ou antécédents de tumeurs hépatiques (bénignes ou malignes).
· Présence ou suspicion d’affections malignes dépendant de stéroïdes sexuels (par exemple des organes génitaux ou des seins).
· Hyperplasie endométriale.
· Hémorragie vaginale non diagnostiquée.
· Jaunisse cholestatique de la grossesse ou jaunisse liée à une utilisation antérieure de la pilule.
· Grossesse connue ou suspectée.
· Cardiopathie valvulaire avec complications.
· Présence ou antécédents de pancréatite associée à une sévère hypertriglycéridémie.
· Hypersensibilité aux substances actives ou à l’un des excipients mentionnés à la rubrique 6.1.
TRIAFEMI est contre-indiqué lors de l’utilisation concomitante de médicaments contenant de l’ombitasvir/paritaprévir/ritonavir et dasabuvir ou de médicaments contenant du glécaprévir/pibrentasvir ou sofosbuvir/velpatasvir/voxilaprévir (voir 4.5).
4.4. Mises en garde spéciales et précautions d'emploi
Si la patiente présente l’un des troubles ou l’un des facteurs de risque mentionnés ci-dessous, la pertinence du traitement par TRIAFEMI doit être discutée avec elle.
En cas d’aggravation ou de survenue de l’un de ces troubles ou facteurs de risque, la nécessité d’interrompre l’utilisation de TRIAFEMI doit être discutée entre le médecin et la patiente.
Consultation/examen médical
Avant l’instauration ou la reprise d’un traitement par TRIAFEMI, une recherche complète des antécédents médicaux (y compris les antécédents familiaux) doit être effectuée et la présence d’une grossesse doit être exclue. La pression artérielle doit être mesurée et un examen physique doit être réalisé, en ayant à l’esprit les contre-indications (voir rubrique 4.3) et les mises en garde (voir rubrique 4.4).
Il est important d’attirer l’attention des patientes sur les informations relatives à la thrombose veineuse et artérielle, y compris le risque associé à TRIAFEMI comparé à celui associé aux autres CHC, les symptômes de la TEV et de la TEA, les facteurs de risque connus et la conduite à tenir en cas de suspicion de thrombose.
Il doit également être indiqué aux patientes de lire attentivement la notice et de suivre les conseils fournis.
La fréquence et la nature des examens doivent être définies sur la base des recommandations en vigueur et adaptées à chaque patiente.
Les patientes doivent être averties que les contraceptifs hormonaux ne protègent pas contre l’infection par le VIH (SIDA) et les autres maladies sexuellement transmissibles.
· Troubles circulatoires
Risque de thrombo-embolie veineuse (TEV)
Le risque de TEV est augmenté chez les femmes qui utilisent un contraceptif hormonal combiné (CHC) en comparaison aux femmes qui n’en utilisent pas. Les CHC contenant du lévonorgestrel, du norgestimate (incluant TRIAFEMI) ou de la noréthistérone sont associés au risque de TEV le plus faible. La décision d’utiliser TRIAFEMI doit être prise uniquement après concertation avec la patiente afin de s’assurer qu’elle comprend le risque de TEV associé à TRIAFEMI l’influence de ses facteurs de risque actuels sur ce risque et le fait que le risque de TEV est maximal pendant la première année d’utilisation. Certaines données indiquent également une augmentation du risque lors de la reprise d’un CHC après une interruption de 4 semaines ou plus.
Parmi les femmes qui n’utilisent pas de CHC et qui ne sont pas enceintes, environ 2 sur 10 000 développeront une TEV sur une période d’un an. Cependant, chez une femme donnée, le risque peut être considérablement plus élevé, selon les facteurs de risque qu’elle présente (voir ci-dessous).
On estime que sur 10 000 femmes qui utilisent un CHC contenant du lévonorgestrel, environ 61 développeront une TEV sur une période d’un an.
Les données disponibles à ce jour suggèrent que le risque de TEV associé à l’utilisation de CHC contenant du norgestimate est similaire à celui associé à l’utilisation de CHC contenant du lévonorgestrel.
Ce nombre de TEV par année est inférieur à celui attendu pendant la grossesse ou en période post-partum.
La TEV peut être fatale dans 1 à 2 % des cas.
1. Point central de l’intervalle de 5‑7 pour 10 000 années-femmes sur la base d’un risque relatif, pour les CHC contenant du lévonorgestrel par rapport à la non-utilisation d’un CHC, d’environ 2,3 à 3,6
Nombre de cas de TEV pour 10 000 femmes sur une période d’un an

De façon extrêmement rare, des cas de thrombose ont été signalés chez des utilisatrices de CHC dans d’autres vaisseaux sanguins, p. ex. les veines et artères hépatiques, mésentériques, rénales ou rétiniennes.
Facteurs de risque de TEV
Le risque de complications thrombo-emboliques veineuses chez les utilisatrices de CHC peut être considérablement accru si d’autres facteurs de risque sont présents, en particulier si ceux-ci sont multiples (voir le tableau ci-dessous).
TRIAFEMI est contre-indiqué chez les femmes présentant de multiples facteurs de risque, ceux-ci les exposant à un risque élevé de thrombose veineuse (voir rubrique 4.3). Lorsqu’une femme présente plus d’un facteur de risque, il est possible que l’augmentation du risque soit supérieure à la somme des risques associés à chaque facteur pris individuellement – dans ce cas, le risque global de TEV doit être pris en compte. Si le rapport bénéfice/risque est jugé défavorable, le CHC ne doit pas être prescrit (voir rubrique 4.3).
Tableau : Facteurs de risque de TEV
|
Facteur de risque |
Commentaire |
|
Obésité (indice de masse corporelle supérieur à 30 kg/m²). |
L’élévation de l’IMC augmente considérablement le risque. Ceci est particulièrement important à prendre en compte si d’autres facteurs de risque sont présents. |
|
Immobilisation prolongée, intervention chirurgicale majeure, toute intervention chirurgicale sur les jambes ou le bassin, neurochirurgie ou traumatisme majeur. Remarque : l’immobilisation temporaire, y compris les trajets aériens > 4 heures, peut également constituer un facteur de risque de TEV, en particulier chez les femmes présentant d’autres facteurs de risque. |
Dans ces situations, il est conseillé de suspendre l’utilisation de la pilule (au moins quatre semaines à l’avance en cas de chirurgie programmée) et de ne reprendre le CHC que deux semaines au moins après la complète remobilisation. Une autre méthode de contraception doit être utilisée afin d’éviter une grossesse non désirée. Un traitement anti-thrombotique devra être envisagé si TRIAFEMI n’a pas été interrompu à l’avance. |
|
Antécédents familiaux (thrombo-embolie veineuse survenue dans la fratrie ou chez un parent, en particulier à un âge relativement jeune, c.-à-d. avant 50 ans). |
En cas de prédisposition héréditaire suspectée, la femme devra être adressée à un spécialiste pour avis avant toute décision concernant l’utilisation de CHC. |
|
Autres affections médicales associées à la TEV. |
Cancer, lupus érythémateux disséminé, syndrome hémolytique et urémique, maladies inflammatoires chroniques intestinales (maladie de Crohn ou rectocolite hémorragique) et drépanocytose. |
|
Âge. |
En particulier au-delà de 35 ans. |
Il n’existe aucun consensus quant au rôle éventuel joué par les varices et les thrombophlébites superficielles dans l’apparition ou la progression d’une thrombose veineuse.
L’augmentation du risque de thrombo-embolie pendant la grossesse, et en particulier pendant les 6 semaines de la période puerpérale, doit être prise en compte (pour des informations concernant « Grossesse et allaitement », voir rubrique 4.6).
Symptômes de TEV (thrombose veineuse profonde et embolie pulmonaire)
Les femmes doivent être informées qu’en cas d’apparition de ces symptômes, elles doivent consulter un médecin en urgence et lui indiquer qu’elles utilisent un CHC. Les symptômes de la thrombose veineuse profonde (TVP) peuvent inclure :
· Gonflement unilatéral d’une jambe et/ou d’un pied ou le long d’une veine de la jambe.
· Douleur ou sensibilité dans une jambe, pouvant n’être ressentie qu’en position debout ou lors de la marche.
· Sensation de chaleur, rougeur ou changement de la coloration cutanée de la jambe affectée.
Les symptômes de l’embolie pulmonaire (EP) peuvent inclure :
· Apparition soudaine et inexpliquée d’un essoufflement ou d’une accélération de la respiration.
· Toux soudaine, pouvant être associée à une hémoptysie.
· Douleur thoracique aiguë.
· Étourdissements ou sensations vertigineuses sévères.
· Battements cardiaques rapides ou irréguliers.
Certains de ces symptômes (p. ex. « essoufflement », « toux ») ne sont pas spécifiques et peuvent être interprétés à tort comme des signes d’événements plus fréquents ou moins sévères (infections respiratoires, p. ex.).
Les autres signes d’une occlusion vasculaire peuvent inclure : douleur soudaine, gonflement et coloration légèrement bleutée d’une extrémité.
Si l’occlusion se produit dans l’œil, les symptômes peuvent débuter sous la forme d’une vision trouble indolore pouvant évoluer vers une perte de la vision. Dans certains cas, la perte de la vision peut survenir presque immédiatement.
Risque de thrombo-embolie artérielle (TEA)
Des études épidémiologiques ont montré une association entre l’utilisation de CHC et l’augmentation du risque de thrombo-embolie artérielle (infarctus du myocarde) ou d’accident cérébrovasculaire (p. ex. accident ischémique transitoire, AVC). Les événements thrombo-emboliques artériels peuvent être fatals.
Facteurs de risque de TEA
Le risque de complications thrombo-emboliques artérielles ou d’accident cérébrovasculaire chez les utilisatrices de CHC augmente avec la présence de facteurs de risque (voir le tableau).TRIAFEMI est contre-indiqué chez les femmes présentant un facteur de risque sévère ou de multiples facteurs de risque de TEA qui les exposent à un risque élevé de thrombose artérielle (voir rubrique 4.3).
Lorsqu’une femme présente plus d’un facteur de risque, il est possible que l’augmentation du risque soit supérieure à la somme des risques associés à chaque facteur pris individuellement – dans ce cas, le risque global doit être pris en compte. Si le rapport bénéfice/risque est jugé défavorable, le CHC ne doit pas être prescrit (voir rubrique 4.3).
Tableau : Facteurs de risque de TEA
|
Facteur de risque |
Commentaire |
|
Âge |
En particulier au-delà de 35 ans |
|
Tabagisme |
Il doit être conseillé aux femmes de ne pas fumer si elles souhaitent utiliser un CHC. Il doit être fortement conseillé aux femmes de plus de 35 ans qui continuent de fumer d’utiliser une méthode de contraception différente. |
|
Hypertension artérielle |
|
|
Obésité (indice de masse corporelle supérieur à 30 kg/m2) |
L’élévation de l’IMC augmente considérablement le risque. Ceci est particulièrement important à prendre en compte chez les femmes présentant d’autres facteurs de risque. |
|
Antécédents familiaux (thrombo-embolie artérielle survenue dans la fratrie ou chez un parent, en particulier à un âge relativement jeune, c.-à-d. avant 50 ans) |
En cas de prédisposition héréditaire suspectée, la femme devra être adressée à un spécialiste pour avis avant toute décision concernant l’utilisation de CHC. |
|
Migraine |
L’accroissement de la fréquence ou de la sévérité des migraines lors de l’utilisation d’un CHC (qui peut être le prodrome d’un événement cérébrovasculaire) peut constituer un motif d’arrêt immédiat du CHC. |
|
Autres affections médicales associées à des événements indésirables vasculaires |
Diabète, hyperhomocystéinémie, valvulopathie cardiaque et fibrillation auriculaire, dyslipoprotéinémie et lupus érythémateux disséminé |
Symptômes de TEA
Les femmes doivent être informées qu’en cas d’apparition de ces symptômes, elles doivent consulter un médecin en urgence et lui indiquer qu’elles utilisent un CHC.
Les symptômes d’un accident cérébrovasculaire peuvent inclure :
· Apparition soudaine d’un engourdissement ou d’une faiblesse du visage, d’un bras ou d’une jambe, en particulier d’un côté du corps.
· Apparition soudaine de difficultés à marcher, de sensations vertigineuses, d’une perte d’équilibre ou de coordination.
· Apparition soudaine d’une confusion, de difficultés à parler ou à comprendre.
· Apparition soudaine de difficultés à voir d’un œil ou des deux yeux.
· Céphalée soudaine, sévère ou prolongée, sans cause connue.
· Perte de conscience ou évanouissement avec ou sans crise convulsive.
· Des symptômes temporaires suggèrent qu’il s’agit d’un accident ischémique transitoire (AIT).
Les symptômes de l’infarctus du myocarde (IM) peuvent inclure :
· Douleur, gêne, pression, lourdeur, sensation d’oppression ou d’encombrement dans la poitrine, le bras ou sous le sternum.
· Sensation de gêne irradiant vers le dos, la mâchoire, la gorge, le bras, l’estomac.
· Sensation d’encombrement, d’indigestion ou de suffocation.
· Transpiration, nausées, vomissements ou sensations vertigineuses.
· Faiblesse, anxiété ou essoufflement extrêmes.
· Battements cardiaques rapides ou irréguliers.
Dans de rares cas, des tumeurs bénignes du foie, et dans de plus rares cas encore, des tumeurs malignes du foie ont été rapportées chez les utilisatrices de COC.
Dans des cas isolés, ces tumeurs ont conduit à des hémorragies intra-abdominales mettant en jeu le pronostic vital. Chez les femmes sous COC, la survenue de douleurs de la partie supérieure de l’abdomen, d’augmentation du volume du foie ou de signes d’hémorragie intra-abdominale doit faire évoquer une tumeur hépatique.
Cancer du sein
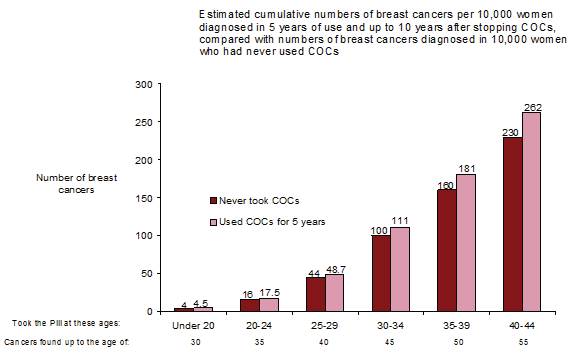
Une méta-analyse de 54 études épidémiologiques a conclu qu’il existe une légère augmentation du risque relatif (RR = 1,24) de cancer du sein chez les femmes sous contraception orale. Cette majoration du risque disparaît progressivement au cours des 10 années qui suivent l’arrêt du COC.
Le cancer du sein étant rare chez les femmes de moins de 40 ans, le nombre plus élevé de cancers diagnostiqués chez les utilisatrices habituelles ou nouvelles utilisatrices de COC reste faible par rapport au risque global de cancer du sein.
Le cancer du sein est rare chez les femmes de moins de 40 ans, qu'elles prennent ou non des COC. Bien que ce risque de fond augmente avec l'âge, le nombre excessif de diagnostics de cancer du sein chez les utilisatrices actuelles et récentes de COC est faible par rapport au risque global de cancer du sein (voir le diagramme à barres).
Le facteur de risque le plus important du cancer du sein chez les utilisatrices de COC est l'âge auquel les femmes arrêtent le COC; plus l'âge d'arrêt est élevé, plus le nombre de cancers du sein diagnostiqués est élevé. La durée d'utilisation est moins importante et l'excès de risque disparaît progressivement au cours des 10 années suivant l'arrêt de l'utilisation des COC, de sorte qu'à 10 ans, il ne semble plus y avoir d'excès.
L’augmentation possible du risque de cancer du sein doit être discutée avec l’utilisatrice et comparée aux avantages des COC, en tenant compte des preuves qu’ils offrent une protection substantielle contre le risque de développer certains autres cancers (par exemple les cancers de l’ovaire et de l’endomètre).
|
|
Cancer du col de l’utérus
Certaines études épidémiologiques suggèrent que les COC pourraient être associés à une augmentation du risque de cancer du col de l’utérus chez les utilisatrices de COC à long terme (> 5 ans). Il n’est cependant pas établi si ces pathologies sont dues à d’autres facteurs comme le comportement sexuel et le papillomavirus humain (HPV).
Autres tumeurs
L’utilisation de COC plus fortement dosés (50 µg éthinylestradiol) diminue le risque de cancer de l’endomètre et de l’ovaire. Ceci demande à être confirmé avec les COC plus faiblement dosés.
Troubles du cycle
Des saignements irréguliers (« spottings » ou métrorragies) peuvent survenir sous COC en particulier au cours des premiers mois. Ces saignements irréguliers seront considérés comme significatifs s’ils persistent après environ 3 cycles.
Si les saignements irréguliers persistent ou surviennent après des cycles réguliers, une recherche étiologique non hormonale doit alors être effectuée ; des examens diagnostiques appropriés doivent être pratiqués afin d’exclure une tumeur maligne ou une grossesse. Ces examens peuvent comporter un curetage.
Chez certaines femmes, les hémorragies de privation peuvent ne pas survenir au cours de l’intervalle libre. Si le COC a été pris tel que décrit dans la rubrique 4.2, il est peu probable que la femme soit enceinte. Cependant, si le COC n’a pas été pris correctement avant l’absence de la première hémorragie de privation ou si deux hémorragies de privation successives ne se produisent pas, il convient de s’assurer de l’absence de grossesse avant de poursuivre le COC.
Autres affections
Chez les femmes atteintes d’hypertriglycéridémie ou ayant des antécédents familiaux d’hypertriglycéridémie, un risque accru de pancréatite peut survenir en cas d’utilisation d’un COC.
Une augmentation modérée de la pression artérielle a été observée chez de nombreuses femmes sous COC mais elle était rarement cliniquement significative. Une interruption immédiate du COC n’est justifiée que dans ces rares cas. La persistance de chiffres tensionnels élevés ou le non contrôle des chiffres tensionnels par un traitement antihypertenseur chez une femme hypertendue prenant un COC doit faire interrompre celui-ci. Le COC pourra éventuellement être repris après normalisation des chiffres tensionnels par un traitement antihypertenseur.
La survenue ou l’aggravation des pathologies suivantes a été observée au cours de la grossesse ou chez des femmes prenant des COC, bien que la responsabilité des COC n’ait pu être établie : ictère et/ou prurit dû à une cholestase, lithiase biliaire, porphyrie, lupus érythémateux disséminé, syndrome hémolytico-urémique, chorée de Sydenham, herpès gravidique, hypoacousie par otosclérose.
Les œstrogènes exogènes peuvent déclencher ou aggraver les symptômes d’un angio-oedème héréditaire ou acquis.
La survenue d’anomalie hépatiques aiguës ou chroniques peut nécessiter l’arrêt du COC jusqu’à la normalisation des paramètres hépatiques. La récidive d’un ictère cholestatique et/ou d’un prurit liée à une cholestase survenue au cours d’une grossesse précédente ou de la prise antérieure d’hormones stéroïdiennes doit faire arrêter le COC.
Les COC peuvent avoir un effet sur la résistance périphérique à l’insuline et la tolérance au glucose ; il n’est cependant pas obligatoire de modifier le traitement chez des diabétiques utilisant un CHC faiblement dosé (contenant moins de 0,05 mg d’éthinylestradiol). Les femmes diabétiques doivent cependant être étroitement surveillées, en particulier lors de l’instauration d’un COC.
Des cas d’aggravation de dépression endogène, d’épilepsie, de maladie de Crohn et de rectocolite hémorragique ont été observés sous COC.
Un chloasma peut survenir, en particulier chez les femmes ayant des antécédents de chloasma gravidique. Les femmes ayant une prédisposition au chloasma sous COC doivent éviter de s’exposer au soleil ou aux rayons ultra-violets.
L’état dépressif et la dépression sont des effets indésirables bien connus liés à l’utilisation de contraceptifs hormonaux (voir rubrique 4.8). La dépression peut être grave et constitue un facteur de risque bien connu de comportement suicidaire et de suicide. Il convient de conseiller aux femmes de contacter leur médecin en cas de changements d’humeur et de symptômes dépressifs, y compris peu de temps après le début du traitement.
Excipients
Ce médicament contient du lactose. Les patients présentant une intolérance au galactose, un déficit total en lactase ou un syndrome de malabsorption du glucose ou du galactose (maladies héréditaires rares) ne doivent pas prendre ce médicament.
4.5. Interactions avec d'autres médicaments et autres formes d'interactions
Réduction potentielle de l’effet contraceptif associé à la co-administration d’autres médicaments
Remarque : Les informations sur la prescription de médicaments concomitants doivent être consultées pour identifier les interactions potentielles.
Effets d’autres médicaments sur TRIAFEMI
Des interactions peuvent survenir avec les médicaments inducteurs des enzymes microsomales, pouvant induire une augmentation de l’élimination des hormones sexuelles et conduire à des hémorragies de rupture et/ou à un échec de la contraception.
Prise en charge
L’induction enzymatique peut déjà s’observer après quelques jours de traitement. L’induction enzymatique maximale est observée généralement en quelques semaines. Après arrêt du traitement médicamenteux l’induction enzymatique peut se maintenir pendant environ 4 semaines.
Traitement à court terme
Les femmes sous traitement par des médicaments inducteurs enzymatiques doivent transitoirement utiliser une méthode barrière ou une autre méthode de contraception en plus du COC. La méthode barrière doit être utilisée pendant toute la durée du traitement concomitant et pendant les 28 jours après son arrêt. Si le traitement médicamenteux se poursuit au-delà de la fin des comprimés actifs de la plaquette de COC, la plaquette suivante est à commencer immédiatement après la précédente sans l’intervalle habituel sans comprimés.
Traitement à long terme
Chez les femmes sous traitement à long terme par des substances actives inducteurs enzymatiques, une autre méthode contraceptive non hormonale fiable est recommandée.
Les interactions suivantes ont été rapportées dans la littérature :
Substances augmentant la clairance des COC (diminution de l’efficacité des COC par induction enzymatique), par exemple :
Barbituriques, bosentan, carbamazépine, phénytoïne, primidone, rifampicine, et médicaments du VIH ritonavir, névirapine et éfavirenz et potentiellement aussi felbamate, griséofulvine, oxcarbazépine, topiramate et produits contenant le remède à base de plantes millepertuis (Hypericum perforatum).
Substances induisant des effets variables sur la clairance des COC :
Quand co-administrées avec les COC, plusieurs associations d’inhibiteurs de la protéase du VIH et inhibiteurs non nucléosidiques de la transcriptase inverse, incluant les associations avec des inhibiteurs du VHC, peuvent augmenter ou diminuer les concentrations plasmatiques en estrogènes ou progestatifs. L’effet net de ces modifications peut s’avérer cliniquement significatif dans certains cas.
Ainsi, les informations de prescription de médicaments concomitants du VIH/VHC doivent être consultées pour identifier les interactions potentielles et toute recommandation s’y rapportant. En cas de doute, une méthode contraceptive barrière supplémentaire doit être utilisée par les femmes sous traitement par un inhibiteur de la protéase ou inhibiteur non nucléosidique de la transcriptase inverse.
Substances diminuant la clairance des COC (inhibiteurs enzymatiques) :
La pertinence clinique d’interactions potentielles avec les inhibiteurs enzymatiques reste inconnue.
L’administration concomitante de puissants inhibiteurs du cytochrome CYP3A4 peut augmenter les concentrations plasmatiques de l’estrogène ou du progestatif ou des deux.
Des doses d’Etoricoxib de 60 à 120 mg/jour ont montré une augmentation des concentrations plasmatiques d’éthinylestradiol de 1,4 à 1,6 respectivement lorsqu’elles sont prises concomitamment avec un contraceptif hormonal combiné contenant 0,035 mg d’éthinylestradiol.
Effets de TRIAFEMI sur d’autres médicaments
Les contraceptifs oraux peuvent influencer le métabolisme de certaines autres substances actives. Par conséquent, les concentrations plasmatiques et tissulaires peuvent augmenter (par exemple ciclosporine) ou diminuer (par exemple lamotrigine).
Les données cliniques suggèrent que l’éthinylestradiol inhibe l’élimination des substrats du cytochrome CYP1A2, entraînant une augmentation faible (par exemple théophylline) ou modérée (par exemple tizanidine) de leur concentration sérique.
Interactions pharmacodynamiques :
Au cours des essais cliniques menés chez des patients traités pour une infection par le virus de l'hépatite C (VHC) et recevant des médicaments contenant ombitasvir/paritaprevir/ritonavir et dasabuvir, avec ou sans ribavirine, il a été observé des augmentations de transaminase (ALAT) supérieures à 5 fois la limite supérieure de la normale (LSN), significativement plus fréquentes chez les femmes utilisant des médicaments contenant de l'éthynylestradiol, tel que les contraceptifs hormonaux combinés (CHC). De plus, chez des patients traités par le glecaprevir/pibrentasvir ou sofosbuvir/velpatasvir/voxilaprevir, des augmentations des ALAT ont été observées chez les femmes utilisant des médicaments contenant de l'éthinylestradiol tels que les CHC (voir la rubrique 4.3). Par conséquent, les utilisatrices de TRIAFEMI doivent passer à une méthode alternative de contraception (par exemple une contraception progestative seule ou des méthodes non hormonales) avant de commencer un traitement avec ces schémas thérapeutique combinés. TRIAFEMI pourra être réutilisé 2 semaines après la fin du traitement avec ces combinaisons de médicaments.
Analyses biologiques
L’utilisation de stéroïdes contraceptifs peut influencer les résultats de certains tests biologiques, y compris les paramètres biochimiques de la fonction hépatique, thyroïdienne, surrénale ou rénale, les concentrations plasmatiques de protéines (de transport), par exemple la globuline de la liaison aux corticostéroîdes, et les fractions de lipides/lipoprotéines, les paramètres des glucides et les paramètres de coagulation et de fibrinolyse. Les modifications restent généralement comprises dans les valeurs normales de laboratoire.
Les concentrations sériques en acide folique peuvent être diminuées par les COC. Si une femme est enceinte rapidement après l’arrêt des COC, ceci est cliniquement important.
4.6. Fertilité, grossesse et allaitement
Grossesse
L’augmentation du risque de TEV en période post-partum doit être prise en compte lors de la reprise de TRIAFEMI, comprimé (voir rubriques 4.2 et 4.4).
Allaitement
Les COC peuvent influer sur l'allaitement, car ils peuvent en réduire la quantité et modifier la composition du lait maternel.
Par conséquent, l'utilisation de COC ne devrait généralement pas être recommandée avant que la mère qui allaite ait complètement sevré son enfant. De petites quantités de stéroïdes contraceptifs et/ou de leurs métabolites peuvent être excrétées avec le lait lors de l'utilisation de COC. Ces quantités peuvent affecter l'enfant.
Fertilité
TRIAFEMI est indiqué pour la prévention de la grossesse. Pour des informations sur le retour à la fertilité, voir rubrique 5.1.
4.7. Effets sur l'aptitude à conduire des véhicules et à utiliser des machines
Description de certains effets indésirables particuliers
Une augmentation du risque d'événement thrombotique et thrombo-embolique artériel et veineux, incluant l’infarctus du myocarde, l’AVC, les accidents ischémiques transitoires, la thrombose veineuse et l’embolie pulmonaire, a été observée chez les femmes utilisant des CHC ; ceci est abordé plus en détails en rubrique 4.4.
Le tableau ci-dessous répertorie tous les effets indésirables signalés lors de l’utilisation de TRIAFEMI au cours d’essais cliniques ou d’expériences post-commercialisation avec des comprimés de norgestimate et d’éthinylestradiol.
Les fréquences attribuées aux effets indésirables ont été établies sur la base d’une évaluation des incidences observées notamment dans 6 essais cliniques portant sur 4 826 femmes et 49 800 cycles. La fréquence des effets indésirables rapportés uniquement après commercialisation est inconnue (ne peut pas être estimée sur la base des données disponibles).
Les catégories de fréquence affichées utilisent la convention suivante : très fréquent (> 1/10); fréquent (> 1/100 à < 1/10); peu fréquent (> 1/1000 à < 1/100); rare (> 1/10 000 à < 1/1 000); très rare (< 1/10 000); et indéterminé (ne peut pas être estimé à partir des données disponibles).
|
Infections et infestations |
|
|
Fréquent |
Infection urinaire, infection vaginale |
|
Tumeurs bénignes, malignes et non précisées (inclus kystes et polypes) |
|
|
Rare |
Kyste mammaire |
|
Fréquence indéterminée1 |
Adénome hépatique2, cancer du sein2, tumeur bénigne du sein2, hyperplasie nodulaire focale2, fibroadénome du sein2 |
|
Affections du système immunitaire |
|
|
Fréquent |
Hypersensibilité |
|
Fréquence indéterminée |
Aggravation des symptômes de l’angioœdème héréditaire ou acquis. |
|
Troubles du métabolisme et de la nutrition |
|
|
Fréquent |
Rétention hydrique |
|
Peu fréquent |
Augmentation et diminution de l’appétit, modification du poids |
|
Rare |
Troubles de l’appétit |
|
Fréquence indéterminée1 |
Dyslipidémie1 |
|
Affections psychiatriques |
|
|
Fréquent |
Modification de l’humeur, dépression, nervosité, insomnie |
|
Peu fréquent |
Anxiété, modifications de la libido |
|
Affections du système nerveux |
|
|
Très fréquent |
Céphalées |
|
Fréquent |
Migraines, étourdissement |
|
Peu fréquent |
Syncope, paresthésie |
|
Fréquence indéterminée1 |
convulsion1 |
|
Affections oculaires |
|
|
Peu fréquent |
Gêne visuelle, sécheresse oculaire |
|
Fréquence indéterminée1 |
Intolérance aux lentilles de contact1, thrombose veineuse rétinienne |
|
Affections de l’oreille et du labyrinthe |
|
|
Rare |
Vertiges |
|
Affections cardiaques |
|
|
Peu fréquent |
Palpitations |
|
Rare |
Tachycardie |
|
Affections vasculaires |
|
|
Peu fréquent |
Thrombose2, hypertension, bouffées de chaleur |
|
Rare |
Thrombo-embolies veineuse et artérielle2 incluant accident vasculaire cérébral, thrombose d’un vaisseau sanguin, infarctus du myocarde, thrombose veineuse profonde, embolie pulmonaire |
|
Fréquence indéterminée |
Syndrome de Budd-Chiari1,8 |
|
Affections respiratoires, thoraciques, médiastinales |
|
|
Peu fréquent |
Dyspnée |
|
Affections gastro-intestinales |
|
|
Très fréquent |
Troubles gastro-intestinaux3,4, vomissements5, diarrhées5, nausées4 |
|
Fréquent |
Douleurs gastro-intestinales, douleurs abdominales, distension abdominale, constipation, flatulence |
|
Rare |
Pancréatite |
|
Affections hépatobiliaires |
|
|
Rare |
Hépatite2 |
|
Affections de la peau et du tissu sous-cutané |
|
|
Fréquent |
Acné, rash |
|
Peu fréquent |
Alopécie, hirsutisme, urticaire, prurit, érythème, décoloration cutanée |
|
Rare |
Hyperhidrose, réaction de photosensibilité |
|
Fréquence indéterminée1 |
Angioedeme1, érythème noueux1, sueurs nocturnes1 |
|
Affections musculo-squelettiques et des tissus conjonctifs |
|
|
Fréquent |
Spasmes musculaires, douleurs dans les extrémités, douleurs dorsales6 |
|
Peu fréquent |
Myalgie |
|
Affections des organes de reproduction et du sein |
|
|
Très fréquent |
Dysménorrhée4, métrorragies4, hémorragies de privation anormales4 |
|
Fréquent |
Aménorrhée4, pertes génitales, douleurs mammaires |
|
Peu fréquent |
Ecoulement mammaire, gonflement mammaire, kystes ovariens, sécheresse vulvovaginale, dysplasie cervicale2 |
|
Rare |
Pertes vaginales |
|
Fréquence indéterminée1 |
Suppression de la lactation1 |
|
Troubles généraux et anomalies au site d’administration |
|
|
Fréquent |
Douleur à la poitrine, œdème, asthénie7 |
|
Investigations |
|
|
Fréquent |
Augmentation du poids |
|
Peu fréquent |
Diminution du poids |
1. Ces effets indésirables post-marketing n’ont pas été observés durant les essais cliniques. Par conséquent, basée sur les données disponibles, la réelle incidence de ces effets ne peut être estimée.
2 Voir section 4.4 Mises en garde spéciales et précautions d’emploi.
3 Rapporté comme nausée ou vomissement.
4 Incidence des effets indésirables rapportée par cycle ; catégorie de fréquence basée sur l’incidence combinée la plus élevée au cours du cycle de traitement 1.
5 Incidence des effets indésirables rapportée par cycle ; catégorie de fréquence basée sur l’incidence combinée la plus élevée au cours du cycle de traitement 12.
6 Cette valeur d’incidence calculée peut être légèrement supérieure à l’incidence réelle, dans la mesure où plus d’un terme d’évènement rapporté dans le même essai est codé selon le terme préféré de MedDRA « Dorsalgie ». Il est possible que le même sujet ait signalé plus d’un terme de l’évènement et que, par conséquent, il soit compté plus d’une fois pour le terme préféré « Dorsalgie ».
7 Terme de niveau supérieur ; catégorie de fréquence basée sur l’incidence du terme préféré le plus courant parmi les symptômes asthéniques de niveau supérieur à partir de données d’essais cliniques regroupées, à savoir la fatigue.
8 Y compris thrombose veineuse hépatique.
Description des effets indésirables sélectionnés
Un risque accru d'événements thrombotiques et thrombo-emboliques artériels et veineux, y compris infarctus du myocarde, accidents vasculaires cérébraux, accidents ischémiques transitoires, thrombose veineuse et embolie pulmonaire a été observé chez les femmes utilisant des CHC, qui sont décrits plus en détail à la section 4.4.
Les effets indésirables graves suivants ont été rapportés chez les femmes utilisant des CHC, qui sont décrits dans la rubrique 4.4 Mises en garde spéciales et précautions d'emploi :
· Troubles thromboemboliques veineux ;
· Troubles thromboemboliques artériels ;
· L'hypertension artérielle ;
· Tumeurs du foie ;
· Occurrence ou détérioration d’affections dont l’association avec l’utilisation de CHC n’est pas concluante : maladie de Crohn, colite ulcéreuse, épilepsie, myome utérin, porphyrie, lupus érythémateux systémique, herpès gestationis, chorée de Sydenham, syndrome hémolytique et urémique, jaunisse cholestatique ;
· Chloasma ;
· Les perturbations aiguës ou chroniques de la fonction hépatique peuvent nécessiter l'arrêt de l'utilisation des CHC jusqu'à ce que les marqueurs de la fonction hépatique reviennent à la normale.
La fréquence du diagnostic de cancer du sein est très légèrement accrue chez les utilisatrices de CHC. Le cancer du sein étant rare chez les femmes de moins de 40 ans, le nombre de patients en excès est faible par rapport au risque global de cancer du sein. La causalité avec l'utilisation des CHC est inconnue. Pour plus d'informations, voir sections 4.3 et 4.4.
Interactions
Les saignements et/ ou l'échec contraceptif peuvent résulter des interactions d'autres médicaments (inducteurs enzymatiques) avec des contraceptifs oraux (voir la section 4.5).
Déclaration des effets indésirables suspectés
La déclaration des effets indésirables suspectés après autorisation du médicament est importante. Elle permet une surveillance continue du rapport bénéfice/risque du médicament. Les professionnels de santé déclarent tout effet indésirable suspecté via le système national de déclaration : Agence nationale de sécurité du médicament et des produits de santé (ANSM) et réseau des Centres Régionaux de Pharmacovigilance - Site internet : www.signalement-sante.gouv.fr.
Aucun cas de surdosage n'a mis en évidence de problèmes graves de santé. Les symptômes pouvant survenir sont : nausées, vomissements et saignements vaginaux. Il n’existe pas d’antidote et le traitement doit être purement symptomatique.
5. PROPRIETES PHARMACOLOGIQUES
5.1. Propriétés pharmacodynamiques
Estroprogestatif combiné minidosé, triphasique.
Le norgestimate est un progestatif de 3ème génération appartenant à la classe thérapeutique des gonanes.
Mécanisme d’action
TRIAFEMI agit par inhibition des gonadotrophines grâce aux actions oestrogéniques et progestatives de l’éthinylestradiol et du norgestimate. Le principal mécanisme d'action est l'inhibition de l'ovulation. Des modifications au niveau de la glaire cervicale, de la motilité des trompes de Fallope et de l'endomètre peuvent également contribuer à l'efficacité du produit.
Effets pharmacodynamiques
Des études de liaison de la SHBG (globuline liant les hormones sexuelles) ainsi que des études chez l’animal et chez l’homme ont montré que le norgestimate et la norelgestromine, principal métabolite sérique du norgestimate après administration orale, exercent une activité progestative élevée avec une androgénicité intrinsèque minimale, qui illustre l’action sélective de TRIAFEMI. Le norgestimate, en association avec l’éthinylestradiol, ne neutralise pas l’augmentation de la SHBG induite par les œstrogènes, ce qui entraine une baisse des taux de testostérone libre dans le sérum par rapport aux valeurs initiales.
Efficacité et sécurité clinique
L'indice de Pearl de cet estroprogestatif (nombre de grossesses pour 100 années femmes) est de 0 à 0,77 % en fonction des études (48 598 cycles). L’indice de Pearl tient compte de toutes les grossesses survenues au cours des études y compris chez les femmes n’ayant pas pris correctement les comprimés (oublis).
TRIAFEMI a été testé contre un placebo au cours de 2 études cliniques randomisées et en double aveugle chez des femmes présentant une acné légère à modérée. TRIAFEMI n’a pas été testé contre un autre traitement hormonal antiacnéique.
A partir de travaux in vitro sur des lignées cellulaires humaines transfectées exprimant le récepteur aux androgènes, les propriétés suivantes ont été démontrées mais n'ont pas fait l'objet d'études in vivo:
· le norgestimate et le déacetyl-norgestimate présentent une inhibition compétitive avec les androgènes pour la liaison au récepteur (sur des cellules PALM de cancer prostatique: PC3 Androgen Luciferase MMTV);
· le norgestimate et le déacetyl-norgestimate réduisent le transfert cyto-nucléaire des complexes androgènes-récepteur des androgènes (sur des cellules de mammifères COS7);
· le norgestimate et le déacetyl-norgestimate inhibent faiblement l'activité transcriptionnelle de la luciférase induite par les androgènes (sur des cellules humaines de cancer prostatique PALM) à faible concentration.
5.2. Propriétés pharmacocinétiques
Absorption
Le norgestimate est absorbé rapidement après administration orale. Après une administration unique ou multiple (trois cycles) de Norgestimate associé à l’Ethinylestradiol, les concentrations sériques de norgestimate restent inférieures à la limite de quantification des métabolites (0,1 ng/mL) du norgestimate, la norelgestromine et le norgestrel, se trouvent dans des concentrations mesurables en circulation, atteignant des niveaux sériques maximaux environ 1,5 heure après la dose. L'augmentation de la Cmax et de l'ASC pour la norelgestromine est proportionnelle à la dose après administration de 0,180 à 0,250 mg de norgestimate.
Distribution
La norelgestromine et le norgestrel sont fortement liés (> 97%) aux protéines sériques. La norelgestromine est liée à l'albumine mais pas à la SHBG, tandis que le norgestrel est lié principalement au SHBG et, dans une moindre mesure, à l'albumine.
Des études ont montré que le manque de liaison de la norelgestromine à la SHBG est unique par rapport à d'autres progestatifs dans les contraceptifs oraux et joue un rôle clé dans la potentialisation de son activité biologique. En revanche, le norgestrel formé du norgestimate est largement lié à SHBG, ce qui limite son activité biologique. Ces résultats, ainsi que la sélectivité de la norelgestromine pour le récepteur de la progestérone, indiquent que ce métabolite pourrait expliquer le profil clinique unique de norgestimate.
Biotransformation
Le norgestimate est rapidement métabolisé par des mécanismes de premier passage (intestinal et/ ou hépatique) en norelgestromine (concentrations sériques maximales observées dans les 2 heures) et en norgestrel, tous deux progestatifs pharmacologiquement actifs.
Élimination
La norelgestromine et le norgestrel sont tous les deux ensuite métabolisés et leurs métabolites sont éliminés par voie rénale et fécale. Les valeurs de demi-vie d'élimination à l'état stationnaire étaient de 10 à 15 heures pour l'éthinylestradiol, 24,9 heures pour la norelgestromine et 45 heures pour le norgestrel. Après l'administration de 14C-norgestimate, 47% de la radioactivité administrée a été éliminée dans l'urine et 37% dans les fèces.
Relations pharmacocinétique/pharmacodynamique
Après l'administration de 0,250 mg de norgestimate associé à 0, 035 mg d'éthinylestradiol, l'ASC0-24h moyen à l'état d'équilibre, basé sur des taux sériques non liés à SHBG, était de 18,1 h ng / mL pour la norelgestromine et 3,64 h ng / mL pour le norgestrel. L'ASC pour le norgestrel suite à l'administration de 0,250 mg de norgestimate/ 0, 035 mg d'éthinylestradiol, correspond à l'exposition après une dose de lévonorgestrel d'environ 30 microgrammes en association avec l'éthinylestradiol.
ETHINYLESTRADIOL
Absorption
L’éthinylestradiol administré par voie orale est absorbé rapidement et totalement. Les concentrations sériques d'éthinylestradiol sont mesurables dans les 0,5 heure qui suivent l'administration, atteignant leur maximum environ 1 à 2 heures après la dose.
Distribution
L’éthinylestradiol est fortement (environ 98%) mais non spécifiquement lié à l’albumine sérique.
Biotransformation
L'éthinylestradiol est métabolisé en divers métabolites hydroxylés et en leurs conjugués glucuronide et sulfate.
Élimination
L'éthinylestradiol est ensuite métabolisé et les métabolites sont éliminés par les voies rénale et fécale. La demi-vie d’élimination à l’état d’équilibre était de 10 à 15 heures pour l’éthinylestradiol.
5.3. Données de sécurité préclinique
Comprimé blanc: lactose anhydre, amidon prégélatinisé, crospovidone, stéarate de magnésium.
Comprimé bleu ciel et bleu foncé: lactose anhydre, amidon prégélatinisé, crospovidone, stéarate de magnésium, laque aluminique d'indigotine.
2 ans.
6.4. Précautions particulières de conservation
A conserver à une température ne dépassant pas +25°C.
6.5. Nature et contenu de l'emballage extérieur
21 comprimés (7 comprimés blancs, 7 comprimés bleu ciel, 7 comprimés bleu foncé) sous plaquette thermoformée (PVC/PVDC/Aluminium); boîte de 1 ; 3 ; 6 plaquettes.
6.6. Précautions particulières d’élimination et de manipulation
Pas d’exigences particulières.
7. TITULAIRE DE L’AUTORISATION DE MISE SUR LE MARCHE
9/11, RUE JEANNE BRACONNIER
BATIMENT "LE NEWTON"
92366 MEUDON-LA-FORET
8. NUMERO(S) D’AUTORISATION DE MISE SUR LE MARCHE
· 358 995-0: 21 comprimés sous plaquette (PVC/PVDC/Aluminium). Boîte de 1 plaquette.
· 358 996-7: 21 comprimés sous plaquette (PVC/PVDC/Aluminium). Boîte de 3 plaquettes.
· 34009 301 887 1 2 : 6 plaquette(s) PVC-Aluminium de 21 comprimés (7 comprimés blancs, 7 comprimés bleu ciel et 7 comprimés bleu foncé).
9. DATE DE PREMIERE AUTORISATION/DE RENOUVELLEMENT DE L’AUTORISATION
[à compléter ultérieurement par le titulaire]
10. DATE DE MISE A JOUR DU TEXTE
[à compléter ultérieurement par le titulaire]
Sans objet.
12. INSTRUCTIONS POUR LA PREPARATION DES RADIOPHARMACEUTIQUES
Liste I
ANSM - Mis à jour le : 19/02/2026
Norgestimate/Ethinylestradiol
· Gardez cette notice. Vous pourriez avoir besoin de la relire.
· Si vous avez d’autres questions, interrogez votre médecin, ou votre pharmacien ou votre sage-femme.
· Ce médicament vous a été personnellement prescrit. Ne le donnez pas à d’autres personnes. Il pourrait leur être nocif, même si les signes de leur maladie sont identiques aux vôtres.
· Si vous ressentez un quelconque effet indésirable, parlez-en à votre médecin ou votre pharmacien ou votre sage-femme. Ceci s’applique aussi à tout effet indésirable qui ne serait pas mentionné dans cette notice. Voir rubrique 4.
Points importants à connaître concernant les contraceptifs hormonaux combinés (CHC) :
· Ils comptent parmi les méthodes de contraception réversibles les plus fiables lorsqu’ils sont utilisés correctement.
· Ils augmentent légèrement le risque de formation d’un caillot sanguin dans les veines et les artères, en particulier pendant la première année de leur utilisation ou lorsque le contraceptif hormonal combiné est repris après une interruption de 4 semaines ou plus.
· Soyez vigilante et consultez votre médecin ou votre sage-femme si vous pensez présenter les symptômes évocateurs d’un caillot sanguin (voir rubrique 2 « Caillots sanguins »).
1. Qu'est-ce que TRIAFEMI, comprimé et dans quels cas est-il utilisé?
2. Quelles sont les informations à connaître avant de prendre TRIAFEMI, comprimé?
3. Comment prendre TRIAFEMI, comprimé ?
4. Quels sont les effets indésirables éventuels ?
5. Comment conserver TRIAFEMI, comprimé ?
6. Contenu de l’emballage et autres informations.
1. QU’EST-CE QUE TRIAFEMI, comprimé ET DANS QUELS CAS EST-IL UTILISE ?
Ce médicament est un contraceptif et un antiacnéique.
Il empêche l’ovulation. Il est préconisé dans le but d’éviter une grossesse, chez la femme ayant une acné légère à modérée.
Ce contraceptif contient deux types d’hormones féminines : un estrogène et un progestatif.
Ces hormones agissent en empêchant la libération de l’ovule (ovulation) de vos ovaires ce qui vous empêche d’être enceinte. Par ailleurs, TRIAFEMI rend plus épaisse la glaire (mucus) du col de l’utérus ce qui rend plus difficile l’entrée des spermatozoïdes dans l’utérus. L’effet contraceptif commence à la prise du premier comprimé.
2. QUELLES SONT LES INFORMATIONS A CONNAITRE AVANT DE PRENDRE TRIAFEMI, comprimé ?
Avant de commencer à utiliser TRIAFEMI, vous devez lire les informations concernant les caillots sanguins (thrombose) dans cette rubrique « Caillots sanguins ». Il est particulièrement important de lire la description des symptômes d’un caillot sanguin dans cette rubrique « Caillots sanguins »).
Avant que vous ne commenciez à prendre TRIAFEMI, votre médecin vous posera certaines questions concernant vos antécédents médicaux personnels et ceux de vos parents proches.
Le médecin mesurera également votre tension artérielle, et en fonction de votre situation personnelle, il peut également effectuer certains autres tests.
Dans cette notice, plusieurs situations sont décrites où vous devez arrêter de prendre TRIAFEMI, où l’efficacité contraceptive de TRIAFEMI peut être réduite. Dans ces situations, vous devez soit ne pas avoir de rapports sexuels ou vous devez prendre des précautions contraceptives non hormonales supplémentaires, par exemple utiliser un préservatif ou une autre méthode barrière. N’utilisez pas les méthodes du calendrier ou de la prise de la température. Ces méthodes peuvent être non fiables parce que TRIAFEMI modifie les variations mensuelles de la température corporelle et de la glaire cervicale.
TRIAFEMI, comme d’autres contraceptifs hormonaux, ne protège pas contre l’infection par le VIH (sida) ou tout autre infection sexuellement transmissible.
Ne prenez jamais TRIAFEMI, comprimé :
Dans quels cas ne devez-vous jamais utiliser TRIAFEMI, comprimé ?
Vous ne devez pas utiliser TRIAFEMI si vous êtes dans l’une des situations listées ci-dessous. Si tel est le cas, vous devez en informer votre médecin ou votre sage-femme. Votre médecin ou votre sage-femme discutera avec vous d’autres méthodes de contraception qui seraient plus adaptées.
· si vous avez (ou avez déjà eu) un caillot dans un vaisseau sanguin d’une jambe (thrombose veineuse profonde [TVP]), d’un poumon (embolie pulmonaire [EP]) ou d’autres organes ;
· si vous vous savez atteinte d’un trouble affectant la coagulation sanguine – par exemple, un déficit en protéine C, un déficit en protéine S, un déficit en antithrombine III, une mutation du facteur V de Leiden ou la présence d’anticorps anti-phospholipides ;
· si vous devez être opérée ou si vous êtes alitée pendant une durée prolongée (voir la rubrique « Caillots sanguins ») ;
· si vous avez déjà eu une crise cardiaque ou un accident vasculaire cérébral (AVC) ;
· si vous avez (ou avez déjà eu) une angine de poitrine (une maladie provoquant des douleurs intenses dans la poitrine et pouvant être le signe précurseur d’une crise cardiaque) ou un accident ischémique transitoire (AIT - symptômes temporaires d’AVC) ;
· si vous avez l’une des maladies suivantes pouvant augmenter le risque de caillot dans les artères :
o diabète sévère avec atteinte des vaisseaux sanguins,
o pression artérielle très élevée,
o taux très élevé de graisses dans le sang (cholestérol ou triglycérides),
o maladie appelée hyperhomocystéinémie.
· si vous avez (ou avez déjà eu) un type de migraine appelé « migraine avec aura » ;
· si vous avez un trouble valvulaire cardiaque qui a causé des complications ;
· si vous avez (ou avez déjà eu) une maladie sévère du foie et votre fonction hépatique n’est pas normalisée ;
· si vous avez (ou avez déjà eu) une tumeur du foie (bénigne ou maligne) ;
· en cas de tumeurs malignes du sein ou des organes génitaux ;
· si vous avez des saignements vaginaux inexpliqués ;
· si vous avez eu une jaunisse pendant votre grossesse causée par un canal biliaire ou si vous avez déjà eu une jaunisse lors de l'utilisation de la pilule ;
· si vous présentez un épaississement anormal de la muqueuse utérine ;
· si vous êtes allergique au norgestimate ou à l’éthinylestradiol ou à l'un des autres composants contenus dans ce médicament (mentionnés dans la rubrique 6).
· si vous avez (ou avez déjà eu) une pancréatite (inflammation du pancréas) associée à une hypertriglycéridémie sévère ;
· Si vous avez une hépatite C et que vous prenez des médicaments contenant du dasabuvir, de l’ombitasvir, du paritaprévir et du ritonavir ou du glécaprévir/pibrentasvir ou sofosbuvir/velpatasvir/voxilaprévir (voir aussi rubrique « Autres médicaments et TRIAFEMI »).
· Si vous êtes enceinte ou pensez l’être
· En cas de post-ménopause
Avertissements et précautions
Dans quels cas devez-vous faire attention avec TRIAFEMI, comprimé ?
Dans quels cas devez-vous contacter votre médecin ?
Consultez un médecin de toute urgence
- si vous remarquez de possibles signes d’un caillot sanguin, qui pourraient indiquer que vous avez un caillot sanguin dans une jambe (thrombose veineuse profonde), que vous avez un caillot sanguin dans un poumon (embolie pulmonaire) ou que vous faites une crise cardiaque ou un AVC (voir la rubrique « Caillots sanguins » ci-dessous).
Pour la description des symptômes de ces effets indésirables graves, reportez-vous à la rubrique « Comment reconnaître un caillot sanguin ».
Si vous êtes dans l’une des situations suivantes, informez-en votre médecin ou votre sage-femme.
Parlez à votre médecin ou votre sage-femme avant de prendre TRIAFEMI.
Si le problème apparaît ou s’aggrave pendant l’utilisation de TRIAFEMI, vous devez également en informer votre médecin.
· si vous êtes atteinte de la maladie de Crohn ou de rectocolite hémorragique (maladies inflammatoires chroniques des intestins) ;
· si vous avez un lupus érythémateux disséminé (LED) (une maladie qui affecte votre système de défenses naturelles, dite auto-immune) ;
· si vous avez un syndrome hémolytique et urémique (SHU) (un trouble de la coagulation sanguine qui entraîne une défaillance des reins) ;
· si vous avez une drépanocytose (une maladie héréditaire touchant les globules rouges) ;
· si avez des taux élevés de graisses dans le sang (hypertriglycéridémie) ou des antécédents familiaux de ce trouble. L’hypertriglycéridémie a été associée à une augmentation du risque de pancréatite (inflammation du pancréas) ;
· si vous devez être opérée ou si vous êtes alitée pendant une durée prolongée (reportez-vous à la rubrique 2, « Caillots sanguins ») ;
· si vous venez juste d’accoucher, vous êtes exposée à un risque augmenté de caillots sanguins. Vous devez demander à votre médecin combien de temps après l’accouchement vous pouvez commencer à prendre TRIAFEMI;
· si vous avez une inflammation des veines situées sous la peau (thrombophlébite superficielle) ;
· si vous avez des varices.
· si vous développez des symptômes d’angio-oedème, tels qu’un gonflement du visage, de la langue et/ou de la gorge et/ou des difficultés à avaler ou un urticaire avec éventuellement des difficultés à respirer, consultez immédiatement votre médecin. Les produits contenant des œstrogènes peuvent déclencher ou aggraver les symptômes d’un angio-oedème héréditaire ou acquis
· si vous souffrez d'otosclérose (perte auditive) ;
· si vous avez ou avez déjà eu un chloasma (taches pigmentées cutanées jaunes-marrons, essentiellement sur le visage ou le cou, aussi appelée masque de grossesse). Dans ce cas, évitez l’exposition directe au soleil et aux rayons ultraviolets ;
· si vous présentez une éruption cutanée accompagnée de cloques pendant la grossesse (herpès gestationnel) survenue la première fois pendant une grossesse ;
· si vous avez (ou avez déjà eu) des calculs biliaires ou une inflammation de la vésicule biliaire ;
· si vous avez une maladie du sang appelée porphyrie (augmentation de la sécrétion de pigment sanguin) ;
· si vous souffrez d’épilepsie ;
· si vous avez une maladie des nerfs dans laquelle se produisent des mouvements brusques du corps (chorée de Sydenham) ;
· si un parent proche a ou a déjà eu un cancer du sein;
· si vous avez une dépression;
· certaines femmes qui utilisent des contraceptifs hormonaux dont TRIAFEMI ont fait état d’une dépression ou d’un état dépressif. La dépression peut être grave et peut parfois donner lieu à des idées suicidaires. Si vous présentez des changements d’humeur et des symptômes dépressifs, sollicitez les conseils de votre médecin dès que possible.
· si vous avez une maladie du foie.
CAILLOTS SANGUINS
L’utilisation d’un contraceptif hormonal combiné tel que TRIAFEMI augmente le risque d’apparition d’un caillot sanguin en comparaison à une non-utilisation. Dans de rares cas, un caillot sanguin peut bloquer des vaisseaux sanguins et provoquer de graves problèmes.
Les caillots sanguins peuvent se former :
· dans les veines (on parle alors de « thrombose veineuse » ou de « thrombo-embolie veineuse » [TEV]) ;
· dans les artères (on parle alors de « thrombose artérielle » ou de « thrombo-embolie artérielle » [TEA]).
Le rétablissement, suite à des caillots sanguins, n’est pas toujours complet. Dans de rares cas, ils peuvent entraîner des séquelles graves et durables et, dans de très rares cas, ils peuvent être fatals.
Il est important de garder à l’esprit que le risque global de caillot sanguin dû à TRIAFEMI est faible.
COMMENT RECONNAÎTRE UN CAILLOT SANGUIN
Consultez un médecin de toute urgence si vous remarquez l’un des signes ou symptômes suivants.
|
Présentez-vous l’un de ces signes ? |
Il peut éventuellement s’agir de : |
|
· Gonflement d’une jambe ou le long d’une veine de la jambe ou du pied, en particulier s’il s’accompagne de : o douleur ou sensibilité dans la jambe, pouvant n’être ressentie qu’en position debout ou lors de la marche ; o chaleur dans la jambe affectée ; o changement de couleur de la peau de la jambe, devenant p. ex. pâle, rouge ou bleue. |
Thrombose veineuse profonde. |
|
· apparition soudaine et inexpliquée d’un essoufflement ou d’une respiration rapide ; · toux soudaine sans cause apparente, avec parfois des crachats de sang ; · douleur aiguë dans la poitrine, qui peut s’accentuer en cas de respiration profonde ; · étourdissements ou sensations vertigineuses sévères ; · battements de cœur rapides ou irréguliers ; · douleur intense dans l’estomac. En cas de doute, consultez un médecin car certains de ces symptômes, comme la toux ou l’essoufflement, peuvent être pris à tort pour les signes d’une maladie moins sévère telle qu’une infection respiratoire (p.ex. un simple rhume). |
Embolie pulmonaire. |
|
Symptômes apparaissant le plus souvent dans un seul œil : · perte immédiate de la vision ou vision trouble sans douleur pouvant évoluer vers une perte de la vision. |
Thrombose veineuse rétinienne (caillot sanguin dans l’œil). |
|
· douleur, gêne, pression, lourdeur dans la poitrine ; · sensation d’oppression ou d’encombrement dans la poitrine, le bras ou derrière le sternum ; · sensation d’encombrement, d’indigestion ou de suffocation ; · sensation de gêne dans le haut du corps irradiant vers le dos, la mâchoire, la gorge, le bras et l’estomac ; · transpiration, nausées, vomissements ou sensations vertigineuses ; · faiblesse, anxiété ou essoufflement extrêmes ; · battements de cœur rapides ou irréguliers. |
Crise cardiaque. |
|
· apparition soudaine d’une faiblesse ou d’un engourdissement au niveau du visage, d’un bras ou d’une jambe, en particulier d’un côté du corps ; · apparition soudaine d’une confusion, de difficultés à parler ou à comprendre ; · apparition soudaine de difficultés à voir d’un œil ou des deux yeux ; · apparition soudaine de difficultés à marcher, de sensations vertigineuses, d’une perte d’équilibre ou de coordination ; · maux de tête soudains, sévères ou prolongés, sans cause connue ; · perte de conscience ou évanouissement avec ou sans crise convulsive. · Parfois, les symptômes de l’AVC peuvent être de courte durée, avec un rétablissement presque immédiat et complet, mais vous devez tout de même consulter un médecin de toute urgence car vous pourriez être exposée au risque d’un nouvel AVC. |
Accident vasculaire cérébral (AVC). |
|
· gonflement et coloration légèrement bleutée d’une extrémité ; · douleur intense dans l’estomac (« abdomen aigu »). |
Caillots sanguins bloquant d’autres vaisseaux sanguins. |
CAILLOTS SANGUINS DANS UNE VEINE
Que peut-il se passer si un caillot sanguin se forme dans une veine ?
· Un lien a été établi entre l’utilisation de contraceptifs hormonaux combinés et l’augmentation du risque de caillots sanguins dans les veines (thrombose veineuse). Cependant, ces effets indésirables sont rares. Le plus souvent, ils surviennent pendant la première année d’utilisation d’un contraceptif hormonal combiné.
· Lorsqu’un caillot sanguin se forme dans une veine d’une jambe ou d’un pied, il peut provoquer une thrombose veineuse profonde (TVP).
· Si le caillot sanguin migre de la jambe vers le poumon, il peut provoquer une embolie pulmonaire.
· Dans de très rares cas, un caillot peut se former dans une veine d’un autre organe, comme l’œil (thrombose veineuse rétinienne).
À quel moment le risque d’apparition d’un caillot sanguin dans une veine est-il le plus élevé ?
Le risque d’apparition d’un caillot sanguin dans une veine est maximal pendant la première année d’utilisation d’un contraceptif hormonal combiné pris pour la première fois. Le risque peut également être augmenté lorsque vous recommencez à prendre un contraceptif hormonal combiné (le même produit ou un produit différent) après une interruption de 4 semaines ou plus.
Après la première année, le risque diminue mais reste toujours légèrement plus élevé que si vous n’utilisiez pas de contraceptif hormonal combiné.
Lorsque vous arrêtez de prendre TRIAFEMI, le risque d’apparition de caillot sanguin revient à la normale en quelques semaines.
Quel est le risque d’apparition d’un caillot sanguin ?
Le risque dépend de votre risque de base de TEV et du type de contraceptif hormonal combiné que vous prenez.
Le risque global de caillot sanguin dans une jambe ou un poumon (TVP ou EP) associé à TRIAFEMI, comprimé est faible.
· Sur 10 000 femmes qui n’utilisent aucun contraceptif hormonal combiné et qui ne sont pas enceintes, environ 2 développeront un caillot sanguin sur une période d’un an.
· Sur 10 000 femmes qui utilisent un contraceptif hormonal combiné contenant du lévonorgestrel ou de la noréthistérone, ou du norgestimate comme TRIAFEMI, environ 5 à 7 développeront un caillot sanguin sur une période d’un an.
· Le risque d’apparition d’un caillot sanguin variera selon vos antécédents médicaux personnels (voir « Facteurs augmentant le risque de caillot sanguin » ci-dessous).
|
|
Risque d’apparition d’un caillot sanguin sur une période d’un an |
|
Femmes qui n’utilisent pas de contraceptif hormonal combiné (pilule/patch/anneau) et qui ne sont pas enceintes |
Environ 2 femmes sur 10 000 |
|
Femmes qui utilisent une pilule contraceptive hormonale combinée contenant du lévonorgestrel, de la noréthistérone ou du norgestimate |
Environ 5 à 7 femmes sur 10 000 |
|
Femmes qui utilisent TRIAFEMI |
Environ 5 à 7 femmes sur 10 000 |
Facteurs augmentant le risque de caillot sanguin dans une veine
Le risque de caillot sanguin associé à TRIAFEMI, comprimé est faible mais certaines situations peuvent augmenter ce risque. Le risque sera plus élevé :
· si vous avez un surpoids important (indice de masse corporelle [IMC] supérieur à 30 kg/m2) ;
· si l’un des membres de votre famille proche a eu un caillot sanguin dans une jambe, un poumon ou un autre organe à un âge relativement jeune (p. ex. avant l’âge de 50 ans). Si tel est le cas, vous pourriez être atteinte d’un trouble héréditaire de la coagulation sanguine ;
· si vous devez être opérée ou si vous êtes alitée pendant une période prolongée en raison d’une blessure ou d’une maladie, ou si votre jambe est immobilisée (p. ex. plâtre). Il pourra être nécessaire d’interrompre l’utilisation de TRIAFEMI plusieurs semaines avant l’opération chirurgicale et/ou tant que votre mobilité est réduite. Si vous devez arrêter d’utiliser TRIAFEMI, demandez à votre médecin ou votre sage-femme à quel moment vous pourrez recommencer à l’utiliser ;
· avec l’âge (en particulier au-delà de 35 ans) ;
· si vous avez accouché dans les semaines précédentes.
Plus vous cumulez ces situations, plus le risque d’apparition d’un caillot sanguin augmente.
Les voyages en avion (de plus de 4 heures) peuvent augmenter temporairement le risque de caillot sanguin, en particulier si vous présentez déjà certains des autres facteurs listés.
Il est important de prévenir votre médecin ou votre sage-femme si vous êtes concernée par l’une de ces situations, même si vous n’en êtes pas certaine. Votre médecin ou votre sage-femme pourra décider s’il est nécessaire d’arrêter le traitement par TRIAFEMI.
Si l’une des situations ci-dessus évolue pendant la période où vous utilisez TRIAFEMI, par exemple si un membre de votre famille proche développe une thrombose sans raison connue ou si vous prenez beaucoup de poids, parlez-en à votre médecin ou votre sage-femme.
CAILLOTS SANGUINS DANS UNE ARTÈRE
Que peut-il se passer si un caillot sanguin se forme dans une artère ?
Comme un caillot sanguin dans une veine, un caillot dans une artère peut provoquer de graves problèmes. Par exemple, il peut provoquer une crise cardiaque ou un accident vasculaire cérébral (AVC).
Facteurs augmentant le risque de caillot sanguin dans une artère
Il est important de noter que le risque de crise cardiaque ou d’AVC lié à l’utilisation de TRIAFEMI, comprimé est très faible mais peut augmenter :
· avec l’âge (au-delà de 35 ans) ;
· si vous fumez. Lors de l’utilisation d’un contraceptif hormonal combiné tel que TRIAFEMI, comprimé, il est conseillé d’arrêter de fumer. Si vous ne parvenez pas à arrêter de fumer et si vous êtes âgée de plus de 35 ans, votre médecin ou votre sage-femme pourra vous conseiller d’utiliser une méthode de contraception différente ;
· si vous êtes en surpoids ;
· si vous avez une pression artérielle élevée ;
· si un membre de votre famille proche a déjà eu une crise cardiaque ou un AVC à un âge relativement jeune (avant l’âge de 50 ans). Si tel est le cas, le risque que vous ayez une crise cardiaque ou un AVC pourrait également être plus élevé ;
· si vous, ou un membre de votre famille proche, avez un taux de graisses élevé dans le sang (cholestérol ou triglycérides) ;
· si vous avez des migraines, en particulier des migraines avec aura ;
· si vous avez des problèmes cardiaques (maladie de la valve cardiaque, trouble du rythme appelé fibrillation auriculaire) ;
· si vous êtes diabétique.
Si vous cumulez plusieurs de ces situations ou si l’une d’entre elles est particulièrement sévère, le risque d’apparition d’un caillot sanguin peut être encore plus élevé.
Si l’une des situations ci-dessus évolue pendant la période où vous utilisez TRIAFEMI, comprimé, par exemple si vous commencez à fumer, si un membre de votre famille proche développe une thrombose sans raison connue ou si vous prenez beaucoup de poids, parlez-en à votre médecin ou votre sage-femme.
TRIAFEMI, comprimé et cancer
Le risque de cancer du sein augmente avec l’âge. Le risque de cancer de sein augmente légèrement avec les contraceptifs oraux. Comparé au risque de cancer du sein à un moment donné de la vie, le risque accru associé à l’utilisation de contraceptifs oraux est faible. L’excès de risque du cancer de sein diminue progressivement au cours des 10 années qui suivent l’arrêt de l’utilisation des contraceptifs oraux. Les cancers du sein diagnostiqués chez les femmes utilisant des contraceptifs oraux n’étaient pas à un stade aussi avancé que chez les non utilisatrices.. Il est important de régulièrement vérifier vos seins et vous devez contacter votre médecin si vous ressentez la moindre grosseur.
Dans de rares cas, des tumeurs hépatiques bénignes, et dans des cas encore plus rares des tumeurs hépatiques malignes ont été rapportées chez les utilisatrices de pilules. Contactez votre médecin si vous ressentez une douleur abdominale sévère et inhabituelle.
Le cancer du col de l'utérus est un peu plus fréquent lorsque les contraceptifs oraux sont utilisés depuis longtemps. Ce n’est pas nécessairement dû à l’utilisation de contraceptifs oraux et cela peut être lié au comportement sexuel ou à d’autres facteurs.
Saignements intermenstruels
Pendant les premiers mois où vous prenez TRIAFEMI, vous pouvez avoir des pertes de sang inattendues (saignements en dehors de la semaine sans prise). Si ces saignements persistent pendant plus de quelques mois, ou s’ils débutent après quelques mois, votre médecin ou votre sage-femme doit en rechercher la cause.
Que faire si aucun saignement ne se produit pendant la semaine sans prise
Si vous avez pris correctement tous les comprimés, et n’avez pas eu de vomissements ni de forte diarrhée et si vous n’avez pris aucun autre médicament, il est très improbable que vous soyez enceinte.
Si l’hémorragie de privation ne survient pas deux mois de suite, vous pourriez être enceinte. Contactez immédiatement votre médecin ou votre sage-femme. Ne commencez pas la plaquette suivante jusqu’à ce que vous soyez sûre que vous n’êtes pas enceinte.
Le taux sanguin de folate peut être plus faible chez les femmes qui utilisent des contraceptifs oraux. Cela peut être important pour les femmes qui tombent enceintes peu après l’arrêt des contraceptifs oraux.
Enfants et adolescents
Sans objet.
Autres médicaments et TRIAFEMI, comprimé
|
Informez toujours votre médecin des médicaments ou des produits à base de plantes que vous utilisez déjà. Indiquez également à tout autre médecin ou sage-femme qui vous prescrit un autre médicament (ou au pharmacien) que vous prenez TRIAFEMI. Ils peuvent vous dire si vous devez prendre des précautions contraceptives supplémentaires (par exemple des préservatifs) et, dans l'affirmative, pendant combien de temps ou si l'utilisation d'un autre médicament dont vous avez besoin doit être modifiée. |
Certains médicaments et des produits à base de plantes
· peuvent avoir une influence sur les taux sanguins de TRIAFEMI
· peuvent le rendre moins efficace pour prévenir une grossesse
· peuvent provoquer des saignements inattendus.
Ceux-ci inclus :
· les médicaments utilisés pour le traitement :
o de l’épilepsie (par ex. hydantoïnes, primidone, phénytoïne, barbituriques, carbamazépine, oxcarbazépine, topiramate et felbamate);
o de la tuberculose (par ex. rifampicine);
o des infections par le VIH et par le virus de l'hépatite C (appelés aussi inhibiteurs de la protéase et inhibiteurs non nucléosidiques de la transcriptase inverse, tels que le ritonavir, la névirapine, l'efavirenz, atazanavir, indinavir, etravirine)
o des infections fongiques (par ex. griséofulvine);
o de l’arthrite, arthrose (étoricoxib);
o de l’hypertension artérielle pulmonaire (bosentan);
o les remèdes à base de millepertuis.
TRIAFEMI peut influencer l’effet d’autres médicaments, par ex. :
· les médicaments contenant de la ciclosporine (un immunosuppresseur empêchant le rejet de greffe après une transplantation);
· l’antiépileptique lamotrigine (ce qui pourrait entraîner une augmentation de la fréquence des convulsions);
· la théophylline (utilisée pour traiter les problèmes respiratoires);
· la tizanidine (utilisée pour traiter les douleurs musculaires et/ou les crampes musculaires);
Ne prenez pas TRIAFEMI si vous avez une hépatite C et prenez les médicaments contenant de l’ombitasvir/paritaprévir/ritonavir et dasabuvir ou glécaprévir/pribentasvir ou sofosbuvir/velpatasvir/voxilaprévir car ces produits pourraient entraîner des élévations aux tests sanguins de la fonction hépatique (élévation de l’enzyme hépatique ALAT).
Votre médecin ou votre sage-femme prescrira une autre méthode de contraception avant le début du traitement avec ces médicaments.
TRIAFEMI peut être recommencé environ 2 semaines après la fin de ce traitement. Voir rubrique « Ne prenez jamais TRIAFEMI ».
Analyse biologique
Si vous devez faire une analyse de sang, dites à votre médecin ou au personnel du laboratoire que vous prenez la pilule, car les contraceptifs hormonaux peuvent perturber les résultats de certains tests.
TRIAFEMI avec des aliments, boissons et de l’alcool
TRIAFEMI peut être pris avec ou sans nourriture, éventuellement avec une petite quantité d’eau.
Grossesse, allaitement et fertilité
Grossesse
Si vous êtes enceinte, vous ne devez pas prendre TRIAFEMI. Si vous tombez enceinte alors que vous prenez TRIAFEMI, vous devez arrêter immédiatement et contacter votre médecin ou votre sage-femme.
Si vous souhaitez être enceinte, vous pouvez arrêter de prendre TRIAFEMI à tout moment (voir également « Si vous arrêtez de prendre TRIAFEMI »).
Demandez conseil à votre médecin ou à votre pharmacien avant de prendre tout médicament.
Allaitement
Prendre TRIAFEMI n’est généralement pas conseillé pendant l’allaitement. Si vous souhaitez prendre la pilule alors que vous allaitez, veuillez consulter votre médecin ou votre sage-femme.
Demandez conseil à votre médecin, ou à votre sage-femme ou à votre pharmacien avant de prendre tout médicament.
Fertilité
TRIAFEMI est indiqué pour prévenir une grossesse.
Conduite de véhicules et utilisation de machines
Il n’existe pas de données suggérant que l’utilisation de TRIAFEMI influence la capacité à conduire un véhicule ou à utiliser une machine.
TRIAFEMI, comprimé contient du lactose.
Si votre médecin vous a informée d’une intolérance à certains sucres, contactez-le avant de prendre ce médicament.
3. COMMENT PRENDRE TRIAFEMI, comprimé ?
Veillez à toujours prendre ce médicament en suivant exactement les indications de votre médecin, votre sage-femme ou de votre pharmacien. Vérifiez auprès de votre médecin, de votre sage-femme ou de votre pharmacien en cas de doute.
Posologie
Le 1er jour d’utilisation de votre plaquette, collez l’étiquette* du jour de la semaine correspondant et prenez le comprimé numéro 1. Les jours suivants prenez les comprimés environ à la même heure de la journée, dans l’ordre chronologique jusqu’au 21ème jour.
Ensuite, vous ne prendrez plus de comprimés pendant 7 jours. Une hémorragie de privation doit apparaître pendant cet intervalle, généralement le 2ème ou le 3ème jour après la prise du dernier comprimé.
Commencez la plaquette suivante dès le 8ème jour, même si l’hémorragie de privation persiste. Vous devez toujours commencer la plaquette suivante le même jour de la semaine.
*Les étiquettes sont disponibles dans votre boite.

Mode d’administration
Voie orale.
Prendre régulièrement et sans oubli 1 comprimé par jour au même moment de la journée (celui où vous risquez le moins d'oublier) avec ou sans nourriture, si nécessaire avec une petite quantité d'eau et sans mâcher, pendant 21 jours consécutifs avec un arrêt de 7 jours entre deux plaquettes. L’effet contraceptif commence dès la prise du premier comprimé.
Après avoir pris les 21 comprimés de la plaquette, vous avez un intervalle de 7 jours sans prise de comprimés. Si vous avez pris le dernier comprimé de la première plaquette un vendredi, vous prendrez le premier comprimé de votre plaquette suivante le samedi de la semaine suivante.
Deux ou trois jours après l’arrêt du dernier comprimé de la plaquette, vous devriez avoir une hémorragie de privation similaire à des règles. Cette hémorragie de privation peut ne pas être terminée au moment de commencer la plaquette suivante de comprimés. Vous n’avez pas besoin d’utiliser d’autre méthode contraceptive pendant ces 7 jours d’absence de prise de comprimés, à condition que vous ayez pris correctement vos comprimés et démarré la prochaine plaquette au bon moment.
Puis commencer la plaquette suivante
Commencer une nouvelle plaquette après un intervalle de 7 jours sans comprimés.
Si vous utilisez TRIAFEMI de cette façon, vous serez ainsi protégée contre une grossesse pendant les 7 jours où vous ne prenez pas de comprimés.
Quand devez-vous commencer la plaquette
· Début de contraception orale : Si vous n’avez pas utilisé de contraceptif avec des hormones au cours du mois précédent
1er comprimé à prendre le 1er jour des règles.
Commencez TRIAFEMI le premier jour du cycle c'est-à-dire le premier jour des règles. Si vous commencez TRIAFEMI le premier jour des règles, vous êtes immédiatement protégée. Vous pouvez aussi commencer TRIAFEMI entre le 2ème et le 5ème jour de votre cycle, mais alors vous devrez utiliser une autre méthode de contraception, comme les préservatifs, pendant les 7 premiers jours.
· Si vous passez d’une contraception hormonale combinée, ou anneau vaginal hormonal combiné ou dispositif transdermique hormonal combiné
Vous pouvez commencer TRIAFEMI de préférence le lendemain de la prise du dernier comprimé actif du contraceptif oral combiné précédent ou au plus tard le jour suivant la période habituelle d’arrêt des comprimés (ou encore après le dernier comprimé inactif du contraceptif oral combiné précédent). Pour prendre le relais d’un anneau vaginal ou d’un dispositif transdermique, suivez l’avis de votre médecin.
· Si vous passez d’une méthode uniquement progestative (pilule uniquement progestative, injectable, implant à progestatif seul, ou dispositif intra-utérin libérant un progestatif)
Vous pouvez prendre le relais de la pilule microdosée le jour que vous souhaitez. Vous pouvez prendre le relais d’un implant ou d’un dispositif intra-utérin le jour du retrait. Vous pouvez prendre le relais d’un contraceptif injectable le jour où la nouvelle injection était prévue). Dans tous ces cas vous devez utiliser une autre méthode de contraception, comme les préservatifs, pendant les 7 premiers jours.
· Après une fausse couche
Demandez conseil à votre médecin ou votre sage-femme.
· Après avoir eu un bébé
Commencez à prendre TRIAFEMI entre le 21ème et le 28ème jour après l’accouchement. Si vous commencez après le 28ème jour, vous devrez utiliser une autre méthode de contraception, comme les préservatifs, pendant les 7 premiers jours. Toutefois, si vous avez déjà eu des rapports sexuels, il faut écarter toute probabilité de grossesse avant de commencer à utiliser TRIAFEMI ou attendre vos premières règles.
· Si vous allaitez ou que vous souhaitez commencer TRIAFEMI après la naissance du bébé
Veuillez lire la rubrique « Allaitement ».
Suivez les indications de votre médecin si vous ne savez pas quand démarrer votre contraception.
Durée du traitement
Selon l’avis médical.
Le traitement devra être poursuivi plusieurs mois. Les premiers signes d'amélioration de l’acné se manifestent au bout de 2 mois parfois plus.
Si vous avez pris plus de TRIAFEMI que vous n’auriez dû
Il n’a pas de signalement d’effets indésirables graves en cas de prise excessive de TRIAFEMI. Si vous prenez plusieurs comprimés à la fois alors vous pouvez avoir des symptômes de nausées ou vomissements. Les jeunes filles peuvent avoir des saignements vaginaux.
Si vous avez pris trop de comprimés de TRIAFEMI, ou vous découvrez qu’un enfant en a pris, prenez immédiatement contact avec votre médecin, votre pharmacien, un hôpital ou un centre anti-poison notamment en cas de prise accidentelle par un enfant, pour évaluer les risques et être informée de ce qu’il faut faire.
Si vous oubliez de prendre TRIAFEMI
Si l'oubli est constaté dans les 12 heures qui suivent l'heure habituelle de la prise, l'effet contraceptif n'est pas diminué. Prenez immédiatement le comprimé oublié, et poursuivez le traitement normalement en prenant le comprimé suivant à l'heure habituelle.
Si l'oubli est constaté plus de 12 heures après l'heure habituelle de la prise, il existe un risque de grossesse. Plus le nombre de comprimés oubliés est important, plus le risque de grossesse est important.
Le risque de grossesse est particulièrement important si vous oubliez de prendre des comprimés au début ou à la fin de la plaquette. Par conséquent, vous devez respecter les règles suivantes (voir aussi le schéma ci-dessous) :
· Plus d’1 comprimé oublié dans cette plaquette
Consultez votre médecin.
· Un comprimé oublié entre les jours 1 à 7 (première rangée)
Prenez le comprimé oublié dès que vous vous en souvenez, même si cela signifie que vous devez prendre deux comprimés en même temps. Continuez à prendre les comprimés à l’heure habituelle et utilisez des précautions supplémentaires les 7 jours suivants, par exemple un préservatif. Si vous avez eu des relations sexuelles la semaine précédant l’oubli du comprimé, vous pouvez être enceinte. Dans ce cas, veuillez consulter votre médecin.
· Un comprimé oublié entre les jours 8 à 14 (seconde rangée)
Prenez le comprimé oublié dès que vous vous en souvenez, même si cela signifie que vous devez prendre deux comprimés en même temps Continuez à prendre les comprimés à l’heure habituelle. La protection contre une grossesse n’est pas réduite et vous n’avez pas besoin de prendre des précautions supplémentaires.
· Un comprimé oublié entre les jours 15 à 21 (troisième rangée)
Vous pouvoir choisir entre deux options :
1. Prenez le comprimé oublié dès que vous vous en souvenez, même si cela signifie que vous devez prendre deux comprimés en même temps. Continuez à prendre les comprimés à l’heure habituelle. Au lieu de suivre l’intervalle sans comprimés commencez la plaquette suivante.
Très probablement, vous aurez des règles à la fin de la seconde plaquette mais vous pouvez avoir des saignements légers ou comme des règles pendant la seconde plaquette.
2. Vous pouvez aussi arrêter la plaquette et passer directement à l’intervalle sans comprimés de 7 jours (noter le jour où vous avez oublié votre comprimé). Si vous voulez commencer une nouvelle plaquette le jour où vous commencez habituellement, respectez un intervalle sans comprimés de moins de 7 jours.
En suivant une de ces recommandations, vous resterez protégée contre une grossesse.
Si vous avez oublié un comprimé d’une plaquette et que vous n’avez pas de règles pendant l’intervalle sans comprimés, vous pouvez être enceinte. Vous devez contacter votre médecin avant de commencer la plaquette suivante.
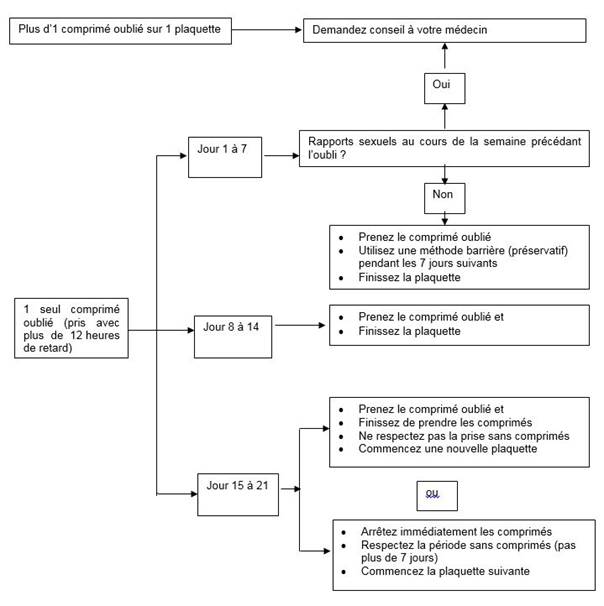
En cas de vomissements ou de diarrhée sévère
Si vous vomissez dans les 3 ou 4 heures qui suivent la prise d’un comprimé ou si vous avez une diarrhée sévère, l’absorption du comprimé ne peut pas être complète. La situation est identique à un oubli de comprimé. Après des vomissements ou une diarrhée, prenez un autre comprimé d’une plaquette de réserve dès que possible et si possible dans les 12 heures qui suivent l’heure habituelle de prise. Si vous ne pouvez pas respecter de délai ou si plus de 12 heures se sont écoulées, les mêmes consignes que celles données pour les oublis de comprimés de plus de 12 heures s’appliquent. Reportez-vous au paragraphe « Si vous oubliez de prendre TRIAFEMI ».
Retarder vos règles : ce que vous devez savoir
Même si ce n’est pas recommandé, vous pouvez retarder vos règles en commençant immédiatement une nouvelle plaquette de TRIAFEMI sans débuter ou finir la période sans comprimés.
Pendant la prise des comprimés de la deuxième plaquette, des petits saignements ou des spottings peuvent survenir.
Commencez ensuite à prendre les comprimés de la plaquette suivante après la période habituelle des 7 jours sans comprimés.
Vous pouvez demander conseil à votre médecin ou votre sage-femme avant de décider de retarder vos règles.
Changer le premier jour de vos règles : ce que vous devez savoir
Si vous prenez les comprimés conformément aux instructions, vos règles commenceront pendant la semaine sans comprimés. Si vous souhaitez changer ce jour, réduisez le nombre de jours sans comprimés (mais ne les augmentez jamais, 7 est le maximum).
Par exemple, si vos journées sans comprimés commencent normalement un vendredi et que vous souhaitez le remplacer par un mardi (3 jours plus tôt), démarrez une nouvelle plaquette 3 jours plus tôt que d’habitude.
Si l'intervalle sans comprimés est très court (par exemple, 3 jours ou moins), il est possible que vous ne saigniez pas pendant ces jours. Vous pouvez alors avoir des saignements légers ou similaires à ceux de la menstruation.
Si vous ne savez pas comment procéder, contactez votre médecin pour obtenir des conseils.
Si vous arrêtez de prendre TRIAFEMI
Vous pouvez arrêter de prendre TRIAFEMI quand vous le souhaitez.
Si vous ne souhaitez pas tomber enceinte, demandez conseil à votre médecin ou votre sage-femme sur d’autres méthodes de contraception plus sures.
Si vous souhaitez être enceinte, arrêtez de prendre TRIAFEMI et attendez pendant un temps avant d’essayer. Il vous sera ainsi plus facile de calculer la date attendue de délivrance.
Si vous avez d’autres questions sur l’utilisation de ce médicament, demandez conseil à votre médecin, votre sage-femme ou votre pharmacien.
4. QUELS SONT LES EFFETS INDESIRABLES EVENTUELS ?
Il existe une augmentation du risque de caillots sanguins dans les veines (thrombo-embolie veineuse [TEV]) ou dans les artères (thrombo-embolie artérielle [TEA]) chez toutes les femmes qui prennent des contraceptifs hormonaux combinés. Pour plus de précisions sur les différents risques associés à la prise de contraceptifs hormonaux combinés, reportez-vous à la rubrique 2, « Quelles sont les informations à connaître avant d’utiliser TRIAFEMI, comprimé »
Les effets indésirables suivants ont été décrits avec l’usage de TRIAFEMI :
Très fréquent : concerne plus de 1 femme sur 10
· Maux de tête (mais si ceux-ci sont inhabituels ou de longue durée, consulter un médecin le plus rapidement possible)
· Problème d’estomac comme des maux de ventre, nausée, vomissement, des diarrhées
· Saignements et spottings entre les menstruations pendant les premiers mois (bien que cela cesse habituellement lorsque votre corps s'adapte à TRIAFEMI - voir section 2, les saignements entre les règles ne devraient pas durer longtemps.
· Dysménorrhées et métrorragies.
Fréquent : concerne jusqu’à 1 femme sur 10
· Infections des voies urinaires
· Infections vaginales
· Dépression, des changements d'humeur, se sentir nerveux
· Migraine (consultez un médecin dès que possible s'il s'agit de votre première migraine ou si elle est pire que d'habitude)
· l'acné, rash cutané
· Réactions allergiques (hypersensibilité)
· Rétention hydrique
· Seins douloureux
· Douleur dans la poitrine
· Spasmes musculaires, douleur dans les jambes, les bras et le dos
· Absence de règles
· Prise de poids
· Sensation de fatigue
· Etourdissements
· Maux d'estomac et ballonnements, constipation, flatulence
· Mains, chevilles ou pieds enflés
· Difficulté à dormir (insomnie)
Peu fréquent : concerne jusqu’à 1 femme sur 100
· Problèmes mammaires, tels que des seins plus volumineux,; produisant du liquide à partir des mamelons
· Cellules anormales dans le col de l'utérus (identifiées par un frottis)
· Etat anxieux ou se sentir s’évanouir
· Sensation de picotement ou d'engourdissement
· Changements de couleur de peau
· Problèmes de peau tels que rougeurs et démangeaisons ou décoloration
· Amincissement des cheveux (alopécie), croissance excessive des cheveux
· Changements dans l'appétit, le poids peut varier, perte de poids
· Changement de libido
· Yeux secs
· Changements de vision
· Palpitations (sentir votre cœur battre)
· Bouffées de chaleur
· Douleur musculaire
· Sécheresse vaginale et vulvaire
· Kystes ovariens (peuvent causer des douleurs et un gonflement de l'abdomen, des changements de règles).
· Hypertension artérielle
· Essoufflement
Rare : concerne jusqu’à une femme sur 1000
· Seins grumeleux
· Avoir le vertige
· Battements du cœur plus rapides
· Pancréatite (inflammation du pancréas causant de vives douleurs à l'abdomen et au dos)
· Transpiration accrue
· Sensibilité à la lumière.
· Hépatite (inflammation du foie causant de vives douleurs à l'abdomen et au dos)
· Pertes vaginales (modification du liquide vaginal)
Fréquence indéterminée : la fréquence ne peut être déterminée à partir des données disponibles
· Quantité réduite de lait maternel (si vous allaitez)
· Les lentilles de contact peuvent être inconfortables
· Gonflement grumeleux douloureux rouge sur les jambes
· Modifications des taux de graisse dans le sang (constatées par des tests sanguins)
· Sueurs nocturnes
· Adénomes hépatiques (tumeurs bénignes du foie généralement induites par une hormone)
· Cancer du sein
· Tumeurs bénignes du sein
· Hyperplasie nodulaire focale (tumeur bénigne)
· Fibroadénome du sein
· Accident vasculaire cérébral (AVC)
· Contractions anormales violentes et involontaires ou série de contractions musculaires (convulsions)
· Attaque cardiaque
· Angioœdème (gonflement dans les couches profondes de la peau).
Les effets indésirables graves suivants ont été signalés un peu plus souvent chez les femmes utilisant des pilules contraceptives (voir rubrique 2 « Dans quels cas devez-vous faire attention avec TRIAFEMI, comprimé ? »)
· Pression artérielle élevée
· Tumeurs du foie ou cancer du sein
· Perturbations de la fonction hépatique
· Des caillots sanguins nocifs dans une veine ou une artère, par exemple :
o dans une jambe ou un pied (à savoirThrombose veineuse profonde TVP)
o dans un poumon (à savoir embolie pulmonaire EP)
o crise cardiaque
o accident vasculaire cérébral
o mini-accident vasculaire cérébral ou symptômes ressemblant à un accident vasculaire cérébral temporaire, appelé attaque ischémique transitoire (AIT) ou caillots sanguins dans le foie, l'estomac /l'intestin, les reins ou les yeux.
Le risque d'avoir un caillot sanguin peut être plus élevé si d'autres conditions augmentent ce risque (voir rubrique 2 pour plus d'informations sur les conditions augmentant le risque de caillot sanguin et les symptômes d'un caillot sanguin).
Les conditions suivantes peuvent survenir ou s'aggraver avec les contraceptifs oraux combinés: maladie de Crohn, colite ulcéreuse, épilepsie, myome utérin, porphyrie (trouble du métabolisme entraînant des douleurs abdominales et des troubles mentaux), lupus érythémateux disséminé (le corps attaque ses organes et tissus), l'herpès en fin de grossesse, la chorée de Sydenham (mouvements saccadés rapides), le syndrome hémolytique et urémique (une affection qui survient après une diarrhée causée par E. coli), des problèmes de foie révélés par la jaunisse, des troubles de la vésicule biliaire ou la formation de calculs biliaires.
o Consultez immédiatement un médecin, si vous développez l’un des symptômes d’angio-œdème suivants : gonflement du visage, de la langue et/ou de la gorge et/ou des difficultés à avaler ou un urticaire avec éventuellement des difficultés à respirer (voir aussi « Avertissements et précautions »).
Déclaration des effets secondaires
Si vous ressentez un quelconque effet indésirable, parlez-en à votre médecin, votre sage-femme ou votre pharmacien. Ceci s’applique aussi à tout effet indésirable qui ne serait pas mentionné dans cette notice. Vous pouvez également déclarer les effets indésirables directement via le système national de déclaration : Agence nationale de sécurité du médicament et des produits de santé (ANSM) et réseau des Centres Régionaux de Pharmacovigilance - Site internet: www.signalement-sante.gouv.fr
En signalant les effets indésirables, vous contribuez à fournir davantage d’informations sur la sécurité du médicament.
5. COMMENT CONSERVER TRIAFEMI, comprimé ?
Tenir ce médicament hors de la vue et de la portée des enfants.
N’utilisez pas ce médicament après la date de péremption indiquée sur la boîte.
A conserver à une température ne dépassant pas +25°C.
N’utilisez pas ce médicament si vous remarquez des signes visibles de détérioration.
Ne jetez aucun médicament au tout à l’égout ou avec les ordures ménagères. Demandez à votre pharmacien d’éliminer les médicaments que vous n’utilisez plus. Ces mesures contribueront à protéger l’environnement.
6. CONTENU DE L’EMBALLAGE ET AUTRES INFORMATIONS
Ce que contient TRIAFEMI, comprimé
· Les substances actives sont :
Norgestimate ................................................................................................................... 0,180 mg
Ethinylestradiol ................................................................................................................ 0,035 mg
pour un comprimé blanc
Norgestimate .................................................................................................................. 0,215 mg
Ethinylestradiol ................................................................................................................ 0,035 mg
pour un comprimé bleu ciel
Norgestimate ................................................................................................................... 0,250 mg
Ethinylestradiol ................................................................................................................ 0,035 mg
pour un comprimé bleu foncé
· Les autres composants sont :
· Comprimé blanc : lactose anhydre, amidon prégélatinisé, crospovidone, stéarate de magnésium
· Comprimé bleu ciel et bleu foncé : lactose anhydre, amidon prégélatinisé, crospovidone, stéarate de magnésium, laque aluminique d'indigotine
Qu’est-ce que TRIAFEMI, comprimé et contenu de l’emballage extérieur
Ce médicament se présente sous forme de comprimé. Boîte de 1, 3 ou 6 plaquettes de 21 comprimés.
Titulaire de l’autorisation de mise sur le marché
BATIMENT "LE NEWTON"
9/11, RUE JEANNE BRACONNIER
92366 MEUDON-LA-FORET
Exploitant de l’autorisation de mise sur le marché
BÄTIMENT « LE NEWTON »
11 RUE JEANNE BRACONNIER
92360 MEUDON
BATIMENT "LE NEWTON"
9/11, RUE JEANNE BRACONNIER
92366 MEUDON-LA-FORET
ou
LABORATOIRE DELPHARM
PARC D’ACTIVITES ROUBAIX-EST
22 RUE DE TOUFFLERS
CS 50070
59452 LYS-LEZ-LANNOY
Noms du médicament dans les Etats membres de l'Espace Economique Européen
Sans objet.
La dernière date à laquelle cette notice a été révisée est :
[à compléter ultérieurement par le titulaire]
{MM/AAAA} {mois AAAA}.
Des informations détaillées sur ce médicament sont disponibles sur le site Internet de l’ANSM (France).