Dernière mise à jour le 01/06/2026
DASATINIB EG 100 mg, comprimé pelliculé
Indications thérapeutiques
DASATINIB EG contient la substance active dasatinib. Ce traitement est utilisé pour traiter la leucémie myéloïde chronique (LMC), chez les patients adultes, adolescents et les enfants âgés d'au moins 1 an. La leucémie est un cancer touchant les globules blancs du sang. Ces cellules permettent habituellement à l'organisme de lutter contre les infections. Les personnes atteintes de LMC présentent une prolifération incontrôlée des globules blancs appelés granulocytes. DASATINIB EG permet de lutter contre la prolifération de ces cellules leucémiques.
DASATINIB EG est utilisé pour traiter les leucémies aiguës lymphoblastiques (LAL) à chromosome Philadelphie (Ph+) chez les patients adultes, les adolescents et les enfants âgés d’au moins 1 an et des LMC en phase blastique lymphoïde, chez les patients adultes pour lesquels les traitements antérieurs ne sont pas bénéfiques. La leucémie est un cancer touchant les globules blancs du sang. Ces cellules permettent habituellement à l’organisme de lutter contre les infections. Chez les personnes atteintes de LAL, les globules blancs appelés lymphocytes ont une croissance trop rapide et une durée de vie trop longue. DASATINIB EG permet de lutter contre la prolifération de ces cellules leucémiques.
Si vous avez des questions sur la manière dont agit DASATINIB EG ou sur les raisons pour lesquelles il vous a été prescrit, veuillez interroger votre médecin.
Présentations
> 30 plaquettes prédécoupées unitaires en aluminium OPA : polyamide orienté PVC de 1 comprimé
Code CIP : 34009 301 774 6 4
Déclaration de commercialisation : 11/12/2019
Cette présentation est agréée aux collectivités
- Prix hors honoraire de dispensation : 1379,01 €
- Honoraire de dispensation : 1,02 €
- Prix honoraire compris : 1380,03 €
- Taux de remboursement :100%
Service médical rendu (SMR)
Amélioration du service médical rendu (ASMR)
Ce médicament étant un générique, l'ASMR n'a pas été évalué par la commission de la transparence (CT), il est possible de se référer à la /aux spécialité(s) de référence du groupe générique auquel appartient ce médicament (cliquez ici pour aller à la rubrique des groupes génériques)
Autres informations
- Titulaire de l'autorisation : EG LABO - Laboratoires EuroGenerics
- Conditions de prescription et de délivrance :
- liste I
- médicament nécessitant une surveillance particulière pendant le traitement
- prescription initiale hospitalière semestrielle
- prescription initiale réservée à certains spécialistes
- renouvellement de la prescription réservé aux médecins compétents en CANCEROLOGIE
- renouvellement de la prescription réservé aux spécialistes en HEMATOLOGIE
- renouvellement de la prescription réservé aux spécialistes en ONCOLOGIE MEDICALE
- Statut de l'autorisation : Valide
- Type de procédure : Procédure de reconnaissance mutuelle
- Code CIS : 6 813 577 3
ANSM - Mis à jour le : 10/03/2026
DASATINIB EG 100 mg, comprimé pelliculé
2. COMPOSITION QUALITATIVE ET QUANTITATIVE
Dasatinib............................................................................................................................. 100 mg
Pour un comprimé pelliculé.
Excipients à effet notoire :
Chaque comprimé pelliculé contient 138 mg de lactose monohydraté.
Pour la liste complète des excipients, voir rubrique 6.1.
Comprimé pelliculé (comprimé).
Comprimé pelliculé blanc à blanc cassé, biconvexe, ovale, comportant la mention « 100 » gravée sur une face, l’autre face étant lisse, et mesurant 14,8 mm × 7,2 mm.
4.1. Indications thérapeutiques
DASATINIB EG est indiqué chez des patients adultes atteints de :
· Leucémie myéloïde chronique (LMC) à Chromosome Philadelphie (Ph+) en phase chronique nouvellement diagnostiquée.
· LMC en phase chronique, accélérée ou blastique en cas de résistance ou d’intolérance à un traitement antérieur incluant l'imatinib.
· Leucémie aiguë lymphoblastique (LAL) et de LMC en phase blastique lymphoïde Ph+ en cas de résistance ou intolérance à un traitement antérieur.
DASATINIB EG est indiqué chez des patients pédiatriques atteints de :
· LMC Ph+ en phase chronique (LMC Ph+ PC) nouvellement diagnostiquée ou LMC Ph+ PC en cas de résistance ou d’intolérance à un traitement antérieur incluant l'imatinib.
· Leucémie aiguë lymphoblastique (LAL Ph+) nouvellement diagnostiquée en association avec de la chimiothérapie
4.2. Posologie et mode d'administration
Le traitement doit être instauré par un médecin expérimenté dans le diagnostic et le traitement des leucémies.
Patients adultes
La posologie initiale recommandée pour la phase chronique de LMC est de 100 mg de dasatinib une fois par jour.
La posologie initiale recommandée pour la phase accélérée de LMC, la phase blastique myéloïde ou blastique lymphoïde (phase avancée) de LMC ou la LAL Ph+ est de 140 mg une fois par jour (voir rubrique 4.4).
Population pédiatrique (LMC Ph+ PC et LAL Ph+)
La posologie chez les enfants et les adolescents est établie en fonction du poids (tableau 1). Le dasatinib est administré par voie orale une fois par jour sous la forme de DASATINIB EG, comprimé pelliculé ou d’une poudre pour suspension buvable (voir le Résumé des Caractéristiques Produit pour Dasatinib, poudre pour suspension buvable). La posologie doit être recalculée tous les 3 mois en fonction des changements de poids corporel, ou plus souvent si nécessaire.
Le comprimé n’est pas recommandé chez les patients de moins de 10 kg ; la poudre pour suspension buvable doit être utilisée chez ces patients. Une augmentation ou une réduction de la posologie sont recommandées selon la réponse et la tolérance individuelle des patients. Aucune donnée de traitement par DASATINIB EG chez l'enfant âgé de moins de 1 an n'est disponible.
DASANTINIB comprimé et DASATINIB poudre pour suspension ne sont pas bioéquivalents. Les patients pouvant avaler des comprimés et souhaitant passer du dasatinib en poudre pour suspension buvable au DASATINIB EG, comprimé pelliculé, ou les patients ne pouvant pas avaler des comprimés et souhaitant passer à la suspension buvable, peuvent le faire, sous réserve que les recommandations relatives à la posologie soient suivies.
La posologie quotidienne initiale de DASATINIB EG, comprimé recommandée chez les patients pédiatriques est indiquée dans le tableau 1.
|
Tableau 1 : Posologie de DASATINIB EG, comprimé pour les patients pédiatriques atteints de LMC Ph+ PC ou LAL Ph+ |
||
|
Poids corporel (kg) a |
Dose quotidienne (mg) |
|
|
de 10 à moins de 20 kg |
40 mg |
|
|
de 20 à moins de 30 kg |
60 mg |
|
|
de 30 à moins de 45 kg |
70 mg |
|
|
au moins 45 kg |
100 mg |
|
a Le comprimé n’est pas recommandé pour les patients de moins de 10 kg ; la poudre pour suspension buvable doit être
utilisée chez ces patients.
Durée du traitement
Dans les études cliniques, le traitement par dasatinib chez les patients adultes atteints de LMC Ph+ PC, de LMC en phase accélérée, en phase blastique myéloïde ou blastique lymphoïde (phase avancée) ou de LAL Ph+ et chez les patients pédiatriques atteints de LMC Ph+ PC était poursuivi jusqu'à progression de la maladie ou intolérance. L'effet de l'arrêt du traitement sur l’issue de la maladie à long terme après obtention d'une réponse moléculaire ou cytogénétique [incluant une réponse cytogénétique complète (RCyC), une réponse moléculaire majeure (RMM) et une RM 4.5] n'a pas été étudié.
Dans les études cliniques, le traitement par dasatinib chez les patients pédiatriques atteints de LAL Ph+ était administré en continu et associé à des cures successives de chimiothérapie, pendant une durée maximale de 2 ans. Chez les patients qui reçoivent ensuite une greffe de cellules souches, le dasatinib peut être administré pendant une année supplémentaire après la transplantation.
Pour atteindre la posologie recommandée, DASATINIB EG est disponible en comprimés pelliculés de 20 mg, 50 mg, 70 mg, 80 mg, 100 mg et 140 mg et en poudre pour suspension buvable. Une augmentation ou une réduction de dose est recommandée en fonction de la réponse et de la tolérance au traitement.
Augmentation de la posologie
Dans les études cliniques conduites chez des patients adultes atteints de LMC ou de LAL Ph+, des augmentations de dose à 140 mg une fois par jour (LMC en phase chronique) ou à 180 mg une fois par jour (phase avancée de LMC ou LAL Ph+) étaient autorisées chez des patients n'ayant pas obtenu de réponse hématologique ou cytogénétique à la dose initiale recommandée.
Les augmentations de posologie indiquées dans le Tableau 2 sont recommandées chez les patients pédiatriques atteints de LMC Ph+ PC qui n’obtiennent pas une réponse hématologique, cytogénétique et moléculaire aux moments recommandés, selon les directives de traitement actuelles, et qui tolèrent le traitement.
Tableau 2 : Augmentation de posologie pour les patients pédiatriques atteints de LMC Ph+ PC
|
|
Posologie (dose maximale par jour) |
|
|
|
Dose initiale |
Augmentation de dose |
|
Comprimés |
40 mg |
50 mg |
|
60 mg |
70 mg |
|
|
70 mg |
90 mg |
|
|
100 mg |
120 mg |
|
DASATINIB EG étant administré en association avec une chimiothérapie chez les patients pédiatriques atteints de LAL Ph+, une augmentation de la posologie n’est pas recommandée chez ces patients.
Adaptation de la posologie en cas d'effets indésirables
Myélosuppression
Dans les études cliniques, les cas de myélosuppression ont nécessité un arrêt de traitement, une réduction de dose ou une sortie d'étude. Parfois, le recours à un support transfusionnel en plaquettes et en globules rouges était nécessaire. Pour les myélosupressions persistantes, le recours aux facteurs de croissance hématopoïétiques était nécessaire.
Les recommandations relatives aux adaptations de la posologie chez l’adulte sont résumées dans le tableau 3 et chez les patients pédiatriques atteints de LMC Ph+ PC dans le Tableau 4. Les recommandations pour les patients pédiatriques atteints de LAL Ph+ traités en association avec une chimiothérapie figurent dans un paragraphe distinct après les tableaux.
Tableau 3 : Ajustement de la posologie en cas de neutropénie et de thrombocytopénie chez l’adulte
|
Adultes atteints de LMC en phase chronique (dose initiale 100 mg une fois par jour) |
PNN < 0,5 x 109/L et/ou plaquettes < 50 x 109/L |
1. Arrêt du traitement jusqu'à ce que PNN ≥ 1,0 x 109/L et plaquettes ≥ 50 x 109/L. 2 Reprendre le traitement à la dose initiale. 3. Si les plaquettes sont < 25 x 109/L et/ou si récidive de PNN < 0,5 x 109/L pendant une durée > 7 jours, recommencer à l'étape 1 et reprendre le traitement à une dose réduite de 80 mg une fois par jour pour le second épisode. Pour le troisième épisode, réduire encore la dose à 50 mg une fois par jour (pour les patients nouvellement diagnostiqués) ou arrêter (pour les patients résistants ou intolérants à un traitement antérieur incluant l'imatinib). |
|
Adultes atteints de LMC en phase accélérée ou blastique et LAL Ph+ (dose initiale 140 mg une fois par jour) |
PNN < 0,5 × 109/L et/ou plaquettes < 10 × 109/L |
1. Vérifier si la cytopénie est imputable à la leucémie (ponction ou biopsie médullaire). 2. Si la cytopénie n'est pas imputable à la leucémie, arrêter le traitement jusqu'à ce que PNN ≥ 1,0 × 109/L et plaquettes ≥ 20 × 109/L et reprendre le traitement à la dose initiale. 3. En cas de récidive de la cytopénie, recommencer à l'étape 1 et reprendre le traitement à la dose réduite de 100 mg une fois par jour (second épisode) ou de 80 mg une fois par jour (troisième épisode). 4. Si la cytopénie est imputable à la leucémie, envisager une augmentation posologique à 180 mg une fois par jour. |
PNN : polynucléaires neutrophiles
Tableau 4 : Ajustement de la posologie en cas de neutropénie et de thrombocytopénie chez les patients pédiatriques atteints de LMC Ph+ PC
|
1. Si la cytopénie persiste plus de 3 semaines, vérifier si la cytopénie est imputable à la leucémie (ponction ou biopsie médullaire).
2. Si la cytopénie n’est pas imputable à la leucémie, arrêter le traitement jusqu’à ce que PNN ≥ 1,0 × 109/L et plaquettes ≥ 75 × 109/L et reprendre le traitement à la dose initiale.
3. En cas de récidive de la cytopénie, recommencer une ponction/biopsie médullaire et reprendre le traitement à la dose réduite |
Posologie (dose maximale par jour) |
|||
|
|
Dose initiale d’origine |
Réduction de dose à un niveau |
Réduction de dose à deux niveaux |
|
|
Comprimés |
40 mg |
20 mg |
* |
|
|
60 mg |
40 mg |
20 mg |
||
|
70 mg |
60 mg |
50 mg |
||
|
100 mg |
80 mg |
70 mg |
||
PNN : polynucléaires neutrophiles
* dose de comprimé plus faible non disponible
Pour les patients pédiatriques atteints de LMC Ph+ PC, en cas de récurrence de neutropénie ou de thrombocytopénie de grade ≥ 3 durant une réponse hématologique complète (RHC), DASATINIB EG doit être interrompu, et pourra être repris ultérieurement à une dose réduite. Des réductions temporaires de dose pour des degrés intermédiaires de cytopénie et de réponse de la maladie doivent être instaurées si nécessaire.
Pour les patients pédiatriques atteints de LAL Ph+, aucune modification posologique n’est recommandée en cas de toxicités hématologiques de grade 1 à 4.
Si la neutropénie et/ou la thrombocytopénie imposent de reporter de plus de 14 jours la cure de traitement suivante, le dasatinib doit être interrompu et repris, à la même dose au début de la cure suivante. Si la neutropénie et/ou la thrombocytopénie persistent et que la cure de traitement suivante est reportée de 7 jours supplémentaires, un examen de la moelle osseuse devra être réalisé afin de déterminer la cellularité et le pourcentage de blastes. Si la cellularité médullaire est < 10 %, le traitement par dasatinib doit être interrompu jusqu’à PNN > 500/μL (0,5 x 109/L) ; le traitement pourra alors être repris à une dose complète. Si la cellularité médullaire est > 10 %, la reprise du traitement par dasatinib pourra être envisagée.
Effets indésirables extra-hématologiques
En cas de survenue d'un effet indésirable extra-hématologique modéré de grade 2 sous dasatinib, le traitement doit être interrompu jusqu'à résolution de l'effet indésirable ou un retour à l'état d'origine. Le traitement doit être repris à la même posologie si l'effet indésirable survient pour la première fois et la posologie doit être réduite s'il s'agit d'un effet indésirable récurrent. En cas de survenue d'un effet indésirable grave extra-hématologique de grade 3 ou 4 sous dasatinib, le traitement doit être interrompu jusqu'à résolution de l'effet indésirable. Le traitement peut ensuite être repris, de manière appropriée, à une posologie réduite en fonction de la sévérité initiale de l'effet indésirable. Pour les patients atteints de LMC en phase chronique qui ont reçu 100 mg une fois par jour, une réduction de posologie à 80 mg une fois par jour est recommandée avec, si nécessaire, une réduction supplémentaire de 80 mg une fois par jour à 50 mg une fois par jour. Pour les patients en phase avancée de LMC ou LAL Ph+ qui ont reçu 140 mg une fois par jour, une réduction de posologie à 100 mg une fois par jour est recommandée avec, si nécessaire, une réduction supplémentaire de 100 mg une fois par jour à 50 mg une fois par jour. Chez les patients pédiatriques atteints de LMC PC présentant des effets indésirables extra hématologiques, les recommandations relatives à la réduction de posologie pour les effets indésirables hématologiques décrits ci-dessus doivent être suivies. Chez les patients pédiatriques atteints de LAL Ph+ présentant des effets indésirables extra hématologiques, une réduction d’un niveau de dose de la posologie doit être suivi, si nécessaire, selon les recommandations relatives à la réduction posologique pour les effets indésirables hématologiques décrits ci-dessus.
Epanchement pleural
En cas de diagnostic d'épanchement pleural, le dasatinib doit être interrompu jusqu'au moment où le patient est asymptomatique ou a retrouvé son état d'origine. Si l'épisode ne s'améliore pas après environ une semaine, un traitement par diurétiques ou corticostéroïdes ou une utilisation concomitante des deux doit être envisagé (voir rubriques 4.4 et 4.8). Suite à la résolution du premier épisode, la reprise du dasatinib à la même dose doit être envisagée. Suite à la résolution d'un épisode ultérieur, la reprise du dasatinib avec une posologie inférieure doit être envisagée. Suite à la résolution d'un épisode sévère (grade 3 ou 4), le traitement peut être repris d'une manière appropriée à une posologie réduite selon la sévérité initiale de l'effet indésirable.
Réduction posologique en cas d’utilisation concomitante d’inhibiteurs puissants du CYP3A4
L’utilisation concomitante d’inhibiteurs puissants du CYP3A4 et de jus de pamplemousse avec du dasatinib doit être évitée (voir rubrique 4.5). Dans la mesure du possible, il faut choisir un autre médicament concomitant sans potentiel d’inhibition enzymatique ou avec un potentiel d’inhibition enzymatique minimal. Si le dasatinib doit être administré avec un puissant inhibiteur du CYP3A4, envisagez de réduire la dose à :
· 40 mg tous les jours pour les patients prenant quotidiennement Dasatinib 140 mg, comprimé.
· 20 mg tous les jours pour les patients prenant quotidiennement Dasatinib 100 mg, comprimé.
· 20 mg tous les jours pour les patients prenant quotidiennement Dasatinib 70 mg, comprimé.
Pour les patients prenant tous les jours 60 mg ou 40 mg de dasatinib, envisagez d’interrompre la dose de dasatinib jusqu’à l’arrêt de l’inhibiteur du CYP3A4 ou de passer à une dose inférieure avec la formulation en poudre pour suspension buvable (voir Résumé des Caractéristiques du Produit dasatinib poudre pour suspension buvable). Avant la réinstauration du dasatinib, prévoyez une période d'élimination d’environ 1 semaine après l’arrêt de l’inhibiteur.
Ces doses réduites de dasatinib devraient ajuster l’aire sous la courbe (ASC) au taux observé sans inhibiteurs du CYP3A4 ; cependant, aucune donnée clinique n’est disponible avec ces ajustements posologiques chez les patients recevant des inhibiteurs puissants du CYP3A4. Dans le cas où le dasatinib n’est pas toléré après la réduction posologique, l’inhibiteur puissant du CYP3A4 doit être arrêté ou le dasatinib doit être interrompu jusqu’à l’arrêt de l’inhibiteur. Avant d’augmenter la dose de dasatinib, prévoyez une période d'élimination d’environ 1 semaine après l’arrêt de l’inhibiteur.
Populations particulières
Sujets âgés
Aucune différence pharmacocinétique liée à l'âge cliniquement significative n'a été observée chez ces patients. Aucune adaptation de la posologie n’est recommandée chez le sujet âgé.
Insuffisance hépatique
Les patients présentant une altération légère, modérée ou sévère de la fonction hépatique peuvent recevoir le traitement à la posologie initiale recommandée. Cependant, le dasatinib doit être utilisé avec précaution chez les patients ayant une insuffisance hépatique (voir rubrique 5.2).
Insuffisance rénale
Aucune étude clinique n'a été menée avec le dasatinib chez des patients présentant une diminution de la fonction rénale (l'étude menée chez les patients atteints de LMC en phase chronique nouvellement diagnostiquée a exclu les patients présentant un taux sérique de créatinine > 3 fois la limite supérieure de la normale et les études menées chez les patients présentant une LMC, résistants ou intolérants à un traitement antérieur par imatinib, ont exclu les patients présentant un taux sérique de la créatinine > à 1,5 fois la limite supérieure de la normale). Dans la mesure où la clairance rénale du dasatinib et de ses métabolites est < 4 %, une diminution de la clairance totale chez les insuffisants rénaux n’est pas attendue.
Mode d'administration
DASATINIB EG doit être administré par voie orale.
Les comprimés pelliculés ne doivent pas être écrasés, coupés ou mâchés afin de préserver la constance du dosage et minimiser les risques d'exposition cutanée ; ils doivent être avalés tels quels. Les comprimés pelliculés ne doivent pas être délités car l’exposition des patients recevant un comprimé délité est plus faible que chez ceux qui avalent un comprimé tel quel. Dasatinib poudre pour suspension buvable est également disponible pour les patients pédiatriques atteints de LMC Ph+ PC ou de LAL Ph+ et chez les adultes atteints de LMC PC ne pouvant pas avaler de comprimés.
Dasatinib EG peut être pris pendant ou en dehors des repas et doivent l'être de manière régulière, soit le matin, soit le soir. Dasatinib EG ne doit pas être pris avec du pamplemousse ou du jus de pamplemousse (voir la rubrique 4.5).
Hypersensibilité à la substance active ou à l’un des excipients mentionnés à la rubrique 6.1.
4.4. Mises en garde spéciales et précautions d'emploi
Interactions médicamenteuses
Le dasatinib est un substrat et un inhibiteur du cytochrome P450 (CYP) 3A4. Par conséquent, il existe un risque potentiel d'interaction avec d'autres médicaments co-administrés, qui sont principalement métabolisés par le CYP3A4 ou qui modulent son activité (voir rubrique 4.5).
L'utilisation concomitante de dasatinib et de médicaments ou substances puissants inhibiteurs du CYP3A4 (exemple : kétoconazole, itraconazole, érythromycine, clarithromycine, ritonavir, télithromycine, jus de pamplemousse) peut augmenter l'exposition au dasatinib. Par conséquent, chez les patients traités par dasatinib, l'administration concomitante d'inhibiteurs puissants du CYP3A4 n'est pas recommandée (voir rubrique 4.5).
L'utilisation concomitante de dasatinib et de médicaments inducteurs du CYP3A4 (exemple : dexaméthasone, phénytoïne, carbamazépine, rifampicine, phénobarbital ou préparations à base de plantes contenant de l'Hypericum perforatum connu sous le nom de millepertuis) peut réduire de manière substantielle l'exposition au dasatinib et ainsi potentiellement augmenter le risque d'échec thérapeutique. Par conséquent, chez les patients recevant du dasatinib, les médicaments administrés de manière concomitante devront être choisis pour avoir un faible potentiel d'induction du CYP3A4 (voir rubrique 4.5).
L'utilisation concomitante de dasatinib et d'un substrat du CYP3A4 pourrait augmenter l'exposition à ce dernier. Par conséquent, la prudence est recommandée lors de la co-administration de dasatinib et d'un substrat du CYP3A4 à faible index thérapeutique, comme notamment astémizole, terfénadine, cisapride, pimozide, quinidine, bépridil ou alcaloïdes de l'ergot de seigle (ergotamine, dihydroergotamine) (voir rubrique 4.5).
L'utilisation concomitante de dasatinib et d'un antihistaminique H2 (exemple : famotidine), d'un inhibiteur de pompe à protons (exemple : oméprazole) ou d'hydroxyde d'aluminium/hydroxyde de magnésium peut réduire l'exposition au dasatinib. Les antihistaminiques H2 et les inhibiteurs de pompe à protons ne sont pas recommandés.
Les produits à base d'hydroxyde d'aluminium et/ou d'hydroxyde de magnésium doivent être administrés au moins 2 heures avant ou 2 heures après la prise de dasatinib (voir rubrique 4.5).
Populations particulières
D’après les résultats d’une étude de pharmacocinétique en dose unique, les patients présentant une altération légère, modérée ou sévère de la fonction hépatique peuvent recevoir le traitement à la posologie initiale recommandée (voir rubriques 4.2 et 5.2). Etant donné les limites de cette étude, le dasatinib doit être administré avec précaution chez les patients atteints d'insuffisance hépatique.
Principaux effets indésirables
Myélosuppression
Le traitement par dasatinib est associé à des anémies, des neutropénies et des thrombocytopénies. Leur survenue est plus précoce et plus fréquente dans les phases avancées de LMC ou dans la LAL Ph+, que dans les phases chroniques de LMC. Chez les patients adultes atteints de LMC en phase avancée ou de LAL Ph+ traités par dasatinib en monothérapie, des numérations de la formule sanguine (NFS) doivent être effectuées une fois par semaine durant les deux premiers mois, puis une fois par mois, et en fonction de l'état clinique. Chez les patients adultes et pédiatriques atteints de LMC en phase chronique, des numérations de la formule sanguine doivent être effectuées toutes les 2 semaines pendant 12 semaines, puis tous les 3 mois, ou selon la clinique. Chez les patients pédiatriques atteints de LAL Ph+ traités par dasatinib en association à une chimiothérapie, des NFS doivent être effectuées avant de débuter chaque cure de chimiothérapie et en fonction de l’état clinique. Pendant les cures de chimiothérapie de consolidation, les NFS doivent être effectuées tous les 2 jours jusqu’au retour à la normale (voir rubriques 4.2 et 4.8). Les cas de myélosuppression sont généralement réversibles et le plus souvent pris en charge par un arrêt temporaire de dasatinib ou par une réduction de dose.
Accidents hémorragiques
Chez les patients atteints de LMC en phase chronique (n=548), 5 patients (1%) ayant reçu du dasatinib ont présenté une hémorragie de grade 3 ou 4. Dans des études cliniques menées chez les patients atteints de LMC en phase avancée, ayant reçu la dose recommandée de dasatinib (n=304), une hémorragie sévère du système nerveux central (SNC) est survenue chez 1% des patients. Un cas d'évolution fatale était associé à une thrombocytopénie de grade 4 selon le Common Toxicity Criteria (CTC). Une hémorragie gastro-intestinale de grade 3 ou 4 est survenue chez 6% des patients atteints de LMC en phase avancée et a généralement nécessité l'interruption du traitement et des transfusions. D'autres hémorragies de grade 3 ou 4 sont survenues chez 2% des patients atteints de LMC en phase avancée. La plupart de ces effets indésirables hémorragiques liés au traitement chez ces patients ont été associés à une thrombocytopénie de grade 3 ou 4 (voir rubrique 4.8). De plus, des essais in vitro et in vivo suggèrent un effet réversible du traitement par dasatinib sur l'activation des plaquettes.
Des précautions doivent être prises chez les patients nécessitant un traitement par des médicaments antiagrégants plaquettaires ou par anticoagulants.
Rétention hydrique
Le dasatinib est associé à des rétentions hydriques. Dans l'étude de Phase III menée chez les patients atteints de LMC en phase chronique nouvellement diagnostiquée, des cas de rétention hydrique de grade 3 ou 4 ont été rapportés chez 13 patients (5%) du groupe traité par dasatinib et chez 2 patients (1%) du groupe traité par imatinib, après un minimum de 60 mois de suivi (voir rubrique 4.8). Chez tous les patients atteints de LMC en phase chronique traités par dasatinib, des rétentions hydriques sévères sont survenues chez 32 patients (6%) recevant du dasatinib à la dose recommandée (n=548). Dans les études cliniques menées chez des patients atteints de LMC en phase avancée ou LAL Ph+ recevant du dasatinib à la dose recommandée (n=304), des rétentions hydriques de grade 3 ou 4 ont été rapportées chez 8% des patients, incluant des épanchements pleuraux et péricardiques de grade 3 ou 4, rapportés respectivement chez 7% et 1% des patients. Chez ces patients, des oedèmes pulmonaires ainsi que des hypertensions pulmonaires de grade 3 ou 4 ont chacun été rapportés chez 1% des patients.
Les patients développant des symptômes évoquant un épanchement pleural, tels qu'une dyspnée ou une toux sèche, doivent être explorés par des radiographies pulmonaires. Les épanchements pleuraux de grade 3 ou 4 peuvent nécessiter des ponctions évacuatrices et la mise sous oxygène. Les effets indésirables de rétentions hydriques ont généralement été pris en charge par des traitements symptomatiques dont des diurétiques et des cures courtes de stéroïdes (voir rubriques 4.2 et 4.8). Les patients âgés de 65 ans et plus, comparés à des patients plus jeunes, sont plus susceptibles de développer un épanchement pleural, une dyspnée, une toux, un épanchement péricardique et une insuffisance cardiaque congestive, et doivent être étroitement surveillés. Des cas de chylothorax ont également été rapportés chez des patients présentant un épanchement pleural (voir rubrique 4.8).
Hypertension artérielle pulmonaire (HTAP)
Des cas d'HTAP (hypertension artérielle pulmonaire pré-capillaire confirmée par cathétérisme cardiaque droit) ont été rapportés en association avec le traitement par dasatinib (voir rubrique 4.8). Dans ces cas, l'HTAP a été rapportée après initiation du traitement par dasatinib, y compris après plus d'un an de traitement.
Avant d'initier un traitement par dasatinib, les signes ou symptômes de maladie cardio-pulmonaire sous-jacente doivent être recherchés. Une échographie cardiaque doit être effectuée à l'initiation du traitement chez tout patient qui présente des symptômes de maladie cardiaque et doit être envisagée chez les patients ayant des facteurs de risque de maladie pulmonaire ou cardiaque. Chez les patients qui développent une dyspnée et une fatigue après instauration du traitement, les étiologies fréquentes doivent être recherchées, y compris épanchement pleural, œdème pulmonaire, anémie, ou infiltration pulmonaire. Conformément aux recommandations de prise en charge des effets indésirables extra-hématologiques (voir rubrique 4.2), la dose de dasatinib doit être réduite ou le traitement interrompu pendant cette évaluation. Si aucune explication n'est trouvée, ou s'il n'y a aucune amélioration après réduction de dose ou arrêt du traitement, le diagnostic d'HTAP doit être envisagé. L'approche diagnostique doit suivre les recommandations.
Si l'HTAP est confirmée, le traitement par dasatinib doit être arrêté définitivement. Le suivi doit être effectué en accord avec les recommandations. Une amélioration des paramètres clinique et hémodynamique a été observée après arrêt du traitement chez les patients traités par dasatinib présentant une HTAP.
Allongement de l'intervalle QT
Les données in vitro suggèrent que le dasatinib peut potentiellement entraîner un allongement de la repolarisation ventriculaire cardiaque (intervalle QT) (voir rubrique 5.3). Chez 258 patients traités par dasatinib et 258 patients traités par imatinib dans l'étude de Phase III menée dans la LMC en phase chronique nouvellement diagnostiquée après un minimum de 60 mois de suivi, 1 patient (< 1%) dans chaque groupe présentait, comme effet indésirable, un allongement de l'intervalle QTc. Les médianes de l'allongement de l'intervalle QTcF par rapport à la valeur initiale étaient de 3,0 msec chez les patients traités par dasatinib contre 8,2 msec chez les patients traités par imatinib. Un patient (< 1%) dans chaque groupe a présenté un allongement de l'intervalle QTcF > 500 msec. Chez 865 patients atteints de leucémie traités par dasatinib dans les essais cliniques de phase II, les variations moyennes de l'intervalle QTc par rapport à la valeur initiale (utilisant la méthode de Fredericia QTcF), étaient de 4 à 6 msec ; la valeur maximale de la limite supérieure des intervalles de confiance à 95 % des variations moyennes était < 7 msec (voir rubrique 4.8).
Parmi les 2 182 patients résistants ou intolérants à un traitement antérieur par imatinib ayant reçu du dasatinib dans les études cliniques, 15 (1 %) ont eu une prolongation de l'intervalle QTc rapportée comme un effet indésirable. Vingt et un de ces patients (1 %) ont eu un QTcF > 500 msec.
Le dasatinib doit être administré avec précaution chez les patients présentant ou susceptibles de développer un allongement de l'intervalle QTc. Cela inclut les patients présentant une hypokaliémie ou une hypomagnésémie, les patients présentant un syndrome d’allongement congénital du QT, les patients traités par des médicaments antiarythmiques ou d'autres médicaments susceptibles d'entraîner un allongement de l'intervalle QT et les patients ayant reçu des doses cumulatives d’anthracyclines élevées. L'hypokaliémie et l'hypomagnésémie doivent être corrigées avant administration du dasatinib.
Effets indésirables cardiaques
Le dasatinib a été étudié lors d'un essai clinique randomisé chez 519 patients atteints de LMC en phase chronique nouvellement diagnostiquée, dont certains présentaient une maladie cardiaque antérieure. Des effets indésirables cardiaques tels qu'insuffisance cardiaque congestive/dysfonctionnement cardiaque, épanchement péricardique, arythmie, palpitations, allongement de l'intervalle QT et infarctus du myocarde (y compris d’issue fatale) ont été rapportés chez les patients traités par dasatinib. Les effets indésirables cardiaques étaient plus fréquents chez les patients présentant des facteurs de risque ou des antécédents de maladie cardiaque. Les patients présentant des facteurs de risque (par exemple, hypertension, hyperlipidémie, diabète) ou des antécédents de maladie cardiaque (par exemple, intervention coronaire percutanée antérieure, maladie documentée des artères coronaires) doivent être étroitement surveillés pour des signes ou symptômes cliniques indiquant un dysfonctionnement cardiaque tels que douleurs de poitrine, essoufflement et diaphorèse.
En cas de survenue de ces signes ou symptômes cliniques, il est conseillé aux médecins d'arrêter l'administration du dasatinib et d’envisager la nécessité d’un traitement alternatif spécifique à la LMC. Après résolution, une évaluation fonctionnelle sera pratiquée avant de reprendre le traitement par dasatinib. Le traitement par dasatinib peut reprendre à la posologie d'origine en cas d'effets indésirables légers ou modérés (≤ grade 2) et à une posologie inférieure en cas d'effets indésirables sévères (≥ grade 3) (voir rubrique 4.2). Les patients poursuivant le traitement doivent être soumis à une surveillance périodique.
Les patients présentant des maladies cardiovasculaires incontrôlées ou importantes n'ont pas été inclus dans les études cliniques.
Microangiopathie thrombotique (MAT)
Les inhibiteurs de la tyrosine kinase BCR-ABL ont été associés à une microangiopathie thrombotique (MAT), incluant des cas individuels rapportés pour le dasatinib (voir rubrique 4.8). Si des résultats biologiques ou cliniques associés à une MAT surviennent chez un patient traité par dasatinib, le traitement par dasatinib doit être interrompu et une évaluation approfondie de la MAT doit être effectuée, incluant l'activité de l'ADAMTS13 et le dosage des anticorps anti-ADAMTS13. Si la concentration des anticorps anti-ADAMTS13 est élevée et associée à une faible activité d'ADAMTS13, le traitement par dasatinib ne doit pas être repris.
Réactivation de l'hépatite B
Des cas de réactivation du virus de l’hépatite B ont été rapportés chez des patients porteurs chroniques du virus et traités par des inhibiteurs de la tyrosine kinase BCR-ABL. Certains de ces cas ont évolué vers une insuffisance hépatique aiguë ou une hépatite fulminante requérant une transplantation hépatique ou dont l’issue a été fatale.
Tous les patients doivent faire l’objet d’un dépistage d’une infection par le VHB avant l’initiation d’un traitement par dasatinib. Un médecin spécialisé en hépatologie doit être consulté avant instauration du traitement chez les patients porteurs de marqueurs sérologiques positifs (y compris ceux ayant une hépatite B active) et chez les patients dont la sérologie devient positive au cours du traitement. Les patients porteurs du VHB doivent être étroitement suivis tout au long du traitement par dasatinib et plusieurs mois après la fin du traitement (voir rubrique 4.8).
Effets sur la croissance et le développement chez les patients pédiatriques
Dans les essais pédiatriques du dasatinib menés chez des patients pédiatriques atteints de LMC Ph+ PC résistants/intolérants à l’imatinib et des patients pédiatriques atteints de LMC Ph+ PC naïfs de traitement après au moins 2 ans de traitement, des événements indésirables liés au traitement associés à la croissance osseuse et au développement ont été rapportés chez 6 patients (4,6%), dont l’un pour qui l’intensité de l’événement était sévère (retard de croissance de grade 3). Ces 6 cas comprenaient des cas de fusion retardée des épiphyses, d’ostéopénie, de retard de croissance et de gynécomastie (voir rubrique 5.1). Ces résultats sont difficiles à interpréter dans le contexte de maladies chroniques, telles que la LMC, et nécessitent un suivi à long terme.
Dans les essais pédiatriques du dasatinib en association avec une chimiothérapie chez des patients pédiatriques atteints de LAL Ph+ nouvellement diagnostiquée après au maximum 2 ans de traitement, des événements indésirables liés au traitement associé à la croissance osseuse et au développement ont été rapportés chez 1 patient (0,6%). Ce cas était une ostéopénie de grade 1.
Un retard de croissance a été observé chez des patients pédiatriques traités par dasatinib dans les essais cliniques (voir rubrique 4.8). Après une durée maximale de 2 ans de traitement, une tendance à la baisse de la taille attendue a été observée, au même degré que celle observée avec l'utilisation de la chimiothérapie seule, sans impact sur le poids et l'IMC attendus et sans association à des anomalies hormonales ou d'autres paramètres de laboratoire. Une surveillance de la croissance osseuse et du développement chez les patients pédiatriques est recommandée.
Lactose
Ce médicament contient du lactose monohydraté. Les patients présentant une intolérance au galactose, un déficit total en lactase ou un syndrome de malabsorption du glucose et du galactose (maladies héréditaires rares) ne doivent pas prendre ce médicament.
Ce médicament contient moins de 1 mmol (23 mg) de sodium par comprimé, c.-à-d. qu’il est essentiellement « sans sodium ».
4.5. Interactions avec d'autres médicaments et autres formes d'interactions
Substances actives susceptibles d'augmenter les concentrations plasmatiques de dasatinib
Les études in vitro montrent que le dasatinib est un substrat du CYP3A4. L'utilisation concomitante de dasatinib et de médicaments ou substances puissants inhibiteurs du CYP3A4 (exemple : kétoconazole, itraconazole, érythromycine, clarithromycine, ritonavir, télithromycine, jus de pamplemousse) peut augmenter l'exposition au dasatinib. Par conséquent, chez les patients recevant du dasatinib, l'administration systémique d'un inhibiteur puissant du CYP3A4 n'est pas recommandée (voir rubrique 4.2).
Aux concentrations cliniquement significatives, la liaison du dasatinib aux protéines plasmatiques est approximativement de 96 % sur la base des expériences in vitro. Aucune étude d'évaluation de l'interaction du dasatinib avec d'autres médicaments liés aux protéines n'a été menée. L'altération de cette interaction et sa signification clinique ne sont pas connues.
Substances actives susceptibles de diminuer les concentrations plasmatiques de dasatinib
L’administration de dasatinib après 8 jours d'administration quotidienne, le soir, de 600 mg de rifampicine, puissant inducteur du CYP3A4, réduit l'ASC (aire sous la courbe) de dasatinib de 82 %. D'autres médicaments inducteurs du CYP3A4 (exemple : dexaméthasone, phénytoïne, carbamazépine, phénobarbital ou préparations à base de plante contenant de l'Hypericum perforatum connu sous le nom de millepertuis) peuvent également augmenter le métabolisme et diminuer les concentrations plasmatiques de dasatinib. En conséquence, l'utilisation concomitante de dasatinib et de puissants inducteurs du CYP3A4, n'est pas recommandée. Chez les patients pour lesquels la rifampicine ou d'autres inducteurs du CYP3A4 sont indiqués, des thérapeutiques alternatives entraînant une induction enzymatique plus faible doivent être envisagées. L’utilisation concomitante de dexaméthasone, un inducteur faible du CYP3A4, est autorisée avec le dasatinib ; l’ASC du dasatinib devrait diminuer d’environ 25% avec l’utilisation concomitante de dexaméthasone, ce qui est peu susceptible d’être cliniquement significatif.
Antihistaminiques H2 et inhibiteurs de pompe à protons
Une inhibition prolongée de la sécrétion acide gastrique par des antihistaminiques H2 ou des inhibiteurs de pompe à protons (par exemple : famotidine, oméprazole) risque de réduire l'exposition au dasatinib. Dans une étude en dose unique conduite chez des volontaires sains, l'administration de famotidine 10 heures avant une dose unique de dasatinib a réduit l'exposition au dasatinib de 61 %. Lors d'une étude chez 14 sujets sains, l'administration d'une dose unique de 100 mg de dasatinib, 22 heures après administration de 40 mg d'oméprazole pendant 4 jours à l'état d'équilibre, a réduit l'ASC du dasatinib de 43 % et la Cmax du dasatinib de 42 %. L'utilisation d'antiacides doit être envisagée en remplacement des antihistaminiques H2 ou des inhibiteurs de pompe à protons chez les patients traités par dasatinib (voir rubrique 4.4).
Antiacides
Des données non cliniques montrent que la solubilité du dasatinib est pH-dépendante. Chez des sujets sains, l'utilisation concomitante d’antiacides à base d'hydroxyde d'aluminium/d'hydroxyde de magnésium avec le dasatinib a réduit l'ASC d'une dose unique de dasatinib de 55 % et la Cmax de 58 %. Cependant, lorsque les antiacides ont été administrés deux heures avant une dose unique de dasatinib, aucun changement significatif de la concentration ou de l'exposition au dasatinib n’a été observé. Par conséquent, les antiacides peuvent être administrés au moins 2 heures avant ou 2 heures après la prise du dasatinib (voir rubrique 4.4).
Substances actives dont les concentrations plasmatiques sont susceptibles d’être modifiées par le dasatinib
L'utilisation concomitante du dasatinib et d'un substrat du CYP3A4 peut augmenter l'exposition au substrat du CYP3A4. Dans une étude chez des sujets sains, une dose unique de 100 mg de dasatinib a augmenté l'ASC et la Cmax de la simvastatine, substrat connu du CYP3A4, de 20 % et 37 % respectivement. On ne peut pas exclure une augmentation de cet effet suite à des doses répétées de dasatinib. Par conséquent, les substrats du CYP3A4 connus pour avoir un faible index thérapeutique (exemple : astémizole, terfénadine, cisapride, pimozide, quinidine, bépridil ou alcaloïdes de l'ergot de seigle [ergotamine, dihydroergotamine]) doivent être administrés avec précaution chez les patients recevant dasatinib (voir rubrique 4.4).
Les données in vitro montrent un risque potentiel d'interaction avec les substrats du CYP2C8, tels les glitazones.
Population pédiatrique
Les études d’interaction n’ont été réalisées que chez l’adulte.
4.6. Fertilité, grossesse et allaitement
Femmes en âge de procréer/contraception chez les hommes et les femmes
Les hommes sexuellement actifs et les femmes en âge de procréer doivent utiliser une méthode de contraception efficace pendant le traitement.
Grossesse
Sur la base de l'expérience d’utilisation chez l’Homme, le dasatinib est suspecté de provoquer des malformations congénitales incluant des malformations du tube neural, et des effets pharmacologiques nocifs sur le fœtus, lorsqu'il est administré pendant la grossesse. Des études effectuées chez l'animal ont mis en évidence une toxicité sur la reproduction (voir rubrique 5.3.).
DASATINIB EG ne doit pas être utilisé au cours de la grossesse sauf si l'état clinique de la patiente nécessite un traitement par dasatinib. Si DASATINIB EG est utilisé pendant la grossesse, la patiente doit être informée du risque potentiel pour le fœtus.
Allaitement
Les données sur l'excrétion du dasatinib dans le lait maternel humain ou animal sont limitées/insuffisantes. Les données physico-chimiques et les données pharmacodynamiques/toxicologiques disponibles orientent vers une excrétion de dasatinib dans le lait maternel et le risque pour l'enfant allaité ne peut être exclu.
L'allaitement doit être interrompu durant le traitement par DASATINIB EG.
Fertilité
Dans les études effectuées chez l’animal, la fertilité des rats mâles et femelles n’a pas été affectée par le traitement par dasatinib (voir rubrique 5.3). Les médecins et autres professionnels de santé devraient informer les patients de sexe masculin d’âge approprié sur les effets éventuels du dasatinib sur la fertilité, notamment sur l’éventualité de la préservation de sperme.
4.7. Effets sur l'aptitude à conduire des véhicules et à utiliser des machines
Résumé du profil de tolérance
Les données décrites ci-dessous reflètent l'exposition au dasatinib, en monothérapie, à toutes les doses évaluées dans les études cliniques (N=2 900), incluant 324 patients adultes atteints d’une LMC en phase chronique nouvellement diagnostiquée, 2 388 patients atteints de LMC ou de LAL Ph+ résistants ou intolérants à l'imatinib, et 188 patients pédiatriques.
Chez les patients adultes atteints de LMC en phase chronique, de LMC en phase avancée ou de LAL Ph+, la durée médiane du traitement par dasatinib a été de 19,2 mois (de 0 à 93,2 mois). Dans un essai randomisé mené chez des patients atteints de LMC en phase chronique nouvellement diagnostiquée, la durée médiane du traitement était d’environ 60 mois. Chez les 1 618 patients adultes atteints de LMC en phase chronique, la durée médiane du traitement était de 29 mois (intervalle de 0 à 92,9 mois). Chez les 1 094 patients adultes atteints de LMC ou de LAL Ph+ en phase avancée, la durée médiane du traitement par dasatinib a été de 6.2 mois (de 0 à 93,2 mois). Parmi les 188 patients inclus dans les études pédiatriques, la durée médiane du traitement était de 26.3 mois (intervalle de 0 à 99.6 mois). Dans le sous-groupe de 130 patients pédiatriques atteints de LMC en phase chronique traités par dasatinib, la durée médiane du traitement était de 42,3 mois (intervalle de 0,1 à 99,6 mois).
La majorité des patients traités par dasatinib ont présenté des effets indésirables au cours du traitement. Dans la population totale des 2 712 patients adultes traités par dasatinib, 520 (19 %) ont présenté des effets indésirables ayant conduit à l'arrêt du traitement.
Le profil général de tolérance du dasatinib dans la population pédiatrique atteinte de LMC Ph+ PC était similaire à celui de la population adulte, indépendamment de la formulation, à l’exception de l’absence de cas rapporté d’épanchement péricardique, d’épanchement pleural, d’œdème pulmonaire ou hypertension pulmonaire dans la population pédiatrique. Parmi les 130 sujets pédiatriques atteints de LMC PC traités par dasatinib, 2 (1,5 %) ont présenté des effets indésirables entraînant l’arrêt du traitement.
Liste tabulée des effets indésirables
Les effets indésirables suivants, en dehors des anomalies biologiques, ont été observés chez les patients ayant participé aux études cliniques portant sur le dasatinib en monothérapie dans les études cliniques et lors du suivi post-commercialisation (tableau 5). Ces effets sont présentés par classe de systèmes d’organes et par fréquence. Les fréquences sont définies comme suit : très fréquent (≥ 1/10) ; fréquent (≥ 1/100, < 1/10) ; peu fréquent (≥ 1/1 000, < 1/100) ; rare (≥ 1/10 000, < 1/1 000) : fréquence indéterminée (ne peut être estimée sur la base des données post-commercialisation disponibles).
Au sein de chaque groupe de fréquence, les effets indésirables sont présentés suivant un ordre décroissant de gravité.
Tableau 5 : Résumé tabulé des effets indésirables
|
Infections et infestations |
|
|
Très fréquent |
infection (dont infection bactérienne, virale, fongique, non-spécifiée) |
|
Fréquent |
pneumonie (dont infection bactérienne, virale et fongique), infection/inflammation des voies respiratoires hautes, infection virale herpétique (y compris cytomégalovirus-CMV), entérocolite, septicémie (y compris des cas peu fréquents d'issue fatale) |
|
Fréquence indéterminée |
réactivation de l'hépatite B |
|
Affections hématologiques et du système lymphatique |
|
|
Très fréquent |
myélosuppression (y compris anémie, neutropénie, thrombocytopénie) |
|
Fréquent |
neutropénie fébrile |
|
Peu fréquent |
adénopathie, lymphocytopénie |
|
Rare |
érythroblastopénie |
|
Affections du système immunitaire |
|
|
Peu fréquent |
hypersensibilité (dont érythème noueux)
|
|
Rare |
choc anaphylactique |
|
Affections endocriniennes |
|
|
Peu fréquent |
hypothyroïdie |
|
Rare |
hyperthyroïdie, thyroïdite |
|
Troubles du métabolisme et de la nutrition |
|
|
Fréquent |
troubles de l'appétita, hyperuricémie |
|
Peu fréquent |
syndrome de lyse tumorale, déshydratation, hypoalbuminémie, hypercholestérolémie |
|
Rare |
diabète |
|
Affections psychiatriques |
|
|
Fréquent |
dépression, insomnie |
|
Peu fréquent |
anxiété, état confusionnel, affection de l'humeur, diminution de la libido |
|
Affections du système nerveux |
|
|
Très fréquent |
maux de tête |
|
Fréquent |
neuropathie (dont neuropathie périphérique), étourdissement, dysgueusie, somnolence |
|
Peu fréquent |
hémorragie du système nerveux central*b, syncope, tremblements, amnésie, trouble de l’équilibre |
|
Rare |
accident vasculaire cérébral, accident ischémique transitoire, convulsion, névrite optique, paralysie faciale, démence, ataxie |
|
Affections oculaires |
|
|
Fréquent |
trouble visuel (dont perturbation de la vue, vision trouble et réduction de l'acuité visuelle), sécheresse oculaire |
|
Peu fréquent |
Atteinte visuelle, conjonctivite, photophobie, larmoiement |
|
Affections de l'oreille et du labyrinthe |
|
|
Fréquent |
acouphènes |
|
Peu fréquent |
perte d’audition, vertige |
|
Affections cardiaques |
|
|
Fréquent |
insuffisance cardiaque congestive/dysfonctionnement cardiaque*c, épanchement péricardique*, arythmie (dont tachycardie), palpitations |
|
Peu fréquent |
infarctus du myocarde (y compris d'issue fatale)*, allongement de l'intervalle QT* à l'électrocardiogramme, péricardite, arythmie ventriculaire (dont tachycardie ventriculaire), angine de poitrine, cardiomégalie, onde T anormale à l’électrocardiogramme, augmentation de la troponine |
|
Rare |
cœur pulmonaire, myocardite, syndrome coronaire aigu, arrêt cardiaque, allongement de l’intervalle PR à l'électrocardiogramme, coronaropathie, pleuropéricardite |
|
Fréquence indéterminée |
fibrillation auriculaire/flutter atrial |
|
Affections vasculaires |
|
|
Très fréquent |
hémorragie*d |
|
Fréquent |
hypertension, flush |
|
Peu fréquent |
hypotension, thrombophlébite, thrombose |
|
Rare |
thrombose veineuse profonde, embolie, livedo réticulaire
|
|
Fréquence indeterminée |
microangiopathie thrombotique |
|
Affections respiratoires, thoraciques et médiastinales |
|
|
Très fréquent |
épanchement pleural*, dyspnée |
|
Fréquent |
œdème pulmonaire*, hypertension pulmonaire*, infiltration pulmonaire, pneumonie, toux |
|
Peu fréquent |
hypertension artérielle pulmonaire, bronchospasme, asthme, chylothorax* |
|
Rare |
embolie pulmonaire, syndrome de détresse respiratoire aigu |
|
Fréquence indéterminée |
maladie pulmonaire interstitielle |
|
Affections gastro-intestinales |
|
|
Très fréquent |
diarrhée, vomissement, nausée, douleurs abdominales |
|
Fréquent |
saignement gastro-intestinal*, colite (dont colite neutropénique), gastrite, inflammation des muqueuses (dont mucite/stomatites), dyspepsie, distension abdominale, constipation, troubles des tissus mous de la bouche |
|
Peu fréquent |
pancréatite (y compris pancréatite aiguë), ulcère gastro-intestinal haut, œsophagite, ascites*, fissure anale, dysphagie, reflux gastro-œsophagien |
|
Rare |
gastro-entéropathie exsudative, iléus, fistule anale |
|
Fréquence indéterminée |
hémorragie gastro-intestinale fatale* |
|
Affections hépatobiliaires |
|
|
Peu fréquent |
hépatite, cholécystite, cholestase |
|
Affections de la peau et du tissu sous-cutané |
|
|
Très fréquent |
rash cutanée |
|
Fréquent |
alopécie, dermatite (dont eczéma), prurit, acné, sécheresse cutanée, urticaire, hyperhidrose |
|
Peu fréquent |
dermatose neutrophilique, photosensibilité, trouble pigmentaire, panniculite, ulcère cutané, affections bulleuses, trouble unguéal, syndrome d'érythrodysesthésie palmo-plantaire, troubles capillaires |
|
Rare |
vascularite leucocytoclasique, fibrose de la peau |
|
Fréquence indéterminée |
syndrome de Stevens-Johnsonf |
|
Affections musculo-squelettiques et systémiques |
|
|
Très fréquent |
douleur musculo-squelettiqueg |
|
Fréquent |
arthralgie, myalgie, faiblesse musculaire, raideur musculo-squelettique, spasme musculaire |
|
Peu fréquent |
rhabdomyolyse, ostéonécrose, inflammation musculaire, tendinite, arthrite
|
|
Rare |
fusion des épiphyses retardéeh, retard de croissanceh |
|
Affections du rein et des voies urinaires |
|
|
Peu fréquent |
atteinte rénale (y compris insuffisance rénale), pollakiurie, protéinurie |
|
Fréquence indéterminée |
syndrome néphrotique |
|
Affections gravidiques, puerpérales et périnatales |
|
|
Rare |
avortement |
|
Affections des organes de reproduction et du sein |
|
|
Peu fréquent |
gynécomastie, troubles menstruels |
|
Troubles généraux et anomalies au site d'administration |
|
|
Très fréquent |
œdème périphériquei, fatigue, pyrexie, œdème du visagej |
|
Fréquent |
asthénie, douleur, douleur dans la poitrine, œdème généralisé*k, frissons |
|
Peu fréquent |
malaise, autre œdème superficiell |
|
Rare |
troubles de la démarche |
|
Investigations |
|
|
Fréquent |
perte de poids, prise de poids |
|
Peu fréquent |
augmentation de la phospho-créatine kinase sanguine, augmentation des gamma-glutamyltransférases |
|
Lésions, intoxications et complications liées aux procédures |
|
|
Fréquent |
contusion |
a Inclut diminution de l'appétit, satiété précoce, augmentation de l'appétit.
b Inclut hémorragie du système nerveux central, hématome cérébral, hémorragie cérébrale, hématome extra-dural, hémorragie intracrânienne, accident vasculaire cérébral hémorragique, hémorragie subarachnoïdienne, hématome sous-dural et hémorragie sous-durale.
c Inclut augmentation du peptide natriurétique cérébral, dysfonctionnement ventriculaire, dysfonctionnement ventriculaire gauche, dysfonctionnement ventriculaire droit, insuffisance cardiaque, insuffisance cardiaque aiguë, insuffisance cardiaque chronique, insuffisance cardiaque congestive, cardiomyopathie, cardiomyopathie congestive, dysfonctionnement diastolique, diminution de la fraction d'éjection et insuffisance ventriculaire, insuffisance ventriculaire gauche, insuffisance ventriculaire droite et hypokinésie ventriculaire.
d Exclut les hémorragies gastro-intestinales et les hémorragies du système nerveux central ; ces effets indésirables sont reportés dans la classe de systèmes d’organes des affections gastro-intestinales et des affections du système nerveux respectivement.
e Inclut éruption cutanée liée au médicament, érythème, érythème polymorphe, érythrose, rash exfoliant, érythème généralisé, rash génital, rash cutané à la chaleur, milium, miliaire, psoriasis pustuleux, rash, rash érythémateux, rash folliculaire, rash généralisé, rash maculaire, rash maculo-papuleux, rash papuleux, rash prurigineux, rash pustuleux, rash vésiculaire, exfoliation cutanée, irritation cutanée, éruption cutanée toxique, urticaire vésiculeux et rash vasculitique.
f Depuis la commercialisation, des cas de syndrome de Stevens-Johnson ont été rapportés. Il n'a pas pu être déterminé si ces effets indésirables cutanéo-muqueux étaient directement liés au dasatinib ou aux médicaments concomitants.
g Douleur musculo-squelettique rapportée pendant ou après l’arrêt du traitement
h Fréquence rapportée comme étant fréquente dans les études pédiatriques.
i Œdème gravitationnel, œdème localisé, œdème périphérique.
j Œdème conjonctival, œdème des yeux, gonflement des yeux, œdème des paupières, œdème du visage, des lèvres, œdème maculaire, œdème buccal, œdème orbital, œdème périorbital, gonflement du visage.
k Surcharge liquidienne, rétention hydrique, œdème gastro-intestinal, œdème généralisé, gonflement périphérique, œdème, œdème dû à une maladie cardiaque, épanchement périphérique, œdème après une procédure, œdème viscéral.
l Gonflement des organes génitaux, œdème du site d'incision, œdème génital, œdème du pénis, gonflement du pénis, œdème du scrotum, gonflement de la peau, gonflement des testicules, gonflement vulvo-vaginal.
* Pour plus de précisions, voir section « Description d'effets indésirables sélectionnés »
Description d'effets indésirables sélectionnés
Myélosuppression
Le traitement par dasatinib est associé à des anémies, des neutropénies et des thrombocytopénies. Leur survenue est plus précoce et plus fréquente chez les patients en phase avancée de LMC ou avec une LAL Ph+ qu’en phase chronique de LMC (voir rubrique 4.4).
Hémorragie
Des effets indésirables hémorragiques liés au médicament, allant des pétéchies et épistaxis aux hémorragies gastro-intestinales de grade 3 ou 4 et hémorragies du système nerveux central (SNC) de grade 3 ou 4 ont été rapportés chez les patients traités par dasatinib (voir rubrique 4.4).
Rétention hydrique
Divers effets indésirables, tels qu'épanchement pleural, ascite, œdème pulmonaire et épanchement péricardique avec ou sans œdème superficiel peuvent être décrits sous le terme général de « rétention hydrique ». Dans l’étude chez les patients LMC en phase chronique nouvellement diagnostiquée, après un minimum de 60 mois de suivi, les effets indésirables de rétention hydrique liés au traitement par dasatinib incluaient : épanchement pleural (28 %), œdème superficiel (14 %), hypertension pulmonaire (5 %), œdème généralisé (4 %), et épanchement péricardique (4 %). Une insuffisance cardiaque congestive/un dysfonctionnement cardiaque et un œdème pulmonaire ont été rapportés chez < 2 % des patients.
Le taux cumulé d’épanchement pleural (tous grades) lié au traitement par dasatinib au cours du temps a été de 10 % à 12 mois, 14 % à 24 mois, 19 % à 36 mois, 24 % à 48 mois et 28 % à 60 mois. Un total de 46 patients traités par dasatinib ont présenté des épanchements pleuraux récurrents. Dix-sept patients ont présenté 2 effets indésirables distincts, 6 ont présenté 3 effets indésirables, 18 ont présenté de 4 à 8 effets indésirables, et 5 patients ont présenté > 8 épisodes d’épanchements pleuraux.
Le délai médian de survenue du premier épanchement pleural de grade 1 ou 2 lié au traitement par dasatinib a été de 114 semaines (de 4 à 299 semaines). Moins de 10 % des patients avec un épanchement pleural ont présenté des épanchements pleuraux sévères (grade 3 ou 4) liés au traitement par dasatinib. Le délai médian d'apparition du premier épanchement pleural de grade ≥ 3 lié au traitement par dasatinib a été de 175 semaines (de 114 à 274 semaines). La durée médiane des épanchements pleuraux (tous grades) liés au traitement par dasatinib a été de 283 jours (~40 semaines).
Les épanchements pleuraux ont été généralement réversibles et pris en charge par une interruption du traitement par dasatinib, l'utilisation de diurétiques ou d'autres soins médicaux appropriés (voir rubriques 4.2 et 4.4). Parmi les patients traités par dasatinib présentant des épanchements pleuraux liés au traitement (n = 73), 45 (62 %) ont interrompu le traitement, et 30 (41 %) ont eu des réductions de doses. En outre, 34 (47 %) ont reçu des diurétiques, 23 (32 %) ont reçu des corticoïdes et 20 (27 %) ont reçu à la fois des corticoïdes et des diurétiques. Neuf patients (12 %) ont subi une thoracocentèse thérapeutique.
Six pour cent des patients traités par dasatinib ont arrêté le traitement en raison d'un épanchement pleural lié au médicament.
Les épanchements pleuraux n'ont pas affecté la capacité des patients à obtenir une réponse. Parmi les patients traités par dasatinib présentant un épanchement pleural, 96 % ont obtenu une RCyCc, 82 % ont obtenu une RMM et 50 % ont obtenu une RM4.5 malgré des interruptions ou des ajustements de dose. Voir la rubrique 4.4 pour des informations complémentaires concernant les patients atteints de LMC en phase chronique et de LMC en phase avancée ou de LAL Ph +.
Des cas de chylothorax ont été signalés chez des patients présentant un épanchement pleural. Certains cas de chylothorax se sont résolus après l'arrêt, l'interruption ou la réduction de la dose de dasatinib, mais la plupart des cas ont également nécessité un traitement supplémentaire.
Hypertension artérielle pulmonaire (HTAP)
Des cas d'HTAP (hypertension artérielle pulmonaire pré-capillaire confirmée par cathétérisme cardiaque droit) ont été rapportés en association avec le traitement par dasatinib. Dans ces cas, l'HTAP a été rapportée après initiation du traitement par dasatinib, y compris après plus d'un an de traitement.
Les patients ayant présenté une HTAP pendant le traitement par dasatinib prenaient souvent des médicaments concomitants ou présentaient des co-morbidités en plus de la pathologie cancéreuse sous-jacente. Une amélioration des paramètres clinique et hémodynamique a été observée après arrêt du traitement chez les patients traités par dasatinib présentant une HTAP.
Allongement de l'intervalle QT
Dans l’étude de phase III chez des patients atteints de LMC en phase chronique nouvellement diagnostiquée, un patient (< 1%) parmi les patients traités par dasatinib a présenté un allongement de l’intervalle QTcF > 500 msec, après un minimum de 12 mois de suivi (voir rubrique 4.4). Aucun autre patient n’a présenté un QTcF > 500 msec après un minimum de 60 mois de suivi.
Dans cinq études de phase II chez des patients résistants ou intolérants à un traitement antérieur par imatinib, des ECG répétés à valeur initiale et pendant le traitement ont été obtenus à des moments préétablis et lus de façon centralisée pour 865 patients recevant du dasatinib à 70 mg deux fois par jour. L'intervalle QT a été corrigé pour le rythme cardiaque selon la méthode de Fridericia. A tous les points suivant l'administration au jour 8, les modifications moyennes de l'intervalle QTcF par rapport aux valeurs initiales étaient de 4 à 6 msec, les limites supérieures des intervalles de confiance à 95 % étant < 7 msec. Parmi les 2 182 patients résistants ou intolérants à un traitement antérieur par imatinib traités par dasatinib lors des études cliniques, un allongement de l'intervalle QTc a été rapporté en tant qu'effet indésirable chez 15 d'entre eux (1 %). Vingt et un patients (1 %) ont présenté un allongement de l'intervalle QTcF > 500 msec (voir rubrique 4.4).
Effets indésirables cardiaques
Les patients présentant des facteurs de risque ou des antécédents de maladie cardiaque doivent être étroitement surveillés quant aux signes ou symptômes indiquant un dysfonctionnement cardiaque et doivent être évalués et traités d'une manière appropriée (voir rubrique 4.4).
Réactivation de l'hépatite B
Des cas de réactivation du virus de l’hépatite B ont été rapportés chez des patients traités par des inhibiteurs de la tyrosine kinase BCR-ABL. Certains de ces cas ont évolué vers une insuffisance hépatique aiguë ou une hépatite fulminante requérant une transplantation hépatique ou dont l’issue a été fatale (voir rubrique 4.4).
Dans l’étude de Phase III d’optimisation de dose chez les patients en phase chronique de LMC résistants ou intolérants à un traitement antérieur par imatinib (durée médiane de traitement : 30 mois), la fréquence de l’épanchement pleural et de l’insuffisance cardiaque congestive/dysfonctionnement cardiaque était plus basse chez les patients traités par dasatinib 100 mg une fois par jour que chez ceux traités par dasatinib 70 mg deux fois par jour. La myélosuppression a aussi été rapportée moins fréquemment dans le groupe de traitement de 100 mg une fois par jour (voir Anomalies des paramètres biologiques ci-dessous).
La durée médiane de traitement dans le groupe 100 mg une fois par jour a été de 37 mois (de 1-91 mois). Les taux cumulés des effets indésirables sélectionnés rapportés avec la dose initiale recommandée de 100 mg une fois par jour sont indiqués dans le Tableau 6a.
Tableau 6a : Effets indésirables sélectionnés rapportés dans l’étude de phase 3 d’optimisation de dose (patients atteints de LMC en phase chronique intolérants ou résistants à l’imatinib)a
|
|
Suivi minimum de 2 ans |
Suivi minimum de 5 ans |
Suivi minimum de 7 ans |
|||
|
|
Tous grades |
Grades 3/4 |
Tous grades |
Grades 3/4 |
Tous grades |
Grades 3/4 |
|
Terme préférentiel |
Pourcentage (%) de patients |
|||||
|
Diarrhée |
27 |
2 |
28 |
2 |
28 |
2 |
|
Rétention hydrique |
34 |
4 |
42 |
6 |
48 |
7 |
|
Œdème superficiel |
18 |
0 |
21 |
0 |
22 |
0 |
|
Epanchement pleural |
18 |
2 |
24 |
4 |
28 |
5 |
|
Œdème généralisé |
3 |
0 |
4 |
0 |
4 |
0 |
|
Epanchement péricardique |
2 |
1 |
2 |
1 |
3 |
1 |
|
Hypertension pulmonaire |
0 |
0 |
0 |
0 |
2 |
1 |
|
Hémorragie |
11 |
1 |
11 |
1 |
12 |
1 |
|
Saignement gastro-intestinal |
2 |
1 |
2 |
1 |
2 |
1 |
a Résultats de l’étude de Phase 3 d’optimisation de dose rapportés dans la population recevant la dose initiale recommandée de 100 mg une fois par jour (n = 165)
Dans l'étude de phase III d'optimisation de dose chez les patients en phase avancée de LMC et les patients atteints de LAL Ph+, la durée médiane de traitement a été de 14 mois pour les phases accélérées de LMC, 3 mois pour les phases blastiques myéloïdes de LMC, 4 mois pour les phases blastiques lymphoïdes et 3 mois pour les patients atteints de LAL Ph+. Les effets indésirables sélectionnés, qui ont été rapportés avec la dose initiale recommandée de 140 mg une fois par jour sont présentés dans le tableau 6b. Une dose de 70 mg deux fois par jour a également été étudiée. La dose de 140 mg une fois par jour a montré un profil d'efficacité comparable à celui de la dose de 70 mg deux fois par jour, mais un profil de tolérance plus favorable.
Tableau 6b : Effets indésirables sélectionnés rapportés dans l'étude de phase III d'optimisation de dose : phase avancée de LMC et LAL Ph+a
|
|
140 mg une fois par jour n = 304 |
|
|
|
Tous grades |
Grades 3/4 |
|
Terme préférentiel |
Pourcentage (%) de patients |
|
|
Diarrhée |
28 |
3 |
|
Rétention hydrique |
33 |
7 |
|
Œdème superficiel |
15 |
< 1 |
|
Epanchement pleural |
20 |
6 |
|
Œdème généralisé |
2 |
0 |
|
Insuffisance cardiaque congestive/dysfonctionnement cardiaqueb |
1 |
0 |
|
Epanchement péricardique |
2 |
1 |
|
Œdème pulmonaire |
1 |
1 |
|
Hémorragie |
23 |
8 |
|
Saignement gastro-intestinal |
8 |
6 |
a Résultats de l’étude de phase 3 d'optimisation de dose rapportés dans la population recevant la dose initiale recommandée de 140 mg une fois par jour (n = 304) ; suivi final de l’étude à 2 ans.
b Inclut dysfonctionnement ventriculaire, insuffisance cardiaque, insuffisance cardiaque congestive, cardiomyopathie, cardiomyopathie congestive, dysfonctionnement diastolique, diminution de la fraction d’éjection ventriculaire, et insuffisance ventriculaire.
De plus, le dasatinib a été administré en association avec une chimiothérapie dans deux études menées auprès de 161 patients pédiatriques atteints d’une LAL Ph+. Dans l’étude pivot, 106 patients pédiatriques ont reçu du dasatinib en association avec une chimiothérapie selon un schéma posologique continu. Dans l’étude support, sur 55 patients pédiatriques, 35 ont reçu du dasatinib en association avec une chimiothérapie dans un schéma posologique discontinu (deux semaines de traitement suivies d’une à deux semaines sans traitement), et 20 patients ont reçu du dasatinib en association avec une chimiothérapie selon un schéma posologique continu. Parmi les 126 patients pédiatriques atteints de LAL Ph+ traités par le dasatinib selon un schéma posologique continu, la durée médiane du traitement était de 23.6 mois (intervalle de 1.4 à 33 mois).
Parmi les 126 patients pédiatriques atteints de LAL Ph+ recevant le traitement selon le schéma posologique continu, 2 patients (1.6%) ont présenté des effets indésirables entraînant l’arrêt du traitement. Les effets indésirables rapportés à une fréquence ≥ 10% dans ces deux études pédiatriques chez des patients recevant le traitement selon un schéma posologique continu sont présentés dans le tableau 7. Il est à noter que l’épanchement pleural a été rapporté chez 7 patients (5.6%) de ce groupe, et n’est donc pas inclus dans le tableau.
|
Tableau 7 : Effets indésirables rapportés chez ≥10% des patients pédiatriques atteints d’une LAL Ph+ traités par le dasatinib selon un schéma posologique continu en association avec une chimiothérapie (N=126)a |
||
|
|
Pourcentage (%) de patients |
|
|
Effets indésirables |
Tous grades |
Grade ¾ |
|
Neutropénie febrile |
27.0 |
26.2 |
|
Nausée |
20.6 |
5.6 |
|
Vomissement |
20.6 |
4.8 |
|
Douleur abdominale |
14.3 |
3.2 |
|
Diarrhée |
12.7 |
4.8 |
|
Pyrexie |
12.7 |
5.6 |
|
Maux de tête |
11.1 |
4.8 |
|
Diminution de l’appétit |
10.3 |
4.8 |
|
Fatigue |
10.3 |
0 |
a dans l’étude pivot, sur un total de 106 patients, 24 patients ont pris la poudre pour suspension orale au moins une fois, dont 8 ayant pris exclusivement la formulation en poudre pour suspension orale
Anomalies des paramètres biologiques
Hématologie
Dans l’étude de Phase III de LMC en phase chronique nouvellement diagnostiquée, après un minimum de 12 mois de suivi, les anomalies biologiques suivantes de grade 3 et 4 ont été rapportées chez des patients prenant du dasatinib : neutropénie (21%), thrombocytopénie (19%) et anémie (10%). Après un minimum de 60 mois de suivi, les taux cumulatifs de neutropénie, thrombocytopénie et anémie ont été de 29%, 22% et 13%, respectivement.
Chez les patients traités par dasatinib, atteints de la LMC en phase chronique nouvellement diagnostiquée et qui ont présenté une myélosuppression de grade 3 ou 4, la résolution est généralement survenue suite à de brèves interruptions d’administration et/ou des réductions de dose ; le traitement a été arrêté définitivement chez 1,6% des patients après un minimum de 12 mois de suivi. Après un minimum de 60 mois de suivi, le taux cumulatif d'arrêt définitif de traitement dû à des myélosuppressions de grade 3 ou 4 a été de 2,3%.
Chez les patients atteints de LMC résistants ou intolérants à un traitement antérieur par imatinib, des cytopénies (thrombocytopénie, neutropénie et anémie) ont été rapportés constamment. Cependant, la survenue des cytopénies s'est montrée clairement dépendante de la phase de la maladie. La fréquence des anomalies hématologiques de grade 3 ou 4 est présentée dans le Tableau 8.
Tableau 8 : Anomalies biologiques hématologiques de grade CTC 3/4 dans les études cliniques chez les patients résistants ou intolérants à un traitement antérieur par imatiniba
|
|
Phase chronique (n = 165)b |
Phase accélérée (n = 157)c |
Phase blastique myéloïde (n = 74)c |
Phase blastique lymphoïde et LAL (n = 165)c |
|
|
Pourcentage (%) de patients |
|||
|
Paramètres hématologiques |
|
|
|
|
|
Neutropénie |
36 |
58 |
77 |
76 |
|
Thrombocytopénie |
23 |
63 |
78 |
74 |
|
Anémie |
13 |
47 |
74 |
44 |
a Résultats de l'étude de Phase 3 d'optimisation de dose rapportés après 2 ans de suivi.
b Résultats de l'étude CA180-034 avec la dose initiale recommandée de 100 mg une fois par jour.
c Résultats de l'étude CA180-035 avec la dose initiale recommandée de 140 mg une fois par jour. Grades CTC : neutropénie (Grade 3 ≥ 0,5– < 1,0 × 109 /L, Grade 4 < 0,5 × 109/L) ; thrombocytopénie (Grade 3 ≥ 25– < 50 × 109/L, Grade 4 < 25 × 109/L) ; anémie (hémoglobine Grade 3 ≥ 65 – < 80 g/L, Grade 4 < 65 g/L).
Le taux cumulatif des cytopénies de grade 3 ou 4 parmi les patients traités à 100 mg une fois par jour
était similaire à 2 et 5 ans, dont neutropénie (35% vs. 36%), thrombocytopénie (23% vs. 24%) et
anémie (13% vs. 13%).
Chez les patients ayant présenté des myélosuppressions de grade 3 ou 4, le retour à la normale est généralement survenu après de brèves interruptions et/ou réductions de dose et après arrêt définitif du traitement dans 5 % des cas. La plupart des patients ont poursuivi le traitement sans récidive d’autres signes de myélosuppression.
Biochimie
Dans l'étude de Phase III de LMC en phase chronique nouvellement diagnostiquée, après un minimum de 12 mois de suivi, des cas d'hypophosphatémie de grade 3 ou 4 ont été rapportées chez 4 % des patients traités par dasatinib, et des augmentations des transaminases, de créatinine et de bilirubine de grades 3 ou 4 ont été rapportées chez ≤ 1 % des patients. Après un minimum de 60 mois de suivi, le taux cumulatif d'hypophosphatémie de grade 3 ou 4 a été de 7 %, d'augmentation de créatinine et de bilirubine de grades 3 ou 4 a été de 1 %, d'augmentation des transaminases est resté à 1 %. Les anomalies portant sur ces paramètres biochimiques ne sont pas à l'origine d'arrêts de traitement par dasatinib.
Suivi à 2 ans
Des augmentations des transaminases ou de la bilirubine à un grade 3 ou 4 ont été rapportées chez 1% des patients atteints de LMC (résistants ou intolérants à l'imatinib) en phase chronique, mais cette fréquence a été supérieure chez les patients atteints de LMC en phase avancées et dans les LAL Ph+ atteignant 1 % à 7% des patients. Cet événement a été habituellement traité par réduction de dose ou par interruption de traitement. Dans l'étude de phase III d'optimisation de dose dans le traitement de la LMC en phase chronique, des élévations de transaminases ou de bilirubine de grades 3 ou 4 ont été rapportées chez ≤ 1% des patients avec une incidence faible, similaire dans les quatre groupes de traitement. Dans l'étude de Phase III d'optimisation de dose dans le traitement de la LMC en phase avancée et de la LAL Ph+, des élévations de transaminases ou de bilirubine de grades 3 ou 4 ont été rapportées chez 1 à 5 % des patients dans tous les groupes de traitement.
Environ 5 % des patients traités par dasatinib qui avaient une calcémie normale ont présenté une hypocalcémie transitoire de grade 3 ou 4 durant l'étude. En général, la survenue d’une hypocalcémie n'était pas associée à des symptômes cliniques. Les patients ayant développé une hypocalcémie de grade 3 ou 4 ont vu leur taux revenir à la normale après supplémentation orale en calcium.
Des hypocalcémies, hypokaliémies et hypophosphatémies de grades 3 et 4 ont été rapportées dans toutes les phases de LMC mais ont été rapportées à une fréquence plus élevée chez les patients en phase blastique myéloïde ou blastique lymphoïde de LMC et LAL Ph+. Des élévations de la créatinine de grade 3 ou 4 ont été rapportées chez moins de 1% des patients en phase chronique de LMC et cette fréquence était augmentée de 1 à 4% chez les patients en phase avancée de LMC.
Population pédiatrique
Le profil de tolérance du dasatinib administré en monothérapie chez les patients pédiatriques atteints de LMC Ph+ PC était similaire au profil de tolérance chez les adultes. Le profil de tolérance du dasatinib administré en association avec une chimiothérapie chez les patients pédiatriques d’une LAL Ph+ était cohérent avec le profil de tolérance connu du dasatinib chez l’adulte et avec les effets attendus de la chimiothérapie, à l’exception du taux d’épanchement pleural inférieur chez les patients pédiatriques par rapport aux adultes.
Dans les études pédiatriques portant sur la LMC, les taux d’anomalies biologiques étaient cohérents avec les valeurs biologiques attendues chez l'adulte.
Dans les études pédiatriques portant sur la LAL, les taux d’anomalies biologiques étaient cohérents avec le profil connu de paramètres biologiques chez l’adulte, dans le contexte d’un patient atteint d’une leucémie aiguë recevant une chimiothérapie.
Autre population spéciale
Bien que le profil de tolérance du dasatinib dans la population âgée soit similaire à celui dans la population plus jeune, les patients âgés de 65 ans et plus sont plus sujets aux effets indésirables fréquemment rapportés tels que fatigue, épanchement pleural, dyspnée, toux, hémorragie gastro-intestinale basse et perturbation de l'appétit ; ils sont plus susceptibles de développer des effets indésirables moins fréquemment rapportés tels que distension abdominale, sensations vertigineuses, épanchement péricardique, insuffisance cardiaque congestive, perte de poids, et ils doivent être étroitement surveillés (voir rubrique 4.4).
Déclaration des effets indésirables suspectés
La déclaration des effets indésirables suspectés après autorisation du médicament est importante. Elle permet une surveillance continue du rapport bénéfice/risque du médicament. Les professionnels de santé déclarent tout effet indésirable suspecté via le système national de déclaration : Agence nationale de sécurité du médicament et des produits de santé (ANSM) et réseau des Centres Régionaux de Pharmacovigilance - Site internet : https://signalement.social-sante.gouv.fr.
Les observations de surdosage de dasatinib dans les études cliniques se limitent à des cas isolés. Le surdosage le plus élevé, à 280 mg par jour pendant une semaine, a été rapporté chez deux patients et les deux ont développé une diminution significative du nombre de plaquettes. Compte tenu que le dasatinib est associé à des myélosuppressions de grade 3 ou 4 (voir rubrique 4.4), les patients ayant absorbé une dose plus importante que la dose recommandée doivent être étroitement surveillés pour la myélosuppression et un traitement symptomatique approprié doit être donné.
5. PROPRIETES PHARMACOLOGIQUES
5.1. Propriétés pharmacodynamiques
Pharmacodynamique
Le dasatinib inhibe l'activité de la kinase BCR-ABL, des kinases de la famille SRC, d'un certain nombre d'autres kinases oncogènes sélectives dont le c-KIT, des récepteurs de l'éphrine (EPH), et du récepteur β du PDGF. Le dasatinib est un inhibiteur puissant de la kinase BCR-ABL agissant à des concentrations sub-nanomolaires de 0,6-0,8 nM. Il se lie aussi bien à la forme active qu’à la forme inactive de l'enzyme BCR-ABL.
Mécanisme d’action
In vitro, le dasatinib est actif sur différentes lignées cellulaires leucémiques sensibles et résistantes à l'imatinib. Ces études non cliniques ont montré que dasatinib peut surmonter les résistances à l'imatinib provoquées par l'hyperexpression de BCR-ABL, les mutations du domaine de la kinase BCR-ABL, l'activation de voies de signalisation alternatives impliquant les kinases de la famille SRC (LYN, HCK) et l'hyperexpression du gène MDR (« Multi Drug Resistance »). De plus, le dasatinib inhibe les kinases de la famille SRC à une concentration subnanomolaire.
In vivo sur des modèles murins, dasatinib a empêché la progression de la LMC de la phase chronique à la phase blastique et a prolongé la survie des souris porteuses de lignées cellulaires de LMC humaines de multiples origines dont le système nerveux central.
Efficacité et sécurité clinique
Dans l'étude de phase I, des réponses hématologiques et cytogénétiques ont été observées dans toutes les phases de LMC et dans les LAL Ph+ chez les premiers patients traités et suivi jusqu’à 27 mois. Ces réponses ont été durables dans toutes les phases de LMC et dans les LAL Ph+.
Quatre études cliniques de Phase II, bras-unique, non contrôlées et ouvertes ont été menées dans le but de déterminer la tolérance et l'efficacité de dasatinib chez des patients atteints de LMC en phase chronique, accélérée ou blastique myéloïde, résistants ou intolérants à l'imatinib. Une étude randomisée, non-comparative a été conduite chez des patients en phase chronique, en échec à un traitement initial de 400 ou 600 mg d'imatinib. La dose initiale de dasatinib était de 70 mg deux fois par jour. Des adaptations posologiques ont été autorisées dans le but d'améliorer l'efficacité du traitement ou d’en réduire la toxicité (voir rubrique 4.2). Deux études randomisées, ouvertes, de Phase III ont été menées afin d'évaluer l'efficacité de dasatinib administré une fois par jour comparé à dasatinib administré deux fois par jour. De plus, une étude randomisée, ouverte, comparative, de Phase III a été menée chez des patients adultes atteints de LMC en phase chronique nouvellement diagnostiquée.
L'efficacité du dasatinib est jugée sur les taux de réponse hématologique ou cytogénétique.
La durabilité de la réponse et les taux de survie estimés fournissent une preuve supplémentaire du bénéfice clinique du dasatinib.
Au total, 2 712 patients ont été inclus dans les essais cliniques dont 23% étaient âgés de ≥ 65 ans et 5% de ≥ 75 ans.
LMC en phase chronique nouvellement diagnostiquée.
Une étude comparative, randomisée, multicentrique, ouverte, internationale de Phase III a été menée chez des patients adultes atteints de LMC en phase chronique nouvellement diagnostiquée. Les patients ont été randomisés afin de recevoir soit dasatinib 100 mg une fois par jour soit imatinib 400 mg une fois par jour. Le critère principal était le taux de Réponse Cytogénétique Complète confirmée (RCyCc) dans les 12 mois. Les critères secondaires incluaient le temps en RCyCc (mesure de la durabilité de la réponse), le temps jusqu'à RCyCc, le taux de réponse moléculaire majeure (RMM), le temps jusqu'à RMM, la survie sans progression (SSP) et la survie globale (SG). D'autres résultats pertinents d'efficacité incluaient les taux de RCyC et de réponse moléculaire (RMC) complète. L'étude est en cours.
519 patients au total ont été randomisés dans un groupe de traitement: 259 dans le groupe dasatinib et 260 dans le groupe imatinib. Les caractéristiques à l'inclusion étaient bien équilibrées entre les deux groupes de traitement quant à l'âge (l'âge médian était de 46 ans pour le groupe dasatinib et de 49 ans pour le groupe imatinib avec 10% et 11% de patients âgés de 65 ans ou plus, respectivement), au sexe (femmes 44% et 37%, respectivement), et race (caucasiens 51% et 55% ; asiatiques 42% et 37%, respectivement). A l'inclusion, la distribution du score de Hasford était similaire dans les groupes de traitement dasatinib et imatinib (risque bas : 33% et 34%; risque intermédiaire 48% et 47%; risque élevé: 19% et 19%, respectivement).
Avec un minimum de 12 mois de suivi, 85% des patients randomisés dans le groupe dasatinib et 81% des patients randomisés dans le groupe imatinib recevaient toujours le traitement de première intention. Un arrêt de traitement dans les 12 mois pour cause de progression de la maladie est survenu chez 3% des patients traités par dasatinib et chez 5% des patients traités par imatinib.
Après un minimum de 60 mois de suivi, 60% des patients randomisés dans le groupe dasatinib et 63% des patients randomisés dans le groupe imatinib recevaient toujours le traitement de première intention. Un arrêt de traitement dans les 60 mois pour cause de progression de la maladie est survenu chez 11% des patients traités par dasatinib et chez 14% des patients traités par imatinib.
Les résultats d'efficacité sont présentés dans le Tableau 9. Dans les 12 premiers mois du traitement, une proportion statistiquement significative plus grande de patients dans le groupe dasatinib a atteint une RCyCc comparés aux patients dans le groupe imatinib. L'efficacité du dasatinib a été constamment démontrée dans les divers sous-groupes, y compris âge, sexe et score de Hasford à l'inclusion.
Tableau 9 : Résultats d'efficacité dans une étude de phase 3 chez des patients présentant une LMC en phase chronique nouvellement diagnostiquée
|
|
Dasatinib n = 259 |
Imatinib n =260 |
p-value |
|
|
Taux de réponse (95% IC) |
||
|
Réponse cytogénétique dans les 12 mois |
|
|
|
|
RCyCca |
76,8% (71,2– 81,8) |
66,2% (60,1-71,9) |
p< 0,007* |
|
RCyCb |
85,3% (80,4-89,4) |
73,5% (67,7-78,7) |
- |
|
Dans les 24 mois |
|
|
|
|
RCyCca |
80,3% |
74,2% |
- |
|
RCyCb |
87,3% |
82,3% |
- |
|
Dans les 36 mois |
|
|
|
|
RCyCca |
82,6% |
77,3% |
- |
|
RCyCb |
88,0% |
83,5% |
- |
|
Dans les 48 mois |
|
|
|
|
RCyCca |
82,6% |
78,5% |
- |
|
RCyCb |
87,6% |
83,8% |
- |
|
Dans les 60 mois |
|
|
|
|
RCyCca |
83,0% |
78,5% |
- |
|
RCyCb |
88,0% |
83,8% |
- |
|
Réponse moléculaire majeurec |
|
|
|
|
12 mois |
52,1% (45,9-58,3) |
33,8% (28,1-39,9) |
p< 0,00003* |
|
24 mois |
64,5% (58,3-70,3) |
50% (43,8-56,2) |
- |
|
36 mois |
69,1% (63,1-74,7) |
56,2% (49,9-62,3) |
- |
|
48 mois |
75,7% (70,0-80,8) |
62,7% (56,5-68,6) |
- |
|
60 mois |
76,4% (70,8-81,5) |
64,2% (58,1-70,1) |
p=0,0021 |
|
|
Hazard ratio (HR) |
|
|
|
|
dans les 12 mois (99,99% IC) |
|
|
|
Temps jusqu'à RCyCc |
1,55 (1,0-2,3) |
p< 0,0001* |
|
|
Temps jusqu'à RMM |
2,01 (1,2-3,4) |
p< 0,0001* |
|
|
Durabilité de la RCyCc |
0,7 (0,4-1,4) |
p< 0,035 |
|
|
|
dans les 24 mois (95 % IC) |
|
|
|
Temps jusqu'à RCyCc |
1,49 (1,22-1,82) |
- |
|
|
Temps jusqu'à RMM |
1,69 (1,34-2,12) |
- |
|
|
Durabilité de la RCyCc |
0,77 (0,55-1,10) |
- |
|
|
|
dans les 36 mois (95 % IC) |
|
|
|
Temps jusqu'à RCyCc |
1,48 (1,22-1,80) |
- |
|
|
Temps jusqu'à RMM |
1,59 (1,28-1,99) |
- |
|
|
Durabilité de la RCyCc |
0,77 (0,53-1,11) |
- |
|
|
|
dans les 48 mois (95 % IC) |
|
|
|
Temps jusqu'à RCyCc |
1,45 (1,20-1,77) |
- |
|
|
Temps jusqu'à RMM |
1,55 (1,26-1,91) |
- |
|
|
Durabilité de la RCyCc |
0,81 (0,56-1,17) |
- |
|
|
|
dans les 60 mois (95 % IC) |
|
|
|
Temps jusqu'à RCyCc |
1,46 (1,20-1,77) |
p=0,0001 |
|
|
Temps jusqu'à RMM |
1,54 (1,25-1,89) |
p < 0,0001 |
|
|
Durabilité de la RCyCc |
0,79 (0,55-1,13) |
p=0,1983 |
|
a Une réponse cytogénétique complète confirmée (RCyCc) est définie comme une réponse enregistrée à deux occasions consécutives (à un intervalle d'au moins 28 jours).
b Une réponse cytogénétique complète (RCyC) est basée sur une seule évaluation cytogénétique de moelle osseuse.
c Une réponse moléculaire majeure (quelle que soit la date de mesure) a été définie comme un rapport BCR-ABL ≤ 0,1% par RQ-PCR dans les prélèvements sanguins périphériques, quantification standardisée selon l'échelle internationale. Il s'agit de taux cumulatifs représentant un suivi minimum pour la période spécifiée. *Ajusté pour le score de Hasford et indique une significativité statistique à un degré de significativité nominal prédéfini.
IC = intervalle de confiance.
Après 60 mois de suivi, le temps médian jusqu'à RCyCc était de 3,1 mois dans le groupe dasatinib et de 5,8 mois dans le groupe imatinib chez les patients avec une RCyC confirmée. Le temps médian jusqu'à RMM après 60 mois de suivi était de 9,3 mois dans le groupe dasatinib et de 15,0 mois dans le groupe imatinib chez les patients avec une RMM. Ces résultats sont cohérents avec ceux obtenus à 12 mois, 24 mois et 36 mois.
Le temps jusqu'à RMM est représenté graphiquement à la Figure 1. Le temps jusqu'à RMM est toujours plus court chez les patients traités par dasatinib que chez les patients traités par imatinib.
Figure 1 : Estimation Kaplan-Meier du temps jusqu'à réponse moléculaire majeure (RMM)
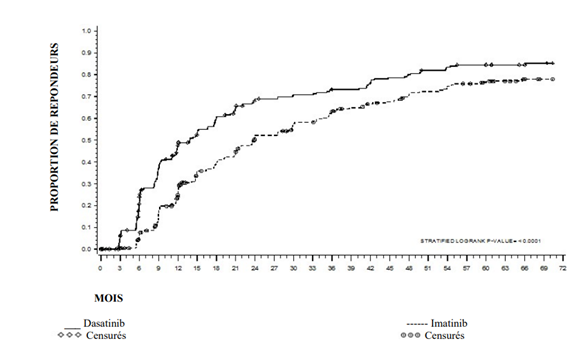
|
GROUPE |
# REPONDEURS / # RANDOMISES |
HAZARD RATIO (IC95%) |
|
Dasatinib |
198/259 |
|
|
Imatinib |
167/260 |
|
|
Dasatinib sur imatinib |
|
1,54 (1,25 - 1,89) |
Les taux de RCyCc dans les groupes de traitement dasatinib et imatinib, respectivement, à 3 mois (54% et 30%), 6 mois (70% et 56%), 9 mois (75% et 63%), 24 mois (80% et 74%), 36 mois (83% et 77%), 48 mois (83% et 79%) et 60 mois (83% et 79%) étaient en cohérence avec le critère primaire. Les taux de RMM dans les groupes de traitement dasatinib et imatinib, respectivement, à 3 mois (8% et 0,4%), 6 mois (27% et 8%), 9 mois (39% et 18%), 12 mois (46% et 28%), 24 mois (64% et 46%), 36 mois (67% et 55%), 48 mois (73% et 60%) et 60 mois (76% et 64%) étaient aussi en cohérence avec le critère primaire.
Les taux de RMM à des temps spécifiques sont représentés graphiquement à la Figure 2. Les taux de RMM étaient systématiquement plus élevés chez les patients traités par dasatinib que chez les patients traités par imatinib.
Figure 2 : Taux de RMM au cours du temps - tous les patients atteints de LMC en phase chronique nouvellement diagnostiquée, randomisés dans une étude de Phase 3
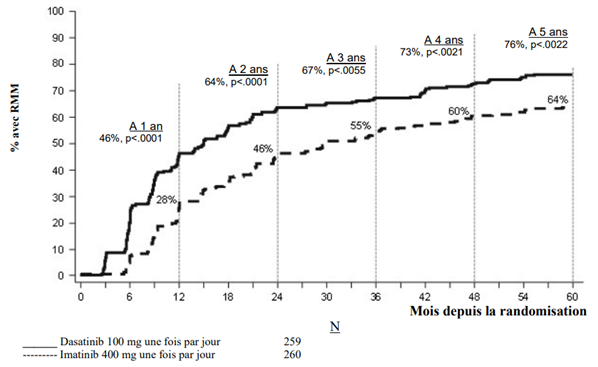
La proportion de patients ayant obtenu un ratio BCR-ABL ≤ 0,01% (diminution de 4-log) à tout moment était plus élevée dans le groupe dasatinib que dans le groupe imatinib (54,1% versus 45%). La proportion de patients obtenant un ratio BCR-ABL ≤ 0,0032% (diminution de 4,5-log) à tout moment était plus élevée dans le groupe dasatinib que dans le groupe imatinib (44% versus 34%).
Les taux de RM4.5 au cours du temps sont représentés graphiquement à la Figure 3. Ces taux étaient toujours plus élevés chez les patients traités par dasatinib que chez les patients traités par imatinib.
Figure 3 : Taux de RM4.5 au cours du temps - tous les patients atteints de LMC en phase chronique nouvellement diagnostiqués, randomisés dans une étude de Phase 3
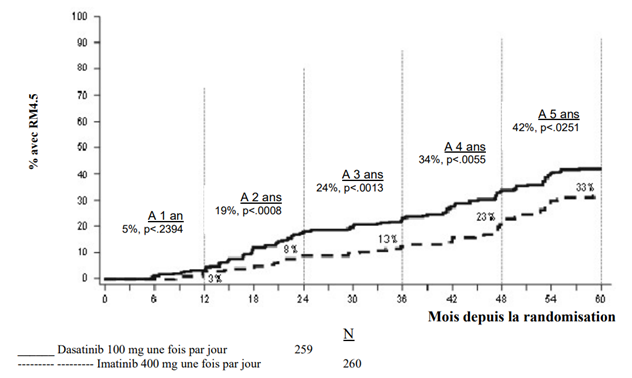
Le taux de RMM, à tout moment, dans chaque groupe de risque, déterminé par le score de Hasford, était plus élevé dans le groupe dasatinib que dans le groupe imatinib (risque faible : 90% et 69% ; risque intermédiaire : 71% et 65% ; risque élevé : 67% et 54%, respectivement).
Dans une analyse supplémentaire, plus de patients traités par dasatinib (84%) ont obtenu une réponse moléculaire précoce (définie par des niveaux BCR-ABL ≤ 10% à 3 mois) comparativement aux patients traités par imatinib (64%). Les patients ayant obtenu une réponse moléculaire précoce présentaient un risque inférieur de transformation, un taux plus élevé de survie sans progression (SSP) et un taux plus élevé de survie globale (SG), comme présenté dans le Tableau 10.
Tableau 10 : Patients sous dasatinib avec BCR-ABL 10% et > 10% à 3 mois
|
Dasatinib N = 235 |
Patients avec BCR-ABL ≤ 10% à 3 mois |
Patients avec BCR-ABL > 10% à 3 mois |
|
Nombre de patients (%) |
198 (84,3) |
37 (15,7) |
|
Transformation à 60 mois, n/N (%) |
6/198 (3,0) |
5/37 (13,5) |
|
Taux de SSP à 60 mois (95% IC) |
92,0% (89,6, 95,2) |
73,8% (52,0, 86,8) |
|
Taux de SG à 60 mois (95% IC) |
93,8% (89,3, 96,4) |
80,6% (63,5, 90,2) |
Le taux de SG à des temps spécifiques est représenté graphiquement à la Figure 4. Le taux de SG était systématiquement plus élevé dans le groupe de patients sous dasatinib ayant atteint un niveau de BCR-ABL ≤ 10% à 3 mois, par rapport à ceux ne l'ayant pas atteint.
Figure 4 : Courbe de survie globale pour dasatinib en fonction du taux de BCR-ABL (≤ 10% ou > 10%) à 3 mois - étude de Phase 3 chez des patients atteints de LMC en phase chronique nouvellement diagnostiquée
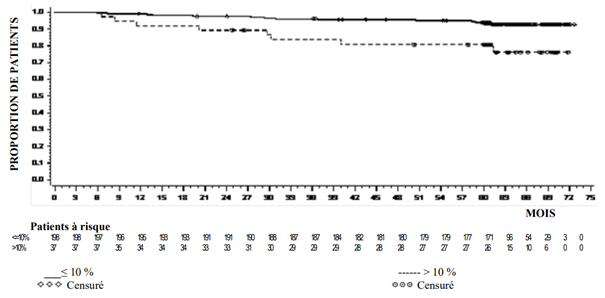
|
GROUPE |
# DECES / # patients du graphique |
MÉDIANE (IC 95 %) |
HAZARD RATIO (IC 95%) |
|
≤ 10 % |
14/198 |
.(. - .) |
|
|
> 10 % |
8/37 |
.(. - .) |
|
|
|
|
|
0,29 (0,12 – 0,69) |
La progression de la maladie était définie comme une augmentation des globules blancs malgré une prise en charge thérapeutique appropriée, une perte de RHC, de RCyC ou RCy partielle, une évolution vers la phase accélérée ou la phase blastique ou le décès. A 60 mois, le taux estimé de SSP était de 88,9% (IC : 84% - 92,4%) pour les deux groupes de traitement dasatinib et imatinib. A 60 mois, une transformation en phase accélérée ou blastique est survenue chez moins de patients traités par dasatinib (n = 8; 3%) que de patients traités par imatinib (n = 15 ; 5,8%). Les taux estimés de survie à 60 mois pour les patients traités par dasatinib et imatinib étaient de 90,9% (IC: 86,6% - 93,8%) et 89,6% (IC : 85,2% - 92,8%), respectivement. Il n'y avait pas de différence sur la SG (HR 1,01, IC 95% : 0,58 à 1,73, p=0,9800) et la SSP (HR 1,00, IC 95% : 0,58 à 1,72, p=0,9998) entre dasatinib et imatinib.
Chez les patients ayant présenté une progression de la maladie ou ayant arrêté le traitement par dasatinib ou imatinib, un séquençage de BCR-ABL a été effectué sur les échantillons sanguins lorsqu'ils étaient disponibles. Des taux similaires de mutation ont été observés dans les deux bras de traitement. Les mutations détectées chez les patients traités par dasatinib étaient: T315I, F317I/L et V299L.
Le spectre de mutations observé dans le bras de traitement imatinib était différent. Sur la base de données in-vitro, il apparaît que dasatinib n'est pas actif contre la mutation T315I.
LMC en phase chronique - résistance ou intolérance à un traitement antérieur par l'imatinib
Deux études cliniques ont été menées chez des patients résistants ou intolérants à l'imatinib; l'objectif principal de ces études en termes d'efficacité était la réponse cytogénétique majeure (RCyM):
Etude 1
Une étude ouverte, randomisée, non comparative, multicentrique a été menée chez des patients en échec d’un traitement préalable par imatinib à 400 ou 600 mg. Les patients ont été randomisés (2 :1) pour recevoir respectivement soit dasatinib (70 mg deux fois par jour) soit imatinib (400 mg deux fois par jour). Un changement de bras de traitement était autorisé en cas de progression de la maladie ou d'intolérance au traitement non contrôlables par une modification de dose. Le critère principal était la RCyM à 12 semaines. Les résultats sont disponibles pour 150 patients: 101 étaient randomisés dans le groupe dasatinib et 49 dans le groupe imatinib (tous résistants à l'imatinib). Le délai médian entre le diagnostic et la randomisation était de 64 mois dans le bras dasatinib et de 52 mois dans le bras imatinib. Tous les patients étaient largement prétraités. Sous imatinib une réponse hématologique complète (RHC) antérieure avait été obtenue chez 93% de l'ensemble de cette population. Une RCyM à l'imatinib avait été obtenue antérieurement chez respectivement 28% et 29% des patients randomisés dans les bras dasatinib et imatinib.
La durée médiane du traitement a été de 23 mois dans le bras dasatinib (avec 44% des patients traités pendant plus de 24 mois à ce jour) et 3 mois pour imatinib (avec 10% des patients traités pendant plus de 24 mois à ce jour). Quatre-vingt-treize pour cent des patients dans le bras dasatinib et 82% des patients dans le bras imatinib ont atteint une RHC avant tout changement de bras (cross-over).
Après 3 mois, une RCyM a été observée plus souvent dans le bras dasatinib (36%) que dans le bras imatinib (29%). Notamment, 22% des patients ont atteint une Réponse Cytogénétique Complète (RCyC) dans le bras dasatinib contre seulement 8% dans le bras imatinib. Avec un traitement et un suivi plus long (médian de 24 mois), une RCyM a été obtenue chez 53% des patients traités par dasatinib (RCyC obtenue chez 44% d’entre eux) et chez 33% des patients traités par imatinib (RCyC obtenue chez 18% d’entre eux) avant changement de bras. Parmi les patients ayant reçu imatinib 400 mg avant l'entrée dans l'étude, une RCyM a été obtenue chez 61% des patients dans le bras dasatinib et chez 50% des patients dans le bras imatinib.
Selon l'estimation de Kaplan-Meier, la proportion de patients ayant conservé une RCyM pendant 1 an était de 92% (IC 95% : [85%-100%]) pour dasatinib (97% de RCyC, IC 95% : [92%-100%]) et de 74% (IC 95% : [49%-100%]) pour imatinib (100% de RCyC). La proportion de patients ayant conservé une RCyM pendant 18 mois était de 90% (IC 95% : [82%-98%]) pour dasatinib (94% de RCyC, IC 95% : [87%-100%]) et de 74% (IC 95% : [49%-100%]) pour imatinib (100% de RCyC).
Selon l'estimation de Kaplan-Meier, la proportion de patients en survie sans progression (SSP) à 1 an était de 91% (IC 95% : [85%-97%]) pour dasatinib et de 73% (IC 95% : [54%-91%]) pour imatinib. La proportion de patient en SSP à 2 ans était de 86% (IC 95% : [78%-93%]) avec dasatinib et de 65% (IC 95% : [43%-87%]) pour imatinib.
Un total de 43% des patients du bras dasatinib, et 82% du bras imatinib ont été en échec de traitement, échec défini par une progression de la maladie ou un changement vers un autre traitement (réponse insuffisante, intolérance aux traitements de l'étude, etc.).
Le taux de réponse moléculaire majeur (défini par un rapport BCR-ABL/transcrits contrôles ≤ 0,1% mesuré par RQ-PCR dans des échantillons de sang périphérique), avant changement de bras de traitement était de 29% avec dasatinib et de 12% avec imatinib.
Etude 2
Une étude ouverte, bras unique, multicentrique a été menée chez des patients résistants ou intolérants à l'imatinib (i.e., les patients qui ont présenté une toxicité significative pendant le traitement par imatinib, empêchant la poursuite du traitement).
Un total de 387 patients a été traité par dasatinib à la dose de 70 mg deux fois par jour (288 patients résistants et 99 intolérants). Le délai médian entre le diagnostic et le début du traitement était de 61 mois. La majorité des patients (53%) a reçu un traitement antérieur par imatinib pendant plus de trois ans. La majorité des patients résistants (72%) a reçu de l'imatinib à une dose > 600 mg. En plus de l'imatinib, 35% des patients avaient déjà reçu une chimiothérapie, 65% de l'interféron, et 10% ont déjà eu une transplantation médullaire. Trente-huit pour cent des patients présentaient en prétraitement des mutations connues pour induire des résistances à l'imatinib.
La durée médiane du traitement sous dasatinib était de 24 mois avec 51% de patients traités pendant plus de 24 mois à ce jour. Les résultats d'efficacité sont présentés dans le Tableau 11. Une RCyM a été obtenue chez 55% des patients résistants à l'imatinib et chez 82% des patients intolérants à l'imatinib. Avec un suivi minimal de 24 mois, 21 des 240 patients qui ont obtenu une RCyM ont progressé et la durée médiane de RCyM n'a pas été atteinte.
Selon l'estimation de Kaplan-Meier, 95% (IC 95% : [92%-98%]) des patients ont conservé une RCyM à 1 an et 88% (IC 95% : [83%-93%]) ont conservé une RCyM à 2 ans. La proportion de patients ayant conservé une RCyC à 1 an était de 97% (IC 95% : [94%-99%]) et la proportion de patients ayant conservé une RCyC à 2 ans était de 90% (IC 95% : [86%-95%]). Quarante deux pour cent des patients résistants à l'imatinib n’ayant jamais eu de RCyM antérieure sous imatinib (n= 188) ont obtenu une RCyM avec dasatinib.
Il y avait 45 mutations BCR-ABL différentes chez 38% des patients inclus dans cette étude clinique. Une réponse hématologique complète ou une RCyM a pu être obtenue pour tous les types de mutation BCR-ABL associés à une résistance à l'imatinib sauf la mutation T315I. Les taux de RCyM à 2 ans étaient semblables quelque soit le statut mutationnel initial: toute mutation BCR-ABL, mutation de la boucle P ou absence de mutation (63%, 61% et 62%, respectivement).
Parmi les patients résistants à l'imatinib, le taux estimé de SSP était de 88% (IC 95% : [84%-92%]) à 1 an et de 75% (IC 95% : [69%-81%]) à 2 ans. Parmi les patients intolérants à l'imatinib, le taux estimé de SSP était de 98% (IC 95% : [95%-100%]) à 1 an et de 94% (IC 95% : [88%-99%]) à 2 ans.
Le taux de réponse moléculaire majeure à 24 mois était de 45% (35% chez les patients résistants à l'imatinib et 74% chez les patients intolérants à l'imatinib).
LMC en phase accélérée
Une étude ouverte, bras unique, multicentrique a été menée chez des patients intolérants ou résistants à l'imatinib. Un total de 174 patients a reçu dasatinib à la dose de 70 mg deux fois par jour (161 patients résistants et 13 intolérants à l'imatinib). Le délai médian entre le diagnostic et le début du traitement était de 82 mois. La durée médiane du traitement par dasatinib a été de 14 mois avec 31% de patients traités pendant > 24 mois à ce jour. Le taux de réponse moléculaire majeure (évaluée chez 41 patients avec une RCyC) était de 46% à 24 mois. Des résultats complémentaires d'efficacité sont présentés dans le Tableau 11.
LMC en phase blastique myéloïde
Une étude ouverte, bras unique, multicentrique a été menée chez des patients intolérants ou résistants à l'imatinib. Un total de 109 patients a reçu dasatinib à la dose de 70 mg deux fois par jour (99 patients résistants et 10 intolérants à l'imatinib). Le délai médian entre le diagnostic et le début du traitement a été de 48 mois. La durée médiane du traitement par dasatinib a été de 3,5 mois avec 12% de patients traités pendant > 24 mois à ce jour. Le taux de réponse moléculaire majeure (évaluée chez 19 patients avec une RCyC) était de 68% à 24 mois. Des résultats complémentaires d'efficacité sont présentés dans le Tableau 11.
LMC en phase blastique lymphoïde et LAL Ph+
Une étude en ouvert, bras unique, multicentrique a été menée chez des patients atteints de LMC en phase blastique lymphoïde ou de LAL Ph+, résistants ou intolérants à un traitement antérieur par imatinib. Un total de 48 patients en phase blastique lymphoïde de LMC a reçu dasatinib à la dose de 70 mg deux fois par jour (42 patients résistants et 6 intolérants à l'imatinib). Le délai médian entre le diagnostic et le début du traitement était de 28 mois. La durée médiane du traitement par dasatinib était de 3 mois avec 2% des patients traités pendant > 24 mois à ce jour. Le taux de réponse moléculaire majeure (l'ensemble des 22 patients traités avec une RCyC) était de 50% à 24 mois. De plus, 46 patients atteints de LAL Ph+ ont reçu du dasatinib à la dose de 70 mg deux fois par jour (44 patients résistants et 2 intolérants à l’imatinib). Le délai médian entre le diagnostic et le début du traitement était de 18 mois. La durée médiane du traitement par dasatinib a été de 3 mois avec 7 % de patients traités pendant > 24 mois à ce jour. Le taux de réponse moléculaire majeure (l'ensemble des patients traités avec une RCyC) était de 52 % à 24 mois. Des résultats complémentaires d'efficacité sont présentés dans le tableau 11. On note que des réponses hématologiques majeures (RHMa) ont été obtenues rapidement (pour la plupart en 35 jours à partir de la première administration de dasatinib pour les patients atteints de LMC en phase blastique lymphoïde, et en 55 jours pour les patients atteints de LAL Ph+).
Tableau 11 : Efficacité du dasatinib dans les études cliniques de phase II à bras uniquea
|
|
Chronique (n= 387) |
Accéléré (n= 174) |
Blastique myéloïde (n= 109) |
Blastique lymphoïde (n= 48) |
LAL Ph+ (n = 46) |
|
Taux de réponse hématologiqueb (%) |
|||||
|
RHMa (IC à 95 %) |
N/A |
64% (57-72) |
33% (24-43) |
35% (22-51) |
41 % (27-57) |
|
RHC (IC à 95 %) |
91% (88-94) |
50% (42-58) |
26% (18-35) |
29% (17-44) |
35 % (21-50) |
|
NEL (IC à 95 %) |
N/A |
14% (10-21) |
7% (3-14) |
6% (1-17) |
7 % (1-18) |
|
Durée de la RHMa (%, selon l'estimation de Kaplan-Meier) |
|||||
|
A 1 an |
N/A |
79% (71-87) |
71% (55-87) |
29% (3-56) |
32 % (8-56) |
|
A 2 ans |
N/A |
60% (50-70) |
41% (21-60) |
10% (0-28) |
24 % (2-47) |
|
Réponse cytogénétiquec (%) |
|||||
|
RCyM (IC à 95 %) |
62% (57-67) |
40% (33-48) |
34% (25-44) |
52% (37-67) |
57 % (41-71) |
|
RCyC (IC à 95 %) |
54% (48-59) |
33% (26-41) |
27% (19-36) |
46% (31-61) |
54 % (39-69) |
|
Survie (%, selon les estimations de Kaplan-Meier) |
|||||
|
Survie sans progression |
|||||
|
A 1 an |
97% (88-94) |
64% (57-72) |
35% (25-44) |
14% (3-25) |
21 % (9-34) |
|
A 2 ans |
80% (75-84) |
46% (38-54) |
20% (11-29) |
5% (0-13) |
12 % (2-23) |
|
Globale |
|||||
|
A 1 an |
97% (95-99) |
83% (77-89) |
48% (38-59) |
30% (14-47) |
35 % (20-51) |
|
A 2 ans |
94% (91-97) |
72% (64-79) |
38% (27-50) |
26% (10-42) |
31 % (16-47) |
Les données présentées dans ce tableau proviennent d’études utilisant une dose initiale de 70 mg deux fois par jour. Voir rubrique 4.2 pour la dose initiale recommandée.
a Les chiffres en gras correspondent aux résultats des critères primaires.
b Critère de réponse hématologique (toutes les réponses confirmées après 4 semaines) : Réponse hématologique majeure : (RHMa) = Réponse Hématologique Complète (RHC) + absence de signe de leucémie (NEL)
RHC (LMC chronique) : numération leucocytaire ≤ LSN institutionnel, plaquettes < 450 000/mm3 , absence de blastes ou de promyélocytes dans le sang périphérique, myélocytes + métamyélocytes dans le sang périphérique < 5%, basophiles circulants dans le sang périphérique < 20%, et absence de toute localisation extramédullaire.
RHC (LMC avancée/LAL Ph+) : numération leucocytaire ≤ LSN institutionnel, PN (polynucléaires neutrophiles) ≥ 1 000/mm3, plaquettes ≥ 100 000/mm3, absence de blaste ou de promyélocyte dans le sang périphérique, blastes médullaires ≤ 5 %, myélocytes + métamyélocytes dans le sang périphérique < 5 %, basophiles circulants dans le sang périphérique < 20 % et absence de toute localisation extramédullaire.
NEL (absence de signe de leucémie) : mêmes critères que pour la RHC mais PN ≥ 500/mm3 et < 1 000/mm3, et plaquettes ≥ 20 000/mm3 et ≤ 100 000/mm3.
c Critère de réponse cytogénétique : complète (0 % de métaphases Ph+) ou partielle (> 0 % - 35 %). Réponse Cytogénétique Majeure (RCyM) (0 % - 35 %) associant les réponses complète et partielle.
N/A : Non applicable ; IC = intervalle de confiance ; LSN = limite supérieure de la normale
L'effet d’une transplantation médullaire après traitement par dasatinib n’a pas été complètement évalué.
Etude clinique de phase III chez les patients atteints de LMC en phase chronique, accélérée, ou blastique lymphoïde et de LAL Ph+ et résistants ou intolérants à l'imatinib
Des études randomisées, en ouvert, ont été menées afin d'évaluer l'efficacité du dasatinib administré une fois par jour comparé à une administration deux fois par jour. Les résultats décrits ci-dessous sont basés sur un suivi minimum de 2 ans et 7 ans après le début du traitement par dasatinib.
Etude 1
Dans l'étude en phase chronique de LMC, le critère primaire était la RCyM chez les patients résistants à l'imatinib. Le principal critère secondaire était la RCyM par niveau de dose totale quotidienne chez les patients résistants à l'imatinib. Les autres critères secondaires comprenaient la durée de la RCyM, la SSP et la survie globale. Un total de 670 patients, dont 497 étaient résistants à l'imatinib, ont été randomisés dans les groupes dasatinib 100 mg une fois par jour, 140 mg une fois par jour, 50 mg deux fois par jour ou 70 mg deux fois par jour. La durée médiane de traitement pour tous les patients encore sous traitement avec un suivi minimum de 5 ans (n=205) était de 59 mois (de 28 à 66 mois). La durée médiane de traitement pour tous les patients avec un suivi de 7 ans était de 29,8 mois (de < 1 à 92,9 mois).
L'efficacité a été obtenue dans tous les groupes de traitement par le dasatinib, avec en ce qui concerne le critère primaire, un schéma une prise par jour démontrant une efficacité comparable (non-infériorité) à celle du schéma deux prises par jour (différence dans la RCyM 1,9%; intervalle de confiance à 95% [-6,8% - 10,6%]) ; toutefois, la dose de 100 mg une fois par jour a démontré une amélioration de la sécurité et de la tolérance. Les résultats d'efficacité sont présentés dans les Tableaux 12 et 13.
Tableau 12 : Efficacité du dasatinib dans l'étude de phase III d’optimisation de dose : phase chronique de LMC chez les patients résistants ou intolérants à l’imatinib (résultats à 2 ans)a
|
Tous les patients |
n=167 |
|
Patients résistants à l'imatinib |
n=124 |
|
Taux de Réponse hématologiqueb (%) (IC 95 %) |
|
|
RHC |
92% (86-95) |
|
Réponse cytogénétiquec (%) (IC 95%) |
|
|
RCyM |
|
|
Tous les patients |
63% (56-71) |
|
Patients résistants à l’imatinib |
59% (50-68) |
|
RCyC |
|
|
Tous les patients |
50% (42-58) |
|
Patients résistants à l’imatinib |
44% (35-53) |
|
Réponse moléculaire majeure chez les patients ayant obtenu une RCyCd (%) (IC 95%) |
|
|
Tous les patients |
69% (58-79) |
|
Patients résistants à l’imatinib |
72% (58-83) |
a Résultats rapportés avec la dose initiale recommandée de 100 mg une fois par jour.
b Critères de réponse hématologique (toutes les réponses confirmées après 4 semaines) : Réponse hématologique complète (RHC) (LMC chronique) : GB ≤ LSN, plaquettes < 450 000/mm3 , pas de blastes ou de promyélocytes dans le sang périphérique, < 5% myelocytes + métamyélocytes dans le sang périphérique, basophiles < 20% dans le sang périphérique, et aucune participation extra-médullaire.
c Critères de réponse cytogénétique : complète (0% de métaphases Ph +) ou partielle (> 0% - 35 %). RCyM (0% - 35%) associant les réponses complète et partielle.
d Critères de réponse moléculaire majeure : définie par un taux de transcrits de BCR-ABL/transcrits contrôles ≤ 0,1% par RQ-PCR dans les échantillons de sang périphérique.
Tableau 13 : Efficacité à long terme du dasatinib dans l'étude de phase III d’optimisation de dose : patients en phase chronique de LMC résistants ou intolérants à l’imatiniba
|
|
Période de suivi minimum |
|||
|
|
1 an |
2 ans |
5 ans |
7 ans |
|
Réponse moléculaire majeure |
||||
|
Tous les patients |
NA |
37 % (57/154) |
44 % (71/160) |
46 % (73/160) |
|
Patients résistants à l’imatinib |
NA |
35 % (41/117) |
42 % (50/120) |
43 % (51/120) |
|
Patients intolérants à l’imatinib |
NA |
43 % (16/37) |
53 % (21/40) |
55 % (22/40) |
|
Survie sans progressionb |
||||
|
Tous les patients |
90 % (86, 95) |
80 % (73, 87) |
51 % (41, 60) |
42 % (33, 51) |
|
Patients résistants à l’imatinib |
88 % (82, 94) |
77 % (68, 85) |
49 % (39, 59) |
39 % (29, 49) |
|
Patients intolérants à l’imatinib |
97 % (92, 100) |
87 % (76, 99) |
56 % (37, 76) |
51 % (32, 67) |
|
Survie globale |
||||
|
Tous les patients |
96 % (93, 99) |
91 % (86, 96) |
78 % (72, 85) |
65 % (56, 72) |
|
Patients résistants à l’imatinib |
94 % (90, 98) |
89 % (84, 95) |
77 % (69, 85) |
63 % (53, 71) |
|
Patients intolérants à l’imatinib |
100 % (100, 100) |
95 % (88, 100) |
82 % (70, 94) |
70 % (52, 82) |
a Résultats rapportés pour la dose initiale recommandée de 100 mg une fois par jour.
b La progression a été définie par une élévation du taux de GB, une perte de RHC ou de RCyM, une augmentation ≥ 30% des métaphases Ph+, un passage en PA/PB de la maladie ou un décès. La SSP a été analysée en intention de traiter et les patients ont été suivis pour les évènements en incluant les traitements ultérieurs.
Selon les estimations de Kaplan-Meier, la proportion de patients traités avec dasatinib 100 mg une fois par jour qui ont maintenu une RCyM pendant 18 mois était de 93% (IC à 95% [88%-98%]).
L'efficacité a également été évaluée chez les patients intolérants à l'imatinib. Dans cette population de patients qui ont reçu 100 mg une fois par jour, une RCyM a été obtenue dans 77% des cas et une RCyC dans 67% des cas.
Etude 2
Dans l'étude en phase avancée de LMC et LAL Ph+, le critère primaire était la RHMa. Un total de 611 patients ont été randomisés dans les groupes dasatinib 140 mg une fois par jour ou 70 mg deux fois par jour. La durée médiane de traitement était approximativement de 6 mois (intervalle 0,03-31 mois).
Sur le critère primaire d'efficacité, le schéma une prise par jour a démontré une efficacité comparable (non-infériorité) à celle du schéma deux prises par jour (différence dans la RHMa de 0,8 % ; intervalle de confiance [-7,1 % - 8,7 %]) ; toutefois, la dose de 140 mg une fois par jour a démontré une amélioration de la sécurité et de la tolérance. Les taux de réponse sont présentés dans le tableau 14.
Tableau 14 : Efficacité du dasatinib dans l'étude de phase III d'optimisation de dose : phase avancée de LMC et LAL Ph+ (résultats à 2 ans)a
|
|
Accéléré (n= 158) |
Blastique myéloïde (n= 75) |
Blastique lymphoïde (n= 33) |
LAL Ph+ (n = 40) |
|
RHMab (IC à 95 %) |
66% (59-74) |
28% (18-40) |
42% (26-61) |
38% (23-54) |
|
RHCb (IC à 95 %) |
47% (40-56) |
17% (10-28) |
21% (9-39) |
33% (19-49) |
|
NELb (IC à 95 %) |
19% (13-26) |
11% (5-20) |
21% (9-39) |
5% (1-17) |
|
RCyMc (IC à 95 %) |
39% (31-47) |
28% (18-40) |
52% (34-69) |
70% (54-83) |
|
RCyC (IC à 95 %) |
32% (25-40) |
17% (10-28) |
39% (23-58) |
50% (34-66) |
a Résultats rapportés avec la dose initiale recommandée de 140 mg une fois par jour (voir rubrique 4.2).
b Critère de réponse hématologique (toutes les réponses confirmées après 4 semaines) : Réponse hématologique majeure (RHMa) = réponse hématologique complète (RHC) + pas de preuve de leucémie (NEL).
RHC : numération leucocytaire ≤ LSN, PNN ≥ 1 000/mm3, plaquettes ≥ 100 000/mm3, absence de blastes ou de promyélocytes dans le sang périphérique, blastes myéloïdes et sang ≤ 5 %, myélocytes plus métamyélocytes dans le sang périphérique < 5 %, basophiles dans le sang périphérique < 20 %, et pas d'envahissement extra-médullaire.
NEL : mêmes critères que pour la RHC mais ANC ≥ 500/mm3 et < 1 000/mm3, ou plaquettes ≥ 20 000/mm3 et ≤ 100 000/mm3.
c RCyM associe les réponses complète (0 % Ph+ en métaphase) et partielle (> 0 %-35 %)
IC = Intervalle de Confiance LNS : limite normale supérieure
Chez les patients en phase accélérée de LMC traités par la dose de 140 mg une fois par jour, la durée médiane de RHMa et la survie globale médiane n'ont pas été atteintes et la médiane de SSP était de 25 mois.
Chez les patients en phase blastique myéloïde de LMC sous traitement par la dose de 140 mg une fois par jour, la durée médiane de RHMa était de 8 mois; la médiane de SSP était de 4 mois et la médiane de survie globale était de 8 mois. Chez les patients en phase blastique lymphoïde de LMC sous traitement par la dose de 140 mg une fois par jour, la durée médiane de RHMa était de 5 mois; la médiane de SSP était de 5 mois et la médiane de survie globale était de 11 mois.
Chez les patients LAL Ph+ sous traitement par la dose de 140 mg une fois par jour, la durée médiane de RHMa était de 5 mois ; la médiane de SSP était de 4 mois et la médiane de survie globale était de 7 mois.
Population pédiatrique
Patients pédiatriques atteints de LMC
Parmi les 130 patients atteints de LMC en phase chronique (LMC PC) traités dans les deux études pédiatriques, un essai de recherche de dose de Phase I, non randomisé, en ouvert et un essai de Phase II non randomisé en ouvert, 84 patients (exclusivement inclus dans l’essai de Phase II) étaient atteints d’une LMC PC nouvellement diagnostiquée et 46 patients (17 dans l’essai de Phase I et 29 dans l’essai de Phase II) étaient résistants ou intolérants au précédent traitement par imatinib. Quatre-vingt-dix-sept des 130 patients pédiatriques atteints de LMC PC ont été traités avec dasatinib comprimés à la dose de 60 mg/m2 une fois par jour (dose maximale de 100 mg une fois par jour pour les patients ayant une surface corporelle importante). Tous les patients ont été traités jusqu’à une progression de la maladie ou l’atteinte d’un niveau de toxicité inacceptable.
Les principaux critères d’évaluation de l’efficacité étaient les suivants : réponse cytogénétique complète (RCyC), réponse cytogénétique majeure (RCyM) et réponse moléculaire majeure (RMM). Les résultats sont indiqués dans le Tableau 15.
Tableau 15 : Efficacité du dasatinib chez les patients pédiatriques atteints de LMC PC Réponse cumulée au fil du temps selon une période de suivi minimale
|
|
3 mois |
6 mois |
12 mois |
24 mois |
|
RCyC (IC à 95 %) |
|
|
|
|
|
Nouvellement diagnostiqué (n = 51)a |
43.1 % (29.3, 57.8) |
66.7 % (52.1, 79.2) |
96.1 % (86.5, 99.5) |
96.1 % (86.5, 99.5) |
|
Avant imatinib (n=46)b |
45.7 % (30.9, 61.0) |
71.7 % (56.5, 84.0) |
78.3 % (63.6, 89.1) |
82.6 % (68.6, 92.2) |
|
RCyM (IC à 95%) |
|
|
|
|
|
Nouvellement diagnostiqué (n = 51)a |
60.8 % (46.1, 74.2) |
90.2 % (78.6, 96.7) |
98.0 % (89.6, 100) |
98.0 % (89.6, 100) |
|
Avant imatinib (n=46)b |
60.9 % (45.4, 74.9) |
82.6 % (68.6, 92.2) |
89.1 % (76.4, 96.4) |
89.1 % (76.4, 96.4) |
|
RMM (IC à 95 %) |
|
|
|
|
|
Nouvellement diagnostiqué (n = 51)a |
7.8 % (2.2, 18.9) |
31.4 % (19.1, 45.9) |
56.9 % (42.2, 70.7) |
74.5 % (60.4, 85.7) |
|
Avant imatinib (n=46)b |
15.2 % (6.3, 28.9) |
26.1 % (14.3, 41.1) |
39.1 % (25.1, 54.6) |
52.2 % (36.9, 67.1) |
a Patients de l’étude pédiatrique de Phase II sur la LMC PC nouvellement diagnostiquée recevant la formulation en comprimés par voie orale
b Patients des études pédiatriques de Phase I et Phase II sur la LMC PC résistants ou intolérants à l’imatinib recevant la formulation en comprimés par voie orale
Dans l’étude pédiatrique de Phase I, après un suivi minimal de 7 ans, parmi les 17 patients atteints de LMC PC résistants ou intolérants à l’imatinib, la durée médiane de la SSP était 53,6 mois et le taux de SG était de 82,4 %.
Dans l’étude pédiatrique de Phase II, chez les patients recevant la formulation en comprimés, le taux de SSP à 24 mois évalué chez les 51 patients atteints de LMC PC nouvellement diagnostiquée était de 94,0% (82,6 ; 98,0), et de 81,7% (61,4 ; 92,0) parmi les 29 patients atteints de LMC PC résistants ou intolérants à l’imatinib. Après un suivi de 24 mois, la SG chez les patients nouvellement diagnostiqués était de 100%, et de 96,6% chez les patients résistants ou intolérants à l’imatinib.
Dans l’étude pédiatrique de Phase II, la LMC a progressé vers la phase blastique chez 1 patient nouvellement diagnostiqué et 2 patients intolérants ou résistants à l’imatinib.
Trente-trois patients pédiatriques atteints de LMC PC nouvellement diagnostiquée ont reçu du dasatinib poudre pour suspension buvable à une dose de 72 mg/m2. Cette dose correspond à une exposition 30% plus faible comparativement à la dose recommandée (voir rubrique 5.2 du Résumé des caractéristiques du produit du dasatinib poudre en suspension buvable). Chez ces patients, la RCyC et la RMM étaient, RCyC : 87,9 % IC à 95 % : (71,8-96,6)] et la RMM : 45,5 % [IC à 95 % : (28,1-63,6)] à 12 mois.
Parmi les patients pédiatriques atteints de LMC PC traités par dasatinib et précédemment exposés à l’imatinib, les mutations observées à la fin du traitement étaient les suivantes : T315A, E255K et F317L. Cependant, les mutations E255K et F317L avaient également été observées avant le traitement. Aucune mutation n’a été observée à la fin du traitement chez les patients atteints de LMC PC nouvellement diagnostiquée.
Patients pédiatriques atteints de LAL
L’efficacité du dasatinib en association à une chimiothérapie a été évaluée dans une étude pivot chez des patients pédiatriques âgés de plus d’un an atteint de LAL Ph+ nouvellement diagnostiquée.
Dans cette étude de phase II, multicentrique, historiquement contrôlée, portant sur le dasatinib associé à une chimiothérapie standard, 106 patients pédiatriques atteints de LAL Ph+ nouvellement diagnostiquée, dont 104 patients avaient une LAL Ph+ confirmée, les patients ont reçu du dasatinib à une dose quotidienne de 60mg/m² selon un schéma posologique continu pendant 24 mois au maximum, en association avec une chimiothérapie. Quatre-vingt-deux patients ont pris exclusivement des comprimés de dasatinib et 24 patients ont pris du dasatinib en poudre pour suspension buvable au moins une fois, dont 8 ayant pris exclusivement le dasatinib en poudre pour suspension buvable. La chimiothérapie de base était la même que celle utilisée dans l’essai AIEOP-BFM ALL 2000 (protocole de chimiothérapie avec plusieurs agents chimiothérapeutiques standards). Le principal critère d’évaluation de l’efficacité était : la survie sans événement (SSE) à 3 ans qui était de 65.5% (55,5 ; 73,7).
Le taux de maladie résiduelle minimale (MRD) négative évalué par réarrangement Ig/TCR était de 71,7% à la fin de la consolidation chez tous les patients traités. Lorsque ce taux était fondé sur les 85 patients ayant des évaluations Ig/TCR évaluables, l’estimation était de 89,4%. Les taux de MRD négative à la fin de l’induction et de la consolidation, mesurés par cytométrie en flux, étaient de 66,0% et 84,0%, respectivement.
5.2. Propriétés pharmacocinétiques
Les propriétés pharmacocinétiques du dasatinib ont été évaluées chez 229 adultes sains et chez 84 patients.
Absorption
Le dasatinib est rapidement absorbé chez les patients après administration orale, avec un pic de concentration entre 0,5 et 3 heures. Après administration orale, l'augmentation de l'exposition moyenne (ASCτ) est approximativement proportionnelle à l'augmentation de la dose pour des posologies allant de 25 mg à 120 mg deux fois par jour. La demi-vie moyenne globale du dasatinib est approximativement de 5 à 6 heures chez les patients.
Les données obtenues chez les sujets sains après une dose unique de 100 mg de dasatinib 30 minutes après un repas riche en graisses ont révélé une augmentation de 14 % de l'ASC moyenne du dasatinib. L'absorption d'un repas pauvre en graisses 30 minutes avant l'administration du dasatinib a entraîné une augmentation moyenne de 21 % de l'ASC du dasatinib.
Les effets de la prise alimentaire sur l'exposition au dasatinib ne sont pas cliniquement significatifs. La variabilité de l’exposition au dasatinib est plus élevée à jeun (CV 47%) par rapport aux repas pauvres en graisses (CV 39%) et aux repas riches en graisses (CV 32%).
Sur la base de l'analyse pharmacocinétique de population, la variabilité de l'exposition au dasatinib a été estimée comme étant principalement due à la variabilité inter-occasion de la biodisponibilité (CV 44%) et, dans une moindre mesure, à la variabilité interindividuelle de la biodisponibilité et de la variabilité interindividuelle de la clairance (respectivement CV 30% et 32%). La variabilité interoccasion aléatoire de l'exposition ne devrait affecter ni l'exposition cumulative ni l’efficacité ou la sécurité.
Distribution
Chez les patients, le dasatinib a un volume de distribution apparent important (2 505 L), coefficient de variation (CV%93%), suggérant que le médicament est fortement distribué dans l'espace extravasculaire. A des concentrations de dasatinib cliniquement significatives, la liaison aux protéines plasmatiques était de 96 % approximativement sur la base d'expériences in vitro.
Biotransformation
Le dasatinib est fortement métabolisé chez l'Homme par de multiples enzymes impliquées dans la production de métabolites. Chez des sujets sains ayant reçu une dose de 100 mg de dasatinib marqué au 14C, le dasatinib sous forme inchangée représentait 29 % de la radioactivité circulante dans le plasma. Les concentrations plasmatiques et l'activité mesurée in vitro indiquent que les métabolites de dasatinib ne semblent pas jouer un rôle majeur dans la pharmacologie du produit. Le CYP3A4 est une enzyme majeure responsable du métabolisme du dasatinib.
Elimination
La demi-vie terminale moyenne du dasatinib est de 3 heures à 5 heures. La clairance orale apparente moyenne est de 363,8 L/h (CV% 81,3%).
L'élimination s'effectue principalement dans les fèces, en grande partie sous forme de métabolites. Après une dose orale unique de dasatinib marqué au 14C, approximativement 89 % de la dose a été éliminée dans les 10 jours, 4 % et 85 % de la radioactivité étant retrouvés respectivement dans les urines et les fèces. Le dasatinib sous forme inchangée représente respectivement 0,1 % et 19 % de la dose dans les urines et les fèces, le reste étant éliminé sous forme de métabolites.
Altérations des fonctions hépatiques et rénale
Les effets de l'insuffisance hépatique sur la pharmacocinétique du dasatinib en dose unique ont été évalués chez 8 patients atteints d’insuffisance hépatique modérée ayant reçu une dose de 50 mg et chez 5 patients atteints d’insuffisance hépatique sévère ayant reçu une dose de 20 mg, comparativement à des sujets sains ayant reçu 70 mg de dasatinib. La Cmax moyenne et l'ASC du dasatinib ajustée pour la dose de 70 mg ont diminué de 47 % et 8 % respectivement chez les patients présentant une insuffisance hépatique modérée comparativement aux sujets ayant une fonction hépatique normale. Chez les patients présentant une insuffisance hépatique sévère, la Cmax moyenne et l'ASC du dasatinib ajustée pour la dose de 70 mg ont diminué de 43 % et 28 %, respectivement, comparativement aux sujets ayant une fonction hépatique normale (voir rubriques 4.2 et 4.4).
Le dasatinib et ses métabolites sont peu excrétés par le rein.
Population pédiatrique
La pharmacocinétique du dasatinib a été évaluée chez 104 patients pédiatriques atteints de leucémie ou de tumeurs solides (72 ayant reçu la formulation en comprimés et 32 ayant reçu la poudre pour suspension buvable).
Dans une étude pédiatrique de pharmacocinétique, l’exposition au dasatinib à des doses normalisées (Cmoy, Cmin et Cmax) est similaire entre les 21 patients atteints de LMC PC et les 16 patients atteints de LAL Ph+.
La pharmacocinétique de la formulation en comprimés du dasatinib a été évaluée chez 72 patients pédiatriques atteints de leucémie ou de tumeurs solides récidivantes ou réfractaires à des posologies par voie orale allant de 60 à 120 mg/m2 une fois par jour et de 50 à 110 mg/m2 deux fois par jour. Les données ont été groupées à travers deux études et indiquent que le dasatinib était rapidement absorbé. La Tmax moyenne a été observée entre 0,5 et 6 heures et la demi-vie moyenne se situait entre 2 et 5 heures à travers tous les niveaux de doses et de groupes d’âges. La PK du dasatinib a montré une proportionnalité de dose avec une augmentation de l’exposition liée à la dose observée chez les patients pédiatriques. Il n’y a pas eu de différence significative de la PK du dasatinib entre les enfants et les adolescents. Les moyennes géométriques de la Cmax, ASC (0-T), et l’ASC (INF) normalisées en fonction de la dose ont semblé similaires entre les enfants et les adolescents à différents niveaux de dose. Une simulation basée sur un modèle pharmacocinétique de la population (PPK) a permis de prédire que la recommandation de dosage selon des intervalles de poids décrite pour le comprimé, dans la rubrique 4.2, devrait fournir une exposition similaire à celle d’un comprimé à la dose de 60 mg/m2. Ces données doivent être prises en compte si des patients doivent passer du comprimé à la poudre pour suspension buvable, ou vice-versa.
5.3. Données de sécurité préclinique
Les principales toxicités sont survenues dans les systèmes gastro-intestinal, hématopoïétique et lymphoïde. La toxicité gastro-intestinale était dose-limitante chez le rat et le singe, l'intestin ayant été constamment un organe cible. Chez le rat, des diminutions minimes à modérées des composantes érythrocytaires ont été accompagnées de modifications de la moelle osseuse. Des changements similaires ont été observés chez le singe avec une incidence moindre. Une toxicité lymphoïde s'est traduite chez le rat par une déplétion lymphoïde des ganglions lymphatiques, de la rate et du thymus, et par une diminution du poids des organes lymphoïdes. Ces modifications des systèmes gastro-intestinal, hématopoïétique et lymphoïde ont été réversibles après l'arrêt du traitement.
Des modifications rénales ont été observées chez les singes traités pendant une période allant jusqu’à 9 mois, et se sont limitées à une augmentation de la minéralisation rénale. Des hémorragies cutanées ont été observées dans une étude de toxicité aiguë à dose orale unique chez le singe, mais n'ont pas été observées dans des études de doses répétées chez le singe comme chez le rat. Chez le rat, le dasatinib a inhibé l'agrégation plaquettaire in vitro et a prolongé le temps de saignement in vivo, mais n'a pas provoqué d'hémorragie spontanée.
L'activité in vitro du dasatinib sur hERG et sur les fibres de Purkinje suggèrent un potentiel prolongement de la repolarisation ventriculaire cardiaque (intervalle QT). Cependant, dans une étude de dose unique in vivo sur des singes conscients soumis à des mesures télémétriques, il n’a pas été constaté de changement de l'intervalle QT ou de l'aspect des ondes de l'ECG.
Le dasatinib n'a pas montré de pouvoir mutagène dans les tests in vitro sur les cellules bactériennes (test d’Ames), ni de pouvoir génotoxique dans le test du micronoyau effectué in vivo chez le rat. Le dasatinib a montré un pouvoir clastogène in vitro sur des cellules d’Ovaire de Hamster Chinois (OHC) en division.
Dans une étude conventionnelle évaluant la fertilité et le développement embryonnaire précoce chez le rat, le dasatinib n'a pas affecté la fertilité des mâles ou des femelles, mais a induit une létalité embryonnaire à des niveaux de doses avoisinant l'exposition humaine en clinique. Dans les études de développement embryo-fœtal, le dasatinib a également induit chez le rat une létalité embryonnaire associée à une diminution de la taille des portées, ainsi qu'une altération du squelette fœtal chez le rat et le lapin. Ces effets sont survenus à des doses qui n'induisaient pas de toxicité maternelle indiquant que le dasatinib est un toxique sélectif de la reproduction depuis l'implantation jusqu'à l'achèvement de l'organogenèse.
Chez la souris, l'immunosuppression induite par le dasatinib était liée à la dose, et a pu être efficacement contrôlée par une réduction de dose et/ou des changements du schéma thérapeutique. Le dasatinib a montré in vitro un potentiel phototoxique dans un test de relargage du rouge neutre dans des fibroblastes de souris. Le dasatinib est considéré comme non-phototoxique in vivo après une administration unique orale à des souris glabres femelles, à des expositions allant jusqu'à 3 fois l'exposition chez l'Homme après une administration de la dose thérapeutique recommandée (basée sur l'ASC).
Dans une étude de cancérogénicité menée sur 2 ans, des doses de dasatinib à 0,3, 1 et 3 mg/kg/jour ont été administrées à des rats par voie orale. L'exposition à la plus forte dose a entraîné un niveau plasmatique (ASC) généralement équivalent à l'exposition chez l'Homme aux doses initiales recommandées allant de 100 mg à 140 mg par jour. Une augmentation statistiquement significative de la fréquence groupée des carcinomes épidermoïdes, des papillomes de l'utérus et du col de l'utérus chez les femelles traitées par de fortes doses, et des adénomes de la prostate chez les males traités avec de faibles doses a été observée. La pertinence de ces données chez l'Homme n'est pas connue.
Noyau du comprimé :
Cellulose microcristalline (E460), lactose monohydraté, croscarmellose sodique, hydroxypropylcellulose (E463), stéarate de magnésium (E470b)
Pelliculage du comprimé :
Poly(alcool vinylique) (E1203), dioxyde de titane (E171), talc (E553b), monostéarate de glycéryle (E471), laurilsulfate de sodium
4 ans
6.4. Précautions particulières de conservation
Ce médicament ne nécessite pas de précautions particulières de conservation.
6.5. Nature et contenu de l'emballage extérieur
Plaquettes prédécoupées unitaires (Aluminium-OPA/Aluminium/PVC).
Flacon en polyéthylène haute densité (PEHD) muni d'une fermeture « sécurité enfant » en polypropylène (PP) et d’une cartouche en plastique (PEHD) contenant du gel de silice.
Boîte contenant 30 × 1, 60 × 1 ou 100 × 1 comprimé pelliculé sous plaquettes prédécoupées unitaires.
Boîte contenant un flacon de 30 comprimés pelliculés.
Toutes les présentations peuvent ne pas être commercialisées.
6.6. Précautions particulières d’élimination et de manipulation
Les comprimés pelliculés sont constitués d'un comprimé noyau entouré d'un film pelliculé visant à éviter que les professionnels de santé soient exposés à la substance active. Cependant, les professionnels de santé doivent porter des gants de chimiothérapie jetables, dans le cas où les comprimés pelliculés sont involontairement écrasés ou cassés, ceci de manière à minimiser les risques d'exposition dermique.
Tout médicament non utilisé ou déchet doit être éliminé conformément à la réglementation en vigueur.
7. TITULAIRE DE L’AUTORISATION DE MISE SUR LE MARCHE
EG LABO – LABORATOIRES EUROGENERICS
CENTRAL PARK
9-15 RUE MAURICE MALLET
92130 ISSY-LES-MOULINEAUX
8. NUMERO(S) D’AUTORISATION DE MISE SUR LE MARCHE
· 34009 301 774 6 4 : Comprimés pelliculés sous plaquettes prédécoupées unitaires (Aluminium-OPA/Aluminium/PVC) ; boîte de 30x1.
· 34009 301 774 7 1 : Comprimés pelliculés en flacon (PEHD) avec fermeture de sécurité-enfant (PP) ; boîte de 30.
· 34009 550 646 5 7 : Comprimés pelliculés sous plaquettes prédécoupées unitaires (Aluminium-OPA/Aluminium/PVC) ; boîte de 60x1.
· 34009 550 646 6 4 : Comprimés pelliculés sous plaquettes prédécoupées unitaires (Aluminium-OPA/Aluminium/PVC) ; boîte de 100x1.
9. DATE DE PREMIERE AUTORISATION/DE RENOUVELLEMENT DE L’AUTORISATION
[à compléter ultérieurement par le titulaire]
10. DATE DE MISE A JOUR DU TEXTE
[à compléter ultérieurement par le titulaire]
Sans objet.
12. INSTRUCTIONS POUR LA PREPARATION DES RADIOPHARMACEUTIQUES
Liste I.
Médicament soumis à prescription initiale hospitalière de 6 mois.
Prescription initiale et renouvellement réservés aux spécialistes en oncologie ou en hématologie ou aux médecins compétents en cancérologie.
Médicament nécessitant une surveillance particulière pendant le traitement.
ANSM - Mis à jour le : 10/03/2026
DASATINIB EG 100 mg, comprimé pelliculé
Dasatinib
· Gardez cette notice. Vous pourriez avoir besoin de la relire.
· Si vous avez d’autres questions, interrogez votre médecin ou votre pharmacien.
· Ce médicament vous a été personnellement prescrit. Ne le donnez pas à d’autres personnes. Il pourrait leur être nocif, même si les signes de leur maladie sont identiques aux vôtres.
· Si vous ressentez un quelconque effet indésirable, parlez-en à votre médecin ou votre pharmacien. Ceci s’applique aussi à tout effet indésirable qui ne serait pas mentionné dans cette notice. Voir rubrique 4.
1. Qu'est-ce que DASATINIB EG 100 mg, comprimé pelliculé et dans quels cas est-il utilisé ?
2. Quelles sont les informations à connaître avant de prendre DASATINIB EG 100 mg, comprimé pelliculé ?
3. Comment prendre DASATINIB EG 100 mg, comprimé pelliculé ?
4. Quels sont les effets indésirables éventuels ?
5. Comment conserver DASATINIB EG 100 mg, comprimé pelliculé ?
6. Contenu de l’emballage et autres informations.
1. QU’EST-CE QUE DASATINIB EG 100 mg, comprimé pelliculé ET DANS QUELS CAS EST-IL UTILISE ?
DASATINIB EG contient la substance active dasatinib. Ce traitement est utilisé pour traiter la leucémie myéloïde chronique (LMC), chez les patients adultes, adolescents et les enfants âgés d'au moins 1 an. La leucémie est un cancer touchant les globules blancs du sang. Ces cellules permettent habituellement à l'organisme de lutter contre les infections. Les personnes atteintes de LMC présentent une prolifération incontrôlée des globules blancs appelés granulocytes. DASATINIB EG permet de lutter contre la prolifération de ces cellules leucémiques.
DASATINIB EG est utilisé pour traiter les leucémies aiguës lymphoblastiques (LAL) à chromosome Philadelphie (Ph+) chez les patients adultes, les adolescents et les enfants âgés d’au moins 1 an et des LMC en phase blastique lymphoïde, chez les patients adultes pour lesquels les traitements antérieurs ne sont pas bénéfiques. La leucémie est un cancer touchant les globules blancs du sang. Ces cellules permettent habituellement à l’organisme de lutter contre les infections. Chez les personnes atteintes de LAL, les globules blancs appelés lymphocytes ont une croissance trop rapide et une durée de vie trop longue. DASATINIB EG permet de lutter contre la prolifération de ces cellules leucémiques.
Si vous avez des questions sur la manière dont agit DASATINIB EG ou sur les raisons pour lesquelles il vous a été prescrit, veuillez interroger votre médecin.
2. QUELLES SONT LES INFORMATIONS A CONNAITRE AVANT DE PRENDRE DASATINIB EG 100 mg, comprimé pelliculé ?
Ne prenez jamais DASATINIB EG 100 mg, comprimé pelliculé :
· si vous êtes allergique au dasatinib ou à l’un des autres composants contenus dans ce médicament, mentionnés dans la rubrique 6.
Si vous pensez être allergique, demandez conseil à votre médecin.
Avertissements et précautions
Adressez-vous à votre médecin ou pharmacien avant de prendre DASATINIB EG.
· si vous prenez des médicaments fluidifiant le sang ou des médicaments pour prévenir la formation de caillot sanguin (voir « Autres médicaments et DASATINIB EG 100 mg, comprimé pelliculé ») ;
· si vous présentez des problèmes de foie, des problèmes cardiaques, ou si vous en avez déjà eu ;
· si vous avez des difficultés à respirer, des douleurs dans la poitrine ou si vous toussez lorsque vous prenez du dasatinib : ceci peut être un signe évoquant la présence de liquide dans les poumons ou dans le thorax (peut être plus fréquent chez les patients âgés de 65 ans et plus), ou un signe de changement dans les vaisseaux sanguins qui amènent le sang aux poumons ;
· si vous avez déjà eu ou pourriez avoir actuellement une hépatite B. En effet, le dasatinib pourrait réactiver votre hépatite B, ce qui peut être fatal dans certains cas. Les patients seront étroitement surveillés par leur médecin afin de détecter tout signe d’infection avant l’instauration du traitement.
· si vous présentez des ecchymoses (bleus sur la peau), des saignements, de la fièvre, de la fatigue et de la confusion lors de la prise de DASATINIB EG, contactez votre médecin. Cela peut être un signe de lésion des vaisseaux sanguins appelée microangiopathie thrombotique (MAT).
Votre médecin vous surveillera régulièrement de manière à vérifier que le traitement par dasatinib produit les effets escomptés. Vous aurez également des contrôles sanguins réguliers durant toute la durée du traitement par DASATINIB EG.
Enfants et adolescents
Ne pas administrer ce médicament à des enfants âgés de moins de un an. L’expérience de son utilisation est limitée dans ce groupe d’âge. La croissance osseuse et le développement seront étroitement surveillés chez les enfants prenant du DASATINIB EG.
Autres médicaments et DASATINIB EG 100 mg, comprimé pelliculé
Informez votre médecin si vous prenez, avez récemment pris ou pourriez prendre tout autre médicament.
Le dasatinib est principalement éliminé par le foie. Certains médicaments peuvent interférer avec l'effet du dasatinib lorsqu'ils sont pris ensemble.
Ces médicaments ne doivent pas être administrés avec le dasatinib :
· kétoconazole, itraconazole - ce sont des médicaments antifongiques
· érythromycine, clarithromycine, télithromycine - ce sont des antibiotiques
· ritonavir - c'est un médicament antiviral
· phénytoïne, carbamazépine, phénobarbital - ce sont des traitements de l'épilepsie
· rifampicine - c'est un traitement contre la tuberculose
· famotidine, oméprazole - ce sont des médicaments bloquant la sécrétion d'acide gastrique
· millepertuis - une préparation à base de plante obtenue sans ordonnance, utilisée pour traiter la dépression et d'autres états (également connue sous le nom de Hypericum perforatum).
Ne prenez pas de médicaments neutralisant les acides gastriques (antiacides tels que hydroxyde d'aluminium/hydroxyde de magnésium) dans les 2 heures précédant ou suivant la prise de dasatinib.
Informez votre médecin si vous prenez des médicaments fluidifiant le sang ou prévenant la formation de caillot sanguin.
DASATINIB EG 100 mg, comprimé pelliculé avec des aliments et boissons
Ne pas prendre de dasatinib avec un pamplemousse ou du jus de pamplemousse.
Si vous êtes enceinte ou si vous pensez être enceinte, veuillez en informer votre médecin immédiatement. Le dasatinib ne doit pas être utilisé pendant la grossesse à moins que cela ne soit clairement nécessaire. Votre médecin discutera avec vous des risques potentiels de la prise de dasatinib pendant la grossesse.
Il est conseillé aux hommes et aux femmes traités par dasatinib d'utiliser une méthode de contraception efficace durant toute la durée du traitement.
Si vous allaitez, informez votre médecin. Vous devez arrêter l'allaitement durant le traitement par dasatinib.
Conduite de véhicules et utilisation de machines
Faites tout particulièrement attention à la conduite de véhicules et à l'utilisation de machines si vous souffrez d'effets secondaires tels que des étourdissements ou une vision trouble.
DASATINIB EG 100 mg, comprimé pelliculé contient :
· Lactose
Si votre médecin vous a informé(e) d’une intolérance à certains sucres, contactez-le avant de prendre ce médicament.
· Sodium
Ce médicament contient moins de 1 mmol (23 mg) de sodium par comprimé, c.-à-d. qu’il est essentiellement « sans sodium ».
3. COMMENT PRENDRE DASATINIB EG 100 mg, comprimé pelliculé ?
Veillez à toujours prendre ce médicament en suivant exactement les indications de votre médecin. Vérifiez auprès de votre médecin ou pharmacien en cas de doute. DASATINIB EG est prescrit chez l'adulte, les adolescents, et les enfants âgés au moins de un an. .
La dose initiale recommandée pour les patients adultes en phase chronique de LMC est de 100 mg une fois par jour.
La dose initiale recommandée pour les patients adultes en phase accélérée ou en crise blastique de LMC ou pour les patients atteints de LAL Ph+ est de 140 mg une fois par jour.
Le dosage chez les enfants atteints de LMC ou LAL Ph+ en phase chronique s’effectue en fonction du poids.
DASATINIB EG est administré par voie orale une fois par jour sous la forme de DASATINIB EG comprimés ou dasatinib poudre pour suspension buvable. DASATINIB EG comprimés n’est pas recommandé pour les patients pesant moins de 10 kg. La poudre pour suspension buvable doit être utilisée pour les patients pesant moins de 10 kg et les patients ne pouvant pas avaler les comprimés. Un changement de posologie peut être nécessaire lors du passage d'une présentation à une autre (comprimés et poudre pour suspension buvable), donc vous ne devez pas passer de l'un à l'autre.
Le médecin décidera de la bonne présentation et de la posologie en fonction de votre poids, des éventuels effets indésirables et de la réponse au traitement. La posologie initiale de DASATINIB EG pour les enfants est calculée en fonction du poids corporel comme indiqué ci-dessous :
|
Poids corporel (kg) a |
Posologie quotidienne (mg) |
|
de 10 à moins de 20 kg |
40 mg |
|
de 20 à moins de 30 kg |
60 mg |
|
de 30 à moins de 45 kg |
70 mg |
|
au moins 45 kg |
100 mg |
a Le comprimé n’est pas recommandé pour les patients pesant moins de 10 kg, la poudre pour suspension buvable doit être utilisée chez ces patients.
Il n'y a pas de recommandation de posologie pour Dasatinib EG chez les enfants âgés de moins de 1 an.
En fonction de votre réponse au traitement, votre médecin peut suggérer d'augmenter, de diminuer les doses ou de suspendre brièvement le traitement. Pour des doses plus élevées ou plus faibles, vous pourrez être amené à prendre une association de différents dosages.
Comment prendre DASATINIB EG 100 mg, comprimé pelliculé
Prenez vos comprimés à la même heure tous les jours. Avalez les comprimés tels quels. Ne pas les écraser, les couper ou les mâcher. Ne pas prendre des comprimés dispersés. Vous ne pourrez pas être sur de recevoir la bonne dose si vous écrasez, coupez ou dispersez les comprimés. DASATINIB EG. Ils peuvent être pris pendant ou en dehors des repas.
Instructions particulières de manipulation pour DASATINIB EG 100 mg, comprimé pelliculé
Il est peu probable que les comprimés viennent à se casser. Cependant, si cela se produit, les personnes autres que le patient lui-même doivent porter des gants lors de la manipulation de DASATINIB EG.
Durée du traitement par DASATINIB EG 100 mg, comprimé pelliculé
Prenez DASATINIB EG quotidiennement jusqu'à ce que votre médecin vous dise d'arrêter. Veillez à prendre DASATINIB EG aussi longtemps qu'il vous sera prescrit.
Si vous avez pris plus de DASATINIB EG 100 mg, comprimé pelliculé que vous n’auriez dû
Si vous avez accidentellement pris plus de comprimés qu'il ne le faut, veuillez en informer votre médecin immédiatement. Vous pourriez avoir besoin d'une surveillance médicale.
Si vous oubliez de prendre DASATINIB EG 100 mg, comprimé pelliculé
Si vous avez d’autres questions sur l’utilisation de ce médicament, demandez plus d’informations à votre médecin ou à votre pharmacien.
4. QUELS SONT LES EFFETS INDESIRABLES EVENTUELS ?
Les symptômes suivants peuvent être les manifestations d'effets indésirables graves :
· si vous avez des douleurs thoraciques, des difficultés à respirer, si vous toussez ou si vous vous évanouissez ;
· si vous saignez de manière inattendue ou si vous avez des bleus sans avoir de blessure ;
· si vous constatez du sang dans les vomissements, les selles ou les urines, ou si vous avez des selles noires ;
· si vous présentez des signes d'infection tels que fièvre, ou frissons importants ;
· si vous avez de la fièvre, des douleurs à la bouche ou à la gorge, des cloques ou un décollement de la peau et/ou des muqueuses.
Informez immédiatement votre médecin si vous observez l'un de ces effets.
Effets indésirables très fréquents (pouvant affecter plus de 1 personne sur 10)
· Infections (dont infections bactérienne, virale et fongique)
· Cœur et poumons : difficulté respiratoire
· Troubles digestifs : diarrhée, sensation nauséeuse (nausée, vomissements)
· Peau, cheveux, yeux, général : éruption cutanée, fièvre, gonflement du visage, des mains et des pieds, maux de tête, sensations de fatigue ou de faiblesse, saignement
· Douleur : douleurs dans les muscles (pendant ou après l’arrêt du traitement), douleur au ventre (abdominale)
· Les examens peuvent révéler : taux de plaquettes bas, taux de globules blancs bas (neutropénie), anémie, liquide autour des poumons.
Effets indésirables fréquents (pouvant affecter jusqu'à 1 personne sur 10)
· Infections : pneumonie, infection herpétique virale (y compris cytomégalovirus - CMV), infection des voies respiratoires hautes, infection sévère du sang ou des tissus (dont certains cas peu fréquents d'issue fatale)
· Cœur et poumon : palpitations, rythme cardiaque irrégulier, insuffisance cardiaque congestive, faiblesse du muscle cardiaque, pression sanguine élevée, augmentation de la pression sanguine dans les poumons, toux
· Problèmes digestifs : troubles de l'appétit, altération du goût, gonflement et distension du ventre (abdomen), inflammation du côlon, constipation, brûlure d'estomac, ulcération de la bouche, prise de poids, perte de poids, gastrite
· Peau, cheveux, yeux, général : picotement de la peau, démangeaisons, sècheresse de la peau, acné, inflammation de la peau, bruit persistant dans les oreilles, perte des cheveux, transpiration excessive, troubles visuels (dont vision trouble et gêne visuelle), sècheresse des yeux, hématome, dépression, insomnie, bouffées de chaleur, étourdissement, contusion (bleu), anorexie, somnolence, œdème généralisé
· Douleur : douleurs des articulations, faiblesse musculaire, douleurs dans la poitrine, douleurs autour des mains et des pieds, frissons, raideur des muscles ou des articulations, spasme musculaire
· Les examens peuvent révéler : liquide autour du cœur, liquide dans les poumons, arythmie, neutropénie fébrile, saignement gastro-intestinal, taux élevés d'acide urique dans le sang
Effets indésirables peu fréquents (pouvant affecter jusqu'à 1 personne sur 100)
· Cœur et poumon : attaque cardiaque (certaines d'issue fatale), inflammation de la membrane entourant le cœur (poche fibreuse), rythme cardiaque irrégulier, douleur dans la poitrine due au manque d'approvisionnement sanguin du cœur (angine de poitrine), pression sanguine basse, rétrécissement des bronches pouvant entraîner des difficultés respiratoires, asthme, augmentation de la pression sanguine dans les artères (vaisseaux sanguins) des poumons
· Problèmes digestifs : inflammation du pancréas, ulcère peptique, inflammation du tube digestif, ballonnement du ventre (abdomen), déchirement de la peau du canal anal, difficulté à avaler, inflammation de la vésicule biliaire, obstruction des conduits biliaires, reflux gastro-œsophagien (remontée d'acide et d'autres contenus de l’estomac dans la gorge)
· Peau, cheveux, yeux, général : réaction allergique incluant une douleur à la pression, nodules sensibles et rouges sur la peau (érythème noueux), anxiété, confusion, variation de l'humeur, baisse de la libido, évanouissement, tremblement, inflammation des yeux pouvant entraîner des rougeurs ou des douleurs, maladie de peau caractérisée par sensibilité, rougeur, et taches bien délimitées avec apparition brutale de fièvre et augmentation des taux de globules blancs (dermatose neutrophile), perte d’audition, sensibilité à la lumière, troubles visuels, larmoiement, modification de la couleur de la peau, inflammation des tissus graisseux sous la peau, ulcère de la peau, cloque sur la peau, modification des ongles, troubles capillaires, syndrome main-pied, atteinte rénale, urines fréquentes, augmentation du volume des seins chez les hommes, troubles menstruels, malaise et faiblesse générale, baisse de la fonction thyroïdienne, perte de l’équilibre durant la marche, ostéonécrose (maladie qui réduit le flux sanguin des os, pouvant entraîner une perte osseuse et la mort du tissu osseux), arthrite, gonflement de la peau n'importe où sur le corps
· Douleur : inflammation des veines pouvant entraîner rougeurs, sensibilité et gonflement, inflammation du tendon
· Cerveau : perte de mémoire
· Les examens peuvent révéler : résultats anormaux des tests sanguins et possible altération de la fonction rénale due aux déchets produits par la tumeur mourante (syndrome de lyse tumorale), taux faible d'albumine dans le sang, taux faible de lymphocytes (type de globule blanc) dans le sang, taux élevé de cholestérol dans le sang, ganglions lymphatiques gonflés, saignements dans le cerveau, irrégularité de l'activité électrique du cœur, augmentation de la taille du cœur, inflammation du foie, protéine dans les urines, augmentation de la créatine phosphokinase (une enzyme principalement trouvée dans le cœur, le cerveau et les muscles squelettiques), augmentation de la troponine (une enzyme présente essentiellement dans le cœur et les muscles squelettiques), augmentation du taux de gamma-glutamyltransférase (une enzyme présente principalement dans le foie), liquide d'apparence laiteuse autour des poumons (chylothorax)
Effets indésirables rares (pouvant affecter jusqu'à 1 personne sur 1 000)
· Cœur et poumon : augmentation du volume du ventricule droit du cœur, inflammation du muscle cardiaque, ensemble d'évènements conduisant au blocage de l'approvisionnement sanguin du muscle cardiaque (syndrome coronaire aigu), arrêt cardiaque (arrêt de la circulation sanguine du cœur), maladie des artères coronaires (cœur), inflammation du tissu recouvrant le cœur et les poumons, caillots de sang, caillots de sang dans les poumons
· Problème digestif : perte de nutriments vitaux tels que des protéines via le tube digestif, occlusion intestinale, fistule anale (ouverture anormale de l'anus vers la peau autour de l'anus), trouble de la fonction rénale, diabète
· Peau, cheveux, yeux, général : convulsion, inflammation du nerf optique pouvant entraîner une perte partielle ou complète de la vision, taches bleu-pourpré de la peau, fonction thyroïdienne anormalement élevée, inflammation de la thyroïde, ataxie (état associé à un manque de coordination musculaire), difficulté à marcher, fausse couche, inflammation des vaisseaux sanguins de la peau, fibrose
· Cerveau : attaque cérébrale, épisode temporaire de dysfonctionnement neurologique causé par la diminution du flux sanguin, paralysie du nerf facial, démence
· Système immunitaire : réaction allergique sévère
· Tissu musculosquelettique et sous-cutané : fusion retardée des extrémités arrondies qui forment les articulations (épiphyses) ; croissance plus lente ou retardée.
Les autres effets indésirables qui ont été rapportés (fréquence indéterminée) incluaient :
· Inflammation pulmonaire
· Saignement dans l’estomac ou dans les intestins, pouvant être fatal
· Réapparition (réactivation) de l’hépatite B si vous avez déjà eu une hépatite B dans le passé (infection hépatique)
· Une réaction avec de la fièvre, des cloques sur la peau et une ulcération des muqueuses
· Maladie des reins avec des symptômes comprenant un œdème et des résultats anormaux de tests de laboratoire tels que des protéines dans l'urine et de faible taux de protéines dans le sang
· Lésion des vaisseaux sanguins appelée microangiopathie thrombotique (MAT), notamment diminution du nombre de globules rouges, diminution du nombre de plaquettes et formation de caillots sanguins.
Votre médecin surveillera l'apparition de certains de ces effets pendant votre traitement.
Déclaration des effets secondaires
Si vous ressentez un quelconque effet indésirable, parlez-en à votre médecin ou à votre pharmacien. Ceci s’applique aussi à tout effet indésirable qui ne serait pas mentionné dans cette notice. Vous pouvez également déclarer les effets indésirables directement via le système national de déclaration : Agence nationale de sécurité du médicament et des produits de santé (ANSM) et réseau des Centres Régionaux de Pharmacovigilance - Site internet : https://signalement.social-sante.gouv.fr
En signalant les effets indésirables, vous contribuez à fournir davantage d’informations sur la sécurité du médicament.
5. COMMENT CONSERVER DASATINIB EG 100 mg, comprimé pelliculé ?
Tenir ce médicament hors de la vue et de la portée des enfants.
N’utilisez pas ce médicament après la date de péremption indiquée sur l'étiquette du flacon, la plaquette ou la boîte après EXP. La date de péremption fait référence au dernier jour de ce mois.
Ce médicament ne nécessite pas de précautions particulières de conservation.
Ne jetez aucun médicament au tout-à-l’égout ou avec les ordures ménagères. Demandez à votre pharmacien d’éliminer les médicaments que vous n’utilisez plus. Ces mesures contribueront à protéger l’environnement.
6. CONTENU DE L’EMBALLAGE ET AUTRES INFORMATIONS
Ce que contient DASATINIB EG 100 mg, comprimé pelliculé
· La substance active est :
Dasatinib............................................................................................................................. 100 mg
Pour un comprimé pelliculé.
· Les autres composants sont :
Noyau du comprimé : cellulose microcristalline (E460), lactose monohydraté (voir rubrique 2 « Quelles sont les informations à connaître avant de prendre DASATINIB EG 100 mg, comprimé pelliculé ? »), croscarmellose sodique, hydroxypropylcellulose (E463), stéarate de magnésium (E470b)
Pelliculage du comprimé : poly(alcool vinylique) (E1203), dioxyde de titane (E171), talc (E553b), monostéarate de glycéryle (E471), laurilsulfate de sodium.
Qu’est-ce que DASATINIB EG 100 mg, comprimé pelliculé et contenu de l’emballage extérieur
Dasatinib EG 100mg : Comprimé pelliculé blanc à blanc cassé, biconvexe, ovale, comportant la mention « 100 » gravée sur une face, l’autre face étant lisse, et mesurant 14,8 mm × 7,2 mm.
Plaquettes prédécoupées unitaires (Aluminium-OPA/Aluminium/PVC).
Flacon en polyéthylène haute densité (PEHD) muni d'une fermeture « sécurité enfant » en polypropylène (PP) et d’une cartouche en plastique (PEHD) contenant du gel de silice.
Boîte contenant 30 × 1, 60 × 1 ou 100 × 1 comprimé pelliculé sous plaquettes prédécoupées unitaires.
Boîte contenant un flacon de 30 comprimés pelliculés.
Toutes les présentations peuvent ne pas être commercialisées.
Titulaire de l’autorisation de mise sur le marché
EG LABO – LABORATOIRES EUROGENERICS
CENTRAL PARK
9-15 RUE MAURICE MALLET
92130 ISSY-LES-MOULINEAUX
Exploitant de l’autorisation de mise sur le marché
EG LABO – LABORATOIRES EUROGENERICS
CENTRAL PARK
9-15 RUE MAURICE MALLET
92130 ISSY-LES-MOULINEAUX
AHARNON STREET, LIMASSOL INDUSTRIAL ESTATE
3056 LIMASSOL
CHYPRE
OU
STADA ARZNEIMITTEL AG
STADASTRASSE 2 - 18
61118 BAD VILBEL
ALLEMAGNE
OU
CENTRAFARM SERVICES B.V.
VAN DE REIJTSTRAAT 31-E
4814 NE BREDA
PAYS-BAS
OU
STADAPHARM GMBH
FEODOR-LYNEN-STRASSE 35
30625 HANOVRE
ALLEMAGNE
Noms du médicament dans les Etats membres de l'Espace Economique Européen
Ce médicament est autorisé dans les Etats membres de l'Espace Economique Européen sous les noms suivants : Conformément à la réglementation en vigueur.
[À compléter ultérieurement par le titulaire]
La dernière date à laquelle cette notice a été révisée est :
[à compléter ultérieurement par le titulaire]
< {MM/AAAA}>< {mois AAAA}.>
Des informations détaillées sur ce médicament sont disponibles sur le site Internet de l’ANSM (France).