Dernière mise à jour le 01/06/2026
TRAVOPROST ZENTIVA 40 microgrammes/mL, collyre en solution
Présentations
> 1 flacon compte-gouttes polypropylène de 2,5 mL
Code CIP : 34009 300 845 8 8
Déclaration de commercialisation : 07/11/2017
Cette présentation est agréée aux collectivités
- Prix hors honoraire de dispensation : 5,56 €
- Honoraire de dispensation : 1,02 €
- Prix honoraire compris : 6,58 €
- Taux de remboursement :65%
Service médical rendu (SMR)
Amélioration du service médical rendu (ASMR)
Ce médicament étant un générique, l'ASMR n'a pas été évalué par la commission de la transparence (CT), il est possible de se référer à la /aux spécialité(s) de référence du groupe générique auquel appartient ce médicament (cliquez ici pour aller à la rubrique des groupes génériques)
ANSM - Mis à jour le : 03/06/2019
TRAVOPROST ZENTIVA 40 microgrammes/mL, collyre en solution
2. COMPOSITION QUALITATIVE ET QUANTITATIVE
Chaque mL de solution contient 40 microgrammes de travoprost.
Quantité moyenne de substance active/goutte : 0.97-1.4 µg.
Excipient(s) à effet notoire : Chaque mL de solution contient 150 microgrammes de chlorure de benzalkonium et 5 mg d’hydroxystéarate de macrogolglycérol 40 (voir rubrique 4.4).
Pour la liste complète des excipients, voir rubrique 6.1.
Solution incolore et limpide.
pH : 5,5 – 7,0.
Osmolalité : 266-294 mOsm/Kg.
4.1. Indications thérapeutiques
Réduction de la pression intraoculaire élevée chez les patients pédiatriques âgés de 2 mois à 18 ans atteints d’hypertonie intraoculaire ou de glaucome pédiatrique (voir rubrique 5.1).
4.2. Posologie et mode d'administration
Utilisation chez les adultes et les sujets âgés.
La posologie est d’une goutte de TRAVOPROST ZENTIVA dans le cul de sac conjonctival de l’œil ou des yeux atteint (s) une fois par jour. L’effet est optimal si le traitement est administré le soir.
Une occlusion nasolacrymale ou une fermeture douce des paupières après administration est recommandée. Ceci peut réduire l’absorption systémique des médicaments administrés par voie oculaire et conduire à une diminution des effets indésirables systémiques.
En cas d’utilisation de plusieurs collyres, les médicaments doivent être administrés avec au moins 5 minutes d’écart (voir rubrique 4.5).
Si une instillation est oubliée, le traitement doit être poursuivi avec l'instillation suivante comme prévu. La posologie ne doit pas dépasser une goutte par jour dans l’œil ou les yeux atteint(s).
En cas de remplacement d’un autre médicament antiglaucomateux ophtalmique par TRAVOPROST ZENTIVA, l’autre médicament doit être interrompu et TRAVOPROST ZENTIVA doit être commencé le jour suivant.
Insuffisants hépatiques et rénaux
Le travoprost a été étudié chez les insuffisants hépatiques modérés à sévères et chez les insuffisants rénaux modérés à sévères (clairance de la créatinine jusqu’à 14 mL/min). Aucune adaptation de la posologie n’est nécessaire chez ces patients (voir rubrique 5.2).
Population pédiatrique
Le travoprost peut être utilisé chez les patients pédiatriques âgés de 2 mois à 18 ans à la même posologie que chez l’adulte. Cependant, les données pour le groupe d’âge 2 mois à 3 ans (9 patients) sont limitées (voir rubrique 5.1).
La tolérance et l’efficacité de travoprost chez les enfants de moins de 2 mois n’ont pas été établies. Aucune donnée n’est disponible.
Mode d’administration
Voie oculaire
Les patients portant des lentilles de contact doivent se référer à la rubrique 4.4.
Le patient doit retirer le sachet protecteur juste avant la première utilisation. Pour éviter la contamination de l’embout compte-gouttes et de la solution, il faut faire attention de ne pas toucher les paupières, les surfaces voisines ou d’autres surfaces avec l’embout compte-gouttes du flacon.
Hypersensibilité au principe actif ou à l’un des excipients listés à la rubrique 6.1.
4.4. Mises en garde spéciales et précautions d'emploi
Modification de la couleur des yeux
Le travoprost peut modifier progressivement la couleur des yeux en augmentant le nombre de mélanosomes (granules pigmentaires) dans les mélanocytes. Avant l’instauration du traitement, les patients doivent être informés du risque de changement permanent de la couleur de l’œil. Un traitement unilatéral peut avoir pour conséquence une hétérochromie définitive. Les effets à long terme sur les mélanocytes et leurs conséquences ne sont actuellement pas connus. La modification de couleur de l’iris évolue lentement et peut passer inaperçue pendant des mois, voire des années. Le changement de la couleur des yeux a surtout été observé chez des patients ayant l’iris bicolore, c’est-à-dire bleu-marron, gris-marron, jaune-marron et vert-marron ; cependant il a également été observé chez des patients ayant des yeux marron. Généralement, pour l’œil concerné, la pigmentation brune entourant la pupille s’étend de façon concentrique vers la périphérie, et l’iris peut devenir, en partie ou en totalité, brun plus foncé. Après l’arrêt du traitement, aucune augmentation ultérieure de la pigmentation brune de l’iris n’a été observée.
Modification de la peau périorbitaire et palpébrale
Au cours des essais cliniques contrôlés, un assombrissement de la peau périorbitaire et/ou palpébrale associé à l’utilisation du travoprost a été rapporté chez 0,4% des patients.
Des changements périorbitaires et ciliaires se traduisant par un approfondissement du sillon palpébral ont été observés avec les analogues de prostaglandines.
Le travoprost peut modifier progressivement les cils de l’œil ou des yeux traité(s). Au cours des essais cliniques ces changements, représentés par une augmentation de la longueur, de l’épaisseur, de la pigmentation et/ou du nombre de cils, ont été observés chez la moitié des patients environ. Le mécanisme de modifications des cils et ses conséquences à long terme sont actuellement inconnus.
Lors des études chez le singe, le travoprost a entraîné une légère augmentation de la fente palpébrale. Cependant, cet effet n’a pas été observé au cours des essais cliniques et est considéré comme étant spécifique à l’espèce.
Il n’y a pas de données concernant l’utilisation du travoprost sur un œil inflammatoire, dans les glaucomes néovasculaire, congénital, à angle étroit ou à angle fermé. L’expérience est encore limitée en ce qui concerne les atteintes oculaires d’origine thyroïdienne, le glaucome à angle ouvert des patients pseudophaques et les glaucomes pigmentaire ou pseudoexfoliatif. Par conséquent, le travoprost doit être utilisé avec précaution chez les patients présentant une inflammation intraoculaire active.
Patients aphaques
Des œdèmes maculaires ont été rapportés durant le traitement avec les analogues de la prostaglandine F2α. Il est recommandé d’utiliser le travoprost avec précaution chez des patients aphaques, pseudophaques présentant une rupture capsulaire du cristallin ou porteurs d’implant de chambre antérieure ou chez des patients présentant des facteurs de risques connus d’œdème maculaire cystoïde.
Iritis/uvéites
Chez les patients présentant des facteurs de risque connus aux iritis/uvéites, le travoprost devra être utilisé avec précaution.
Contact avec la peau
Le contact cutané avec le travoprost doit être évité étant donné que l’absorption transdermique a été démontrée chez le lapin.
Des cas de kératite ponctuée et/ou de kératopathie ulcérative toxique ont été rapportés avec l’utilisation de chlorure de benzalkonium, utilisé communément comme conservateur dans les médicaments ophtalmiques. TRAVOPROST ZENTIVA contenant du chlorure de benzalkonium, un suivi rapproché est demandé en cas d’utilisation fréquente ou prolongée.
Les prostaglandines et les analogues de prostaglandines sont des substances biologiquement actives pouvant être absorbées par la peau. Les femmes enceintes ou souhaitant le devenir devront prendre des précautions appropriées pour éviter une exposition directe au contenu du flacon. En cas de contact accidentel avec le contenu du flacon, laver immédiatement et soigneusement la zone concernée.
Lentilles de contact
Les patients doivent être informés d’enlever leurs lentilles de contact avant instillation de TRAVOPROST ZENTIVA et d’attendre 15 minutes après l’instillation de la dose avant de poser des lentilles de contact.
Excipients
Ce médicament
contient du chlorure de benzalkonium qui est connu pour provoquer une irritation des yeux, des symptômes du syndrome de l’œil sec et peut affecter le film lacrymal et la surface de la cornée. Il doit être utilisé avec prudence chez les patients atteints d’œil sec et ceux présentant un risque d’endommagement de la cornée. Les patients doivent être surveillés en cas d’utilisation prolongée.Ce médicament contient de l’hydroxystéarate de macrogolglycérol 40 qui peut provoquer une irritation de la peau.
Sujets pédiatriques
L’efficacité et la tolérance dans le groupe d’âge 2 mois à 3 ans (9 patients) sont limitées (voir rubrique 5.1). Aucune donnée n’est disponible pour les enfants de moins de 2 mois.
Pour les enfants de moins de 3 ans qui souffrent principalement de GCP (glaucome congénital primaire), la chirurgie, (en particulier la trabéculotomie/goniotomie) reste le traitement de première intention.
Aucune donnée de tolérance à long terme n’est disponible pour les sujets pédiatriques.
4.5. Interactions avec d'autres médicaments et autres formes d'interactions
Aucune étude d’interaction n’a été réalisée.
4.6. Fertilité, grossesse et allaitement
Femmes susceptibles d’être enceintes/contraception
Le travoprost ne doit pas être utilisé chez la femme susceptible d'être enceinte à moins qu’une contraception adéquate ne soit mise en place (voir rubrique 5.3).
Grossesse
Le travoprost a des effets pharmacologiques puissants sur la grossesse et/ou le fœtus/nouveau-né. Le travoprost ne doit pas être utilisé au cours de la grossesse sauf en cas de nécessité absolue.
Allaitement
Il n'existe pas de données concernant l'excrétion du travoprost dans le lait maternel humain. Des études chez l'animal ont démontré une excrétion du travoprost et de ses métabolites dans le lait maternel. L’utilisation du travoprost n’est pas recommandée pendant l'allaitement.
Fécondité
Il n’existe pas de donnée sur les effets du travoprost sur la fécondité humaine. Les études chez l’animal n’ont pas montré d’effet du travoprost sur la fécondité à des doses 250 fois supérieures aux doses oculaires maximales recommandées chez l’homme.
4.7. Effets sur l'aptitude à conduire des véhicules et à utiliser des machines
Le travoprost n’a pas ou n’a qu’une influence négligeable sur la capacité à conduire et à utiliser des machines, cependant, comme avec tout collyre, une vision floue provisoire ou d’autres troubles visuels peuvent diminuer l’aptitude à conduire des véhicules ou à utiliser des machines. En cas de vision floue survenant lors de l’instillation, le patient doit attendre que sa vision redevienne normale avant de conduire un véhicule ou d’utiliser une machine.
Résumé du profil de sécurité
Au cours des études cliniques avec le travoprost, les effets indésirables les plus fréquents étaient l’hyperémie oculaire et une hyperpigmentation de l’iris, survenant respectivement chez environ 20 % et 6 % des patients.
Liste des effets indésirables
Les effets indésirables suivants sont classés de la façon suivante : très fréquents (≥1/10), fréquents (≥1/100, <1/10), peu fréquents (≥1/1 000, <1/100), rares (≥1/10 000, <1/1 000), très rares (<1/10 000) ou inconnu (la fréquence ne peut pas être estimée à partir des données disponibles). Dans chaque groupe de fréquence, les effets indésirables sont présentés dans l’ordre décroissant de gravité. Les effets indésirables ont été obtenus à partir d’études cliniques et de données post marketing sur le travoprost.
|
Classe de systèmes d’organes |
Fréquence |
Effets indésirables |
|
Affections du système immunitaire |
Peu fréquente |
hypersensibilité, allergie saisonnière |
|
Affections psychiatriques |
Inconnue |
dépression, anxiété, insomnie |
|
Affections du système nerveux |
Peu fréquente |
céphalées |
|
Rare |
sensation vertigineuse, anomalie du champ visuel, dysgueusie |
|
|
Affections oculaires |
Très fréquente |
hyperémie oculaire |
|
Fréquente
|
hyperpigmentation de l’iris, douleur oculaire, gêne oculaire, sécheresse oculaire, prurit oculaire, irritation de l’œil |
|
|
Peu fréquente |
érosion de la cornée, uvéite, iritis, inflammation de la chambre antérieure, kératite, kératite ponctuée, photophobie, sécrétions oculaires anormales, blépharite, érythème des paupières, œdème périorbital, prurit des paupières, baisse de l’acuité visuelle, vision trouble, augmentation de la sécrétion lacrymale, conjonctivite, ectropion, cataracte, bord de la paupière croûteux, croissance des cils |
|
|
Rare |
iridocyclite, herpès simplex ophtalmique, inflammation de l’œil, photopsie, eczéma des paupières, œdème conjonctival, effet de halo, follicules conjonctivaux, hypoesthésie oculaire, trichiasis, meibomite, pigmentation de la chambre antérieure, mydriase, asthénopie, hyperpigmentation des cils, épaississement des cils |
|
|
Inconnue |
œdème maculaire, sillon palpébral approfondi |
|
|
Affections de l’oreille et du labyrinthe |
Inconnue |
vertige, acouphènes |
|
Affections cardiaques |
Peu fréquente |
Palpitations |
|
Rare |
fréquence cardiaque irrégulière, fréquence cardiaque diminuée |
|
|
Inconnue |
douleur thoracique, bradycardie, tachycardia, arythmie |
|
|
Affections vasculaires |
Rare |
diminution de la pression artérielle diastolique ou augmentation de la pression artérielle systolique, hypotension, hypertension |
|
Affections respiratoires, thoraciques et médiastinales |
Peu fréquente |
toux, congestion nasale, irritation de la gorge |
|
Rare |
dyspnée, asthme, trouble respiratoire, douleur oropharyngée, dysphonie, rhinite allergique, sécheresse nasale |
|
|
Inconnue |
asthme aggravé, épistaxis |
|
|
Affections gastro-intestinales |
Rare
|
réactivation d’ulcère gastro-duodénal, affection gastro-intestinale, constipation, bouche sèche |
|
Inconnue |
diarrhée, douleur abdominale, nausée, vomissement |
|
|
Affections de la peau et du tissu sous-cutané |
Peu fréquente |
hyperpigmentation cutanée (péri-oculaire), coloration de la peau, texture anormale des cheveux, hypertrichose
|
|
Rare |
dermatite allergique, dermatite de contact, érythème, rash, modification de la couleur des cheveux, madarose |
|
|
Inconnue |
prurit, croissance anormale des cheveux |
|
|
Affections musculo-squelettiques, et systémiques |
Rare |
douleur musculo-squelettique, arthralgie |
|
|
|
|
|
Affections rénales et urinaires |
Inconnue |
dysurie, incontinence urinaire |
|
Troubles généraux et anomalies au site d’administration |
Rare |
asthénie |
|
Exploration |
Inconnue |
augmentation d’un antigène prostatique spécifique |
Sujets pédiatriques
Lors d’un essai clinique de phase 3 d’une durée de 3 mois et d’une étude pharmacocinétique d’une durée de 7 jours, chez les 102 patients pédiatriques exposés au travoprost, le type et les caractéristiques des effets indésirables déclarés ont été similaires à ce qui a été observé pour les patients adultes. Les profils de tolérance à court terme pour les différents sous-groupes pédiatriques ont également été similaires (voir rubrique 5.1). Les effets indésirables les plus souvent déclarés chez les sujets pédiatriques ont été l’hyperémie oculaire (16,9 %) et la croissance des cils (6,5 %). Dans une étude similaire de 3 mois chez des patients adultes, ces événements sont survenus avec une incidence de 11,4 % et 0,0 % respectivement.
Les effets indésirables supplémentaires déclarés pour les enfants dans l’étude pédiatrique sur 3 mois (n = 77) comparés à une étude similaire chez l’adulte (n = 185) ont inclus érythème des paupières, kératite, augmentation de la sécrétion lacrymale, et photophobie. Ces effets ont tous été déclarés avec une incidence de 1,3 % alors qu’aucun n’avait été observé chez l’adulte.
Déclaration des effets indésirables suspectés
La déclaration des effets indésirables suspectés après autorisation du médicament est importante. Elle permet une surveillance continue du rapport bénéfice/risque du médicament. Les professionnels de santé déclarent tout effet indésirable suspecté via le système national de déclaration : Agence nationale de sécurité du médicament et des produits de santé (ANSM) et réseau des Centres Régionaux de Pharmacovigilance - Site internet : www.signalement-sante.gouv.fr.
5. PROPRIETES PHARMACOLOGIQUES
5.1. Propriétés pharmacodynamiques
Mécanisme d’action
Le travoprost, analogue de la prostaglandine F2α, est un agoniste complet hautement sélectif ayant une haute affinité pour le récepteur FP aux prostaglandines et diminue la pression intraoculaire en augmentant l’écoulement de l’humeur aqueuse par le système trabéculaire et les voies uvéosclérales.
Chez l’homme, la diminution de la pression intraoculaire débute environ 2 heures après l’administration et l’effet maximal est atteint au bout de 12 heures. Après une administration unique, une baisse significative de la pression intraoculaire peut être maintenue pour des périodes excédant 24 heures.
Efficacité et sécurité clinique
Lors d’un essai clinique, une réduction de la pression intraoculaire de 8 à 9 mmHg (environ 33%) par rapport à la pression intraoculaire de base de 24 à 26 mmHg a été obtenue chez des patients qui présentaient un glaucome à angle ouvert ou une hypertension oculaire et qui avaient été traités par du travoprost (conservé avec du polyquaternium) à raison d’une fois par jour, le soir. Les données relatives à l’administration conjointe de travoprost avec du timolol 0,5% et les données limitées de l’administration conjointe avec la brimonidine 0,2% ont été rassemblées au cours des essais cliniques montrant un effet additif de travoprost avec ces médicaments antiglaucomateux. Aucune donnée clinique sur l’utilisation conjointe avec les autres médicaments oculaires hypotensifs n’est disponible.
Pharmacologie secondaire
Chez le lapin, le travoprost augmente significativement le flux sanguin à la tête du nerf optique dans les 7 jours suivant l’instillation oculaire (1,4 microgrammes, une fois par jour).
Le travoprost conservé avec le polyquaternium-1 a entrainé une toxicité minimale pour la surface oculaire par rapport aux collyres conservés avec du chlorure de benzalkonium, lors de l’application sur les cellules cornéennes humaines ou après administration locale oculaire chez le lapin.
Population pédiatrique
L’efficacité du travoprost chez l’enfant âgé de 2 mois à moins de 18 ans a été démontrée lors d’une étude clinique de 12 semaines, en double aveugle de travoprost comparé à timolol chez 152 patients présentant une hypertension oculaire ou un glaucome pédiatrique. Les patients ont reçu soit du travoprost à 0,004 % une fois par jour, soit du timolol 0,5 % (ou 0,25 % pour les patients de moins de 3 ans) deux fois par jour. Le critère principal d’efficacité était le changement de la pression intraoculaire (PIO) par rapport à la valeur initiale à la Semaine 12 de l’étude. Les réductions moyennes de la pression intraoculaire dans les groupes travoprost et timolol ont été équivalentes (voir Tableau 1).
Dans les groupes d’âge 3 ans à ˂ 12 ans (n=36) et 12 ans à ˂ 18 ans (n=26), la réduction moyenne de la PIO à la Semaine 12 dans le groupe travoprost était similaire à celle du groupe timolol. La réduction moyenne de la PIO à la Semaine 12 dans le groupe d’âge 2 mois à ˂ 3 ans était de 1,8 mmHg dans le groupe travoprost et de 7,3 mmHg dans le groupe timolol. Les réductions de la PIO pour ce groupe d’âge étaient observées pour 6 patients seulement dans le groupe timolol et 9 patients dans le groupe travoprost où 4 patients du groupe travoprost par rapport à 0 patients dans le groupe timolol avaient une réduction moyenne de la PIO non significative à la Semaine 12. Aucune donnée n’est disponible pour les enfants âgés de moins de 2 mois.
L’effet sur la PIO a été observé après la seconde semaine de traitement et a été constamment maintenu tout au long de la période d’étude de 12 semaines pour tous les groupes d’âge.
Table 1 – Comparaison du changement de la PIO moyenne par rapport à la valeur initiale (mmHg) à la Semaine 12
|
N |
Travoprost Moyenne (SE) |
N |
Timolol Moyenne (SE) |
Différence moyennea |
(IC 95%) |
|
53 |
-6,4 (1.05) |
60 |
-5,8 (0.96) |
-0,5 |
(-2,1 ; 1,0) |
SE = Ecart-Type ; IC = Intervalle de Confiance;
a La différence moyenne est Travoprost - Timolol. Les estimations sont fondées sur les moyennes des moindres carrés dérivées d'un modèle statistique qui rend compte des mesures corrélées de la PIO parmi les patients dont le diagnostic de PIO primaire et de base sont dans le modèle.
5.2. Propriétés pharmacocinétiques
Le travoprost est une prodrogue de type ester. Il est absorbé à travers la cornée où l’ester d’isopropyl devient actif après hydrolyse en acide. Des études chez le lapin ont montré que des pics de concentration, dans l’humeur aqueuse, de 20 ng/mL en acide libre ont été atteints une à deux heures après instillation oculaire de travoprost. Les concentrations dans l’humeur aqueuse déclinent avec une demi-vie de 1,5 heure environ.
Distribution
Suite à l’administration locale oculaire de TRAVOPROST ZENTIVA chez des volontaires sains, une faible exposition systémique à la forme active (acide libre) a été démontrée. Des pics de concentrations plasmatiques de forme active (acide libre) de 25 pg/mL ou moins ont été observés entre 10 à 30 minutes après administration. Ainsi, les niveaux plasmatiques diminuent rapidement au-dessous de la valeur quantifiable de 10 pg/mL une heure après administration. En raison de la faible concentration plasmatique et de l’élimination rapide après administration locale, la demi-vie d’élimination de la forme active (acide libre) n’a pas pu être déterminée.
Biotransformation
Le métabolisme est la principale voie d’élimination du travoprost et de sa forme active (acide libre). Les mécanismes métaboliques systémiques sont équivalents à ceux de la prostaglandine F2α endogène, qui sont caractérisés par la réduction de la double liaison 13-14, l’oxydation de la fonction 15-hydroxyle ainsi que la scission par oxydation β de la partie supérieure de la chaîne.
Élimination
Le travoprost sous forme d’acide libre et ses métabolites sont principalement excrétés par les reins.
Le travoprost a été étudié chez les insuffisants hépatiques modérés à sévères et chez les insuffisants rénaux modérés à sévères (clairance de la créatinine jusqu’à 14 mL/min). Aucune adaptation de la posologie n’est nécessaire chez ces patients.
Population pédiatrique
Une étude pharmacocinétique chez des patients âgés de 2 mois à 18 ans a démontré une exposition plasmatique au travoprost sous forme d’acide libre très faible, avec des concentrations allant d’une valeur inférieure à la limite de dosage de 10 pg/mL à 52,0 pg/mL. Tandis que la majorité des valeurs plasmatiques dans toutes les études était non-quantifiable, rendant les comparaisons statistiques de l’exposition systémique à travers les groupes d’âge irréalisables, la tendance générale montre que l’exposition plasmatique au travoprost sous forme d’acide libre après une administration topique de travoprost est extrêmement faible à travers tous les groupes d’âge évalués.
5.3. Données de sécurité préclinique
Des études de toxicité de reproduction du travoprost par voie systémique ont été réalisées chez le rat, la souris et le lapin. Les résultats sont liés à l’activité agoniste du récepteur FP de l’utérus avec létalité embryonnaire précoce, perte post implantation, fœtotoxicité. Chez la rate gravide, l’administration systémique de travoprost à des doses plus de 200 fois supérieures à la dose thérapeutique pendant la période d’organogenèse, a conduit à une augmentation de l’incidence des malformations. Après administration de 3H-travoprost chez des rates gravides, une faible radioactivité a été mesurée dans le liquide amniotique et dans les tissus fœtaux. Les études de reproduction et de développement ont démontré un effet abortif potentiel avec un taux élevé chez le rat et la souris (respectivement de 180 pg/mL et 30 pg/mL dans le plasma) avec une exposition 1,2 à 6 fois supérieures à l’exposition thérapeutique (jusqu’à 25 pg/mL).
Des études spécifiques d’interactions in vitro avec le travoprost et des médicaments contenant du thiomersal ont été effectuées. Aucune précipitation n’a été observée.
Jetez 4 semaines après la première ouverture.
6.4. Précautions particulières de conservation
Avant ouverture, conserver le flacon dans le sachet à l’abri de l’humidité.
Après première ouverture, ce médicament ne nécessite pas de précautions particulières de conservation.
6.5. Nature et contenu de l'emballage extérieur
Flacon transparent en polypropylène (PP) de 5 mL muni d’un compte-gouttes transparent en polyéthylène de basse densité (PEBD) et d’un bouchon à vis blanc avec fermeture inviolable en polyéthylène de haute densité (PEHD) présenté dans un sachet en polyéthylène téréphtalate/aluminium/polyéthylène (PET/Alu/PE).
Chaque flacon contient 2,5 mL de solution.
Boîte de 1 ou 3 flacons.
Toutes les présentations peuvent ne pas être commercialisées.
6.6. Précautions particulières d’élimination et de manipulation
Pas d’exigences particulières.
7. TITULAIRE DE L’AUTORISATION DE MISE SUR LE MARCHE
35 RUE DU VAL DE MARNE
75013 PARIS
8. NUMERO(S) D’AUTORISATION DE MISE SUR LE MARCHE
· 34009 300 845 8 8 : Flacon (PP) de 2.5 ml. Boite de 1.
· 34009 550 295 9 5 : Flacon (PP) de 2.5 ml. Boite de 3.
9. DATE DE PREMIERE AUTORISATION/DE RENOUVELLEMENT DE L’AUTORISATION
[à compléter ultérieurement par le titulaire]
10. DATE DE MISE A JOUR DU TEXTE
[à compléter ultérieurement par le titulaire]
Sans objet.
12. INSTRUCTIONS POUR LA PREPARATION DES RADIOPHARMACEUTIQUES
Liste I.
ANSM - Mis à jour le : 03/06/2019
TRAVOPROST ZENTIVA 40 microgrammes/mL, collyre en solution
Travoprost
· Gardez cette notice. Vous pourriez avoir besoin de la relire.
· Si vous avez d’autres questions, interrogez votre médecin ou votre pharmacien.
· Ce médicament vous a été personnellement prescrit. Ne le donnez pas à d’autres personnes. Il pourrait leur être nocif, même si les signes de leur maladie sont identiques aux vôtres.
· Si vous ressentez un quelconque effet indésirable, parlez-en à votre médecin, votre pharmacien ou votre infirmier/ère. Ceci s’applique aussi à tout effet indésirable qui ne serait pas mentionné dans cette notice. Voir rubrique 4.
1. Qu'est-ce que TRAVOPROST ZENTIVA 40 microgrammes/mL, collyre en solution et dans quels cas est-il utilisé ?
2. Quelles sont les informations à connaître avant d'utiliser TRAVOPROST ZENTIVA 40 microgrammes/mL, collyre en solution ?
3. Comment utiliser TRAVOPROST ZENTIVA 40 microgrammes/mL, collyre en solution?
4. Quels sont les effets indésirables éventuels ?
5. Comment conserver TRAVOPROST ZENTIVA 40 microgrammes/mL, collyre en solution ?
6. Contenu de l’emballage et autres informations.
1. QU’EST-CE QUE TRAVOPROST ZENTIVA 40 microgrammes/mL, collyre en solution ET DANS QUELS CAS EST-IL UTILISE ?
TRAVOPROST ZENTIVA est utilisé pour réduire une pression élevée à l’intérieur de l’œil chez l’adulte, les adolescents et les enfants âgés de 2 mois et plus. Cette pression peut conduire à une maladie appelée glaucome.
2. QUELLES SONT LES INFORMATIONS A CONNAITRE AVANT D’UTILISER TRAVOPROST ZENTIVA 40 microgrammes/mL, collyre en solution?
N’utilisez jamais TRAVOPROST ZENTIVA 40 microgrammes/mL, collyre en solution :
· si vous êtes allergique au travoprost ou à l’un des autres composants de ce médicament (listés en section 6).
Demandez l’avis de votre médecin si vous êtes atteint de ces troubles.
Avertissements et précautions
· TRAVOPROST ZENTIVA peut augmenter la longueur, l’épaisseur, la couleur et/ou le nombre de vos cils. Des modifications au niveau des paupières, y compris une pilosité ciliaire anormale et des anomalies au niveau des tissus autour de l’œil, ont aussi été observées.
· TRAVOPROST ZENTIVA peut modifier la couleur de votre iris (partie colorée de votre œil). Ce changement peut être permanent. Il peut également survenir une modification de la couleur de la peau autour de l’oeil.
· Si vous avez eu une chirurgie de la cataracte, parlez-en à votre médecin avant d’utiliser TRAVOPROST ZENTIVA.
· Si vous avez ou avez déjà eu une inflammation de l’œil (iritis ou uvéite), parlez-en à votre médecin avant d’utiliser TRAVOPROST ZENTIVA.
· TRAVOPROST ZENTIVA peut, à de rares occasions, provoquer des essoufflements ou une respiration sifflante ou augmenter les symptômes d'asthme. Si vous observez des changements dans votre manière de respirer lorsque vous utilisez TRAVOPROST ZENTIVA, prévenez immédiatement votre médecin.
· Le travoprost peut être absorbé par la peau. Si ce médicament vient en contact avec la peau, il doit être enlevé aussitôt par lavage. Ceci est particulièrement important pour les femmes enceintes ou souhaitant le devenir.
· Si vous êtes porteur de lentilles de contact souples, n’utilisez pas le collyre lorsque vous portez vos lentilles. Après avoir utilisé le collyre, attendez 15 minutes avant de remettre vos lentilles.
Enfants et adolescents
TRAVOPROST ZENTIVA peut être utilisé chez les enfants de 2 mois à < 18 ans à la même posologie que chez l’adulte. L’utilisation du TRAVOPROST ZENTIVA n’est pas recommandée pour les enfants âgés de moins de 2 mois.
Autres médicaments et TRAVOPROST ZENTIVA 40 microgrammes/mL, collyre en solution
Si vous prenez ou avez pris ou utilisé récemment un autre médicament, parlez-en à votre médecin ou à votre pharmacien.
TRAVOPROST ZENTIVA 40 microgrammes/mL, collyre en solution avec des aliments, boissons et de l’alcool
Sans objet.
Grossesse, allaitement et fertilité
Si vous êtes enceinte ou que vous allaitez, si vous pensez être enceinte ou planifiez une grossesse, demandez conseil à votre médecin avant de prendre ce médicament.
Conduite de véhicules et utilisation de machines
Vous pouvez trouver que votre vision est trouble juste après avoir utilisé TRAVOPROST ZENTIVA. Ne conduisez pas de voiture et n’utilisez pas de machines avant que cet effet ait disparu.
TRAVOPROST ZENTIVA 40 microgrammes/mL, collyre en solution contient du chlorure de benzalkonium.
Si vous portez des lentilles de contact souples, n’utilisez pas ce collyre avec vos lentilles. Attendez 15 minutes après l’utilisation du collyre avant de remettre vos lentilles. Il y a un conservateur dans ce médicament (chlorure de benzalkonium) qui peut colorer les lentilles de contact souples.
Ce conservateur peut également provoquer une irritation des yeux, surtout si vous souffrez du syndrôme de l’œil sec ou de troubles de la cornée (couche transparente à l’avant de l’œil). En cas de sensation anormale, de picotements ou de douleur dans les yeux après avoir utilisé ce médicament, contactez votre médecin.
TRAVOPROST ZENTIVA 40 microgrammes/mL, collyre en solution contient du l’hydroxystéarate de macrogolglycérol 40 qui peut provoquer une irritation de la peau.
3. COMMENT UTILISER TRAVOPROST ZENTIVA 40 microgrammes/mL, collyre en solution ?
La posologie recommandée est :
Une goutte dans l’œil ou les yeux malades, une fois par jour - le soir.
Utilisez TRAVOPROST ZENTIVA dans les deux yeux uniquement si votre médecin vous a dit de le faire. Utilisez-le aussi longtemps que votre médecin ou le médecin traitant votre enfant vous l’a dit.
Utilisez TRAVOPROST ZENTIVA seulement en gouttes dans vos yeux ou ceux de votre enfant.
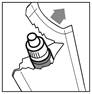
|
1 |
· Ouvrez le sachet d’emballage juste avant d’utiliser le flacon pour la première fois (figure 1). Sortez-en le flacon et inscrivez la date d’ouverture sur le carton dans l’espace prévu à cet effet. |
|
· Prenez le flacon et un miroir. |
|
|
· Lavez-vous les mains. |
|
|
· Enlevez le bouchon. |
|
|
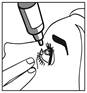
2 |
· Tenez le flacon tête en bas, entre le pouce et vos autres doigts. |
|
· Penchez votre tête ou celle de votre enfant doucement en arrière. Avec un doigt propre, tirez doucement la paupière vers le bas pour créer un sillon entre la paupière et l’oeil. La goutte sera déposée à cet endroit (figure 2). |
|
|
· Rapprochez l’embout du flacon de l’oeil. Utilisez un miroir pour vous aider. |
|
|
3 |
· Ne touchez pas l’œil, les paupières, les surfaces voisines ou d’autres surfaces avec le compte-gouttes. Cela peut infecter le collyre. |
|
· Appuyez légèrement sur le flacon pour libérer une goutte du médicament à la fois (figure 3). |
|
|
· Après avoir utilisé ce médicament, gardez la paupière fermée, maintenez une légère pression en appuyant avec un doigt sur le coin de l’œil près du nez (figure 4). Ceci permet d’empêcher le médicament d’aller dans le reste du corps. |
|
|
4 |
· Si vous devez traiter les deux yeux, recommencez ces étapes pour l’autre œil. |
|
· Refermez bien le flacon immédiatement après usage. |
|
|
· Utilisez un seul flacon à la fois. Ne pas ouvrir le sachet tant que vous n’avez pas besoin du flacon. |
|
|
· Vous devez jeter le flacon 4 semaines après l’avoir ouvert pour la première fois, pour prévenir les infections, et utiliser un nouveau flacon. |


Si une goutte tombe à côté de l’œil, recommencez.
Si vous ou votre enfant utilisez d’autres collyres ou pommades ophtalmiques en même temps, attendez au moins 5 minutes entre l’utilisation de TRAVOPROST ZENTIVA et l’autre traitement.
Si vous ou votre enfant avez reçu trop de TRAVOPROST ZENTIVA 40 microgrammes/mL, collyre en solution dans les yeux
Rincez-les avec de l’eau tiède. Ne mettez pas d’autres gouttes jusqu’à ce que le moment soit venu de prendre la goutte suivante.
Si vous oubliez d’utiliser TRAVOPROST ZENTIVA 40 microgrammes/mL, collyre en solution
Continuez avec la goutte suivante comme prévu. Ne mettez pas de dose double pour compenser une dose oubliée. N’utilisez jamais plus d’une goutte par jour dans l’œil (les yeux) atteint(s).
Si vous arrêtez d’utiliser TRAVOPROST ZENTIVA 40 microgrammes/mL, collyre en solution
N’arrêtez pas de prendre ce médicament sans en avoir préalablement parlé à votre médecin ou au médecin traitant votre enfant, la pression de votre œil ou de celui de votre enfant ne sera pas contrôlée ce qui pourrait provoquer une perte de la vue.
Si vous avez d’autres questions sur l’utilisation de ce médicament, demandez plus d’informations à votre médecin, au médecin traitant votre enfant ou à votre pharmacien.
4. QUELS SONT LES EFFETS INDESIRABLES EVENTUELS ?
Vous pouvez continuer à utiliser le collyre, à moins que les effets indésirables soient graves. Si vous vous inquiétez, contactez votre médecin ou votre pharmacien. Ne pas interrompre l’administration de TRAVOPROST ZENTIVA sans en parler à votre médecin.
Les effets indésirables suivants ont été observés avec TRAVOPROST ZENTIVA :
Effets indésirables très fréquents (peut survenir chez plus d’1 personne sur 10)
· Rougeur oculaire.
Effets indésirables fréquents (peut survenir au maximum chez 1 personne sur 10)
Effets oculaires :
· Changement de la couleur de l’iris (partie colorée de l’œil), douleur oculaire, inconfort oculaire,œil sec, démangeaisons oculaires, irritations oculaires.
Effets indésirables peu fréquents (peut concerner jusqu’à 1 personne sur 100)
Effets oculaires :
· Troubles de la cornée, inflammation de l’œil, inflammation de l’iris, inflammation dans l’œil,
inflammation de la surface de l’œil avec ou sans lésion de surface, sensibilité à la lumière, écoulement de l’œil, inflammation de la paupière, rougeur de paupière, gonflement autour de l’œil, démangeaison de la paupière, vision trouble, augmentation de la production de larme, infection ou inflammation de la conjonctive (fine membrane couvrant la face interne de la paupière et la partie blanche du globe oculaire)(conjonctivite), renversement externe anormal de la paupière inférieure, opacification de l’œil, croûte sur le bord de la paupière, croissance des cils.
Effets secondaires généraux :
· Augmentation des symptômes allergiques, mal de tête, rythme cardiaque irrégulier, toux, nez bouché, irritation de la gorge, assombrissement de la peau autour des yeux, assombrissement de la peau, texture de cheveux anormale, croissance capillaire excessive.
Effets indésirables rares (peut concerner jusqu’à 1 personne sur 1000)
Effets oculaires :
· Perception de flashs, eczéma des paupières, positionnement anormal des cils qui repoussent vers l’œil, gonflement de l’œil, vision réduite, effet de halo, sensation oculaire diminuée, inflammation des glandes des paupières, pigmentation dans l’œil, augmentation de la taille de la pupille, épaississement des cils, changement de la couleur des cils, fatigue des yeux, infection oculaire virale.
Effets secondaires généraux :
· Vertiges, mauvais gout, rythme cardiaque diminué ou irrégulier, tension artérielle diminuée ou augmentée, diminution du souffle, asthme, allergie ou inflammation nasale, sécheresse nasale, dysphonie, inconfort gastro-intestinal ou ulcère, constipation, sécheresse buccale, rougeur ou démangeaison de la peau, éruption, changement de la couleur des cheveux, perte des cils, douleurs articulaires, douleurs musculo-squelettiques, fatigue généralisée.
Fréquence indéterminée : la fréquence ne peut être estimée sur la base des données disponibles
Effets oculaires :
· Inflammation de l’arrière de l’œil, yeux apparaissent plus enfoncés.
Effets secondaires généraux :
· Dépression, anxiété, insomnie, sensation de faux mouvement, acouphènes, douleur thoracique, rythme cardiaque anormal, battements cardiaques accélérés, aggravation de l’asthme, diarrhées, saignements de nez, douleur abdominale, nausée, vomissements, démangeaisons, croissance capillaire anormale, mictions involontaires ou douloureuses, augmentation de l’antigène de la prostate.
Chez les enfants et les adolescents, les effets indésirables le plus souvent observés avec le travoprost sont la rougeur de l’œil et la croissance des cils. Ces deux effets indésirables ont été observés avec une incidence plus élevée chez les enfants et les adolescents que chez les adultes.
Déclaration des effets secondaires
Si vous ressentez un quelconque effet indésirable, parlez-en à votre médecin, votre pharmacien ou à votre infirmier/ère. Ceci s’applique aussi à tout effet indésirable qui ne serait pas mentionné dans cette notice. Vous pouvez également déclarer les effets indésirables directement via le système national de déclaration : Agence nationale de sécurité du médicament et des produits de santé (ANSM) et réseau des Centres Régionaux de Pharmacovigilance - Site internet: www.signalement-sante.gouv.fr.
En signalant les effets indésirables, vous contribuez à fournir davantage d’informations sur la sécurité du médicament.
5. COMMENT CONSERVER TRAVOPROST ZENTIVA 40 microgrammes/mL, collyre en solution ?
Tenir ce médicament hors de la vue et de la portée des enfants.
Ne pas utiliser ce médicament après la date de péremption (« EXP ») mentionnée sur le flacon et la boîte. La date de péremption fait référence au dernier jour de ce mois.
N’utilisez pas ce médicament si vous remarquez que la fermeture inviolable est brisée ou endommagée avant la première ouverture.
Avant ouverture, garder le flacon dans le sachet à l’abri de l’humidité.
Après première ouverture, ce médicament ne nécessite pas de précautions particulières de conservation.
Vous devez jeter le flacon 4 semaines après l’avoir ouvert pour la première fois, pour prévenir les infections, et utiliser un nouveau flacon. Inscrivez la date à laquelle vous l’avez ouvert dans l’espace prévu à cet effet sur chaque étiquette du flacon et sur la boîte.
Ne jetez aucun médicament au tout-à-l’égout ou avec les ordures ménagères. Demandez à votre pharmacien d’éliminer les médicaments que vous n’utilisez plus. Ces mesures contribueront à protéger l’environnement.
6. CONTENU DE L’EMBALLAGE ET AUTRES INFORMATIONS
Ce que contient TRAVOPROST ZENTIVA 40 microgrammes/mL, collyre en solution
· La substance active est : le travoprost.
Chaque ml de solution contient 40 microgrammes de travoprost.
· Les autres composants sont :
Chlorure de benzalkonium, hydroxystéarate de macrogolglycérol 40, trométamol, édétate disodique, acide borique (E284), mannitol (E421), hydroxyde de sodium, eau pour préparations injectables ou eau purifiée.
Ce médicament est une solution incolore et limpide, fourni dans un étui contenant un flacon transparent de 5 mL en polypropylène (PP) muni d’un compte-goutte transparent en polyéthylène de basse densité (PEBD) et d’un bouchon à vis blanc avec fermeture inviolable en polyéthylène de haute densité (PEHD) présenté dans un sachet. Chaque flacon contient 2,5 mL de solution.
Ce produit est disponible dans les conditionnements suivants : Boîte de 1 ou 3 flacons.
Toutes les présentations peuvent ne pas être commercialisées.
Titulaire de l’autorisation de mise sur le marché
35 RUE DU VAL DE MARNE
75013 PARIS
Exploitant de l’autorisation de mise sur le marché
35 RUE DU VAL DE MARNE
75013 PARIS
6 DERVENAKION STR.
15351 PALLINI ATTIKI
GRECE
OU
BALKANPHARMA-RAZGRAD AD
68 APRILSKO VASTANIE BLVD
7200 RAZGRAD
BULGARIE
OU
JADRAN - GALENSKI LABORATORIJ d.d.
Svilno 20,
Rijeka 51000
CroatiE
Noms du médicament dans les Etats membres de l'Espace Economique Européen
Ce médicament est autorisé dans les Etats membres de l'Espace Economique Européen sous les noms suivants : Conformément à la réglementation en vigueur.
[À compléter ultérieurement par le titulaire]
La dernière date à laquelle cette notice a été révisée est :
[à compléter ultérieurement par le titulaire]
Sans objet.