Dernière mise à jour le 01/06/2026
PALEXIA 20 mg/ml, solution buvable
Ce médicament n'est ou ne sera bientôt plus disponible sur le marché.
Si vous prenez actuellement ce médicament, il vous est recommandé d'en parler avec votre médecin
ou avec votre pharmacien qui pourra vous orienter vers un autre traitement.
Indications thérapeutiques
Classe pharmacothérapeutique : Autres antalgiques opioïdes, code ATC : N02AX06
Le tapentadol - la substance active contenue dans PALEXIA - est un antalgique puissant qui agit contre la douleur et qui appartient à la classe des opioïdes. PALEXIA 20 mg/ml, solution buvable est utilisé pour le traitement des douleurs aiguës modérées à sévères de l’enfant à partir de 2 ans et avec un poids supérieur à 16 kg et de l'adulte qui peuvent être traitées seulement par des antalgiques opioïdes.
Service médical rendu (SMR)
Les libellés affichés ci-dessous ne sont que des résumés ou extraits issus des avis rendus par la Commission de la Transparence. Seul l'avis complet de la Commission de la Transparence fait référence.
Cet avis est consultable à partir du lien `Avis du jj/mm/aaaa` ou encore sur demande auprès de la HAS (plus d'informations dans l'aide). Les avis et synthèses d'avis contiennent un paragraphe sur la place du médicament dans la stratégie thérapeutique.
| Valeur du SMR | Avis | Motif de l'évaluation | Résumé de l'avis |
|---|---|---|---|
| Faible | Avis du 11/12/2019 | Inscription (CT) | Le service médical rendu par PALEXIA, solution buvable est faible dans l’indication de l’AMM chez l’adulte. |
| Important | Avis du 11/12/2019 | Inscription (CT) | Le service médical rendu par PALEXIA, solution buvable est important dans l’indication de l’AMM chez l’enfant. |
Amélioration du service médical rendu (ASMR)
Les libellés affichés ci-dessous ne sont que des résumés ou extraits issus des avis rendus par la Commission de la Transparence. Seul l'avis complet de la Commission de la Transparence fait référence.
Cet avis est consultable à partir du lien `Avis du jj/mm/aaaa` ou encore sur demande auprés de la HAS (plus d'informations dans l'aide). Les avis et synthéses d'avis contiennent un paragraphe sur la place du médicament dans la stratégie thérapeutique.
| Valeur de l'ASMR | Avis | Motif de l'évaluation | Résumé de l'avis |
|---|---|---|---|
| V (Inexistant) | Avis du 11/12/2019 | Inscription (CT) | Compte tenu : • de l’efficacité du tapentadol démontrée versus placebo dans la douleur aiguë modérée à sévère principalement en post-opératoire, • de l’absence d’étude contrôlée versus comparateur actif chez l’enfant, • de la difficulté d’évaluer, chez l’adulte, l’efficacité du tapentadol versus comparateur actif compte tenu de la méthodologie des études réalisées, la commission de la Transparence considère que PALEXIA, solution buvable n’apporte pas d’amélioration du service médical rendu (ASMR V) dans la prise en charge des douleurs aiguës modérées à sévères de l’enfant et de l'adulte. |
ANSM - Mis à jour le : 21/02/2020
PALEXIA 20 mg/ml, solution buvable
2. COMPOSITION QUALITATIVE ET QUANTITATIVE
Tapentadol............................................................................................................................. 20 mg
Sous forme de chlorhydrate de tapentadol
Pour 1 ml
Excipients à effet notoire :
PALEXIA 20 mg/ml, solution buvable contient du propylène glycol et du sodium. Voir rubrique 4.4.
Pour la liste complète des excipients, voir rubrique 6.1.
Solution limpide et incolore
pH 3,5 à 4,5
4.1. Indications thérapeutiques
PALEXIA 20 mg/ml, solution buvable est indiqué dans le traitement des douleurs aiguës modérées à sévères de l’enfant à partir de 2 ans avec un poids supérieur à 16 kg et de l'adulte, qui ne peuvent être correctement traitées que par des antalgiques opioïdes.
4.2. Posologie et mode d'administration
L’utilisation chez l’enfant est réservée à l’hôpital où un équipement approprié pour permettre une assistance respiratoire est disponible.
La posologie doit être adaptée à l'intensité de la douleur, au traitement antalgique pris antérieurement et à la possibilité d’assurer un suivi du patient.
Adultes :
Les patients doivent débuter leur traitement avec des doses uniques de 50 mg de tapentadol en solution buvable, prises toutes les 4 à 6 heures. Des doses d’initiation plus élevées peuvent être nécessaires en fonction de l’intensité de la douleur et traitements antalgiques précédemment administrés.
Le 1er jour du traitement, une dose supplémentaire peut être prise 1 heure après la 1ère dose, si la douleur n’est pas contrôlée. La dose peut être ensuite augmentée au cas par cas jusqu’à une posologie qui assure une antalgie suffisante tout en minimisant le risque d’effets indésirables, sous étroite surveillance du médecin traitant.
Des doses quotidiennes totales supérieures à 700 mg/jour de tapentadol le premier jour de traitement ainsi que des doses quotidiennes d’entretien supérieures à 600 mg/jour de tapentadol n’ont pas été étudiées et ne sont donc pas recommandées.
Tableau de calcul de dose / volume pour PALEXIA 20 mg/ml solution buvable
|
Dose unitaire de tapentadol prescrite |
Volume (ml) à administrer |
|
25 mg |
1,25 ml |
|
50 mg |
2,5 ml |
|
75 mg |
3,75 ml |
|
100 mg |
5 ml |
Durée de traitement
La solution buvable est destinée au traitement de douleurs aiguës. Si une prolongation du traitement est envisagée ou devient nécessaire chez l’adulte et que PALEXIA a permis le soulagement satisfaisant de la douleur sans survenue d’effet indésirable intolérable, la possibilité de remplacer le traitement par PALEXIA, comprimés à libération prolongée doit être envisagée.
Comme pour tous les traitements symptomatiques, l’utilisation au long cours du tapentadol doit être évaluée régulièrement.
Arrêt du traitement
Des symptômes de sevrage peuvent apparaître après un arrêt brutal du traitement par tapentadol (voir rubrique 4.8). Si un patient n’a plus besoin d’être traité par du tapentadol, il est préférable de diminuer progressivement la posologie afin de prévenir les symptômes de sevrage.
Insuffisance rénale
Chez les patients présentant une insuffisance rénale légère à modérée, un ajustement des doses n’est pas nécessaire (voir rubrique 5.2).
Dans les études contrôlées d’efficacité, PALEXIA n’a pas été évalué chez les patients présentant une insuffisance rénale sévère et n’est donc pas recommandé chez ces patients (voir rubriques 4.4 et 5.2).
Insuffisance hépatique
Chez les patients présentant une insuffisance hépatique légère, une adaptation posologique n’est pas nécessaire (voir rubrique 5.2).
PALEXIA doit être utilisé avec prudence chez les patients présentant une insuffisance hépatique modérée. Chez ces patients, le traitement doit être initié à 25 mg de tapentadol, solution buvable, et doit respecter un délai minimal de 8 heures entre les prises. L’instauration du traitement à une dose journalière supérieure à 150 mg de tapentadol n’est pas recommandée. Le traitement doit assurer le maintien de l’antalgie associé à une tolérance acceptable, en diminuant ou en augmentant l’intervalle entre les prises (voir rubriques 4.4 et 5.2).
PALEXIA n’a pas été étudié chez les patients souffrant d’insuffisance hépatique sévère et, par conséquent, n’est pas recommandé dans cette population (voir rubriques 4.4 et 5.2).
Patients âgés (personnes âgées de 65 ans et plus)
En général, une adaptation posologique n’est pas nécessaire chez les patients âgés. Cependant, les patients âgés étant susceptibles d’avoir une fonction rénale ou hépatique diminuée, une attention particulière doit être portée au choix de la posologie (voir rubriques 4.2 et 5.2).
Population pédiatrique
La posologie recommandée pour les enfants dépend de l’âge et du poids.
Pour les enfants et les adolescents âgés de 2 ans à moins 18 ans, la dose unitaire recommandée est de 1,25 mg/kg toutes les 4 heures.
La dose maximale par jour est 7,5 mg/kg (environ 6 fois la dose unitaire).
Pour les enfants et adolescents ayant un IMC (indice de masse corporelle) élevé, la dose maximale ne doit pas dépasser la dose maximale calculée pour un poids au 97,5 percentile pour l’âge donné.
Des diminutions de la dose peuvent être envisagées au fil du temps en fonction de la diminution de la douleur aigue.
Posologie recommandée pour les enfants de plus de 16 kg (Palexia 20 mg/ml solution buvable)
|
2 ans à moins de 18 ans Poids supérieur à 16 kg |
||||
|
1,25 mg/kg toutes les 4 heures |
||||
|
PALEXIA 20 mg/ml solution buvable (dosage avec une pipette doseuse de 5 ml incluse) |
||||
|
kg (poids) |
ml (Dose Volume) |
|
kg (poids) |
ml (Dose Volume) |
|
16,1 – 17,5 |
1,0 |
|
49,6 – 51,1 |
3,1 |
|
17,6 – 19,1 |
1,1 |
|
51,2 – 52,7 |
3,2 |
|
19,2 – 20,7 |
1,2 |
|
52,8 – 54,3 |
3,3 |
|
20,8 – 22,3 |
1,3 |
|
54,4 – 55,9 |
3,4 |
|
22,4 – 23,9 |
1,4 |
|
56,0 – 57,5 |
3,5 |
|
24,0 -25,5 |
1,5 |
|
57,6 – 59,1 |
3,6 |
|
25,6 -27,1 |
1,6 |
|
59,2 – 60,7 |
3,7 |
|
27,2 -28,7 |
1,7 |
|
60,8 – 62,3 |
3,8 |
|
28,8 – 30,3 |
1,8 |
|
62,4 – 63,9 |
3,9 |
|
30,4 – 31,9 |
1,9 |
|
64,0 – 65,5 |
4,0 |
|
32,0 – 33,5 |
2,0 |
|
65,6 – 67,1 |
4,1 |
|
33,6 – 35,1 |
2,1 |
|
67,2 – 68,7 |
4,2 |
|
35,2 – 36,7 |
2,2 |
|
68,8 – 70,3 |
4,3 |
|
36,8 – 38,3 |
2,3 |
|
70,4 – 71,9 |
4,4 |
|
38,4 – 39,9 |
2,4 |
|
72,0 – 73,5 |
4,5 |
|
40,0 – 41,5 |
2,5 |
|
73,6 – 75,1 |
4,6 |
|
41,6 – 43,1 |
2,6 |
|
75,2 – 76,7 |
4,7 |
|
43,2 – 44,7 |
2,7 |
|
76,8 – 78,3 |
4,8 |
|
44,8 – 46,3 |
2,8 |
|
78,4 – 79,9 |
4,9 |
|
46,4 – 47,9 |
2,9 |
|
≥ 80,0 |
5,0 |
|
48,0 – 49,5 |
3,0 |
|
|
|
Palexia 20 mg/ml solution buvable n’est pas recommandé chez les enfants avec un poids de 16 kg ou moins en raison de la concentration élevée en tapentadol.
La sécurité et l’efficacité de PALEXIA chez les enfants de moins de 2 ans n’ont pas été établies. Les données actuellement disponibles sont décrites en rubriques 5.1 et 5.2 mais aucune recommandation sur la posologie ne peut être faite pour les enfants de moins de 2 ans.
Durée du traitement :
La solution buvable est destinée au traitement de douleurs aigues. Comme pour tous les traitements symptomatiques, l’utilisation continue du tapentadol doit être évaluée régulièrement. Chez les enfants, la durée de traitement ne doit pas dépasser 3 jours car aucune donnée de sécurité et d’efficacité d’un traitement plus long n’est disponible.
Arrêt du traitement :
Des symptômes de sevrage peuvent apparaître après un arrêt brutal du traitement par tapentadol (voir rubrique 4.8). Si un patient n’a plus besoin d’être traité par du tapentadol, il est préférable de diminuer progressivement la posologie afin de prévenir les symptômes de sevrage.
Insuffisance rénale :
PALEXIA n’a pas été étudié chez les enfants et les adolescents insuffisants rénaux, par conséquent l’utilisation dans cette population n’est pas recommandée (voir sections 4.4 et 5.2)
Insuffisance hépatique :
PALEXIA n’a pas été étudié chez les enfants et les adolescents insuffisants hépatiques, par conséquent l’utilisation dans cette population n’est pas recommandée (voir sections 4.4 et 5.2).
Mode d’administration
PALEXIA solution buvable est réservé à la voie orale.
PALEXIA peut être pris au cours ou en dehors d’un repas.
PALEXIA peut être administré soit en l’état, soit diluée dans de l’eau ou toute boisson non alcoolisée. La boite contient une seringue pour administration orale avec un adaptateur qu’il est recommandé d’utiliser afin de prélever du flacon le volume exact correspondant à la dose de tapentadol prescrite.
PALEXIA peut également être administré par sonde nasogastrique en polyuréthane, silicone ou chlorure de polyvinyle (ces matériaux ont été testés et n’ont pas montré d’interaction ou de dégradation du tapentadol).
· chez les patients présentant une hypersensibilité au tapentadol ou à l’un des excipients mentionnés à la rubrique 6.1 ;
· dans les situations cliniques dans lesquelles des principes actifs agonistes du récepteur μ-opioïde sont contre-indiqués, telles que : les patients souffrant de dépression respiratoire sévère (paramètres non contrôlés ou absence de matériel de réanimation), et les patients atteints d’asthme bronchique aigu ou sévère ou d’une hypercapnie ;
· chez les patients qui ont ou sont suspectés d’avoir un iléus paralytique ;
· chez les patients présentant une intoxication aiguë à l’alcool, aux hypnotiques, aux antalgiques d’action centrale ou à des psychotropes (voir rubrique 4.5).
4.4. Mises en garde spéciales et précautions d'emploi
Potentiel d’abus et de toxicomanie/dépendance
PALEXIA présente un risque d’abus et de dépendance. Ceci est à prendre en compte lors de la prescription et/ou la délivrance de PALEXIA, dans les situations où il y a une augmentation du risque de mésusage, d’abus, de dépendance ou d’utilisation détournée.
Les signes de toxicomanie et d’abus doivent être attentivement surveillés chez tous les patients traités par des principes actifs agonistes des récepteurs μ-opioïdes.
L’utilisation concomitante de PALEXIA et de médicaments sédatifs tels que les benzodiazépines et médicaments apparentés, peut entraîner une sédation, une dépression respiratoire, un coma et le décès. Compte-tenu de ces risques, la prescription concomitante avec ces médicaments sédatifs doit être réservée aux patients pour lesquels il n’existe pas d’ alternative thérapeutique.
Si la décision est prise de prescrire PALEXIA de façon concomitante avec des médicaments sédatifs, une diminution de la posologie de l’un ou des deux produit(s) doit être envisagée et la durée du traitement doit être aussi courte que possible.
Les patients doivent être étroitement surveillés afin de détecter des signes et symptômes éventuels de dépression respiratoire et de sédation.
A cet égard, il est fortement recommandé d'informer les patients et leurs soignants de faire attention à ces symptômes (voir rubrique 4.5).
Dépression respiratoire
A fortes doses ou chez les patients sensibles aux agonistes des récepteurs μ-opioïdes, PALEXIA peut provoquer une dépression respiratoire dose-dépendante. PALEXIA doit donc être administré avec précaution chez les patients présentant une fonction respiratoire altérée. D’autres antalgiques non agonistes des récepteurs μ-opioïdes doivent être envisagés et PALEXIA doit être utilisé sous étroite surveillance à la dose efficace la plus faible possible chez ces patients. Si une dépression respiratoire apparaît, elle doit être traitée comme toute autre dépression respiratoire induite par un agoniste des récepteurs μ-opioïdes (voir rubrique 4.9).
Traumatisme crânien et hypertension intracrânienne
PALEXIA ne doit pas être utilisé chez les patients particulièrement sensibles aux effets centraux de la rétention du dioxyde de carbone comme : une augmentation de la pression intracrânienne, une conscience altérée ou un coma. Les antalgiques agonistes des récepteurs μ-opioïdes peuvent masquer l’évolution clinique des patients ayant un traumatisme crânien. PALEXIA doit être utilisé avec précaution chez les patients ayant un traumatisme crânien ou une tumeur cérébrale.
Epilepsies
PALEXIA n’a pas été étudié de façon systématique chez les patients ayant des troubles épileptiques et ces patients ont été exclus des essais cliniques. Cependant, comme les autres antalgiques agonistes des récepteurs µ-opioïdes, PALEXIA doit être administré avec prudence chez les patients ayant des antécédents de troubles épileptiques ou dans des situations à risque de crise d’épilepsie.
PALEXIA peut en outre accroître le risque de convulsions chez les patients prenant d’autres produits abaissant le seuil épileptogène (voir rubrique 4.5).
Insuffisance rénale
Dans les études d’efficacité, PALEXIA n’a pas été étudié chez les patients présentant une insuffisance rénale sévère. L’utilisation dans cette population n’est donc pas recommandée (voir rubriques 4.2 et 5.2).
Insuffisance hépatique
Les patients présentant une insuffisance hépatique légère ou modérée ont montré des augmentations respectives de 2 et 4,5 fois de l’exposition systémique par rapport aux sujets ayant une fonction hépatique normale. PALEXIA doit être utilisé avec précaution chez les patients ayant une insuffisance hépatique modérée (voir rubriques 4.2 et 5.2), en particulier lors de l’initiation du traitement.
PALEXIA n’a pas été étudié chez les patients souffrant d’insuffisance hépatique sévère. L’utilisation dans cette population n’est par conséquent pas recommandée (voir rubriques 4.2 et 5.2).
Maladie du pancréas / des voies biliaires
Des substances actives agonistes des récepteurs μ-opioïdes peuvent provoquer un spasme du sphincter d’Oddi. PALEXIA doit être utilisé avec précaution chez les patients ayant une maladie des voies biliaires, y compris en cas de pancréatite aiguë.
Troubles respiratoires liés au sommeil
Les opioïdes peuvent provoquer des troubles respiratoires liés au sommeil, notamment l’apnée centrale du sommeil (ACS) et l’hypoxémie liée au sommeil. Le risque d’ACS augmente en fonction de la dose d’opioïdes utilisée. Chez les patients présentant une ACS, une diminution de la dose totale d’opioïdes doit être envisagée.
Agonistes / Antagonistes opioïdes mixtes
Une attention particulière doit être portée lors de l’administration de PALEXIA avec des agonistes / antagonistes mixtes des récepteurs µ-opioïdes (tels que la pentazocine, la nalbuphine) ou agonistes partiels µ-opioïdes (tel que la buprénorphine). Chez les patients recevant de la buprénorphine pour le traitement de la dépendance aux opioïdes des alternatives thérapeutiques doivent être envisagées (telles que, par exemple, l’arrêt temporaire de la buprénorphine) si l’administration d’un agoniste µ-opioïde complet (tel que le tapentadol) devient nécessaire pour soulager des douleurs aiguës. Lors de l’utilisation concomitante avec la buprénorphine, la nécessité d’augmenter les doses des agonistes des récepteurs µ-opioïdes a été rapportée, et une surveillance étroite des effets indésirables comme la dépression respiratoire est nécessaire dans certains cas.
Palexia 20 mg/ml solution buvable contient du propylène glycol et du sodium
Ce médicament contient 10 mg de propylène glycol pour 5 ml de solution (dose unitaire maximum) ; ce qui équivaut à 2,0 mg/ml.
Ce médicament contient moins de 1 mmol (23 mg) de sodium par dose unitaire maximum, c.-à-d. qu’il est essentiellement « sans sodium ».
Population pédiatrique
Les mêmes mises en garde et précautions d’emploi de Palexia 20 mg/ml, solution buvable s’appliquent pour les enfants en tenant compte des considérations supplémentaires :
· Palexia 20 mg/ml, solution buvable n’a pas été étudié chez l’enfant et l’adolescent avec une insuffisance rénale ou hépatique, par conséquent, l’utilisation dans cette population n’est pas recommandée (voir sections 4.2 et 5.2).
· Palexia 20 mg/ml, solution buvable n’est pas recommandé chez l’enfant de moins de 2 ans (voir section 4.1).
· Palexia 20 mg/ml, solution buvable n’est pas recommandé chez l’enfant avec un poids inférieur ou égal à 16 kg (voir section 4.2)
· Palexia 20 mg/ml, solution buvable n'a pas été évalué de façon systématique chez les enfants et les adolescents obèses. Par conséquent, les patients pédiatriques souffrant d'obésité doivent faire l'objet d'un suivi approfondi et la dose maximum recommandée pour l'âge ne doit pas être dépassée
· Palexia 20 mg/ml, solution buvable est destiné au traitement de la douleur aigue, et a donc été étudié en traitement de courte durée. Par conséquent, aucune donnée de sécurité à long terme chez l’enfant (par exemple pour la croissance ou le développement) n’est disponible.
Palexia 20 mg/ml, solution buvable est destiné au traitement de la douleur aigue, et a donc été étudié en traitement de courte durée. Par conséquent, aucune donnée de sécurité à long terme chez l’enfant (par exemple pour la croissance ou le développement) n’est disponible.
4.5. Interactions avec d'autres médicaments et autres formes d'interactions
Médicaments sédatifs tels que les benzodiazépines et médicaments apparentés :
L’utilisation concomitante de PALEXIA avec des médicaments sédatifs tels que les benzodiazépines ou d’autres dépresseurs du système respiratoire ou du système nerveux central (autres opioïdes, antitussifs ou traitements de substitution, barbituriques, antipsychotiques, anti-histamine H1, alcool) augmente le risque de sédation, de dépression respiratoire, de coma et de décès en raison de l’effet cumulatif de dépression du SNC. Par conséquent lorsqu’une association de PALEXIA avec un dépresseur du système respiratoire ou du système nerveux central est envisagée, une diminution de la posologie de l’un ou des deux produit(s) doit être envisagée et la durée de l'utilisation concomitante doit être limitée (voir rubrique 4.4).
Agonistes / Antagonistes opioïdes mixtes
Une attention particulière doit être portée lors de l’utilisation concomitante de PAEXIA avec des agonistes / antagonistes mixtes des récepteurs µ-opioïdes (tels que la pentazocine, la nalbuphine) ou agonistes partiels µ-opioïdes (comme la buprénorphine) (voir également rubrique 4.4).
PALEXIA peut provoquer des convulsions et accroître le potentiel épileptogène des inhibiteurs sélectifs de la recapture de la sérotonine (ISRS), des inhibiteurs de la recapture de la sérotonine et de la noradrénaline (IRSN), des antidépresseurs tricycliques, des antipsychotiques et d’autres médicaments abaissant le seuil épileptogène.
Des cas de syndrome sérotoninergique ont été rapportés lors de l'utilisation thérapeutique de tapentadol en association avec d'autres médicaments sérotoninergiques tels que les inhibiteurs sélectifs de la recapture de la sérotonine (IRSS), les inhibiteurs de la recapture de la sérotonine et de la noradrénaline (IRSN) et les antidépresseurs tricycliques. La présence d’un syndrome sérotoninergique est probable si l’un des symptômes suivants est observé :
Clonus spontané,
Clonus inductible ou oculaire accompagné d’agitation ou de diaphorèse,
Tremblement et hyperréflexie,
Hypertonie, température corporelle > 38°C et clonus inductible ou oculaire.
L’arrêt des substances sérotoninergiques permet habituellement d’obtenir une amélioration rapide. Le traitement dépend du type et de la sévérité des symptômes.
La voie principale d’élimination du tapentadol est la conjugaison avec l’acide glucuronique via l’uridine diphosphate transférase (UGT) et principalement les isoformes UGT1A6, UGT1A9 et UGT2B7. En conséquence, une administration concomitante avec des inhibiteurs puissants de ces isoenzymes (telles que ketoconazole, fluconazole, acide meclofénamique) pourrait entrainer une augmentation de l’exposition systémique au tapentadol (voir rubrique 5.2).
La voie majeure d’élimination étant la glucuro-conjugaison, le potentiel d’interaction chez l’adulte est faible.
De plus, in vitro, le tapentadol n’a induit ou inhibé aucune des principales enzymes CYP dont le CYP3A4.
Chez les patients traités par tapentadol, une attention particulière doit être portée en cas d’instauration ou d’arrêt de traitement concomitant par des médicaments inducteurs enzymatiques puissants (ex : rifampicine, phénorbarbital, millepertuis (hypericum perforatum)) car cela peut respectivement entrainer une diminution de l’efficacité ou un risque d’effets indésirables.
Le traitement avec PALEXIA doit être évité chez les patients recevant des inhibiteurs de la monoamine oxydase (IMAO) ou qui en ont pris au cours des 14 derniers jours en raison des effets additifs potentiels sur les concentrations synaptiques de noradrénaline qui peuvent provoquer des effets indésirables cardiovasculaires, comme des crises hypertensives (voir rubrique 4.4).
Population pédiatrique
La voie majeure d’élimination étant la glucuro-conjugaison, le potentiel d’interaction chez l’enfant de moins de 5 mois est faible (voir rubrique 4.2).
4.6. Fertilité, grossesse et allaitement
Grossesse
Il existe très peu de données sur l’utilisation chez la femme enceinte. Les études chez l’animal n’ont pas montré d’effet tératogène. Cependant un retard du développement et une embryotoxicité ont été observés à des doses entraînant des effets pharmacologiques exagérés (effets µ-opioïdes centraux liés à des doses supérieures à la gamme thérapeutique). Des effets sur le développement postnatal ont été observés au seuil maternel de la dose sans effet toxique (NOAEL) (voir rubrique 5.3).
PALEXIA ne doit être utilisé pendant la grossesse que si le bénéfice clinique attendu justifie le risque potentiel pour le fœtus.
Travail et accouchement
Les effets du tapentadol sur le travail et l’accouchement chez l’humain ne sont pas connus. L’utilisation de PALEXIA n’est pas recommandée pendant le travail et l’accouchement. En raison de l’activité agoniste des récepteurs µ-opioïdes du tapentadol, les nouveau-nés de mères qui ont été traitées par tapentadol doivent être suivis compte tenu du risque de dépression respiratoire.
Il n’existe pas de données sur l’excrétion du tapentadol dans le lait humain. D’après une étude chez des ratons allaités par des mères traitées par tapentadol, on peut conclure que le tapentadol est excrété dans le lait (voir rubrique 5.3). Par conséquent, on ne peut exclure un risque pour l’enfant allaité. PALEXIA ne doit pas être utilisé pendant l’allaitement.
4.7. Effets sur l'aptitude à conduire des véhicules et à utiliser des machines
Les effets indésirables du médicament rapportés chez les patients adultes au cours des études contrôlées versus placebo ont été principalement d’intensité légère à modérée. Les effets indésirables les plus fréquents affectaient le système gastro-intestinal (nausées, vomissements) et le système nerveux central (somnolence, vertiges et maux de tête).
Les effets indésirables les plus graves sont la sédation, la dépression respiratoire et les réactions allergiques.
Le tableau ci-dessous liste les effets indésirables identifiés durant les études cliniques réalisées chez l’adulte avec une autre formulation à libération immédiate de tapentadol (PALEXIA, comprimé pelliculé) ou issus des données post-commercialisation chez l’adulte. Ils sont listés par classe et fréquence.
Les fréquences sont définies comme très fréquent : (³1/10); fréquent (³1/100, <1/10); peu fréquent (³1/1,000, <1/100); rare (³1/10,000, <1/1000); très rare (<1/10000) ; fréquence indéterminée (ne peut être estimée d’après les données disponibles).
|
EFFETS INDESIRABLES |
|
||||
|
Système de Classe d’Organe |
Fréquence |
|
|||
|
Trés fréquent
|
Fréquent
|
Peu fréquent
|
Rare
|
Fréquence indéterminée |
|
|
Affections du système immunitaire |
|
|
|
Hypersensibilité médicamenteuse*. |
|
|
Troubles du métabolisme et de la nutrition |
|
Diminution de l’appétit. |
|
|
|
|
Affections psychiatriques |
|
Anxiété, état confusionnel, hallucinations, troubles du sommeil, rêves anormaux. |
Humeur dépressive, désorientation, agitation, nervosité, Impatience, euphorie. |
Pensées anormales. |
Délire** |
|
Affections du système nerveux |
Vertiges, Somnolence, maux de tête. |
Tremblements. |
Troubles de l’attention, Troubles de la mémoire, présyncope, sédation, Ataxie, dysarthrie, hypoesthésie, paresthésie, contractions musculaires involontaires. |
Convulsion, diminution du niveau de conscience. Trouble de coordination. |
|
|
Affections oculaires
|
|
|
Troubles visuels |
|
|
|
Affections cardiaques
|
|
|
Augmentation de la fréquence cardiaque. Palpitations. |
Diminution de la fréquence cardiaque. |
|
|
Affections vasculaires
|
|
Flush. |
Diminution de la pression sanguine. |
|
|
|
Affections respiratoires, thoraciques et mediastinales |
|
|
Dépression respiratoire, diminution de la saturation en oxygène, dyspnée, |
|
|
|
Affections gastro-intestinales |
Nausée, vomissements |
Constipation, diarrhée, dyspepsie, bouche sèche. |
Inconfort abdominal. |
Ralentissement de la vidange gastrique |
|
|
Affections de la peau et du tissu sous-cutané |
|
Prurit, hyperhidrose, éruption cutanée. |
Urticaire. |
|
|
|
Affections musculo-squelettiques et systémique |
|
Spasmes musculaires. |
Sensation de lourdeur. |
|
|
|
Affections du rein des voies urinaires |
|
|
Gêne mictionnelle, pollakiurie. |
|
|
|
Troubles généraux et anomalies au site d’administration |
|
Asthénie, fatigue, Impression de changement de température du corps |
Syndrome de sevrage, œdème, sensation d’étrangeté, sensation ébrieuse, irritabilité, sensation de relâchement |
|
|
|
* De rares cas d’angiœdème, d’anaphylaxie et de choc anaphylactique ont été rapportés post-commercialisation. ** Des cas de délire ont été observés post-commercialisation chez les patients présentant des facteurs de risque supplémentaires tels qu’un cancer et un âge avancé. |
|||||
Les essais cliniques réalisés chez l’adulte avec une autre formulation à libération immédiate de tapentadol (PALEXIA comprimé pelliculé) chez les patients exposés plus de 90 jours ont montré peu de symptômes de sevrage lors de l’arrêt brutal du traitement, symptômes qui ont été qualifiés généralement de légers lorsqu’ils apparaissaient. Néanmoins, les médecins doivent être vigilants sur ces symptômes de sevrage (voir rubrique 4.2) et traiter les patients de façon appropriée lorsque ces symptômes apparaissent.
Le risque d’idées suicidaires et de tentatives de suicide est connu pour être plus élevé chez les patients souffrant de douleurs chroniques. De plus, les substances avec une forte influence sur le système monoaminergique ont été associées à une augmentation du risque de suicide chez les patients souffrant de dépression, surtout en début de traitement. Les données issues des rapports d’études cliniques et post-commercialisation n’ont pas démontré d’augmentation de ce risque avec le tapentadol.
Population pédiatrique :
La fréquence, le type et la sévérité des effets indésirables chez les enfants et les adolescents traités par Palexia 20 mg/ml solution buvable devraient être les mêmes que chez l’adultes traités par Palexia 20 mg/ml solution buvable. Aucune nouvelle donnée de sécurité n’a été identifiée dans les essais pédiatriques complétés, pour chacun des sous-groupes d’âge étudiés.
Aucune donnée sur les symptômes de sevrage chez les enfants utilisant une formulation à libération immédiate de tapentadol, issue des essais cliniques, n’est disponible. Cependant les médecins doivent être vigilants quant aux symptômes de sevrage après administration répétée de tapentadol et son arrêt brutal (voir rubrique 4.2).
Déclaration des effets indésirables suspectés
La déclaration des effets indésirables suspectés après autorisation du médicament est importante. Elle permet une surveillance continue du rapport bénéfice/risque du médicament. Les professionnels de santé déclarent tout effet indésirable suspecté via le système national de déclaration : Agence nationale de sécurité du médicament et des produits de santé (ANSM) et réseau des Centres Régionaux de Pharmacovigilance - Site internet : www.signalement-sante.gouv.fr.
Symptômes
Les cas de surdosage chez l’homme avec le tapentadol sont très limités. Les données pré-cliniques suggèrent que des symptômes similaires à ceux des autres antalgiques d’action centrale agoniste sur les récepteurs µ-opioïdes sont attendus en cas de surdosage avec du tapentadol. En principe, ces symptômes sont les suivants : myosis, vomissements, collapsus cardiovasculaire, troubles de la conscience pouvant aller jusqu’au coma, convulsions et dépression respiratoire pouvant aller jusqu’à l’arrêt respiratoire.
Prise en charge
La prise en charge du surdosage doit être axée sur le traitement des symptômes induits par les agonistes µ-opioïdes. Une attention primordiale doit être accordée au rétablissement des voies respiratoires du patient et à l’instauration d’une ventilation assistée ou contrôlée lorsqu’un surdosage en tapentadol est suspecté.
Les antagonistes purs des récepteurs opioïdes comme la naloxone sont des antidotes spécifiques de la dépression respiratoire due au surdosage en opioïdes. La dépression respiratoire provoquée par un surdosage, peut perdurer au-delà de l’action de l’antagoniste opioïde. L’administration d’un antagoniste opioïde n’exonère pas de la surveillance continue des voies respiratoires, de la respiration et de la non obstruction des voies aériennes. Si la réponse à l’antagoniste opioïde est insuffisante ou de courte durée, une dose additionnelle d’antagoniste (exemple : naloxone) doit être administrée en respectant les indications fournies par le fabricant du produit.
Une décontamination gastro-intestinale peut être envisagée pour éliminer le principe actif non absorbé.
Une décontamination gastro-intestinale au charbon actif ou par lavage gastrique peut être envisagée au cours des 2 heures suivant la prise. Avant de tenter la décontamination gastro-intestinale, il faut prendre soin de sécuriser les voies respiratoires.
5. PROPRIETES PHARMACOLOGIQUES
5.1. Propriétés pharmacodynamiques
Classe pharmacothérapeutique : Autres antalgiques opioïdes, code ATC : N02AX06
Le tapentadol est un opioïde fort avec une activité agoniste µ-opioïde et des propriétés additionnelles d'inhibition de la recapture de la noradrénaline. Le tapentadol exerce ses effets antalgiques directement, sans métabolite pharmacologiquement actif.
Le tapentadol a démontré son efficacité sur des modèles précliniques de douleur nociceptive, neuropathique, viscérale et inflammatoire ; son efficacité a été confirmée dans des essais cliniques réalisés chez l’adulte avec une autre formulation à libération immédiate de tapentadol (PALEXIA comprimé pelliculé) dans la douleur nociceptive parmi lesquelles les douleurs post-opératoires orthopédique et abdominale ainsi que la douleur chronique de l'arthrose de la hanche ou du genou. L'effet antalgique du tapentadol dans les essais sur la douleur nociceptive chez l’adulte était équivalent à celui de l'opioïde fort utilisé comme comparateur.
Effets sur le système cardiovasculaire : dans un essai chez l’adulte visant à mesurer l'intervalle QT chez l'homme, aucun effet n'a été observé, à doses répétées thérapeutiques et supra thérapeutiques. De la même façon, le tapentadol n'a pas d'effet significatif sur les autres paramètres de l'ECG (fréquence cardiaque, intervalle PR, durée du QRS, morphologie de l'onde T ou onde U)
Population pédiatrique
L’efficacité du tapentadol en solution buvable administrée jusqu’à 72 heures a été démontrée chez les enfants et les adolescents (âge compris entre 2 ans et moins de 18 ans) avec une douleur post-chirurgicale.
5.2. Propriétés pharmacocinétiques
Absorption
Le tapentadol est rapidement et complètement absorbé après administration orale de PALEXIA. La biodisponibilité moyenne après administration d’une dose unique (à jeun) de PALEXIA est d’environ 32% en raison de l’important effet de premier passage hépatique. Les concentrations sériques maximales de tapentadol sont obtenues environ 1,25 heures après administration des comprimés pelliculés. Une augmentation dose-dépendante des valeurs de la Cmax et de l’ASC du tapentadol a été observée après administration de comprimés pelliculés aux doses thérapeutiques. Un essai en prises multiples (toutes les 6 heures) avec des doses de 75 mg à 175 mg de tapentadol administrés sous forme de comprimé pelliculé a montré des ratios d’accumulation de 1,4 à 1,7 pour le principe actif et entre 1,7 et 2 pour le métabolite principal tapentadol-O-glucuronide. Ces paramètres sont essentiellement déterminés par l’intervalle de dose et la demi-vie apparente du tapentadol et de ses métabolites. L’état d’équilibre des concentrations sériques de tapentadol est atteint le 2e jour de traitement.
Effets de la nourriture
L’ASC et la Cmax ont augmenté respectivement de 25% et 16%, lorsque les comprimés pelliculés ont été administrés après un petit déjeuner à teneur élevée en graisses et en calories. Dans ces conditions, le pic de concentration plasmatique a été retardé de 1,5 heures. D’après les données d’efficacité aux temps précoces observées dans les études de phase II/III, les effets de l’alimentation ne semblent pas être cliniquement significatifs. PALEXIA peut être administré pendant ou en dehors des repas.
Distribution
Le tapentadol est distribué largement dans tout l’organisme. Après une administration par voie intraveineuse, le volume de distribution (Vd) du tapentadol est de 540 +/- 98 l. La liaison aux protéines sériques est faible et s’élève à environ 20%, principalement lié à l’albumine.
Métabolisme
Environ 97% du tapentadol est métabolisé. La voie principale du métabolisme du tapentadol est la conjugaison avec l’acide glucuronique qui produit des glucuronides. Après administration orale, environ 70% de la dose est excrétée dans les urines sous formes conjuguées (55% sous forme de glucuronide et 15 % sous forme de sulfate de tapentadol). L’uridine diphosphate glucuronyl transferase (UGT) est la principale enzyme impliquée dans la glucuronidation (principalement les isoformes UGT1A6, UGT1A9 et UGT2B7). Au total, 3% du tapentadol est excrété dans l’urine sous forme inchangée. Le tapentadol est, de plus, métabolisé en N-desmethyl tapentadol (13%) par le CYP2C9 et CYP2C19 et en hydroxy-tapentadol (2%) par le CYP2D6, ensuite métabolisés par conjugaison. Par conséquent, le métabolisme de la substance active par le complexe cytochrome P450 est moins important que la glucuroconjugaison.
Aucun métabolite ne contribue à l’activité antalgique.
Élimination
Le tapentadol et ses métabolites sont excrétés quasi exclusivement (99%) par les reins. La clairance totale après administration par voie intraveineuse est de 1530 +/- 177 ml/min.
Populations spécifiques
Patients âgés
Dans un essai clinique, l’exposition moyenne (ASC) du tapentadol était équivalente entre une population âgée (65-78 ans) et une population de jeunes adultes (19 - 43 ans). Une diminution de 16% de la Cmax moyenne a été observée dans le groupe de sujets âgés par rapport aux jeunes adultes.
Insuffisance rénale
L’ASC et la Cmax du tapentadol ont été comparables chez des sujets avec des degrés variés de fonction rénale (de normale à insuffisance rénale sévère). Cependant, on a observé une augmentation de l’exposition moyenne (ASC) au tapentadol-O-glucuronide proportionnelle au degré d’insuffisance rénale. Chez les sujets avec une insuffisance rénale légère, modérée, et sévère, l’ASC du tapentadol-O-glucuronide est respectivement 1,5, -2,5 et 5,5 fois plus élevée que chez des sujets ayant une fonction rénale normale.
Insuffisance hépatique
L’administration du tapentadol a montré une exposition et des taux sériques de tapentadol plus élevés chez les sujets ayant une insuffisance hépatique par rapport aux sujets ayant une fonction hépatique normale. Les paramètres pharmacocinétiques du tapentadol en cas d’insuffisance hépatique légère ou modérée ont été respectivement multipliés par 1,7 et 4,2 pour l’ASC, par 1,4 et 2,5 pour la Cmax et par 1,2 et 1,4 pour la demi-vie par rapport au groupe ayant une fonction hépatique normale. La formation du tapentadol-O-glucuronide diminue avec l’augmentation de la sévérité de l’insuffisance hépatique.
Interactions pharmacocinétiques
Le tapentadol est principalement métabolisé par la glucuroconjugaison et seule une petite quantité est métabolisée par les voies oxydatives.
La glucuroconjugaison est un système de forte activité/faible affinité, qui n’est pas facilement saturé même dans des conditions pathologiques. Les concentrations thérapeutiques des principes actifs sont généralement bien au-dessous des concentrations nécessaires pour une inhibition potentielle de la glucuroconjugaison. Les interactions cliniquement significatives secondaires à la glucuroconjugaison sont donc peu probables.
Dans une série d’essais d’interactions médicamenteuses avec le paracétamol, le naproxène, l’acide acétylsalicylique et le probénécide, une possible influence de ces substances actives sur la glucuroconjugaison du tapentadol a été étudiée. Les essais avec naproxène (500 mg 2 fois par jour pendant 2 jours) et probénicide (500 mg 2 fois par jour pendant 2 jours) ont montré des augmentations de l’ASC du tapentadol respectivement de 17% et 57%. Globalement, aucune modification cliniquement pertinente des concentrations sériques de tapentadol n’a été observée dans ces essais.
Des études d’interaction avec le métoclopramide et l’oméprazole ont été conduites pour étudier une possible influence de ces substances actives sur l’absorption du tapentadol. Ces essais ont également montré qu’il n’y a aucun effet cliniquement pertinent sur les concentrations sériques de tapentadol.
Les études in vitro n’ont pas révélé de potentiel inhibiteur ou inducteur du tapentadol sur les enzymes du cytochrome P450. Par conséquent des interactions cliniquement significatives via le système cytochrome P450 sont peu probables.
La liaison du tapentadol aux protéines plasmatiques est faible (environ 20%). Par conséquent la probabilité d’interaction pharmacocinétique par déplacement du site de liaison aux protéines est faible.
Population pédiatrique
Absorption
Dans la population pédiatrique, les concentrations sériques maximum ont été observées au même moment que pour l’adulte, sans modification liée à l’âge.
Effet de la nourriture
Aucun essai portant spécifiquement sur les effet de la nourriture chez les enfants et les adolescents n’a été réalisé.
Dans l’essai de phase III réalisé chez les enfants et les adolescents, la solution buvable de tapentadol a été donnée sans tenir compte de la prise alimentaire.
Considérant les données d’efficacité obtenues durant l’essai de phase III chez les enfants et les adolescents, l’effet de la nourriture ne semble pas être cliniquement significatif. Palexia 4mg/ml, solution buvable peut être donné pendant ou en dehors de repas.
Distribution
Le volume de distribution par groupe d’âge chez les enfants après administration orale de tapentadol et dérivé de la modélisation de la pharmacocinétique de population (Pop PK) est montrée dans le tableau suivant :
|
Groupe d’âge |
Volume de distribution apparent (V/F) après administration orale (L) Moyenne +/- ET |
|
12 ans à moins de 18 ans |
923 +/- 36 |
|
6 ans à moins de 12 ans |
534 +/- 25 |
|
2 ans à moins de 6 ans |
276 +/- 17 |
Paramètres basés sur un nouveau modèle combiné
Métabolisme
Chez les humains âgés de 5 ans et plus, le métabolisme du tapentadol est important.
Elimination
La clairance pédiatrique du tapentadol après administration orale et dérivée de la modélisation de la Pop PK pour les différents groupes d’âge est indiquée dans le tableau ci-dessous :
|
Groupe d’âge |
Clairance apparente du tapentadol (CL/F) après administration orale (L/h) Moyenne +/- ET |
|
12 ans à moins de 18 ans |
212 +/- 7 |
|
6 ans à moins de 12 ans |
149 +/- 7 |
|
2 ans à moins de 6 ans |
80 +/- 5 |
Paramètres basé sur le nouveau modèle combiné
Populations spécifiques
Insuffisances hépatique et rénale :
Palexia 20 mgl/ml n’a pas été étudié chez les enfants et adolescents souffrant d’insuffisance rénale et hépatique.
Interactions pharmacocinétiques
Aucune étude d’interaction médicamenteuse spécifique n’a été réalisée chez les adultes et les adolescents.
5.3. Données de sécurité préclinique
L’excrétion dans le lait maternel a été étudiée chez le raton allaité par des mères traitées par tapentadol. Les ratons ont été exposés de façon dose dépendante au tapentadol et tapentadol-O-glucuronide. Il en a été conclu que le tapentadol est excrété dans le lait maternel.
Les rats juvéniles ont été traités de J6 à J90 post naissance, ce qui a couvert la période de développement correspondant à la petite enfance, l’enfance et l’adolescence chez les humains. Durant les 3 premiers jours de traitement, une augmentation numériquement plus élevée de l’incidence de la mortalité est survenue à des doses ≥ 25 mg/kg/jour avec une exposition plasmatique au tapentadol à la LOAEL comparable à l’exposition plasmatique prédite pour les enfants.
Le tapendol a été bien toléré chez les chiots de plus de 10 jours. Il n’y a pas eu de signe clinique lié au traitement, d’effets sur le poids, sur la consommation de nourriture, le pré-sevrage ou le développement reproductif, la croissance des os longs, l’activité motrice, le comportement ou l’apprentissage et la mémoire. Le poids des organes et l’évaluation macroscopique ou microscopique n’ont pas montré de modification liée au traitement. Le tapentadol n’a pas eu d’influence sur le développement sexuel, l’accouplement ou les paramètres de grossesse chez les animaux traités.
5 ans.
La durée de conservation après ouverture est de 6 semaines.
6.4. Précautions particulières de conservation
Flacon non ouvert : Pas de précautions particulières de conservation.
Après ouverture : Conserver le flacon à l’endroit, à la verticale.
6.5. Nature et contenu de l'emballage extérieur
Flacons en polyéthylène de haute densité (PEHD) scellés par une feuille d’aluminium et fermé par un capuchon en polyéthylène de haute densité (PEHD) / polypropylène (PP) avec sécurité enfant.
Chaque flacon de solution buvable est fourni avec une seringue pour administration orale et un adaptateur fixé à la seringue.
Le volume de la seringue est de 5 ml, graduée par intervalle de 0,1 ml. De plus, l’échelle droite montre les doses unitaires pour l’adulte. .
Flacons de 100 ml et 200 ml
Toutes les présentations peuvent ne pas être commercialisées.
6.6. Précautions particulières d’élimination et de manipulation
Pas d’exigences particulières pour l’élimination.
Pour les autres manipulations voir rubrique 4.2.
Tout médicament non utilisé ou déchet doit être éliminé conformément à la réglementation en vigueur.
7. TITULAIRE DE L’AUTORISATION DE MISE SUR LE MARCHE
IMMEUBLE EUREKA
19, RUE ERNEST RENAN – CS 90001
92024 NANTERRE CEDEX
8. NUMERO(S) D’AUTORISATION DE MISE SUR LE MARCHE
· 34009 276 927 1 0 : 100 ml en flacon (PEHD) avec bouchon sécurité enfant (PEHD/Polypropylène) + seringue pour administration orale de 5 ml
· 34009 276 928 8 8 : 200 ml en flacon (PEHD) avec bouchon sécurité enfant (PEHD/Polypropylène) + seringue pour administration orale de 5 ml
9. DATE DE PREMIERE AUTORISATION/DE RENOUVELLEMENT DE L’AUTORISATION
[à compléter ultérieurement par le titulaire]
10. DATE DE MISE A JOUR DU TEXTE
[à compléter ultérieurement par le titulaire]
Sans objet.
12. INSTRUCTIONS POUR LA PREPARATION DES RADIOPHARMACEUTIQUES
Stupéfiant
Prescription sur ordonnances sécurisées.
Prescription limitée à 28 jours
ANSM - Mis à jour le : 21/02/2020
PALEXIA 20 mg/ml, solution buvable
Tapentadol
· Gardez cette notice. Vous pourriez avoir besoin de la relire.
· Si vous avez d’autres questions, interrogez votre médecin ou votre pharmacien.
· Ce médicament vous a été personnellement prescrit. Ne le donnez pas à d’autres personnes. Il pourrait leur être nocif, même si les signes de leur maladie sont identiques aux vôtres.
· Si vous ressentez un quelconque effet indésirable, parlez-en à votre médecin ou votre pharmacien. Ceci s’applique aussi à tout effet indésirable qui ne serait pas mentionné dans cette notice. Voir rubrique 4.
1. Qu'est-ce que PALEXIA 20 mg/ml, solution buvable et dans quels cas est-il utilisé ?
2. Quelles sont les informations à connaître avant de prendre PALEXIA 20 mg/ml, solution buvable ?
3. Comment prendre PALEXIA 20 mg/ml, solution buvable ?
4. Quels sont les effets indésirables éventuels ?
5. Comment conserver PALEXIA 20 mg/ml, solution buvable ?
6. Contenu de l’emballage et autres informations.
1. QU’EST-CE QUE PALEXIA 20 mg/ml, solution buvable ET DANS QUELS CAS EST-IL UTILISE ?
Classe pharmacothérapeutique : Autres antalgiques opioïdes, code ATC : N02AX06
Le tapentadol - la substance active contenue dans PALEXIA - est un antalgique puissant qui agit contre la douleur et qui appartient à la classe des opioïdes. PALEXIA 20 mg/ml, solution buvable est utilisé pour le traitement des douleurs aiguës modérées à sévères de l’enfant à partir de 2 ans et avec un poids supérieur à 16 kg et de l'adulte qui peuvent être traitées seulement par des antalgiques opioïdes.
2. QUELLES SONT LES INFORMATIONS A CONNAITRE AVANT DE PRENDRE PALEXIA 20 mg/ml, solution buvable ?
Ne prenez jamais PALEXIA 20 mg/ml, solution buvable :
· si vous êtes allergique (hypersensible) au tapentadol ou à l'un des autres composants contenus dans ce médicament listés à la rubrique 6 (« Informations supplémentaires ») ;
· si vous avez de l'asthme ou si votre respiration est dangereusement lente ou faible (dépression respiratoire, hypercapnie);
· si vous avez une paralysie de l'intestin;
· en cas d'intoxication aigue par l'alcool, des somnifères, d'autres médicaments contre la douleur ou des médicaments psychotropes (médicaments qui agissent sur l'humeur et les émotions) (voir rubrique « Autres médicaments et Palexia 20 mg/ml, solution buvable »).
Avertissements et précautions
Adressez-vous à votre médecin ou pharmacien avant de prendre PALEXIA 20 mg/ml, solution buvable :
· si vous avez une respiration lente ou peu profonde ;
· si vous souffrez d’une augmentation de la pression intracrânienne ou des troubles de la conscience pouvant aller jusqu’au coma ;
· si vous avez eu un traumatisme crânien ou une tumeur du cerveau ;
· si vous souffrez d’une maladie du foie ou des reins (voir rubrique « comment prendre PALEXIA »)
· si vous souffrez d’une maladie du pancréas, dont la pancréatite, ou des voies biliaires ;
· si vous prenez des médicaments appelés agonistes / antagonistes mixtes des récepteurs opioïdes (tels que la pentazocine, la nalbuphine) ou agonistes partiels des récepteurs µ-opioïdes (tels que la buprénorphine)
· Si vous êtes susceptible de souffrir d’épilepsie ou de convulsions, ou si vous prenez d’autres médicaments connus pour augmenter le risque de crises épileptiques, car le risque de convulsions pourrait augmenter.
PALEXIA peut entrainer une dépendance physique et psychique. Si vous avez une tendance à abuser des médicaments ou si vous êtes dépendant à des médicaments, vous ne devez prendre ce médicament que pendant de courtes durées et sous stricte surveillance médicale.
Enfants et adolescents
Palexia 20 mg/ml, solution buvable n'a pas été évalué de façon systématique chez les enfants et les adolescents souffrant d’obésité. Par conséquent, les patients pédiatriques souffrant d'obésité doivent faire l'objet d'un suivi approfondi et la dose maximale recommandée pour l'âge ne doit pas être dépassée.
Ne pas donner ce médicament aux enfants de moins de 2 ans.
Troubles respiratoires liés au sommeil
PALEXIA 20 mg/ml, solution buvable contient une substance active appartenant au groupe des opioïdes. Les opioïdes peuvent provoquer des troubles respiratoires liés au sommeil, tels que l’apnée centrale du sommeil (respiration superficielle/pause respiratoire pendant le sommeil) et l’hypoxémie liée au sommeil (faible taux d’oxygène dans le sang).
Le risque de souffrir d’apnée centrale du sommeil dépend de la dose d’opioïdes. Votre médecin peut envisager de diminuer votre dose totale d’opioïdes si vous souffrez d’apnée centrale du sommeil.
Autres médicaments et PALEXIA 20 mg/ml, solution buvable
Informez votre médecin ou pharmacien si vous prenez, avez récemment pris ou pourriez prendre tout autre médicament.
L'utilisation concomitante de PALEXIA 20 mg/ml, solution buvable avec des médicaments sédatifs tels que les benzodiazépines et médicaments apparentés (certains somnifères ou tranquillisants (ex barbituriques), ou d’autres médicaments qui soulagent la douleur comme les opioïdes, la morphine et la codéine (aussi utilisés pour soigner la toux), des antipsychotiques, des antihistaminiques H1, de l’alcool) augmente le risque de somnolence, de difficultés respiratoires (dépression respiratoire), de coma et peut mettre en jeu le pronostic vital. Compte-tenu de ces risques, l'utilisation concomitante ne doit être envisagée qu’en l’absence d’autre option de traitement.
Toutefois, si votre médecin vous prescrit PALEXIA 20 mg/ml, solution buvable en association avec un médicament sédatif, il devra limiter la dose et la durée du traitement concomitant.
Veuillez informer votre médecin de tous médicaments sédatifs que vous prenez et respectez les doses prescrites par votre médecin. Il peut être utile de demander à vos amis ou parents de faire attention aux signes et symptômes décrits ci-dessus. Contactez votre médecin si vous présentez ce type de symptômes.
Si vous prenez un médicament qui influe sur les niveaux de sérotonine (tels que certains médicaments pour traiter la dépression), consultez votre médecin avant de prendre PALEXIA 20 mg/ml, solution buvable car des cas de syndrome sérotoninergique ont été rapportés. Le syndrome sérotoninergique est rare mais peut menacer le pronostic vital. Les symptômes incluent des contractions rythmiques involontaires des muscles dont les muscles qui contrôlent les mouvements oculaires, l’agitation, une transpiration excessive, tremblements, une exagération des réflexes, une augmentation de la tension musculaire, une température du corps supérieure à 38°C.En cas de survenue, demandez conseil à votre médecin.
L’association de PALEXIA avec d’autres médicaments agonistes/antagonistes mixtes des récepteurs μ-opioïde (exemple : pentazocine, nalbuphine) ou agonistes partiels (exemple : buprénorphine) n’a pas été étudiée. Il est possible que PALEXIA perde de son efficacité s’il est donné avec l’un de ces médicaments. Prévenez votre médecin si vous êtes actuellement traité par l’un de ces médicaments.
La prise de PALEXIA avec des inhibiteurs ou inducteurs puissants (exemples : rifampicine, phénobarbital, le millepertuis) de certaines enzymes nécessaires à l’élimination du tapentadol de votre corps peut avoir une influence sur l’efficacité du tapentadol ou pourrait entrainer des effets indésirables surtout au début ou à l’arrêt de cet autre traitement. Tenez votre médecin informé de tous les médicaments que vous prenez.
PALEXIA 20 mg/ml, solution buvable ne doit pas être associé à des inhibiteurs de la MAO (certains médicaments pour le traitement de la dépression). Dites à votre docteur si vous prenez des inhibiteurs de la MAO ou si vous en avez pris au cours des 14 derniers jours.
PALEXIA 20 mg/ml, solution buvable avec de l’alcool
Ne consommez pas d'alcool pendant le traitement par PALEXIA, car cela pourrait accentuer certains effets indésirables tels que la somnolence. La nourriture n'a pas d'influence sur l'action de ce médicament.
Si vous êtes enceinte ou que vous allaitez, si vous pensez être enceinte ou planifiez une grossesse, demandez conseil à votre médecin ou pharmacien avant de prendre ce médicament.
Ne prenez pas ces comprimés :
· si vous êtes enceinte, sauf si votre médecin vous le recommande.
L’utilisation de Palexia 20 mg/ml, solution buvable n’est pas recommandée :
· au cours de l’accouchement car cela pourrait entrainer une respiration dangereusement lente ou peu profonde (dépression respiratoire) chez le nouveau-né,
· au cours de l’allaitement car le tapentadol pourrait être présent dans le lait maternel.
Conduite de véhicules et utilisation de machines
PALEXIA peut provoquer une somnolence, des sensations de vertige et une vision floue et peut modifier vos réactions. Cela peut arriver notamment lorsque vous commencez à prendre PALEXIA, lorsque votre médecin modifie la posologie ou lorsque vous buvez de l'alcool ou prenez des tranquillisants. Demandez à votre médecin si vous pouvez conduire une voiture ou utiliser des machines.
PALEXIA 20 mg/ml, solution buvable contient du sodium
Ce médicament contient moins de 1 mmol (23 mg) de sodium par dose unitaire maximum, c.-à-d. qu’il est essentiellement « sans sodium ».
PALEXIA 20 mg/ml, solution buvable contient du propylène glycol
Ce médicament contient 48 mg de propylène glycol pour 25 ml de solution (dose unitaire maximale), ce qui équivaut à 2,0 mg/ml.
3. COMMENT PRENDRE PALEXIA 20 mg/ml, solution buvable ?
Votre médecin ajustera la dose en fonction de l'intensité de votre douleur et de votre sensibilité personnelle à la douleur. D'une manière générale, il faut prendre la dose minimale qui soulage la douleur.
Posologie
Adulte
La posologie habituelle est de 50 mg de tapentadol (2,5 ml de solution buvable), 75 mg de tapentadol (3,75 ml de solution orale) ou 100 mg de tapentadol (5 ml de solution buvable) toutes les 4 à 6 heures.
Il n’est pas recommandé de prendre des doses quotidiennes totales supérieures à 700 mg par jour de tapentadol le premier jour de traitement et supérieures 600 mg par jour de tapentadol les jours suivants. Si nécessaire, votre médecin peut vous prescrire un dosage ou un délai entre les prises plus approprié. Si vous sentez que l’effet de ce médicament est trop fort ou trop faible, parlez-en à votre médecin ou votre pharmacien.
Patients âgés
Chez les patients âgés (plus de 65 ans), un ajustement des doses n’est généralement pas nécessaire. Cependant, l’excrétion du tapentadol peut être retardée chez certains patients de ce groupe d’âge. Si vous êtes concerné, votre médecin pourra vous proposer une posologie différente.
Maladie du foie ou des reins (insuffisance)
Les patients qui ont des problèmes sévères du foie ne doivent pas prendre ce médicament. Si vous avez des problèmes hépatiques d’intensité modérée, votre médecin pourra vous recommander une posologie différente. En cas de problèmes légers du foie, un ajustement de la posologie n’est pas nécessaire.
Les patients atteints de problèmes sévères des reins ne doivent pas prendre ce médicament. En cas de problèmes rénaux d’intensité légère à modérée, un ajustement de la posologie n’est pas nécessaire.
Enfants et adolescents
PALEXIA doit être administré à l’enfant à l’hôpital uniquement.
Palexia 20 mg/ml solution buvable doit être utilisé uniquement chez les enfants avec un poids de plus de 16 kg.
La posologie de Palexia 20 mg/ml solution buvable pour les enfants et les adolescents âgés de 2 à moins de 18 ans est de 1,25 mg/kg toutes les 4 heures. La dose exacte vous sera donnée par votre médecin ou votre infirmer(e).
Il faut toujours attendre un délais de 4 heures avant de donner la dose suivante. La dose peut être diminuée en fonction de la diminution de la douleur aiguë.
Mode d’administration
PALEXIA 20 mg/ml solution buvable est à prendre par voie orale.
Vous pouvez prendre la solution buvable l'estomac vide ou avec les repas.
La boite contient une seringue pour administration orale et un adaptateur fixé à la seringue. Ce dispositif doit être utilisé pour prélever dans le flacon, la quantité exacte (volume) correspondant à la dose prescrite de tapentadol.
Instructions pour l’ouverture du flacon et l’utilisation de la seringue pour administration orale :
|
|
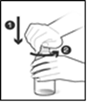
Fig 1 : Le flacon est fermé par un bouchon sécurité-enfant. Pour enlever le bouchon, appuyez dessus et tournez-le dans le sens inverse des aiguilles d’une montre (Fig 1). Enlevez le bouchon et retirez le film de sécurité qui se trouve sur le col du flacon. Si ce film de sécurité est abimé, ne prenez pas ce médicament et parlez-en à votre pharmacien.
|
|
|
Fig 2 : Placez le flacon sur une surface solide et plate. Ouvrez le sachet en plastique contenant la seringue et l’adaptateur au niveau de l’extrémité perforée et sortez la seringue (A) avec l’adaptateur fixé à son extrémité (B). Insérez l’adaptateur fixé à la seringue dans le goulot du flacon (Fig 2).
|
|
|
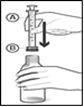
Fig 3 : Pour remplir la seringue, retournez le flacon. Tout en maintenant la seringue en place, tirez doucement sur le piston (C) jusqu'à la ligne qui correspond à la dose prescrite par votre médecin (voir section « Comment prendre PALEXIA »). Ne retirez pas la seringue orale du flacon à cette étape !
|
|
|
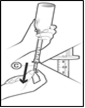
Fig 4 : Remettez le flacon à l’endroit et retirez délicatement la seringue du flacon. Après avoir retiré la seringue, vérifiez attentivement que vous avez prélevé la bonne quantité de solution. L’adaptateur (B) qui était initialement fixé à la seringue devrait maintenant rester dans le flacon (Fig 4).
|
|
|
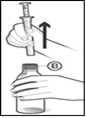
Fig 5 : Prenez votre médicament en mettant la seringue dans votre bouche et en poussant doucement sur le piston. Poussez le piston jusqu’au bout pour vous assurer que toute la solution est utilisée. Si vous préférez, vous pouvez diluer le médicament dans un verre d’eau ou un verre de boisson non alcoolisée ; dans ce cas, buvez tout le contenu du verre pour vous assurer que vous avez pris la bonne dose de produit (Fig 5).
|
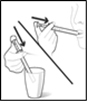
Laissez l’adaptateur dans le flacon, fermez bien le flacon et conservez le dans la position verticale. Rincez la seringue avec de l’eau après chaque utilisation et laissez la sécher. Lors des prises suivantes, placez la seringue dans l’adaptateur fixé au goulot du flacon et suivez les instructions fournies précédemment.
Pendant combien de temps devez-vous prendre Palexia 20 mg/ml, solution buvable ?
Il ne faut pas prendre les médicaments plus longtemps que la période indiquée par le médecin. Chez l’enfant, la durée du traitement ne doit pas dépasser 3 jours.
Si vous avez pris plus de PALEXIA 20 mg/ml, solution buvable que vous n’auriez dû
Après la prise de doses très fortes, vous pourriez présenter les effets suivants :
· un rétrécissement des pupilles, des vomissements, une chute de la pression artérielle, une accélération des battements cardiaques, un état de choc, des troubles de la conscience allant jusqu'au coma (inconscience profonde), des crises d'épilepsie et une respiration dangereusement lente ou faible ou un arrêt respiratoire.
Si cela vous arrive, il faut immédiatement appeler un médecin !
Si vous oubliez de prendre PALEXIA 20 mg/ml, solution buvable
Si vous arrêtez de prendre PALEXIA 20 mg/ml, solution buvable
Si vous interrompez ou arrêtez trop rapidement le traitement, la douleur risque de réapparaître. Si vous désirez arrêter le traitement, consultez votre médecin avant d’arrêter.
L'arrêt du traitement n’entraîne en général pas d’effet. Cependant, dans de rares cas, les personnes ne se sentent pas bien quand elles arrêtent brutalement le traitement après avoir pris ce médicament pendant quelques temps.
Les symptômes peuvent être :
- agitation, larmoiement, nez qui coule, bâillement, transpiration, frissons, douleur musculaire et pupille dilatée.
- Irritabilité, anxiété, mal de dos, douleur articulaire, faiblesse, crampes abdominales, difficulté à dormir, nausées, perte d’appétit, vomissements, diarrhée et augmentations de la pression artérielle, de la fréquence de la respiration ou cardiaque.
Si vous présentez l'un de ces troubles après l'arrêt du traitement, consultez votre médecin.
Vous ne devez pas arrêter votre traitement soudainement sans que votre médecin vous y ait autorisé. Si votre médecin souhaite que vous arrêtiez le traitement, il/elle vous dira comment procéder. Cela pourra inclure une réduction progressive des doses.
Si vous avez d'autres questions à propos de l'utilisation de ce médicament, consultez votre médecin ou votre pharmacien.
4. QUELS SONT LES EFFETS INDESIRABLES EVENTUELS ?
Effets indésirables importants ou symptômes à surveiller et ce qu’il faut faire en cas de survenue :
Ce médicament peut entrainer des réactions allergiques. Les symptômes peuvent être une respiration sifflante, des difficultés à respirer, un gonflement des paupières, du visage ou des lèvres, une éruption cutanée ou des démangeaisons principalement celles touchant tout le corps.
Un autre effet indésirable grave est un état dans lequel vous êtes très somnolent et respirez plus lentement ou moins profondément que d’habitude. Cela apparait plus fréquemment chez les patients âgés ou affaiblis.
Si vous êtes concerné par ces effets indésirables, contactez immédiatement votre médecin.
Autres effets indésirables qui peuvent apparaitre :
Très fréquent (peut concerner plus de 1 patient sur 10) : nausées, vomissements, sensations de vertige, somnolence, maux de tête.
Fréquent (peut concerner 1 à 10 patients sur 100) : diminution de l’appétit, anxiété, confusion, hallucination, troubles du sommeil, rêves anormaux, tremblements, flush (accès de rougeurs), constipation, diarrhée, indigestion, bouche sèche, démangeaisons, augmentation de la transpiration, éruption cutanée, crampes musculaires, sensation de faiblesse, fatigue, impression de changement de la température du corps.
Peu fréquent (peut concerner 1 à 10 patients sur 1 000) : humeur dépressive, désorientation, excitabilité (agitation), nervosité, impatience, euphorie, troubles de l’attention, troubles de la mémoire, état proche de l’évanouissement, sédation, difficulté à contrôler ses mouvements, difficultés à parler, engourdissement, sensations anormales de la peau (ex picotements, fourmillements), secousses musculaires, vision anormale, augmentation du rythme cardiaque, palpitations, diminution de la pression sanguine, respiration dangereusement lente ou faible (dépression respiratoire), diminution de l’oxygène dans le sang, essoufflement, gêne abdominale, urticaire, sensation de lourdeur, gêne pour uriner, urines fréquentes, syndrome de sevrage (voir rubrique « Si vous arrêtez de prendre Palexia 20 mg/ml, solution buvable »), accumulation d’eau dans les tissus (œdème), sensation d’être anormal, impression d’être ivre, irritabilité, sensation de relâchement.
Rare (peut concerner 1 à 10 patients sur 10 000) : réaction allergique aux médicaments (incluant gonflement sous la peau, et urticaire, et dans des cas sévères : difficultés à respirer, une chute de la pression sanguine, perte de connaissance, ou choc), pensée anormale, crise d’épilepsie, diminution du niveau de conscience, coordination anormale des membres, diminution de la fréquence cardiaque, troubles de la vidange gastrique.
Fréquence indéterminée : délire
La probabilité d’avoir des idées et un comportement suicidaires est généralement augmentée chez les patients souffrant de douleur chronique. De plus, certains médicaments pour le traitement de la dépression (qui ont un impact sur le système des neurotransmetteurs dans le cerveau) peuvent augmenter ce risque, surtout au début du traitement. Bien que le tapentadol ait aussi des effets sur les neurotransmetteurs, les données d’utilisation du tapentadol chez l’humain ne mettent pas en évidence d’augmentation du risque de suicide.
Aucun effet indésirable supplémentaire n’a été observé chez les enfants et les adolescents.
Déclaration des effets secondaires
Si vous ressentez un quelconque effet indésirable, parlez-en à votre médecin ou votre pharmacien. Ceci s’applique aussi à tout effet indésirable qui ne serait pas mentionné dans cette notice. Vous pouvez également déclarer les effets indésirables directement via le système national de déclaration : Agence nationale de sécurité du médicament et des produits de santé (ANSM) et réseau des Centres Régionaux de Pharmacovigilance - Site internet: www.signalement-sante.gouv.fr.
En signalant les effets indésirables, vous contribuez à fournir davantage d’informations sur la sécurité du médicament.
5. COMMENT CONSERVER PALEXIA 20 mg/ml, solution buvable ?
Tenir ce médicament hors de la vue et de la portée des enfants.
N’utilisez pas ce médicament après la date de péremption indiquée sur le carton et le flacon après la mention
Flacon non ouvert : Pas de conditions particulières de conservation.
La durée de conservation après ouverture est de 6 semaines.
Après ouverture, conserver le flacon à l’endroit, à la verticale.
Ne jetez aucun médicament au tout-à-l’égout ou avec les ordures ménagères. Demandez à votre pharmacien d’éliminer les médicaments que vous n’utilisez plus. Ces mesures contribueront à protéger l’environnement.
6. CONTENU DE L’EMBALLAGE ET AUTRES INFORMATIONS
Ce que contient PALEXIA 20 mg/ml, solution buvable
· La substance active est :
Tapentadol ..................................................................................................................... 20 mg
Sous forme de chlorhydrate de tapentadol
Pour 1 ml de solution.
· Les autres composants sont : Acide citrique monohydraté, Sucralose (E955), arôme framboise (contenant du propylène glycol [E 1520]), hydroxide de sodium (pour ajustement du pH), eau purifiée.
Qu’est-ce que PALEXIA 20 mg/ml, solution buvable et contenu de l’emballage extérieur
Ce médicament est une solution buvable limpide et incolore.
Palexia 20 mg/ml solution buvable est conditionné dans des flacons en plastique de 100 millilitres ou 200 millilitres, et est fourni avec une seringue pour administration orale avec une graduation de 0,1 ml et un adaptateur fixé à la seringue. De plus, l’échelle droite montre les doses unitaires pour l’adulte.
Toutes les présentations peuvent ne pas être commercialisées.
Titulaire de l’autorisation de mise sur le marché
IMMEUBLE EUREKA
19, RUE ERNEST RENAN – CS 90001
92024 NANTERRE CEDEX
Exploitant de l’autorisation de mise sur le marché
LABORATOIRES GRÜNENTHAL
IMMEUBLE EUREKA
19, RUE ERNEST RENAN – CS 90001
92024 NANTERRE CEDEX
ZIEGLERSTRASS 6
52078 AACHEN
ALLEMAGNE
Noms du médicament dans les Etats membres de l'Espace Economique Européen
Ce médicament est autorisé dans les Etats membres de l'Espace Economique Européen sous les noms suivants : Conformément à la réglementation en vigueur.
[À compléter ultérieurement par le titulaire]
La dernière date à laquelle cette notice a été révisée est :
[à compléter ultérieurement par le titulaire]
< {MM/AAAA}>< {mois AAAA}.>
Des informations détaillées sur ce médicament sont disponibles sur le site Internet de l’ANSM (France).