Dernière mise à jour le 01/06/2026
ANTALYRE, collyre en solution en récipient unidose
Ce médicament n'est ou ne sera bientôt plus disponible sur le marché.
Si vous prenez actuellement ce médicament, il vous est recommandé d'en parler avec votre médecin
ou avec votre pharmacien qui pourra vous orienter vers un autre traitement.
Indications thérapeutiques
Classe pharmacothérapeutique - code ATC : S01BC08
Ce collyre est indiqué dans le traitement des irritations oculaires chroniques non infectées.
Service médical rendu (SMR)
Pas de SMR disponible pour ce médicament ( plus d'informations dans l'aide )
Amélioration du service médical rendu (ASMR)
Pas d'ASMR disponible pour ce médicament (plus d'informations dans l'aide )
ANSM - Mis à jour le : 26/08/2021
ANTALYRE, collyre en solution en récipient unidose
2. COMPOSITION QUALITATIVE ET QUANTITATIVE
Acide salicylique................................................................................................................... 0,4 mg
Pour 0,4 ml de collyre
Pour la liste complète des excipients, voir rubrique 6.1.
Collyre en solution en récipient unidose.
4.1. Indications thérapeutiques
Irritations conjonctivales chroniques non infectieuses.
4.2. Posologie et mode d'administration
Instiller 1 goutte de collyre 2 à 4 fois par jour, dans le cul de sac conjonctival de l’œil malade, en regardant vers le haut, et en tirant légèrement la paupière vers le bas.
Mode d’administration
Voie ophtalmique.
Se laver soigneusement les mains avant de procéder à l’instillation.
Eviter le contact de l’embout avec l’œil ou les paupières.
Chaque récipient unidose contient une quantité suffisante pour traiter les deux yeux.
Jeter l’unidose après utilisation. Ne pas la conserver pour une utilisation ultérieure.
Hypersensibilité à la substance active ou à l’un des excipients mentionnés à la rubrique 6.1.
Grossesse, à partir du début du 6ème mois (au-delà de 24 semaines d’aménorrhée) pour des doses d’aspirine supérieures à 100 mg par jour (voir rubrique 4.6)
4.4. Mises en garde spéciales et précautions d'emploi
En cas d’hypersensibilité, arrêt du traitement.
Ne pas injecter, ne pas avaler.
Le traitement ne dépassera pas 10 jours ; au-delà, la conduite à tenir doit être réévaluée. En l’absence d’amélioration comme en cas de persistance des symptômes, prendre un avis médical.
En cas de traitement concomitant par un autre collyre, attendre 15 minutes entre chaque instillation.
4.5. Interactions avec d'autres médicaments et autres formes d'interactions
L’efficacité d’un collyre peut être affectée par l’instillation simultanée d’un autre collyre.
4.6. Fertilité, grossesse et allaitement
Grossesse
Faibles doses, inférieures ou égales à 100 mg par jour :
Les études cliniques montrent que des doses allant jusqu'à 100 mg/jour semblent être sûres dans le cas d’utilisations obstétricales extrêmement limitées nécessitant une surveillance spécialisée.
Doses comprises entre 100 et 500 mg par jour :
L’expérience clinique concernant l’administration de doses comprises entre 100 mg et 500 mg par jour est insuffisante. En conséquence, les recommandations ci-dessous pour les doses supérieures à 500 mg par jour s’appliquent à ces doses.
Doses supérieures ou égales à 500 mg par jour :
L’inhibition de la synthèse des prostaglandines par les AINS peut affecter le déroulement de la grossesse et/ou le développement de l’embryon ou du foetus.
Risques associés à l’utilisation au cours du 1er trimestre
Les données des études épidémiologiques suggèrent une augmentation du risque de fausse-couche, de malformations cardiaques et de gastroschisis, après traitement par un inhibiteur de la synthèse des prostaglandines en début de grossesse. Le risque absolu de malformation cardiovasculaire est passé de moins de 1% dans la population générale, à approximativement 1,5 % chez les personnes exposées aux AINS. Le risque paraît augmenter en fonction de la dose et de la durée du traitement. Chez l’animal, il a été montré que l’administration d’un inhibiteur de la synthèse des prostaglandines provoquait une perte pré et post-implantatoire accrue et une augmentation de la létalité embryo-foetale. De plus, une incidence supérieure de certaines malformations, y compris cardiovasculaires, a été rapportée chez des animaux ayant reçu un inhibiteur de la synthèse des prostaglandines au cours de la phase d’organogénèse de la gestation.
Risques associés à l’utilisation à partir de la 12ème semaine d’aménorrhée et jusqu’à la naissance:
A partir de la 12ème semaine d’aménorrhée et jusqu’à la naissance, tous les AINS, par l’inhibition de la synthèse des prostaglandines, peuvent exposer le fœtus à une atteinte fonctionnelle rénale :
· in utero pouvant s'observer dès 12 semaines d'aménorrhée (mise en route de la diurèse fœtale) : oligoamnios (le plus souvent réversible à l'arrêt du traitement), voire anamnios en particulier lors d'une exposition prolongée.
· à la naissance, une insuffisance rénale (réversible ou non) peut persister en particulier en cas d'exposition tardive et prolongée (avec un risque d'hyperkaliémie sévère retardée).
Risques associés à l’utilisation au-delà de la 24ème semaine d’aménorrhée et jusqu’à la naissance:
Au-delà de la 24ème semaine d’aménorrhée, les AINS peuvent exposer le fœtus à une toxicité cardio-pulmonaire (fermeture prématurée du canal artériel et hypertension artérielle pulmonaire). La construction du canal artériel peut survenir à partir de 5 mois révolus et peut conduire à une insuffisance cardiaque droite fœtale ou néonatale voire une mort fœtale in utero. Ce risque est d'autant plus important que la prise est proche du terme (moindre réversibilité). Cet effet existe même pour une prise ponctuelle.
En fin de grossesse, la mère et le nouveau-né peuvent présenter :
· un allongement du temps de saignement du fait d’une action anti-agrégante pouvant survenir même après administration de très faibles doses de médicament.
· une inhibition des contractions utérines entraînant un retard de terme ou un accouchement prolongé.
En conséquence pour les doses supérieures à 100 mg/j :
· Sauf nécessité absolue, ce médicament ne doit pas être utilisé chez une femme qui envisage une grossesse ou au cours des 5 premiers mois de grossesse (24 premières semaines d’aménorrhée). Si ce médicament est administré chez une femme souhaitant être enceinte ou enceinte de moins de 6 mois, la dose devra être la plus faible possible et la durée du traitement la plus courte possible. Une prise prolongée est fortement déconseillée.
· A partir du début du 6ème mois (au-delà de 24 semaines : toute prise de ce médicament, même ponctuelle, est contre-indiquée. Une prise par mégarde à partir de cette date justifie une surveillance cardiaque et rénale, fœtale et/ou néonatale selon le terme d'exposition. La durée de cette surveillance sera adaptée à la demi-vie d'élimination de la molécule.
L'acide acétylsalicylique passant dans le lait maternel, ce médicament est déconseillé pendant l’allaitement.
Fertilité
Comme tous les AINS, l'utilisation de ce médicament peut temporairement altérer la fertilité féminine, en agissant sur l’ovulation ; il est donc déconseillé chez les femmes souhaitant concevoir un enfant. Chez les femmes rencontrant des difficultés pour concevoir ou réalisant des tests de fertilité, l'arrêt du traitement doit être envisagé
4.7. Effets sur l'aptitude à conduire des véhicules et à utiliser des machines
· Possibilité de réactions allergiques comme pour tous les autres collyres.
Déclaration des effets indésirables suspectés
La déclaration des effets indésirables suspectés après autorisation du médicament est importante. Elle permet une surveillance continue du rapport bénéfice/risque du médicament. Les professionnels de santé déclarent tout effet indésirable suspecté via le système national de déclaration : Agence nationale de sécurité du médicament et des produits de santé (ANSM) et réseau des Centres Régionaux de Pharmacovigilance - Site internet : www.signalement-sante.gouv.fr.
Sans objet.
5. PROPRIETES PHARMACOLOGIQUES
5.1. Propriétés pharmacodynamiques
Classe pharmacothérapeutique : ANTIINFLAMMATOIRES NON STEROIDIENS, code ATC : S01BC08.
Collyre astringent léger.
5.2. Propriétés pharmacocinétiques
5.3. Données de sécurité préclinique
Borax, acide borique, chlorure de sodium, eau pour préparations injectables.
2 ans.
Après ouverture du récipient unidose : le produit doit être utilisé immédiatement.
Jeter le récipient unidose après utilisation.
Ne pas le conserver pour une utilisation ultérieure.
6.4. Précautions particulières de conservation
A conserver dans le suremballage, à une température ne dépassant pas + 25°C.
6.5. Nature et contenu de l'emballage extérieur
Barrette de 5 récipients unidoses (PEBD) de 0,4 ml conditionnée en sachet. Boîte de 10 récipients unidoses.
6.6. Précautions particulières d’élimination et de manipulation
Pas d’exigences particulières.
7. TITULAIRE DE L’AUTORISATION DE MISE SUR LE MARCHE
LES INDUSTRIES
2 RUE DU GABIAN
98000 MONACO
8. NUMERO(S) D’AUTORISATION DE MISE SUR LE MARCHE
· 34009 364 174 5 8 : 0,4 ml en récipient unidose (PEBD) ; boîte de 10.
9. DATE DE PREMIERE AUTORISATION/DE RENOUVELLEMENT DE L’AUTORISATION
[à compléter ultérieurement par le titulaire]
10. DATE DE MISE A JOUR DU TEXTE
[à compléter ultérieurement par le titulaire]
Sans objet.
12. INSTRUCTIONS POUR LA PREPARATION DES RADIOPHARMACEUTIQUES
Médicament non soumis à prescription médicale.
ANSM - Mis à jour le : 26/08/2021
ANTALYRE, collyre en solution en récipient unidose
Acide salicylique
Vous devez toujours utiliser ce médicament en suivant scrupuleusement les informations fournies dans cette notice ou par votre médecin, votre pharmacien ou votre infirmier/ère.
· Gardez cette notice. Vous pourriez avoir besoin de la relire.
· Adressez-vous à votre pharmacien pour tout conseil ou information.
· Si vous ressentez un quelconque effet indésirable, parlez-en à votre médecin, votre pharmacien ou votre infirmier/ère. Ceci s’applique aussi à tout effet indésirable qui ne serait pas mentionné dans cette notice. Voir rubrique 4.
· Vous devez vous adresser à votre médecin si vous ne ressentez aucune amélioration ou si vous vous sentez moins bien.
1. Qu'est-ce que ANTALYRE, collyre en solution en récipient unidose et dans quels cas est-il utilisé ?
2. Quelles sont les informations à connaître avant d'utiliser ANTALYRE, collyre en solution en récipient unidose ?
3. Comment utiliser ANTALYRE, collyre en solution en récipient unidose ?
4. Quels sont les effets indésirables éventuels ?
5. Comment conserver ANTALYRE, collyre en solution en récipient unidose ?
6. Contenu de l’emballage et autres informations.
1. QU’EST-CE QUE ANTALYRE, collyre en solution en récipient unidose ET DANS QUELS CAS EST-IL UTILISE ?
Classe pharmacothérapeutique - code ATC : S01BC08
Ce collyre est indiqué dans le traitement des irritations oculaires chroniques non infectées.
2. QUELLES SONT LES INFORMATIONS A CONNAITRE AVANT D’UTILISER ANTALYRE, collyre en solution en récipient unidose ?
N’utilisez jamais ANTALYRE, collyre en solution en récipient unidose :
· si vous êtes allergique (hypersensible) à la substance active ou à l’un des autres composants contenus dans ce médicament, mentionnés dans la rubrique 6.
· si vous êtes enceinte, à partir du début du 6ème mois de grossesse (au-delà de 24 semaines d’aménorrhée) pour des doses supérieures à 100 mg par jour.
Avertissements et précautions
Ne pas injecter, ne pas avaler.
Le traitement ne dépassera pas 10 jours ; au-delà, la conduite à tenir devra être réévaluée. En l’absence d’amélioration, comme en cas de persistance des symptômes, prendre un avis médical.
En cas de traitement concomitant par un autre collyre, attendre 15 minutes entre chaque instillation.
Enfants et adolescents
Sans objet.
Autres médicaments et ANTALYRE, collyre en solution en récipient unidose
L’efficacité d’un collyre peut être affectée par l’instillation simultanée d’un autre collyre.
Informez votre médecin ou pharmacien si vous utilisez, avez récemment utilisé ou pourriez utiliser tout autre médicament.
ANTALYRE, collyre en solution en récipient unidose avec des aliments, boissons et de l’alcool
Sans objet.
Grossesse, allaitement et fertilité
Si vous êtes enceinte ou que vous allaitez, si vous pensez être enceinte ou planifiez une grossesse, demandez conseil à votre médecin ou pharmacien avant de prendre tout médicament.
Grossesse
Ce médicament contient de l'aspirine (acide acétylsalicylique). Ne prendre AUCUN AUTRE médicament contenant de l’aspirine (y compris les médicaments sans ordonnance).
· jusqu’à 100 mg par jour : Pendant toute la grossesse, si nécessaire, votre médecin spécialiste peut être amené à vous prescrire de l’aspirine à faibles doses (inférieures ou égales à 100 mg par jour), dans des circonstances exceptionnelles nécessitant une surveillance spécialisée. Si tel est le cas, il est très important de respecter scrupuleusement l’ordonnance de votre médecin, sans dépasser les doses prescrites.
· entre 100 et 500 mg par jour : Par mesure de précaution, les recommandations ci-dessous s’appliquent, sauf prescription contraire de votre médecin spécialiste.
· à partir de 500 mg par jour :
o Avant le début du 6ème mois de grossesse (jusqu’à la 24ème semaine d’aménorrhée), vous ne devez pas prendre ce médicament, sauf en cas d’absolue nécessité déterminée par votre médecin, en raison du risque potentiel de fausses couches ou de malformations. Dans ce cas, la dose devra être la plus faible possible et la durée du traitement la plus courte possible.
o A partir du début du 6ème mois jusqu’à la fin de la grossesse (au-delà de la 24ème semaine d’aménorrhée), ce médicament est contre-indiqué, vous ne devez EN AUCUN CAS prendre ce médicament, car ses effets sur votre enfant peuvent avoir des conséquences graves voire fatales, notamment sur le cœur, les poumons et/ou les reins, et cela même avec une seule prise.
Si vous avez pris ce médicament alors que vous étiez enceinte, parlez-en immédiatement à votre gynécologue obstétricien, afin qu’une surveillance adaptée vous soit proposée si nécessaire.
Allaitement
L’aspirine passant dans le lait maternel, ce médicament est déconseillé pendant l’allaitement. Demandez conseil à votre médecin ou à votre pharmacien avant de prendre tout médicament.
Fertilité
L’aspirine, comme tous les anti-inflammatoires non stéroïdiens (AINS), peut altérer la fertilité des femmes et entraîner des difficultés pour devenir enceinte, de façon réversible à l’arrêt du traitement. Informez votre médecin si vous planifiez une grossesse ou si vous avez des difficultés à concevoir.
Conduite de véhicules et utilisation de machines
Une gêne visuelle passagère peut être ressentie après utilisation du collyre.
ANTALYRE, collyre en solution en récipient unidose contient
Sans objet.
3. COMMENT UTILISER ANTALYRE, collyre en solution en récipient unidose ?
Instillez 1 goutte de collyre 2 à 4 fois par jour, dans le cul de sac conjonctival de l’œil malade.
Chaque unidose contient une quantité suffisante de collyre pour traiter les deux yeux.
Ne pas dépasser la posologie recommandée.
Voie ophtalmique
Lavez-vous soigneusement les mains avant de procéder à l’instillation.
|
|
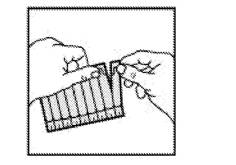
· Sortir la plaquette de son enveloppe protectrice et en détacher une unidose. |
|
|
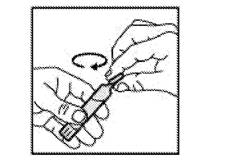
· Ouvrir l’unidose en tournant la partie supérieure. |
|
|
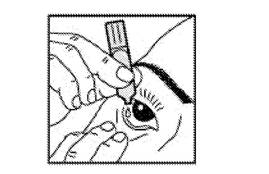
· Instiller 1 goutte dans le cul de sac conjonctival de l’œil malade, en regardant vers le haut, et en tirant légèrement la paupière vers le bas. Evitez le contact de l’embout avec l’œil ou les paupières. |
Jeter l’unidose après utilisation. Ne pas la conserver pour une utilisation ultérieure.
Utilisation chez les enfants et les adolescents
Sans objet.
Si vous avez utilisé plus de ANTALYRE, collyre en solution en récipient unidose que vous n’auriez dû
Consultez immédiatement votre médecin ou votre pharmacien.
Si vous oubliez d’utiliser ANTALYRE, collyre en solution en récipient unidose
Si vous arrêtez d’utiliser ANTALYRE, collyre en solution en récipient unidose
Sans objet.
4. QUELS SONT LES EFFETS INDESIRABLES EVENTUELS ?
· Possibilité de réactions locales : irritation (picotements, brûlures) transitoire, réactions allergiques.
Déclaration des effets secondaires
Si vous ressentez un quelconque effet indésirable, parlez-en à votre médecin, votre pharmacien ou à votre infirmier/ère. Ceci s’applique aussi à tout effet indésirable qui ne serait pas mentionné dans cette notice. Vous pouvez également déclarer les effets indésirables directement via le système national de déclaration : Agence nationale de sécurité du médicament et des produits de santé (ANSM) et réseau des Centres Régionaux de Pharmacovigilance - Site internet: www.signalement-sante.gouv.fr
En signalant les effets indésirables, vous contribuez à fournir davantage d’informations sur la sécurité du médicament.
5. COMMENT CONSERVER ANTALYRE, collyre en solution en récipient unidose ?
Tenir ce médicament hors de la vue et de la portée des enfants.
N’utilisez pas ce médicament après la date de péremption indiquée sur la boite, le récipient unidose. La date de péremption fait référence au dernier jour de ce mois.
A conserver dans le suremballage, à une température ne dépassant pas + 25°C.
Après ouverture du récipient unidose : le produit doit être utilisé immédiatement. Jeter le récipient unidose après utilisation. Ne pas le conserver pour une utilisation ultérieure.
Ne jetez aucun médicament au tout-à-l’égout ou avec les ordures ménagères. Demandez à votre pharmacien d’éliminer les médicaments que vous n’utilisez plus. Ces mesures contribueront à protéger l’environnement.
6. CONTENU DE L’EMBALLAGE ET AUTRES INFORMATIONS
Ce que contient ANTALYRE, collyre en solution en récipient unidose
· La substance active est :
Acide salicylique............................................................................................................ 0,4 mg
Pour un récipient unidose
· Les autres composants sont : borax, acide borique, chlorure de sodium, eau pour préparations injectables.
Qu’est-ce que ANTALYRE, collyre en solution en récipient unidose et contenu de l’emballage extérieur
Ce médicament se présente sous forme de collyre en solution. Barrette de 5 récipients unidoses de 0,4 ml conditionnée en sachet. Boîte de 10 récipients unidoses.
Titulaire de l’autorisation de mise sur le marché
LES INDUSTRIES
2 RUE DU GABIAN
98000 MONACO
Exploitant de l’autorisation de mise sur le marché
LABORATOIRES EUROPHTA
LES INDUSTRIES
2 RUE DU GABIAN
98000 MONACO
RUE DE LA LOMBARDIERE
07100 ANNONAY
Noms du médicament dans les Etats membres de l'Espace Economique Européen
Sans objet.
La dernière date à laquelle cette notice a été révisée est :
[à compléter ultérieurement par le titulaire]
Des informations détaillées sur ce médicament sont disponibles sur le site Internet de l’ANSM (France).