Dernière mise à jour le 28/04/2026
OESCLIM 50 microgrammes/24 heures (10 mg/22 cm2), dispositif transdermique
Ce médicament n'est ou ne sera bientôt plus disponible sur le marché.
Si vous prenez actuellement ce médicament, il vous est recommandé d'en parler avec votre médecin
ou avec votre pharmacien qui pourra vous orienter vers un autre traitement.
Indications thérapeutiques
Classe pharmacothérapeutique :
OESCLIM est un traitement hormonal substitutif (THS). Il contient du 17-b estradiol. OESCLIM est utilisé chez les femmes ménopausées dont les dernières règles datent d’au moins 6 mois.
OESCLIM est utilisé pour :
Soulager les symptômes apparaissant après la ménopause.
Lors de la ménopause, la quantité d’estrogènes produits par l’organisme féminin chute. Chez certaines femmes, cette chute se traduit par des symptômes tels qu’une sensation de chaleur au niveau du visage, du cou et de la poitrine (les « bouffées de chaleur »). OESCLIM soulage ces symptômes après la ménopause.
OESCLIM vous sera prescrit uniquement si vos symptômes altèrent gravement votre vie quotidienne.
Service médical rendu (SMR)
Les libellés affichés ci-dessous ne sont que des résumés ou extraits issus des avis rendus par la Commission de la Transparence. Seul l'avis complet de la Commission de la Transparence fait référence.
Cet avis est consultable à partir du lien `Avis du jj/mm/aaaa` ou encore sur demande auprès de la HAS (plus d'informations dans l'aide). Les avis et synthèses d'avis contiennent un paragraphe sur la place du médicament dans la stratégie thérapeutique.
| Valeur du SMR | Avis | Motif de l'évaluation | Résumé de l'avis |
|---|---|---|---|
| Important | Avis du 25/11/2015 | Renouvellement d'inscription (CT) | Le service médical rendu par les spécialités OESCLIM dans le traitement hormonal substitutif (THS) des symptômes de déficit en estrogènes chez les femmes ménopausées reste important chez les patientes dont les troubles du climatère sont ressentis comme suffisamment gênant pour altérer leur qualité de vie, lorsque ces spécialités sont utilisées selon les préconisations de la Commission. |
Amélioration du service médical rendu (ASMR)
Pas d'ASMR disponible pour ce médicament (plus d'informations dans l'aide )
ANSM - Mis à jour le : 27/09/2023
OESCLIM 50 microgrammes/24 heures (10 mg/22 cm²), dispositif transdermique
2. COMPOSITION QUALITATIVE ET QUANTITATIVE
Estradiol hémihydraté............................................................................................................. 10 mg
Pour un dispositif transdermique de 22 cm²
Un dispositif transdermique libère 50 microgrammes d'estradiol hémihydraté par 24 heures.
Pour la liste complète des excipients, voir rubrique 6.1.
Dispositif transdermique composé d'une matrice polymérique auto-adhésive contenant de l’estradiol hémihydraté, enduite sur un support mousse rectangulaire à angles arrondis. La face adhésive est recouverte d'un feuillet de protection transparent.
4.1. Indications thérapeutiques
L’expérience de ce traitement chez les femmes âgées de plus de 65 ans est limitée.
4.2. Posologie et mode d'administration
Appliquer OESCLIM deux fois par semaine, c'est-à-dire renouveler le dispositif transdermique tous les 3 ou 4 jours.
Pour débuter ou poursuivre un traitement dans l'indication des symptômes post-ménopausiques, la dose minimale efficace doit être utilisée pendant la plus courte durée possible (voir rubrique 4.4).
Ainsi, la posologie usuelle recommandée pour débuter le traitement est d’un dispositif d’OESCLIM 50 microgrammes/24 heures deux fois par semaine.
En fonction de l'évolution clinique, la posologie doit être adaptée aux besoins individuels :
· si la dose choisie n'a pas corrigé les signes et symptômes de déficit estrogénique, un dosage plus fort doit être administré,
· l'apparition d'une sensation de tension des seins, de métrorragies, de rétention d’eau ou ballonnements (persistant pendant plus de 6 semaines) ou d'une irritabilité indique en général que la dose est trop élevée et doit être modifiée.
OESCLIM 50 microgrammes/24 heures peut être utilisé selon le schéma thérapeutique :
· Cyclique (discontinu) pendant 24 à 28 jours, suivis d'un intervalle libre de tout traitement de 2 à 7 jours. Durant cet intervalle, des hémorragies de privation peuvent apparaître.
· Continu, sans aucune période d'arrêt du traitement.
Un traitement continu, non cyclique, peut être indiqué dans le cas où les symptômes de déficit estrogénique se manifestent à nouveau fortement au cours de l'intervalle libre.
S’il s’agit d’une prescription chez une femme ne prenant pas de THS ou d’un relais d’un THS combiné continu, le traitement peut être commencé n’importe quel jour.
Par contre, si le traitement préalable est un THS séquentiel, le cycle de traitement en cours doit être terminé avant de commencer un traitement par OESCLIM 50 microgrammes/24 heures.
Chez les femmes non hystérectomisées, un progestatif doit être ajouté au moins 12 à 14 jours par cycle pour s'opposer au développement d'une hyperplasie endométriale induite par l'estrogène.
Le traitement séquentiel par des progestatifs doit se faire selon le schéma suivant :
Si OESCLIM est administré de façon cyclique (discontinue), le progestatif sera administré durant au moins les 12 à 14 derniers jours du traitement par l'estradiol. Ainsi, il n'y aura aucune administration hormonale pendant l'intervalle libre de chaque cycle.
Si OESCLIM est administré de façon continue (traitement continu séquentiel), il est recommandé de prendre le progestatif durant au moins 12 à 14 jours chaque mois.
Dans les deux cas, des hémorragies de privation peuvent apparaître après l'arrêt du traitement par le progestatif.
Chez les femmes hystérectomisées, il n'est pas recommandé d'associer un progestatif sauf en cas d'antécédent d'endométriose connue.
Mode d’administration
OESCLIM est un dispositif transdermique composé d'une matrice polymérique auto-adhésive contenant du 17β-estradiol, enduite sur un support mousse rectangulaire à angles arrondis. La face adhésive est recouverte d'un feuillet de protection transparent.
Une fois le feuillet de protection détaché, OESCLIM doit immédiatement être appliqué sur la fesse, le tronc ou le haut du bras ou de la cuisse à un endroit ne présentant pas de plis importants et qui ne soit pas le siège de frottements vestimentaires.
La peau doit être sèche, ne doit pas être irritée ou traitée par des produits huileux ou gras.
L’estradiol étant dégradé par les rayons ultraviolets, le dispositif transdermique ne doit pas être exposé directement au soleil.
OESCLIM ne doit pas être appliqué sur les seins. Il est recommandé de ne pas l'appliquer 2 fois de suite au même endroit.
Il est possible de se doucher ou de prendre un bain tout en gardant le dispositif transdermique.
Dans le cas d'un décollement du dispositif transdermique (eau très chaude, hypersudation, frottement anormal de vêtements), il est recommandé de le replacer sur la peau sèche. Si cela n'est pas possible, utilisez un dispositif transdermique neuf qui sera retiré à la date initialement prévue. Reprendre ensuite le rythme de changement du dispositif transdermique conformément au schéma thérapeutique initial.
En cas d’oubli de remplacement d’OESCLIM 50 microgrammes/24 heures, un nouveau dispositif transdermique doit être appliqué dès que possible. Reprendre ensuite le rythme de changement du dispositif transdermique conformément au schéma thérapeutique initial. L'oubli de l’application d’un patch peut favoriser la récurrence des symptômes et la survenue de saignements et de spottings.
· Cancer du sein connu ou suspecté ou antécédents de cancer du sein.
· Tumeurs malignes estrogéno-dépendantes connues ou suspectées (exemple : cancer de l'endomètre).
· Hémorragie génitale non diagnostiquée.
· Hyperplasie endométriale non traitée.
· Antécédent d'accident thrombo-embolique veineux ou accident thrombo-embolique veineux en évolution (thrombose veineuse profonde, embolie pulmonaire).
· Troubles thrombophiliques connus (exemple : déficit en protéine C, protéine S ou antithrombine) (voir rubrique 4.4).
· Accident thrombo-embolique artériel récent ou en évolution (exemple : angor, infarctus du myocarde).
· Affection hépatique aiguë ou antécédents d'affection hépatique, jusqu'à normalisation des tests hépatiques.
· Porphyrie.
4.4. Mises en garde spéciales et précautions d'emploi
Les preuves de l’existence de risques associés à un THS dans le traitement des femmes ménopausées prématurément sont limitées. En raison du faible niveau du risque absolu chez les femmes plus jeunes, le rapport bénéfice/risque pourrait cependant être plus favorable que chez les femmes plus âgées.
Examen clinique et surveillance
Avant de débuter ou de recommencer un traitement hormonal substitutif (THS), il est indispensable d’effectuer un examen clinique et gynécologique complet (y compris le recueil des antécédents médicaux personnels et familiaux), en tenant compte des contre-indications et précautions d’emploi. Pendant toute la durée du traitement, des examens réguliers seront effectués, leur nature et leur fréquence étant adaptées à chaque patiente.
Les femmes doivent être informées du type d’anomalies mammaires pouvant survenir sous traitement ; ces anomalies doivent être signalées au médecin traitant (voir paragraphe « cancer du sein » ci-dessous). Les examens, y compris des examens appropriés par imagerie tels qu’une mammographie, doivent être pratiqués selon les recommandations en vigueur, et adaptés à chaque patiente.
Les femmes doivent être informées qu’OESCLIM 50 microgrammes/24 heures n’est pas un contraceptif et ne restaure pas la fertilité.
Conditions nécessitant une surveillance
Si l’une des affections suivantes survient, est survenue précédemment, et/ou s’est aggravée au cours d’une grossesse ou d’un précédent traitement hormonal, la patiente devra être étroitement surveillée et le rapport bénéfice/risque du THS réévalué individuellement. Les affections suivantes peuvent réapparaître ou s’aggraver au cours du traitement par OESCLIM 50 microgrammes/24 heures, en particulier :
· léiomyome (fibrome utérin) ou endométriose,
· présence de facteurs de risque thrombo-emboliques (voir ci-dessous),
· facteurs de risque de tumeurs estrogéno-dépendantes, par exemple : 1er degré d’hérédité pour le cancer du sein,
· hypertension artérielle,
· troubles hépatiques (par exemple : adénome hépatique),
· diabète avec ou sans atteinte vasculaire,
· lithiase biliaire,
· migraines ou céphalées sévères,
· lupus érythémateux disséminé,
· antécédent d’hyperplasie endométriale (voir ci-dessous),
· épilepsie,
· asthme,
· otospongiose.
Allergie de contact
Comme avec toute formulation locale, bien que cela soit extrêmement rare, une sensibilisation de contact peut survenir. Les femmes qui présentent une sensibilisation de contact à l’un des composants du patch doivent être averties qu’une réaction sévère d’hypersensibilité peut survenir si l’exposition au produit responsable est maintenue.
Le traitement doit être arrêté immédiatement en cas de survenue d’une contre-indication ou dans les cas suivants :
· ictère ou altération de la fonction hépatique,
· augmentation significative de la pression artérielle,
· céphalées de type migraine inhabituelle,
· grossesse.
Hyperplasie endométriale et cancer de l’endomètre
Chez les femmes ayant un utérus intact, le risque d’hyperplasie endométriale et de cancer de l’endomètre augmente en cas d’administration prolongée d’estrogènes seuls. Le risque de cancer de l’endomètre est de 2 à 12 fois supérieur comparé aux femmes ne prenant pas d’estrogènes, selon la durée du traitement et la dose d’estrogènes utilisée (voir rubrique 4.8).
Après arrêt du traitement, le risque peut rester élevé pendant au moins 10 ans.
Chez les femmes non hystérectomisées, l’association d’un progestatif de façon cyclique pendant au moins 12 jours par mois / cycle de 28 jours ou la prise d’un traitement estro-progestatif combiné continu empêche l’augmentation du risque associée aux estrogènes seuls comme THS.
Des métrorragies et des spottings peuvent survenir au cours des premiers mois de traitement. La survenue de saignements irréguliers plusieurs mois après le début du traitement ou la persistance de saignements après l’arrêt du traitement doivent faire rechercher une pathologie sous-jacente. Cette démarche peut nécessiter une biopsie endométriale afin d’éliminer une pathologie maligne.
La stimulation par les estrogènes peut conduire à une transformation maligne ou prémaligne des foyers résiduels d'endométriose. L'association d'un progestatif à l'estrogène doit être envisagée en cas de foyers résiduels d'endométriose chez les femmes qui ont subi une hystérectomie suite à une endométriose.
Cancer du sein
L’ensemble des données disponibles montre un risque accru de cancer du sein chez les femmes prenant un traitement estro-progestatif, ou chez celles prenant un THS à base d’estrogènes seuls, ce risque étant dépendant de la durée du traitement.
Traitement estro-progestatif combiné
· L’essai randomisé contrôlé versus placebo Women’s Health Initiative study (WHI) et une méta-analyse des études épidémiologiques prospectives montrent tous deux une augmentation du risque de survenue de cancer du sein chez les femmes traitées par un THS estro-progestatif combiné, apparaissant au bout d’environ 3 (1-4) ans de traitement (voir rubrique 4.8).
Traitement par des estrogènes seuls
· L’étude WHI n’a pas montré d’augmentation du risque de survenue du cancer du sein chez les femmes hystérectomisées utilisant des estrogènes seuls comme THS. Les études observationnelles ont généralement rapporté une légère augmentation du risque de cancer du sein diagnostiqué, ce risque étant plus faible que chez les utilisatrices d’association estrogènes-progestatifs (voir rubrique 4.8).
Les THS, particulièrement les traitements combinés estrogène/progestatif, augmentent la densité mammaire à la mammographie, ce qui pourrait gêner le diagnostic de cancer du sein.
Accidents thrombo-emboliques veineux
Le THS est associé à un risque 1,3 à 3 fois plus élevé d'accidents thrombo-emboliques veineux (thrombose veineuse profonde ou embolie pulmonaire). Cet événement survient plutôt au cours de la première année de traitement (voir rubrique 4.8).
Les patientes présentant une thrombophilie connue ont un risque accru d’accident thrombo-embolique veineux. Le THS pourrait majorer ce risque. Chez ces patientes, l’utilisation d’un THS est contre-indiquée (voir rubrique 4.3).
Les facteurs de risque reconnus d’accidents thrombo-emboliques veineux sont : utilisation d’estrogènes, âge, chirurgie importante, immobilisation prolongée, obésité (IMC > 30 kg/m²), grossesse/post-partum, lupus érythémateux disséminé (LED), cancer. En revanche, il n’existe aucun consensus sur le rôle possible des varices dans les accidents thrombo-emboliques veineux.
Afin de prévenir tout risque thrombo-embolique veineux post-opératoire, les mesures prophylactiques habituelles doivent être strictement appliquées. En cas d’immobilisation prolongée suite à une intervention chirurgicale programmée, une interruption provisoire du traitement 4 à 6 semaines avant l’intervention est recommandée. Le traitement ne sera réinstauré que lorsque la patiente aura repris une mobilité normale.
Chez les femmes sans antécédents de thrombose veineuse mais avec un membre de la famille proche ayant des antécédents de thrombose à un jeune âge, des examens peuvent être proposés, tout en informant de leurs limites (seuls certains types de troubles thrombophiliques sont identifiés lors de ces examens). Si un trouble thrombophilique lié à des thromboses chez des membres de la famille est identifié ou si le trouble est sévère (par exemple déficits en antithrombine, en protéine S ou protéine C, ou combinaison de troubles), le THS est contre-indiqué.
Chez les femmes suivant déjà un traitement à long terme par anticoagulants, le rapport bénéfice/risque d’un THS doit être évalué avec précaution.
La survenue d’un accident thrombo-embolique impose l’arrêt du THS. Les patientes devront être informées de la nécessité de contacter immédiatement leur médecin en cas de survenue de signes évoquant une thrombose tels que gonflement douloureux d’une jambe, douleurs soudaines dans la poitrine ou dyspnée.
Maladie coronarienne
Les études randomisées contrôlées n’ont pas mis en évidence de protection contre l’infarctus du myocarde chez les femmes avec ou sans maladie coronarienne préexistante traitées par une association d’estro-progestatifs ou par des estrogènes seuls.
Traitement par une association estro-progestative
Le risque relatif de maladie coronarienne est légèrement augmenté lors d’un traitement par une association estro-progestative. Puisque le risque absolu de base de maladie coronarienne dépend fortement de l’âge, le nombre de cas supplémentaire de maladie coronarienne due à l’association estro-progestative est très faible chez les femmes en bonne santé proches de la ménopause, mais augmente avec l’âge.
Traitement par des estrogènes seuls
Les études randomisées contrôlées n’ont pas mis en évidence d’augmentation du risque de maladie coronarienne chez les femmes hystérectomisées utilisant des estrogènes seuls.
Accidents vasculaires cérébraux
Une augmentation jusqu’à 1,5 fois du risque d’accident vasculaire cérébral ischémique a été montrée chez les femmes traitées par une association d’estro-progestatifs ou des estrogènes seuls. Le risque relatif ne change pas avec l’âge ou le temps après la ménopause. Cependant, comme le risque absolu de base d’accident vasculaire cérébral est fortement dépendant de l'âge, le risque global de survenue d’un accident vasculaire cérébral chez la femme utilisant un THS augmentera avec l'âge (voir rubrique 4.8).
Cancer de l’ovaire
Le cancer ovarien est beaucoup plus rare que le cancer du sein.
Les données épidémiologiques provenant d'une importante méta-analyse suggèrent une légère augmentation du risque chez les femmes prenant un THS par oestrogènes seuls ou par une combinaison d'oestrogènes et de progestatifs, qui apparaît dans les cinq ans suivant le début de l'utilisation du produit et diminue progressivement après l'arrêt du traitement.
D'autres études, y compris l'essai WHI (Women's Health Initiative), suggèrent qu'un risque similaire ou légèrement inférieur peut être associé avec une utilisation à long terme de THS combinés (voir rubrique 4.8).
Autres précautions d’emploi
Les estrogènes pouvant provoquer une rétention hydrique, les patientes présentant une insuffisance rénale ou cardiaque doivent être étroitement surveillées.
Les femmes avec une hypertriglycéridémie préexistante doivent être surveillées pendant le traitement hormonal substitutif. De rares cas d’augmentation importante du taux des triglycérides conduisant à une pancréatite ont été observés sous estrogénothérapie.
La fonction thyroïdienne doit être surveillée chez les patientes recevant un traitement substitutif par hormones thyroïdiennes lors d’un traitement par estrogènes (voir rubrique 4.5).
Au cours du traitement par les estrogènes, une augmentation des taux plasmatiques de la TBG (thyroid binding globulin) est observée, elle conduit à une élévation des taux plasmatiques des hormones thyroïdiennes totales mesurées par PBI (protein-bound iodine), de la T4 totale (mesurée sur colonne ou par RIA (radioimmunoassay)) et de la T3 totale (mesurée par RIA). La fixation de la T3 sur la résine est diminuée, reflétant l’augmentation de la TBG. Les concentrations des fractions libres de T4 et de T3 restent inchangées. Les taux sériques d’autres protéines de liaison telles que la CBG (corticoid binding globulin) et la SHBG (sex-hormone binding globulin) peuvent être augmentés, entraînant, respectivement, une augmentation des taux circulants de corticoïdes et de stéroïdes sexuels. Les concentrations des fractions libres ou actives des hormones restent inchangées.
D’autres protéines plasmatiques peuvent également être augmentées (angiotensinogène/substrat de la rénine, alpha-1-antitrypsine, céruloplasmine).
L’utilisation de THS n’améliore pas les fonctions cognitives. Il existe une augmentation du risque de probable démence chez les femmes débutant un traitement combiné continu ou par estrogènes seuls après 65 ans.
Au cours des essais cliniques sur le traitement de l’infection par le virus de l’hépatite C (VHC) par l’association ombitasvir/paritaprévir/ritonavir avec et sans dasabuvir, les élévations du taux d’ALAT au-delà de 5 fois la limite supérieure de la normale (LSN) étaient significativement plus fréquentes chez les femmes utilisant des médicaments contenant de l’éthinylestradiol, tels que des CHC. Des élévations du taux d’ALAT ont également été observées chez les patientes traitées par glécaprévir/pibrentasvir utilisant des médicaments contenant de l’éthinylestradiol, tels que des CHC. Chez les femmes utilisant des médicaments contenant des estrogènes autres que l’éthinylestradiol, comme l’estradiol, la fréquence des élévations du taux d’ALAT était similaire à celle observée chez les femmes n’utilisant aucun estrogène ; cependant, étant donné le nombre limité de femmes prenant ces autres estrogènes, il convient d’être prudent en cas de co-administration avec l’association médicamenteuse ombitasvir/paritaprévir/ritonavir avec ou sans dasabuvir et avec le traitement par glécaprévir/pibrentasvir. Voir rubrique 4.5.
4.5. Interactions avec d'autres médicaments et autres formes d'interactions
Associations faisant l’objet de précautions d’emploi
Le métabolisme des estrogènes peut être augmenté par l'utilisation concomitante de médicaments inducteurs enzymatiques, en particulier des enzymes du cytochrome P450 comme les anticonvulsivants (phénobarbital, phénytoïne, carbamazépine, oxcarbazépine) et les anti-infectieux (rifampicine, rifabutine, névirapine, éfavirenz).
Le ritonavir et le nelfinavir, bien que connus comme de puissants inhibiteurs enzymatiques ont paradoxalement des propriétés inductrices quand ils sont utilisés avec des hormones stéroïdiennes.
Les préparations à base de plante contenant du millepertuis (Hypericum perforatum) pourraient modifier le métabolisme des estrogènes.
Cliniquement, l'augmentation du métabolisme des estrogènes peut conduire à une diminution de l'effet thérapeutique et à une modification du profil des saignements utérins.
L'administration transcutanée évite l'effet de premier passage hépatique, par conséquent le métabolisme des estrogènes administrés par cette voie peut être moins affecté par les inducteurs enzymatiques que par voie orale.
Une surveillance et une adaptation éventuelle de la posologie du THS sont recommandées pendant le traitement par inducteur enzymatique et après son arrêt.
La prise d’estrogènes peut modifier les résultats de certains examens biologiques tels que : les tests fonctionnels hépatiques, thyroïdiens, surrénaliens et rénaux, le taux plasmatique des protéines (porteuses) comme la corticosteroid-binding globulin (CBG) et des fractions lipidiques/lipoprotéiniques, les paramètres du métabolisme glucidique, les paramètres de la coagulation et de la fibrinolyse. Les modifications restent en général dans les limites de la normale.
Effet des THS œstrogéniques sur d'autres médicaments
Il a été démontré que les contraceptifs hormonaux contenant des œstrogènes diminuent significativement les concentrations plasmatiques de lamotrigine en cas d'administration concomitante, en raison de l'induction de la glucuronidation de la lamotrigine. Cela peut réduire le contrôle des crises. Bien que l'interaction potentielle entre le traitement hormonal substitutif et la lamotrigine n'ait pas été étudiée, on s'attend à ce qu'une interaction similaire existe, ce qui pourrait conduire à une réduction du contrôle des crises chez les femmes prenant les deux médicaments en même temps.
Interactions pharmacodynamiques
Au cours des essais cliniques sur le traitement de l’infection par le VHC par l’association médicamenteuse ombitasvir/paritaprévir/ritonavir avec et sans dasabuvir, les élévations du taux d’ALAT au-delà de 5 fois la limite supérieure de la normale (LSN) étaient significativement plus fréquentes chez les femmes utilisant des médicaments contenant de l’éthinylestradiol, tels que des CHC. Chez les femmes utilisant des médicaments contenant des estrogènes autres que l’éthinylestradiol, comme l’estradiol, la fréquence des élévations du taux d’ALAT était similaire à celle observée chez les femmes n’utilisant aucun estrogène ; cependant, étant donné le nombre limité de femmes prenant ces autres estrogènes, il convient d’être prudent en cas de co-administration avec l’association médicamenteuse ombitasvir/paritaprévir/ritonavir avec ou sans dasabuvir et avec le traitement par glécaprévir/pibrentasvir (voir rubrique 4.4).
4.6. Fertilité, grossesse et allaitement
Grossesse
OESCLIM n'a pas d'indication au cours de la grossesse.
La découverte d'une grossesse au cours du traitement par OESCLIM 50 microgrammes/24 heures, impose l'arrêt immédiat du traitement.
A ce jour, la plupart des études épidémiologiques n'ont pas mis en évidence d'effet tératogène ou fœtotoxique chez les femmes enceintes exposées par mégarde à des doses thérapeutiques d'estrogènes.
Allaitement
OESCLIM n'a pas d'indication au cours de l'allaitement.
4.7. Effets sur l'aptitude à conduire des véhicules et à utiliser des machines
OESCLIM n’a aucun effet connu sur l’aptitude à conduire des véhicules et à utiliser des machines.
Dans les études cliniques et pharmacologiques, les événements indésirables suivants sont survenus chez plus de 10 % des patientes traitées par OESCLIM :
· Réaction au site d'application avec une incidence de 32 %, considérée comme sévère par seulement 4 % des patientes, modérée par 19 % et légère (rougeur ou démangeaisons) par 77 %.
· Symptômes d'hyperestrogénie incluant tension mammaire avec une incidence de 22 % (considérée comme légère chez 45 % des patientes, modérée chez 50 % et sévère chez 5 %) et métrorragies avec une incidence de 24,6 %.
Le tableau ci-après mentionne l’incidence des effets indésirables rapportés chez les utilisatrices de THS selon la classification MedDRA par systèmes d’organes (SOC).
|
Système classe organe MedDRA |
Effets indésirables fréquents |
Effets indésirables peu fréquents |
Effets indésirables rares |
Effets indésirables de fréquence indéterminée (ne peut être estimée sur la base des données disponibles) |
|
Infections et infestations |
|
Vaginite, candidose vaginale |
Affections vulvo-vaginale, vulvo-vaginite |
|
|
Tumeurs bénignes, malignes et non précisées (incl kystes et polypes) |
|
Tumeur bénigne mammaire, tumeur du col utérin, tumeur de l'utérus |
|
Augmentation de la taille d’un léiomyome |
|
Affections hématologiques et du système lymphatique |
|
Anémie |
Eosinophilie |
|
|
Affections du système immunitaire |
|
Hypersensibilité |
|
|
|
Troubles du métabolisme et de la nutrition |
Augmentation ou diminution du poids |
Hyperlipémie, hypercholestéro-lémie |
|
|
|
Affections psychiatriques |
|
Labilité émotionnelle, nervosité, humeur dépressive |
Anxiété, irritabilité, diminution de la libido ou augmen-tation de la libido |
|
|
Affections du système nerveux |
Céphalée |
Somnolence, insomnie, paresthésie, sensations vertigineuses |
Hyperkinésie, troubles du sommeil, vertige, migraine |
Exacerbation d’une épilepsie, chorée |
|
Affections oculaires |
|
Anomalie visuelle |
Blépharite, intolérance aux lentilles de contact |
|
|
Affections cardiaques |
|
Hypertension artérielle, palpitations |
|
|
|
Affections vasculaires |
Varices, vasodilatation |
|
Phlébite |
Maladie thrombo-embolique artérielle, par exemple angore et infarctus du myocardee. Pour plus d’information, voir rubriques 4.3 et 4.4 |
|
Affections respiratoires, thoraciques et médiastinales |
|
|
Laryngite, pharyngite, sinusite |
|
|
Affections gastro-intestinales |
Nausées, douleurs abdominales, gonflement abdominal |
Flatulence, diarrhée, dyspepsie |
Vomissements, constipation, augmentation de l'appétit, affections rectales, gastrite, ballonnements |
Pancréatite (chez les femmes ayant une hyper-triglycéridémie pré-existante), reflux gastro-intestinal |
|
Affections hépatobiliaires |
|
Troubles de la vésicule biliaire |
Perturbation du bilan hépatique |
|
|
Affections de la peau et du tissu sous-cutané |
Prurit, rash |
Erythème noueux, urticaire |
Eruption, acné, œdème de Quincke, dermatite fongique, mélanose, hirsutisme |
|
|
Affections musculo-squelettiques et systémiques |
|
Dorsalgies |
Arthralgies, myalgies, crampes musculaires, ténosynovite |
|
|
Affections des reins et des voies urinaires |
|
Incontinence urinaire |
|
|
|
Affections des organes de reproduction et du sein |
Hyperplasie endométriale, ménorragie, saignements vaginaux/ utérins, incluant des spottings, développe-ment de fibromes utérins |
Trouble ovarien, douleurs pelviennes, douleur mammaire, tension mammaire |
Trouble de l'endomètre, galactorrhée, abcès mammaire, anomalies du cycle menstruel, hypertrophie utérine, dysménorrhée, pertes vaginales, syndrome prémenstruel, hypertrophie mammaire |
Maladie fibrokystique des seins |
|
Troubles généraux et anomalies au site d’administration |
|
Œdème |
Fatigue, frissons, syndrome pseudo-grippal, douleurs, manque d'efficacité du dispositif |
|
a Risque de cancer du sein
· Une augmentation jusqu’à 2 fois du risque de cancer du sein a été rapportée chez les femmes ayant pris une association estro-progestative pendant plus de 5 ans.
· L’augmentation du risque est plus faible chez les utilisatrices d'estrogènes seuls comparativement aux utilisatrices d’associations estro-progestatives.
· Le niveau de risque est dépendant de la durée du traitement (voir rubrique 4.4).
· Les estimations du risque absolu basées sur les résultats du plus large essai randomisé contrôlé versus placebo (étude WHI) et de la plus large méta-analyse des études épidémiologiques prospectives sont présentées ci-après.
Plus importante méta-analyse d’études épidémiologiques prospectives
Estimation du risque additionnel de cancer du sein après 5 ans de traitement chez des femmes ayant un IMC de 27 (kg/m²)
|
Age au début du THS (ans) |
Incidence pour 1000 patientes n’ayant jamais pris de THS sur une période de 5 ans (50-54 ans)* |
Risque relatif |
Nombre de cas supplémentaires pour 1000 utilisatrices de THS après 5 ans |
|
THS par estrogènes seuls |
|||
|
50 |
13,3 |
1,2 |
2,7 |
|
Association estro-progestative |
|||
|
50 |
13,3 |
1,6 |
8,0 |
*Issu des taux d’incidence de base en Angleterre en 2015 chez des femmes ayant un IMC de 27 (kg/m²)
Remarque : étant donné que l’incidence de base du cancer du sein diffère selon les pays de l’Union européenne (UE), le nombre de cas supplémentaires de cancer du sein variera proportionnellement.
|
Age au début du THS (ans) |
Incidence pour 1000 patientes n’ayant jamais pris de THS sur une période de 10 ans (50-59 ans)* |
Risque relatif |
Nombre de cas supplémentaires pour 1000 utilisatrices de THS après 10 ans |
|
THS par estrogènes seuls |
|||
|
50 |
26,6 |
1,3 |
7,1 |
|
Association estro-progestative |
|||
|
50 |
26,6 |
1,8 |
20,8 |
*Issu des taux d’incidence de base en Angleterre en 2015 chez des femmes ayant un IMC de 27 (kg/m²)
Remarque : étant donné que l’incidence de base du cancer du sein diffère selon les pays de l’UE, le nombre de cas supplémentaires de cancer du sein variera proportionnellement.
Etude WHI aux Etats-Unis – Risque additionnel de cancer du sein sur 5 ans de traitement
|
Age (ans) |
Incidence pour 1000 femmes dans le bras placebo sur 5 ans |
Risque relatif (IC 95%) |
Nombre de cas supplémentaires pour 1000 utilisatrices de THS sur 5 ans (IC 95%) |
|
Estrogènes seuls (estrogènes conjugués équins) |
|||
|
50 - 79 |
21 |
0,8 (0,7 – 1,0) |
-4 (-6 – 0)* |
|
Associations estro-progestatives (CEE + MPA)# |
|||
|
50 - 79 |
17 |
1,2 (1,0 – 1,5) |
+4 (0 – 9) |
#Lorsque l’analyse était limitée aux femmes n’ayant pas utilisé de THS avant l’étude, il n’était pas observé d’augmentation du risque au cours de 5 premières années de traitement : après 5 ans, le risque était plus élevé que chez les non utilisatrices.
*Etude WHI chez les femmes hystérectomisées n’ayant pas montré d’augmentation du risque de cancer du sein.
b Risque de cancer de l’endomètre
Le risque de cancer de l’endomètre est d’environ 5 pour 1000 femmes ayant un utérus intact et n’utilisant pas de THS.
Chez les femmes ayant un utérus intact, l’utilisation d’un THS à base d’estrogènes seuls n’est pas recommandée en raison de l’augmentation du risque de cancer de l’endomètre (voir rubrique 4.4).
Dans les études épidémiologiques, l’augmentation du risque de cancer de l’endomètre dépendait de la durée de traitement à base d’estrogènes seuls et de la dose d’estrogène et variait entre 5 et 55 cas supplémentaires diagnostiqués pour 1000 femmes âgées de 50 à 65 ans.
L’ajout d’un progestatif au traitement par estrogènes seuls pendant au moins 12 jours par cycle permet de prévenir l’augmentation du risque. Dans l’étude MWS, l’utilisation pendant 5 ans d’un THS combiné (séquentiel ou continu) n’a pas augmenté le risque de cancer de l’endomètre (RR de 1,0 (0,8 – 1,2)).
c Risque de cancer ovarien
L’utilisation d’un THS par estrogènes seuls ou par une combinaison d’estrogènes et de progestatifs a été associée à une légère augmentation du risque de cancer ovarien diagnostiqué (voir rubrique 4.4).
Une méta-analyse portant sur 52 études épidémiologiques a rapporté un risque accru de cancer ovarien chez les femmes prenant actuellement un THS par rapport aux femmes n’en ayant jamais pris (RR 1.43, IC 95% 1.31-1.56). Chez les femmes âgées de 50 à 54 ans, prendre un THS pendant cinq ans entraîne l’apparition d’un cas supplémentaire pour 2000 utilisatrices. Chez les femmes âgées entre 50 et 54 ans qui ne prennent pas de THS, un diagnostic de cancer ovarien sera posé chez environ 2 femmes sur 2000 sur une période de cinq ans.
d Risque d’accident thromboembolique veineux
Le THS est associé à une augmentation de 1,3 à 3 fois du risque relatif de survenue d'un accident thromboembolique veineux, c'est-à-dire thrombose veineuse profonde ou embolie pulmonaire. La probabilité de survenue d’un tel événement est plus élevée au cours de la première année d’utilisation du THS (voir rubrique 4.4). Les résultats des études WHI sont présentés ci-après :
Etudes WHI – Risque supplémentaire d’accident thromboembolique veineux sur 5 ans de traitement
|
Age (ans) |
Incidence pour 1000 femmes dans le bras placebo sur 5 ans |
Risque relatif (IC 95%) |
Nombre de cas supplémentaires pour 1000 utilisatrices de THS |
|
Estrogènes seuls par voie orale* |
|||
|
50 - 59 |
7 |
1,2 (0,6 – 2,4) |
1 (-3 – 10) |
|
Associations estro-progestatives par voie orale |
|||
|
50 - 59 |
4 |
2,3 (1,2 – 4,3) |
5 (1 – 13) |
*Etude chez les femmes hystérectomisées
e Risque de maladie coronarienne
Le risque de maladie coronarienne est légèrement augmenté chez les utilisatrices de THS estro-progestatifs au-delà de 60 ans (voir rubrique 4.4).
f Risque d’accident vasculaire cérébral ischémique
L’utilisation d’un THS à base d’estrogènes seuls ou d’une association estro-progestative est associée à une augmentation jusqu’à 1,5 fois du risque relatif d’AVC ischémique. Le risque d’AVC hémorragique n’est pas augmenté lors de l’utilisation d’un THS.
Ce risque relatif ne dépend pas de l’âge ni de la durée du traitement, mais comme le risque de base est fortement âge-dépendant, le risque global d'AVC chez les femmes utilisant un THS augmente avec l'âge (voir rubrique 4.4).
Etudes WHI combinées – Risque additionnel d’AVC* sur 5 ans de traitement
|
Age (ans) |
Incidence pour 1000 femmes dans le bras placebo sur 5 ans |
Risque relatif (IC 95%) |
Nombre de cas supplémentaires pour 1000 utilisatrices de THS sur 5 ans |
|
50 - 59 |
8 |
1,3 (1,1 – 1,6) |
3 (1 – 5) |
* il n’a pas été fait de distinction entre les AVC ischémiques et hémorragiques.
D’autres effets indésirables ont été rapportés lors de l’association avec un traitement estro-progestatif:
· Troubles cutanés ou sous-cutanés : chloasma, érythème polymorphe,
· Purpura vasculaire.
· Démence probable au-delà de l’âge de 65 ans (voir rubrique 4.4).
Déclaration des effets indésirables suspectés
La déclaration des effets indésirables suspectés après autorisation du médicament est importante. Elle permet une surveillance continue du rapport bénéfice/risque du médicament. Les professionnels de santé déclarent tout effet indésirable suspecté via le système national de déclaration : Agence nationale de sécurité du médicament et des produits de santé (ANSM) et réseau des Centres Régionaux de Pharmacovigilance - Site internet : https://signalement.social-sante.gouv.fr
Le risque de surdosage lors d’une administration transdermique est peu probable.
Nausées, vomissements, somnolence, sensations vertigineuses, saignements intercurrents, sensation de tension mammaire, gonflement abdomino-pelvien, anxiété et irritabilité peuvent survenir chez certaines femmes. Il n’existe pas d’antidote spécifique et le traitement doit être symptomatique.
En cas de surdosage, les dispositifs transdermiques doivent être retirés. Ceci est également applicable dans le cas d’une utilisation non-indiquée chez l’enfant.
5. PROPRIETES PHARMACOLOGIQUES
5.1. Propriétés pharmacodynamiques
Estrogène naturel par voie transdermique.
Le principe actif, 17 β-estradiol de synthèse, est chimiquement et biologiquement identique à l'estradiol endogène humain. Il remplace l'arrêt de production des estrogènes chez les femmes ménopausées et soulage les symptômes climatériques de la ménopause.
Soulagement des symptômes liés au déficit estrogénique
Le soulagement des symptômes de la ménopause a été obtenu dès les premières semaines de traitement.
5.2. Propriétés pharmacocinétiques
La substance active dans le système transdermique d'estradiol est le 17β-estradiol (estradiol) sous forme hémihydratée, qui est identique à l'estradiol ovarien humain. Les données pharmacocinétiques des différents dosages du système transdermique d'estradiol ont été obtenues au cours d’études réalisées chez des femmes ménopausées en bonne santé.
Absorption
Lors de l’utilisation des systèmes transdermiques d'estradiol, l'estradiol est absorbé dans le corps par la peau. Environ 24 heures après l'application des dosages à 25 µg/24 heures et 50 µg/24 heures, les concentrations sériques d’estradiol atteignent des pics moyens respectifs de 40 et 72 pg / ml. Les concentrations plasmatiques diminuent ensuite très lentement. Les paramètres pharmacocinétiques du système transdermique au dosage de 50 µg/24 heures, calculées à partir de plusieurs études sont fournis dans le tableau ci-dessous :
|
Système transdermique |
Cmax (pg/mL) |
AUC0-72 (pg*r/mL) |
Cmoy0-72 (pg/mL) |
C72 (pg/mL) |
|
50 μg/24 heures |
72 [43] |
3294 [1473] |
41 [22] |
34 [15] |
Sont présentées les valeurs moyennes de la valeur arithmétique et de l’écart-type (entre crochets) de 4-5 études.
Aucune accumulation du produit n’a été observée après 3 semaines d’applications répétées.
Distribution
Les estrogènes peuvent être retrouvés sous forme liée ou non liée. Environ 98 - 99% de la dose d'estradiol se lie aux protéines plasmatiques, dont environ 30-52% à l'albumine et environ 46 à 69% à la sex-hormone binding globulin (SHBG).
Biotransformation
Les principaux métabolites non conjugués et conjugués d'estradiol sont respectivement l’estrone et le sulfate d'estrone. Ces métabolites peuvent contribuer à l'activité des estrogènes, soit directement, soit après transformation en estradiol. Le sulfate d'estrone peut subir un cycle entéro-hépatique.
Etant donné que l’estradiol absorbé par voie transdermique ne subit pas de premier passage hépatique, le rapport des concentrations plasmatiques entre l’estradiol et l’un ou l’autre de ses métabolites principaux, l'estrone ou le sulfate d’estrone, est plus proche de celui observé chez les femmes pré ménopausées que lorsqu'il est administré par voie orale.
Le rapport moyen E2/E1 (estradiol/estrone) lors de l'utilisation des dispositifs transdermiques d'estradiol est celui observé chez la femme avant la ménopause (voisin de 1).
Élimination
L’estradiol est majoritairement éliminé par voie urinaire. Dans l'urine, les principaux métabolites sont les glucuronates de l'estrone et de l'estradiol. Lorsque le dispositif transdermique est retiré 72 heures après l'application, la concentration plasmatique d'estradiol revient à l’état basal au bout d'environ 8 à 24 heures.
Linéarité/non-linéarité
La pharmacocinétique de l’estradiol (corrigée par rapport à l’état basal) est proportionnelle aux surfaces appliquées des systèmes transdermiques.
5.3. Données de sécurité préclinique
Au cours d'études de tolérance cutanée chez le lapin, espèce particulièrement sensible, le dispositif transdermique s'est révélé légèrement irritant lors d'applications locales pendant une période variant entre 4 jours et 4 semaines. Aucun pouvoir sensibilisant n'a été observé après les applications locales chez le cobaye.
Composition de la matrice adhésive
Poly(éthylène-acétate de vinyle) (EVA) haute et basse viscosité, éthylcellulose, octyldodécanol, dipropylèneglycol.
Composition du film de protection
Mousse beige : poly(éthylène-acétate de vinyle).
Film protecteur : polyester silicone.
2 ans.
6.4. Précautions particulières de conservation
A conserver à une température ne dépassant pas + 25°C.
6.5. Nature et contenu de l'emballage extérieur
Sachet (papier/aluminium) contenant chacun 1 dispositif transdermique dans des boîtes de 1, 2, 4, 6, 8, 9, 18 ou 24 sachets.
Toutes les présentations peuvent ne pas être commercialisées.
6.6. Précautions particulières d’élimination et de manipulation
Les dispositifs transdermiques non utilisés ne doivent pas être jetés au tout à l’égout ou avec les ordures ménagères. Tout produit non utilisé ou déchet doit être éliminé conformément à la réglementation en vigueur ou rapporté à la pharmacie.
Après utilisation, le dispositif transdermique contient encore des quantités non négligeables de substance active. Ces substances actives hormonales peuvent avoir des effets néfastes si elles atteignent le milieu aquatique. Par conséquent, le dispositif transdermique usagé doit être plié en deux de sorte que les côtés adhésifs collent ensemble et ensuite jeté avec soin. Il ne doit pas être jeté dans les toilettes, ni dans le tout à l’égout.
7. TITULAIRE DE L’AUTORISATION DE MISE SUR LE MARCHE
1 BIS PLACE DE LA DEFENSE – TOUR TRINITY
92400 COURBEVOIE
8. NUMERO(S) D’AUTORISATION DE MISE SUR LE MARCHE
· 34009 338 115-56 : dispositif transdermique en sachet (papier/aluminium). Boîte de 1 sachet
· 34009 345 508-98 : dispositif transdermique en sachet (papier/aluminium). Boîte de 2 sachets
· 34009 338 116-17 : dispositif transdermique en sachet (papier/aluminium). Boîte de 4 sachets
· 34009 338 117-85 : dispositif transdermique en sachet (papier/aluminium). Boîte de 6 sachets
· 34009 338 118-46 : dispositif transdermique en sachet (papier/aluminium). Boîte de 8 sachets
· 34009 338 119-07 : dispositif transdermique en sachet (papier/aluminium). Boîte de 9 sachets
· 34009 345 509-59 : dispositif transdermique en sachet (papier/aluminium). Boîte de 18 sachets
· 34009 338 120-96 : dispositif transdermique en sachet (papier/aluminium). Boîte de 24 sachets
9. DATE DE PREMIERE AUTORISATION/DE RENOUVELLEMENT DE L’AUTORISATION
[à compléter ultérieurement par le titulaire]
10. DATE DE MISE A JOUR DU TEXTE
[à compléter ultérieurement par le titulaire]
Sans objet.
12. INSTRUCTIONS POUR LA PREPARATION DES RADIOPHARMACEUTIQUES
Liste II.
ANSM - Mis à jour le : 27/09/2023
OESCLIM 50 microgrammes/24 heures (10 mg/22 cm²), dispositif transdermique
Estradiol
· Gardez cette notice. Vous pourriez avoir besoin de la relire.
· Si vous avez d’autres questions, interrogez votre médecin ou votre pharmacien.
· Ce médicament vous a été personnellement prescrit. Ne le donnez pas à d’autres personnes. Il pourrait leur être nocif, même si les signes de leur maladie sont identiques aux vôtres.
· Si vous ressentez un quelconque effet indésirable, parlez-en à votre médecin ou votre pharmacien. Ceci s’applique aussi à tout effet indésirable qui ne serait pas mentionné dans cette notice. Voir rubrique 4.
1. Qu'est-ce que OESCLIM 50 microgrammes/24 heures (10 mg/22 cm²), dispositif transdermique et dans quels cas est-il utilisé ?
2. Quelles sont les informations à connaître avant d'utiliser OESCLIM 50 microgrammes/24 heures (10 mg/22 cm²), dispositif transdermique ?
3. Comment utiliser OESCLIM 50 microgrammes/24 heures (10 mg/22 cm²), dispositif transdermique ?
4. Quels sont les effets indésirables éventuels ?
5. Comment conserver OESCLIM 50 microgrammes/24 heures (10 mg/22 cm²), dispositif transdermique ?
6. Contenu de l’emballage et autres informations.
1. QU’EST-CE QUE OESCLIM 50 microgrammes/24 heures (10 mg/22 cm²), dispositif transdermique ET DANS QUELS CAS EST-IL UTILISE ?
Classe pharmacothérapeutique :
OESCLIM est un traitement hormonal substitutif (THS). Il contient du 17-b estradiol. OESCLIM est utilisé chez les femmes ménopausées dont les dernières règles datent d’au moins 6 mois.
OESCLIM est utilisé pour :
Soulager les symptômes apparaissant après la ménopause.
Lors de la ménopause, la quantité d’estrogènes produits par l’organisme féminin chute. Chez certaines femmes, cette chute se traduit par des symptômes tels qu’une sensation de chaleur au niveau du visage, du cou et de la poitrine (les « bouffées de chaleur »). OESCLIM soulage ces symptômes après la ménopause.
OESCLIM vous sera prescrit uniquement si vos symptômes altèrent gravement votre vie quotidienne.
2. QUELLES SONT LES INFORMATIONS A CONNAITRE AVANT D’UTILISER OESCLIM 50 microgrammes/24 heures (10 mg/22 cm²), dispositif transdermique ?
Antécédents médicaux et examens réguliers
L’utilisation d’un THS entraîne des risques qui doivent être pris en considération lorsque vous décidez de commencer ce traitement ou de le continuer.
L’expérience chez les femmes avec une ménopause précoce (liée à une insuffisance ovarienne ou à une chirurgie) est limitée. Si vous avez une ménopause précoce, les risques liés à l’utilisation d’un THS peuvent être différents. Parlez-en à votre médecin.
Avant de commencer (ou recommencer) un THS, votre médecin vous interrogera sur vos antécédents médicaux personnels et familiaux. Votre médecin peut décider de pratiquer un examen physique. Cet examen peut inclure un examen de vos seins et, si nécessaire, un examen gynécologique.
Dès que vous commencez OESCLIM, consultez votre médecin pour des examens réguliers (au moins une fois par an). Lors de ces examens, celui-ci pourra aborder avec vous les bénéfices et les risques liés à la poursuite du traitement par OESCLIM.
Faites régulièrement une mammographie en suivant les recommandations de votre médecin.
N’utilisez jamais OESCLIM si l’une des conditions suivantes s’applique à vous :
Si vous avez des doutes sur un des points ci-dessous, parlez-en à votre médecin avant de prendre OESCLIM.
N’utilisez jamais OESCLIM :
· si vous êtes allergique (hypersensible) à l’estradiol ou à l'un des autres composants contenus dans OESCLIM (listés en rubrique 6),
· si vous avez ou avez eu un cancer du sein ou, s’il existe une suspicion que vous en ayez un,
· si vous avez un cancer sensible aux estrogènes tel qu’un cancer de la paroi de l’utérus (endomètre), ou s’il existe une suspicion que vous en ayez un,
· si vous avez des saignements vaginaux non expliqués,
· si vous avez un développement exagéré de la paroi de l’utérus (hyperplasie de l’endomètre) qui n’est pas traité,
· si vous avez ou avez eu un caillot sanguin dans une veine (thrombose) tel que dans les jambes (thrombose veineuse profonde), ou dans les poumons (embolie pulmonaire),
· si vous avez des troubles de la coagulation sanguine (tels qu’un déficit en protéine C, protéine S ou en antithrombine),
· si vous avez ou avez eu récemment une maladie causée par des caillots sanguins dans les artères, telle qu’une crise cardiaque, un accident vasculaire cérébral ou de l’angine de poitrine,
· si vous avez ou avez eu une maladie du foie, et que vos tests de la fonction hépatique ne sont pas retournés à la normale,
· si vous avez une maladie héréditaire rare du sang appelé « porphyrie ».
Si l’une de ces pathologies apparaît pour la première fois lors du traitement avec OESCLIM, arrêtez le traitement et consultez immédiatement votre médecin.
Avertissements et précautions
Faites attention avec OESCLIM
Signalez à votre médecin avant de débuter votre traitement, si vous avez déjà eu un des signes suivants car ils peuvent revenir ou s’aggraver pendant le traitement par OESCLIM. Si c’est le cas, consultez votre médecin pour des examens plus réguliers :
· fibromes dans votre utérus,
· présence de muqueuse utérine en dehors de l’utérus (endométriose) ou antécédent de développement exagéré de votre muqueuse utérine (hyperplasie endométriale),
· risque augmenté de développer des caillots sanguins (voir « caillots de sang dans une veine (thromboses) »),
· risque augmenté d’avoir un cancer dépendant des estrogènes (par exemple si votre mère, votre sœur ou votre grand-mère a eu un cancer du sein),
· hypertension artérielle,
· maladie du foie, telle qu’une tumeur bénigne du foie,
· diabète,
· calcul biliaire,
· migraine ou maux de tête sévères,
· maladie du système immunitaire qui peut affecter plusieurs parties du corps (lupus érythémateux disséminé, LED),
· épilepsie,
· asthme,
· maladie affectant les tympans ou l’audition (otosclérose),
· niveau élevé de graisses dans votre sang (triglycérides),
· rétention d’eau liée à des troubles cardiaques ou rénaux,
· angioœdème héréditaire ou acquis.
Arrêtez votre traitement et prévenez immédiatement votre médecin
Si vous notez l’apparition des signes suivants :
· une des pathologies signalées en rubrique « N’utilisez jamais OESCLIM »,
· un jaunissement de votre peau ou du blanc de vos yeux. C’est peut-être un signe d’une maladie du foie,
· un gonflement du visage, de la langue et/ou de la gorge, et/ou des difficultés à déglutir ou une urticaire accompagnée de difficultés à respirer qui suggèrent un angioœdème,
· une augmentation importante de votre pression artérielle (les symptômes peuvent être mal de tête, fatigue, sensations vertigineuses),
· des maux de tête tels qu’une migraine, qui apparaissent pour la première fois,
· si vous devenez enceinte,
· si vous remarquez des signes possibles d’un caillot sanguin, tels que :
o gonflement douloureux dans vos jambes,
o douleur brutale à la poitrine,
o difficulté à respirer.
Pour plus d’information, voir « Caillots de sang dans une veine ».
Note : OESCLIM n’est pas un contraceptif. S’il s’est écoulé moins d’un an depuis vos dernières règles, ou si vous avez moins de 50 ans, vous pouvez avoir besoin d’une contraception complémentaire pour éviter une grossesse. Demandez conseil à votre médecin.
THS et cancer
Développement exagéré de la muqueuse utérine (hyperplasie endométriale) et cancer de la paroi de l’utérus (cancer de l’endomètre)
La prise d’un THS à base d’estrogènes seuls augmentera le risque de développement exagéré de la muqueuse utérine (hyperplasie endométriale) et de cancer de la muqueuse utérine (cancer de l’endomètre).
La prise d’un progestatif en association à OESCLIM pendant au moins 12 jours sur chaque cycle de 28 jours vous protège de ce risque supplémentaire. Si vous avez toujours votre utérus, votre médecin vous prescrira donc un progestatif à prendre séparément, en plus d’OESCLIM. Si vous n’avez plus votre utérus (si vous avez eu une hystérectomie), votre médecin vous dira si vous pouvez prendre OESCLIM en toute sécurité sans y associer un progestatif.
Comparaison
Chez les femmes qui ont toujours leur utérus et qui ne prennent pas de THS, 5 sur 1000 auront un cancer de l’endomètre diagnostiqué entre 50 et 65 ans.
Chez les femmes de 50 à 65 ans qui ont toujours leur utérus et qui prennent un THS à base d’estrogènes seuls, le nombre de cas supplémentaires peut varier de 5 à 55 sur 1000 utilisatrices selon la dose et la durée du traitement.
Saignements irréguliers
Lors d’un traitement cyclique ou continu séquentiel avec OESCLIM, vous aurez des saignements une fois par mois (appelés saignements de privation). Mais vous pouvez avoir des saignements irréguliers ou des petites pertes sanguines (spottings), en plus des saignements mensuels. Si ces saignements irréguliers :
· persistent au-delà des 6 premiers mois,
· débutent alors que vous prenez OESCLIM depuis plus de 6 mois,
· persistent après l’arrêt du traitement par OESCLIM,
consultez votre médecin dès que possible.
Cancer du sein
Les données disponibles montrent que la prise d’un traitement hormonal de substitution (THS) estro-progestatif combiné ou d’un THS à base d’estrogènes seuls augmente le risque de cancer du sein. Ce risque supplémentaire dépend de la durée de suivi du THS. Le risque additionnel devient évident au bout de 3 ans d’utilisation. Après avoir arrêté le THS, le risque additionnel diminuera dans le temps, mais pourra perdurer 10 ans ou plus si vous avez suivi un THS pendant plus de 5 ans.
Pour comparaison
Chez les femmes de 50 à 54 ans qui ne prennent pas de THS, un diagnostic de cancer du sein sera posé, en moyenne, chez environ 13 à 17 femmes sur 1 000 après une période de cinq ans.
Chez les femmes âgées de 50 ans qui débutent un THS estro-progestatif pour 5 ans, on dénombrera 21 cas sur 1 000 utilisatrices (soit 4 à 8 cas supplémentaires).
Chez les femmes âgées de 50 ans qui débutent un THS à base d'estrogènes seuls pour 10 ans, on dénombrera 34 cas sur 1 000 utilisatrices (soit 7 cas supplémentaires).
Chez les femmes âgées de 50 ans qui débutent un THS estro-progestatif pour 10 ans, on dénombrera 48 cas sur 1 000 utilisatrices (soit 21 cas supplémentaires).
Vérifiez régulièrement vos seins. Consultez votre médecin si vous remarquez des changements tels que :
· capitons au niveau de la peau,
· modifications au niveau du mamelon,
· boules éventuelles que vous pouvez voir ou sentir.
Cancer ovarien
Le cancer de l’ovaire est rare (beaucoup plus rare que le cancer du sein). L’utilisation d’un THS par estrogènes seuls ou par une combinaison d’estrogènes et de progestatifs a été associée à une légère augmentation du risque de cancer ovarien.
Comparaison
Le risque de cancer ovarien varie en fonction de l’âge. Par exemple, chez les femmes âgées entre 50 et 54 ans qui ne prennent pas de THS, un diagnostic de cancer ovarien sera posé chez 2 femmes sur 2000 en moyenne sur une période de 5 ans. Chez les femmes ayant pris un THS pendant 5 ans, il y aura environ 3 cas sur 2000 utilisatrices (soit environ un cas supplémentaire).
Effets des THS sur le cœur et la circulation
Caillots de sang dans une veine (thrombose veineuse)
Le risque de caillots sanguins dans les veines est environ de 1,3 à 3 fois supérieur chez les utilisatrices de THS par rapport aux non-utilisatrices, particulièrement pendant la première année de traitement.
Ces caillots de sang peuvent être graves, et si l’un d’eux migre vers les poumons, cela peut causer des douleurs dans la poitrine, un essoufflement, un malaise, voire parfois le décès.
Vous avez plus de risque d’avoir un caillot sanguin lorsque vous vieillissez, et si l’une des situations suivantes s’applique à vous. Signalez à votre médecin si l’une de ces situations s’applique à vous :
· vous ne pouvez pas marcher pendant une longue période en raison d’une chirurgie, blessure ou maladie grave (voir également section 3 « Si vous devez subir une opération chirurgicale »),
· vous êtes en surpoids sévère (IMC > 30 kg/m²),
· vous avez des problèmes de coagulation sanguine qui nécessitent un traitement à long terme avec un médicament utilisé pour prévenir les caillots sanguins,
· un de vos parents proches a déjà eu un caillot de sang dans la jambe, le poumon ou un autre organe,
· vous avez un lupus érythémateux disséminé (LED),
· vous avez un cancer.
Pour les signes de caillot sanguin, voir rubrique « Arrêtez votre traitement et prévenez immédiatement votre médecin ».
Comparaison
Chez les femmes de la cinquantaine ne prenant pas de THS, un caillot sanguin veineux survient en moyenne chez 4 à 7 femmes sur 1000 après une période de 5 ans.
Chez les femmes de la cinquantaine prenant un THS estro-progestatif après une période de 5 ans, il y aura 9 à 12 cas sur 1000 utilisatrices (c’est-à-dire 5 cas supplémentaires).
Chez les femmes de la cinquantaine qui n’ont plus leur utérus et qui ont pris un THS contenant uniquement un estrogène pendant plus de 5 ans, il y aura 5 à 8 cas sur 1000 utilisatrices (c’est-à-dire un cas supplémentaire).
Maladie cardiaque (crise cardiaque)
Il n’y a pas de preuves que le THS participe à la prévention d’une crise cardiaque.
Les femmes de plus de 60 ans utilisatrices de THS estro-progestatif ont un risque légèrement plus augmenté de développer une maladie cardiaque que celles qui ne prennent pas de THS.
Pour les femmes qui n’ont plus leur utérus et qui prennent un THS contenant uniquement un estrogène, le risque de développer une maladie cardiaque n’est pas augmenté.
Accident vasculaire cérébral (AVC)
Le risque d’avoir un accident vasculaire cérébral est environ 1,5 fois supérieur chez les utilisatrices de THS par rapport aux non-utilisatrices.
Le nombre de cas supplémentaires d’AVC liés à l’utilisation d’un THS augmente avec l'âge.
Comparaison
Chez les femmes de la cinquantaine ne prenant pas de THS, un AVC est attendu en moyenne chez 8 femmes sur 1000 sur une période de plus de 5 ans.
Pour les femmes de la cinquantaine prenant un THS, il y aura 11 cas sur 1000 utilisatrices sur une période de plus de 5 ans (c’est-à-dire 3 cas supplémentaires).
Autres pathologies
Le THS ne prévient pas la perte de mémoire. Le risque de perte de mémoire pourrait être toutefois plus élevé chez les femmes qui commencent à utiliser un THS après l’âge de 65 ans. Demandez conseil à votre médecin.
Enfants
Sans objet.
Autres médicaments et OESCLIM 50 microgrammes/24 heures (10 mg/22 cm²), dispositif transdermique
Certains médicaments peuvent interférer avec les effets d’OESCLIM. Cette interférence peut entraîner des saignements irréguliers. Cela concerne les médicaments suivants :
· les médicaments utilisés dans le traitement de l’épilepsie (par exemple phénobarbital, phénytoïne, carbamazépine),
· les médicaments utilisés dans le traitement de la tuberculose (par exemple rifampicine et rifabutine),
· les médicaments utilisés dans le traitement des infections par le VIH (par exemple névirapine, éfavirenz, ritonavir et nelfinavir),
· les préparations à base de plantes contenant du millepertuis (Hypericum perforatum).
Le THS peut modifier le mode d'action de certains autres médicaments :
· un médicament contre l'épilepsie (lamotrigine), car il pourrait augmenter la fréquence des crises,
· les médicaments utilisés dans le traitement des infections par le virus de l’hépatite C (VHC) (tels que l’association ombitasvir/paritaprévir/ritonavir avec ou sans dasabuvir, et le glécaprévir/pibrentasvir), qui peuvent entraîner une augmentation de certains paramètres du fonctionnement du foie (augmentation du taux d’ALAT, une enzyme du foie) chez les femmes utilisant des contraceptifs contenant de l’éthinylestradiol. OESCLIM contient de l’estradiol à la place de l’éthinylestradiol. On ne sait pas si une augmentation du taux d’ALAT peut se produire lors de l’utilisation d’OESCLIM avec cette association contre le VHC.
Veuillez informer votre médecin ou votre pharmacien si vous prenez ou avez récemment pris d’autres médicaments incluant des médicaments obtenus sans ordonnance, des médicaments à base de plantes ou d’autres produits naturels. Votre médecin vous conseillera.
Analyses en laboratoire
Si vous devez faire une prise de sang, signalez à votre médecin ou au personnel du laboratoire d’analyse que vous prenez OESCLIM, car ce médicament peut modifier les résultats de certaines analyses.
OESCLIM 50 microgrammes/24 heures (10 mg/22 cm²), dispositif transdermique avec des aliments, boissons et de l’alcool
Sans objet.
OESCLIM doit être uniquement utilisé chez les femmes ménopausées. Si vous devenez enceinte, interrompez le traitement par OESCLIM et parlez-en à votre médecin.
Vous ne devez pas utiliser OESCLIM si vous allaitez.
Si vous êtes enceinte ou que vous allaitez, si vous pensez être enceinte ou planifiez une grossesse, demandez conseil à votre médecin avant de prendre ce médicament.
Conduite de véhicules et utilisation de machines
Il n’y a pas de données qui indiquent qu’OESCLIM peut avoir un effet sur l’aptitude à conduire des véhicules ou à utiliser des machines.
OESCLIM 50 microgrammes/24 heures (10 mg/22 cm²), dispositif transdermique contient <{nommer le/les excipient (s)}>
Sans objet.
3. COMMENT UTILISER OESCLIM 50 microgrammes/24 heures (10 mg/22 cm²), dispositif transdermique ?
Votre médecin veillera à vous prescrire la dose la plus faible pendant une durée la plus courte possible pour traiter vos symptômes. Si vous avez l’impression que la dose est trop forte ou trop faible, parlez-en à votre médecin.
Posologie
OESCLIM 50 microgrammes/24 heures, dispositif transdermique est habituellement appliqué 2 fois par semaine, c'est-à-dire renouvelé tous les 3 à 4 jours.
Votre médecin pourra vous prescrire le traitement selon 2 modalités :
· Traitement cyclique : le dispositif transdermique doit être appliqué deux fois par semaine pendant une durée de 24 à 28 jours, suivi d’un intervalle de 2 à 7 jours sans traitement.
· Traitement continu : le dispositif transdermique doit être appliqué en traitement continu deux fois par semaine.
Si votre médecin associe à ce traitement un autre médicament hormonal (progestatif) durant au moins les 12 à 14 derniers jours du traitement par OESCLIM de chaque cycle de 28 jours, ce traitement ne devra pas être oublié.
Des saignements évoquant les règles peuvent survenir pendant la période d'interruption. Ces saignements sont normaux et peu abondants. Si des saignements abondants ou irréguliers surviennent, consultez votre médecin.
Pour vous aider, nous avons réalisé un calendrier indiquant les jours de la semaine où vous devrez changer le dispositif et le jour où vous devrez retirer le 7ème dispositif (25ème jour du cycle).
Quand faut-il démarrer le traitement ?
Vous pouvez commencer le traitement par OESCLIM au moment qui vous convient si vous n’êtes pas sous traitement à base d’estrogènes.
Si vous suivez actuellement un traitement estro-progestatif cyclique ou séquentiel, vous devez terminer votre cycle de traitement en cours avant de débuter le traitement par OESCLIM ; le moment approprié pour commencer le traitement par OESCLIM est le premier jour de l’hémorragie de privation (saignements évoquant les règles).
Si vous utilisez déjà un traitement estro-progestatif continu, vous pouvez passer directement au traitement par OESCLIM.
Comment prendre un progestatif avec OESCLIM ?
Si vous avez toujours votre utérus, autrement dit si vous n’avez pas subi d’hystérectomie, votre médecin vous prescrira peut-être un progestatif à utiliser en même temps que le dispositif OESCLIM pour éviter tout problème dû à un épaississement de la paroi de l’utérus, c’est-à-dire une hyperplasie de l’endomètre (voir les mises en garde spéciales concernant le cancer de l’endomètre). Il y a deux façons de le prendre :
· Traitement cyclique
OESCLIM est généralement administré pendant 24 à 28 jours, suivis d’une période de 2 à 7 jours sans traitement. En général, le médecin prescrit le progestatif pendant au moins 12 à 14 jours de chaque cycle. Il est possible que vous présentiez une « hémorragie de privation » (saignements évoquant les règles) pendant les derniers jours du traitement par progestatif, voire après l’arrêt.
· Traitement séquentiel continu
OESCLIM est administré sans interruption et il n’y a donc pas de période sans traitement. Le progestatif est généralement prescrit pendant au moins 12 à 14 jours de chaque cycle de 28 jours. Votre médecin vous recommandera cette forme de traitement si vous ressentez des symptômes de ménopause au cours de la période sans traitement. Il est possible que vous présentiez une « hémorragie de privation » (saignements évoquant les règles) pendant les derniers jours du traitement par progestatif, voire après l’arrêt.
Mode et voie d'administration
Comment appliquer le dispositif ?
· Chaque sachet contient un dispositif transdermique.
· Déchirez le sachet le long d'un bord.
· N'utilisez jamais de ciseaux pour ouvrir le sachet, vous risqueriez d'endommager le dispositif transdermique.
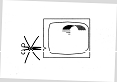
· Sortez le dispositif transdermique du sachet.
· Le dispositif transdermique est composé d'une partie adhésive qui renferme le produit actif et d'un feuillet de protection transparent.
· Retirez le feuillet de protection en évitant de toucher la face adhésive du dispositif transdermique avec les doigts.
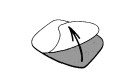
· Une fois le feuillet de protection détaché, OESCLIM doit immédiatement être appliqué sur la fesse, le tronc ou le haut du bras ou de la cuisse à un endroit ne présentant pas de plis importants et qui ne soit pas le siège de frottements vestimentaires et sur une peau sèche, non irritée et non recouverte de crème ou de lotion.
OESCLIM ne doit pas être appliqué sur les seins.
Il est recommandé de ne pas l'appliquer 2 fois de suite au même endroit.
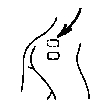
Assurez-vous qu’OESCLIM colle convenablement sur toute sa surface en insistant bien avec la paume de la main, pendant quelques secondes.
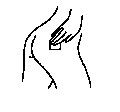
Comment enlever OESCLIM 50 microgrammes/24 heures, dispositif transdermique ?
Pour enlever OESCLIM, il vous suffit de détacher un bord et de tirer doucement sur le dispositif jusqu’à ce qu’il se détache.
Après emploi, le dispositif contient encore des estrogènes, mais en quantité insuffisante pour être encore efficace. Les dispositifs utilisés seront repliés, côté adhésif à l'intérieur, avant d'être jetés.
Précautions particulières
Il est possible de se doucher ou de prendre un bain tout en gardant le dispositif transdermique. En cas de décollement prématuré du dispositif, il faut tenter de le replacer sur une peau sèche.
Si cela n'est pas possible, utilisez un dispositif neuf qui sera retiré à la date initialement prévue. Reprendre ensuite le rythme de changement du dispositif conformément au schéma thérapeutique initial.
Une fois appliqué, le dispositif transdermique ne doit pas être directement exposé au soleil.
Dans tous les cas se conformer strictement à l’ordonnance de votre médecin.
Si vous devez subir une intervention chirurgicale
Si vous devez subir une intervention chirurgicale, informez votre chirurgien que vous prenez OESCLIM. Il sera peut-être nécessaire d’arrêter le traitement environ 4 à 6 semaines avant l’opération afin de réduire le risque de caillots sanguins (voir rubrique 2. Caillots sanguins dans une veine). Demandez à votre médecin quand vous pourrez reprendre OESCLIM.
Le dispositif transdermique provoque-t-il des irritations cutanées ?
Vous pouvez constater des démangeaisons pendant le port ou une certaine rougeur lors de son retrait.
Ces manifestations sont sans gravité et disparaîtront rapidement.
En cas de gêne, placez le dispositif transdermique à un autre endroit (sauf sur les seins).
Si vous avez utilisé plus d’OESCLIM 50 microgrammes/24 heures (10 mg/22 cm²), dispositif transdermique que vous n’auriez dû
Les signes de surdosage sont habituellement une sensation de douleur au niveau des seins et/ou des saignements vaginaux, une irritabilité, une anxiété, des nausées, des vomissements, un gonflement au niveau de l’abdomen ou du bassin, des flatulences, une rétention d’eau et une sensation de lourdeur au niveau des jambes. Ces signes disparaissent lorsque le traitement est arrêté.
Si ces signes persistent, demandez l'avis de votre médecin.
Si vous (ou quelqu’un d’autre) utilise trop de dispositifs transdermiques d’OESCLIM, il est peu probable que cela vous soit néfaste. Vous pouvez avoir la sensation d’être malade (être nauséeuse), ressentir une somnolence, des sensations vertigineuses ou être malade (avoir des vomissements). Aucun traitement n’est nécessaire mais si vous êtes inquiète, demandez conseil à votre médecin.
Dans ce cas, les dispositifs transdermiques doivent être retirés. Ceci est également applicable dans le cas d’une utilisation non-indiquée chez l’enfant.
Si vous oubliez d’utiliser OESCLIM 50 microgrammes/24 heures (10 mg/22 cm²), dispositif transdermique
Si vous avez oublié d’appliquer un nouveau dispositif au jour prévu, faites-le le plus tôt possible et reprenez votre traitement en cours en appliquant les dispositifs suivants aux jours initialement prévus.
Ne prenez pas de dose double pour compenser la dose simple que vous avez oublié de prendre.
Si vous n'avez pas de traitement pendant plusieurs jours de suite, des saignements irréguliers peuvent apparaître.
En cas de doute, consultez votre médecin.
Si vous arrêtez d’utiliser OESCLIM 50 microgrammes/24 heures (10 mg/22 cm²), dispositif transdermique
A l'arrêt du traitement, les signes de déficit en estrogènes liés à la ménopause peuvent réapparaître.
Si vous avez d’autres questions sur l’utilisation de ce médicament, demandez plus d’informations à votre médecin ou à votre pharmacien.
4. QUELS SONT LES EFFETS INDESIRABLES EVENTUELS ?
Les pathologies suivantes sont rapportées plus fréquemment chez les femmes prenant un THS que chez les femmes qui n’en prennent pas :
· cancer du sein,
· épaississement anormal ou cancer de la paroi de l’utérus (hyperplasie endométriale ou cancer),
· cancer de l’ovaire,
· caillots sanguins dans les veines des jambes ou des poumons (thrombo-embolie veineuse),
· maladie cardiaque,
· accident vasculaire cérébral,
· probable perte de mémoire si le THS est commencé après l’âge de 65 ans.
Pour plus d’informations concernant ces effets indésirables, voir rubrique 2.
Les effets indésirables suivants ont été signalés lors de l’utilisation d’OESCLIM :
Effets indésirables très fréquents (survenant chez plus d’une patiente sur 10) :
· réactions cutanées au site d’application (rougeurs, démangeaisons),
· tension mammaire,
· métrorragies.
Effets indésirables fréquents (survenant chez moins d’une patiente sur 10 mais plus d’une patiente sur 100) :
· maux de tête. Si vous avez des maux de tête de type migraine qui apparaissent pour la première fois, arrêtez de prendre OESCLIM et consultez votre médecin immédiatement,
· nausées, douleurs et gonflements abdominaux,
· démangeaisons, boutons,
· développement exagéré de la muqueuse utérine (hyperplasie de l’endomètre), règles abondantes (ménorragie), développement de grosseurs (fibromes) dans l’utérus,
· augmentation ou diminution du poids,
· saignements vaginaux entre les règles,
· varices, dilatation des vaisseaux.
Effets indésirables peu fréquents (survenant chez moins d’une patiente sur 100 mais plus d’une patiente sur 1000) :
· réactions allergiques,
· augmentation de la pression sanguine (hypertension), palpitations,
· nervosité, troubles de l’humeur, humeur dépressive,
· gaz (flatulences), diarrhées, digestion difficile (dyspepsie),
· troubles de la vésicule biliaire,
· plaques sur la peau (érythème noueux), urticaire,
· anémie,
· excès de graisses dans le sang (dont cholestérol),
· douleur du dos,
· somnolence, insomnie, sensation de fourmillements, de picotements (paresthésie), sensations vertigineuses,
· troubles de la vision,
· incontinence urinaire,
· tumeur du sein, du col de l’utérus et de l’utérus,
· inflammation ou infection du vagin (due à un champignon appelé Candida albicans),
· trouble des ovaires, douleur pelvienne, tension au niveau des seins,
· gonflement des chevilles, des pieds ou des doigts (œdème).
Effets indésirables rares (survenant chez moins d’une patiente sur 1000 mais plus d’une patiente sur 10 000) :
· fatigue, frissons, syndrome pseudo-grippal, douleurs, manque d’efficacité du traitement,
· inflammation des veines (phlébite),
· anxiété, irritabilité, diminution ou augmentation de la libido,
· vomissements, constipation, augmentation de l’appétit, troubles au niveau du rectum, gastrites, ballonnements,
· perturbation du bilan hépatique,
· quantité excessive de certains globules blancs dans le sang (éosinophilie),
· douleurs articulaires, douleurs musculaires, crampes musculaires, inflammation du tendon (ténosynovite),
· mouvements involontaires (hyperkinésie),
· troubles du sommeil, vertiges, migraine,
· laryngite, pharyngite, sinusite,
· éruption cutanée, acné, gonflement de la peau autour du visage et du cou pouvant entraîner une difficulté à respirer (angioœdème), inflammation de la peau due à un champignon, augmentation de la pigmentation de la peau, hirsutisme (augmentation de la pilosité),
· blépharite (inflammation de la paupière), intolérance aux lentilles de contact,
· affection ou inflammation de la vulve et du vagin,
· troubles de la muqueuse utérine, règles douloureuses, abcès au niveau des seins, écoulement anormal de lait, troubles des règles, augmentation de la taille de l’utérus, augmentation du volume des seins, pertes vaginales, troubles dans la période qui précède les règles.
Effets indésirables de fréquence indéterminée (ne peut être estimée sur la base des données disponibles) :
· augmentation de la taille des grosseurs dans l’utérus (fibromes),
· mouvements musculaires anormaux (chorée) ; aggravation d’une épilepsie,
· maladie thrombo-embolique artérielle,
· inflammation du pancréas (pancréatite) chez les femmes ayant des taux élevés de certaines graisses dans le sang (hypertriglycéridémie),
· reflux gastro-œsophagien,
· maladie polykystique des seins.
Les effets indésirables suivants ont été rapportés avec d’autres THS :
· divers troubles cutanés :
o décoloration de la peau, notamment du visage ou du cou, connue sous le nom de « taches de grossesse » (chloasma),
o éruption avec des lésions rouges en forme de cibles ou des plaies (érythème polymorphe),
o taches ou points violacés sur la peau (purpura vasculaire),
· baisse de la mémoire ou des capacités mentales (démence possible).
Déclaration des effets secondaires
Si vous ressentez un quelconque effet indésirable, parlez-en à votre médecin ou votre pharmacien. Ceci s’applique aussi à tout effet indésirable qui ne serait pas mentionné dans cette notice. Vous pouvez également déclarer les effets indésirables directement via le système national de déclaration : Agence nationale de sécurité du médicament et des produits de santé (ANSM) et réseau des Centres Régionaux de Pharmacovigilance - Site internet : https://signalement.social-sante.gouv.fr
En signalant les effets indésirables, vous contribuez à fournir davantage d’informations sur la sécurité du médicament.
5. COMMENT CONSERVER OESCLIM 50 microgrammes/24 heures (10 mg/22 cm²), dispositif transdermique ?
Tenir ce médicament hors de la vue et de la portée des enfants.
N’utilisez pas ce médicament après la date de péremption indiquée sur la boîte après EXP. La date de péremption fait référence au dernier jour de ce mois.
A conserver à une température ne dépassant pas + 25°C.
N’utilisez pas ce médicament si vous remarquez des signes de détérioration au niveau de l'emballage.
Ne jetez aucun médicament au tout-à-l’égout ou avec les ordures ménagères. Demandez à votre pharmacien d’éliminer les médicaments que vous n’utilisez plus. Ces mesures contribueront à protéger l’environnement.
6. CONTENU DE L’EMBALLAGE ET AUTRES INFORMATIONS
Ce que contient OESCLIM 50 microgrammes/24 heures (10 mg/22 cm²), dispositif transdermique
· La substance active est :
Estradiol hémihydraté............................................................................................................. 10 mg
Pour un dispositif transdermique de 22 cm²
Un dispositif transdermique libère 50 microgrammes d'estradiol hémihydraté par 24 heures.
· Les autres composants sont :
Composition de la matrice adhésive : poly(éthylène-acétate de vinyle) (EVA) haute et basse viscosité, éthylcellulose, octyldodécanol, dipropylèneglycol.
Composition du film de protection : mousse beige : poly(éthylène-acétate de vinyle).
Film protecteur : polyester silicone.
Titulaire de l’autorisation de mise sur le marché
1 BIS PLACE DE LA DEFENSE – TOUR TRINITY
92400 COURBEVOIE
Exploitant de l’autorisation de mise sur le marché
1 BIS PLACE DE LA DEFENSE – TOUR TRINITY
92400 COURBEVOIE
42/44 RUE DE LONGVIC
21300 CHENOVE
Noms du médicament dans les Etats membres de l'Espace Economique Européen
Ce médicament est autorisé dans les Etats membres de l'Espace Economique Européen sous les noms suivants : Conformément à la réglementation en vigueur.
[À compléter ultérieurement par le titulaire]
La dernière date à laquelle cette notice a été révisée est :
[à compléter ultérieurement par le titulaire]
Des informations détaillées sur ce médicament sont disponibles sur le site Internet de l’ANSM (France).
[1] Indiquer le libellé détaillé qui figure dans l’application form avec le code de la modification selon les lignes directrices https://ec.europa.eu/health//sites/health/files/files/eudralex/vol-2/c_2013_2008/c_2013_2008_pdf/c_2013_2804_fr.pdf