Dernière mise à jour le 01/06/2026
LIPTRUZET 10 mg/10 mg, comprimé pelliculé
Indications thérapeutiques
Classe pharmacothérapeutique : Inhibiteurs de la HMG-CoA réductase en association avec d’autres hypolipidémiants - code ATC : C10BA05
LIPTRUZET est un médicament utilisé pour baisser les taux de cholestérol élevés. LIPTRUZET contient de l’ézétimibe et de l’atorvastatine.
LIPTRUZET est un médicament utilisé chez l’adulte pour diminuer les taux de cholestérol total, de « mauvais » cholestérol (LDL-cholestérol) et les substances grasses appelées triglycérides dans le sang. De plus, LIPTRUZET augmente le taux de « bon » cholestérol (HDL-cholestérol).
LIPTRUZET agit pour réduire votre cholestérol de deux façons. Il réduit le cholestérol absorbé par votre tube digestif ainsi que le cholestérol fabriqué par votre organisme.
Le cholestérol est une des nombreuses substances grasses trouvées dans le système sanguin. Votre cholestérol total est composé principalement de cholestérol LDL et de cholestérol HDL.
Le LDL-cholestérol est souvent appelé le « mauvais » cholestérol parce qu’il peut s’agglomérer sur les parois de vos artères en formant une plaque. Finalement, la création de cette plaque peut mener à un rétrécissement des artères. Ce rétrécissement peut ralentir ou bloquer le flux sanguin vers les organes vitaux tels que le cœur et le cerveau. Ce blocage du flux sanguin peut entraîner une crise cardiaque ou une attaque cérébrale.
Le HDL-cholestérol est souvent appelé le « bon » cholestérol parce qu’il aide à empêcher le mauvais cholestérol de s’agglomérer dans les artères et protège contre les maladies cardiaques.
Les triglycérides sont une autre sorte de graisse dans votre sang qui peuvent augmenter votre risque de maladie cardiaque.
LIPTRUZET est utilisé chez les patients chez qui le régime seul ne contrôle pas les taux de cholestérol. Vous devez continuer votre régime faisant baisser le cholestérol en prenant ce médicament.
LIPTRUZET est utilisé en complément de votre régime faisant baisser le cholestérol si vous avez :
· un taux élevé de cholestérol dans votre sang (hypercholestérolémie primaire [familiale hétérozygote et non familiale]) ou des taux élevés de graisse dans le sang (dyslipidémie mixte) :
o qui ne sont pas contrôlés de façon appropriée par une statine seule,
o pour lesquels vous avez déjà été traité(e) par l’association d’une statine et de l’ézétimibe pris sous forme de comprimés séparés.
· une maladie héréditaire (hypercholestérolémie familiale homozygote) qui augmente le taux de cholestérol sanguin. Vous pourrez également recevoir d’autres traitements.
· une maladie cardiaque. LIPTRUZET réduit le risque de crise cardiaque, d’accident vasculaire cérébral, d’intervention chirurgicale pour augmenter le flux sanguin cardiaque ou d’hospitalisation pour des douleurs thoraciques.
LIPTRUZET ne vous aide pas à perdre du poids.
Présentations
> plaquette(s) aluminium de 30 comprimé(s)
Code CIP : 280 048-9 ou 34009 280 048 9 5
Déclaration de commercialisation : 12/11/2015
Cette présentation est agréée aux collectivités
- Prix hors honoraire de dispensation : 19,60 €
- Honoraire de dispensation : 1,02 €
- Prix honoraire compris : 20,62 €
- Taux de remboursement :65%
- chez les adultes avec hypercholestérolémie (certains types uniquement) ou dyslipidémie mixte si elle ne peut être contrôlée par une statine utilisée seule
- chez les patients recevant déjà un traitement avec les mêmes principes actifs que ceux contenus dans ce médicament, dans le cadre d'un traitement de substitution. ; JOURNAL OFFICIEL ; 03/11/15
> plaquette(s) aluminium de 90 comprimé(s)
Code CIP : 280 049-5 ou 34009 280 049 5 6
Déclaration de commercialisation : 30/09/2019
Cette présentation est agréée aux collectivités
- Prix hors honoraire de dispensation : 58,26 €
- Honoraire de dispensation : 2,76 €
- Prix honoraire compris : 61,02 €
- Taux de remboursement :65%
- chez les adultes avec hypercholestérolémie (certains types uniquement) ou dyslipidémie mixte si elle ne peut être contrôlée par une statine utilisée seule
- chez les patients recevant déjà un traitement avec les mêmes principes actifs que ceux contenus dans ce médicament, dans le cadre d'un traitement de substitution. ; JOURNAL OFFICIEL ; 03/11/15
Service médical rendu (SMR)
Les libellés affichés ci-dessous ne sont que des résumés ou extraits issus des avis rendus par la Commission de la Transparence. Seul l'avis complet de la Commission de la Transparence fait référence.
Cet avis est consultable à partir du lien `Avis du jj/mm/aaaa` ou encore sur demande auprès de la HAS (plus d'informations dans l'aide). Les avis et synthèses d'avis contiennent un paragraphe sur la place du médicament dans la stratégie thérapeutique.
| Valeur du SMR | Avis | Motif de l'évaluation | Résumé de l'avis |
|---|---|---|---|
| Non précisé | Avis du 07/12/2016 | Extension d'indication non sollicitée | Le laboratoire ne demande pas l’inscription des spécialités LIPTRUZET dans cette indication et rappelle que de ce fait ces spécialités ne sont ni remboursables ni agréées aux collectivités dans l’indication : « Prévention des événements cardiovasculaires : LIPTRUZET est indiqué pour réduire le risque d’événements cardiovasculaires (voir la rubrique 5.1) chez les patients présentant une maladie coronaire avec un antécédent de syndrome coronarien aigu (SCA), précédemment traités ou non par statine. » |
| Important | Avis du 18/02/2015 | Inscription (CT) | Le service médical rendu par LIPTRUZET est important : • chez les adultes avec hypercholestérolémie (familiale hétérozygote, homozygote ou non familiale) non contrôlée par un traitement bien conduit par une statine en monothérapie, • chez les patients recevant déjà de l’atorvastatine et de l’ézétimibe aux mêmes doses, dans le cadre d’un traitement de substitution. |
Amélioration du service médical rendu (ASMR)
Les libellés affichés ci-dessous ne sont que des résumés ou extraits issus des avis rendus par la Commission de la Transparence. Seul l'avis complet de la Commission de la Transparence fait référence.
Cet avis est consultable à partir du lien `Avis du jj/mm/aaaa` ou encore sur demande auprés de la HAS (plus d'informations dans l'aide). Les avis et synthéses d'avis contiennent un paragraphe sur la place du médicament dans la stratégie thérapeutique.
| Valeur de l'ASMR | Avis | Motif de l'évaluation | Résumé de l'avis |
|---|---|---|---|
| V (Inexistant) | Avis du 18/02/2015 | Inscription (CT) | LIPTRUZET (association fixe d’ézetimibe 10 mg et d’atorvastatine 10, 20, 40 ou 80 mg) n’apporte pas d’amélioration du service médical rendu (ASMR V) par rapport à la prise séparée des deux principes aux mêmes doses. |
ANSM - Mis à jour le : 14/04/2025
LIPTRUZET 10 mg/10 mg, comprimé pelliculé
2. COMPOSITION QUALITATIVE ET QUANTITATIVE
Chaque comprimé pelliculé contient :
ézétimibe.............................................................................................................................. 10 mg
atorvastatine (sous forme d’atorvastatine calcique trihydratée)................................................. 10 mg
Excipients à effet notoire :
Chaque comprimé pelliculé 10 mg/10 mg contient 153 mg de lactose.
Pour la liste complète des excipients, voir rubrique 6.1.
Comprimé 10 mg/10 mg : comprimé pelliculé biconvexe en forme de gélule, blanc à blanc cassé, mesurant 12,74 mm x 5,10 mm, portant la mention « 257 » gravée sur une face.
4.1. Indications thérapeutiques
Prévention des événements cardiovasculaires
LIPTRUZET est indiqué pour réduire le risque d’événements cardiovasculaires (voir rubrique 5.1) chez les patients présentant une maladie coronaire avec un antécédent de syndrome coronarien aigu (SCA), précédemment traités ou non par une statine.
Hypercholestérolémie
LIPTRUZET est indiqué comme traitement adjuvant au régime chez les patients adultes ayant une hypercholestérolémie primaire (familiale hétérozygote et non familiale) ou une dyslipidémie mixte lorsque l’utilisation d’une association est appropriée :
· patients non contrôlés de façon appropriée par une statine seule,
· patients recevant déjà une statine et de l’ézétimibe.
Hypercholestérolémie familiale homozygote (HFHo)
LIPTRUZET est indiqué comme traitement adjuvant au régime chez les patients adultes ayant une HFHo. Ces patients peuvent recevoir également des traitements adjuvants (par ex. aphérèse des LDL).
4.2. Posologie et mode d'administration
Hypercholestérolémie et/ou maladie coronaire (avec antécédent de syndrome coronarien aigu)
Pendant toute la durée du traitement par LIPTRUZET, le patient devra suivre un régime hypolipidémiant adapté.
La posologie de LIPTRUZET est de 10/10 mg par jour à 10/80 mg par jour. La dose habituelle est de 10/10 mg une fois par jour. Le taux de cholestérol lié aux lipoprotéines de faible densité (LDL‑C), les facteurs de risque de maladie coronaire, et la réponse au traitement hypocholestérolémiant habituel du patient seront pris en compte à l’instauration du traitement ou en cas d’ajustement de la posologie.
La posologie de LIPTRUZET doit être individualisée et tenir compte de l’efficacité connue des différents dosages de LIPTRUZET (voir rubrique 5.1, tableau 4) ainsi que de la réponse au traitement hypolipidémiant en cours. Les ajustements posologiques, si nécessaire, doivent être effectués à intervalles de 4 semaines ou plus.
Hypercholestérolémie familiale homozygote
La posologie de LIPTRUZET chez les patients présentant une HF homozygote est de 10/10 mg à 10/80 mg par jour. Chez ces patients, LIPTRUZET peut être utilisé comme adjuvant d’un autre traitement hypocholestérolémiant (par ex. aphérèse des LDL) ou quand ces traitements ne sont pas disponibles.
Co-administration avec d’autres médicaments
L’administration de LIPTRUZET se fera soit ≥ 2 heures avant ou ≥ 4 heures après l’administration d’un chélateur des acides biliaires.
Chez les patients prenant les médicaments antiviraux contre l'hépatite C elbasvir/grazoprévir de façon concomitante avec LIPTRUZET, la dose de LIPTRUZET ne doit pas dépasser 10 mg/20 mg par jour (voir rubriques 4.4 et 4.5).
Sujets âgés
Aucun ajustement posologique n’est nécessaire chez les patients âgés (voir rubrique 5.2).
Population pédiatrique
La sécurité et l’efficacité de LIPTRUZET chez les enfants n’ont pas été établies (voir rubrique 5.2). Aucune donnée n’est disponible.
Insuffisance hépatique
LIPTRUZET doit être utilisé avec prudence chez les patients présentant une insuffisance hépatique (voir rubriques 4.4 et 5.2). LIPTRUZET est contre-indiqué chez les patients présentant une hépatopathie évolutive (voir rubrique 4.3).
Insuffisance rénale
Aucune adaptation posologique n’est nécessaire chez les patients présentant une insuffisance rénale (voir rubrique 5.2).
Mode d’administration
Voie orale. LIPTRUZET peut être administré en une prise unique à tout moment de la journée, au cours ou en dehors des repas.
Hypersensibilité aux substances actives ou à l’un des excipients mentionnés à la rubrique 6.1.
LIPTRUZET est contre-indiqué pendant la grossesse et l’allaitement et chez les femmes en âge de procréer n’utilisant pas de méthodes contraceptives appropriées (voir rubrique 4.6).
LIPTRUZET est contre-indiqué chez les patients présentant une hépatopathie évolutive ou des élévations persistantes inexpliquées des transaminases sériques supérieures à 3 fois la limite supérieure de la normale (LSN).
LIPTRUZET est contre-indiqué chez les patients traités par les antiviraux contre l’hépatite C glécaprévir/pibrentasvir.
4.4. Mises en garde spéciales et précautions d'emploi
Depuis la mise sur le marché de l’ézétimibe, des cas de myopathie et de rhabdomyolyse ont été rapportés. La plupart des patients qui ont présenté une rhabdomyolyse prenaient également une statine en association à l’ézétimibe. Cependant, des cas de rhabdomyolyse ont été très rarement rapportés avec l’ézétimibe en monothérapie, ou lorsque l’ézétimibe était associé à d’autres médicaments connus comme étant liés à un risque accru de rhabdomyolyse.
LIPTRUZET contient de l’atorvastatine. Comme les autres inhibiteurs de la HMG-CoA réductase, l’atorvastatine peut dans de rares cas entraîner une atteinte des muscles squelettiques et provoquer des myalgies, une myosite et une myopathie pouvant progresser en rhabdomyolyse, une affection susceptible d’engager le pronostic vital caractérisée par un taux élevé de créatine phosphokinase (CPK) (> 10 x LSN), une myoglobinémie et une myoglobinurie pouvant entraîner une insuffisance rénale.
Avant le traitement
LIPTRUZET doit être prescrit avec prudence chez les patients présentant des facteurs de risque de rhabdomyolyse. Un dosage de la CPK doit être effectué avant le début du traitement dans les cas suivants :
· insuffisance rénale,
· hypothyroïdie,
· antécédents personnels ou familiaux de myopathie héréditaire,
· antécédents de toxicité musculaire lors du traitement par une statine ou un fibrate,
· antécédents d’hépatopathie et/ou consommation excessive d’alcool,
· chez les patients âgés (> 70 ans), la nécessité du dosage de la CPK doit être envisagée en fonction de la présence d’autres facteurs de risque de rhabdomyolyse,
· situations dans lesquelles les concentrations plasmatiques peuvent être augmentées, par exemple en raison d'interactions (voir rubrique 4.5) et dans des populations particulières incluant les polymorphismes génétiques (voir rubrique 5.2).
Dans de telles situations, le risque du traitement doit être évalué par rapport au bénéfice potentiel et une surveillance clinique est recommandée.
Si la valeur basale de CPK est significativement élevée (˃ 5 x LSN), le traitement ne devra pas être initié.
Dosage de la créatine phosphokinase
La créatine phosphokinase (CPK) ne doit pas être dosée après un effort intense ou en présence de toute autre cause possible d’élévation de la CPK car cela rendrait l’interprétation des résultats difficile. Si le taux de CPK à l’état basal est significativement élevé par rapport à la normale (˃ 5 x LSN), il doit être contrôlé de nouveau 5 à 7 jours plus tard pour confirmer les résultats.
Pendant le traitement
· Les patients doivent être incités à signaler sans délai toutes douleurs, crampes ou faiblesses musculaires, en particulier si elles sont accompagnées de malaise ou de fièvre ou si les signes et symptômes musculaires persistent après l'arrêt de LIPTRUZET.
· En cas de survenue de ces symptômes chez un patient pendant le traitement par LIPTRUZET, un dosage de la CPK doit être effectué. Si le taux est significativement élevé (> 5 x LSN), le traitement doit être arrêté.
· Si les symptômes musculaires sont sévères et entraînent une gêne quotidienne, l’arrêt du traitement doit être envisagé, même si le taux de CPK est ≤ 5 x LSN.
· En cas de résolution des symptômes et de normalisation du taux de CPK, la reprise du traitement par LIPTRUZET ou par un autre médicament contenant une statine peut être envisagée à la dose la plus faible et sous surveillance attentive.
· Le traitement par LIPTRUZET doit être arrêté en cas d’élévation cliniquement significative du taux de CPK (> 10 x LSN) ou de diagnostic ou suspicion de rhabdomyolyse.
· De très rares cas de myopathie nécrosante à médiation auto-immune (IMNM) ont été signalés pendant ou après le traitement par certaines statines. La myopathie nécrosante à médiation auto-immune (IMNM) est caractérisée cliniquement par une faiblesse musculaire proximale et une élévation de la créatinine kinase sérique, qui persistent malgré l’arrêt du traitement par la statine.
Du fait de l’atorvastatine contenue dans LIPTRUZET, le risque de rhabdomyolyse est majoré lorsque LIPTRUZET est administré en association avec certains médicaments qui peuvent augmenter la concentration plasmatique de l’atorvastatine, tels que les inhibiteurs puissants du CYP3A4 ou des transporteurs protéiques (par exemple ciclosporine, télithromycine, clarithromycine, délavirdine, stiripentol, kétoconazole, voriconazole, itraconazole, posaconazole et inhibiteurs de protéase du VIH incluant ritonavir, lopinavir, atazanavir, indinavir, darunavir, tipranavir/ritonavir, etc.). Le risque de myopathie peut être également augmenté en association avec le gemfibrozil et les autres fibrates, les antiviraux utilisés dans le traitement de l'hépatite C (VHC) (bocéprévir, télaprévir, elbasvir/grazoprévir), l’érythromycine ou la niacine. Des alternatives thérapeutiques (ne présentant pas ces interactions) devront être envisagées dans la mesure du possible (voir rubrique 4.8).
Si l’association de ces médicaments avec LIPTRUZET est nécessaire, le rapport bénéfice/risque du traitement concomitant doit être évalué soigneusement. Chez les patients recevant des médicaments qui augmentent la concentration plasmatique d’atorvastatine, une dose maximale plus faible de LIPTRUZET est recommandée. De plus, en cas d’administration d’inhibiteurs puissants du CYP3A4, une dose initiale plus faible de LIPTRUZET doit être envisagée et une surveillance clinique appropriée de ces patients est recommandée (voir rubrique 4.5).
LIPTRUZET ne doit pas être administré simultanément à l’acide fusidique sous forme systémique, et jusque dans les 7 jours suivant l’arrêt d’un traitement par acide fusidique. Chez les patients où l’utilisation d’acide fusidique systémique est considérée comme essentielle, le traitement par statine doit être interrompu pendant toute la durée du traitement par l’acide fusidique. Des cas de rhabdomyolyses (dont certains fatals) ont été rapportés chez des patients recevant de l’acide fusidique et une statine en association (voir rubrique 4.5). Les patients doivent être informés de la nécessité de consulter immédiatement un médecin s’ils présentent des symptômes de faiblesse musculaire, douleur ou sensibilité.
Le traitement par statine peut être réintroduit sept jours après la dernière dose d’acide fusidique.
Dans des circonstances exceptionnelles, où un traitement prolongé par acide fusidique systémique est nécessaire, par exemple pour le traitement d’infections graves, la nécessité d’une co-administration de LIPTRUZET et d’acide fusidique ne doit être considérée qu’au cas par cas et sous surveillance médicale étroite.
Des cas de myopathie et/ou de rhabdomyolyse ont été rapportés lors de l’administration concomitante d’inhibiteurs de l'HMG-CoA réductase (tels que atorvastatine et ézétimibe/atorvastatine) avec la daptomycine. La prudence est recommandée lors de la prescription d'inhibiteurs de l'HMG-CoA réductase avec la daptomycine, car l'un ou l'autre de ces médicaments peut entrainer une myopathie et/ou une rhabdomyolyse lorsqu'ils sont administrés seuls. Un arrêt temporaire de LIPTRUZET doit être envisagé chez les patients traités par la daptomycine à moins que les avantages d’une administration concomitante prévalent sur les risques. Consulter les informations de prescription de la daptomycine pour obtenir plus d’informations sur cette interaction potentielle avec les inhibiteurs de l'HMG-CoA réductase (tels que atorvastatine et ézétimibe/ atorvastatine) et pour plus de conseils sur la surveillance (voir rubrique 4.5).
Myasthénie et myasthénie oculaire
Dans quelques cas, il a été rapporté que les statines induisaient de novo ou aggravaient une myasthénie préexistante ou une myasthénie oculaire (voir rubrique 4.8). LIPTRUZET doit être arrêté en cas d’aggravation des symptômes. Des récurrences ont été rapportées lorsque la même statine ou une statine différente a été (ré)administrée.
Enzymes hépatiques
Dans les études cliniques, des élévations des transaminases sériques (> 3 x LSN) sont survenues à plusieurs reprises chez des patients recevant de l’ézétimibe et de l’atorvastatine (voir rubrique 4.8).
Un bilan hépatique doit être réalisé avant l’instauration du traitement et régulièrement ensuite. La fonction hépatique doit être contrôlée chez les patients qui développent des signes ou symptômes évocateurs d’une atteinte hépatique. Les patients présentant une élévation des taux de transaminases doivent être surveillés jusqu’à la résolution des anomalies. En cas d’augmentation persistante des transaminases > 3 x LSN, il est recommandé de diminuer la dose ou d’arrêter le traitement par LIPTRUZET.
LIPTRUZET doit être utilisé avec précaution chez les patients consommant d’importantes quantités d’alcool et/ou ayant des antécédents d’atteinte hépatique.
Insuffisance hépatique
Chez les patients ayant une insuffisance hépatique modérée ou sévère, les effets d’une exposition accrue à l’ézétimibe n’étant pas connus, LIPTRUZET n’est pas recommandé (voir rubrique 5.2).
Fibrates
L’efficacité et la sécurité d’emploi de l’ézétimibe administré avec des fibrates n’ont pas été établies ; par conséquent, l’association de LIPTRUZET avec des fibrates n’est pas recommandée (voir rubrique 4.5).
Ciclosporine
La prudence s’impose en cas d’instauration de LIPTRUZET pendant un traitement par la ciclosporine.
Les concentrations de ciclosporine doivent être surveillées chez les patients recevant LIPTRUZET en association avec la ciclosporine (voir rubrique 4.5).
Anticoagulants
Si LIPTRUZET est associé à la warfarine, ou à un autre anticoagulant anti-vitamine K ou à la fluindione, le taux de prothrombine exprimé en INR doit être surveillé de façon appropriée (voir rubrique 4.5).
Etude SPARCL (Stroke Prevention by Aggressive Reduction in Cholesterol Levels)
Dans une analyse post hoc des sous-types d’accidents vasculaires cérébraux chez des patients non coronariens qui présentaient des antécédents récents d’accident vasculaire cérébral (AVC) ou d’accident ischémique transitoire (AIT), l’incidence d’AVC hémorragique était plus élevée chez les patients traités par l’atorvastatine 80 mg que chez les patients recevant le placebo. L’augmentation du risque a été observée en particulier chez les patients qui avaient des antécédents d’accident vasculaire cérébral hémorragique ou d’infarctus lacunaire lors de l’inclusion dans l’étude.
Chez ces patients, le rapport bénéfice/risque de l’atorvastatine 80 mg est incertain et le risque potentiel d’AVC hémorragique doit être soigneusement pris en compte avant l’instauration du traitement (voir rubrique 5.1).
Pneumopathie interstitielle
Des cas exceptionnels de pneumopathie interstitielle ont été rapportés lors de la prise de certaines statines, en particulier en cas de traitement à long terme (voir rubrique 4.8). Les symptômes se caractérisent par une dyspnée, une toux non productive et une altération de l’état de santé général (fatigue, perte de poids et fièvre). En cas de suspicion de pneumopathie interstitielle chez un patient, le traitement par statine doit être interrompu.
Diabète
Certaines données suggèrent que les statines, en tant que classe pharmacologique, augmenteraient la glycémie. Chez certains patients à risque élevé de survenue d’un diabète, les statines peuvent entraîner une hyperglycémie nécessitant l’instauration d’un traitement antidiabétique. Ce risque est néanmoins compensé par la réduction du risque vasculaire sous statines et par conséquent, il ne doit pas être un motif d’arrêt des statines. Les patients à risque (glycémie à jeun comprise entre 5,6 et 6,9 mmol/L, IMC > 30 kg/m2, augmentation du taux des triglycérides, hypertension artérielle) devront faire l’objet d’une surveillance clinique et biologique conformément aux recommandations nationales.
Excipients
LIPTRUZET contient du lactose. Les patients présentant une intolérance au galactose, un déficit total en lactase ou un syndrome de malabsorption du glucose et du galactose (maladies héréditaires rares) ne doivent pas prendre ce médicament.
LIPTRUZET contient moins de 1 mmol (23 mg) de sodium par comprimé et est considéré comme étant essentiellement exempt de sodium.
4.5. Interactions avec d'autres médicaments et autres formes d'interactions
De nombreux mécanismes peuvent contribuer à des interactions potentielles avec les inhibiteurs de la HMG-CoA réductase. Des médicaments ou produits à base de plantes qui inhibent certains enzymes (par ex., le CYP3A4) et/ou transporteurs (par ex., l’OATP1B) peuvent augmenter les concentrations plasmatiques de l’atorvastatine et entraîner un risque accru de myopathie/rhabdomyolyse.
Consulter les informations de prescription de tous les autres médicaments co-administrés pour obtenir plus d’informations sur leurs interactions potentielles avec l’atorvastatine et/ou les modifications potentielles enzymatiques ou des transporteurs ainsi que sur les ajustements possibles de la posologie et du schéma thérapeutique.
Interactions pharmacodynamiques
L’atorvastatine est métabolisée par le cytochrome P450 3A4 (CYP3A4) et est un substrat des transporteurs hépatiques, polypeptides transporteurs d’anions organiques 1B1 (OATP1B1) et 1B3 (OATP1B3). Les métabolites de l’atorvastatine sont des substrats de l’OATP1B1. L’atorvastatine est également identifiée comme étant un substrat de la protéine de multirésistance aux médicaments (MDR1) et de la protéine de résistance au cancer du sein (BCRP), ce qui peut limiter l’absorption intestinale et la clairance biliaire de l’atorvastatine (voir rubrique 5.2). L’administration concomitante d’inhibiteurs du CYP3A4 ou des transporteurs protéiques peut augmenter les concentrations plasmatiques d’atorvastatine et entraîner un risque majoré de myopathie. Le risque peut aussi être augmenté lors de l’administration concomitante de LIPTRUZET avec d’autres médicaments susceptibles d’induire des myopathies tels que les fibrates et l’ézétimibe (voir rubrique 4.4).
Interactions pharmacocinétiques
LIPTRUZET
Aucune interaction pharmacocinétique cliniquement significative n’a été observée lors de l’administration concomitante d’ézétimibe et d’atorvastatine.
Effets d’autres médicaments sur LIPTRUZET
Ezétimibe
Antiacides : l’administration simultanée d'antiacides diminue le taux d'absorption d'ézétimibe mais n'a aucun effet sur la biodisponibilité d’ézétimibe. Cette diminution du taux d’absorption d’ézétimibe n’est pas considérée comme cliniquement significative.
Cholestyramine : l’administration simultanée de cholestyramine diminue d’environ 55 % l’aire sous la courbe (ASC) moyenne de l’ézétimibe total (ézétimibe + glycuronide d’ézétimibe). La diminution supplémentaire du LDL-Cholestérol observée liée à l’association LIPTRUZET et cholestyramine pourrait être réduite par cette interaction (voir rubrique 4.2).
Ciclosporine : une étude réalisée chez 8 patients transplantés rénaux ayant une clairance de la créatinine > 50 mL/min, recevant une dose stable de ciclosporine et une dose unique de 10 mg d’ézétimibe a montré une augmentation de l’ASC moyenne de l’ézétimibe total de 3,4 fois (2,3 à 7,9 fois) par rapport à des volontaires sains d’une autre étude (n = 17) recevant de l’ézétimibe seul. Une autre étude a montré que, chez un patient transplanté rénal ayant une insuffisance rénale sévère et recevant de la ciclosporine et de nombreux médicaments, l’exposition totale à l’ézétimibe était 12 fois supérieure à celle des témoins recevant de l’ézétimibe seul. Dans une étude en cross-over, de deux périodes, chez douze sujets sains, l’administration quotidienne de 20 mg d’ézétimibe pendant 8 jours avec une seule dose de 100 mg de ciclosporine au 7ème jour a entraîné une augmentation moyenne de 15 % de l’ASC de la ciclosporine (avec une variation allant d’une diminution de 10 % à une augmentation de 51 %) comparée à l’administration d’une dose unique de ciclosporine seule. Aucune étude contrôlée de l’effet de l’association ézétimibe/ciclosporine n’a été effectuée chez les patients transplantés rénaux.
La prudence s’impose en cas d’instauration de LIPTRUZET pendant un traitement par la ciclosporine. Les concentrations de ciclosporine doivent être surveillées chez les patients recevant LIPTRUZET en association avec la ciclosporine (voir rubrique 4.4).
Fibrates : l’administration simultanée de fénofibrate ou de gemfibrozil augmente les concentrations totales d’ézétimibe, respectivement d’environ 1,5 et 1,7 fois ; bien que ces augmentations ne soient pas considérées comme cliniquement significatives, l’association de LIPTRUZET avec les fibrates est déconseillée (voir rubrique 4.4).
Atorvastatine
Inhibiteurs du CYP3A4 : les concentrations plasmatiques d’atorvastatine sont augmentées de façon importante lors de l’association avec les inhibiteurs puissants du CYP3A4 (voir le tableau 1 et les informations spécifiques ci-dessous). L’association d’inhibiteurs puissants du CYP3A4 (tels que ciclosporine, télithromycine, clarithromycine, délavirdine, stiripentol, kétoconazole, voriconazole, itraconazole, posaconazole, certains antiviraux utilisés dans le traitement du VHC (par exemple, elbasvir/grazoprévir) et d’inhibiteurs de protéase du VIH incluant ritonavir, lopinavir, atazanavir, indinavir, darunavir, etc.) doit être évitée dans la mesure du possible. Dans les cas où l’association de ces médicaments avec LIPTRUZET s’avère nécessaire, une dose initiale plus faible et une dose maximale plus faible de LIPTRUZET doivent être envisagées et une surveillance clinique appropriée du patient est recommandée (voir tableau 1).
Les inhibiteurs modérés du CYP3A4 (par exemple érythromycine, diltiazem, vérapamil et fluconazole) peuvent augmenter les concentrations plasmatiques d’atorvastatine (voir tableau 1). Un risque accru de myopathie a été observé en cas d’association d’érythromycine avec des statines. Il n’a pas été réalisé d’études d’interactions évaluant les effets de l’amiodarone ou du vérapamil sur l’atorvastatine. L’amiodarone et le vérapamil sont des inhibiteurs connus du CYP3A4 et l’association avec LIPTRUZET peut augmenter l’exposition à l’atorvastatine. Par conséquent, une dose maximale plus faible de LIPTRUZET doit être envisagée et une surveillance clinique du patient est recommandée en cas d’administration concomitante avec des inhibiteurs modérés du CYP3A4. Une surveillance clinique appropriée est recommandée après l’instauration ou une adaptation posologique de l'inhibiteur.
Inhibiteurs de la protéine de résistance du cancer du sein (BCRP) : l'administration concomitante de médicaments inhibiteurs de la BCRP (tels que elbasvir et grazoprevir) peut entraîner une augmentation des concentrations plasmatiques d’atorvastatine et un risque accru de myopathie ; par conséquent, un ajustement posologique de l’atorvastatine doit être envisagé en fonction de la dose prescrite. L'administration concomitante d'élbasvir et de grazoprevir avec l'atorvastatine augmente les concentrations plasmatiques d'atorvastatine de 1,9 fois (voir Tableau 1) ; par conséquent, la dose de LIPTRUZET ne doit pas dépasser 10 mg/20 mg par jour chez les patients recevant de façon concomitante des médicaments contenant de l'elbasvir ou du grazoprevir (voir rubriques 4.2 et 4.4).
Inducteurs du cytochrome P450 3A4 : l’administration concomitante d’atorvastatine avec des inducteurs du cytochrome P450 3A4 (par exemple éfavirenz, rifampicine, millepertuis) peut entraîner des diminutions variables des concentrations plasmatiques d’atorvastatine. Du fait du double mécanisme d’interaction de la rifampicine (induction du cytochrome P450 3A4 et inhibition du transporteur d’influx hépatique OATP1B1), l’administration simultanée de LIPTRUZET et de la rifampicine est conseillée, car l’administration d’atorvastatine décalée dans le temps avec celle de la rifampicine a été associée à une réduction significative des concentrations plasmatiques d’atorvastatine. L’effet de la rifampicine sur les concentrations hépatocytaires d’atorvastatine n’est toutefois pas connu et si l’association ne peut être évitée, l’efficacité du traitement doit être étroitement surveillée.
Inhibiteurs des transporteurs : les inhibiteurs des transporteurs protéiques (tels que la ciclosporine) peuvent augmenter l’exposition systémique à l’atorvastatine (voir tableau 1). L’effet de l’inhibition des transporteurs hépatocytaires sur les concentrations hépatocytaires d’atorvastatine est inconnu. Si l’association s’avère nécessaire, la dose de LIPTRUZET doit être diminuée et l’efficacité du traitement doit être surveillée (voir tableau 1).
Gemfibrozil/fibrates : le traitement par des fibrates seuls peut occasionnellement provoquer des événements indésirables musculaires, incluant une rhabdomyolyse. Le risque de ces événements peut être majoré en cas d’association de fibrates et d’atorvastatine.
Ezétimibe : l’utilisation d’ézétimibe en monothérapie est associée à des effets secondaires musculaires incluant des rhabdomyolyses. Le risque de survenue de ces effets secondaires peut donc être accru en cas d’association d’ézétimibe et d’atorvastatine. Une surveillance clinique appropriée de ces patients est recommandée.
Colestipol : les concentrations plasmatiques d’atorvastatine et de son métabolite actif sont diminuées (d’environ 25 %) en cas d’administration concomitante de colestipol et d’atorvastatine. Cependant les effets hypolipidémiants sont plus importants lorsque l’atorvastatine et le colestipol sont administrés en association que lorsque chaque médicament est administré seul.
Acide fusidique : le risque de myopathie, y compris la rhabdomyolyse, peut être augmenté par la co-administration d’acide fusidique systémique et de statine. Ce mécanisme d’interaction (qu’il soit pharmacodynamique, pharmacocinétique ou les deux) est encore inconnu. Des cas de rhabdomyolyse (dont certains fatals) ont été rapportés chez des patients prenant cette association.
Si un traitement systémique par l’acide fusidique est nécessaire, le traitement par atorvastatine doit être interrompu pendant la durée du traitement par l’acide fusidique (Voir également la rubrique 4.4).
Colchicine : bien qu’il n’ait pas été réalisé d’études d’interactions entre l’atorvastatine et la colchicine, des cas de myopathie ont été rapportés lors de l’administration concomitante d'atorvastatine et de colchicine et la prudence s’impose en cas de prescription d’atorvastatine avec la colchicine.
Daptomycine : le risque de myopathie et/ou de rhabdomyolyse peut être plus élevé lors de l'administration concomitante d'inhibiteurs de l'HMG-CoA réductase avec la daptomycine. Un arrêt temporaire de LIPTRUZET doit être envisagé chez les patients traités par la daptomycine à moins que les avantages d’une administration concomitante prévalent sur les risques (voir rubrique 4.4).
Bocéprévir : l’exposition à l’atorvastatine est augmentée en cas d’administration avec le bocéprévir. Si l’association avec LIPTRUZET est nécessaire, il est recommandé d’instaurer le traitement par LIPTRUZET à la dose la plus faible possible et d’augmenter ensuite la posologie sous surveillance étroite jusqu’à l’obtention de l’effet clinique souhaité, sans dépasser une dose quotidienne de 10/20 mg. Chez les patients recevant déjà LIPTRUZET, la dose quotidienne de LIPTRUZET ne doit pas dépasser 10/20 mg lors d’un traitement concomitant par le bocéprévir.
Effets de LIPTRUZET sur la pharmacocinétique d’autres médicaments
Ezétimibe
Des études précliniques ont montré que l’ézétimibe n’induit pas les enzymes du cytochrome P450 responsables du métabolisme des médicaments. Aucune interaction pharmacocinétique cliniquement significative n’a été observée entre l’ézétimibe et les médicaments connus pour être métabolisés par les cytochromes P450 1A2, 2D6, 2C8, 2C9, et 3A4 ou la N-acétyltransférase.
Anticoagulants : dans une étude chez 12 volontaires sains de sexe masculin, l’administration concomitante d’ézétimibe (10 mg une fois par jour) n’a pas eu d’effet significatif sur la biodisponibilité de la warfarine et sur le temps de prothrombine. Cependant, depuis la mise sur le marché, une augmentation de l'INR a été rapportée chez des patients prenant de l’ézétimibe en association à la warfarine ou à la fluindione. Si LIPTRUZET est associé à la warfarine ou à un autre anticoagulant dérivé de la coumarine (AVK), ou à la fluindione, l’INR doit être surveillé de façon appropriée (voir rubrique 4.4).
Atorvastatine
Digoxine : après administration concomitante de doses répétées de digoxine et d’atorvastatine 10 mg, les concentrations à l’état d’équilibre de la digoxine sont légèrement augmentées. Une surveillance appropriée s’impose chez les patients traités par la digoxine.
Contraceptifs oraux : l’administration concomitante d’atorvastatine avec un contraceptif oral a augmenté les concentrations plasmatiques de noréthistérone et d’éthinylestradiol.
Warfarine : dans une étude clinique menée chez des patients recevant un traitement au long cours par la warfarine, l’administration concomitante d’atorvastatine 80 mg par jour avec la warfarine a entraîné une faible diminution d’environ 1,7 seconde du temps de Quick pendant les 4 premiers jours de traitement ; la valeur s’est normalisée dans les 15 jours suivant le début du traitement par l’atorvastatine. Bien que seulement de très rares cas d’interactions cliniquement significatives avec les anticoagulants aient été rapportés, le temps de Quick doit être déterminé avant l’instauration du traitement par LIPTRUZET chez les patients recevant des anticoagulants coumariniques, et assez fréquemment en début de traitement pour vérifier l’absence de modification significative de sa valeur. Une fois la stabilité du temps de Quick documentée, les contrôles peuvent être effectués aux intervalles recommandés habituellement pour les patients sous anticoagulants coumariniques. La même procédure doit être appliquée en cas de modification de la dose de LIPTRUZET ou d’arrêt du traitement. Le traitement par l’atorvastatine n’a pas été associé à des saignements ou à des modifications du temps de Quick chez les patients ne recevant pas d’anticoagulants.
Tableau 1
Effet des médicaments co-administrés sur la pharmacocinétique de l’atorvastatine
|
Médicament co-administré et schéma posologique |
Atorvastatine |
LIPTRUZET |
|
|
Dose (mg) |
Modification de l’ASC& |
Recommandation clinique# |
|
|
Tipranavir 500 mg 2 fois par jour ritonavir 200 mg 2 fois par jour, 8 jours (jours 14 à 21) |
40 mg le jour 1, 10 mg le jour 20 |
↑ x 9,4 |
Si l’association avec LIPTRUZET est nécessaire, ne pas dépasser 10/10 mg de LIPTRUZET par jour. Une surveillance clinique de ces patients est recommandée. |
|
Ciclosporine 5,2 mg/kg/jour, dose stable |
10 mg 1 fois par jour pendant 28 jours |
↑ x 8,7 |
|
|
Lopinavir 400 mg 2 fois par jour/ritonavir 100 mg 2 fois par jour, 14 jours |
20 mg 1 fois par jour pendant 4 jours |
↑ x 5,9 |
Si l’association avec LIPTRUZET est nécessaire, des doses d’entretien plus faibles de LIPTRUZET sont recommandées. Aux doses de LIPTRUZET supérieures à 10/20 mg, une surveillance clinique est recommandée chez ces patients. |
|
Clarithromycine 500 mg 2 fois par jour, 9 jours |
80 mg 1 fois par jour pendant 8 jours |
↑ x 4,4 |
|
|
Saquinavir 400 mg 2 fois par jour/ritonavir 300 mg 2 fois par jour des jours 5 à 7, dose augmentée à 400 mg 2 fois par jour le jour 8), jours 5 à 18, 30 min après l’administration d’atorvastatine |
40 mg 1 fois par jour pendant 4 jours |
↑ x 3,9 |
Si l’association avec LIPTRUZET est nécessaire, des doses d’entretien plus faibles de LIPTRUZET sont recommandées. Aux doses de LIPTRUZET supérieures à 10/40 mg, une surveillance clinique est recommandée chez ces patients. |
|
Darunavir 300 mg 2 fois par jour/ ritonavir 100 mg 2 fois par jour, 9 jours |
10 mg 1 fois par jour pendant 4 jours |
↑ x 3,3 |
|
|
Itraconazole 200 mg 1 fois par jour, 4 jours |
40 mg DU |
↑ x 3,3 |
|
|
Fosamprénavir 700 mg 2 fois par jour/ritonavir 100 mg 2 fois par jour, 14 jours |
10 mg 1 fois par jour pendant 4 jours |
↑ x 2,5 |
|
|
Fosamprénavir 1 400 mg 2 fois par jour, 14 jours |
10 mg 1 fois par jour pendant 4 jours |
↑ x 2,3 |
|
|
Nelfinavir 1 250 mg 2 fois par jour, 14 jours |
10 mg 1 fois par jour pendant 28 jours |
↑ x 1,7^ |
Pas de recommandation particulière. |
|
Jus de pamplemousse, 240 mL 1 fois par jour* |
40 mg DU |
↑ 37 % |
La consommation de quantités importantes de jus de pamplemousse est déconseillée pendant le traitement par LIPTRUZET. |
|
Diltiazem 240 mg 1 fois par jour, 28 jours |
40 mg DU |
↑ 51 % |
Une surveillance clinique appropriée des patients est recommandée après l’instauration ou une adaptation posologique du diltiazem. |
|
Erythromycine 500 mg 4 fois par jour, 7 jours |
10 mg DU |
↑ 33 %^ |
Une dose maximale plus faible et une surveillance clinique de ces patients sont recommandées. |
|
Amlodipine 10 mg, dose unique |
80 mg DU |
↑ 18 % |
Pas de recommandation particulière. |
|
Cimétidine 300 mg 4 fois par jour, 2 semaines |
10 mg 1 fois par jour pendant 4 semaines |
↓ inférieure à 1 %^ |
Pas de recommandation particulière. |
|
Suspension antiacide d’hydroxydes de magnésium et d’aluminium, 30 mL 4 fois par jour, 2 semaines |
10 mg 1 fois par jour pendant 4 semaines |
↓ 35 %^ |
Pas de recommandation particulière. |
|
Efavirenz 600 mg 1 fois par jour, 14 jours |
10 mg pendant 3 jours |
↓ 41 % |
Pas de recommandation particulière. |
|
Rifampicine 600 mg 1 fois par jour, 7 jours (administrations simultanées) |
40 mg DU |
↑ 30 % |
Si l’association ne peut être évitée, l’administration simultanée de LIPTRUZET et de rifampicine est recommandée, avec une surveillance clinique. |
|
Rifampicine 600 mg 1 fois par jour, 5 jours (administrations séparées) |
40 mg DU |
↓ 80 % |
|
|
Gemfibrozil 600 mg 2 fois par jour, 7 jours |
40 mg DU |
↑ 35 % |
Association déconseillée. |
|
Fénofibrate 160 mg 1 fois par jour, 7 jours |
40 mg DU |
↑ 3 % |
Association déconseillée. |
|
Bocéprévir 800 mg 3 fois par jour, 7 jours |
40 mg DU |
↑ x 2,3 |
Une dose initiale plus faible et une surveillance clinique des patients sont recommandées. La dose quotidienne de LIPTRUZET ne doit pas dépasser 10/20 mg pendant un traitement concomitant par le bocéprévir. |
|
Elbasvir 50 mg 1 fois par jour/Grazoprévir 200 mg 1 fois par jour, 13 jours |
10 mg, DU |
↑ x 1,94 |
La dose de LIPTRUZET ne doit pas dépasser 10 mg/20 mg par jour lors de l’association avec des produits contenant de l'elbasvir ou du grazoprévir. |
|
Glécaprévir 400 mg 1 fois par jour/Pibrentasvir 120 mg 1 fois par jour, 7 jours |
10 mg 1 fois par jour pendant 7 jours |
↑ x 8,3 |
L’association avec des produits contenant du glécaprévir ou du pibrentasvir est contre-indiquée (voir rubrique 4.3) |
|
& Les données présentées sous forme « x » représentent un simple rapport entre l'administration concomitante des deux médicaments et l'atorvastatine seule (par ex, x 1 = pas de modification). Les données présentées sous forme de % de variation représentent la différence en % par rapport à l’atorvastatine seule (par ex, 0 % = pas de modification). # Voir les rubriques 4.4 et 4.5 pour la pertinence clinique. * Contient un ou plusieurs composants qui inhibent le CYP3A4 et peuvent augmenter les concentrations plasmatiques des médicaments métabolisés par cette isoenzyme. La consommation d’un verre de 240 mL de jus de pamplemousse a également entraîné une diminution de 20,4 % de l’ASC du métabolite actif orthohydroxylé. Des quantités importantes de jus de pamplemousse (plus de 1,2 litre par jour pendant 5 jours) ont augmenté l’ASC de l’atorvastatine de 2,5 fois et l’ASC des substances actives (atorvastatine et métabolites). ^ Activité totale en équivalent d’atorvastatine. Une augmentation est indiquée par « ↑ », une diminution par « ↓ ». DU = dose unique. |
|||
Tableau 2
Effet de l’atorvastatine sur la pharmacocinétique des médicaments co-administrés
|
Atorvastatine |
Médicament co-administré |
LIPTRUZET |
|
|
et schéma posologique |
Médicament/dose (mg) |
Modification de l’ASC& |
Recommandation clinique |
|
80 mg 1 fois par jour pendant 10 jours |
Digoxine 0,25 mg 1 fois par jour, 20 jours |
↑ 15 % |
Les patients traités par la digoxine doivent être surveillés de façon appropriée. |
|
40 mg 1 fois par jour pendant 22 jours |
Contraceptif oral 1 fois par jour, 2 mois - noréthistérone 1 mg - éthinylestradiol 35 μg |
↑ 28 % ↑ 19 % |
Pas de recommandation particulière. |
|
80 mg 1 fois par jour pendant 15 jours |
* Phénazone, 600 mg DU |
↑ 3 % |
Pas de recommandation particulière. |
|
10 mg 1 fois par jour pendant 4 jours |
Fosamprénavir 1 400 mg 2 fois par jour, 14 jours |
↓ 27 % |
Pas de recommandation particulière. |
|
& Les données présentées sous forme de % de variation représentent la différence en % par rapport à l’atorvastatine seule (par ex, 0 % = pas de modification). * L’administration concomitante de doses répétées d’atorvastatine et de phénazone n’a eu que peu ou pas d’effet détectable sur la clairance de la phénazone. Une augmentation est indiquée par « ↑ », une diminution par « ↓ ». DU = dose unique. |
|||
4.6. Fertilité, grossesse et allaitement
Femmes en âge de procréer
Les femmes en âge de procréer doivent utiliser des méthodes contraceptives appropriées pendant le traitement (voir rubrique 4.3).
Grossesse
L’athérosclérose est une maladie chronique et en général, l’arrêt des médicaments hypolipidémiants au cours de la grossesse devrait avoir peu d’impact sur le risque à long terme associé à l’hypercholestérolémie primaire.
LIPTRUZET
LIPTRUZET est contre-indiqué pendant la grossesse (voir rubrique 4.3). Aucune donnée clinique n’est disponible sur l’utilisation de LIPTRUZET pendant la grossesse. LIPTRUZET ne doit pas être utilisé pendant la grossesse, ni chez une femme envisageant une grossesse ou chez laquelle une grossesse est suspectée. Le traitement par LIPTRUZET doit être suspendu pendant la grossesse ou tant qu'il n’a pas été établi que la femme n’est pas enceinte (voir rubrique 4.3).
L’administration concomitante d’ézétimibe et d’atorvastatine chez des rates en gestation a montré une augmentation liée au médicament de l’anomalie squelettique « diminution de l’ossification des sternèbres » dans le groupe recevant la dose élevée d’ézétimibe/atorvastatine. Cela peut être lié à la diminution du poids des fœtus observée. Chez des lapines en gestation, une faible incidence de malformations squelettiques (sternèbres soudées, vertèbres caudales soudées et modification asymétrique des sternèbres) a été observée.
Atorvastatine
La sécurité d’emploi n’a pas été établie chez la femme enceinte. Aucune étude clinique contrôlée de l’atorvastatine n’a été menée chez la femme enceinte. De rares cas d’anomalies congénitales ont été rapportés après une exposition intra-utérine à des inhibiteurs de la HMG-CoA réductase. Les études effectuées chez l’animal ont mis en évidence une toxicité sur la reproduction (voir rubrique 5.3). Le traitement de la mère par l’atorvastatine peut réduire le taux fœtal de mévalonate qui est un précurseur de la biosynthèse du cholestérol.
Ezétimibe
Aucune donnée clinique n’est disponible sur l’utilisation de l’ézétimibe pendant la grossesse. Les études chez l'animal sur l'utilisation de l'ézétimibe en monothérapie n'ont pas mis en évidence d'effet délétère direct ou indirect sur la grossesse, le développement embryonnaire ou fœtal, la naissance ou le développement post-natal (voir rubrique 5.3).
LIPTRUZET est contre-indiqué pendant l’allaitement. Du fait du risque potentiel d’effets indésirables graves, les femmes traitées par LIPTRUZET ne doivent pas allaiter. Des études chez la rate ont montré que l’ézétimibe est excrété dans le lait maternel. Chez la rate, les concentrations plasmatiques d’atorvastatine et de ses métabolites actifs sont comparables à celles observées dans le lait. On ne sait pas si les substances actives de LIPTRUZET sont excrétées dans le lait maternel humain (voir rubrique 4.3).
Fertilité
Il n’a pas été réalisé d’études de fertilité avec LIPTRUZET.
Atorvastatine
Dans les études effectuées chez l’animal, l’atorvastatine n'a pas eu d'effet sur la fertilité mâle ou femelle.
Ezétimibe
Chez le rat, l’ézétimibe n’a pas eu d’effet sur la fertilité mâle ou femelle.
4.7. Effets sur l'aptitude à conduire des véhicules et à utiliser des machines
La sécurité d’emploi de LIPTRUZET (ou de l’association d’ézétimibe et d’atorvastatine équivalente à LIPTRUZET) a été évaluée chez plus de 2 400 patients dans 7 études cliniques.
Tableau des effets indésirables
Les effets indésirables observés au cours des études cliniques de LIPTRUZET (ou lors de la co-administration d’ézétimibe et d’atorvastatine équivalent au LIPTRUZET) ou d’ézétimibe ou d’atorvastatine ou qui ont été rapportés depuis la commercialisation de LIPTRUZET ou d’ézétimibe ou d’atorvastatine sont listés dans le tableau 3. Ces effets indésirables sont présentés par classe de systèmes d’organes et par fréquence. Les fréquences sont définies comme suit : très fréquent (≥ 1/10), fréquent (≥ 1/100, < 1/10), peu fréquent (≥ 1/1 000, < 1/100), rare (≥ 1/10 000, < 1/1 000) ; très rare (< 1/10 000) et fréquence indéterminée (ne peut être estimée à partir des données disponibles).
Effets indésirables
|
Classe de système d’organes Fréquence |
Effet indésirable |
|
Infections et infestations |
|
|
Peu fréquent |
grippe |
|
Fréquence indéterminée |
rhinopharyngite |
|
Affections hématologiques et du système lymphatique |
|
|
Fréquence indéterminée |
thrombopénie |
|
Affections du système immunitaire |
|
|
Fréquence indéterminée |
hypersensibilité incluant anaphylaxie, angio-œdème, rash et urticaire |
|
Troubles du métabolisme et de la nutrition |
|
|
Fréquence indéterminée |
diminution de l’appétit, anorexie, hyperglycémie, hypoglycémie |
|
Affections psychiatriques |
|
|
Peu fréquent |
dépression, insomnie, troubles du sommeil |
|
Fréquence indéterminée |
cauchemars |
|
Affections du système nerveux |
|
|
Peu fréquent |
étourdissements, dysgueusie, maux de tête, paresthésies |
|
Fréquence indéterminée |
hypoesthésie, amnésie, neuropathie périphérique, myasthénie |
|
Affections oculaires |
|
|
Fréquence indéterminée |
vision floue, troubles visuels, myasthénie oculaire |
|
Affections de l'oreille et du labyrinthe |
|
|
Fréquence indéterminée |
acouphènes, perte d’audition |
|
Affections cardiaques |
|
|
Peu fréquent |
bradycardie sinusale |
|
Affections vasculaires |
|
|
Peu fréquent |
bouffées vasomotrices |
|
Fréquence indéterminée |
hypertension |
|
Affections respiratoires, thoraciques et médiastinales |
|
|
Peu fréquent |
dyspnée |
|
Fréquence indéterminée |
toux, douleur laryngo-pharyngée, épistaxis |
|
Affections gastro-intestinales |
|
|
Fréquent |
diarrhée |
|
Peu fréquent |
gêne abdominale, météorisme, douleur abdominale, douleur abdominale basse, douleur abdominale haute, constipation, dyspepsie, flatulences, selles fréquentes, gastrite, nausées, gêne gastrique |
|
Fréquence indéterminée |
pancréatite, reflux gastro-œsophagien, éructations, vomissements, sécheresse buccale |
|
Affections hépatobiliaires |
|
|
Fréquence indéterminée |
hépatite, cholélithiase, cholécystite, cholestase, insuffisance hépatique fatale et non fatale |
|
Affections de la peau et du tissu sous-cutané |
|
|
Peu fréquent |
acné, urticaire |
|
Fréquence indéterminée |
alopécie, éruption cutanée, prurit, érythème polymorphe, angio-œdème, dermatose bulleuse incluant érythème polymorphe, syndrome de Stevens-Johnson et nécrolyse épidermique toxique |
|
Affections musculo-squelettiques et du tissu conjonctif |
|
|
Fréquent |
myalgies |
|
Peu fréquent |
arthralgies, dorsalgies, fatigue musculaire, spasmes musculaires, faiblesse musculaire, douleurs dans les extrémités |
|
Fréquence indéterminée |
myopathie/rhabdomyolyse, rupture musculaire, tendinopathie parfois compliquée de rupture du tendon, douleur de la nuque, gonflement articulaire, myosite, syndrome pseudo-lupique, myopathie nécrosante à médiation auto-immune (voir rubrique 4.4) |
|
Affections des organes de reproduction et du sein |
|
|
Fréquence indéterminée |
gynécomastie |
|
Troubles généraux et anomalies au site d’administration |
|
|
Peu fréquent |
asthénie, fatigue, malaise, œdème |
|
Fréquence indéterminée |
douleur thoracique, algies, œdème périphérique, pyrexie |
|
Investigations |
|
|
Peu fréquent |
augmentation des ALAT et/ou ASAT, augmentation de la phosphatase alcaline, augmentation de la créatine phosphokinase sanguine (CPK), augmentation de la gamma-glutamyltransférase, augmentation des enzymes hépatiques, anomalies du bilan hépatique, prise de poids |
|
Fréquence indéterminée |
leucocyturie |
Paramètres biologiques
Dans les études cliniques contrôlées, les augmentations cliniquement significatives des transaminases sériques (ALAT et/ou ASAT ≥ 3 x LSN, consécutives) étaient de 0,6 % pour les patients traités par LIPTRUZET. Ces augmentations sont généralement asymptomatiques, non associées à une cholestase et, les valeurs reviennent à leur valeur initiale spontanément ou après l’arrêt du traitement (voir rubrique 4.4).
Les événements indésirables suivants ont été rapportés avec certaines statines :
· dysfonction sexuelle ;
· cas exceptionnels de pneumopathie interstitielle, en particulier lors d’un traitement au long cours (voir rubrique 4.4) ;
· diabète de type II : la fréquence dépend de la présence ou absence de facteurs de risque (glycémie à jeun ≥ 5,6 mmol/L, IMC > 30 kg/m2, hypertriglycéridémie, antécédents d’hypertension artérielle).
Déclaration des effets indésirables suspectés
La déclaration des effets indésirables suspectés après autorisation du médicament est importante. Elle permet une surveillance continue du rapport bénéfice/risque du médicament. Les professionnels de santé déclarent tout effet indésirable suspecté via le système national de déclaration : Agence nationale de sécurité du médicament et des produits de santé (ANSM) et réseau des Centres Régionaux de Pharmacovigilance - Site internet : https://signalement.social-sante.gouv.fr/.
En cas de surdosage, un traitement symptomatique voire des mesures complémentaires peuvent être utilisées avec surveillance de la fonction hépatique et du taux de CPK sérique.
Ezétimibe
Dans les études cliniques, l’administration d’ézétimibe à la dose de 50 mg/jour chez 15 sujets sains sur une période allant jusqu’à 14 jours ou 40 mg/jour chez 18 patients ayant une hyperlipidémie primaire sur une période allant jusqu’à 56 jours a été généralement bien tolérée. Quelques cas de surdosage ont été rapportés avec l’ézétimibe ; la plupart d’entre eux n’ont pas été associés à des événements indésirables. Il n’a pas été rapporté d’effet indésirable grave. Chez l’animal, aucune toxicité n’a été observée après des doses orales uniques de 5 000 mg/kg d'ézétimibe chez le rat et la souris et de 3 000 mg/kg chez le chien.
Atorvastatine
Du fait de la liaison importante de l’atorvastatine aux protéines plasmatiques, l’hémodialyse ne devrait pas augmenter significativement la clairance de l’atorvastatine.
5. PROPRIETES PHARMACOLOGIQUES
5.1. Propriétés pharmacodynamiques
LIPTRUZET (ézétimibe/atorvastatine) est un agent hypolipidémiant qui inhibe de façon sélective l’absorption intestinale du cholestérol et des phytostérols apparentés et inhibe la synthèse endogène de cholestérol.
Mécanisme d’action
LIPTRUZET
Le cholestérol plasmatique provient de l’absorption intestinale et de la synthèse endogène. LIPTRUZET contient de l’ézétimibe et de l’atorvastatine, deux hypolipidémiants avec des mécanismes d’action complémentaires. LIPTRUZET réduit le cholestérol total (C-total) élevé, le LDL-cholestérol, les apolipoprotéines B (Apo B), les triglycérides (TG) et le cholestérol non lié aux lipoprotéines de basse densité (non HDL-C) et augmente le cholestérol lié aux lipoprotéines de haute densité (HDL-C) par la double inhibition de l’absorption et de la synthèse du cholestérol.
Ezétimibe
L’ézétimibe inhibe l’absorption intestinale du cholestérol. L’ézétimibe est actif par voie orale et a un mécanisme d’action qui diffère de celui des autres classes d’hypocholestérolémiants (statines, résines échangeuses d’ions, fibrates et stérols végétaux). La cible moléculaire de l’ézétimibe est le transporteur de stérols NPC1L1 (Niemann-Pick C1-Like 1), qui est responsable de l’absorption intestinale du cholestérol et des phytostérols.
L’ézétimibe se localise au niveau de la bordure en brosse de l’intestin grêle et inhibe l’absorption du cholestérol, ce qui entraîne une diminution des apports au foie du cholestérol intestinal alors que les statines diminuent la synthèse du cholestérol hépatique. Ainsi, ces deux molécules administrées simultanément entraînent, avec des mécanismes distincts, une diminution complémentaire du cholestérol. Une étude clinique de deux semaines réalisée chez 18 patients hypercholestérolémiques a montré que l’ézétimibe inhibe l'absorption intestinale du cholestérol de 54 % par rapport à un placebo.
Plusieurs études précliniques visant à déterminer la sélectivité de l’ézétimibe sur l’inhibition de l’absorption du cholestérol ont été réalisées. L’ézétimibe inhibe l’absorption du [14C]-cholestérol mais n’a pas d’effet sur l’absorption des triglycérides, des acides gras, des acides biliaires, de la progestérone, de l’éthinylestradiol ou des vitamines liposolubles A et D.
Atorvastatine
L’atorvastatine est un inhibiteur compétitif sélectif de la HMG-CoA réductase, l’enzyme de l’étape limitante responsable de la conversion de la 3-hydroxy-3-méthyl-glutaryl-coenzyme A en mévalonate, un précurseur des stérols, dont le cholestérol. Dans le foie, les triglycérides et le cholestérol sont incorporés dans les lipoprotéines de très faible densité (VLDL) et libérés dans le plasma pour être transportés vers les tissus périphériques. Les lipoprotéines de faible densité (LDL) sont formées à partir des VLDL et catabolisées essentiellement par l’intermédiaire de récepteurs ayant une affinité élevée pour les LDL (récepteurs des LDL).
L’atorvastatine diminue le cholestérol plasmatique et les taux sériques de lipoprotéines en inhibant la HMG-CoA réductase et par conséquent, la biosynthèse de cholestérol dans le foie, et augmente le nombre de récepteurs des LDL à la surface des hépatocytes pour amplifier la capture et le catabolisme des LDL.
L’atorvastatine diminue la synthèse des LDL et le nombre de particules de LDL. Elle induit une augmentation importante et prolongée de l’activité des récepteurs des LDL accompagnée d’une augmentation de la qualité des particules de LDL en circulation. L’atorvastatine est efficace pour réduire le LDL-C chez les patients présentant une hypercholestérolémie familiale homozygote, une population difficile à contrôler par les médicaments hypolipidémiants.
Une étude de dose-réponse a montré que l’atorvastatine réduit les taux de Cholestérol Total (CT) (de 30 % à 46 %), de LDL-C (de 41 % à 61 %), d’apolipoprotéine B (de 34 % à 50 %) et de triglycérides (de 14 % à 33 %) tout en induisant des augmentations variables du HDL-C et de l’apolipoprotéine A1. Ces résultats sont également observés chez les patients présentant une hypercholestérolémie familiale hétérozygote, des formes non familiales d’hypercholestérolémie ou une dyslipidémie mixte, y compris les patients présentant un diabète non insulinodépendant.
Efficacité et sécurité clinique
Dans les études cliniques contrôlées, LIPTRUZET a entraîné des réductions significatives des taux de CT, LDL-C, Apo B et TG et augmenté le HDL-C chez les patients présentant une hypercholestérolémie.
Hypercholestérolémie primaire
Dans une étude contrôlée versus placebo, 628 patients présentant une dyslipidémie ont été randomisés pour recevoir le placebo, l’ézétimibe (10 mg), l’atorvastatine (10 mg, 20 mg, 40 mg ou 80 mg) ou une association d’ézétimibe et d’atorvastatine équivalente à LIPTRUZET (10/10 mg, 10/20 mg, 10/40 mg et 10/80 mg) pendant une durée allant jusqu’à 12 semaines.
Les patients recevant toute dose de LIPTRUZET ont été comparés à ceux recevant toute dose d’atorvastatine. LIPTRUZET a induit des diminutions significativement plus importantes du CT, du LDL‑C, de l’ApoB, des TG et du non-HDL-C et une augmentation significativement plus importante du HDL-C que l’atorvastatine en monothérapie (voir tableau 4).
Tableau 4
Réponse à LIPTRUZET chez des patients présentant une dyslipidémie primaire
(Variation moyennea en % à la semaine 12 par rapport à la valeur initiale avant le traitementb)
|
Traitement (dose quotidienne) |
N |
CT |
LDL-C |
Apo B |
TGa |
HDL‑C |
Non-HDL-C |
|
Données combinées (toutes doses de LIPTRUZET)c |
255 |
-41 |
-56 |
-45 |
-33 |
+7 |
-52 |
|
Données combinées (toutes doses d’atorvastatine)c |
248 |
-32 |
-44 |
-36 |
-24 |
+4 |
-41 |
|
Ezétimibe 10 mg |
65 |
-14 |
-20 |
-15 |
-5 |
+4 |
-18 |
|
Placebo |
60 |
+4 |
+4 |
+3 |
-6 |
+4 |
+4 |
|
LIPTRUZET, par dose |
|
|
|
|
|
|
|
|
10/10 |
65 |
-38 |
-53 |
-43 |
-31 |
+9 |
-49 |
|
10/20 |
62 |
-39 |
-54 |
-44 |
-30 |
+9 |
-50 |
|
10/40 |
65 |
-42 |
-56 |
-45 |
-34 |
+5 |
-52 |
|
10/80 |
63 |
-46 |
-61 |
-50 |
-40 |
+7 |
-58 |
|
Atorvastatine, par dose |
|
|
|
|
|
|
|
|
10 mg |
60 |
-26 |
-37 |
-28 |
-21 |
+6 |
-34 |
|
20 mg |
60 |
-30 |
-42 |
-34 |
-23 |
+4 |
-39 |
|
40 mg |
66 |
-32 |
-45 |
-37 |
-24 |
+4 |
-41 |
|
80 mg |
62 |
-40 |
-54 |
-46 |
-31 |
+3 |
-51 |
a Pour les triglycérides, variation en % médiane par rapport à la valeur initiale.
b Valeur initiale, sans traitement hypolipidémiant.
c LIPTRUZET (toutes doses combinées, 10 mg/10 mg à 10 mg/80 mg) a entraîné une réduction significative du CT, du LDL-C, de l’Apo B, des TG et du non- HDL-C et une augmentation significative du HDL-C par rapport à l’atorvastatine (toutes doses combinées, 10 à 80 mg).
Dans une étude contrôlée, l’étude TEMPO (Titration of Atorvastatin vs Ezetimibe Add-On to Atorvastatin in Patients with Hypercholesterolaemia), 184 patients ayant un taux de LDL-C ≥ 2,6 mmol/L et ≤ 4,1 mmol/l et un risque modéré à élevé de maladie coronarienne ont reçu l’atorvastatine 20 mg pendant une durée minimale de 4 semaines avant la randomisation. Les patients qui n’avaient pas obtenu un taux de LDL-C < 2,6 mmol/L ont été randomisés pour recevoir une association d’ézétimibe et d’atorvastatine (équivalente à LIPTRUZET 10/20 mg) ou l’atorvastatine 40 mg pendant 6 semaines.
LIPTRUZET 10/20 mg a été significativement plus efficace que le doublement de la dose d’atorvastatine à 40 mg pour induire des réductions supplémentaires du cholestérol total (‑20% versus ‑7%), du LDL-C (‑31% versus ‑11%), de l’Apo B (‑21% versus ‑8%) et du non-HDL-C (‑27% versus ‑10%). Pour le HDL-C et les TG, les résultats n’ont pas été significativement différents entre les deux groupes de traitement. De même, le nombre de patients ayant atteint un taux de LDL-C < 2,6 mmol/L a été significativement plus élevé dans le groupe recevant LIPTRUZET 10/20 mg que dans celui recevant l’atorvastatine 40 mg (84 % versus 49 %).
Dans une étude contrôlée, l’étude EZ-PATH (Ezetimibe Plus Atorvastatin vs Atorvastatin Titration in Achieving Lower LDL-C Targets in Hypercholesterolaemic Patients), 556 patients à risque cardiovasculaire élevé ayant un taux de LDL-C ≥ 1,8 mmol/L et ≤ 4,1 mmol/L ont reçu l’atorvastatine 40 mg pendant une durée minimale de 4 semaines avant la randomisation. Les patients qui n’avaient pas obtenu un taux de LDL-C < 1,8 mmol/L ont été randomisés pour recevoir une association d’ézétimibe et d’atorvastatine (équivalente à LIPTRUZET 10/40 mg) ou l’atorvastatine 80 mg pendant 6 semaines.
LIPTRUZET 10/40 mg a été significativement plus efficace que le doublement de la dose d’atorvastatine à 80 mg pour induire des réductions supplémentaires du cholestérol total (‑17% versus ‑7%), du LDL-C (‑27% versus ‑11%), de l’Apo B (‑18% versus ‑8%), des TG (‑12% versus ‑6%) et du non-HDL-C (‑23% versus ‑9%).
Pour le HDL-C, les résultats n’ont pas été significativement différents entre les deux groupes de traitement. De même, le nombre de patients ayant atteint un taux de LDL-C < 1,8 mmol/L a été significativement plus élevé dans le groupe recevant LIPTRUZET 10/40 mg que dans celui recevant l’atorvastatine 80 mg (74 % versus 32 %).
Dans une étude contrôlée de 8 semaines, 308 patients présentant une hypercholestérolémie traités par l’atorvastatine et qui n’avaient pas atteint l’objectif de LDL-C du National Cholesterol Education Program (NCEP) (objectif de LDL-C basé sur la valeur initiale du LDL-C et le statut de risque coronarien) ont été randomisés pour recevoir l’ézétimibe 10 mg ou le placebo en plus de leur traitement en cours par l’atorvastatine.
Parmi les patients qui n’avaient pas le taux cible de LDL-C lors de l’inclusion (~83 %), le nombre de patients ayant atteint leur objectif de LDL-C a été significativement plus élevé chez les patients recevant l’ézétimibe en association avec l’atorvastatine que chez ceux qui recevaient le placebo en association avec l’atorvastatine (67 % versus 19 %). L’ézétimibe associé à l'atorvastatine a induit une diminution significativement plus importante du LDL-C que le placebo associé à l’atorvastatine (25 % versus 4 %). L’ézétimibe associé à l'atorvastatine a également diminué significativement les taux de CT, d’Apo B et de TG par rapport au placebo associé à l’atorvastatine.
Dans une étude de phase II contrôlée de 12 semaines, 1 539 patients à haut risque cardiovasculaire ayant un taux de LDL-C compris entre 2,6 et 4,1 mmol/L traités par l'atorvastatine 10 mg par jour ont été randomisés pour recevoir : atorvastatine 20 mg, rosuvastatine 10 mg ou LIPTRUZET 10/10 mg. Après 6 semaines de traitement (période I), les patients qui n’avaient pas obtenu un taux de LDL-C < 2,6 mmol/L avec l’atorvastatine 20 mg ont permuté pour recevoir l’atorvastatine 40 mg ou LIPTRUZET 10/20 mg pendant 6 semaines (période II) et les patients recevant la rosuvastatine 10 mg pendant la période I ont permuté pour recevoir la rosuvastatine 20 mg ou LIPTRUZET 10/20 mg. Les réductions du LDL-C et les comparaisons entre le groupe LIPTRUZET et les autres groupes de traitement sont présentées dans le tableau 5.
Tableau 5
Réponse à LIPTRUZET* chez des patients à risque élevé ayant un taux de LDL-C lors de l’inclusion compris entre 2,6 et 4,1 mmol/L sous atorvastatine 10 mg par jour
|
Traitement |
N |
Variation en pourcentage par rapport aux valeurs initiales |
||||||
|
|
|
CT |
LDL-C |
Apo B |
TG |
HDL-C |
Non-HDL-C |
|
|
Période I Relais de l’atorvastatine 10 mg |
|
|
|
|
|
|
|
|
|
LIPTRUZET 10/10 mg |
120 |
-13,5 |
-22,2 |
-11,3 |
-6,0 |
+0,6 |
-18,3 |
|
|
Atorvastatine 20 mg |
480 |
-6,4§ |
-9,5§ |
-6,0¶ |
-3,9 |
-1,1 |
-8,1§ |
|
|
Rosuvastatine 10 mg |
939 |
-7,7§ |
-13,0§ |
-6,9# |
-1,1 |
+1,1 |
-10,6§ |
|
|
Période II Relais de l’atorvastatine 20 mg |
|
|
|
|
|
|
|
|
|
LIPTRUZET 10/20 mg |
124 |
-10,7 |
-17,4 |
-9,8 |
-5,9 |
+0,7 |
-15,1 |
|
|
Atorvastatine 40 mg |
124 |
-3,8 Þ |
-6,9 Þ |
-5,4 |
-3,1 |
+1,7 |
-5,8 Þ |
|
|
Relais de la rosuvastatine 10 mg |
|
|
|
|
|
|
|
|
|
LIPTRUZET 10/20 mg |
231 |
-11,8 |
-17,1 |
-11,9 |
-10,2 |
+0,1 |
-16,2 |
|
|
Rosuvastatine 20 mg |
205 |
-4,5Þ |
-7,5 Þ |
-4,1Þ |
-3,2ß |
+0,8 |
-6,4 Þ |
|
* Association d’ézétimibe et d’atorvastatine équivalente à LIPTRUZET 10/10 mg ou LIPTRUZET 10/20 mg.
Les M-estimations (basées sur la méthode de Huber), l’IC à 95 % et la valeur p ont été déterminés par ajustement d’un modèle de régression robuste avec des termes pour le traitement et la valeur initiale.
Les variations en pourcentage de la moyenne géométrique des taux de TG par rapport aux valeurs initiales ont été calculées sur la base d’une rétrotransformation par exponentiation des moyennes des moindres carrés (MC) du modèle et exprimées sous forme de (moyenne géométrique – 1) multipliée par 100.
§ p < 0,001 vs LIPTRUZET 10/10
¶ p < 0,01 vs LIPTRUZET 10/10
# p < 0,05 vs LIPTRUZET 10/10
Þ p < 0,001 vs LIPTRUZET 10/20
ß p < 0,05 vs LIPTRUZET 10/20
Le tableau 5 ne comporte pas de données comparant les effets de LIPTRUZET 10/10 mg ou 10/20 mg à des doses supérieures à 40 mg d’atorvastatine ou 20 mg de rosuvastatine.
Dans une étude contrôlée versus placebo, l’étude MIRACL (Myocardial Ischaemia Reduction with Aggressive Cholesterol-Lowering), des patients présentant un syndrome coronarien aigu (IDM sans onde Q ou angor instable) ont été randomisés pour recevoir l’atorvastatine 80 mg/jour (n = 1 538) ou le placebo (n = 1 548). Le traitement a été instauré pendant la phase aiguë après l’hospitalisation et a duré 16 semaines. L’atorvastatine 80 mg/jour a induit une réduction de 16 % (p = 0,048) du risque du critère principal composite : décès pour toute cause, IDM non fatal, arrêt cardiaque avec réanimation ou angor avec signes d’ischémie myocardique nécessitant une hospitalisation. Ce résultat a été dû principalement à une réduction de 26 % des réhospitalisations pour angor avec signes d’ischémie myocardique (p = 0,018).
LIPTRUZET contient de l’atorvastatine. Dans une étude contrôlée versus placebo, l'étude ASCOTT-LLA (Anglo-Scandinavian Cardiac Outcomes Trial Lipid-Lowering Arm), l’effet de l’atorvastatine 10 mg sur les événements coronariens fatals et non fatals a été évalué chez 10 305 patients hypertendus âgés de 40 à 80 ans ayant un taux de CT ≤ 6,5 mmol/L et au moins trois facteurs de risque cardiovasculaire. Les patients ont été suivis pendant une durée médiane de 3,3 ans. L’atorvastatine 10 mg a réduit significativement (p < 0,001) le risque relatif de : événements coronariens fatals plus IDM non fatals de 36 % (réduction du risque absolu : 1,1 %), événements cardiovasculaires totaux et procédures de revascularisation de 20 % (réduction du risque absolu : 1,9 %) et événements coronariens totaux de 29 % (réduction du risque absolu : 1,4 %).
Dans une étude contrôlée versus placebo, l’étude CARDS (Collaborative Atorvastatin Diabetes Study), l’effet de l’atorvastatine 10 mg sur les événements cardiovasculaires a été évalué chez 2 838 patients âgés de 40 à 75 ans diabétiques de type 2 ayant un ou plusieurs facteurs de risque cardiovasculaire et des taux de LDL-C ≤ 4,1 mmol/L et de TG ≤ 6,8 mmol/L. Les patients ont été suivis pendant une durée médiane de 3,9 ans. L’atorvastatine 10 mg a réduit significativement (P < 0,05) : le taux d’événements cardiovasculaires majeurs de 37 % (réduction du risque absolu : 3,2 %), le risque d’accident vasculaire cérébral de 48 % (réduction du risque absolu : 1,3 %) et le risque d’infarctus du myocarde de 42 % (réduction du risque absolu : 1,9 %).
Prévention des événements cardiovasculaires
Dans une étude multicentrique, randomisée, en double-insu, versus comparateur actif, évaluant l’association ézétimibe/simvastatine, 18 144 patients ont été inclus dans les 10 jours suivant une hospitalisation pour syndrome coronarien aigu (SCA ; infarctus du myocarde aigu ou angor instable). Tous les patients ont été randomisés selon un ratio 1 : 1 pour recevoir soit ézétimibe/simvastatine 10 mg/40 mg (n = 9 067) soit simvastatine 40 mg (n = 9 077) et ont été suivis pendant une période médiane de 6,0 ans.
Les patients avaient un âge moyen de 63,6 ans ; 76 % étaient des hommes, 84 % étaient d’origine caucasienne, et 27 % étaient diabétiques. La valeur moyenne du LDL-C au moment de l’événement qualifiant l’entrée dans l’étude était de 80 mg/dL (2,1 mmol/L) pour ceux déjà sous traitement hypolipidémiant (n = 6 390) et de 101 mg/dL (2,6 mmol/L) pour ceux sans traitement hypolipidémiant antérieur (n = 11 594). Avant l’hospitalisation pour le SCA, qualifiant l’inclusion dans l’étude, 34 % des patients étaient sous traitement par statine. Après un an, le LDL-C moyen des patients toujours sous traitement était de 53,2 mg/dL (1,4 mmol/L) dans le groupe ézétimibe/simvastatine et 69,9 mg/dL (1,8 mmol/L) dans le groupe simvastatine en monothérapie.
Le critère d'évaluation principal était un critère composite comprenant le décès d’origine cardiovasculaire, l’événement coronarien majeur (défini comme infarctus du myocarde non fatal, angor instable documenté nécessitant une hospitalisation, ou intervention de revascularisation coronaire intervenant au moins 30 jours après la randomisation des groupes de traitement) et l’AVC non fatal. L’étude a démontré que le traitement par ézétimibe ajouté à la simvastatine apporte un bénéfice supplémentaire en réduisant le critère composite principal comprenant le décès d’origine cardiovasculaire, l’événement coronarien majeur et l’AVC non fatal, comparé au traitement par simvastatine seule (réduction du risque relatif de 6,4 %, p = 0,016). Le critère d’évaluation principal est survenu chez 2 572 des 9 067 patients (taux de survie à 7 ans selon Kaplan-Meier [KM] de 32,72 %) dans le groupe ézétimibe/simvastatine et chez 2 742 des 9 077 patients (taux de survie à 7 ans selon KM de 34,67 %) dans le groupe simvastatine seule (Voir Figure 1 et Tableau 6). Ce bénéfice supplémentaire devrait être similaire avec la co-administration d’ézétimibe et d’atorvastatine. La mortalité totale est restée inchangée dans ce groupe à haut risque.
Un bénéfice global a été observé pour tous les AVC ; cependant il y a eu une petite augmentation non-significative d’AVC hémorragique dans le groupe ézétimibe/simvastatine en comparaison avec le groupe simvastatine seule. Le risque d’AVC hémorragique lorsqu’ézétimibe est co-administré avec des statines plus puissantes n’a pas été évalué dans des études à long-terme.
L’effet du traitement de l’association ézétimibe/simvastatine était généralement cohérent sur l’ensemble des résultats recueillis dans de nombreux sous-groupes, incluant sexe, âge, origine ethnique, antécédent de diabète, taux de lipides à l’inclusion, traitement antérieur par statine, antécédent d’AVC, et hypertension.
Figure 1 : Effet d’ézétimibe/simvastatine sur le critère composite principal comprenant le décès d’origine cardiovasculaire, l’événement coronarien majeur ou l’AVC non fatal.
|
|
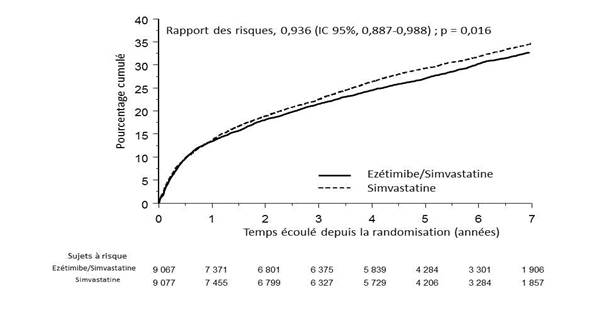
Tableau 6
Evénements cardiovasculaires majeurs par groupe de traitement chez tous les patients randomisés d’IMPROVE-IT
|
Résultat |
Ezétimibe/Simvastatine |
Simvastatine |
Hazard Ratio |
Valeur |
||
|
|
n |
K-M % |
n |
K-M % |
|
|
|
Critère d’efficacité composite principal |
||||||
|
(décès cardiovasculaire, événements coronariens majeurs et AVC non‑fatal) |
2 572 |
32.72% |
2 742 |
34.67% |
0.936 (0.887, 0.988) |
0.016 |
|
Composants du critère d’efficacité composite principal et choix des critères d’efficacité (premières apparitions d’un événement défini pouvant survenir à tout moment) |
||||||
|
Décès cardiovasculaire |
537 |
6,89% |
538 |
6,84% |
1,000 (0,887, 1,127) |
0,997 |
|
Evénement coronarien majeur : |
|
|
|
|
|
|
|
Infarctus non fatal |
945 |
12,77% |
1 083 |
14,41% |
0,871 (0,798, 0,950) |
0,002 |
|
Angor instable exigeant une hospitalisation |
156 |
2,06% |
148 |
1,92% |
1,059 (0.846, 1,326) |
0,618 |
|
Intervention de revascularisation coronaire après 30 jours |
1 690 |
21,84% |
1 793 |
23,36% |
0,947 (0,886, 1,012) |
0,107 |
|
AVC non fatal |
245 |
3,49% |
305 |
4,24% |
0,802 (0,678, 0,949) |
0,010 |
* 6% ont reçu des doses plus fortes d’ézétimibe/simvastatine 10/80 mg.
27% ont reçu des doses plus fortes de simvastatine 80 mg.
Taux de survie à 7 ans selon Kaplan-Meier.
Hypercholestérolémie familiale homozygote (HFHo)
Une étude randomisée, en double aveugle d’une durée de 12 semaines a été réalisée chez des patients présentant une hypercholestérolémie familiale homozygote HFHo (diagnostic clinique et/ou génotypique). Les résultats ont été analysés à partir d’un sous-groupe de patients (n = 36) recevant 40 mg d’atorvastatine comme dose initiale. L’augmentation de la dose d’atorvastatine de 40 à 80 mg (n = 12) a entraîné une réduction du LDL-cholestérol de 2 % par rapport à la valeur initiale avec 40 mg d’atorvastatine. L’association d’ézétimibe et d'atorvastatine à doses équivalentes à LIPTRUZET (doses de 10/40 mg et 10/80 mg combinées, n = 24) a entraîné une réduction du LDL-cholestérol de 19 % par rapport à la valeur initiale avec 40 mg d’atorvastatine. Chez ces patients, l’association d’ézétimibe et d'atorvastatine à doses équivalentes à LIPTRUZET (10/80 mg, n = 12), a entraîné une réduction du LDL-cholestérol de 25 % par rapport à la valeur initiale avec l’atorvastatine 40 mg.
Après avoir terminé l’étude de 12 semaines, les patients éligibles (n = 35) qui recevaient l’atorvastatine 40 mg lors de l’inclusion ont été affectés pour recevoir l’association d’ézétimibe et d'atorvastatine équivalente à LIPTRUZET 10/40 mg pendant 24 mois supplémentaires. Après au moins 4 semaines de traitement, la dose d’atorvastatine pouvait être doublée à une dose maximale de 80 mg. À la fin des 24 mois, LIPTRUZET (doses de 10/40 mg et 10/80 mg combinées) avait induit une diminution du LDL-C concordant avec celle observée dans l’étude de 12 semaines.
L’Agence européenne des médicaments a accordé une dérogation à l’obligation de soumettre les résultats d’études réalisées avec LIPTRUZET dans tous les sous-groupes de la population pédiatrique dans l’indication d’hypercholestérolémie et de dyslipidémie mixte (voir rubrique 4.2 pour les informations concernant l’usage pédiatrique).
5.2. Propriétés pharmacocinétiques
La bioéquivalence de LIPTRUZET et de l’administration concomitante de doses correspondantes d’ézétimibe et d’atorvastatine sous forme de comprimés séparés a été démontrée.
Absorption
Les effets d’un repas riche en graisses sur la pharmacocinétique de l’ézétimibe et de l’atorvastatine administrés sous forme de comprimés de LIPTRUZET sont comparables à ceux rapportés pour chacun des deux médicaments.
Ezétimibe
Après administration orale, l’ézétimibe est rapidement absorbé et subit une importante glucuroconjugaison conduisant à la formation d’un composé glycuronide phénolique pharmacologiquement actif (ézétimibe-glycuronide). Les concentrations plasmatiques maximales moyennes (Cmax) sont atteintes en 1 à 2 heures pour l’ézétimibe-glycuronide et en 4 à 12 heures pour l’ézétimibe. La biodisponibilité absolue de l’ézétimibe ne peut être déterminée car le produit est pratiquement insoluble dans les milieux aqueux adaptés aux injections.
L’administration concomitante d’aliments (repas riches en graisses ou repas sans graisse) n’a pas d’effet sur la biodisponibilité orale de l’ézétimibe administré sous forme de comprimés de 10 mg.
Atorvastatine
Après administration orale, l’atorvastatine est rapidement absorbée ; la concentration plasmatique maximale (Cmax) est atteinte en 1 à 2 heures. L’importance de l’absorption est dose-dépendante. Après administration orale, la biodisponibilité des comprimés pelliculés d’atorvastatine est de 95 % à 99 % par rapport à la solution buvable. La biodisponibilité absolue de l’atorvastatine est d’environ 12 % et la biodisponibilité systémique de l’activité inhibitrice de la HMG-CoA réductase est d’environ 30 %. La faible biodisponibilité systémique est due à la clairance présystémique dans la muqueuse gastro-intestinale et/ou à l’effet de premier passage hépatique.
Distribution
Ezétimibe
La liaison aux protéines plasmatiques humaines est de 99,7 % pour l’ézétimibe et de 88 à 92 % pour l’ézétimibe-glycuronide.
Atorvastatine
Le volume de distribution moyen de l’atorvastatine est d’environ 381 litres. La liaison de l’atorvastatine aux protéines plasmatiques est ≥ 98 %.
Biotransformation
Ezétimibe
L’ézétimibe est principalement métabolisé dans l’intestin grêle et le foie par glucuroconjugaison (réaction de phase II) suivie d’une excrétion biliaire. Un métabolisme oxydatif minimal (réaction de phase I) a été observé dans toutes les espèces étudiées. L’ézétimibe et l’ézétimibe-glycuronide sont les principaux produits dérivés détectés dans le plasma, représentant respectivement 10 à 20 % et 80 à 90 % du produit total dans le plasma.
L’ézétimibe et l’ézétimibe-glycuronide sont tous deux éliminés lentement du plasma ; il existe un recyclage entéro-hépatique significatif. La demi-vie de l’ézétimibe et de l’ézétimibe-glycuronide est d’environ 22 heures.
Atorvastatine
L’atorvastatine est métabolisée par le cytochrome P450 3A4 en dérivés ortho- et parahydroxylés et en plusieurs produits de bêta-oxydation. En plus d’autres voies, ces produits sont ensuite métabolisés par glucuroconjugaison. In vitro, l’inhibition de la HMG-CoA réductase par les métabolites ortho- et parahydroxylés est équivalente à celle exercée par l’atorvastatine. Environ 70 % de l’activité d’inhibition de la HMG-CoA réductase circulante sont attribués aux métabolites actifs.
Elimination
Ezétimibe
Chez l’homme, après administration orale 14C-ézétimibe (20 mg), l’ézétimibe total représente environ 93 % de la radioactivité totale du plasma. Environ 78 % de la radioactivité est retrouvée dans les selles et 11 % dans les urines sur une période de recueil de 10 jours. Après 48 heures, aucune radioactivité n’était détectable dans le plasma.
Atorvastatine
L’atorvastatine est éliminée essentiellement par voie biliaire après un métabolisme hépatique et/ou extra-hépatique. Cependant, le médicament ne semble pas subir de cycle entéro-hépatique significatif. Chez l’homme, la demi-vie d’élimination plasmatique moyenne de l’atorvastatine est d’environ 14 heures. La demi-vie de l’activité inhibitrice de la HMG‑CoA réductase est d’environ 20 à 30 heures en raison de la contribution des métabolites actifs.
L’atorvastatine est un substrat des transporteurs hépatiques, polypeptides transporteurs d’anions organiques 1B1 (OATP1B1) et 1B3 (OATP1B3). Les métabolites de l’atorvastatine sont des substrats de l’OATP1B1. L’atorvastatine est également identifiée comme étant un substrat des transporteurs d’efflux de type protéine de multirésistance aux médicaments (MDR1) et protéine de résistance au cancer du sein (BCRP), ce qui peut limiter l’absorption intestinale et la clairance biliaire de l’atorvastatine.
Population pédiatrique
Ezétimibe
Les propriétés pharmacocinétiques de l’ézétimibe sont similaires chez l’enfant ≥ 6 ans et chez l’adulte. Aucune donnée pharmacocinétique chez l’enfant de moins de 6 ans n’est disponible. L’expérience clinique concernant les enfants et adolescents est limitée aux patients avec HFHo, HFHe ou sitostérolémie.
Atorvastatine
Dans une étude en ouvert de 8 semaines, des patients pédiatriques (âgés de 6 à 17 ans) au stade 1 (n = 15) ou au stade 2 (n = 24) de Tanner présentant une hypercholestérolémie familiale hétérozygote et un taux initial de LDL-C ≥ 4 mmol/L ont été traités respectivement par 5 mg ou 10 mg d’atorvastatine comprimés à croquer ou 10 mg ou 20 mg d’atorvastatine comprimés pelliculés une fois par jour. Le poids corporel était la seule covariable significative dans le modèle de PK de population de l’atorvastatine. Chez les patients pédiatriques, la clairance orale apparente de l’atorvastatine a semblé comparable à celle observée chez les adultes après mise à l’échelle allométrique par le poids corporel. Des diminutions régulières des taux de LDL-C et de CT ont été observées sur l’éventail d’expositions à l’atorvastatine et à la o-hydroxy-atorvastatine.
Sujets âgés
Ezétimibe
Les concentrations plasmatiques d’ézétimibe total sont environ 2 fois plus élevées chez le sujet âgé (≥ 65 ans) que chez le sujet jeune (18 à 45 ans). La diminution du LDL‑cholestérol et la sécurité d’emploi sont comparables chez les sujets âgés et jeunes traités par l’ézétimibe.
Atorvastatine
Les concentrations plasmatiques de l’atorvastatine et de ses métabolites actifs sont plus élevées chez les volontaires sains âgés que chez les adultes jeunes, tandis que les effets hypolipidémiants sont comparables à ceux observés dans les populations de patients plus jeunes.
Insuffisance hépatique
Ezétimibe
Après administration d’une dose unique de 10 mg d’ézétimibe, l’ASC moyenne de l’ézétimibe total augmente d’environ 1,7 fois chez les patients présentant une insuffisance hépatique légère (score de Child-Pugh de 5 ou 6) par rapport aux sujets sains. Une étude de doses répétées (10 mg par jour) d’une durée de 14 jours réalisée chez des patients présentant une insuffisance hépatique modérée (score de Child-Pugh de 7 à 9), montre que l’ASC moyenne de l’ézétimibe total augmente d’environ 4 fois le jour 1 et le jour 14, par rapport aux volontaires sains. Aucun ajustement posologique n’est nécessaire chez les patients présentant une insuffisance hépatique légère. Chez les patients présentant une insuffisance hépatique modérée ou sévère (score de Child-Pugh >9), compte tenu des effets inconnus d’une exposition accrue, l’ézétimibe n’est pas recommandé (voir rubriques 4.2 et 4.4).
Atorvastatine
Les concentrations plasmatiques d’atorvastatine et de ses métabolites actifs sont considérablement augmentées (augmentation d’environ 16 fois pour la Cmax et d’environ 11 fois pour l’ASC) chez les patients présentant une insuffisance hépatique chronique d’origine alcoolique (grade B de Child-Pugh).
Insuffisance rénale
Ezétimibe
Chez des patients présentant une insuffisance rénale sévère (n = 8, clairance moyenne de la créatinine ≤ 30 mL/min/1,73 m2), l’administration d’une dose unique de 10 mg d’ézétimibe a entraîné une augmentation de l'ASC de l’ézétimibe total, d’environ 1,5 fois par rapport aux volontaires sains (n = 9).
Dans cette étude, chez un autre patient transplanté rénal recevant de nombreux médicaments dont la ciclosporine, l’exposition à l’ézétimibe total était 12 fois supérieure.
Atorvastatine
L’insuffisance rénale n’a pas d’effet sur les concentrations plasmatiques ou les effets hypolipidémiants de l’atorvastatine et de ses métabolites actifs.
Sexe
Ezétimibe
Les concentrations plasmatiques de l’ézétimibe total sont légèrement plus élevées (approximativement 20 %) chez la femme que chez l’homme. La diminution du LDL‑C et la sécurité d’emploi sont comparables chez l’homme et la femme traités par ézétimibe.
Atorvastatine
Les concentrations d’atorvastatine et de ses métabolites actifs sont différentes entre les femmes et les hommes (femmes : Cmax plus élevée d’environ 20 % et ASC plus basse d’environ 10 %). Ces différences n’ont pas de pertinence clinique et n’entraînent pas de différences cliniquement significatives des effets hypolipidémiants entre les hommes et les femmes.
Polymorphisme du gène SLCO1B1
Atorvastatine
La captation hépatique de tous les inhibiteurs de la HMG-CoA réductase, y compris l’atorvastatine, implique le transporteur OATP1B1. Chez les patients présentant un polymorphisme de SLCO1B1, l’exposition à l’atorvastatine peut être augmentée, ce qui peut entraîner un risque accru de rhabdomyolyse (voir rubrique 4.4). Un polymorphisme du gène codant pour OATP1B1 (SLCO1B1 c.521CC) est associé à une exposition à l’atorvastatine (ASC) 2,4 fois plus élevée que chez les sujets non porteurs de ce variant génotypique (génotype c.521TT). Une diminution de la captation hépatique d’atorvastatine d’origine génétique est également possible chez ces patients. Les conséquences possibles sur l’efficacité ne sont pas connues.
5.3. Données de sécurité préclinique
Dans les études de trois mois associant ézétimibe et atorvastatine chez le rat et le chien, les effets toxiques observés ont été essentiellement ceux typiquement associés aux statines. Les anomalies histopathologiques caractéristiques des statines étaient limitées au foie. Certains effets toxiques étaient plus prononcés que ceux observés lors du traitement par des statines seules. Cela est imputé à des interactions pharmacocinétiques et/ou pharmacodynamiques observées suite à l’administration de l’association.
L’administration concomitante d’ézétimibe et d’atorvastatine chez des rates gestantes a montré une augmentation liée au médicament de l’anomalie squelettique « diminution de l’ossification des sternèbres » dans le groupe recevant la dose élevée d’ézétimibe/atorvastatine (1 000/108,6 mg/kg). Cela peut être lié à la diminution du poids des fœtus observée. Chez des lapines en gestation, une faible incidence de malformations squelettiques (sternèbres soudées, vertèbres caudales soudées et modification asymétrique des sternèbres) a été observée.
Une série de tests in vivo et in vitro n’a pas montré de potentiel génotoxique de l’ézétimibe administré seul ou en association avec l’atorvastatine.
Ezétimibe
Les études de toxicité chronique de l’ézétimibe réalisées chez l’animal n’ont pas montré d’organes cibles. Chez le chien traité pendant 4 semaines par ézétimibe (≥ 0,03 mg/kg/jour), la concentration de cholestérol dans la bile vésiculaire a été multiplié par un facteur de 2,5 à 3,5. En revanche, une étude d’un an réalisée chez le chien recevant des doses allant jusqu’à 300 mg/kg/jour n’a pas montré d’augmentation de l’incidence de lithiase biliaire ni d’autre effet hépatobiliaire. L’interprétation de ces données chez l’Homme n’est pas connue. Un risque lithogène chez des patients traités par ézétimibe ne peut être exclu.
Les tests de cancérogénèse à long terme de l’ézétimibe ont été négatifs.
L’ézétimibe n’a pas d’effet sur la fertilité du rat mâle ou femelle, ni d’effet tératogène chez le rat et le lapin, ni sur le développement pré- ou post-natal. L’ézétimibe franchit la barrière placentaire chez la rate et la lapine gravides recevant des doses répétées de 1 000 mg/kg/jour.
Atorvastatine
L’atorvastatine n’a pas montré de potentiel mutagène et clastogène dans une série de quatre tests in vitro et d’un test in vivo. L’atorvastatine n’a pas été cancérogène chez le rat, mais après administration de doses élevées chez la souris (entraînant une ASC0-24h 6 à 11 fois supérieure à celle observée chez l’homme à la dose maximale recommandée), il a été observé des adénomes hépatocellulaires chez les mâles et des carcinomes hépatocellulaires chez les femelles. Des données issues des études expérimentales chez l’animal indiquent que les inhibiteurs de la HMG-CoA réductase peuvent affecter le développement embryonnaire ou fœtal. Chez le rat, le lapin et le chien, l’atorvastatine n’a pas eu d’effets sur la fertilité et n’a pas été tératogène ; cependant, une toxicité fœtale a été observée chez le rat et le lapin aux doses maternotoxiques. Chez le rat, un retard de développement des petits et une réduction de la survie postnatale ont été observés pendant l’exposition des mères à des doses élevées d’atorvastatine. Un passage transplacentaire a été mis en évidence chez la rate. Chez la rate, les concentrations plasmatiques d’atorvastatine sont comparables à celles observées dans le lait. On ne sait pas si l’atorvastatine ou ses métabolites sont excrétés dans le lait maternel humain.
Couche d’ézétimibe : croscarmellose sodique, lactose monohydraté, stéarate de magnésium, cellulose microcristalline, povidone, laurylsulfate de sodium.
Couche d’atorvastatine : cellulose microcristalline, lactose monohydraté, hydroxypropylcellulose, croscarmellose sodique, polysorbate 80, carbonate de calcium, stéarate de magnésium, silice colloïdale anhydre.
Pelliculage
Hypromellose, macrogol 8000, dioxyde de titane (E171), talc.
6.4. Précautions particulières de conservation
A conserver dans l’emballage d’origine à l’abri de l’oxygène.
6.5. Nature et contenu de l'emballage extérieur
10, 30, 90 ou 100 comprimés pelliculés sous forme de plaquettes (aluminium/aluminium) (cavité en oPA/Al/PVC avec opercule en Al) avec purge à l’azote.
30 x 1 et 45 x 1 comprimés pelliculés sous forme de plaquettes unitaires (aluminium/aluminium) (cavité en oPA/Al/PVC avec opercule en Al) avec purge à l’azote.
Toutes les présentations peuvent ne pas être commercialisées.
6.6. Précautions particulières d’élimination et de manipulation
Pas d'exigences particulières.
7. TITULAIRE DE L’AUTORISATION DE MISE SUR LE MARCHE
176 RUE MONTMARTRE
75002 PARIS
8. NUMERO(S) D’AUTORISATION DE MISE SUR LE MARCHE
· 34009 280 048 9 5 : 30 comprimés sous plaquettes (aluminium/aluminium)
· 34009 280 049 5 6 : 90 comprimés sous plaquettes (aluminium/aluminium)
· 34009 587 184 0 3 : 45 comprimés sous plaquettes unitaires (aluminium/aluminium)
9. DATE DE PREMIERE AUTORISATION/DE RENOUVELLEMENT DE L’AUTORISATION
[à compléter ultérieurement par le titulaire]
10. DATE DE MISE A JOUR DU TEXTE
[à compléter ultérieurement par le titulaire]
Sans objet.
12. INSTRUCTIONS POUR LA PREPARATION DES RADIOPHARMACEUTIQUES
Liste I
ANSM - Mis à jour le : 14/04/2025
LIPTRUZET 10 mg/10 mg, comprimé pelliculé
Ezétimibe/atorvastatine
· Gardez cette notice. Vous pourriez avoir besoin de la relire.
· Si vous avez d’autres questions, interrogez votre médecin ou votre pharmacien.
· Ce médicament vous a été personnellement prescrit. Ne le donnez pas à d’autres personnes. Il pourrait leur être nocif, même si les signes de leur maladie sont identiques aux vôtres.
· Si vous ressentez un quelconque effet indésirable, parlez-en à votre médecin ou votre pharmacien. Ceci s’applique aussi à tout effet indésirable qui ne serait pas mentionné dans cette notice. Voir rubrique 4.
1. Qu'est-ce que LIPTRUZET 10 mg/10 mg, comprimé pelliculé et dans quels cas est-il utilisé ?
2. Quelles sont les informations à connaître avant de prendre LIPTRUZET 10 mg/10 mg, comprimé pelliculé ?
3. Comment prendre LIPTRUZET 10 mg/10 mg, comprimé pelliculé ?
4. Quels sont les effets indésirables éventuels ?
5. Comment conserver LIPTRUZET 10 mg/10 mg, comprimé pelliculé ?
6. Contenu de l’emballage et autres informations.
1. QU’EST-CE QUE LIPTRUZET 10 mg/10 mg, comprimé pelliculé ET DANS QUELS CAS EST-IL UTILISE ?
Classe pharmacothérapeutique : Inhibiteurs de la HMG-CoA réductase en association avec d’autres hypolipidémiants - code ATC : C10BA05
LIPTRUZET est un médicament utilisé pour baisser les taux de cholestérol élevés. LIPTRUZET contient de l’ézétimibe et de l’atorvastatine.
LIPTRUZET est un médicament utilisé chez l’adulte pour diminuer les taux de cholestérol total, de « mauvais » cholestérol (LDL-cholestérol) et les substances grasses appelées triglycérides dans le sang. De plus, LIPTRUZET augmente le taux de « bon » cholestérol (HDL-cholestérol).
LIPTRUZET agit pour réduire votre cholestérol de deux façons. Il réduit le cholestérol absorbé par votre tube digestif ainsi que le cholestérol fabriqué par votre organisme.
Le cholestérol est une des nombreuses substances grasses trouvées dans le système sanguin. Votre cholestérol total est composé principalement de cholestérol LDL et de cholestérol HDL.
Le LDL-cholestérol est souvent appelé le « mauvais » cholestérol parce qu’il peut s’agglomérer sur les parois de vos artères en formant une plaque. Finalement, la création de cette plaque peut mener à un rétrécissement des artères. Ce rétrécissement peut ralentir ou bloquer le flux sanguin vers les organes vitaux tels que le cœur et le cerveau. Ce blocage du flux sanguin peut entraîner une crise cardiaque ou une attaque cérébrale.
Le HDL-cholestérol est souvent appelé le « bon » cholestérol parce qu’il aide à empêcher le mauvais cholestérol de s’agglomérer dans les artères et protège contre les maladies cardiaques.
Les triglycérides sont une autre sorte de graisse dans votre sang qui peuvent augmenter votre risque de maladie cardiaque.
LIPTRUZET est utilisé chez les patients chez qui le régime seul ne contrôle pas les taux de cholestérol. Vous devez continuer votre régime faisant baisser le cholestérol en prenant ce médicament.
LIPTRUZET est utilisé en complément de votre régime faisant baisser le cholestérol si vous avez :
· un taux élevé de cholestérol dans votre sang (hypercholestérolémie primaire [familiale hétérozygote et non familiale]) ou des taux élevés de graisse dans le sang (dyslipidémie mixte) :
o qui ne sont pas contrôlés de façon appropriée par une statine seule,
o pour lesquels vous avez déjà été traité(e) par l’association d’une statine et de l’ézétimibe pris sous forme de comprimés séparés.
· une maladie héréditaire (hypercholestérolémie familiale homozygote) qui augmente le taux de cholestérol sanguin. Vous pourrez également recevoir d’autres traitements.
· une maladie cardiaque. LIPTRUZET réduit le risque de crise cardiaque, d’accident vasculaire cérébral, d’intervention chirurgicale pour augmenter le flux sanguin cardiaque ou d’hospitalisation pour des douleurs thoraciques.
LIPTRUZET ne vous aide pas à perdre du poids.
2. QUELLES SONT LES INFORMATIONS A CONNAITRE AVANT DE PRENDRE LIPTRUZET 10 mg/10 mg, comprimé pelliculé ?
Ne prenez jamais LIPTRUZET 10 mg/10 mg, comprimé pelliculé :
· si vous êtes allergique (hypersensible) à l’ézétimibe, à l'atorvastatine ou à l’un des autres composants contenus dans ce médicament mentionnés dans la rubrique 6,
· si vous avez ou avez eu dans le passé une maladie du foie,
· si vous présentez des résultats anormaux inexpliqués du bilan hépatique,
· si vous êtes une femme en âge d’avoir des enfants et que vous n’utilisez pas une méthode contraceptive fiable,
· si vous êtes enceinte, si vous planifiez une grossesse ou si vous allaitez
· si vous utilisez l’association glécaprévir/pibrentasvir dans le traitement de l’hépatite C.
Avertissements et précautions
Adressez-vous à votre médecin ou pharmacien avant de prendre LIPTRUZET :
· si vous avez eu dans le passé un accident vasculaire cérébral avec des saignements dans le cerveau ou si vous avez des petites poches de liquide dans le cerveau à la suite d’un accident vasculaire cérébral,
· si vous avez des problèmes rénaux,
· si votre glande thyroïde n’est pas suffisamment active (hypothyroïdie),
· si vous avez présenté dans le passé des courbatures ou des douleurs musculaires répétées ou inexpliquées, ou si vous avez des antécédents personnels ou familiaux de problèmes musculaires,
· si vous avez eu antérieurement des problèmes musculaires pendant un traitement par d’autres médicaments diminuant les taux de lipides (« hypolipidémiants ») (par exemple une autre « statine » ou un « fibrate »),
· si vous buvez régulièrement d’importantes quantités d’alcool,
· si vous avez des antécédents de maladie de foie,
· si vous avez plus de 70 ans,
· si votre médecin vous a informé(e) d'une intolérance à certains sucres, contactez-le avant de prendre ce médicament,
· si vous prenez ou avez pris dans les 7 derniers jours un médicament appelé acide fusidique (pour traiter les infections bactériennes) par voie orale ou injectable. L’association d’acide fusidique et de LIPTRUZET peut entraîner des problèmes musculaires graves (rhabdomyolyse),
Contactez immédiatement votre médecin si vous ressentez de façon inexpliquée des douleurs, une sensibilité ou une faiblesse musculaire en prenant LIPTRUZET. En effet, dans de rares cas, les problèmes musculaires peuvent être graves, notamment en cas de dégradation des muscles pouvant endommager les reins. L’atorvastatine peut entrainer des problèmes musculaires, et des cas de problèmes musculaires ont également été rapportés avec l’ézétimibe.
Prévenez également votre médecin ou votre pharmacien si vous avez une faiblesse musculaire constante. Des examens complémentaires et un traitement peuvent être nécessaires pour la diagnostiquer et la traiter.
Adressez-vous à votre médecin ou pharmacien avant de prendre LIPTRUZET :
− si vous avez une insuffisance respiratoire sévère.
Si l’un de ces cas vous concerne (ou en cas de doute), demandez conseil à votre médecin ou pharmacien avant de prendre LIPTRUZET ; votre médecin vous prescrira une analyse de sang avant, et éventuellement pendant votre traitement par LIPTRUZET afin de déterminer votre risque d'effets indésirables musculaires. Le risque d’effets indésirables musculaires, par exemple de rhabdomyolyse, augmente lorsque certains médicaments sont pris au même moment (voir rubrique 2 « Autres médicaments et LIPTRUZET 10 mg/10 mg, comprimé pelliculé »).
Au cours de votre traitement avec ce médicament, si vous êtes diabétique ou si vous présentez un risque de survenue d’un diabète, vous serez suivi attentivement par votre médecin. Vous pouvez débuter un diabète si vous avez un taux de sucre (glycémie) et de graisse élevés dans le sang, si vous êtes en surpoids et si vous avez une pression artérielle (tension) élevée.
Informez votre médecin de tous vos problèmes médicaux, y compris les allergies.
L’association de LIPTRUZET avec les fibrates (médicaments qui réduisent le cholestérol) doit être évitée car aucune étude de cette association n’a été effectuée.
Enfants
L’utilisation de LIPTRUZET chez les enfants et adolescents n’est pas recommandée.
Autres médicaments et LIPTRUZET 10 mg/10 mg, comprimé pelliculé
Certains médicaments peuvent modifier l’effet de LIPTRUZET ou LIPTRUZET peut modifier l’effet de certains médicaments (voir rubrique 3). Ce type d’interaction peut diminuer l'efficacité de l'un ou des deux médicaments. Elle peut également augmenter le risque de survenue ou la sévérité des effets indésirables, dont celui consistant en la dégradation importante des muscles appelée « rhabdomyolyse », décrite à la rubrique 4 :
· ciclosporine (souvent utilisé chez les patients transplantés),
· érythromycine, clarithromycine, télithromycine, acide fusidique**, rifampicine (utilisés pour traiter les infections bactériennes),
· kétoconazole, itraconazole, voriconazole, fluconazole, posaconazole (utilisés pour traiter les infections fongiques [mycoses]),
· gemfibrozil, autres fibrates, dérivés de l’acide nicotinique, colestipol, cholestyramine (utilisés pour réguler les taux de lipides),
· certains inhibiteurs calciques utilisés pour traiter l’angine de poitrine ou l’hypertension artérielle, par exemple amlodipine, diltiazem,
· digoxine, vérapamil, amiodarone (utilisés pour réguler le rythme cardiaque),
· des médicaments utilisés dans le traitement de l’infection par le VIH, par exemple ritonavir, lopinavir, atazanavir, indinavir, darunavir, association de tipranavir/ritonavir, etc. (traitements du SIDA),
· certains médicaments utilisés dans le traitement de l’hépatite C, comme le télaprévir, le bocéprévir et l’association elbasvir/grazoprévir,
**Si vous devez prendre de l’acide fusidique par voie orale pour traiter une infection bactérienne vous devrez interrompre temporairement la prise de ce médicament. Votre médecin vous indiquera quand vous pourrez reprendre LIPTRUZET en toute sécurité. L’association de LIPTRUZET et d’acide fusidique peut de manière rare entraîner une faiblesse, une sensibilité ou une douleur musculaire (rhabdomyolyse). Pour plus d’informations concernant la rhabdomyolyse, voir rubrique 4.
Autres médicaments connus pour interagir avec LIPTRUZET
· contraceptifs oraux (médicaments utilisés pour prévenir une grossesse),
· stiripentol (médicament anticonvulsivant de l’épilepsie),
· cimétidine (médicament utilisé pour traiter les brûlures d’estomac et les ulcères gastro-duodénaux),
· phénazone (antalgique, utilisé pour soulager la douleur),
· antiacides (médicaments contenant de l’aluminium ou du magnésium utilisés pour soulager les maux d’estomac),
· warfarine, phenprocoumone, acénocoumarol ou fluindione (médicaments utilisés pour empêcher la formation de caillots sanguins),
· colchicine (utilisée pour traiter la goutte),
· millepertuis (produit en vente libre utilisé pour traiter la dépression).
LIPTRUZET 10 mg/10 mg, comprimé pelliculé avec des aliments, boissons et de l’alcool
Voir la rubrique 3 pour les instructions concernant la façon de prendre LIPTRUZET. Veuillez noter ce qui suit :
Jus de pamplemousse
Ne consommez pas plus d’un ou deux petits verres de jus de pamplemousse par jour, car le jus de pamplemousse, en quantités importantes, peut modifier les effets de LIPTRUZET.
Alcool
Evitez de boire des quantités excessives d’alcool pendant le traitement par ce médicament. Pour plus d’informations, voir la rubrique 2 « Avertissements et précautions ».
Grossesse et allaitement
Vous ne devez pas prendre LIPTRUZET si vous êtes enceinte, si vous pensez être enceinte ou si vous planifiez une grossesse. Vous ne devez pas prendre LIPTRUZET si vous êtes en âge de procréer et que vous n’utilisez pas une méthode contraceptive fiable. Si vous découvrez que vous êtes enceinte pendant un traitement avec LIPTRUZET, arrêtez-le immédiatement et informez votre médecin.
Vous ne devez pas prendre LIPTRUZET si vous allaitez.
La sécurité d’emploi de LIPTRUZET pendant la grossesse et l’allaitement n’a pas encore été établie.
Demandez conseil à votre médecin ou pharmacien avant de prendre ce médicament.
Conduite de véhicules et utilisation de machines
Aucun effet sur l’aptitude à conduire des véhicules ou à utiliser des machines n’est attendu avec LIPTRUZET. Cependant, il est à noter que des cas d’étourdissements ont été observés chez les patients traités.
LIPTRUZET 10 mg/10 mg, comprimé pelliculé contient du lactose.
Les comprimés de LIPTRUZET contiennent un sucre appelé lactose. Si votre médecin vous a informé(e) d'une intolérance à certains sucres, contactez-le avant de prendre ce médicament.
LIPTRUZET 10 mg/10 mg, comprimé pelliculé contient du sodium.
Ce médicament contient moins de 1 mmol (23 mg) de sodium par comprimé, c’est-à-dire qu’il est essentiellement « sans sodium ».
3. COMMENT PRENDRE LIPTRUZET 10 mg/10 mg, comprimé pelliculé ?
· Vous devez suivre un régime hypocholestérolémiant avant de débuter le traitement par LIPTRUZET.
· Vous devez poursuivre ce régime hypocholestérolémiant pendant toute la durée du traitement par LIPTRUZET.
Posologie
Quelle est la dose à prendre ?
La dose recommandée est de un comprimé de LIPTRUZET par voie orale une fois par jour.
A quel moment prendre la dose ?
LIPTRUZET peut être pris à tout moment de la journée. Il peut être pris au cours ou en dehors des repas.
Si LIPTRUZET vous est prescrit en association avec de la cholestyramine ou un autre chélateur des acides biliaires (médicaments baissant le cholestérol), vous devez prendre LIPTRUZET au moins 2 heures avant ou 4 heures après le chélateur des acides biliaires.
Si vous avez pris plus de LIPTRUZET 10 mg/10 mg, comprimé pelliculé que vous n’auriez dû
Consultez immédiatement votre médecin ou votre pharmacien.
Si vous oubliez de prendre LIPTRUZET 10 mg/10 mg, comprimé pelliculé
Ne prenez pas de dose double pour compenser le comprimé que vous avez oublié de prendre. Prenez votre dose habituelle de LIPTRUZET le lendemain à l’horaire habituel.
Si vous arrêtez de prendre LIPTRUZET 10 mg/10 mg, comprimé pelliculé
Sans objet.
Si vous avez d’autres questions sur l’utilisation de ce médicament, demandez plus d’informations à votre médecin ou à votre pharmacien.
4. QUELS SONT LES EFFETS INDESIRABLES EVENTUELS ?
Si vous présentez l’un des effets indésirables graves ou symptômes suivants, arrêtez de prendre vos comprimés et contactez immédiatement votre médecin ou allez au service des urgences de l’hôpital le plus proche.
· Réaction allergique grave provoquant un gonflement du visage, de la langue et de la gorge pouvant entraîner d’importantes difficultés pour respirer.
· Maladie grave se manifestant par une desquamation sévère et un gonflement de la peau, la formation de vésicules sur la peau, dans la bouche, la zone génitale et autour des yeux et une fièvre ; éruption cutanée caractérisée par des taches roses à rouges, en particulier sur la paume des mains ou la plante des pieds, pouvant former des cloques.
· Faiblesse musculaire, sensibilité musculaire, douleurs musculaires ou rupture musculaire, coloration rouge-brunâtre des urines, en particulier si elles sont accompagnées d’une sensation de malaise ou de fièvre, car cela peut être causé par une dégradation anormale des muscles pouvant entraîner une insuffisance rénale et engager le pronostic vital.
· Manifestations pseudo-lupiques (incluant éruption cutanée, troubles articulaires et effets sur les cellules sanguines).
Si vous présentez des saignements ou ecchymoses (« bleus ») inattendus ou inhabituels, consultez votre médecin le plus tôt possible car cela peut être un signe d’atteinte hépatique.
Les effets indésirables fréquents suivants ont été rapportés (peuvent affecter jusqu’à 1 patient sur 10) :
· diarrhée,
· douleurs musculaires.
Les effets indésirables peu fréquents suivants ont été rapportés (peuvent affecter jusqu’à 1 patient sur 100) :
· grippe,
· dépression, difficulté d’endormissement, troubles du sommeil,
· étourdissements, maux de tête, sensations de fourmillements,
· rythme cardiaque lent,
· bouffées de chaleur,
· essoufflement,
· douleur abdominale, ballonnement abdominal, constipation, indigestion, flatulences (« gaz »), selles fréquentes, gastrite (inflammation de l’estomac), nausées, gêne gastrique, maux d’estomac,
· acné, urticaire,
· douleurs articulaires, douleurs dans le dos, crampes dans les jambes, fatigue musculaire, spasmes ou faiblesse musculaires, douleurs dans les bras et les jambes,
· faiblesse inhabituelle, sensation de fatigue ou de ne pas se sentir bien, gonflement notamment des chevilles (œdème),
· élévations de certains paramètres des analyses sanguines de la fonction hépatique ou musculaire (créatine phosphokinase, CPK),
· prise de poids.
Les effets indésirables suivants ont été rapportés avec une fréquence indéterminée (fréquence ne pouvant être estimée sur la base des données disponibles) :
· myasthénie oculaire (maladie provoquant une faiblesse des muscles oculaires).
Adressez-vous à votre médecin si vous présentez une faiblesse dans vos bras ou vos jambes qui s’aggrave après des périodes d’activité, une vision double ou des paupières tombantes, des difficultés à avaler ou un essoufflement.
De plus, les effets indésirables ci-dessous ont été rapportés chez des patients prenant soit LIPTRUZET soit des médicaments contenant de l’ézétimibe ou de l’atorvastatine :
· réactions allergiques avec gonflement du visage, des lèvres, de la langue et/ou de la gorge pouvant entraîner des difficultés pour respirer ou avaler (qui nécessitent un traitement immédiat),
· éruption cutanée en relief, avec parfois des lésions « en cocarde »,
· problèmes de foie,
· toux,
· brûlures d’estomac,
· diminution de l’appétit, perte d’appétit,
· hypertension artérielle,
· éruption cutanée accompagnée de démangeaisons, réactions allergiques incluant éruption cutanée et urticaire,
· lésion d'un tendon,
· calculs biliaires ou inflammation de la vésicule biliaire (pouvant provoquer des douleurs abdominales, des nausées, des vomissements),
· inflammation du pancréas, souvent accompagnée de douleurs abdominales sévères,
· diminution du nombre de cellules sanguines, pouvant entraîner des ecchymoses ou saignements (thrombopénie),
· inflammation des voies nasales, saignements de nez,
· douleur à la nuque, douleur, douleur thoracique, douleur dans la gorge,
· augmentations et diminutions du taux de sucre dans le sang (si vous êtes diabétique, vous devez continuer à surveiller attentivement votre glycémie),
· cauchemars,
· engourdissements ou fourmillements dans les doigts et les orteils,
· diminution de la sensibilité à la douleur ou du sens du toucher,
· modifications du goût, bouche sèche,
· perte de mémoire,
· tintements ou bourdonnements dans les oreilles et/ou la tête, perte d’audition,
· vomissements,
· éructations,
· chute de cheveux,
· augmentation de la température,
· présence de globules blancs dans les urines,
· vision floue, troubles visuels,
· gynécomastie (hypertrophie des seins chez les hommes).
Autres effets indésirables possibles, rapportés avec certaines statines :
· troubles sexuels,
· dépression,
· problèmes respiratoires incluant toux persistante et/ou essoufflement ou fièvre,
· diabète. Vous pouvez débuter un diabète si vous avez des taux de sucre (glycémie) et de graisse élevés dans le sang, si vous êtes en surpoids et si vous avez une pression artérielle (tension) élevée. Vous serez suivi attentivement par votre médecin au cours de votre traitement avec ce médicament,
· douleur, sensibilité ou faiblesse musculaires incessantes en particulier si, en même temps, vous ne vous sentez pas bien ou avez une température élevée qui peuvent ne pas disparaître après l’arrêt de LIPTRUZET (fréquence indéterminée).
Déclaration des effets secondaires
Si vous ressentez un quelconque effet indésirable, parlez-en à votre médecin ou votre pharmacien. Ceci s’applique aussi à tout effet indésirable qui ne serait pas mentionné dans cette notice. Vous pouvez également déclarer les effets indésirables directement via le système national de déclaration : Agence nationale de sécurité du médicament et des produits de santé (ANSM) et réseau des Centres Régionaux de Pharmacovigilance - Site internet : https://signalement.social-sante.gouv.fr/
En signalant les effets indésirables, vous contribuez à fournir davantage d’informations sur la sécurité du médicament.
5. COMMENT CONSERVER LIPTRUZET 10 mg/10 mg, comprimé pelliculé ?
Tenir ce médicament hors de la vue et de la portée des enfants.
A conserver dans l’emballage d’origine, à l’abri de l’oxygène.
Ne jetez aucun médicament au tout-à-l’égout ou avec les ordures ménagères. Demandez à votre pharmacien d’éliminer les médicaments que vous n’utilisez plus. Ces mesures contribueront à protéger l’environnement.
6. CONTENU DE L’EMBALLAGE ET AUTRES INFORMATIONS
Ce que contient LIPTRUZET 10 mg/10 mg, comprimé pelliculé
· Les substances actives sont :
ézétimibe........................................................................................................................ 10 mg
atorvastatine (sous forme d’atorvastatine calcique trihydratée)........................................... 10 mg
· Les autres composants sont :
carbonate de calcium, silice colloïdale anhydre, croscarmellose sodique, hydroxypropylcellulose, lactose monohydraté, stéarate de magnésium, cellulose microcristalline, polysorbate 80, povidone, laurylsulfate de sodium.
Le pelliculage contient : hypromellose, macrogol 8000, dioxyde de titane (E171), talc.
Qu’est-ce que LIPTRUZET 10 mg/10 mg, comprimé pelliculé et contenu de l’emballage extérieur
Présentations :
10, 30, 90 et 100 comprimés pelliculés sous plaquettes (aluminium/aluminium) (cavité en oPA/Al/PVC avec opercule en Al) avec purge à l’azote.
30 x 1 et 45 x 1 comprimés pelliculés sous plaquettes unitaires (aluminium/aluminium) (cavité en oPA/Al/PVC avec opercule en Al) avec purge à l’azote.
Toutes les présentations peuvent ne pas être commercialisées.
Titulaire de l’autorisation de mise sur le marché
176 RUE MONTMARTRE
75002 PARIS
Exploitant de l’autorisation de mise sur le marché
176 RUE MONTMARTRE
75002 PARIS
WAARDERWEG 39
2031 BN HAARLEM
PAYS-BAS
Ou
ORGANON HEIST BV
INDUSTRIEPARK 30
2220 HEIST-OP-DEN-BERG
BELGIQUE
Ou
ORGANON N.V.
KLOOSTERSTRAAT 6
5349 AB OSS
PAYS BAS
Noms du médicament dans les Etats membres de l'Espace Economique Européen
[à compléter ultérieurement par le titulaire]
La dernière date à laquelle cette notice a été révisée est :
[à compléter ultérieurement par le titulaire]
Des informations détaillées sur ce médicament sont disponibles sur le site Internet de l’ANSM (France).