Dernière mise à jour le 29/06/2026
ACARIZAX 12 SQ-HDM, lyophilisat sublingual
Indications thérapeutiques
Classe pharmacothérapeutique : extraits allergéniques, acariens, code ATC : V01AA03.
ACARIZAX contient un extrait allergénique d’acariens. Il se présente sous la forme d'un lyophilisat sublingual qui est similaire à un comprimé mais plus fragile. L’extrait est absorbé dans l’organisme en plaçant le lyophilisat sous la langue.
ACARIZAX est utilisé dans le traitement de la rhinite allergique (inflammation de la muqueuse du nez) chez les adultes et les enfants (âgés de 5 à 65 ans) et de l’asthme allergique chez les adultes (âgés de 18 à 65 ans), provoqués par les acariens. ACARIZAX agit en améliorant votre tolérance immunitaire (aptitude de votre organisme à se défendre) aux acariens. Il peut être nécessaire de prendre le traitement pendant 8 à 14 semaines avant de remarquer une amélioration des symptômes.
Le médecin évaluera vos symptômes allergiques et réalisera un test cutané (prick test) et/ou un prélèvement sanguin afin de confirmer le bien-fondé d’un traitement par ACARIZAX.
Il est recommandé de prendre la première dose d’ACARIZAX sous la surveillance du médecin. Vous devrez rester sous surveillance médicale pendant au moins 30 minutes après la prise de la première dose, afin de surveiller votre tolérance au médicament. Cela vous donnera aussi la possibilité de discuter avec votre médecin de tout effet secondaire que vous pourriez ressentir.
ACARIZAX doit être prescrit par des médecins expérimentés dans le traitement des allergies.
Présentations
> 3 plaquette(s) aluminium de 10 lyophilisat(s)
Code CIP : 34009 300 453 4 3
Déclaration de commercialisation : 15/02/2018
Cette présentation est agréée aux collectivités
- Prix hors honoraire de dispensation : 69,55 €
- Honoraire de dispensation : 1,02 €
- Prix honoraire compris : 70,57 €
- Taux de remboursement :15%
Service médical rendu (SMR)
Les libellés affichés ci-dessous ne sont que des résumés ou extraits issus des avis rendus par la Commission de la Transparence. Seul l'avis complet de la Commission de la Transparence fait référence.
Cet avis est consultable à partir du lien `Avis du jj/mm/aaaa` ou encore sur demande auprès de la HAS (plus d'informations dans l'aide). Les avis et synthèses d'avis contiennent un paragraphe sur la place du médicament dans la stratégie thérapeutique.
| Valeur du SMR | Avis | Motif de l'évaluation | Résumé de l'avis |
|---|---|---|---|
| Faible | Avis du 01/04/2026 | Extension d'indication | Le service médical rendu par ACARIZAX est faible dans l’indication de l’AMM. |
| Faible | Avis du 09/01/2019 | Extension d'indication | Le service médical rendu par ACARIZAX 12 SQ-HDM, lyophilisat oral, est faible chez les adolescents (âgés de 12 à 17 ans) qui, sur la base d'un diagnostic réunissant une histoire clinique évocatrice et la positivité d’un test de sensibilisation aux acariens de la poussière de maison (prick test cutané et/ou présence d’IgE spécifiques), présentent une rhinite allergique aux acariens persistante modérée à sévère insuffisamment contrôlée par les traitements symptomatiques. |
| Faible | Avis du 22/02/2017 | Inscription (CT) | Le service médical rendu par ACARIZAX 12 SQ-HDM, lyophilisat oral, est faible chez l’adulte ayant une rhinite allergique persistante aux acariens, modérée à sévère, insuffisamment contrôlée par les traitements symptomatiques ainsi que chez l’adulte ayant un asthme allergique aux acariens, insuffisamment contrôlé par les corticostéroïdes inhalés et associé à une rhinite allergique légère à sévère aux acariens. |
Amélioration du service médical rendu (ASMR)
Les libellés affichés ci-dessous ne sont que des résumés ou extraits issus des avis rendus par la Commission de la Transparence. Seul l'avis complet de la Commission de la Transparence fait référence.
Cet avis est consultable à partir du lien `Avis du jj/mm/aaaa` ou encore sur demande auprés de la HAS (plus d'informations dans l'aide). Les avis et synthéses d'avis contiennent un paragraphe sur la place du médicament dans la stratégie thérapeutique.
| Valeur de l'ASMR | Avis | Motif de l'évaluation | Résumé de l'avis |
|---|---|---|---|
| V (Inexistant) | Avis du 01/04/2026 | Extension d'indication | ACARIZAX 12 SQ-HDM, lyophilisat oral, n’apporte pas d’amélioration du service médical rendu (ASMR V) chez les enfants (âgés de 5 à 11 ans) qui, sur la base d’un diagnostic réunissant une histoire clinique évocatrice et la positivité d’un test de sensibilisation aux acariens de la poussière de maison (prick test cutané et/ou présence d’IgE spécifiques), présentent une rhinite allergique aux acariens persistante modérée à sévère insuffisamment contrôlée par les traitements symptomatiques par rapport aux comparateurs pertinents (APSI). |
| V (Inexistant) | Avis du 09/01/2019 | Extension d'indication | Compte tenu : • de la faible qualité de la démonstration de la supériorité d’ACARIZAX par rapport au placebo chez l’adolescent dans la mesure où les résultats sont issus d’analyses post-hoc en sous-groupes sur de faibles effectifs, • de la faible quantité d’effet en termes d’amélioration de la symptomatologie et de réduction de la consommation de traitements symptomatiques, • de l’absence de démonstration d’un impact en termes de qualité de vie, • de l’absence de données d’efficacité et de tolérance à long termes (au-delà d’un an), • de l’absence de comparaison aux APSI, • du besoin médical, ACARIZAX 12 SQ-HDM, lyophilisat oral, n’apporte pas d’amélioration du service médical rendu (ASMR V) chez les adolescents (âgés de 12 à 17 ans) qui, sur la base d'un diagnostic réunissant une histoire clinique évocatrice et la positivité d’un test de sensibilisation aux acariens de la poussière de maison (prick test cutané et/ou présence d’IgE spécifiques), présentent une rhinite allergique aux acariens persistante modérée à sévère insuffisamment contrôlée par les traitements symptomatiques. |
| V (Inexistant) | Avis du 22/02/2017 | Inscription (CT) | ACARIZAX 12 SQ-HDM, lyophilisat oral, n’apporte pas d’amélioration du service médical rendu (ASMR V) dans la prise en charge, chez l’adulte : • de la rhinite allergique persistante aux acariens, modérée à sévère, insuffisamment contrôlée par les médicaments symptomatiques, • de l’asthme allergique aux acariens, insuffisamment contrôlé par les corticostéroïdes inhalés et associé à une rhinite allergique légère à sévère aux acariens. |
ANSM - Mis à jour le : 16/02/2026
ACARIZAX 12 SQ-HDM, lyophilisat sublingual
2. COMPOSITION QUALITATIVE ET QUANTITATIVE
Extrait allergénique standardisé d’acariens de la poussière de maison Dermatophagoides pteronyssinus et Dermatophagoides farinae 12 SQ-HDM* par lyophilisat sublingual.
Pour la liste complète des excipients, voir rubrique 6.1.
* SQ-HDM est l’unité de dose pour ACARIZAX. La méthode de standardisation SQ est basée sur l’activité biologique, la teneur en allergènes majeurs et la composition de l’extrait allergénique. HDM est l’abréviation de house dust mite (acarien de la poussière de maison).
Lyophilisat sublingual rond blanc à blanc cassé en creux.
4.1. Indications thérapeutiques
· une rhinite allergique aux acariens persistante modérée à sévère insuffisamment contrôlée par les traitements symptomatiques
et/ou
· un asthme allergique aux acariens insuffisamment contrôlé par les corticostéroïdes inhalés et associé à une rhinite allergique légère à sévère aux acariens. L’asthme du patient doit être soigneusement évalué avant l’instauration du traitement (voir rubrique 4.3).
ACARIZAX est indiqué chez les enfants (âgés de 5 à 17 ans) qui, sur la base d’un diagnostic réunissant une histoire clinique évocatrice et la positivité d’un test de sensibilisation aux acariens de la poussière de maison (prick test cutané et/ou présence d’IgE spécifiques), présentent une rhinite allergique aux acariens persistante modérée à sévère insuffisamment contrôlée par les traitements symptomatiques.
4.2. Posologie et mode d'administration
La posologie recommandée chez les enfants et les adultes (âgés de 5 à 65 ans) est d’un lyophilisat sublingual (12 SQ-HDM) par jour.
L’apparition de l’effet clinique est attendue 8 à 14 semaines après l’instauration du traitement. Les recommandations thérapeutiques internationales préconisent une durée d’immunothérapie allergénique d’environ 3 ans pour modifier l’évolution de la maladie. Les données d’efficacité avec ACARIZAX sont disponibles sur une période de 18 mois de traitement chez l’adulte ; aucune donnée n’est disponible sur une période de 3 ans de traitement (voir rubrique 5.1). S’il n’est pas observé d’amélioration pendant la première année de traitement par ACARIZAX, la poursuite du traitement n’est pas justifiée.
Population pédiatrique
Rhinite allergique : la posologie à utiliser chez les enfants (5-17 ans) est la même que pour les adultes. L’expérience clinique dans le traitement de la rhinite allergique par ACARIZAX n’a pas été établie chez les enfants de moins de 5 ans.
ACARIZAX n’est pas indiqué pour le traitement de la rhinite allergique chez les enfants de moins de 5 ans. Les données actuellement disponibles sont décrites aux rubriques 4.8 et 5.1.
Asthme allergique : l’efficacité dans le traitement de l’asthme allergique par ACARIZAX n’a pas été établie chez les enfants de moins de 18 ans. ACARIZAX n’est pas indiqué dans le traitement de l’asthme allergique chez les enfants de moins de 18 ans. Les données actuellement disponibles sont décrites aux rubriques 4.8 et 5.1.
Patients âgés
L’expérience clinique d’une immunothérapie par ACARIZAX n’a pas été établie chez le sujet de plus de 65 ans. ACARIZAX n’est pas indiqué chez le sujet de plus de 65 ans (voir rubrique 5.1).
Mode d’administration
Le traitement par ACARIZAX doit être instauré par des médecins expérimentés dans le traitement des allergies.
La première prise de lyophilisat sublingual devra être réalisée sous surveillance médicale pendant au moins 30 minutes afin d’évaluer et traiter les éventuels effets indésirables d’apparition immédiate.
ACARIZAX est un lyophilisat sublingual. L’alvéole de la plaquette sera ouverte avec des doigts bien secs pour libérer le lyophilisat sublingual. Immédiatement après sa libération de la plaquette, le lyophilisat sublingual doit être placé sous la langue où il se dissout. Ne pas déglutir pendant environ 1 minute. Ne pas absorber d’aliment ni de boisson dans les 5 minutes qui suivent la prise du médicament.
Si le traitement par ACARIZAX est interrompu pendant une durée allant jusqu’à 7 jours, le patient peut reprendre le traitement de lui-même. Si le traitement est interrompu pendant plus de 7 jours, un avis médical est recommandé pour la reprise éventuelle du traitement.
· Hypersensibilité à l’un des excipients mentionnés à la rubrique 6.1.
· VEMS < 70% de la valeur théorique (après un traitement médicamenteux adapté) lors de l’initiation du traitement.
· Exacerbation sévère d’asthme au cours des 3 derniers mois.
· Infection aiguë des voies respiratoires chez les sujets asthmatiques ; l’initiation du traitement par ACARIZAX doit être différée jusqu’à la guérison de l’infection respiratoire.
· Maladies auto-immunes évolutives ou mal contrôlées, déficits immunitaires, immunodépression ou maladies néoplasiques malignes évolutives.
· Inflammation buccale aiguë sévère ou plaies de la muqueuse buccale (voir rubrique 4.4).
4.4. Mises en garde spéciales et précautions d'emploi
L’asthme est un facteur de risque connu dans la survenue de réactions allergiques systémiques sévères.
Les patients seront informés du fait qu’ACARIZAX n’est pas indiqué pour le traitement des exacerbations aiguës d’asthme. Dans ce cas, un bronchodilatateur de courte durée d’action doit être utilisé. Un avis médical est requis en cas de perte d’efficacité des bronchodilatateurs ou d’augmentation de leur consommation par le patient.
Les patients doivent être informés de la nécessité de consulter immédiatement un médecin en cas d’aggravation soudaine de leur asthme.
ACARIZAX doit être initialement utilisé en complément du traitement antiasthmatique en cours et non en remplacement de celui-ci.
Les traitements en cours pour le contrôle de l’asthme ne doivent pas être arrêtés brutalement après l’instauration d’un traitement par ACARIZAX. La diminution des doses des médicaments administrés pour le contrôle de l’asthme ne doit être envisagée que de façon progressive et sous contrôle médical selon les recommandations de prise en charge de l’asthme.
Réactions allergiques systémiques sévères
Le traitement doit être interrompu et un avis médical immédiat est requis en cas de réaction allergique systémique sévère, d'exacerbation sévère d’asthme, d’angio-œdème, de dysphagie, de dyspnée, de dysphonie, d’hypotension ou de sensation de constriction pharyngée.
Les symptômes annonciateurs d'une réaction systémique peuvent inclure des flushs (bouffées vasomotrices), un prurit, une sensation de chaleur, un malaise général et une agitation ou une anxiété.
L'adrénaline peut être nécessaire pour traiter des réactions allergiques systémiques sévères. Les traitements concomitants par antidépresseurs tricycliques, inhibiteurs de la monoamine oxydase (IMAO) et/ou inhibiteurs de la catéchol-O-méthyltransférase (COMT), peuvent potentialiser les effets de l’adrénaline et mettre en jeu le pronostic vital. Les effets de l’adrénaline peuvent être diminués chez les patients traités par bêta-bloquants.
Les patients présentant une cardiopathie peuvent être exposés à un risque plus important en cas de réactions allergiques systémiques. L’expérience clinique du traitement par ACARIZAX chez les patients présentant une cardiopathie est limitée.
Ceci doit être pris en considération avant d’instaurer une immunothérapie allergénique.
L’instauration d’un traitement par ACARIZAX chez les patients ayant déjà présenté une réaction allergique systémique au cours d'une immunothérapie aux acariens par voie sous-cutanée doit être envisagée avec précaution, en ayant à disposition les traitements nécessaires en cas de survenue de réactions indésirables. L’expérience acquise depuis la commercialisation d’un comprimé sublingual similaire destiné au traitement de l'allergie aux pollens de graminées montre que le risque de réaction allergique sévère peut être plus important chez les patients ayant déjà présenté une réaction allergique systémique au cours d'une immunothérapie aux pollens de graminées par voie sous-cutanée.
Inflammation de la muqueuse buccale
En cas d'inflammation sévère de la muqueuse buccale (exemple : lichen plan buccal, ulcérations ou mycose), de plaies dans la bouche ou de chirurgie buccopharyngée, y compris une extraction dentaire ou la perte d’une dent, l’instauration du traitement par ACARIZAX sera différée, ou le traitement en cours temporairement interrompu, jusqu'à la cicatrisation.
Réactions allergiques locales
Un traitement par ACARIZAX expose le patient aux allergènes auxquels il est allergique. De ce fait, la survenue de réactions allergiques locales est attendue au cours du traitement. Ces réactions sont généralement légères ou modérées mais des réactions oropharyngées plus sévères peuvent survenir. En cas d'apparition de réactions indésirables locales significatives induites par l'administration d'ACARIZAX, l'utilisation d'un antihistaminique doit être envisagée.
Œsophagite à éosinophiles
Des cas d’œsophagite à éosinophiles ont été rapportés au cours d'un traitement par ACARIZAX. Chez les patients présentant des symptômes gastro-œsophagiens sévères ou persistants, tels que dysphagie ou dyspepsie, ACARIZAX doit être interrompu et un avis médical est requis.
Maladies auto-immunes en rémission
Les données disponibles concernant un traitement par immunothérapie allergénique chez des patients présentant des maladies auto-immunes en rémission sont limitées. ACARIZAX doit ainsi être prescrit avec précaution chez ces patients.
Allergie alimentaire
ACARIZAX peut contenir des traces de protéines de poisson. Les données disponibles n’ont pas mis en évidence de risque accru de réactions allergiques chez les patients présentant une allergie au poisson.
Ce médicament contient moins de 1 mmol (23 mg) de sodium par dose, c’est-à-dire qu’il est essentiellement « sans sodium ».
4.5. Interactions avec d'autres médicaments et autres formes d'interactions
4.6. Fertilité, grossesse et allaitement
Grossesse
Aucune donnée clinique n’est disponible concernant l’utilisation d’ACARIZAX chez la femme enceinte. Les études réalisées chez l’animal n’indiquent pas de risque accru pour le fœtus. Le traitement par ACARIZAX ne doit pas être instauré au cours de la grossesse. Si une grossesse survient en cours de traitement, la décision de poursuite ou non de l'immunothérapie allergénique devra prendre en considération l’état clinique de la patiente (incluant la fonction respiratoire) ainsi que ses antécédents de réactions apparues lors des prises précédentes d’ACARIZAX. En cas d’asthme préexistant, une surveillance étroite est recommandée pendant la grossesse.
Aucune donnée clinique n’est disponible concernant l’utilisation d’ACARIZAX au cours de l’allaitement. Il n’est pas attendu d’effets particuliers chez les enfants allaités.
Fertilité
Aucune donnée clinique n’est disponible concernant l’effet d’ACARIZAX sur la fertilité. Lors d’une étude de toxicité en administrations réitérées chez la souris, aucun effet n’a été observé sur les organes de reproduction des animaux des deux sexes.
4.7. Effets sur l'aptitude à conduire des véhicules et à utiliser des machines
Résumé du profil de tolérance
Les effets indésirables attendus au cours du traitement par ACARIZAX sont la survenue de réactions allergiques locales légères à modérées au cours des premiers jours du traitement, disparaissant en 1 à 3 mois avec la poursuite du traitement (voir rubrique 4.4). Dans la majorité des cas, les réactions apparaissent toujours dans les 5 minutes suivant la prise d’ACARIZAX et disparaissent en quelques minutes ou plusieurs heures. Des réactions allergiques oropharyngées plus sévères peuvent apparaître (voir rubrique 4.4).
Des cas isolés d’aggravation aiguë sévère des symptômes d’asthme ont été rapportés. Le traitement par ACARIZAX ne doit pas être instauré chez les patients ayant des facteurs de risque connus (voir rubrique 4.3).
Tableau des effets indésirables
Le tableau des effets indésirables mentionné ci-dessous est établi à partir de données issues d'essais cliniques contrôlés contre placebo réalisés chez des adultes et adolescents (plus de 2 100 patients traités par ACARIZAX) atteints de rhinite et/ou d’asthme allergique aux acariens et de déclarations spontanées.
Les réactions indésirables sont regroupées selon leur fréquence de survenue et conformément à la classification MedDRA : très fréquent (≥ 1/10), fréquent (≥ 1/100 à < 1/10), peu fréquent (≥ 1/1 000 à < 1/100), rare (≥ 1/10 000 à < 1/1 000), très rare (< 1/10 000).
|
Classes organiques |
Fréquence |
Effets indésirables |
|
Infections et infestations |
Très fréquent |
Rhinopharyngite |
|
Fréquent |
Bronchite, pharyngite, rhinite, sinusite |
|
|
Peu fréquent |
Laryngite |
|
|
Affections du système immunitaire |
Peu fréquent |
Réaction anaphylactique |
|
Affections du système nerveux |
Fréquent |
Dysgueusie |
|
Peu fréquent |
Sensation vertigineuse, paresthésie |
|
|
Affections oculaires |
Fréquent |
Prurit oculaire |
|
Peu fréquent |
Conjonctivite allergique |
|
|
Affections de l’oreille et du labyrinthe |
Très fréquent |
Prurit auriculaire |
|
Peu fréquent |
Gêne auriculaire |
|
|
Affections cardiaques |
Peu fréquent |
Palpitations |
|
Affections respiratoires, thoraciques et médiastinales |
Très fréquent |
Irritation de la gorge |
|
Fréquent |
Asthme, toux*, dysphonie, dyspnée, douleur oropharyngée, œdème pharyngé |
|
|
Peu fréquent |
Congestion nasale, gêne nasale, œdème nasal, érythème pharyngé, rhinorrhée, éternuement, sensation de constriction pharyngée, hypertrophie des amygdales |
|
|
Rare |
Œdème laryngé, obstruction nasale, œdème de la trachée |
|
|
Affections gastro-intestinales |
Très fréquent |
Œdème labial, œdème buccal, prurit oral |
|
Fréquent |
Douleur abdominale, diarrhée, dysphagie, dyspepsie, reflux gastro-œsophagien, glossodynie, glossite, prurit labial, ulcération buccale, douleur buccale, prurit lingual, nausées, gêne buccale, érythème de la muqueuse buccale, paresthésie buccale, stomatite, œdème lingual, vomissements |
|
|
Peu fréquent |
Sécheresse buccale, douleur labiale, ulcération labiale, irritation œsophagienne, vésicules buccales, hypertrophie des glandes salivaires, hypersécrétion salivaire |
|
|
Rare |
Œsophagite à éosinophiles |
|
|
Affections de la peau et du tissu sous-cutané |
Fréquent |
Prurit, urticaire |
|
Peu fréquent |
Erythème, éruption cutanée |
|
|
Rare |
Angio-œdème |
|
|
Troubles généraux et anomalies au site d’administration |
Fréquent |
Gêne thoracique, fatigue |
|
Peu fréquent |
Malaise, sensation de corps étranger |
Description de certaines réactions indésirables
En cas d'apparition d'effets indésirables significatifs liés à l'administration d’ACARIZAX, le recours à un traitement symptomatique de l’allergie doit être envisagé.
Des cas de réactions allergiques systémiques graves, y compris d’anaphylaxie, ont été rapportés depuis la commercialisation. La première administration du lyophilisat sublingual doit donc être réalisée sous surveillance médicale (voir rubrique 4.2). Cependant, des cas de réaction allergique systémique grave se sont aussi produits à des prises ultérieures à la première prise.
En cas d’aggravation aiguë des symptômes d’asthme ou de réactions allergiques systémiques sévères, d’angio-œdème, de dysphagie, de dyspnée, de dysphonie, d’hypotension ou de sensation de constriction pharyngée, un avis médical immédiat est requis. Des crises d’hypertension ont été rapportées à la suite d’une détresse respiratoire peu après la prise d'ACARIZAX. Le traitement doit être interrompu définitivement ou jusqu’à avis contraire du médecin.
*Dans les essais cliniques, la toux a été observée avec la même fréquence pour ACARIZAX et le placebo.
Population pédiatrique
Adolescents âgés de 12 à 17 ans
Les effets indésirables rapportés chez les adolescents ont été similaires à ceux observés chez les adultes en termes de fréquence, de type de réactions et de sévérité.
Enfants âgés de 5 à 11 ans
Dans l'ensemble, le profil de tolérance chez les enfants traités par ACARIZAX a été similaire à celui observé chez les adultes et les adolescents. La majorité des effets indésirables étaient légers à modérés et ont été observés à une fréquence similaire chez les enfants et les adultes/adolescents. Le profil de tolérance global chez les enfants asthmatiques était similaire à celui observé chez les enfants non asthmatiques. Le profil de tolérance d'ACARIZAX chez les enfants repose principalement sur des données issues d'essais cliniques internationaux en double aveugle, contrôlés versus placebo (environ 900 enfants traités avec ACARIZAX), avec sollicitation active au cours des 28 premiers jours de traitement des effets indésirables locaux prédéfinis.
Les effets indésirables suivants ont été observés plus fréquemment dans les essais pédiatriques que dans les essais chez les adultes et les adolescents :
Douleur abdominale, diarrhée, dysgueusie, glossodynie, ulcération buccale, nausées, œdème pharyngé et œdème lingual ont été catégorisés comme très fréquent (≥ 1/10). Tous faisaient partie des effets indésirables sollicités prédéfinis.
Des symptômes de conjonctivite allergique ont été catégorisés comme fréquent (≥ 1/100 à < 1/10).
Angio-œdème et œsophagite à éosinophiles ont été catégorisés comme peu fréquent (≥ 1/1 000 à < 1/100).
Enfants âgés de moins de 5 ans
Aucune donnée n’est disponible chez les enfants de moins de 5 ans traités par ACARIZAX.
Déclaration des effets indésirables suspectés
La déclaration des effets indésirables suspectés après autorisation du médicament est importante. Elle permet une surveillance continue du rapport bénéfice/risque du médicament. Les professionnels de santé déclarent tout effet indésirable suspecté via le système national de déclaration : Agence nationale de sécurité du médicament et des produits de santé (ANSM) et réseau des Centres Régionaux de Pharmacovigilance - Site internet : https://signalement.social-sante.gouv.fr/.
Lors des essais de phase I, des adultes allergiques aux acariens ont été exposés à des doses allant jusqu’à 32 SQ-HDM. Il n’y a pas de donnée relative à une exposition à des doses supérieures à la dose journalière recommandée de 12 SQ-HDM chez les enfants (5 – 17 ans).
Si des doses supérieures à la dose quotidienne recommandée sont prises, le risque d’effets indésirables augmente, y compris le risque de réactions allergiques systémiques ou de réactions allergiques locales sévères. En cas de réaction sévère telle qu’un angio-œdème, une dysphagie, une dyspnée, une modification de la voix ou une sensation de constriction pharyngée, un avis médical immédiat est requis. Un traitement symptomatique adapté est préconisé.
5. PROPRIETES PHARMACOLOGIQUES
5.1. Propriétés pharmacodynamiques
Classe pharmacothérapeutique : extraits allergéniques, acariens, code ATC : V01AA03.
Mécanisme d’action
ACARIZAX est une immunothérapie allergénique. L’immunothérapie allergénique consiste en l’administration répétée d’allergènes à un individu allergique dans le but de modifier sa réponse immunitaire à ces allergènes.
L’activité pharmacodynamique de l'immunothérapie allergénique a pour cible le système immunitaire mais le mécanisme d'action exact à l'origine de l’effet clinique n’est pas totalement connu. Les travaux ont mis en évidence une augmentation de la production d'IgG4 spécifiques anti-acariens induits par l'administration d'ACARIZAX et la production d’anticorps circulants pouvant entrer en compétition au niveau de la liaison des IgE avec les allergènes d’acariens. Cet effet a été observé dès quatre semaines de traitement.
ACARIZAX agit sur la cause de l’allergie respiratoire aux acariens et son effet clinique au cours du traitement a été démontré au niveau des voies respiratoires supérieures et inférieures. La protection induite par ACARIZAX a entraîné une amélioration du contrôle des symptômes et de la qualité de vie, liés à un soulagement des symptômes, une diminution du recours aux autres médicaments et la réduction du risque d'exacerbations.
Efficacité clinique chez l’adulte
L’efficacité d’ACARIZAX 12 SQ-HDM dans le traitement des allergies respiratoires aux acariens a été évaluée au cours de deux essais cliniques randomisés, en double aveugle, contrôlés contre placebo, menés dans des populations différentes et utilisant des critères primaires différents. Deux tiers des patients inclus dans ces essais étaient sensibilisés aux acariens et à d'autres allergènes. Les résultats cliniques n'ont pas été influencés par le fait que les patients soient mono ou polysensibilisés. Des données complémentaires provenant d’un essai réalisé en chambre d’exposition aux allergènes et d’un essai mené à de plus faibles doses sont également présentées.
Rhinite allergique :
Essai MERIT (MT-06)
· L’essai MERIT a inclus 992 adultes présentant une rhinite allergique modérée à sévère aux acariens malgré un traitement symptomatique de la rhinite. Ces patients ont été randomisés pour recevoir une prise quotidienne de 12 SQ-HDM, de 6 SQ-HDM ou d’un placebo pendant environ 1 an et pouvaient recourir librement à un traitement symptomatique standardisé de la rhinite. Les patients ont été examinés par un spécialiste environ tous les deux mois durant la totalité de l’essai.
· Le critère primaire était le score total combiné quotidien moyen de rhinite (daily total combined rhinitis score, TCRS) évalué durant les 8 dernières semaines de traitement.
o Le TCRS était la somme du score des symptômes de rhinite et du score médicamenteux de rhinite. Le score des symptômes de rhinite évaluait chacun des quatre symptômes nasaux (rhinorrhée, obstruction nasale, prurit nasal, éternuements) quotidiennement sur une échelle de 0 à 3 (symptômes absents, légers, modérés ou sévères) ; soit une échelle de score allant de 0 à 12. Le score médicamenteux de rhinite était la somme du score de la prise d’un corticostéroïde nasal (2 points par bouffée, maximum 4 bouffées par jour) et du score de la prise d’un antihistaminique oral (4 points par comprimé, maximum 1 comprimé par jour) ; soit une échelle de score allant de 0 à 12. L’échelle de score du TCRS allait ainsi de 0 à 24.
· Les principaux critères secondaires prédéfinis étaient le score total combiné de rhinoconjonctivite et de qualité de vie liée à la rhinoconjonctivite (rhinoconjunctivitis quality of life, RQLQ).
· Des analyses des jours avec exacerbation de rhinite ont été également réalisées a posteriori pour illustrer davantage la pertinence clinique des résultats.
o Une exacerbation de rhinite était définie comme un jour où les symptômes étaient revenus au niveau élevé requis pour l’inclusion des patients dans l’essai : score de symptômes de rhinite d’au moins 6 ou d’au moins 5 en présence d'un symptôme côté comme sévère.
|
|
Essai MERIT : évolution du score total combiné de rhinite au cours du temps TCRS : score total combiné de rhinite (score des symptômes + score médicamenteux). Le critère primaire était le TCRS quotidien moyen au cours des 8 dernières semaines de traitement environ (semaines 44 à 52 environ). Moyennes ajustées du TCRS moyen au cours du temps. Les lignes verticales indiquent l’erreur standard pour ces moyennes ajustées. Les intervalles ne se chevauchant pas indiquent une différence statistiquement significative. |
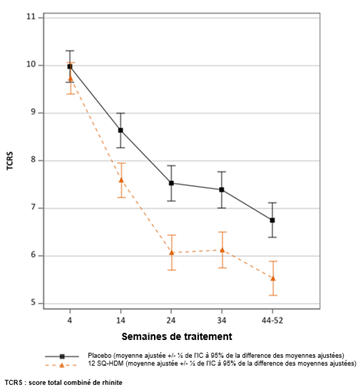
Résultats de l'essai MERIT |
12 SQ-HDM |
Placebo |
Efficacité |
|
|||
|
Critère primaire |
N |
Score |
N |
Score |
Différence absolue c |
Différence relative d |
Valeur de p |
|
Score total combiné de rhinite |
|||||||
|
FAS-MI a (moyenne ajustée) |
318 |
5,71 |
338 |
6,81 |
1,09 [0,35 ; 1,84] |
- |
0,004 |
|
FAS b (moyenne ajustée) |
284 |
5,53 |
298 |
6,76 |
1,22 [0,49 ; 1,96] |
18% |
0,001 |
|
FAS b (médiane) |
284 |
5,88 |
298 |
7,54 |
1,66 |
22% |
- |
|
Principaux critères secondaires prédéfinis |
N |
Score |
N |
Score |
Différence absolue c |
Différence relative d |
Valeur de p |
|
Score de symptômes de rhinite |
|||||||
|
FAS b (moyenne ajustée) |
284 |
2,76 |
298 |
3,30 |
0,54 [0,18 ; 0,89] |
16% |
0,003 |
|
FAS b (médiane) |
284 |
2,98 |
298 |
3,98 |
1,00 |
25% |
- |
|
Score médicamenteux de rhinite |
|||||||
|
FAS b (moyenne ajustée) |
284 |
2,22 |
298 |
2,83 |
0,60 [0,08 ; 1,13] |
21% |
0,024 |
|
FAS b (médiane) |
284 |
2,83 |
298 |
4,00 |
1,17 |
29% |
- |
|
Score total combiné de rhinoconjonctivite |
|||||||
|
FAS b (moyenne ajustée) |
241 |
7,91 |
257 |
9,12 |
1,21 [0,13 ; 2,28] |
13% |
0,029 |
|
FAS b (médiane) |
241 |
8,38 |
257 |
10,05 |
1,67 |
17% |
- |
|
Questionnaire de qualité de vie relatif à la rhinoconjonctivite (RQLQ) |
|||||||
|
FAS b (moyenne ajustée) |
229 |
1,38 |
240 |
1,58 |
0,19 e [0,02 ; 0,37] |
12% |
0,031 |
|
FAS b (médiane) |
229 |
1,25 |
240 |
1,46 |
0,21 |
14% |
- |
|
Critères post hoc |
N |
Proportion |
N |
Proportion |
Odds Ratio f (IC à 95%) |
Valeur de p |
|
|
Pourcentage de jours avec exacerbation de rhinite |
|||||||
|
FAS (estimation) b |
284 |
5,33% |
298 |
11,14% |
0,45 [0,28 ; 0,72] |
0,001 |
|
|
Pourcentage de jours avec exacerbation de rhinite malgré la prise de médicaments symptomatiques de la rhinite |
|||||||
|
FAS (estimation) b |
284 |
3,43% |
298 |
6,50% |
0,51 [0,32 ; 0,81] |
0,005 |
|
|
N : nombre de patients dans le groupe de traitement pour qui des données étaient disponibles pour l’analyse IC : intervalle de confiance a FAS-MI : analyse sur l'ensemble des données disponibles (full analysis set) avec imputations multiples. Pour l’analyse, les patients sortis de l’étude avant la période d’évaluation de l’efficacité ont été considérés comme des patients sous placebo. Pour l’analyse principale (FAS-MI), seule la différence absolue a été pré-spécifiée. b FAS : analyse sur l'ensemble des données disponibles (full analysis set). Toutes les données disponibles utilisées dans leur intégralité (c’est-à-dire conduites chez tous les patients disposant de données au cours de la période d’évaluation de l’efficacité). c Différence absolue : placebo moins 12 SQ-HDM, intervalle de confiance à 95%. d Différence relative par rapport au placebo : placebo moins 12 SQ-HDM divisé par le placebo. e La différence entre les groupes 12 SQ-HDM et placebo a été principalement régie par des différences dans les trois domaines suivants : troubles du sommeil, problèmes pratiques et symptômes nasaux. f Odds ratio pour la survenue d’une exacerbation de rhinite : 12 SQ-HDM par rapport au placebo. |
|||||||
Données complémentaires - rhinite allergique
Un essai de phase II randomisé, en double aveugle et contrôlé contre placebo a été mené en chambre d’exposition aux allergènes chez 124 adultes présentant une rhinite allergique aux acariens. Les sujets ont été sevrés de tout traitement symptomatique anti-allergique avant chaque exposition à l’allergène. Lors de l’exposition allergénique de fin d’essai après 24 semaines de traitement par 12 SQ-HDM, 6 SQ-HDM ou placebo, le score moyen des symptômes de rhinite a été de 7,45 (IC95% : [6,57 ; 8,33]) dans le groupe placebo et de 3,83 (IC95% : [2,94 ; 4,72]) dans le groupe 12 SQ-HDM, correspondant à une différence absolue de 3,62 et à une différence relative de 49% (IC95% : [35% ; 60%] ; p < 0,001). La différence entre les groupes 12 SQ-HDM et placebo a été également statistiquement significative à 16 semaines (scores moyens de 4,82 et 6,90, différence absolue de 2,08, différence relative de 30%, IC95% : [17% ; 42%], p < 0,001) et à 8 semaines (scores moyens de 5,34 et 6,71, différence absolue de 1,37, différence relative de 20%, IC95% : [7% ; 33%], p = 0,007).
Asthme allergique :
Essai MITRA (MT-04)
· L’essai MITRA a inclus 834 adultes présentant un asthme allergique aux acariens et insuffisamment contrôlés par la prise quotidienne d’un corticostéroïde inhalé (CSI) correspondant à 400 à 1 200 μg de budésonide. Tous les patients ont reçu un traitement de 7 à 12 mois par 12 SQ-HDM, 6 SQ-HDM ou placebo en complément du corticostéroïde inhalé et d'un bêta-agoniste de courte durée d’action avant une réduction de la dose du corticostéroïde inhalé. Le protocole n'a pas inclus de phase initiale de recherche de la dose minimale efficace de la corticothérapie inhalée. L’efficacité a été évaluée par le délai de survenue de la première exacerbation d’asthme modérée à sévère lors de la période de réduction du corticostéroïde inhalé sur les 6 derniers mois des 13 à 18 mois de traitement.
· Une exacerbation d’asthme modérée était définie par au moins un des critères suivants et la nécessité de modifier son traitement :
o Réveil(s) nocturne(s) ou augmentation des symptômes : un ou plusieurs réveils nocturnes dus à l’asthme et nécessitant la prise d’un ꞵ2-agoniste de courte durée d'action (SABA) lors de deux nuits consécutives ou augmentation du score quotidien des symptômes ≥ 0,75 pendant deux jours consécutifs par rapport à la valeur initiale à l’entrée dans l’essai.
o Augmentation du nombre de prises d’un ꞵ2-agoniste de courte durée d'action : augmentation du nombre de prises d’un ꞵ2-agoniste de courte durée d'action pendant deux jours consécutifs par rapport à la valeur initiale à l’entrée dans l’essai (augmentation minimale : 4 bouffées/jour).
o Dégradation de la fonction respiratoire : diminution du DEP ≥ 20% par rapport à la valeur initiale à l’entrée dans l’étude lors de deux matins ou soirs consécutifs ou diminution du VEMS ≥ 20% par rapport à la valeur initiale à l’entrée dans l’essai.
o Consultation médicale : consultation d’un service d’urgences ou d’un centre investigateur pour traitement de l’asthme ne requérant pas une corticothérapie systémique.
· Une exacerbation d’asthme sévère était définie par la présence d’au moins un des deux critères suivants :
o Nécessité d’une corticothérapie systémique pendant ≥ 3 jours.
o Consultation d’un service d’urgences entraînant une corticothérapie systémique ou une hospitalisation pendant ≥ 12 h.
|
|
Essai MITRA – représentation graphique du critère primaire d'efficacité : évolution en fonction du temps du risque d’exacerbation d’asthme modérée à sévère au cours de la période de réduction ou de sevrage du CSI. Dans le graphique, temps = 0 représente le moment de la réduction de 50% de la dose du CSI. Après environ 3 mois (soit 90 jours), le traitement par CSI était complétement arrêté chez les patients qui n'avaient pas fait d’exacerbation. |
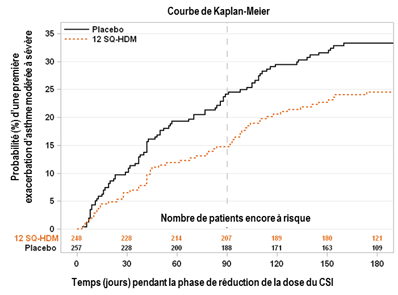
|
Résultats de l’essai MITRA |
12 SQ-HDM |
Placebo |
Efficacité 12 SQ-HDM par rapport au placebo |
Valeur de p |
|||
|
N |
n (%) |
N |
n (%) |
Risque relatif [IC à 95%] |
Réduction du risque a |
||
|
Critère primaire |
|||||||
|
Toute exacerbation modérée ou sévère (FAS-MI) b |
282 |
59 (21%) |
277 |
83 (30%) |
0,69 [0,50 ; 0,96] |
31% |
0,027 |
|
Toute exacerbation modérée ou sévère (FAS) c |
248 |
59 (24%) |
257 |
83 (32%) |
0,66 [0,47 ; 0,93] |
34% |
0,017 |
|
Analyses prédéfinies des composantes du critère primaire |
|||||||
|
Réveil(s) nocturne(s) ou augmentation des symptômes c |
248 |
39 (16%) |
257 |
57 (22%) |
0,64 [0,42 ; 0,96] |
36% |
0,031 |
|
Augmentation de l’utilisation d’un SABA c |
248 |
18 (7%) |
257 |
32 (12%) |
0,52 [0,29 ; 0,94] |
48% |
0,029 |
|
Dégradation de la fonction respiratoire c |
248 |
30 (12%) |
257 |
45 (18%) |
0,58 [0,36 ; 0,93] |
42% |
0,022 |
|
Exacerbation sévère c |
248 |
10 (4%) |
257 |
18 (7%) |
0,49 [0,23 ; 1,08] |
51% |
0,076 |
|
N : nombre de patients dans le groupe de traitement pour qui des données étaient disponibles pour l’analyse. n (%) : nombre et pourcentage de patients du groupe de traitement répondant au critère. IC : intervalle de confiance a Estimation par le risque relatif. b FAS-MI : analyse sur l'ensemble des données disponibles (full analysis set) avec imputations multiples. Pour l’analyse, les patients sortis de l’essai avant la période d’évaluation de l’efficacité ont été considérés comme des patients sous placebo. c FAS : analyse sur l'ensemble des données disponibles (full analysis set). Toutes les données disponibles utilisées dans leur intégralité (tous les patients chez qui des données avaient été recueillies au cours de la période d’évaluation de l’efficacité). |
|||||||
Des analyses des symptômes d’asthme et de la prise de médicaments symptomatiques au cours des 4 dernières semaines de la période de traitement avant la réduction de la dose du corticostéroïde inhalé ont été également menées a posteriori afin d’évaluer l’effet d’ACARIZAX en complément d’un corticostéroïde inhalé. Ces analyses ont porté sur les scores des symptômes d'asthme diurnes et nocturnes, les réveils nocturnes et les prises d’un ꞵ2-agoniste de courte durée d'action. Elles ont montré des différences numériques constamment en faveur du groupe 12 SQ-HDM comparativement au groupe placebo pour tous les paramètres évalués durant les 4 semaines précédant la réduction de la dose du corticostéroïde inhalé. Les différences ont été statistiquement significatives uniquement pour le score des symptômes d'asthme diurnes (p = 0,0450) et l’odds radio pour l’absence de réveil nocturne (p = 0,0409).
Données complémentaires – asthme allergique
Lors d’un essai de phase II randomisé, en double aveugle et contrôlé contre placebo, 604 patients ≥ 14 ans présentant un asthme allergique aux acariens contrôlé par la prise d’un corticostéroïde inhalé (100 à 800 μg de budésonide) et ayant des antécédents de rhinite allergique aux acariens ont été randomisés pour recevoir 1, 3 ou 6 SQ-HDM ou un placebo pendant environ un an. Lors de la période d’évaluation de l’efficacité au cours des 4 dernières semaines de l'essai, la modification moyenne de la dose du corticostéroïde inhalé par rapport à la valeur initiale à l’entrée dans l'essai a été de 207,6 μg de budésonide dans le groupe 6 SQ-HDM et 126,3 μg dans le groupe placebo, correspondant à une différence absolue de 81 μg de budésonide par jour (IC95% : [27 ; 136], p = 0,004). Les réductions relatives moyennes et médianes de la dose du corticostéroïde inhalé par rapport à la valeur initiale à l’entrée dans l’essai ont été de 42% et 50% dans le groupe 6 SQ-HDM et de 15% et 25% dans le groupe placebo. Une analyse d’un sous-groupe (N = 108) de patients chez qui le contrôle de l’asthme était moindre et qui prenaient un corticostéroïde inhalé à raison de ≥ 400 μg de budésonide a été conduite a posteriori et a montré que la modification moyenne de la dose quotidienne du corticostéroïde inhalé par rapport à la valeur initiale à l’entrée dans l’essai était de 384,4 μg de budésonide dans le groupe 6 SQ-HDM et de 57,8 μg dans le groupe placebo, correspondant à une différence absolue de 327 μg de budésonide par jour (IC95% : [182 ; 471], p < 0,0001, analyse a posteriori).
Population pédiatrique
Efficacité clinique chez l’enfant
L’efficacité d’ACARIZAX 12 SQ-HDM dans le traitement des allergies respiratoires aux acariens chez l’enfant a été évaluée au cours de deux essais cliniques en double aveugle, randomisés, contrôlés versus placebo. L’objectif principal était d’évaluer l’efficacité dans la rhinite allergique dans l’essai MT-12 et dans l’asthme allergique dans l’essai MT-11.
Rhinite allergique :
Enfants âgés de 5 à 11 ans
Essai MATIC (MT-12)
L'efficacité d’ACARIZAX 12 SQ-HDM dans le traitement de la rhinite allergique aux acariens chez les enfants de 5 à 11 ans a été évaluée dans un essai en double aveugle, randomisé, contrôlé versus placebo (essai MATIC (MT-12)).
· L'essai MATIC (MT-12) a inclus 1 458 enfants (âgés de 5 à 11 ans) présentant une rhinite/rhinoconjonctivite allergique aux acariens modérée à sévère (score total combiné quotidien de rhinite (TCRS) moyen au début de l’essai de 18,3). Environ 40% de la population de l’essai présentait un asthme concomitant au début de l’essai. Les sujets ont été randomisés pour recevoir une prise quotidienne de 12 SQ-HDM ou d’un placebo pendant environ 1 an et pouvaient recourir librement à un traitement symptomatique standardisé de la rhinite et de la conjonctivite.
· Le critère primaire était le score total combiné quotidien moyen de rhinite (TCRS) évalué durant les 8 dernières semaines de traitement.
o Le TCRS est la somme du score quotidien des symptômes de rhinite (DSS) et du score quotidien médicamenteux de rhinite (DMS). Le score des symptômes de rhinite évaluait chacun des 4 symptômes nasaux (rhinorrhée, obstruction nasale, éternuements, prurit nasal) quotidiennement sur une échelle de 0 à 3 (symptômes absents, légers, modérés ou sévères) ; soit une échelle de score allant de 0 à 12. Le score médicamenteux de rhinite était la somme du score de la prise d’un corticostéroïde nasal (maximum 8 points/jour) et du score de la prise d’un antihistaminique oral (maximum 4 points/jour) ; soit une échelle de score allant de 0 à 12. L’échelle de score du TCRS allait ainsi de 0 à 24.
· Après 1 an de traitement avec 12 SQ-HDM, une différence absolue des moyennes ajustées de 0,97 (intervalle de confiance à 95 % [0,50 ; 1,44]) et une différence relative de 22 % (p < 0,0001) par rapport au placebo ont été observées. L’effet du traitement peut varier entre les patients selon leurs pathologies allergiques.
· L’apparition d’un effet clinique a été observée après 8 semaines de traitement (p = 0,01).
|
|
Essai MATIC : évolution du score total combiné de rhinite au cours du temps TCRS : score total combiné de rhinite (score des symptômes + score médicamenteux). Le TCRS a été mesuré comme une moyenne sur 2 semaines d'évaluation à partir de la semaine 8 et de la semaine 16. Le critère primaire était le TCRS quotidien moyen au cours des 8 dernières semaines de traitement environ (~ semaines 44 à 52). Moyennes ajustées du TCRS moyen au cours du temps. Les lignes verticales indiquent l’erreur standard pour ces moyennes ajustées. Les intervalles ne se chevauchant pas indiquent une différence statistiquement significative. |
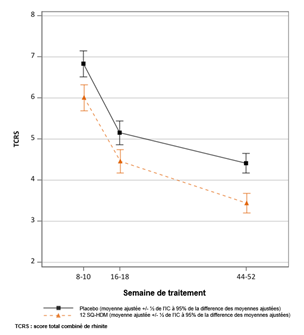
Une analyse en sous-groupe du critère primaire (TCRS) selon la présence ou non d’un asthme à l’état initial a montré une différence absolue des moyennes ajustées de 1,26 (intervalle de confiance à 95% [0,46 ; 2,06]) chez les enfants avec un asthme concomitant et de 0,77 (intervalle de confiance à 95% [0,19 ; 1,36]) chez les enfants sans asthme concomitant. Une analyse groupée du TCRS dans 5 essais de phase III chez des patients atteints de rhinite allergique aux acariens traités par 12 SQ-HDM ou placebo a montré une différence absolue des moyennes ajustées de 1,27 (intervalle de confiance à 95% [0,82 ; 1,72]) chez les patients avec un asthme concomitant (N = 1 450) et de 0,81 (intervalle de confiance à 95% [0,49 ; 1,13]) chez les patients sans asthme concomitant (N = 2 595).
Essai MATIC : graphique en « forest plots » de la différence de traitement du TCRS quotidien moyen dans les sous-groupes d’asthme à l’état initial – cas observé (FAS)
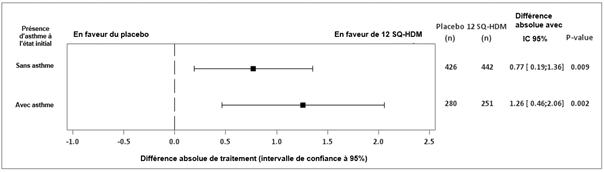
IC : Intervalle de confiance, FAS : analyse sur l'ensemble des données disponibles (full analysis set), n : nombre de sujets avec des observations contribuant à l'analyse, TCRS : score total combiné quotidien de rhinite.
Les analyses pré-définies des critères de jugement pour l'asthme ont évalué le score quotidien des symptômes d'asthme, la prise de bêta-agonistes de courte durée d'action, les jours sans prise de bêta-agonistes de courte durée d'action et les réveils nocturnes nécessitant la prise de bêta-agonistes de courte durée d'action. Les résultats ont montré des différences constamment en faveur de 12 SQ-HDM par rapport au placebo pour les 4 paramètres. Les différences étaient statistiquement significatives pour le score quotidien des symptômes d'asthme (p = 0,0259) et pour les réveils nocturnes nécessitant la prise d'un bêta-agoniste de courte durée d'action (p = 0,0279).
Enfants âgés de 5 à 17 ans
Essai MAPIT (MT-11)
· L'objectif principal était de démontrer l'efficacité d'ACARIZAX 12 SQ-HDM versus placebo chez les enfants et les adolescents (5-17 ans) atteints d'asthme allergique aux acariens ayant eu des exacerbations d'asthme cliniquement pertinentes après au moins 4 mois de traitement. ACARIZAX 12 SQ-HDM a été administré en complément du traitement de fond de l'asthme (CSI à faible dose associé avec β2-agonistes à longue durée d'action ou CSI à forte/moyenne dose avec ou sans β2-agonistes à longue durée d'action). La population de l'essai présentait également des antécédents cliniques de rhinite allergique aux acariens de toute sévérité (score total combiné de rhinite (TCRS) > 0 au début de l'essai ; TCRS moyen de 9,0 au début de l'essai). L'essai MT-11 n'a pas été conçu pour évaluer l'effet clinique sur la rhinite allergique. Les résultats des critères d’évaluation sur la rhinite (TCRS, DSS et DMS) sont présentés dans le tableau ci-dessous.
Résultats de l’essai MATIC |
12 SQ‑HDM |
Placebo |
Efficacité |
|
|||
|
Critères secondaires additionnels prédéfinisa |
N |
Score |
N |
Score |
Différence absolueb |
Différence relativec |
Valeur de pd |
|
Score total combiné de rhinite |
|||||||
|
FASe (moyenne ajustée) |
253 |
2,16 |
259 |
2,46 |
0,30 [-0,22; 0,81] |
12,1% |
0,2597 |
|
Score de symptômes de rhinite |
|||||||
|
FAS (moyenne ajustée) |
253 |
0,55 |
259 |
0,67 |
0,12 [-0,04; 0,28] |
18,2% |
0,1349 |
|
Score médicamenteux de rhinite |
|||||||
|
FAS (moyenne ajustée) |
253 |
1,27 |
259 |
1,40 |
0,12 [-0,24; 0,48] |
8,8% |
0,5071 |
FAS : analyse sur l’ensemble des données disponibles (full analysis set). N: nombre de sujets avec des observations contribuant à l’analyse.
aCritères d’évaluation de rhinite.
bDifférence absolue : placebo moins 12 SQ-HDM, intervalle de confiance à 95%.
cDifférence relative par rapport au placebo : placebo moins 12 SQ-HDM divisé par le placebo.
dLes valeurs de p n’ont pas été ajustées pour tenir compte de la multiplicité. Ainsi, les analyses sont à considérées comme exploratoires.
eToutes les données disponibles ont été utilisées dans leur intégralité, i.e. sujets ayant fourni des données durant la période d’évaluation d’efficacité.
Adolescents âgés de 12 à 17 ans
L’efficacité d’ACARIZAX 12 SQ-HDM dans le traitement de la rhinite allergique aux acariens chez les adolescents a été évaluée au cours de 2 essais cliniques randomisés, en double aveugle, contrôlés contre placebo (P001 et TO-203-3-2). Dans ces essais, une proportion de patients était des adolescents.
· L’essai P001 a inclus 189 adolescents (sur les 1 482 patients randomisés au total) présentant une rhinite/rhinoconjonctivite allergique aux acariens modérée à sévère avec ou sans asthme. Ces patients ont été randomisés pour recevoir une prise quotidienne de 12 SQ-HDM ou d’un placebo pendant environ 1 an et pouvaient recourir librement à un traitement symptomatique standardisé de la rhinite.
Le critère primaire était le score total combiné quotidien moyen de rhinite (total combined rhinitis score, TCRS) évalué durant les 8 dernières semaines de traitement.
Après 1 an de traitement par 12 SQ-HDM, la différence absolue des médianes a été de 1,0 (IC95% : [0,1 ; 2,0]) et la différence relative a été de 22% (p = 0,024) par rapport au placebo dans le groupe des adolescents.
· L’essai TO-203-3-2 a inclus 278 adolescents (sur les 851 patients randomisés au total) présentant une rhinite allergique aux acariens persistante modérée à sévère. Ces patients ont été randomisés pour recevoir une prise quotidienne de 12 SQ-HDM, de 6 SQ-HDM ou d’un placebo pendant environ 1 an et pouvaient recourir librement à un traitement symptomatique standardisé de la rhinite.
Le critère primaire était le score total combiné quotidien moyen de rhinite (total combined rhinitis score, TCRS) évalué durant les 8 dernières semaines de traitement.
A la fin de l’essai, après 1 an de traitement par 12 SQ-HDM, la différence absolue des moyennes a été de 1,0 (IC95% : [0,1 ; 1,9], p = 0,037) et la différence relative a été de 20% par rapport au placebo dans le groupe des adolescents.
Sous-groupe d’adolescents |
12 SQ-HDM |
Placebo |
Efficacité |
|
|||
|
Critère primaire : score TCRS |
N |
Score |
N |
Score |
Différence absolue |
Différence relative d |
Valeur de p |
|
P001 |
|||||||
|
FAS (moyenne ajustée) |
76 |
3,6 |
84 |
4,8 |
1,2 a [0,1 ; 2,3] |
25% |
<0,05 |
|
FAS (médiane) |
76 |
3,3 |
84 |
4,3 |
1,0 b [0,1 ; 2,0] |
22% |
0,024 |
|
TO-203-3-2 |
|||||||
|
FAS (moyenne ajustée) |
99 |
4,1 |
92 |
5,1 |
1,0 c [0,1 ; 1,9] |
20% |
0,037 |
|
FAS (médiane) |
99 |
4,2 |
92 |
5,2 |
1,0 |
19% |
- |
|
TCRS : score total combiné de rhinite a ANCOVA b Estimation selon la méthode Hodges-Lehmann avec un intervalle de confiance à 95% (analyse primaire dans l’essai P001) c Modèle linéaire à effets mixtes (analyse primaire dans l’essai TO-203-3-2) d Différence relative par rapport au placebo : placebo moins 12 SQ-HDM divisé par le placebo. |
|||||||
Asthme allergique :
Enfants âgés de 5 à 17 ans
L'essai MAPIT (MT-11) a inclus 533 enfants et adolescents (5-17 ans) atteints d'asthme allergique aux acariens. Les sujets avaient des antécédents récents d'exacerbations d'asthme alors qu'ils prenaient un traitement de fond de l'asthme (CSI à faible dose avec β2-agonistes de longue durée d'action ou CSI à dose moyenne/élevée avec ou sans β2-agonistes de longue durée d'action). Les sujets ont été randomisés pour recevoir une prise quotidienne de 12 SQ-HDM ou d’un placebo pendant environ 24-30 mois en complément de leur traitement de fond de l’asthme. Le critère primaire était le taux annualisé d'exacerbations d'asthme cliniquement pertinentes, calculé comme le nombre d'exacerbations par an et par sujet pendant la période d'évaluation de l'efficacité.
Le risque relatif d'exacerbations d'asthme (12 SQ-HDM divisé par le placebo) était en faveur de 12 SQ-HDM mais il n'y avait pas de différence statistiquement significative au niveau de l'effet du traitement entre les deux groupes (RR = 0,89, IC à 95 % [0,60 ; 1,31], p = 0,54).
Pour les sujets ayant participé à l'essai MAPIT (MT-11), le taux d'exacerbation d'asthme a été généralement faible dans les deux groupes au cours de l'essai et a diminué d'environ 67 % pendant la pandémie de COVID-19 par rapport au niveau observé avant la pandémie de COVID-19. En raison du faible taux d'exacerbation d'asthme observé dans les deux groupes, il n'a pas été possible de détecter une différence statistiquement significative (voir rubrique 4.2 pour des informations sur l'utilisation chez l’enfant).
L’Agence Européenne des Médicaments a accordé une dérogation à l’obligation de soumettre les résultats d’essais réalisés avec ACARIZAX chez des enfants âgés de moins de 5 ans dans l’allergie respiratoire aux acariens (traitement de la rhinite allergique, traitement de l’asthme).
Patients âgés
ACARIZAX n’est pas indiqué chez les patients de plus de 65 ans (voir rubrique 4.2). Il existe des données limitées sur la sécurité d’emploi et la tolérance chez des patients de plus de 65 ans.
Traitement à long terme
Les recommandations thérapeutiques internationales préconisent une durée d'immunothérapie allergénique d'environ 3 ans pour modifier l’évolution de la maladie. Des données d’efficacité issues de l’essai MITRA sont disponibles pour un traitement de 18 mois par ACARIZAX. L’efficacité à long terme n’a pas été établie.
5.2. Propriétés pharmacocinétiques
Aucune étude clinique du profil pharmacocinétique et du métabolisme d’ACARIZAX n’a été réalisée. L’effet d’une immunothérapie allergénique est médié par des mécanismes immunologiques et les informations disponibles sur les propriétés pharmacocinétiques sont limitées.
Les molécules actives d’un extrait allergénique sont essentiellement composées de protéines. Dans le cas de l’immunothérapie allergénique administrée par voie sublinguale, des études ont montré l’absence d’absorption passive des allergènes à travers la muqueuse buccale.
Des données indiquent que les allergènes seraient captés à travers la muqueuse buccale par les cellules dendritiques, en particulier les cellules de Langerhans. Les allergènes non absorbés de cette façon seraient hydrolysés en acides aminés et en petits polypeptides dans la lumière des voies digestives. Aucune donnée ne suggère que les allergènes présents dans ACARIZAX soient significativement absorbés dans le système vasculaire après administration sublinguale.
5.3. Données de sécurité préclinique
Mannitol
Hydroxyde de sodium (pour ajustement du pH)
4 ans.
6.4. Précautions particulières de conservation
Pas de précaution particulière de conservation.
6.5. Nature et contenu de l'emballage extérieur
Plaquettes (Aluminium/Aluminium) dans un emballage extérieur en carton. Chaque plaquette contient 10 lyophilisats sublinguaux.
Boîtes de 10, 30 et 90.
Toutes les présentations peuvent ne pas être commercialisées.
6.6. Précautions particulières d’élimination et de manipulation
Tout médicament non utilisé ou déchet doit être éliminé conformément à la réglementation en vigueur.
7. TITULAIRE DE L’AUTORISATION DE MISE SUR LE MARCHE
Bøge Alle 6-8
2970 Hørsholm
Danemark
8. NUMERO(S) D’AUTORISATION DE MISE SUR LE MARCHE
· 34009 300 453 3 6 : 10 lyophilisats sous plaquettes (Aluminium/Aluminium).
· 34009 300 453 4 3 : 30 lyophilisats sous plaquettes (Aluminium/Aluminium).
· 34009 302 864 3 2 : 90 lyophilisats sous plaquettes (Aluminium/Aluminium).
9. DATE DE PREMIERE AUTORISATION/DE RENOUVELLEMENT DE L’AUTORISATION
[à compléter ultérieurement par le titulaire]
Date de dernier renouvellement:{JJ mois AAAA}
10. DATE DE MISE A JOUR DU TEXTE
[à compléter ultérieurement par le titulaire]
{JJ mois AAAA}
Sans objet.
12. INSTRUCTIONS POUR LA PREPARATION DES RADIOPHARMACEUTIQUES
Liste I
ANSM - Mis à jour le : 16/02/2026
ACARIZAX 12 SQ-HDM, lyophilisat sublingual
Pour utilisation chez les adultes et les enfants (âgés de 5 à 65 ans)
Extrait allergénique standardisé d’acariens de la poussière de maison
(Dermatophagoides pteronyssinus et Dermatophagoides farinae)
· Gardez cette notice. Vous pourriez avoir besoin de la relire.
· Si vous avez d’autres questions, interrogez votre médecin ou votre pharmacien.
· Ce médicament vous a été personnellement prescrit. Ne le donnez pas à d’autres personnes. Il pourrait leur être nocif, même si les signes de leur maladie sont identiques aux vôtres.
· Si vous ressentez un quelconque effet indésirable, parlez-en à votre médecin ou votre pharmacien. Ceci s’applique aussi à tout effet indésirable qui ne serait pas mentionné dans cette notice. Voir rubrique 4.
1. Qu'est-ce que ACARIZAX 12 SQ-HDM, lyophilisat sublingual et dans quels cas est-il utilisé ?
2. Quelles sont les informations à connaître avant de prendre ACARIZAX 12 SQ-HDM, lyophilisat sublingual ?
3. Comment prendre ACARIZAX 12 SQ-HDM, lyophilisat sublingual ?
4. Quels sont les effets indésirables éventuels ?
5. Comment conserver ACARIZAX 12 SQ-HDM, lyophilisat sublingual ?
6. Contenu de l’emballage et autres informations.
1. QU’EST-CE QUE ACARIZAX 12 SQ-HDM, lyophilisat sublingual ET DANS QUELS CAS EST-IL UTILISE ?
Classe pharmacothérapeutique : extraits allergéniques, acariens, code ATC : V01AA03.
ACARIZAX contient un extrait allergénique d’acariens. Il se présente sous la forme d'un lyophilisat sublingual qui est similaire à un comprimé mais plus fragile. L’extrait est absorbé dans l’organisme en plaçant le lyophilisat sous la langue.
ACARIZAX est utilisé dans le traitement de la rhinite allergique (inflammation de la muqueuse du nez) chez les adultes et les enfants (âgés de 5 à 65 ans) et de l’asthme allergique chez les adultes (âgés de 18 à 65 ans), provoqués par les acariens. ACARIZAX agit en améliorant votre tolérance immunitaire (aptitude de votre organisme à se défendre) aux acariens. Il peut être nécessaire de prendre le traitement pendant 8 à 14 semaines avant de remarquer une amélioration des symptômes.
Le médecin évaluera vos symptômes allergiques et réalisera un test cutané (prick test) et/ou un prélèvement sanguin afin de confirmer le bien-fondé d’un traitement par ACARIZAX.
Il est recommandé de prendre la première dose d’ACARIZAX sous la surveillance du médecin. Vous devrez rester sous surveillance médicale pendant au moins 30 minutes après la prise de la première dose, afin de surveiller votre tolérance au médicament. Cela vous donnera aussi la possibilité de discuter avec votre médecin de tout effet secondaire que vous pourriez ressentir.
ACARIZAX doit être prescrit par des médecins expérimentés dans le traitement des allergies.
2. QUELLES SONT LES INFORMATIONS A CONNAITRE AVANT DE PRENDRE ACARIZAX 12 SQ-HDM, lyophilisat sublingual ?
Ne prenez jamais ACARIZAX 12 SQ-HDM, lyophilisat sublingual :
· Si vous êtes allergique à l’un des autres composants contenus dans ce médicament, mentionnés dans la rubrique 6.
· Si votre fonction respiratoire est altérée (à déterminer avec votre médecin).
· Si vous avez un asthme sévère qui s’est aggravé au cours des trois derniers mois (à déterminer avec votre médecin).
· Si vous avez un asthme et une infection respiratoire en cours d'évolution, tel qu'un rhume, des maux de gorge ou une pneumonie, le jour où vous devez prendre la première dose d’ACARIZAX. Votre médecin reportera le début de votre traitement jusqu’à ce que votre état de santé s’améliore.
· Si vous avez une maladie qui affecte votre système immunitaire, vous prenez des médicaments qui affaiblissent votre système immunitaire ou vous êtes atteint d’un cancer.
· Si vous avez récemment subi une extraction dentaire ou tout autre acte de chirurgie de la bouche, ou vous présentez des aphtes ou une infection de la bouche. Votre médecin pourra reporter le début de votre traitement à une date ultérieure ou interrompre votre traitement jusqu’à la cicatrisation de votre bouche.
Avertissements et précautions
Adressez-vous à votre médecin avant de prendre ACARIZAX :
· Si vous êtes traité pour une dépression par des antidépresseurs tricycliques ou des inhibiteurs de la monoamine-oxydase (IMAO) ou pour une maladie de Parkinson par des inhibiteurs de la catéchol-O-méthyltransférase (COMT).
· Si vous avez déjà eu une réaction allergique sévère à la suite d'une injection d’un extrait allergénique d’acariens.
· Si vous êtes allergique au poisson. ACARIZAX peut contenir des traces de protéines de poisson. Les données disponibles ne montrent pas de risque accru de réactions allergiques chez les patients souffrant d’une allergie au poisson.
· Si vous ressentez des symptômes allergiques sévères, tels qu'une difficulté à avaler ou à respirer, des modifications de votre voix, une hypotension (pression artérielle basse) ou une sensation de corps étranger dans la gorge. Arrêtez le traitement et contactez immédiatement votre médecin.
· Si vos symptômes d’asthme s’aggravent nettement. Arrêtez le traitement et contactez immédiatement votre médecin.
En cas d’asthme, vous devez continuer à prendre vos médicaments antiasthmatiques habituels après avoir commencé votre traitement par ACARIZAX. Votre médecin vous expliquera comment réduire progressivement les traitements antiasthmatiques au cours du temps.
Vous devez arrêter de prendre ACARIZAX et contacter votre médecin si vous ressentez des brûlures d’estomac sévères ou persistantes ou des difficultés à avaler car ces symptômes peuvent être le signe d’une inflammation allergique de l’œsophage.
Certaines réactions allergiques locales légères à modérées sont prévisibles durant votre traitement. Cependant, en cas de réaction sévère, contactez votre médecin, qui déterminera les médicaments antiallergiques dont vous pourriez avoir besoin tels que les antihistaminiques.
Ce médicament contient moins de 1 mmol (23 mg) de sodium par dose, c’est-à-dire qu’il est essentiellement « sans sodium ».
Enfants
Rhinite allergique (inflammation de la muqueuse du nez) :
ACARIZAX n’est pas indiqué chez les enfants de moins de 5 ans.
Asthme allergique :
ACARIZAX n’est pas destiné au traitement de l’asthme allergique chez les enfants de moins de 18 ans.
Autres médicaments et ACARIZAX 12 SQ-HDM, lyophilisat sublingual
Informez votre médecin ou pharmacien si vous prenez ou avez récemment pris tout autre médicament, y compris un médicament obtenu sans ordonnance. Si vous prenez d’autres médicaments pour traiter les symptômes de votre allergie tels que des antihistaminiques, des médicaments pour soulager les symptômes d’asthme ou des corticoïdes, informez-en votre médecin afin qu’il puisse vous indiquer comment les prendre au cours de votre traitement par ACARIZAX. Les effets indésirables provoqués par ACARIZAX pourraient être plus nombreux ou plus intenses si vous arrêtez de prendre ces médicaments pour traiter vos symptômes allergiques.
ACARIZAX 12 SQ-HDM, lyophilisat sublingual avec des aliments et boissons
Ne pas absorber d’aliments ni de boissons dans les 5 minutes suivant la prise de ce médicament.
A l’heure actuelle, aucune donnée concernant l’utilisation d’ACARIZAX pendant la grossesse n’est disponible. Le traitement par ACARIZAX ne devra pas être débuté pendant la grossesse. Si vous devenez enceinte pendant le traitement, parlez-en à votre médecin pour qu’il évalue s’il est adapté ou non de poursuivre le traitement.
A l’heure actuelle, aucune donnée concernant l’utilisation d’ACARIZAX pendant l’allaitement n’est disponible. Il n’est cependant pas attendu d’effets chez les enfants allaités. Demandez à votre médecin si vous pouvez continuer à prendre ACARIZAX pendant l’allaitement.
Conduite de véhicules et utilisation de machines
Le traitement par ACARIZAX n'a pas d'effet ou a un effet négligeable sur la capacité à conduire un véhicule ou à utiliser des machines. Cependant, vous êtes seul apte à évaluer si vous ressentez une influence. Vous devez donc lire toutes les informations contenues dans cette notice, particulièrement la rubrique 4 « Quels sont les effets indésirables éventuels ? ». En cas de doute, demandez conseil à votre médecin ou votre pharmacien.
ACARIZAX 12 SQ-HDM, lyophilisat sublingual contient <{nommer le/les excipient (s)}>
Sans objet.
3. COMMENT PRENDRE ACARIZAX 12 SQ-HDM, lyophilisat sublingual ?
La posologie habituelle est d’un lyophilisat sublingual par jour. Votre médecin vous informera de la durée du traitement par ACARIZAX.
Assurez-vous que vos mains soient sèches avant de manipuler le médicament.
Prenez le médicament de la façon suivante :
|
|
|
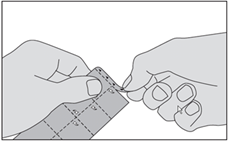
1. Détachez la bande marquée de triangles en haut de la plaquette. |
|
|
|
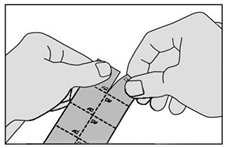
2. Détachez un carré de la plaquette le long des lignes perforées. |
|
|
|
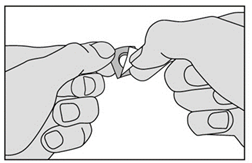
3. Ne faites pas sortir le médicament en le poussant au travers de la pellicule. Ceci pourrait l'endommager car il est friable. Retirez la pellicule en la tirant à partir du coin marqué. |
|
|
|
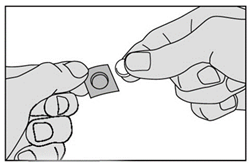
4. Retirez délicatement le médicament de la pellicule et prenez-le immédiatement. |
|
|
|
5. Placez le médicament sous la langue. Laissez-le en place jusqu’à ce qu’il soit dissous. Ne pas avaler pendant 1 minute. Ne pas manger ni boire pendant au moins 5 minutes. |
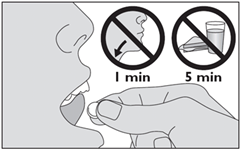
Si vous avez pris plus d’ACARIZAX 12 SQ-HDM, lyophilisat sublingual que vous n’auriez dû
Si vous avez pris trop de lyophilisats sublinguaux, vous pouvez ressentir des symptômes allergiques y compris des symptômes locaux au niveau de la bouche et de la gorge. Si les symptômes qui apparaissent sont de forte intensité, contactez immédiatement un médecin ou un hôpital.
Si vous oubliez de prendre ACARIZAX 12 SQ-HDM, lyophilisat sublingual
Si vous n’avez pas pris ACARIZAX pendant plus de 7 jours, contactez votre médecin avant de reprendre votre traitement par ACARIZAX.
Si vous arrêtez de prendre ACARIZAX 12 SQ-HDM, lyophilisat sublingual
Si vous ne prenez pas ce médicament selon les prescriptions, l’effet du traitement peut ne pas se faire sentir.
Si vous avez d’autres questions sur l’utilisation de ce médicament, demandez plus d’informations à votre médecin ou à votre pharmacien.
4. QUELS SONT LES EFFETS INDESIRABLES EVENTUELS ?
Les effets indésirables peuvent être une réponse allergique à l'allergène avec lequel vous êtes traité. En général, les effets indésirables durent de quelques minutes à plusieurs heures après la prise du médicament et diminuent en un à trois mois après le début du traitement.
Effets indésirables les plus graves :
Peu fréquents (pouvant concerner jusqu’à 1 personne sur 100) :
· Réaction allergique grave
Arrêtez de prendre ACARIZAX et contactez immédiatement votre médecin ou un hôpital si vous présentez l'un des symptômes suivants :
· Aggravation d'un asthme existant
· Gonflement rapide du visage, de la bouche, de la gorge ou de la peau
· Difficultés à avaler
· Difficultés à respirer
· Modifications de la voix
· Pression artérielle basse
· Sensation de gorge obstruée (comme un gonflement)
· Urticaire et démangeaisons de la peau
Autres effets indésirables possibles :
Très fréquents (pouvant concerner plus d’1 personne sur 10) :
· Sensation d’irritation dans la gorge
· Gonflement de la bouche et des lèvres
· Démangeaisons de la bouche et des oreilles
· Infections des voies respiratoires
Fréquents (pouvant concerner jusqu’à 1 personne sur 10) :
· Sensation de fourmillements ou engourdissement de la bouche ou de la langue
· Démangeaisons des yeux
· Démangeaisons de la langue et des lèvres
· Gonflement de la langue ou de la gorge
· Inflammation, gêne ou sensation de brûlure dans la bouche
· Rougeur ou ulcération dans la bouche
· Douleur dans la bouche
· Altération du goût
· Douleur ou inconfort de l’estomac
· Diarrhée
· Sensation d'être malade (nausées) et vomissements
· Douleur en avalant ou difficultés à avaler
· Symptômes d’asthme
· Toux
· Essoufflement
· Gêne thoracique
· Indigestion et brûlures d’estomac
· Enrouement
· Fatigue
· Urticaire et démangeaisons de la peau
Peu fréquents (pouvant concerner jusqu’à 1 personne sur 100) :
· Inflammation des yeux
· Sensation de battements cardiaques rapides, intenses ou irréguliers
· Inconfort au niveau des oreilles
· Sensation de gorge serrée
· Gêne nasale, sensation de nez bouché ou écoulement nasal, éternuements
· Cloques dans la bouche
· Irritation de l’œsophage
· Sensation de corps étranger bloqué dans la gorge
· Sensations vertigineuses
· Sensation de malaise général
· Sécheresse de la bouche
· Sensation de picotements de la peau
· Rougeur de la gorge
· Augmentation de taille des amygdales
· Douleur des lèvres
· Ulcération des lèvres
· Augmentation de taille des glandes salivaires
· Augmentation de la production de salive
· Rougeur de la peau
· Eruption cutanée
Rare (pouvant concerner jusqu’à 1 personne sur 1 000) :
· Gonflement rapide du visage ou de la peau
· Inflammation allergique de l’œsophage (œsophagite à éosinophiles)
Si un effet indésirable vous inquiète ou vous gêne, contactez votre médecin qui déterminera les médicaments dont vous pourriez avoir besoin tels que les antihistaminiques afin de vous soulager.
Effets indésirables chez les enfants
Les effets secondaires attendus chez les enfants sont similaires aux effets indésirables chez les adultes et les adolescents.
Certains effets indésirables peuvent être plus fréquents chez les enfants :
Très fréquent (pouvant concerner plus d’1 personne sur 10) : gonflement de la langue ou de la gorge, ulcération dans la bouche, douleur dans la bouche, altération du goût, douleur à l’estomac, diarrhée, sensation d’être malade (nausées).
Fréquent (pouvant concerner jusqu’à 1 personne sur 10) : inflammation des yeux.
Peu fréquent (pouvant concerner jusqu’à 1 personne sur 100) : gonflement rapide du visage et de la peau et inflammation allergique de l’œsophage (œsophagite à éosinophiles).
Déclaration des effets secondaires
Si vous ressentez un quelconque effet indésirable, parlez-en à votre médecin ou votre pharmacien. Ceci s’applique aussi à tout effet indésirable qui ne serait pas mentionné dans cette notice. Vous pouvez également déclarer les effets indésirables directement via le système national de déclaration : Agence nationale de sécurité du médicament et des produits de santé (ANSM) et réseau des Centres Régionaux de Pharmacovigilance - Site internet: https://signalement.social-sante.gouv.fr/
En signalant les effets indésirables, vous contribuez à fournir davantage d’informations sur la sécurité du médicament.
5. COMMENT CONSERVER ACARIZAX 12 SQ-HDM, lyophilisat sublingual ?
Tenir ce médicament hors de la vue et de la portée des enfants.
N’utilisez pas ce médicament après la date de péremption indiquée sur la plaquette et la boîte après EXP. La date de péremption fait référence au dernier jour de ce mois.
Pas de précautions particulières de conservation.
Ne jetez aucun médicament au tout-à-l’égout ou avec les ordures ménagères. Demandez à votre pharmacien d’éliminer les médicaments que vous n’utilisez plus. Ces mesures contribueront à protéger l’environnement.
6. CONTENU DE L’EMBALLAGE ET AUTRES INFORMATIONS
Ce que contient ACARIZAX 12 SQ-HDM, lyophilisat sublingual
· La substance active est : un extrait allergénique standardisé d’acariens de la poussière de maison Dermatophagoides pteronyssinus et Dermatophagoides farinae. L’activité par lyophilisat sublingual est exprimée en unité SQ-HDM. L’activité d’un lyophilisat sublingual est de 12 SQ-HDM.
· Les autres composants sont : la gélatine (à base de poisson), le mannitol et l’hydroxyde de sodium (pour ajustement du pH).
Qu’est-ce que ACARIZAX 12 SQ-HDM, lyophilisat sublingual et contenu de l’emballage extérieur
Lyophilisat sublingual blanc à blanc cassé circulaire avec image imprimée en creux sur un côté.
Plaquettes thermoformées (aluminium) avec pellicule amovible (aluminium), dans un emballage extérieur en carton. Chaque plaquette contient 10 lyophilisats sublinguaux.
Boîtes de 10, 30 ou 90 lyophilisats sublinguaux.
Toutes les présentations peuvent ne pas être commercialisées.
Titulaire de l’autorisation de mise sur le marché
BØGE ALLÉ 6-8
2970 HØRSHOLM
DANEMARK
Exploitant de l’autorisation de mise sur le marché
ALK
TOUR W
102 TERRASSE BOIELDIEU
92800 PUTEAUX
MIGUEL FLETA 19
28037 MADRID
ESPAGNE
Noms du médicament dans les Etats membres de l'Espace Economique Européen
Ce médicament est autorisé dans les Etats membres de l'Espace Economique Européen sous les noms suivants : Conformément à la réglementation en vigueur.
[À compléter ultérieurement par le titulaire]
La dernière date à laquelle cette notice a été révisée est :
[à compléter ultérieurement par le titulaire]
{MM/AAAA}
Des informations détaillées sur ce médicament sont disponibles sur le site Internet de l’ANSM (France).