Dernière mise à jour le 03/08/2026
OCTAPLEX 1000 UI, poudre et solvant pour solution pour perfusion
Indications thérapeutiques
OCTAPLEX est utilisé pour traiter et prévenir les saignements :
· provoqués par des médicaments appelés anti-vitamine K (comme la Warfarine). Ces médicaments bloquent l’effet de la vitamine K et entraînent une diminution des facteurs de coagulation vitamine K dépendants dans votre organisme. OCTAPLEX est utilisé quand une correction rapide de cette diminution est nécessaire.
· chez les personnes nées avec un déficit en facteurs de coagulation vitamine K dépendants II et X et est utilisé dans le cas où aucun facteur de coagulation spécifique de haute pureté n'est disponible.
Composition en substances actives
-
Poudre ( Composition pour un flacon )
- > facteur II de coagulation humain 560 - 1520 UI
- > facteur VII de coagulation humain 360 - 960 UI
- > facteur IX de coagulation humain 1000 UI
- > facteur X de coagulation humain 720 - 1200 UI
- > protéine C humaine 520 - 1240 UI
- > protéine S 480 - 1280 UI
-
Solvant ( Composition )
- > Pas de substance active.
Présentations
> 1 flacon(s) poudre en verre - 1 flacon(s) solvant en verre de 40 mL avec dispositif(s) de transfert Nextaro®
Code CIP : 34009 550 151 0 9
Déclaration de commercialisation : 11/08/2017
Cette présentation est agréée aux collectivités
Service médical rendu (SMR)
Les libellés affichés ci-dessous ne sont que des résumés ou extraits issus des avis rendus par la Commission de la Transparence. Seul l'avis complet de la Commission de la Transparence fait référence.
Cet avis est consultable à partir du lien `Avis du jj/mm/aaaa` ou encore sur demande auprès de la HAS (plus d'informations dans l'aide). Les avis et synthèses d'avis contiennent un paragraphe sur la place du médicament dans la stratégie thérapeutique.
| Valeur du SMR | Avis | Motif de l'évaluation | Résumé de l'avis |
|---|---|---|---|
| Important | Avis du 25/05/2016 | Inscription (CT) | Le service médical rendu par OCTAPLEX 1000 UI, poudre et solvant pour solution pour perfusion est important dans les indications de l’AMM. |
Amélioration du service médical rendu (ASMR)
Les libellés affichés ci-dessous ne sont que des résumés ou extraits issus des avis rendus par la Commission de la Transparence. Seul l'avis complet de la Commission de la Transparence fait référence.
Cet avis est consultable à partir du lien `Avis du jj/mm/aaaa` ou encore sur demande auprés de la HAS (plus d'informations dans l'aide). Les avis et synthéses d'avis contiennent un paragraphe sur la place du médicament dans la stratégie thérapeutique.
| Valeur de l'ASMR | Avis | Motif de l'évaluation | Résumé de l'avis |
|---|---|---|---|
| V (Inexistant) | Avis du 25/05/2016 | Inscription (CT) | Cette spécialité est un complément de gamme qui n’apporte pas d’amélioration du service médical rendu (ASMR V) par rapport à la présentation déjà inscrite. |
Autres informations
- Titulaire de l'autorisation : OCTAPHARMA France
- Conditions de prescription et de délivrance :
- liste I
- prescription hospitalière
- Statut de l'autorisation : Valide
- Type de procédure : Procédure décentralisée
- Code CIS : 6 787 723 4
ANSM - Mis à jour le : 17/04/2025
OCTAPLEX 1000 UI, poudre et solvant pour solution pour perfusion
2. COMPOSITION QUALITATIVE ET QUANTITATIVE
OCTAPLEX se présente sous forme de poudre et solvant pour solution pour perfusion contenant un complexe prothrombique humain. OCTAPLEX contient nominalement :
|
Nom du composant |
OCTAPLEX 1000 UI (UI) Quantité par flacon de 40 mL |
OCTAPLEX Quantité (UI/mlL) après reconstitution avec l’eau pour préparations injectables |
|
Facteur II de coagulation humain |
560 ‑1520 |
14 ‑ 38 |
|
Facteur VII de coagulation humain |
360 ‑ 960 |
9 ‑ 24 |
|
Facteur IX de coagulation humain |
1000 |
25 |
|
Facteur X de coagulation humain |
720 ‑ 1200 |
18 ‑ 30 |
|
Protéine C |
520 ‑ 1240 |
13 ‑ 31 |
|
Protéine S |
480 ‑ 1280 |
12 ‑ 32 |
Le contenu en protéines totales par flacon est 520 - 1640 mg (flacon de 1 000 UI). L’activité spécifique du produit est ≥ 0,6 UI/mg de protéines, exprimée en activité du facteur IX.
Excipients à effet notoire : sodium (150 - 250 mg/flacon de 1 000 UI); héparine (200 - 500 UI/flacon de 1 000 UI soit 0,2 - 0,5 UI/UI FIX)
Pour la liste complète des excipients, voir rubrique 6.1.
Poudre et solvant pour solution pour perfusion.
La poudre est d’une couleur blanc-bleuté.
Le solvant est un liquide limpide et incolore.
4.1. Indications thérapeutiques
· Traitement des saignements et prophylaxie péri-opératoire des accidents hémorragiques lors d'un déficit acquis en facteurs de coagulation du complexe prothrombique, tel que le déficit induit par un traitement par anti-vitamines K, ou en cas de surdosage en anti-vitamines K, quand une correction urgente du déficit est requise.
· Traitement des saignements et prophylaxie péri-opératoire lors d'un déficit congénital en l’un des facteurs de coagulation vitamine K dépendants II et X, lorsque aucun facteur de coagulation spécifique de haute pureté n’est disponible.
4.2. Posologie et mode d'administration
Posologie
Les posologies recommandées ci-dessous sont données à titre indicatif. Le traitement doit être initié sous la surveillance d’un médecin spécialiste des troubles de la coagulation. La posologie et la durée du traitement de substitution dépendent de la sévérité du trouble, de la localisation et de l'intensité de l'accident hémorragique, ainsi que de l'état clinique du patient.
La dose et la fréquence d’administration doivent être calculées sur la base de chaque cas individuel. L’intervalle entre les administrations doit être adapté aux demi-vies, dans la circulation sanguine, des différents facteurs de coagulation du complexe prothrombique (voir rubrique 5.2).
La posologie individuelle peut uniquement être déterminée sur la base d’évaluations régulières limitées aux taux plasmatiques des facteurs de coagulation à corriger, ou sur la base d’évaluations globales des taux de complexe prothrombique (temps de prothrombine, INR), et en fonction du suivi continu de l’état clinique du patient.
Dans le cas d'interventions chirurgicales majeures, il est essentiel de réaliser un suivi précis du traitement de substitution à l'aide de tests de coagulation (tests spécifiques évaluant les facteurs de coagulation et/ou tests globaux évaluant les taux de complexe prothrombique).
Saignements et prophylaxie péri-opératoire des accidents hémorragiques lors d'un traitement par anti-vitamines K :
La dose nécessaire dépend de l'INR déterminé avant le traitement et du poids corporel.Le tableau suivant donne les doses approximatives (unités de produit reconstitué/kg de poids corporel)
|
2 à < 4 |
4 à 6 |
> 6 |
|
|
Dose d’Octaplex (unitésde facteur IX) / kg de poids corporel |
25 |
35 |
50 |
Les unités font référence aux Unités Internationales.
La dose est définie en fonction du poids jusqu’à un poids corporel de 100 kg maximum.
Pour les patients pesant plus de 100 kg, la dose maximale administrée en une fois (UI de facteur IX) ne devra donc pas dépasser 2 500 UI pour un INR de 2 à < 4, 3 500 UI pour un INR de 4 à 6 et 5 000 UI pour un INR > 6.
La correction de la diminution de l'hémostase induite par les anti-vitamines K dure environ 6 à 8 heures. Toutefois, les effets de la vitamine K, si elle est administrée simultanément, sont habituellement obtenus en 4 à 6 heures. Aussi, un traitement répété par le complexe prothrombique humain n'est généralement pas nécessaire lorsque la vitamine K a été administrée.
Comme ces recommandations sont empiriques et que le taux de récupération et la durée de l'effet peuvent varier, le suivi de l'INR durant le traitement est obligatoire.
Saignements et prophylaxie péri-opératoire lors de déficit congénital des facteurs de coagulation vitamine K dépendants II et X, lorsque aucun facteur de coagulation spécifique n’est disponible :
Le calcul de la dose nécessaire pour le traitement repose sur la notion empirique qu’approximativement 1 UI de facteur II ou de facteur X par kg de poids corporel élève l'activité plasmatique du facteur II ou X de 0,02 et 0,017 UI/mL respectivement.
La posologie d'un facteur spécifique administré est exprimée en Unités Internationales (UI) par rapport au standard de l'OMS en vigueur pour chaque facteur. L'activité plasmatique d’un facteur de coagulation spécifique est exprimée soit en pourcentage (de l’activité normale du plasma humain) soit en Unités Internationales (par rapport au standard international pour le facteur de coagulation spécifique).
Une Unité Internationale (UI) de l'activité d’un facteur de coagulation correspond à la quantité de ce facteur contenue dans un mL de plasma humain normal.
Par exemple, la détermination de la posologie requise en facteur X est basée sur la notion empirique que 1 Unité Internationale (UI) de facteur X par kg de poids corporel augmente l'activité plasmatique de facteur X de 0,017 UI/mL. La posologie requise est déterminée en utilisant la formule suivante :
Unités requises = poids corporel (kg) x augmentation souhaitée en facteur X (UI/mL) x 60
où 60 (mL/kg) correspond à l'inverse du taux de récupération estimé.
Posologie requise pour le facteur II:
Unités requises = poids corporel (kg) x augmentation souhaitée en facteur II (UI/mL) x 50
Si le taux de récupération individuel est connu, cette valeur doit être utilisée pour le calcul.
Population pédiatrique
Aucune donnée n’est disponible concernant l’utilisation d’OCTAPLEX dans la population pédiatrique.
Mode d’administration
OCTAPLEX doit être administré par voie intraveineuse. La perfusion être administrée au rythme de 0,12 mL/kg/min (environ 3 unités/kg/min), sans dépasser un débit maximal de 8 mL/min (environ 210 unités/min), en respectant les conditions d'asepsie.
· Hypersensibilité aux substances actives ou à l’un des excipients mentionnés à la rubrique 6.1.
· Hypersensibilité à I'héparine ou à ses dérivés incluant les HBPM.
· Antécédent de thrombopénie induite par I'héparine (ou TIH) grave de type II.
· Déficit en IgA avec anticorps connus contre les IgA.
4.4. Mises en garde spéciales et précautions d'emploi
Traçabilité
Afin d’améliorer la traçabilité des médicaments biologiques, le nom et le numéro de lot du produit administré doivent être clairement enregistrés.
L'avis d'un spécialiste des troubles de la coagulation doit être demandé.
Chez les patients présentant un déficit acquis en facteurs de coagulation vitamine K dépendants (par exemple induit par un traitement par anti-vitamines K), OCTAPLEX doit être utilisé uniquement lorsqu’une correction rapide du taux de complexe prothrombique est nécessaire, par exemple en cas d’hémorragie majeure ou d’urgence chirurgicale. Dans les autres cas, la diminution de la posologie des anti-vitamines K et/ou l'administration de vitamine K est habituellement suffisante.
Les patients traités par un anti-vitamine K peuvent présenter un état d’hypercoagulabilité sous-jacente qui peut être potentialisé par l’administration de concentré de complexe prothrombique.
En cas de réactions allergiques ou de type anaphylactique, la perfusion doit être immédiatement arrêtée. En cas de choc, le traitement symptomatique actuel de l'état de choc devra être instauré.
Les mesures standards pour prévenir les infections résultant de l'utilisation de médicaments préparés à partir de sang ou de plasma humain incluent la sélection des donneurs, la recherche de marqueurs spécifiques d'infection sur les dons individuels et les mélanges de plasma et l'inclusion d'étapes de fabrication efficaces pour l'inactivation/élimination virale.
Cependant, lorsque des médicaments préparés à partir de sang ou de plasma humain sont administrés, la possibilité de transmission d'agents infectieux ne peut être totalement exclue. Ceci s'applique aussi aux virus inconnus ou émergents et à d'autres agents pathogènes.
Les mesures prises sont considérées comme efficaces vis-à-vis des virus enveloppés tels que le virus de l’immunodéficience humaine (VIH), le virus de l’hépatite B (VHB) et le virus de l’hépatite C (VHC). Les mesures prises peuvent être d’efficacité limitée vis-à-vis de certains virus non-enveloppés tels que le virus de l’hépatite A (VHA) et le parvovirus B19. L’infection par le parvovirus B19 peut être sévère chez la femme enceinte (avec infection du fœtus) et chez les personnes atteintes de déficits immunitaires ou d’une augmentation de l’érythropoïèse (par exemple, l’anémie hémolytique).
Une vaccination appropriée (hépatite A et B) est recommandée pour les patients recevant de façon régulière/répétée des complexes prothrombiques dérivés du plasma humain.
Il est fortement recommandé, à chaque injection de OCTAPLEX, de relever le nom et le numéro de lot du produit afin de conserver la traçabilité entre le patient et le lot du produit.Il existe un risque de thrombose ou de coagulation intravasculaire disséminée lorsque des patients présentant un déficit congénital ou acquis sont traités par du complexe prothrombique humain, particulièrement en cas d’administrations répétées. Les patients traités par un complexe prothrombique humain doivent être surveillés étroitement pour détecter les signes ou symptômes évocateurs d'une coagulation intravasculaire disséminée ou d'une thrombose. En raison du risque de complications thromboemboliques, un suivi étroit doit être réalisé lorsque un complexe prothrombique humain est administré aux patients présentant des antécédents de maladie coronarienne, aux patients atteints de maladie hépatique, aux patients en période péri- ou post-opératoire, aux nouveau-nés ou aux patients présentant un risque de manifestations thromboemboliques ou de coagulation intravasculaire disséminée. Dans chacune de ces situations, le bénéfice potentiel du traitement doit être évalué par rapport aux risques de ces complications.
Aucune donnée n'est disponible sur l'utilisation d'OCTAPLEX lors de saignement périnatal faisant suite à un déficit en vitamine K chez le nouveau-né.
Ce médicament contient 150 à 250 mg de sodium par flacon (flacon de 1 000 UI), ce qui équivaut à 7,5 - 12,5% de l’apport alimentaire quotidien maximal recommandé par l’OMS de 2 g de sodium par adulte.
Ce médicament contient de I'héparine et peut provoquer des réactions allergiques, des thrombopénies immunoallergiques graves de type II (TIH) et des troubles de la coagulation.
En cas de déficit congénital de l’un des facteurs vitamine K dépendants, le facteur de coagulation spécifique doit être utilisé quand il est disponible.
4.5. Interactions avec d'autres médicaments et autres formes d'interactions
Le complexe prothrombique humain neutralise les effets d'un traitement par anti-vitamines K, mais aucune interaction avec d’autres spécialités pharmaceutiques n'est connue.
Associations à prendre en compte
Interférence avec des tests biologiques :
Lorsque des tests de coagulation sensibles à l'héparine sont réalisés chez les patients recevant de hautes doses de complexe prothrombique humain, l'héparine contenue dans le produit administré doit être prise en compte.
4.6. Fertilité, grossesse et allaitement
La sécurité du complexe prothrombique chez la femme enceinte et lors de l’allaitement n’a pas été établie.
Les études chez l’animal ne permettent pas d’établir la sécurité pendant la grossesse, le développement embryo-foetal, l’accouchement ou le développement post-natal. Aussi, le complexe prothrombique humain ne doit être administré au cours de la grossesse et de l’allaitement qu’en cas de nécessité absolue.
4.7. Effets sur l'aptitude à conduire des véhicules et à utiliser des machines
Aucune étude sur les effets sur l’aptitude à conduire des véhicules ou à utiliser des machines n’a été réalisée.
Résumé du profil de sécurité
Le traitement de substitution peut provoquer la formation d’anticorps circulants inhibant un ou plusieurs facteurs du complexe prothrombique humain. La présence de tels inhibiteurs se traduit par une mauvaise réponse clinique.
Des réactions allergiques ou de type anaphylactique peuvent survenir dans de rares cas (≥1/10 000 et <1/1 000), dont des réactions anaphylactiques.
Une augmentation de la température corporelle a été observée dans de très rares cas (<1/10 000).
Il existe un risque de manifestations thromboemboliques après l'administration de complexe prothrombique humain (voir rubrique 4.4).
Liste des réactions indésirables d’Octaplex présentée sous la forme de tableau
Le tableau présenté ci-dessous est conforme à la classification MedDRA par système d’organe (SOC et terme préférentiel). Les fréquences sont basées sur les données issues des études cliniques, en respectant la convention suivante : très fréquent (≥ 1/10) ; fréquent (≥ 1/100 à < 1/10) ; peu fréquent (≥ 1/1 000 à < 1/100) ; rare (≥ 1/10 000 à < 1/1 000) ; très rare (< 1/10 000) ou fréquence indéterminée (ne peut être estimée sur la base des données disponibles).
|
Classe de systèmes d’organes standards MedDRA |
Effets indésirables |
Fréquence |
|
Affections psychiatriques |
Anxiété |
peu fréquent |
|
Affections vasculaires |
Thrombose veineuse profonde |
fréquent |
|
Thrombose |
peu fréquent |
|
|
Hypertension |
peu fréquent |
|
|
Affections respiratoires, thoraciques et médiastinales |
Embolie pulmonaire |
peu fréquent |
|
Bronchospasme |
peu fréquent |
|
|
Hémoptysie |
peu fréquent |
|
|
Épistaxis |
peu fréquent |
|
|
Troubles généraux et anomalies au site d’administration |
Brûlures au site d'injection |
peu fréquent |
|
Investigations |
Augmentation des D-dimères (fibrine) |
peu fréquent |
|
Augmentation de la thrombine dans le sang |
peu fréquent |
|
|
Fonction hépatique anormale |
peu fréquent |
|
|
Lésions, intoxications et complications liées aux procédures |
Thrombose dans le dispositif |
peu fréquent |
Les effets indésirables suivants ont été rapportés lors de l’utilisation d’Octaplex après sa commercialisation. Étant donné que, après la commercialisation du produit, ces réactions indésirables sont rapportées volontairement et par une population dont la taille est incertaine, il n’est pas possible d’estimer de manière fiable la fréquence de ces réactions.
|
Affections du système immunitaire Choc anaphylactique, hypersensibilité |
|
Affections du système nerveux Tremblements |
|
Affections cardiaques Arrêt cardiaque, tachycardie |
|
Affections vasculaires Collapsus circulatoire, hypotension |
|
Affections respiratoires, thoraciques et médiastinales Dyspnée, insuffisance respiratoire |
|
Affections gastro-intestinales Nausées |
|
Affections de la peau et du tissu sous-cutané Urticaire, éruption cutanée |
|
Troubles généraux et anomalies au site d’administration Frissons |
OCTAPLEX contient de l’héparine. Par conséquent, une réaction allergique soudaine, déclenchant une diminution de la numération des plaquettes sanguines à une valeur inférieure à 100 000/µl ou de 50% de la numération initiale (thrombocytopénie de type II), peut être, dans de rares cas, observée. Chez les patients sans antécédents d'hypersensibilité à l'héparine, cette thrombocytopénie peut survenir 6 à 14 jours après le début du traitement. Chez les patients ayant des antécédents d'hypersensibilité à l'héparine, cette réaction peut survenir quelques heures après le début du traitement.
Le traitement avec OCTAPLEX doit être immédiatement interrompu chez les patients présentant cette réaction allergique. Ces patients ne devront plus recevoir des médicaments contenant de l’héparine à l’avenir.
Pour la sécurité virale, voir 4.4.
Population pédiatrique
Aucune donnée n’est disponible concernant l’utilisation d’Octaplex dans la population pédiatrique.
Déclaration des effets indésirables suspectés
La déclaration des effets indésirables suspectés après autorisation du médicament est importante. Elle permet une surveillance continue du rapport bénéfice/risque du médicament. Les professionnels de santé déclarent tout effet indésirable suspecté via le système national de déclaration : Agence nationale de sécurité du médicament et des produits de santé (ANSM) et réseau des Centres Régionaux de Pharmacovigilance - Site internet : https://signalement.social-sante.gouv.fr/.
L'utilisation de doses élevées de complexe prothrombique humain a été associée à des cas d’infarctus du myocarde, de coagulation intravasculaire disséminée, de thrombose veineuse ou d’embolie pulmonaire. Aussi, en cas de surdosage, le risque de développement de complications thromboemboliques ou de coagulation intravasculaire disséminée est augmenté.
5. PROPRIETES PHARMACOLOGIQUES
5.1. Propriétés pharmacodynamiques
Classe pharmacothérapeutique : antihémorragiques, facteurs de coagulation sanguine II, VII, IX et X en association. Code ATC : B02BD01.
Les facteurs de coagulation II, VII, IX et X qui sont synthétisés dans le foie à l'aide de la vitamine K, forment le complexe prothrombique.
Le facteur VII est le zymogène du facteur VIIa (sérine protéase active) par lequel la voie extrinsèque de la coagulation est activée. Le complexe facteur tissulaire-facteur VIIa active les facteurs de coagulation X et IX, entraînant la formation des facteurs Xa et IXa. Suite à d’autres activations de la cascade de coagulation, la prothrombine (facteur II) est activée et transformée en thrombine. Sous l’action de la thrombine, le fibrinogène est converti en fibrine ce qui aboutit à la formation du caillot.
La formation normale de thrombine est aussi d’une importance vitale pour la fonction plaquettaire dans l’hémostase primaire.
Un déficit sévère et isolé en facteur VII entraîne une baisse de la formation de thrombine et une tendance aux hémorragies dues au défaut de formation de fibrine et une diminution de l’hémostase primaire. Un déficit isolé en facteur IX est une des hémophilies classiques (hémophilie B). Les déficits isolés en facteur II ou X sont très rares mais dans les formes sévères se traduisent par des tendances hémorragiques similaires à celles observées dans l’hémophilie classique.
Un déficit acquis en facteurs de coagulation vitamine K dépendants survient lors de traitement par les anti-vitamines K. Quand le déficit devient sévère, une forte tendance hémorragique se développe, caractérisée par des saignements rétro-péritonéaux ou cérébraux, plutôt que des hémorragies musculaires et articulaires. Une insuffisance hépatique sévère peut également provoquer une baisse marquée des facteurs de la coagulation vitamine K dépendants, ainsi qu'un tableau clinique hémorragique qui est souvent complexe, dû à la fois à une coagulation intravasculaire continue, des taux de plaquettes réduits, un déficit en inhibiteurs de la coagulation et une fibrinolyse perturbée.
L’administration de complexe prothrombique humain augmente le taux plasmatique des facteurs de coagulation vitamine K dépendants et peut corriger temporairement le défaut de coagulation chez les patients présentant un déficit d’un ou plusieurs de ces facteurs.
5.2. Propriétés pharmacocinétiques
Absorption
Les demi-vies plasmatiques sont de l’ordre de :
|
Facteur de coagulation |
Demi-vie |
|
Facteur II |
48 - 60 heures |
|
Facteur VII |
1,5 - 6 heures |
|
Facteur IX |
20 - 24 heures |
|
Facteur X |
24 - 48 heures |
Distribution
OCTAPLEX est administré par voie intraveineuse et est donc immédiatement disponible dans l’organisme.
5.3. Données de sécurité préclinique
Il n’existe aucune donnée de sécurité préclinique pertinente pour la sécurité clinique, à l’exception des données présentées dans les autres sections du RCP.
Poudre :
Héparine : 0,2 - 0,5 UI/UI FIX
Citrate de sodium dihydraté
Solvant :
Eau pour préparations injectables
Ce médicament ne doit pas être mélangé avec d’autres produits ou d’autres médicaments a l’exception de ceux mentionnés dans la rubrique 6.6.
3 ans.
La stabilité chimique et physique a été démontrée pendant un maximum de 8 heures à une température de +25°C.
Du point de vue microbiologique, le produit doit être utilisé immédiatement ą moins que les conditions d'ouverture et de reconstitution permettent d'écarter un risque de contamination. S’il n’est pas utilisé immédiatement, la durée et les conditions de conservation en cours d’utilisation sont de la responsabilité de l’utilisateur.
6.4. Précautions particulières de conservation
A conserver à une température ne dépassant pas +25°C.
Ne pas congeler.
A conserver dans l’emballage d’origine à l’abri de la lumière.
Pour les conditions de conservation du médicament après reconstitution, voir la rubrique 6.3.
6.5. Nature et contenu de l'emballage extérieur
Une boîte contient :
· Poudre en flacon (verre de type I) muni d’un bouchon (caoutchouc halobutyle) et d’un sertissage (aluminium) avec un opercule amovible (plastique)
· 40 mL d’eau pour préparations injectables en flacon (verre de type I) muni d’un bouchon (caoutchouc halobutyle) et d’un sertissage (aluminium) avec un opercule amovible (plastique)
· 1 nécessaire de transfert Nextaro®.
6.6. Précautions particulières d’élimination et de manipulation
Lire toutes les instructions et les suivre scrupuleusement !
Durant la procédure décrite ci-dessous, l’asepsie doit être maintenue !
Le produit se reconstitue rapidement à température ambiante.
La solution doit être limpide ou légèrement opalescente. Ne pas utiliser de solution trouble ou présentant un dépôt. Les produits reconstitués doivent être inspectés visuellement pour mettre en évidence la présence de particules et un changement de coloration avant administration.
Après reconstitution, la solution doit être utilisée immédiatement.
Tout produit non utilisé ou déchet doit être éliminé conformément à la réglementation en vigueur.
Instructions pour la reconstitution:
1. Si nécessaire, amenez le solvant (eau pour préparations injectables) et la poudre, dans les flacons fermés, à température ambiante. Cette température doit être maintenue pendant la reconstitution.
Si un bain-marie est utilisé pour chauffer, une attention doit être portée pour que l'eau n’entre pas en contact avec les bouchons en caoutchouc ou les opercules des flacons. La température du bain-marie ne doit pas dépasser 37°C.
2. Retirez les opercules détachables du flacon de poudre et du flacon de solvant et désinfectez les bouchons en caoutchouc de façon appropriée.
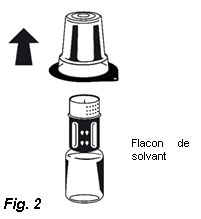
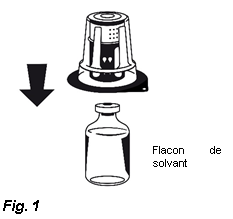
3. Retirez le couvercle de l'emballage externe du Nextaro®. Placez le flacon de solvant sur une surface plane et tenez-le fermement. Sans retirer l’emballage externe, placez la partie bleue du Nextaro® sur le dessus du flacon de solvant et appuyez fermement jusqu'à ce qu'il s'enclenche (Fig. 1). N’effectuez pas de rotation lors de la fixation ! Tout en maintenant le flacon de solvant, retirez soigneusement l'emballage externe du Nextaro® ; attention à bien laisser le Nextaro® fermement fixé au flacon de solvant (Fig. 2).
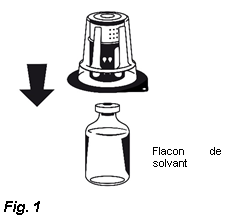 | |||
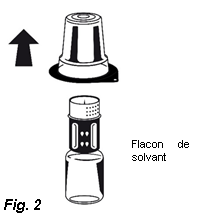 | |||
|
|
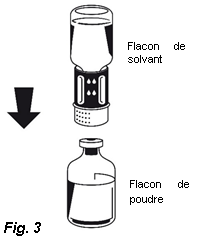
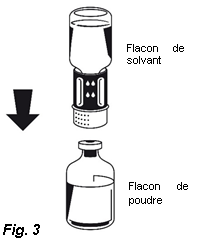
4. Placez le flacon de poudre sur une surface plane et le tenir fermement. Prenez le flacon de solvant avec le Nextaro® fixé et le retourner. Placez la partie blanche du connecteur Nextaro®sur le dessus du flacon de poudre et appuyez fermement jusqu'à ce qu'il s'enclenche (Fig. 3). Ne pas effectuer de rotation lors de la fixation ! Le solvant s'écoule automatiquement dans le flacon de poudre.
|
|
|
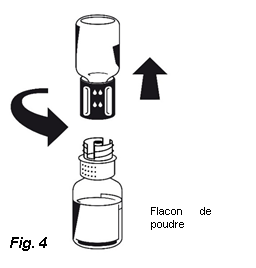
5. Les deux flacons toujours fixés, tournez doucement le flacon de poudre jusqu'à ce que le produit soit dissous. OCTAPLEX se dissout rapidement à température ambiante pour donner une solution incolore à légèrement bleue. Dévissez le Nextaro® en deux parties (Fig. 4).
Eliminez le flacon de solvant vide avec la partie bleue du Nextaro®.
|
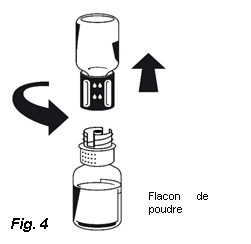
Si la poudre ne se dissout pas complètement ou si un agrégat s'est formé, ne pas utiliser la solution.
Instructions pour la perfusion :
Par précaution, le pouls du patient doit être pris avant et pendant la perfusion. En cas d'augmentation nette du pouls, la vitesse de perfusion doit être réduite ou l'administration doit être interrompue.
Fixez une seringue de 40 mL (1 000 UI) à la sortie Luer Lock sur la partie blanche du Nextaro®. Retournez le flacon et prélevez la solution dans la seringue. Dès que la solution a été transférée, tenez fermement le piston de la seringue (en la tenant tournée vers le bas) et retirez la seringue du Nextaro®. Eliminez le Nextaro® et le flacon vide.
Désinfectez le site d'injection de façon appropriée.
Injectez par voie intraveineuse au rythme de 0,12 mL/kg/min. (environ 3 unités/kg/min.) sans dépasser un débit maximal de 8 mL/min. (environ 210 unités/min.), en respectant les conditions d’asepsie.
Du sang ne doit pas refluer dans la seringue en raison du risque de formation de caillots de fibrine. Le Nextaro® est réservé à un usage unique.
7. TITULAIRE DE L’AUTORISATION DE MISE SUR LE MARCHE
OCTAPHARMA FRANCE
62 BIS AVENUE ANDRE MORIZET
92100 BOULOGNE BILLANCOURT
FRANCE
8. NUMERO(S) D’AUTORISATION DE MISE SUR LE MARCHE
· 34009 550 151 0 9 : Poudre en flacon (verre de type I) muni d'un bouchon (caoutchouc halobutyle) et d’un sertissage (aluminium) avec un opercule amovible (plastique) + 40 mL d’eau pour préparations injectables en flacon (verre de type I) muni d'un bouchon (caoutchouc halobutyle) et d'un sertissage (aluminium) avec un opercule amovible (plastique) + 1 nécessaire de transfert Nextaro®.
9. DATE DE PREMIERE AUTORISATION/DE RENOUVELLEMENT DE L’AUTORISATION
[à compléter ultérieurement par le titulaire]
10. DATE DE MISE A JOUR DU TEXTE
[à compléter ultérieurement par le titulaire]
Sans objet.
12. INSTRUCTIONS POUR LA PREPARATION DES RADIOPHARMACEUTIQUES
Sans objet.
Liste I
Médicament soumis à prescription hospitalière.
ANSM - Mis à jour le : 17/04/2025
OCTAPLEX 1000 UI, poudre et solvant pour solution pour perfusion
Complexe prothrombique humain
Veuillez lire attentivement cette notice avant d’utiliser ce médicament car elle contient des informations importantes pour vous.
· Gardez cette notice. Vous pourriez avoir besoin de la relire.
· Si vous avez d’autres questions, interrogez votre médecin, votre pharmacien ou votre infirmier/ère.
· Ce médicament vous a été personnellement prescrit. Ne le donnez pas à d’autres personnes. II pourrait leur être nocif, même si les signes de leur maladie sont identiques aux vôtres.
· Si vous ressentez un quelconque effet indésirable, parlez-en à votre médecin, votre pharmacien ou votre infirmier/ère. Ceci s’applique aussi à tout effet indésirable qui ne serait pas mentionné dans cette notice. Voir rubrique 4.
2. Quelles sont les informations à connaître avant d'utiliser OCTAPLEX 1000 UI, poudre et solvant pour solution pour perfusion ?
3. Comment utiliser OCTAPLEX 1000 UI, poudre et solvant pour solution pour perfusion ?
4. Quels sont les effets indésirables éventuels ?
5. Comment conserver OCTAPLEX 1000 UI, poudre et solvant pour solution pour perfusion ?
6. Contenu de l’emballage et autres informations.
1. QU’EST-CE QUE OCTAPLEX 1000 UI, poudre et solvant pour solution pour perfusion ET DANS QUELS CAS EST-IL UTILISE ?
OCTAPLEX est utilisé pour traiter et prévenir les saignements :
· provoqués par des médicaments appelés anti-vitamine K (comme la Warfarine). Ces médicaments bloquent l’effet de la vitamine K et entraînent une diminution des facteurs de coagulation vitamine K dépendants dans votre organisme. OCTAPLEX est utilisé quand une correction rapide de cette diminution est nécessaire.
· chez les personnes nées avec un déficit en facteurs de coagulation vitamine K dépendants II et X et est utilisé dans le cas où aucun facteur de coagulation spécifique de haute pureté n'est disponible.
2. QUELLES SONT LES INFORMATIONS A CONNAITRE AVANT D’UTILISER OCTAPLEX 1000 UI, poudre et solvant pour solution pour perfusion ?
N'utilisez jamais OCTAPLEX 1000 UI, poudre et solvant pour solution pour perfusion :
· si vous êtes allergique à l'un des composants contenus dans ce médicament (mentionnés dans la rubrique 6),
· si vous êtes allergique (hypersensible) à l'héparine ou à ses dérivés,
· si vous avez des antécédents de thrombopénie induite par I’héparine,
· si votre médecin vous a informé(e) que vous étiez allergique à I'héparine, contactez-Ie avant de prendre le médicament,
· si vous avez un déficit en IgA, avec des anticorps connus contre les IgA.
Avertissements et précautions
· Prenez conseil auprès d’un médecin spécialiste des troubles de la coagulation lorsque vous recevez OCTAPLEX
· Si vous présentez un déficit acquis en facteurs de coagulation vitamine K dépendants (induit par les antivitamines K par exemple). OCTAPLEX doit être administré uniquement lorsqu’une correction rapide de la diminution du taux de facteurs de coagulation vitamine K dépendants est requise, par exemple en cas d'hémorragie majeure ou d'urgence chirurgicale. Dans les autres cas, la diminution de la posologie des antivitamines K et/ou l'administration de vitamine K est généralement suffisante.
· Si vous êtes traité par un antagoniste de la vitamine K (comme la warfarine), vous avez un risque élevé de formation de caillots dans le sang. Dans ce cas, le traitement par OCTAPLEX peut augmenter ce risque.
· Si vous êtes né avec un déficit héréditaire de l'un des facteurs vitamine K dépendants, le facteur de coagulation spécifique doit être utilisé quand il est disponible.
· En cas de réaction allergique ou de type anaphylactique, votre médecin arrêtera immédiatement la perfusion et vous donnera un traitement approprié.
· Il existe un risque de thrombose ou de coagulation intravasculaire disséminée (maladie grave entraînant la formation de caillots de sang dans tout l’organisme) si vous êtes traité par OCTAPLEX (notamment si vous le recevez régulièrement). Vous devez être étroitement surveillé pour détecter les signes ou symptômes évocateurs d'une coagulation intravasculaire disséminée ou d'une thrombose.
· Ceci est particulièrement important si vous présentez des antécédents de maladie coronarienne, une maladie hépatique, si vous allez subir une opération et également lorsque OCTAPLEX est administré aux nouveau-nés.
· Aucune donnée n'est disponible sur l'utilisation d'OCTAPLEX lors de saignement périnatal faisant suite à un déficit en vitamine K chez le nouveau-né.
· Ce médicament contient de I'héparine et peut provoquer des réactions allergiques et une baisse du nombre de cellules sanguines, pouvant influer sur la coagulation.
· Ce médicament contient du sodium. Ce médicament contient 150 ‑ 250 mg de sodium par flacon de 1 000 UI.
· A prendre en compte chez les patients contrôlant leur apport alimentaire en sodium.
Sécurité virale :
· Lorsque des médicaments sont préparés à partir de sang ou de plasma humain, des mesures sont mises en place afin de prévenir le risque de transmission d’infection aux patients. Celles-ci comprennent une sélection rigoureuse des donneurs de sang et plasma afin d’exclure tout risque d’infection et la réalisation de tests de dépistage des marqueurs viraux/d’infection sur chaque don et sur les mélanges de plasma.
· Les fabricants de ces produits incluent également dans le traitement du sang ou du plasma, des étapes capables d'éliminer et/ou d'inactiver les virus. Malgré ces mesures, lorsque des médicaments préparés à partir de sang ou de plasma humain sont administrés, le risque de transmission d’agents infectieux ne peut être totalement exclu. Ceci s’applique également à des agents viraux inconnus ou émergents ou à d’autres types d’agents infectieux.
· Ces mesures sont considérées comme efficaces vis-à-vis des virus enveloppés tels que le virus de l’immunodéficience humaine (VIH), le virus de l’hépatite B (VHB) et le virus de l’hépatite C (VHC). Les mesures prises peuvent être d’efficacité limitée vis-à-vis des virus non enveloppés tels que le virus de l’hépatite A (VHA) et le parvovirus B19. L’infection par le parvovirus B19 peut être sévère chez la femme enceinte (avec infection du fœtus) et chez les personnes dont le système immunitaire est déficient ou atteints de certains types d’anémie (par exemple l’anémie hémolytique ou la drépanocytose).
· Une vaccination appropriée (hépatite A et B) vous est recommandée si vous recevez du complexe prothrombique dérivé du plasma humain de façon régulière/répétée.
Enfants et adolescents
Aucune donnée n’est disponible concernant l’utilisation d’OCTAPLEX chez les enfants et les adolescents.
Autres médicaments et OCTAPLEX 1000 UI, poudre et solvant pour solution pour perfusion
OCTAPLEX ne doit pas être mélangé à d’autres médicaments.
OCTAPLEX arrête l’effet des médicaments antagonistes de la vitamine K (comme la Warfarine) mais aucune interaction avec d’autres médicaments n’est connue.
OCTAPLEX peut impacter les résultats des tests de coagulation sensibles à l'héparine.
Informez votre médecin ou pharmacien si vous prenez, avez récemment pris ou pourriez prendre tout autre médicament.
OCTAPLEX 1000 UI, poudre et solvant pour solution pour perfusion avec des aliments et boissons
Sans objet.
Grossesse et allaitement
OCTAPLEX ne doit être utilisé pendant la grossesse ou l'allaitement qu’en cas de nécessité absolue.
Demandez conseil à votre médecin et à votre pharmacien avant de prendre tout médicament.
Conduite de véhicules et utilisation de machines
Les effets d’OCTAPLEX concernant l’aptitude à conduire des véhicules ou à utiliser des machines ne sont pas connus.
OCTAPLEX 1000 UI, poudre et solvant pour solution pour perfusion contient du sodium.
L’héparine peut provoquer des réactions allergiques et diminuer le nombre de cellules sanguines risquant ainsi d’affecter le système de coagulation du sang. Les patients ayant des antécédents de réactions allergiques induites par l’héparine doivent éviter d’utiliser des médicaments contenant de l’héparine.
Ce médicament contient 150 – 250 mg de sodium (composant principal du sel de cuisine/table) par flacon (flacon de 1 000 UI). Cela équivaut à 7,5 – 12,5 % de l’apport alimentaire quotidien maximal recommandé de sodium pour un adulte.
3. COMMENT UTILISER OCTAPLEX 1000 UI, poudre et solvant pour solution pour perfusion ?
Veillez à toujours utiliser ce médicament en suivant exactement les indications de votre médecin ou pharmacien. Vérifiez auprès de votre médecin ou pharmacien en cas de doute.
Le traitement par OCTAPLEX doit être initié sous la surveillance d'un médecin spécialiste des troubles de la coagulation.
Dans un premier temps, la solution est dissoute dans de l’eau ;
Puis, la solution est administrée dans une veine (voie intraveineuse).
La posologie et la durée du traitement dépendent :
· de la sévérité de votre pathologie,
· de la localisation et de l'intensité de l'accident hémorragique,
· de votre état clinique général.
Si vous avez utilisé plus de OCTAPLEX 1000 UI, poudre et solvant pour solution pour perfusion que vous n’auriez dû :
En cas de surdosage il y a un risque plus élevé de développer :
· des complications de la coagulation (par exemple, une attaque cardiaque et la formation de caillots dans vos veines ou vos poumons),
· une coagulation intravasculaire disséminée (une maladie grave entraînant la formation de caillots de sang dans tout l’organisme).
Si vous oubliez d’utiliser OCTAPLEX 1000 UI, poudre et solvant pour solution pour perfusion
Sans objet.
Si vous arrêtez d’utiliser OCTAPLEX 1000 UI, poudre et solvant pour solution pour perfusion
Sans objet.
Si vous avez d’autres questions sur l’utilisation de ce médicament, demandez plus d'informations à votre médecin, à votre pharmacien ou à votre infirmier/ère.
4. QUELS SONT LES EFFETS INDESIRABLES EVENTUELS ?
Fréquent (peut toucher jusqu’à 1 personne sur 10)
Caillots dans les vaisseaux sanguins.
Peu fréquent (peut toucher jusqu’à 1 personne sur 100)
Anxiété, augmentation de la pression artérielle, symptômes semblables à de l’asthme, toux avec crachats de sang, saignement de nez, brûlures au site d'injection, caillots dans le dispositif.
Rare (peut toucher jusqu’à 1 personne sur 1 000)
Des réactions de type allergique peuvent survenir. Une élévation transitoire des résultats des tests hépatiques (transaminases) a été observée dans de rares cas.
Les patients traités par OCTAPLEX pour un traitement de substitution peuvent développer des anticorps neutralisants (inhibiteurs) dirigés contre n’importe quel facteur de coagulation du complexe prothrombique. Si de tels inhibiteurs apparaissent, le traitement de substitution ne sera pas très efficace.
Très rare (peut toucher jusqu’à 1 personne sur 10 000)
Une augmentation de la température corporelle (fièvre) a été observée.
Il y a un risque de coagulation sanguine après administration de ce médicament.
Fréquence indéterminée (ne peut être estimée sur la base des données disponibles)
Grave réaction allergique et choc allergique, hypersensibilité, tremblements, insuffisance cardiaque, augmentation du rythme cardiaque, insuffisance circulatoire, chute de la tension artérielle, insuffisance respiratoire, difficultés à respirer, nausées, urticaire, éruption cutanée (rash), frissons.
L’héparine contenue dans la préparation peut provoquer une diminution soudaine du nombre de plaquettes dans le sang. C’est une réaction allergique appelée « thrombocytopénie de type II induite par l’héparine ». Dans de rares cas, chez des patients sans antécédents d'hypersensibilité à l'héparine, la diminution du nombre de plaquettes peut survenir 6 à 14 jours après le début du traitement. Chez les patients ayant des antécédents d'hypersensibilité à l'héparine, cette diminution peut survenir quelques heures après le début du traitement.
Le traitement avec OCTAPLEX doit être immédiatement interrompu chez les patients présentant cette réaction allergique. Ces patients ne devront plus recevoir des médicaments contenant de l’héparine à l’avenir.
Pour toute information sur la sécurité virale, voir la section 2.
Déclaration des effets secondaires
Si vous ressentez un quelconque effet indésirable, parlez-en à votre médecin, votre pharmacien ou à votre infirmier/ère. Ceci s’applique aussi à tout effet indésirable qui ne serait pas mentionné dans cette notice. Vous pouvez également déclarer les effets indésirables directement via le système national de déclaration : Agence nationale de sécurité du médicament et des produits de santé (ANSM) et réseau des Centres Régionaux de Pharmacovigilance - Site internet: https://signalement.social-sante.gouv.fr/.
En signalant les effets indésirables, vous contribuez à fournir davantage d’informations sur la sécurité du médicament.
5. COMMENT CONSERVER OCTAPLEX 1000 UI, poudre et solvant pour solution pour perfusion ?
Tenir hors de la vue et de la portée des enfants.
N’utilisez pas ce médicament après la date de péremption indiquée sur l’étiquette. La date de péremption fait référence au dernier jour de ce mois.
La poudre ne doit être dissoute qu’immédiatement avant l’injection. La stabilité de la solution a été démontrée pendant un maximum de 8 heures à une température comprise +25°C. Afin d’éviter toute contamination, la solution doit toutefois être utilisée immédiatement et une seule fois.
Ne jetez aucun médicament au tout à l’égout ni avec les ordures ménagères. Demandez à votre pharmacien d’éliminer les médicaments que vous n’utilisez plus. Ces mesures contribueront à protéger l’environnement.
6. CONTENU DE L’EMBALLAGE ET AUTRES INFORMATIONS
Ce que contient OCTAPLEX 1000 UI, poudre et solvant pour solution pour perfusion
· Les substances actives sont :
|
Nom du constituant |
OCTAPLEX Quantité par flacon de 40 mL |
OCTAPLEX Quantité par mL de solution reconstituée |
|
Protéines totales |
520 ‑ 1640 mg |
13 ‑ 41 mg/mL |
|
Facteur II de coagulation humain |
560 ‑ 1520 UI |
14 ‑ 38 UI/mL |
|
Facteur VII de coagulation humain |
360 ‑ 960 UI |
9 ‑ 24 UI/mL |
|
Facteur IX de coagulation humain |
1000 UI |
25 UI/mL |
|
Facteur X de coagulation humain |
720 ‑ 1200 UI |
18 ‑ 30 UI/mL |
|
Protéine C |
520 ‑ 1240 UI |
13 ‑ 31 UI/mL |
|
Protéine S |
480 ‑ 1280 UI |
12 ‑ 32 UI/mL |
L'activité spécifique du produit est ≥ 0,6 UI/mg, exprimée en activité du facteur IX.
· Les autres composants sont :
Héparine, citrate de sodium dihydraté, eau pour préparations injectables.
OCTAPLEX se présente sous la forme d’une poudre et d’un solvant pour solution pour perfusion. La poudre est hygroscopique, blanche ou légèrement colorée ou se présente sous la forme d’une masse friable contenu dans un flacon en verre. Le solvant est de l’eau pour préparations injectables et est fourni dans un flacon en verre. La solution reconstituée est claire ou légèrement opalescente et peut être colorée.
OCTAPLEX contient :
· 1 flacon de poudre pour solution pour perfusion,
· 1 flacon de solvant eau pour préparations injectables
· 1 dispositif de transfert Nextaro®
Titulaire de l’autorisation de mise sur le marché
62 BIS AVENUE ANDRE MORIZET
92100 BOULOGNE BILLANCOURT
FRANCE
Exploitant de l’autorisation de mise sur le marché
62 BIS AVENUE ANDRE MORIZET
92100 BOULOGNE BILLANCOURT
FRANCE
OCTAPHARMA PHARMAZEUTIKA PRODUKTIONSGES m.b.H
OBERLAAER STRASSE 235
1100 VIENNE
AUTRICHE
ou
OCTAPHARMA SAS
70-72, RUE DU MARECHAL FOCH
67380 LINGOLSHEIM
FRANCE
Noms du médicament dans les Etats membres de l'Espace Economique Européen
Ce médicament est autorisé dans les Etats membres de l'Espace Economique Européen sous les noms suivants : Conformément à la réglementation en vigueur.
[À compléter ultérieurement par le titulaire]
La dernière date à laquelle cette notice a été révisée est :
[à compléter ultérieurement par le titulaire]
Des informations détaillées sur ce médicament sont disponibles sur le site Internet de l’ANSM (France).
Informations réservées aux professionnels de santé
Les informations suivantes sont destinées exclusivement aux professionnels de santé :
Les informations générales sur l’utilisation d’OCTAPLEX sont décrites en section 3.
Instructions pour le traitement
Lire toutes les instructions et les suivre scrupuleusement.
Durant la procédure décrite ci-dessous, l’asepsie doit être maintenue.
Le produit se reconstitue rapidement à température ambiante.
La solution doit être limpide ou légèrement opalescente. Ne pas utiliser de solution trouble ou présentant un dépôt. Les produits reconstitués doivent être inspectés visuellement, pour mettre en évidence la présence de particules et un changement de coloration avant administration.
Après reconstitution, la solution doit être utilisée immédiatement.
Tout produit non utilisé ou déchet doit être éliminé conformément à la réglementation en vigueur.
Posologie
Saignements et prophylaxie péri-opératoire des accidents hémorragiques lors d'un traitement par anti-vitamines K :
La dose nécessaire dépend de l'INR déterminé avant le traitement et du poids corporel.Le tableau suivant donne les doses approximatives (unités de produit reconstitué/kg de poids corporel)
|
INR avant |
2 à < 4 |
4 à 6 |
> 6 |
|
Dose d’Octaplex (unités de facteur IX) / kg de poids corporel) |
25 |
35 |
50 |
Les unités font référence aux Unités Internationales.
La dose est définie en fonction du poids jusqu’à un poids corporel de 100 kg maximum. Pour les patients pesant plus de 100 kg, la dose maximale administrée en une fois (UI de facteur IX) ne devra pas dépasser 2 500 UI pour un INR de 2 à < 4, 3500 UI pour un INR de 4 à 6 et 5000 UI pour un INR > 6.
Comme ces recommandations sont empiriques et que le taux de récupération et la durée de l'effet peuvent varier, le suivi de l'INR durant le traitement est obligatoire.
Saignements et prophylaxie péri-opératoire lors de déficit congénital des facteurs de coagulation vitamine K dépendants II et X, lorsque aucun facteur de coagulation spécifique n’est disponible :
Le calcul de la posologie pour un traitement repose sur les notions empiriques suivantes :
Approximativement 1 UI de facteur II ou X par kg de poids corporel élève l’activité plasmatique des facteurs II et X de 0,02 et 0.017 UI/mL respectivement.
Unités requises = poids corporel (kg) x augmentation souhaitée en Facteur X (UI/mL) x 60
où 60 (mL/kg) correspond à l’inverse du taux de récupération estimé.
Posologie requise pour le facteur II:
Unités requises = poids corporel (kg) x augmentation souhaitée en facteur II (UI/mL) x 50
Si le taux de récupération individuel est connu, cette valeur doit être utilisée pour le calcul.
Instructions pour la reconstitution :
|
|
1. Si nécessaire, amenez le solvant (eau pour préparations injectables) et la poudre, dans les flacons fermés, à température ambiante. Cette température doit être maintenue pendant la reconstitution. Si un bain-marie est utilisé pour chauffer, une attention doit être portée pour que l'eau n’entre pas en contact avec les bouchons en caoutchouc ou les opercules des flacons. La température du bain-marie ne doit pas dépasser 37 °C.
2. Retirez les opercules détachables du flacon de poudre et du flacon de solvant et désinfecter les bouchons en caoutchouc de façon appropriée.
3. Retirez le couvercle de l'emballage externe du Nextaro®. Placez le flacon de solvant sur une surface plane et le tenir fermement. Sans retirer l’emballage externe, placez la partie bleue du Nextaro® sur le dessus du flacon de solvant et appuyer fermement jusqu'à ce qu'il s'enclenche (Fig. 1). N’effectuez pas de rotation lors de la fixation ! Tout en maintenant le flacon de solvant, retirer soigneusement l'emballage externe du Nextaro® ; attention à bien laisser le Nextaro® fermement fixé au flacon de solvant (Fig. 2).
|
|
|
4. Placez le flacon de poudre sur une surface plane et le tenir fermement. Prenez le flacon de solvant avec le Nextaro® fixé et le retourner. Placez la partie blanche du connecteur Nextaro® sur le dessus du flacon de poudre et appuyer fermement jusqu'à ce qu'il s'enclenche (Fig. 3). N’effectuez pas de rotation lors de la fixation ! Le solvant s'écoule automatiquement dans le flacon de poudre. |
|
|
5. Les deux flacons toujours fixés, tournez doucement le flacon de poudre jusqu'à ce que le produit soit dissous. OCTAPLEX se dissout rapidement à température ambiante pour donner une solution incolore à légèrement bleue. Dévissez le Nextaro® en deux parties (Fig. 4). Eliminez le flacon de solvant vide avec la partie bleue du Nextaro®.
|
Si la poudre ne se dissout pas complètement ou si un agrégat s’est formé, ne pas utiliser la solution.
Instructions pour la perfusion
Par précaution, le pouls du patient doit être pris avant et pendant la perfusion. En cas d'augmentation nette du pouls, la vitesse de perfusion doit être réduite ou l’administration doit être interrompue.
1. Fixez une seringue de 40 mL (pour 1 000 UI) à la sortie Luer Lock sur la partie blanche du Nextaro®. Retournez le flacon et prélevez la solution dans la seringue.
Dès que la solution a été transférée, tenez fermement le piston de la seringue (en la tenant tournée vers le bas) et retirez la seringue du Nextaro®. Eliminez le Nextaro® et le flacon vide.
2. Désinfectez le site d’injection de façon appropriée.
3. Injectez la solution par voie intraveineuse au rythme de 0,12 mL/kg/min. (environ 3 unités/kg/min.) sans dépasser un débit maximal de 8 mL/min. (environ 210 unités/min.), en respectant les conditions d’asepsie.
Du sang ne doit pas refluer dans la seringue en raison du risque de formation de caillots de fibrine. Le Nextaro® est réservé à un usage unique.