Dernière mise à jour le 03/08/2026
EVEROLIMUS EG 10 mg, comprimé
Indications thérapeutiques
EVEROLIMUS EG est utilisé chez les patients adultes pour traiter :
· le cancer du sein avancé avec récepteurs hormonaux positifs chez les femmes ménopausées chez qui d'autres traitements (appelés « inhibiteurs non-stéroïdiens de l'aromatase ») ne permettent plus de contrôler la maladie. Il est administré avec un médicament appelé exémestane, un inhibiteur stéroïdien de l'aromatase, qui est utilisé dans le traitement hormonal du cancer.
· des tumeurs avancées appelées tumeurs neuroendocrines d’origine gastrique, intestinale, pulmonaire ou pancréatique. Il est administré si les tumeurs sont inopérables et si elles ne produisent pas en excès certaines hormones spécifiques ou d’autres substances naturelles apparentées.
Présentations
> plaquette(s) OPA : polyamide orienté aluminium PVC-Aluminium de 30 comprimé(s)
Code CIP : 34009 301 484 6 4
Déclaration de commercialisation : 11/03/2022
Cette présentation est agréée aux collectivités
- Prix hors honoraire de dispensation : 1146,11 €
- Honoraire de dispensation : 1,02 €
- Prix honoraire compris : 1147,13 €
- Taux de remboursement :100%
Service médical rendu (SMR)
Amélioration du service médical rendu (ASMR)
Ce médicament étant un générique, l'ASMR n'a pas été évalué par la commission de la transparence (CT), il est possible de se référer à la /aux spécialité(s) de référence du groupe générique auquel appartient ce médicament (cliquez ici pour aller à la rubrique des groupes génériques)
Autres informations
- Titulaire de l'autorisation : EG LABO - Laboratoires EuroGenerics
- Conditions de prescription et de délivrance :
- liste I
- médicament nécessitant une surveillance particulière pendant le traitement
- prescription hospitalière
- prescription réservée aux médecins compétents en CANCEROLOGIE
- prescription réservée aux spécialistes et services HEMATOLOGIE
- prescription réservée aux spécialistes et services ONCOLOGIE MEDICALE
- Statut de l'autorisation : Valide
- Type de procédure : Procédure décentralisée
- Code CIS : 6 748 917 8
ANSM - Mis à jour le : 31/01/2024
EVEROLIMUS EG 10 mg comprimé
2. COMPOSITION QUALITATIVE ET QUANTITATIVE
Évérolimus............................................................................................................................. 10 mg
Pour un comprimé
Excipient à effet notoire : chaque comprimé contient 262 mg de lactose.
Pour la liste complète des excipients, voir rubrique 6.1.
Comprimé ovale, plat, blanc à blanc cassé, d’environ 15 mm de long et 6 mm de large, portant la mention « EVR » sur une face et « NAT » sur l’autre face.
4.1. Indications thérapeutiques
Cancer du sein avancé avec récepteurs hormonaux positifs
EVEROLIMUS EG est indiqué dans le traitement du cancer du sein avancé avec récepteurs hormonaux positifs, HER2/neu négatif, en association avec l’exémestane, chez les femmes ménopausées sans atteinte viscérale symptomatique en cas de récidive ou de progression de la maladie, après un traitement par un inhibiteur non-stéroïdien de l'aromatase.
Tumeurs neuroendocrines d’origine pancréatique
EVEROLIMUS EG est indiqué dans le traitement de tumeurs neuroendocrines d’origine pancréatique non résécables ou métastatiques bien ou moyennement différenciées avec progression de la maladie chez l’adulte.
Tumeurs neuroendocrines d’origine gastro-intestinale ou pulmonaire
EVEROLIMUS EG est indiqué dans le traitement de tumeurs neuroendocrines d’origine gastro-intestinale ou pulmonaire non résécables ou métastatiques, bien différenciées (Grade 1 ou Grade 2), non fonctionnelles, en progression chez l’adulte (voir rubriques 4.4 et 5.1).
Cancer du rein
EVEROLIMUS EG est indiqué dans le traitement du cancer du rein avancé chez les patients ayant progressé sous ou après une thérapie ciblée anti-VEGF.
4.2. Posologie et mode d'administration
Le traitement par EVEROLIMUS EG doit être instauré et suivi par un médecin ayant l’expérience des traitements anticancéreux.
Pour les différents schémas posologiques, EVEROLIMUS EG est disponible en comprimés de 2,5 mg, 5 mg et 10 mg.
La dose recommandée d’évérolimus est de 10 mg une fois par jour. Le traitement doit être poursuivi aussi longtemps qu’un bénéfice clinique est observé ou jusqu’à la survenue d’une toxicité inacceptable.
Si une dose est oubliée, le patient ne doit pas prendre une dose supplémentaire, mais prendre la prochaine dose prescrite comme d’habitude.
Ajustement de la posologie en cas d’effets indésirables
La prise en charge d’effets indésirables sévères et/ou intolérables suspectés d’être liés au traitement peut nécessiter une réduction de dose et/ou une interruption temporaire du traitement par EVEROLIMUS EG. Pour les effets indésirables de Grade 1, une adaptation de la dose n’est généralement pas nécessaire. S’il est nécessaire de diminuer la posologie, la dose recommandée est de 5 mg par jour et elle ne doit pas être inférieure à 5 mg par jour.
Le tableau 1 résume les recommandations pour l’adaptation de la posologie en cas d’effets indésirables spécifiques (voir également rubrique 4.4).
Tableau 1 Recommandations pour l’adaptation de la posologie d’EVEROLIMUS EG
|
Effet indésirable |
Sévérité1 |
Adaptation de la posologie d’EVEROLIMUS EG |
|
Pneumopathie non infectieuse |
Grade 2 |
Envisager l'interruption du traitement jusqu'à l'amélioration des symptômes à un Grade ≤ 1. Réintroduire le traitement à 5 mg par jour. Arrêter le traitement en l'absence de récupération dans un délai de 4 semaines. |
|
Grade 3 |
Interrompre le traitement jusqu’à la résolution des symptômes à un Grade ≤ 1. Envisager la reprise du traitement à 5 mg par jour. Si une toxicité de Grade 3 réapparait, envisager l’arrêt du traitement. |
|
|
Grade 4 |
Arrêter le traitement. |
|
|
Stomatite |
Grade 2 |
Interrompre temporairement le traitement jusqu'à rétablissement à un Grade ≤ 1. Réintroduire le traitement à la même dose. En cas de récidive de la stomatite à un Grade 2, interrompre le traitement jusqu'à rétablissement à un Grade ≤ 1. Réintroduire le traitement à 5 mg par jour. |
|
Grade 3 |
Interrompre temporairement le traitement jusqu'à rétablissement à un Grade ≤ 1. Réintroduire le traitement à 5 mg par jour. |
|
|
Grade 4 |
Arrêter le traitement. |
|
|
Autres toxicités non hématologiques (sauf événements métaboliques) |
Grade 2 |
Si la toxicité est tolérable, aucune adaptation de la posologie n'est nécessaire. Si la toxicité devient intolérable, interrompre temporairement le traitement jusqu'à rétablissement à un Grade ≤ 1. Réintroduire le traitement à la même dose. En cas de récidive de la toxicité à un Grade 2, interrompre le traitement jusqu'à rétablissement à un Grade ≤ 1. Réintroduire le traitement à 5 mg par jour. |
|
Grade 3 |
Interrompre temporairement le traitement jusqu'à rétablissement à un Grade ≤ 1. Envisager de réintroduire le traitement à 5 mg par jour. Si une toxicité de Grade 3 réapparait, envisager l’arrêt du traitement. |
|
|
Grade 4 |
Arrêter le traitement. |
|
|
Événements métaboliques (par exemple, hyperglycémie, dyslipidémie) |
Grade 2 |
Aucune adaptation de la posologie n'est nécessaire. |
|
Grade 3 |
Interrompre temporairement le traitement. Réintroduire le traitement à 5 mg par jour. |
|
|
Grade 4 |
Arrêter le traitement. |
|
|
Thrombopénie |
Grade 2 (< 75, ≥ 50 x 109/l) |
Interrompre temporairement le traitement jusqu'à rétablissement à un Grade ≤ 1 (≥ 75 x 109/l). Réintroduire le traitement à la même dose. |
|
Grade 3 & 4 (< 50 x 109/l) |
Interrompre temporairement le traitement jusqu'à rétablissement à un Grade ≤ 1 (≥ 75 x 109/l). Réintroduire le traitement à 5 mg par jour. |
|
|
Neutropénie |
Grade 2 (≥ 1x109/l) |
Aucune adaptation de la posologie n'est nécessaire. |
|
Grade 3 (< 1, ≥ 0,5 x 109/l) |
Interrompre temporairement le traitement jusqu'à rétablissement à un Grade ≤ 2 (≥ 1 x 109/l). Réintroduire le traitement à la même dose. |
|
|
Grade 4 (< 0,5 x 109/l) |
Interrompre temporairement le traitement jusqu'à rétablissement à un Grade ≤ 2 (≥ 1 x 109/l). Réintroduire le traitement à 5 mg par jour. |
|
|
Neutropénie fébrile |
Grade 3 |
Interrompre temporairement le traitement jusqu'à rétablissement à un Grade ≤ 2 (≥ 1,25 x 109/l) et absence de fièvre. Réintroduire le traitement à 5 mg par jour. |
|
Grade 4 |
Arrêter le traitement. |
|
|
1 Grades évalués selon les critères communs de terminologie des événements indésirables (CTCAE, Common Terminology Criteria for Adverse Events), version 3.0, de l’Institut national américain du cancer (NCI, National Cancer Institute) |
||
Populations particulières
Patients âgés (≥ 65 ans)
Aucune adaptation de la posologie n’est nécessaire (voir rubrique 5.2).
Insuffisance rénale
Aucune adaptation de la posologie n’est nécessaire (voir rubrique 5.2).
Insuffisance hépatique
· Insuffisance hépatique légère (classe A de Child-Pugh) - la dose quotidienne recommandée est de 7,5 mg.
· Insuffisance hépatique modérée (classe B de Child-Pugh) - la dose quotidienne recommandée est de 5 mg.
· Insuffisance hépatique sévère (classe C de Child-Pugh) - EVEROLIMUS EG n’est recommandé que si le bénéfice attendu est supérieur au risque. Dans ce cas, la dose quotidienne ne devra pas dépasser 2,5 mg.
Une adaptation posologique devra être effectuée si la fonction hépatique du patient (classe de Child-Pugh) change au cours du traitement (voir également rubriques 4.4 et 5.2).
Population pédiatrique
La sécurité et l’efficacité de l’évérolimus chez les enfants âgés de 0 à 18 ans n’ont pas été établies. Aucune donnée n’est disponible.
Mode d’administration
EVEROLIMUS EG doit être pris par voie orale une fois par jour à la même heure chaque jour, avec ou sans aliments de façon constante (voir rubrique 5.2). Les comprimés d’EVEROLIMUS EG doivent être avalés entiers avec un verre d’eau. Les comprimés ne doivent pas être mâchés ou écrasés.
4.4. Mises en garde spéciales et précautions d'emploi
La pneumopathie non infectieuse est un effet de classe des dérivés de la rapamycine, y compris l’évérolimus. Des pneumopathies non infectieuses (y compris maladie pulmonaire interstitielle) ont été fréquemment rapportées chez des patients traités par l’évérolimus (voir rubrique 4.8).
Certains cas ont été sévères et, en de rares occasions, une issue fatale a été observée. Le diagnostic de pneumopathie non infectieuse doit être envisagé chez les patients présentant des signes et symptômes respiratoires non spécifiques tels qu’hypoxie, épanchement pleural, toux ou dyspnée et lorsqu’une étiologie infectieuse, néoplasique ou toute autre cause non médicamenteuse ont été exclues par les investigations appropriées. Les infections opportunistes telles que la pneumonie à Pneumocystis jirovecii (carinii) (PPJ/PPC) devraient être exclues dans le cadre du diagnostic différentiel de pneumopathie non infectieuse (voir « Infections » ci-dessous). Il convient de recommander aux patients de signaler sans attendre tout symptôme respiratoire nouveau ou qui s’aggrave.
Chez les patients qui développent des modifications radiologiques évoquant une pneumopathie non infectieuse sans symptôme ou accompagnées de symptômes mineurs, le traitement par EVEROLIMUS EG peut être poursuivi sans modification de la posologie. En présence d’une symptomatologie modérée (Grade 2) ou sévère (Grade 3), l’administration de corticoïdes peut être indiquée jusqu’à résolution des symptômes cliniques.
Chez les patients qui nécessitent l’utilisation de corticoïdes pour le traitement d’une pneumopathie non infectieuse, une prophylaxie de la PPJ/PPC doit être envisagée.
Infections
L’évérolimus possède des propriétés immunosuppressives et il peut prédisposer les patients aux infections bactériennes, fongiques, virales ou aux infections à protozoaires, comprenant les infections par des agents pathogènes opportunistes (voir rubrique 4.8). Des infections localisées et systémiques, incluant des pneumonies, d’autres infections bactériennes, des infections fongiques invasives telles qu’aspergillose, candidose ou pneumonie à PPJ/PPC et des infections virales, notamment des cas de réactivation du virus de l’hépatite B, ont été décrites chez des patients traités par l’évérolimus. Certaines de ces infections ont été sévères (conduisant par exemple à un sepsis, une insuffisance respiratoire ou hépatique) et parfois d’issue fatale.
Les médecins et les patients doivent être conscients du risque accru d’infection associé à la prise d’EVEROLIMUS EG. Les infections préexistantes doivent être traitées de manière appropriée et la guérison complète doit être obtenue avant l’instauration d’un traitement par EVEROLIMUS EG. Au cours d’un traitement par EVEROLIMUS EG il convient d’être vigilant aux signes et symptômes d’infection : si une infection est diagnostiquée, un traitement approprié doit être instauré dans les plus brefs délais et l’arrêt temporaire ou définitif d’EVEROLIMUS EG doit être envisagé.
Si le diagnostic d’une infection fongique systémique invasive est fait, le traitement par EVEROLIMUS EG doit être arrêté immédiatement et définitivement, et le patient doit être traité par un traitement antifongique approprié.
Des cas de PPJ/PPC, dont certains avec une issue fatale, ont été rapportés chez des patients qui recevaient de l’évérolimus. La PPJ/PPC peut être associée à l'utilisation concomitante de corticostéroïdes ou d'autres agents immunosuppresseurs. Une prophylaxie des PPJ/PPC doit être envisagée lorsque l'utilisation concomitante de corticoïdes ou d'autres agents immunosuppresseurs est nécessaire.
Réactions d’hypersensibilité
Des réactions d’hypersensibilité se manifestent par des symptômes comprenant, de façon non exhaustive, anaphylaxie, dyspnée, bouffée congestive, douleur thoracique et angiœdème (par exemple, gonflement des voies respiratoires et de la langue avec ou sans difficultés respiratoires) ont été observés avec l’évérolimus (voir rubrique 4.3).
Utilisation concomitante avec des inhibiteurs de l’enzyme de conversion de l’angiotensine (ECA)
Les patients traités de manière concomitante par des inhibiteurs de l’ECA (par exemple, ramipril) peuvent être plus à risque de développer un œdème de Quincke (par exemple, gonflement des voies respiratoires ou de la langue, avec ou sans altération de la respiration) (voir rubrique 4.5).
Stomatite
La stomatite, incluant ulcérations buccales et mucite, est l’effet indésirable le plus fréquemment rapporté chez des patients traités par évérolimus (voir rubrique 4.8). La stomatite apparait le plus souvent au cours des 8 premières semaines de traitement. Une étude en simple bras chez des patientes ménopausées atteintes d’un cancer du sein traitées par évérolimus et exémestane a montré qu’une solution orale de corticoïdes sans alcool, administrée en bain de bouche pendant les 8 premières semaines de traitement, peut diminuer l’incidence et la gravité des stomatites (voir rubrique 5.1). La prise en charge des stomatites peut ainsi inclure l’utilisation prophylactique et/ou thérapeutique de traitements topiques, comme une solution orale de corticoïdes sans alcool utilisée en bain de bouche. Cependant, les produits à base d’alcool, d’eau oxygénée, d’iode ou de dérivés du thym doivent être évités car ils peuvent exacerber l’affection. La surveillance et le traitement des infections fongiques sont recommandés, particulièrement chez les patients ayant été traités par des médicaments à base de stéroïdes. Les agents antifongiques ne doivent pas être utilisés, sauf si une infection fongique a été diagnostiquée (voir rubrique 4.5).
Insuffisance rénale
Des cas d’insuffisance rénale (incluant des insuffisances rénales aigues) dont certains d’issue fatale, ont été observés chez des patients traités par l’évérolimus (voir rubrique 4.8). La fonction rénale doit être surveillée particulièrement lorsque les patients présentent des facteurs de risques supplémentaires qui pourraient davantage altérer leur fonction rénale.
Analyses biologiques et surveillance
Fonction rénale
Des augmentations de la créatininémie, généralement légères, et de la protéinurie ont été rapportées (voir rubrique 4.8). Il est recommandé de surveiller la fonction rénale, notamment l’urémie, la protéinurie ou la créatininémie, avant le début du traitement par EVEROLIMUS EG et régulièrement ensuite.
Glycémie
Des cas d’hyperglycémie ont été rapportés (voir rubrique 4.8). La glycémie à jeun doit être contrôlée avant le début du traitement par EVEROLIMUS EG et régulièrement ensuite. Une surveillance plus fréquente est recommandée quand EVEROLIMUS EG est co-administré avec des médicaments pouvant induire une hyperglycémie. Dans la mesure du possible, la glycémie doit être équilibrée de façon optimale avant l’instauration du traitement par EVEROLIMUS EG.
Lipémie
Des cas de dyslipidémie (incluant des cas d’hypercholestérolémie et d’hypertriglycéridémie) ont été rapportés. Il est recommandé de surveiller la cholestérolémie et la triglycéridémie avant le début du traitement par EVEROLIMUS EG et de manière régulière par la suite, et de mettre en place un traitement approprié.
Paramètres hématologiques
Des diminutions de l’hémoglobine, des lymphocytes, des neutrophiles et des plaquettes ont été observées (voir rubrique 4.8). La numération-formule sanguine doit être contrôlée avant l’instauration du traitement par EVEROLIMUS EG et régulièrement ensuite.
Tumeurs carcinoïdes fonctionnelles
Dans un essai randomisé, en double aveugle et multicentrique, réalisé chez des patients avec des tumeurs carcinoïdes fonctionnelles, l’évérolimus en association avec l’octréotide à libération prolongée a été comparé au placebo associé à l’octréotide à libération prolongée. L’étude n’a pas satisfait le critère principal d’efficacité (survie sans progression [SSP]) et l’analyse intermédiaire de la survie globale (SG) était numériquement en faveur du bras placebo associé à l’octréotide à libération prolongée. En conséquence, la sécurité et l’efficacité de l’évérolimus chez les patients atteints de tumeurs carcinoïdes fonctionnelles n’ont pas été démontrées.
Facteurs pronostics des tumeurs neuroendocrines d'origine gastro-intestinale ou pulmonaire
Chez les patients atteints de tumeurs neuroendocrines d’origine gastro-intestinale ou pulmonaire non fonctionnelles et présentant des facteurs de bon pronostic au diagnostic, (par exemple site tumoral primitif localisé à l’iléon et des valeurs normales de la chromogranine A ou absence d’atteinte osseuse) une évaluation individuelle du rapport bénéfice risque doit être réalisée avant de débuter le traitement par évérolimus. Des preuves limitées de bénéfice en termes de SSP ont été rapportées dans le sous-groupe de patients chez lesquels le site tumoral primitif était l’iléon (voir rubrique 5.1).
Interactions
L’administration concomitante avec des inhibiteurs et des inducteurs du CYP3A4 et/ou de la glycoprotéine P (PgP), qui agit comme pompe d’efflux de nombreux médicaments, doit être évitée (voir rubrique 4.5). Si l’administration concomitante avec un inhibiteur ou un inducteur modéré du CYP3A4 et/ou de la PgP ne peut être évitée, l'état clinique du patient doit être étroitement surveillé. Des ajustements de dose d’EVEROLIMUS EG peuvent être envisagés en fonction de l’ASC prévisible (voir rubrique 4.5).
Le traitement concomitant avec des inhibiteurs puissants du CYP3A4 entraîne une augmentation considérable de la concentration plasmatique de l’évérolimus (voir rubrique 4.5). Il n’y a pas actuellement de données suffisantes pour permettre des recommandations posologiques dans cette situation. Ainsi, le traitement concomitant par EVEROLIMUS EG avec des inhibiteurs puissants n’est pas recommandé.
La prudence est requise lorsqu'EVEROLIMUS EG est pris en association avec des substrats du CYP3A4 à marge thérapeutique étroite administrés par voie orale, en raison du risque d’interactions médicamenteuses. Si EVEROLIMUS EG est administré en association avec des substrats du CYP3A4 à marge thérapeutique étroite administrés par voie orale (par exemple : pimozide, terfénadine, astémizole, cisapride, quinidine ou dérivés de l'ergot de seigle), le patient doit être surveillé pour détecter tout effet indésirable décrit dans les mentions légales du substrat du CYP3A4 administré par voie orale (voir rubrique 4.5).
Insuffisance hépatique
L’exposition à l’évérolimus a été augmentée chez les patients présentant une insuffisance hépatique légère (classe A de Child-Pugh), modérée (classe B de Child-Pugh) et sévère (classe C de Child-Pugh) (voir rubrique 5.2).
L’utilisation d’EVEROLIMUS EG est uniquement recommandée chez les patients présentant une insuffisance hépatique sévère (classe C de Child-Pugh) si le bénéfice potentiel est supérieur au risque (voir rubriques 4.2 et 5.2).
Aucune donnée clinique d’efficacité ou de tolérance n’est actuellement disponible pour appuyer des recommandations d’adaptation de posologie pour la prise en charge des effets indésirables chez les patients souffrant d’une insuffisance hépatique.
Vaccinations
L’utilisation de vaccins vivants doit être évitée au cours du traitement par EVEROLIMUS EG (voir rubrique 4.5).
Lactose
Ce médicament contient du lactose. Son utilisation est déconseillée chez les patients présentant une intolérance au galactose, un déficit en lactase de Lapp ou un syndrome de malabsorption du glucose ou du galactose (maladies héréditaires rares).
Complications de la cicatrisation des plaies
Une altération de la cicatrisation des plaies est un effet de la classe des dérivés de la rapamycine, y compris l’évérolimus. La prudence est donc requise avec l’utilisation d’EVEROLIMUS EG dans la phase péri-opératoire.
Complications de la radiothérapie
Des réactions graves et sévères aux radiations (telles que oesophagite radique, pneumopathie radique et lésion cutanée radique), y compris des cas mortels, ont été rapportés lorsque l’évérolimus était pris pendant, ou peu après, une radiothérapie. La prudence est donc requise quant à la potentialisation des toxicités radio-induites chez les patients prenant de l’évérolimus peu de temps après une radiothérapie.
De plus, un phénomène de rappel (PR) a été rapporté chez des patients prenant de l’évérolimus qui avaient préalablement reçu une radiothérapie. Dans le cas d’un PR, l’interruption ou l’arrêt du traitement par évérolimus doit être considéré.
4.5. Interactions avec d'autres médicaments et autres formes d'interactions
Les interactions connues et théoriques avec des inhibiteurs et des inducteurs sélectifs du CYP3A4 et de la PgP sont décrites dans le tableau 2 ci-dessous.
Inhibiteurs du CYP3A4 et de la PgP susceptibles d’augmenter les concentrations d’évérolimus
Les inhibiteurs du CYP3A4 ou de la PgP peuvent augmenter les concentrations sanguines de l’évérolimus en diminuant le métabolisme ou l’efflux de l’évérolimus des cellules intestinales.
Inducteurs du CYP3A4 et de la PgP susceptibles de diminuer les concentrations d’évérolimus
Les substances qui sont des inducteurs du CYP3A4 et de la PgP peuvent diminuer les concentrations sanguines de l’évérolimus en augmentant le métabolisme ou l’efflux de l’évérolimus des cellules intestinales.
Tableau 2 Effets des autres substances actives sur l’évérolimus
|
Substances actives par interaction |
Interaction – Modifications de l’ASC et/ou de la Cmax de l’évérolimus Rapport des moyennes géométriques (intervalle des valeurs observées) |
Recommandations pour l’administration concomitante |
|
Inhibiteurs puissants du CYP3A4 et/ou de la PgP |
||
|
Kétoconazole |
ASC ↑15,3 fois (intervalle 11,2-22,5) Cmax ↑4,1 fois (intervalle 2,6-7,0) |
L’administration concomitante d’évérolimus avec des inhibiteurs puissants n’est pas recommandée. |
|
Itraconazole, posaconazole, voriconazole |
Non étudiée, une augmentation importante des concentrations de l’évérolimus est attendue. |
|
|
Télithromycine, clarithromycine |
||
|
Néfazodone |
||
|
Ritonavir, atazanavir, saquinavir, darunavir, indinavir, nelfinavir |
||
|
Inhibiteurs modérés du CYP3A4 et/ou de la PgP |
||
|
Érythromycine |
ASC ↑4,4 fois (intervalle 2,0-12,6) Cmax ↑2.0 fois (intervalle 0,9-3,5) |
Utilisation avec précaution quand l’administration concomitante avec des inhibiteurs modérés du CYP3A4 ou de la PgP ne peut être évitée. Si l’administration concomitante avec un inhibiteur modéré du CYP3A4 ou de la PgP est requise, une réduction de dose à 5 mg par jour ou 2,5 mg par jour peut être envisagée. Toutefois, il n’y a pas de données cliniques suffisantes sur l’ajustement de dose. En raison de la variabilité entre les sujets, les ajustements de dose recommandés peuvent ne pas être optimaux pour tous les individus ; une surveillance étroite des effets indésirables est donc recommandée (voir les sections 4.2 et 4.4). Si l’inhibiteur modéré est arrêté, envisager une période d’élimination d’au moins 2 à 3 jours (temps moyen d’élimination des inhibiteurs modérés les plus utilisés) avant de reprendre la dose d’EVEROLIMUS EG utilisée avant l’instauration du traitement concomitant. |
|
Imatinib |
ASC ↑ 3,7 fois Cmax ↑ 2,2 fois |
|
|
Vérapamil |
ASC ↑3,5 fois (intervalle 2,2-6,3) Cmax ↑2,3 fois (intervalle 1,3-3,8) |
|
|
Ciclosporine orale |
ASC ↑2,7 fois (intervalle 1,5-4,7) Cmax ↑1,8 fois (intervalle 1,3-2,6) |
|
|
Cannabidiol (inhibiteur de la P-gp |
ASC ↑ 2.5 fois Cmax ↑ 2.5-fois |
|
|
Fluconazole |
Non étudiée. Exposition accrue attendue. |
|
|
Diltiazem |
||
|
Dronédarone |
Non étudiée. Exposition accrue attendue |
|
|
Amprénavir, fosamprénavir |
Non étudiée. Exposition accrue attendue. |
|
|
Jus de pamplemousse ou autres aliments ayant un effet sur le CYP3A4 et/ou la PgP |
Non étudiée. Exposition accrue attendue (variation élevée des effets). |
L’association doit être évitée. |
|
Inducteurs puissants et modérés du CYP3A4 et/ou de la PgP |
||
|
Rifampicine |
ASC ↓63 % (intervalle 0-80 %) Cmax ↓58 % (intervalle 10-70 %) |
Éviter l’utilisation concomitante avec les inducteurs puissants du CYP3A4. Si une administration concomitante avec un inducteur puissant du CYP3A4 est requise, une augmentation de la dose d’EVEROLIMUS EG de 10 mg à 20 mg par jour doit être envisagée en utilisant une augmentation par paliers de 5 mg ou moins le 4e jour et le 8e jour suivant l’instauration du traitement par l’inducteur. Il est prévu que cette dose d’EVEROLIMUS EG permette d’ajuster l’ASC à l’intervalle de valeurs observées sans inducteur. Toutefois il n’existe aucune donnée clinique disponible avec cet ajustement de dose. Lorsque le traitement par l’inducteur est arrêté, envisager une période d’élimination d’au moins 3 à 5 jours (temps raisonnable pour une levée significative de l’induction enzymatique), avant de reprendre la dose d’EVEROLIMUS EG utilisée avant l’instauration du traitement concomitant. |
|
Dexaméthasone |
Non étudiée. Diminution de l’exposition attendue. |
|
|
Carbamazépine, phénobarbital, phénytoïne |
Non étudiée. Diminution de l’exposition attendue. |
|
|
Efavirenz, névirapine |
Non étudiée. Diminution de l’exposition attendue. |
|
|
Millepertuis (Hypericum Perforatum) |
Non étudiée. Diminution importante de l’exposition attendue. |
Les préparations contenant du millepertuis ne doivent pas être utilisées pendant un traitement par l’évérolimus. |
Produits dont la concentration plasmatique peut être altérée par l’évérolimus
En fonction des résultats des études in vitro, au vu des concentrations systémiques obtenues avec des doses de 10 mg par jour par voie orale, l’inhibition de la PgP, du CYP3A4 et du CYP2D6 est peu vraisemblable. Toutefois, l’inhibition du CYP3A4 et de la PgP dans l’intestin ne peut être exclue. Une étude d'interaction chez des sujets sains a montré que l'administration concomitante d'une dose orale de midazolam, un substrat sensible du CYP3A, avec l'évérolimus entraînait une augmentation de 25 % de la Cmax du midazolam et une augmentation de 30 % de l’ASC(0-inf) du midazolam. Cet effet est susceptible d’être dû à l’inhibition du CYP3A4 intestinal par l’évérolimus. L’évérolimus peut donc modifier la biodisponibilité des substrats du CYP3A4 administrés en association par voie orale. Toutefois, un effet cliniquement pertinent sur l'exposition aux substrats du CYP3A4 administrés par voie systémique n'est pas attendu (voir rubrique 4.4).
L’administration concomitante d'évérolimus et d'octréotide à libération prolongée a augmenté la Cmin de l'octréotide avec un rapport des moyennes géométriques (évérolimus/placebo) de 1,47. Un effet cliniquement significatif sur l’efficacité de l'évérolimus chez les patients atteints de tumeurs neuroendocrines avancées n’a pas pu être établi.
L'administration concomitante d'évérolimus et d'exémestane a augmenté la Cmin et la C2h de l'exémestane de 45 % et 64 % respectivement. En revanche, les taux correspondants d'œstradiol à l'état d'équilibre (4 semaines) n'ont pas été différents entre les deux groupes de traitement. Aucune augmentation des effets indésirables liés à l'exémestane n'a été observée chez les patientes atteintes d'un cancer du sein avancé avec récepteurs hormonaux positifs recevant l'association. L'augmentation des taux d'exémestane ne devrait pas avoir d'impact sur l'efficacité ou la tolérance.
Utilisation concomitante avec des inhibiteurs de l’enzyme de conversion de l’angiotensine (ECA)
Les patients traités de manière concomitante par des inhibiteurs de l’ECA (par exemple, ramipril) peuvent être plus à risque de développer un angiœdème (voir rubrique 4.4).
Vaccinations
La réponse immunitaire à la vaccination peut être affectée. Ainsi, la vaccination au cours d’un traitement par EVEROLIMUS EG peut être moins efficace. L’utilisation de vaccins vivants doit être évitée pendant le traitement par EVEROLIMUS EG (voir rubrique 4.4). Exemples de vaccins vivants : vaccin antigrippal intranasal, vaccins anti-rougeoleux, vaccin contre les oreillons, vaccin anti-rubéolique, vaccin antipoliomyélitique oral, vaccin BCG (bacille de Calmette et Guérin), vaccin antiamarile, vaccin contre la varicelle et vaccin antityphoïdique TY21a.
Radiothérapie
Des cas de potentialisation des toxicités radio-induites ont été rapportés chez des patients recevant l’évérolimus (voir rubriques 4.4 et 4.8).
4.6. Fertilité, grossesse et allaitement
Femmes en âge d’avoir des enfants/Contraception chez les hommes et les femmes
Les femmes en âge d’avoir des enfants doivent utiliser une méthode contraceptive hautement efficace (par exemple : méthode contraceptive hormonale ne contenant pas d’œstrogène administrée par voie orale, injectable ou implantable, contraceptifs à base de progestérone, hystérectomie, ligature des trompes, abstinence complète, méthodes barrières, dispositif intra-utérin [DIU], et/ou stérilisation de la femme/de l’homme) pendant le traitement par l’évérolimus et jusqu’à 8 semaines après l’arrêt du traitement. Il n’y a pas lieu d’interdire aux patients masculins de procréer.
Grossesse
Il n'existe pas de données suffisamment pertinentes concernant l'utilisation de l’évérolimus chez la femme enceinte. Les études effectuées chez l'animal ont mis en évidence une toxicité sur la reproduction, incluant une toxicité embryonnaire et fœtale (voir rubrique 5.3). Le risque potentiel en clinique n’est pas connu.
EVEROLIMUS EG n’est pas recommandé chez la femme enceinte et chez la femme en âge de procréer sans méthode contraceptive.
Allaitement
On ne sait pas si l’évérolimus est excrété dans le lait maternel humain. Cependant, les études chez le rat ont montré que l’évérolimus et/ou ses métabolites passaient facilement dans le lait (voir rubrique 5.3). Par conséquent, les femmes traitées par EVEROLIMUS EG ne doivent pas allaiter pendant le traitement et pendant les 2 semaines qui suivent la prise de la dernière dose.
Fertilité
La possibilité que l’évérolimus provoque une stérilité chez les patients hommes et femmes est inconnue, néanmoins on a observé une aménorrhée (aménorrhée secondaire et autres irrégularités menstruelles) et un déséquilibre associé du rapport hormone lutéinisante (LH)/hormone folliculostimulante (FSH) chez les femmes. Les observations précliniques indiquent que le traitement par EVEROLIMUS EG peut diminuer la fertilité masculine et féminine (voir rubrique 5.3).
4.7. Effets sur l'aptitude à conduire des véhicules et à utiliser des machines
Résumé du profil de tolérance
Le profil de tolérance est issu des données regroupées de 2 879 patients traités par l’évérolimus dans onze études cliniques, incluant cinq études de phase III randomisées, en double aveugle, contrôlées contre placebo et six études en ouvert de phase I et de phase II, liées aux indications approuvées.
Les effets indésirables les plus fréquents (fréquence ≥ 1/10) issus des données regroupées de tolérance étaient (par ordre décroissant) : stomatite, rash, fatigue, diarrhée, infection, nausée, diminution de l’appétit, anémie, dysgueusie, pneumopathie inflammatoire, œdème périphérique, hyperglycémie, asthénie, prurit, perte de poids, hypercholestérolémie, épistaxis, toux et céphalées.
Les effets indésirables de Grades 3-4 les plus fréquents (fréquence ≥ 1/100 à < 1/10) ont été : stomatite, anémie, hyperglycémie, infection, fatigue, diarrhée, pneumopathie inflammatoire, asthénie, thrombopénie, neutropénie, dyspnée, protéinurie, lymphopénie, hémorragie, hypophosphatémie, rash, hypertension, pneumonie, augmentation de l’alanine aminotransférase (ALAT), augmentation de l’aspartate aminotransférase (ASAT) et diabète. Les grades suivent la classification CTCAE version 3.0 et 4.03.
Liste des effets indésirables sous forme de tableau
Le tableau 3 montre les catégories de fréquence des effets indésirables rapportés dans les analyses des données regroupées de tolérance. Les effets indésirables sont présentés selon les classes de systèmes d’organes et les catégories de fréquence MedDRA. Les catégories de fréquence sont définies selon la convention suivante : très fréquent (≥ 1/10) ; fréquent (≥ 1/100, < 1/10) ; peu fréquent (≥ 1/1 000, < 1/100) ; rare (≥ 1/10 000, < 1/1 000) ; très rare (< 1/10 000); fréquence indéterminée (ne peut être estimée sur la base des données disponibles). Au sein de chaque groupe de fréquence, les effets indésirables sont présentés suivant un ordre décroissant de gravité.
Tableau 3 Effets indésirables rapportés dans les études cliniques
|
Classe de systèmes d’organes |
Fréquence |
Terme préférentiel |
|
Infections et infestations |
Très fréquent |
Infectionsa,* |
|
Affections hématologiques et du système lymphatique |
Très fréquent |
Anémie |
|
Fréquent |
Thrombopénie, neutropénie, leucopénie, lymphopénie |
|
|
Peu fréquent |
Pancytopénie |
|
|
Rare |
Érythroblastopénie |
|
|
Affections du système immunitaire |
Peu fréquent |
Hypersensibilité |
|
Troubles du métabolisme et de la nutrition |
Très fréquent |
Diminution de l’appétit, hyperglycémie, hypercholestérolémie |
|
Fréquent |
Hypertriglycéridémie, hypophosphatémie, diabète, hyperlipidémie, hypokaliémie, déshydratation, hypocalcémie |
|
|
Affections psychiatriques |
Fréquent |
Insomnie |
|
Affections du système nerveux |
Très fréquent |
Dysgueusie, céphalées |
|
Peu fréquent |
Agueusie |
|
|
Affections oculaires |
Fréquent |
Œdème de la paupière |
|
Peu fréquent |
Conjonctivite |
|
|
Affections cardiaques |
Peu fréquent |
Insuffisance cardiaque congestive |
|
Affections vasculaires |
Fréquent |
Hémorragieb, hypertension, lymphoedèmeg |
|
Peu fréquent |
Bouffées vasomotrices, thrombose veineuse profonde |
|
|
Affections respiratoires, thoraciques et médiastinales |
Très fréquent |
Pneumopathie inflammatoirec, épistaxis, toux |
|
Fréquent |
Dyspnée |
|
|
Peu fréquent |
Hémoptysie, embolie pulmonaire |
|
|
Rare |
Syndrome de détresse respiratoire aiguë |
|
|
Affections gastro-intestinales |
Très fréquent |
Stomatited, diarrhée, nausée |
|
Fréquent |
Vomissements, sécheresse buccale, douleur abdominale, mucite, douleur orale, dyspepsie, dysphagie |
|
|
Affections hépatobiliaires |
Fréquent |
Augmentation de l’aspartate aminotransférase, augmentation de l’alanine aminotransférase |
|
Affections de la peau et du tissu sous-cutané |
Très fréquent |
Rash, prurit |
|
Fréquent |
Sécheresse cutanée, anomalies des ongles, alopécie légère, acné, érythème, onychoclasie, syndrome mains-pieds, exfoliation cutanée, lésion cutanée |
|
|
Rare |
Angiœdème* |
|
|
Affections musculo-squelettiques et systémiques |
Fréquent |
Arthralgie |
|
Affections du rein et des voies urinaires |
Fréquent |
Protéinurie*, augmentation de la créatinémie, insuffisance rénale* |
|
Peu fréquent |
Augmentation des mictions diurnes, insuffisance rénale aiguë* |
|
|
Affections des organes de reproduction et du sein |
Fréquent |
Irrégularités menstruellese |
|
Peu fréquent |
Aménorrhéee |
|
|
Troubles généraux et anomalies au site d’administration |
Très fréquent |
Fatigue, asthénie, œdème périphérique |
|
Fréquent |
Pyrexie |
|
|
Peu fréquent |
Douleur thoracique non-cardiaque, altération de la cicatrisation des plaies |
|
|
Investigations |
Très fréquent |
Perte de poids |
|
Lésion, intoxication et complications liées aux procédures |
Fréquence indéterminéef |
Phénomène de rappel, potentialisation des toxicités radio-induites |
|
* Voir également la sous-rubrique « Description de certains effets indésirables » a Inclut tous les effets de la classe d’organe « infections et infestations » y compris (fréquent) pneumonie, infection des voies urinaires ; (peu fréquent) bronchite, zona, sepsis, abcès, et cas isolés d'infections opportunistes [par exemple : aspergillose, candidose, PPJ/PPC et hépatite B (voir aussi rubrique 4.4)] et (rare) myocardite virale b Inclut différents événements hémorragiques de différents sites non cités individuellement c Inclut (très fréquent) pneumopathie inflammatoire, pneumopathie interstitielle diffuse, (fréquent) infiltration pulmonaire et (rare) hémorragie alvéolaire pulmonaire, toxicité pulmonaire et alvéolite d Inclut (très fréquent) stomatite, (fréquent) stomatite aphteuse, ulcération buccale et de la langue et (peu fréquent) glossodynie, glossite e Fréquence fondée sur le nombre de femmes âgées de 10 à 55 ans dans les données regroupées f Effet indésirable identifié dans le cadre post-commercialisation g Effet indésirable identifié à partir des rapports post-commercialisation. Fréquence définie sur l’ensemble des données de sécurité issues d’études en oncologie. |
||
Description de certains effets indésirables
Dans les cas rapportés au cours des études cliniques et spontanément après la commercialisation, l’évérolimus a été associé à des cas sévères de réactivation d’hépatite B, dont certains d’issue fatale. La réactivation d’infections est un effet attendu pendant les phases d’immunosuppression.
Dans les cas rapportés au cours des études cliniques et spontanément après la commercialisation, l’évérolimus a été associé à des événements d’insuffisance rénale (dont certains d’issue fatale) et de protéinurie. La surveillance de la fonction rénale est recommandée (voir rubrique 4.4).
Dans les cas rapportés au cours des études cliniques et spontanément après la commercialisation, l’évérolimus a été associé à des cas d’aménorrhée (aménorrhée secondaire et autres irrégularités menstruelles).
Dans les cas rapportés au cours des études cliniques et spontanément après la commercialisation, l’évérolimus a été associé à des cas de PPJ /PPC, dont certains d’issue fatale (voir rubrique 4.4).
Dans les cas rapportés au cours des études cliniques et spontanément après la commercialisation, des cas d’angiœdème ont été rapportés avec et sans utilisation concomitante d’inhibiteurs de l’ECA (voir rubrique 4.4).
Personnes âgées
Dans les données de tolérance regroupées, 37 % des patients traités par évérolimus avaient 65 ans ou plus. Le nombre de patients ayant présenté un effet indésirable entrainant l’arrêt du traitement était supérieur chez les patients de 65 ans et plus (20 % versus 13 %). Les effets indésirables les plus fréquents menant à l’arrêt du traitement étaient les pneumopathies inflammatoires (y compris pneumopathies interstitielles diffuses), les stomatites, la fatigue et les dyspnées.
Déclaration des effets indésirables suspectés
La déclaration des effets indésirables suspectés après autorisation du médicament est importante. Elle permet une surveillance continue du rapport bénéfice/risque du médicament. Les professionnels de santé déclarent tout effet indésirable suspecté via le système national de déclaration : Agence nationale de sécurité du médicament et des produits de santé (ANSM) et réseau des Centres Régionaux de Pharmacovigilance - Site internet : https://signalement.social-sante.gouv.fr/.
Les cas rapportés de surdosage chez l’Homme sont très limités. Des doses uniques allant jusqu’à 70 mg ont été administrées avec une tolérance aiguë acceptable. Un traitement symptomatique approprié doit être instauré dans tous les cas de surdosage.
5. PROPRIETES PHARMACOLOGIQUES
5.1. Propriétés pharmacodynamiques
Mécanisme d’action
L’évérolimus est un inhibiteur sélectif de la cible de la rapamycine chez les mammifères (mTOR, mammalian target of rapamycin). mTOR est une sérine-thréonine kinase importante dont l’activité est connue comme étant régulée à la hausse dans un certain nombre de cancers humains. L’évérolimus se lie à la protéine intracellulaire FKBP-12, formant un complexe qui inhibe l’activité du complexe mTOR 1 (mTORC1). L’inhibition de la voie de signalisation du mTORC1 interfère avec la traduction et la synthèse de protéines en réduisant l’activité de la protéine S6 kinase 1 ribosomale (S6K1, S6 ribosomal protein kinase 1) et de la protéine 1 se liant au facteur d’élongation 4E chez les eucaryotes (4EBP-1, eukaryotic elongation factor 4E-binding protein) qui régulent les protéines impliquées dans le cycle cellulaire, l’angiogenèse et la glycolyse. On pense que la protéine S6K1 phosphoryle le domaine contenant la fonction d’activation de la transcription 1 (AF-1, activation function 1) du récepteur aux œstrogènes, qui est responsable de l’activation du récepteur indépendamment du ligand. L’évérolimus réduit les taux de facteur de croissance de l’endothélium vasculaire (VEGF), qui potentialise les processus d’angiogenèse tumorale. L’évérolimus est un inhibiteur puissant de la croissance et de la prolifération des cellules tumorales, des cellules endothéliales, des fibroblastes et des cellules musculaires lisses vasculaires et il a été montré qu’il réduit la glycolyse au sein des tumeurs solides in vitro et in vivo.
Efficacité et sécurité clinique
Cancer du sein avancé avec récepteurs hormonaux positifs
BOLERO-2 (étude CRAD001Y2301) est une étude de phase III, multicentrique, randomisée, en double aveugle de l’évérolimus + exémestane versus placebo + exémestane conduite chez des femmes ménopausées atteintes d'un cancer du sein avancé avec récepteurs aux œstrogènes positifs, HER2/neu négatif, présentant une récidive ou une progression de la maladie après un traitement antérieur par létrozole ou anastrozole. La randomisation a été stratifiée en fonction de la sensibilité documentée à l'hormonothérapie antérieure et en fonction de la présence de métastases viscérales. La sensibilité à l'hormonothérapie antérieure a été définie comme (1) un bénéfice clinique documenté (réponse complète [RC], réponse partielle [RP], stabilisation de la maladie ≥ 24 semaines) après au moins une hormonothérapie antérieure au stade avancé ou (2) au moins 24 mois d'hormonothérapie adjuvante avant la récidive.
Le critère principal d'évaluation de l'étude était la survie sans progression (SSP) évaluée selon les critères d’évaluation de la réponse dans les tumeurs solides (RECIST, Response Evaluation Criteria in Solid Tumors), sur la base de l'évaluation de l'investigateur (évaluation radiologique locale). Les analyses de confirmation de la SSP se sont basées sur une évaluation radiologique centralisée indépendante.
Les critères secondaires d'évaluation étaient la survie globale (SG), le taux de réponse objective, le taux de bénéfice clinique, la tolérance, la modification de la qualité de vie (QdV) et le délai de détérioration de l'indice de performance (PS, Performance Status) ECOG (Eastern Cooperative Oncology Group).
Au total, 724 patientes ont été randomisées selon un rapport de 2:1 pour recevoir l'association évérolimus (10 mg par jour) + exémestane (25 mg par jour) (n = 485) ou l'association placebo + exémestane (25 mg par jour) (n = 239). Au moment de l’analyse finale de la SG, la durée médiane du traitement par évérolimus était de 24,0 semaines (intervalle 1,0-199,1 semaines). La durée médiane du traitement par exémestane était plus longue dans le bras évérolimus + exémestane avec 29,5 semaines (1,0-199,1), contre 14,1 semaines (1,0-156,0) dans le bras placebo + exémestane.
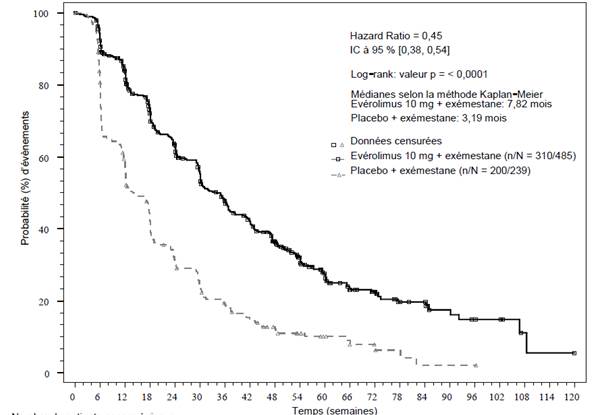
Les résultats d'efficacité pour le critère principal d’évaluation ont été obtenus à partir de l'analyse finale de la SSP (voir tableau 4 et figure 1). Il n’y avait pas de cross over vers le groupe évérolimus + exémestane pour les patientes du groupe placebo + exémestane au moment de la progression de la maladie.
Tableau 4 Résultats d'efficacité de BOLERO-2
|
Analyse |
Évérolimusa n = 485 |
Placeboa n = 239 |
HR (rapport de risque) |
Valeur de p |
|
|
Survie sans progression médiane (mois) (IC à 95 %) |
|||||
|
Évaluation radiologique selon l'investigateur |
7,8 (6,9 à 8,5) |
3,2 (2,8 à 4,1) |
0,45 (0,38 à 0,54) |
< 0,0001 |
|
|
Évaluation radiologique indépendante |
11,0 (9,7 à 15,0) |
4,1 (2,9 à 5,6) |
0,38 (0,31 à 0,48) |
< 0,0001 |
|
|
Survie globale médiane (mois) (IC à 95 %) |
|
||||
|
Survie globale médiane |
31,0 (28,0 - 34,6) |
26,6 (22,6 - 33,1) |
0,89 (0,73 - 1,10) |
0,1426 |
|
|
Meilleure réponse globale (%) (IC à 95 %) |
|
||||
|
Taux de réponse objectiveb |
12,6 % (9,8 à 15,9) |
1,7 % (0,5 à 4,2) |
S/Od |
< 0,0001e |
|
|
Taux de bénéfice cliniquec |
51,3 % (46,8 à 55,9) |
26,4 % (20,9 à 32,4) |
S/Od |
< 0,0001e |
|
|
a Plus exémestane b Taux de réponse objective = proportion de patientes avec une réponse complète ou partielle c Taux de bénéfice clinique = proportion de patientes avec une réponse complète ou partielle ou une stabilisation de la maladie ≥ 24 semaines d Sans objet e La valeur de p est obtenue à partir du test exact de Cochran-Mantel-Haenszel en utilisant une version stratifiée du test de permutation de Cochran-Armitage. |
|
||||
Figure 1 BOLERO-2 - Courbes de Kaplan-Meier de la survie sans progression (évaluation radiologique de l'investigateur)
|
Nombre de patientes encore à risque |
||||||||||||||||||||||
|
Temps (semaines) |
0 |
6 |
12 |
18 |
24 |
30 |
36 |
42 |
48 |
54 |
60 |
66 |
72 |
78 |
84 |
90 |
96 |
102 |
108 |
114 |
120 |
|
|
Évérolimus |
485 |
436 |
366 |
304 |
257 |
221 |
185 |
158 |
124 |
91 |
66 |
50 |
35 |
24 |
22 |
13 |
10 |
8 |
2 |
1 |
0 |
|
|
Placebo |
239 |
190 |
132 |
96 |
67 |
50 |
39 |
30 |
21 |
15 |
10 |
8 |
5 |
3 |
1 |
1 |
1 |
0 |
0 |
0 |
0 |
|
L'effet estimé du traitement sur la SSP a été confirmé par une analyse en sous-groupes programmée de la SSP selon l’évaluation de l'investigateur. Pour tous les sous-groupes analysés (âge, sensibilité à l’hormonothérapie antérieure, nombre d’organes atteints, statut des lésions uniquement osseuses lors de l’inclusion et présence de métastases viscérales, et selon les sous-groupes pronostiques et démographiques majeurs), un effet positif du traitement a été observé sous évérolimus + exémestane avec un rapport de risque (HR) estimé compris entre 0,25 et 0,60 versus placebo + exémestane.
Aucune différence en termes de délai de détérioration ≥ 5 % des scores des domaines fonctionnels et du score global du QLQ-C30 n’a été observée entre les deux groupes de traitement.
L’objectif principal de l’étude était d’estimer le HR de la SSP pour le traitement évérolimus + exémestane versus évérolimus seul. L’objectif secondaire clé était d’estimer le HR de la SSP pour le traitement évérolimus + exémestane versus capécitabine.
Les autres critères secondaires incluaient l’évaluation de la SG, le taux de réponse objective, le taux de bénéfice clinique, la tolérance, le délai de détérioration de l'indice de performance ECOG, le délai de détérioration de la QdV, et la satisfaction au traitement (Treatment Satisfaction Questionnaire for Medication, TSQM). Aucune comparaison statistique formelle n’a été prévue.
Un total de 309 patients ont été randomisés suivant un ratio de 1 :1 :1 pour l’association d’évérolimus (10 mg par jour) + exémestane (25 mg par jour) (n=104), évérolimus seul (10 mg par jour) (n=103), ou capécitabine (une dose de 1 250 mg/m2 deux fois par jour pendant 2 semaines suivi d’une semaine de repos, par cycle de 3 semaines) (n=102). Au moment de la collecte des données, la durée moyenne du traitement était de 27,5 semaines (intervalle 2,0-165,7) dans le bras évérolimus + exémestane, 20 semaines (1,3-145,0) dans le bras évérolimus, et 26,7 semaines (1,4-177,1) dans le bras capécitabine.
Le résultat de l’analyse finale de la SSP avec 154 évènements de SSP observés, basé sur une évaluation locale par les investigateurs, a montré un HR estimé de 0,74 (IC à 90% : 0,57 ; 0,97) en faveur du bras évérolimus + exémestane par rapport au bras évérolimus. La SSP médiane était respectivement de 8,4 mois (IC à 90% : 6,6 ; 9,7) et de 6,8 mois (IC à 90% : 5,5 ; 7,2).
Figure 2 BOLERO-6 – Courbes de Kaplan-Meier de la Survie Sans Progression (évaluation radiologique de l'investigateur)
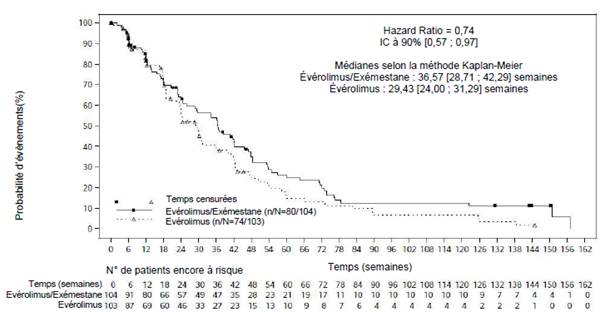
Concernant le critère secondaire clé relatif à la SSP, basé sur un nombre total de 148 évènements de SSP observés, le HR estimé était de1,26 (IC à 90% : 0,96; 1,66) en faveur de la capécitabine par rapport au bras évérolimus+ exémestane en association.
Les résultats du critère secondaire de SG n’étaient pas cohérents avec les résultats observés sur le critère primaire de la SSP, avec une tendance observée en faveur du bras évérolimus seul. Le HR estimé était de 1,27 (IC à 90%: 0,95; 1,70) concernant la comparaison de la SG dans le bras évérolimus seul par rapport au bras évérolimus+exémestane. Le HR estimé pour la comparaison de la SG dans le bras évérolimus+ exémestane en association par rapport au bras capécitabine était de 1,33 (IC à 90%: 0,99; 1,79).
Tumeurs neuroendocrines d’origine pancréatiques (pNET) avancées
RADIANT-3 (étude CRAD001C2324), une étude de phase III, multicentrique, randomisée, en double aveugle de l’évérolimus + meilleur traitement de soutien (BSC, best supportive care) versus placebo + BSC chez des patients atteints de pNET avancées, a démontré un bénéfice clinique statistiquement significatif sous évérolimus versus placebo avec un allongement de 2,4 fois de la survie sans progression (SSP) médiane (11,04 mois versus 4,6 mois) (HR : 0,35 ; IC à 95 % : 0,27, 0,45 ; p < 0,0001) (voir tableau 5 et figure 3).
L'étude RADIANT-3 a été conduite chez des patients atteints de pNET avancées bien et moyennement différenciées dont la maladie avait progressé au cours des 12 mois précédents. Le traitement par analogues de la somatostatine était autorisé dans le cadre du meilleur traitement de soutien (BSC).
Le critère principal d'évaluation de l'étude était la SSP évaluée au moyen des critères d’évaluation de la réponse dans les tumeurs solides (RECIST, Response Evaluation Criteria in Solid Tumors). Après progression radiologique documentée, la levée de l'aveugle pour les patients concernés pouvait être faite par l'investigateur. Les patients randomisés dans le groupe placebo pouvaient alors recevoir de l’évérolimus en ouvert.
Les critères secondaires d'évaluation étaient la tolérance, le taux de réponse objective, la durée de la réponse et la survie globale (SG).
Au total, 410 patients ont été randomisés selon un rapport de 1:1 pour recevoir soit de l’évérolimus 10 mg/jour (n = 207) soit un placebo (n = 203). Les caractéristiques démographiques étaient bien équilibrées (âge médian : 58 ans, hommes : 55 %, Caucasiens : 78,5 %). Cinquante-huit pour cent des patients des deux groupes avaient reçu un traitement systémique antérieur. La durée médiane du traitement en aveugle était de 37,8 semaines (intervalle 1,1-129,9 semaines) pour les patients qui ont reçu l’évérolimus et 16,1 semaines (intervalle 0,4-147,0 semaines) pour ceux qui ont reçu un placebo.
Suite à une progression de la maladie ou après levée de l'aveugle, 172 des 203 patients (84,7 %) initialement randomisés dans le bras placebo ont effectué un cross over pour recevoir l’évérolimus en ouvert.
La durée médiane de traitement en ouvert était de 47,7 semaines pour l’ensemble des patients : 67,1 semaines chez les 53 patients randomisés dans le bras évérolimus qui ont ensuite reçu de l’évérolimus en ouvert et 44,1 semaines chez les 172 patients randomisés dans le bras placebo qui ont ensuite reçu de l’évérolimus en ouvert.
Tableau 5 RADIANT-3 – Résultats en termes d’efficacité
|
Population |
Évérolimus n = 207 |
Placebo n = 203 |
HR (rapport de risque) (IC à 95 %) |
Valeur de p |
|
Survie sans progression médiane (mois) (IC à 95 %) |
||||
|
Évaluation radiologique de l'investigateur |
11,04 (8,41, 13,86) |
4,60 (3,06, 5,39) |
0,35 (0,27, 0,45) |
< 0,0001 |
|
Évaluation radiologique Indépendante |
13,67 (11,17, 18,79) |
5,68 (5,39, 8,31) |
0,38 (0,28, 0,51) |
< 0,0001 |
|
Survie globale médiane (mois) (IC à 95 %) |
||||
|
Survie globale médiane |
44,02 (35,61, 51,75) |
37,68 (29,14, 45,77) |
0,94 (0,73, 1,20) |
0,300 |
Figure 3 RADIANT-3 - Courbes de Kaplan-Meier de la survie sans progression (évaluation radiologique par l’investigateur)

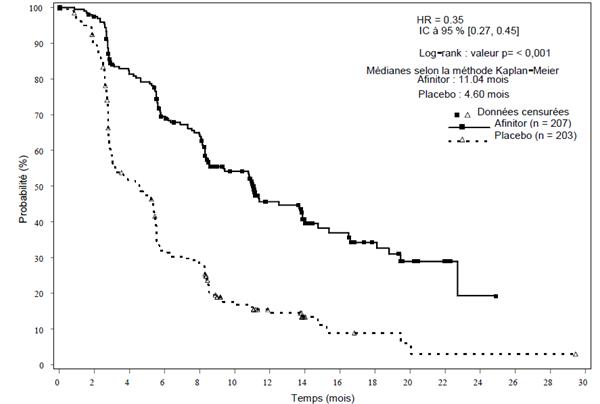
|
Nombre de patients encore à risque |
||||||||||||||||
|
Évérolimus |
207 |
189 |
153 |
126 |
114 |
80 |
49 |
36 |
28 |
21 |
10 |
6 |
2 |
0 |
0 |
0 |
|
Placebo |
203 |
117 |
98 |
59 |
52 |
24 |
16 |
7 |
4 |
3 |
2 |
1 |
1 |
1 |
1 |
0 |
Tumeurs neuroendocrines avancées d’origine gastro-intestinale ou pulmonaire
RADIANT-4 (étude CRAD001T2302), une étude de phase III multicentrique randomisée en double aveugle évaluant l’évérolimus + meilleur traitement de soutien (BSC, best supportive care) versus placebo + BSC qui a été conduite chez des patients atteints de tumeurs neuroendocrines d’origine gastro-intestinale ou pulmonaire avancées, bien différenciées (Grade 1 ou Grade 2), non fonctionnelles, sans syndrome carcinoïde antérieur ou en cours.
Le critère principal de l’étude était la survie sans progression (SSP) sur la base d’une évaluation radiologique indépendante selon les critères d’évaluation de la réponse dans les tumeurs solides (RECIST). Une analyse complémentaire de la SSP était basée sur l’évaluation locale par l’investigateur. Les critères secondaires comprenaient la survie globale (SG), le taux de réponse global, le taux de contrôle de la maladie, la sécurité d’emploi, la modification de la qualité de vie (FACT-G) et le délai de détérioration de l’indice de performance de l’Organisation Mondiale de la Santé (IP OMS).
Au total, 302 patients ont été randomisés selon un rapport de 2:1 pour recevoir soit l’évérolimus (10 mg par jour) (n = 205) soit le placebo (n = 97). Les caractéristiques démographiques et les caractéristiques de la maladie étaient dans l’ensemble équilibrées (âge médian de 63 ans [intervalle 22 à 86], 76 % de Caucasiens, utilisation antérieure d’analogues de la somatostatine [ASS]). La durée médiane du traitement en aveugle était de 40,4 semaines pour les patients ayant reçu de l’évérolimus et de 19,6 semaines pour ceux ayant reçu le placebo. Après l’analyse principale de la SSP, 6 patients du bras placebo ont bénéficié d’un cross over vers le groupe en ouvert évérolimus.
Les résultats d’efficacité pour le critère principal d’évaluation SSP (évaluation indépendante radiologique) ont été obtenus à partir de l’analyse finale de la SSP (voir Tableau 6 et figure 4).
Les résultats d’efficacité pour la SSP (évaluation radiologique par l’investigateur) ont été obtenus à partir de l’analyse finale de la SG (voir Tableau 6).
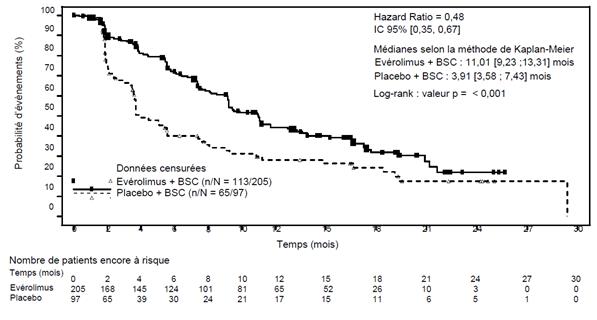
Tableau 6 RADIANT-4 – Résultats en termes de survie sans progression
|
Population |
Évérolimus n = 205 |
Placebo n = 97 |
HR (rapport de risque) (IC à 95 %) |
Valeur de pa |
|
Survie sans progression médiane (mois) (IC à 95 %) |
||||
|
Évaluation radiologique indépendante |
11,01 (9,2-13,3) |
3,91 (3,6-7,4) |
0,48 (0,35-0,67) |
< 0,001 |
|
Évaluation radiologique par l’investigateur |
14,39 (11,24-17,97) |
5,45 (3,71-7,39) |
0,40 (0,29-0,55) |
< 0,001 |
|
a Valeur de p d’après un test du log-rank stratifié unilatéral |
||||
Figure 4 RADIANT-4 – Courbes de Kaplan-Meier de la survie sans progression (revue radiologique indépendante)
Dans les analyses complémentaires, un effet positif du traitement a été observé dans tous les sous-groupes à l’exception du sous-groupe de patients chez lesquels le site tumoral primitif était l’iléon (Iléon : HR = 1,22 [IC à 95 % : 0,56 à 2,65] ; Hors iléon : HR = 0,34 [IC à 95 % : 0,22 à 0,54] ; Poumon : HR = 0,43 [IC à 95 % : 0,24 à 0,79]) (voir figure 5).
Figure 5 RADIANT-4 – Résultats en termes de survie sans progression par sous-groupes de patients prédéfinis (évaluation radiologique indépendante)
Évérolimus + BSC Placebo + BSC En faveur de
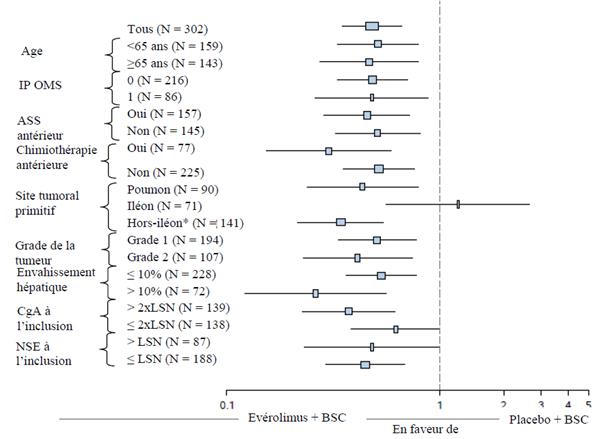
*Hors-iléon : estomac, colon, rectum, appendice, cæcum, duodénum, jéjunum, carcinome d’origine primitive inconnue et autres origines gastro-intestinales
LSN : Limite Supérieure de la Normale
CgA : Chromogranine A
NSE : Énolase neurono-spécifique
Rapport de risque (HR) (IC à 95 %) d’après le modèle de Cox stratifié.
L’analyse finale de la survie globale (SG) n’a pas montré de différence statistiquement significative entre les patients ayant reçu l’évérolimus ou le placebo pendant la période de traitement en aveugle de l’étude (HR=0,90 [95% CI : 0,66 à 1,22])
Aucune différence du délai de détérioration définitive de l’indice de performance de l’OMS (HR=1,02; [95% CI : 0,65, 1,61]) ni du délai de détérioration définitive de la qualité de vie (score FACT-G total HR=0,74; [95% CI : 0,50, 1,10])) n’a été observée entre les deux bras.
Cancer du rein avancéRECORD-1 (étude CRAD001C2240), une étude de phase III multicentrique internationale randomisée en double aveugle comparant l’évérolimus 10 mg/jour au placebo, administrés tous les deux en association avec un traitement symptomatique optimal (BSC, best supportive care) a été menée chez des patients ayant un cancer du rein métastatique dont la maladie avait progressé sous ou après un traitement par un VEGFR-TKI (inhibiteur de la tyrosine kinase du récepteur du facteur de croissance de l’endothélium vasculaire [sunitinib, sorafénib ou sunitinib plussorafénib]). Un traitement antérieur par le bévacizumab et l’interféron-α était également autorisé. Les patients ont été stratifiés en fonction du score pronostique du Memorial Sloan-Kettering Cancer Center (MSKCC) (groupes à risque faible versus intermédiaire versus élevé) et des traitements anticancéreux antérieurs (1 versus 2 VEGFR-TKI).
Le critère principal était la survie sans progression, documentée selon les critères d’évaluation de la réponse dans les tumeurs solides (RECIST) et évaluée en aveugle par un comité d’évaluation centralisé indépendant. Les critères secondaires étaient la tolérance, le taux de réponse tumorale objective, la survie globale, les symptômes liés à la maladie et la qualité de vie.
Après documentation d’une progression radiologique, l’aveugle pouvait être levé par l’investigateur : les patients randomisés dans le groupe placebo ont pu alors recevoir l’évérolimus 10 mg/jour en ouvert. Le comité indépendant de surveillance a recommandé d’arrêter cette étude au moment de la deuxième analyse intermédiaire car le critère principal avait été atteint.
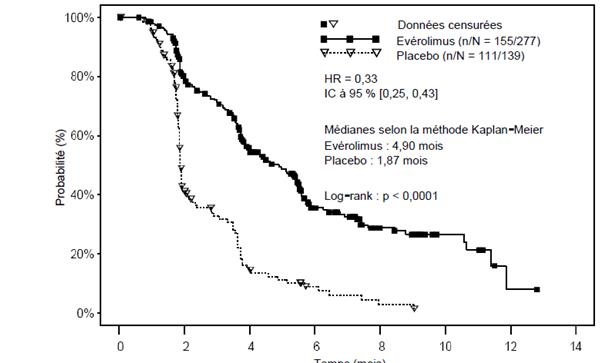
Au total, 416 patients ont été randomisés selon un rapport de 2:1 pour recevoir l’évérolimus (n = 277) ou le placebo (n = 139). Les caractéristiques démographiques étaient équilibrées (âge médian : 61 ans [intervalle 27-85 ans], hommes : 78 %, Caucasiens : 88 %, nombre de traitements antérieurs par un VEGFR-TKI [1 – 74 %, 2 – 26 %]). La durée moyenne du traitement en aveugle était de 141 jours (intervalle : 19-451 jours) pour les patients recevant l’évérolimus et de 60 jours (intervalle : 21-295 jours) pour ceux ayant reçu le placebo.
L’évérolimus s’est révélé supérieur au placebo sur le critère principal de survie sans progression, avec une réduction statistiquement significative de 67 % du risque de progression ou décès (voir tableau 7 et figure 6).
Tableau 7 RECORD-1 – Résultats en termes de survie sans progression
|
Population |
n |
Évérolimus n = 277 |
Placebo n = 139 |
HR (rapport de risque) (IC à 95 %) |
Valeur de pa |
|
Survie sans progression médiane (mois) (IC à 95 %) |
|||||
|
Analyse principale |
|||||
|
Tous patients (évaluation centrale indépendante en aveugle) |
416 |
4,9 (4,0-5,5) |
1,9 (1,8-1,9) |
0,33 (0,25-0,43) |
< 0,0001a |
|
Analyses de confirmation/de sensibilité |
|||||
|
Tous patients (évaluation locale par l’investigateur) |
416 |
5,5 (4,6-5,8) |
1,9 (1,8-2,2) |
0,32 (0,25-0,41) |
< 0,0001a |
|
Score pronostique MSKCC (revue centralisée indépendante en aveugle) |
|||||
|
Risque faible |
120 |
5,8 (4,0-7,4) |
1,9 (1,9-2,8) |
0,31 (0,19-0,50) |
< 0,0001 |
|
Risque intermédiaire |
235 |
4,5 (3,8-5,5) |
1,8 (1,8-1,9) |
0,32 (0,22-0,44) |
< 0,0001 |
|
Haut risque |
61 |
3,6 (1,9-4,6) |
1,8 (1,8-3,6) |
0,44 (0,22-0,85) |
0,007 |
|
a Test log-rank stratifié |
|||||
Figure 6 RECORD-1 – Courbes de survie sans progression Kaplan-Meier (revue centralisée indépendante)
Temps (mois)
|
Nombre de patients encore à risque |
||||||||
|
Temps (mois) |
0 |
2 |
4 |
6 |
8 |
10 |
12 |
14 |
|
Évérolimus |
277 |
192 |
115 |
51 |
26 |
10 |
1 |
0 |
|
Placebo |
139 |
47 |
15 |
6 |
2 |
0 |
0 |
0 |
Les taux de survie sans progression à 6 mois étaient de 36 % dans le groupe évérolimus contre 9 % dans le groupe placebo.
Des réponses tumorales objectives confirmées ont été observées chez 5 patients (2 %) traités par l’évérolimus et chez aucun patient recevant le placebo. Ainsi, le bénéfice en termes de survie sans progression concerne essentiellement la population présentant une stabilisation de la maladie (correspondant à 67 % du groupe de traitement par l’évérolimus).
Aucune différence statistiquement significative entre les traitements n’a été observée sur la survie globale (Hazard ratio: 0,87 ; intervalle de confiance [0,65-1,17] ; p = 0,177). Le cross over vers le traitement par évérolimus en ouvert après progression chez les patients qui avaient reçu préalablement au placebo (cross over) ne permettait pas la mise en évidence d’une différence pour le critère de survie globale entre les deux bras.
Autres études
La stomatite est l’effet indésirable le plus fréquemment rapporté chez les patients traités par évérolimus (voir rubriques 4.4 et 4.8). Dans une étude post-autorisation en simple bras chez les femmes ménopausées atteintes d’un cancer du sein avancé (N = 92), un traitement local avec une solution orale de dexaméthasone 0,5 mg/5 ml sans alcool a été administré en bain de bouche (4 fois par jour pendant les 8 premières semaines de traitement) à partir de l’initiation du traitement par évérolimus (10 mg/jour) plus exémestane (25 mg/jour) pour réduire l’incidence et la gravité des stomatites. L’incidence des stomatites de Grade ≥ 2 après 8 semaines de traitement était de 2,4 % (n = 2/85 patients évaluables) ce qui était inférieur à l’incidence historiquement rapportée. L’incidence des stomatites de Grade 1 était de 18,8 % (n = 16/85) et aucun cas de stomatite de Grade 3 ou 4 n’a été rapporté. Le profil de sécurité global était cohérent avec celui qui avait été établi pour évérolimus en oncologie et dans la sclérose tubéreuse de Bourneville (STB), à l’exception d’une légère augmentation de la fréquence des candidoses buccales rapportées chez 2,2 % (n = 2/92) des patients.
Population pédiatrique
L’Agence européenne des médicaments a accordé une dérogation à l’obligation de soumettre les résultats d’études réalisées avec le médicament de référence dans tous les sous-groupes de la population pédiatrique atteints de tumeurs neuroendocrines d’origine pancréatique, de tumeurs neuroendocrines d’origine thoracique et de cancers du rein (voir rubrique 4.2 pour les informations concernant l’usage pédiatrique).
5.2. Propriétés pharmacocinétiques
Chez les patients ayant des tumeurs solides avancées, les concentrations maximales (Cmax) d’évérolimus sont atteintes à une durée médiane d’une heure après l’administration quotidienne d’une dose orale de 5 à 10 mg à jeun ou avec un repas léger sans matières grasses. La Cmax est proportionnelle à la dose entre 5 mg et 10 mg. L’évérolimus est un substrat et un inhibiteur modéré de la PgP.
Effet des aliments
Chez des volontaires sains, l’exposition systémique à l’évérolimus 10 mg (mesurée par l’ASC) est diminuée de 22 % et le pic plasmatique de la Cmax est diminué de 54 % lorsque le médicament est pris avec un repas riche en graisses. Avec un repas léger, l’ASC est diminuée de 32 % et la Cmax est diminuée de 42 %. Les aliments n’ont toutefois pas eu d’effet apparent sur le profil de concentration en fonction du temps au cours de la phase post-absorption.
Distribution
Le rapport sang-plasma de l’évérolimus, qui est dépendant de la concentration sur l’intervalle de 5 à 5 000 ng/ml, est de 17 % à 73 %. Environ 20 % de la concentration de l’évérolimus dans le sang total est retrouvée dans le compartiment plasmatique chez les patients cancéreux recevant de l’évérolimus 10 mg par jour. La liaison aux protéines plasmatiques est d’environ 74 % chez les sujets sains et chez les patients présentant une insuffisance hépatique modérée. Chez les patients ayant des tumeurs solides à un stade avancé, le volume de distribution Vd était de 191 l dans le compartiment central apparent et de 517 l dans le compartiment périphérique apparent.
Biotransformation
L’évérolimus est un substrat du CYP3A4 et de la PgP. Après administration orale, l’évérolimus est le composé principal circulant dans le sang chez l’homme. Six métabolites principaux de l’évérolimus ont été identifiés dans le sang chez l’homme, incluant trois métabolites monohydroxylés, deux produits à cycle ouvert formés par hydrolyse et un conjugué phosphatidylcholine de l’évérolimus. Ces métabolites ont également été identifiés chez les espèces animales utilisées dans les études de toxicité et ils ont présenté une activité 100 fois plus faible environ que celle de l’évérolimus lui-même. Par conséquent, l’évérolimus est considéré comme responsable de la majorité de l’activité pharmacologique globale.
Elimination
La valeur moyenne de la clairance orale (CL/F) de l’évérolimus chez des patients ayant des tumeurs solides de stade avancé recevant 10 mg par jour était de 24,5 l/h. La valeur moyenne de la demi-vie d’élimination était approximativement de 30 heures.
Il n’a pas été mené d’études d’excrétions spécifiques chez les patients cancéreux, mais des données issues d’études menées chez des patients transplantés sont disponibles. Après administration d’une dose unique d’évérolimus radiomarqué en association avec la ciclosporine, 80 % de la radioactivité ont été retrouvés dans les fèces et 5 % ont été excrétés dans les urines. La molécule mère n’a pas été détectée dans les urines ou dans les fèces.
Pharmacocinétique à l’état d’équilibre
Après l’administration d’évérolimus chez des patients ayant des tumeurs solides de stade avancé, l’ASC0-τ a été dose-proportionnelle dans l’intervalle posologique de 5 mg à 10 mg par jour. L’état d’équilibre a été atteint en deux semaines. La Cmax est proportionnelle à la dose entre 5 mg et 10 mg. Le tmax est atteint 1 à 2 heures après l’administration. Il a été observé une corrélation significative entre l’ASC0-τ et la concentration résiduelle avant administration à l’état d’équilibre.
Populations particulières
Insuffisance hépatique
La sécurité d’emploi, la tolérance et la pharmacocinétique de l’évérolimus ont été évaluées dans deux études avec administration d’évérolimus en comprimé à dose unique par voie orale chez 8 et 34 sujets présentant une insuffisance hépatique comparés à des sujets ayant une fonction hépatique normale.
Dans la première étude, l’ASC moyenne de l’évérolimus chez 8 sujets présentant une insuffisance hépatique modérée (Child-Pugh B) était deux fois plus importante que celle retrouvée chez les 8 sujets présentant une fonction hépatique normale.
Dans la seconde étude réalisée chez 34 sujets présentant différents degrés d’insuffisance hépatique, une augmentation de l’exposition (c.-à-d., ASC0-inf) de 1,6 fois, 3,3 fois et 3,6 fois a été observée chez les sujets ayant une insuffisance hépatique respectivement légère (classe A de Child-Pugh), modérée (classe B de Child-Pugh) et sévère (classe C de Child-Pugh) par rapport aux sujets normaux.
Les simulations de pharmacocinétique en administration répétée viennent à l’appui des recommandations posologiques chez les sujets présentant une insuffisance hépatique déterminée selon leur statut Child-Pugh.
À partir des résultats de ces deux études, une adaptation de la posologie est recommandée chez les patients présentant une insuffisance hépatique (voir rubriques 4.2 et 4.4).
Insuffisance rénale
Dans une analyse de pharmacocinétique de population chez 170 patients ayant des tumeurs solides de stade avancé, aucun effet significatif de la clairance de la créatinine (25-178 ml/min) n’a été observé sur la clairance orale (CL/F) de l’évérolimus. Une insuffisance rénale post-transplantation (clairance de la créatinine comprise entre 11 ml/min et 107 ml/min) n’a pas eu d’effet sur la pharmacocinétique de l’évérolimus chez des patients transplantés.
Patients âgés
Dans une analyse pharmacocinétique de population chez des patients cancéreux, aucun effet significatif de l’âge (27 à 85 ans) sur la clairance orale de l’évérolimus n’a été observé.
Groupe ethnique
La clairance orale (CL/F) est similaire chez les patients cancéreux japonais et caucasiens ayant une fonction hépatique comparable. Selon une analyse de pharmacocinétique de population, la CL/F est supérieure de 20 % en moyenne chez les patients transplantés de race noire.
5.3. Données de sécurité préclinique
L’évérolimus a semblé exacerber spontanément des pathologies préexistantes (myocardite chronique chez le rat, infection du plasma et du cœur par le virus Coxsackie chez le singe, infestation coccidienne du tractus gastro-intestinal chez le cochon nain, lésions cutanées chez la souris et le singe). Ces effets ont été généralement observés à des niveaux d’exposition systémique équivalents ou supérieurs aux niveaux d’exposition thérapeutique, à l’exception des effets observés chez le rat, qui se sont produits à des niveaux inférieurs à l’exposition thérapeutique en raison d’une distribution tissulaire élevée.
Au cours d’une étude de fertilité chez le rat mâle, la morphologie testiculaire a été affectée à des doses de 0,5 mg/kg et plus et la motilité et le nombre de spermatozoïdes et le taux plasmatique de testostérone ont été diminués à la dose de 5 mg/kg, ce qui a entraîné une diminution de la fertilité des mâles. Ces altérations ont été réversibles.
Au cours des études portant sur la fonction de reproduction réalisées chez l’animal, la fertilité des femelles n’a pas été affectée. Cependant, chez le rat femelle, les doses orales d’évérolimus ≥ 0,1 mg/kg (environ 4 % de l’ASC0-24h des patients recevant 10 mg par jour) ont entrainé une augmentation des pertes préimplantatoires.
L’évérolimus a traversé la barrière placentaire et s’est avéré toxique pour le fœtus. Chez le rat, l’évérolimus a provoqué une embryo/fœtotoxicité se manifestant par une mortalité et une réduction du poids fœtal à un niveau d’exposition systémique inférieur au niveau thérapeutique. L’incidence de modifications et de malformations du squelette (fente sternale par exemple) a été augmentée aux doses de 0,3 mg/kg et 0,9 mg/kg. Chez le lapin, l’embryotoxicité s’est manifestée par une augmentation des résorptions tardives.
Les études de génotoxicité évaluant les critères pertinents n’ont pas mis en évidence d’activité clastogène ou mutagène. L’administration d’évérolimus pendant des durées allant jusqu’à 2 ans n’a pas indiqué de potentiel oncogène chez la souris et le rat jusqu’aux doses les plus élevées, correspondant respectivement à 3,9 et 0,2 fois l’exposition clinique estimée.
2 ans
6.4. Précautions particulières de conservation
6.5. Nature et contenu de l'emballage extérieur
Plaquettes en OPA/Aluminium/PVC//Aluminium.
Boîtes de 10, 30 ou 90 comprimés ou 10x1, 30x1 ou 90x1 comprimés (dose unitaire).
Toutes les présentations peuvent ne pas être commercialisées.
6.6. Précautions particulières d’élimination et de manipulation
Tout médicament non utilisé ou déchet doit être éliminé conformément à la réglementation en vigueur.
7. TITULAIRE DE L’AUTORISATION DE MISE SUR LE MARCHE
EG LABO - LABORATOIRES EUROGENERICS
9 – 15 RUE MAURICE MALLET
92130 ISSY LES MOULINEAUX
8. NUMERO(S) D’AUTORISATION DE MISE SUR LE MARCHE
· 34009 301 484 6 4 : 30 comprimés sous plaquettes (OPA/Aluminium/PVC/Aluminium)
· 34009 550 555 3 2 : 90 comprimés sous plaquettes (OPA/Aluminium/PVC/Aluminium).
9. DATE DE PREMIERE AUTORISATION/DE RENOUVELLEMENT DE L’AUTORISATION
[à compléter ultérieurement par le titulaire]
10. DATE DE MISE A JOUR DU TEXTE
[à compléter ultérieurement par le titulaire]
Sans objet.
12. INSTRUCTIONS POUR LA PREPARATION DES RADIOPHARMACEUTIQUES
Liste I.
Médicament nécessitant une surveillance particulière pendant le traitement.
Prescription hospitalière.
Prescription réservée aux spécialistes et services de cancérologie, d’hématologie, et d’oncologie médicale.
ANSM - Mis à jour le : 31/01/2024
Évérolimus
· Gardez cette notice. Vous pourriez avoir besoin de la relire.
· Si vous avez d’autres questions, interrogez votre médecin ou votre pharmacien.
· Ce médicament vous a été personnellement prescrit. Ne le donnez pas à d’autres personnes. Il pourrait leur être nocif, même si les signes de leur maladie sont identiques aux vôtres.
· Si vous ressentez un quelconque effet indésirable, parlez-en à votre médecin ou votre pharmacien. Ceci s’applique aussi à tout effet indésirable qui ne serait pas mentionné dans cette notice. Voir rubrique 4.
1. Qu'est-ce que EVEROLIMUS EG 10 mg, comprimé et dans quels cas est-il utilisé ?
2. Quelles sont les informations à connaître avant de prendre EVEROLIMUS EG 10 mg, comprimé ?
3. Comment prendre EVEROLIMUS EG 10 mg, comprimé ?
4. Quels sont les effets indésirables éventuels ?
5. Comment conserver EVEROLIMUS EG 10 mg, comprimé ?
6. Contenu de l’emballage et autres informations.
1. QU’EST-CE QUE EVEROLIMUS EG 10 mg, comprimé ET DANS QUELS CAS EST-IL UTILISE ?
EVEROLIMUS EG est utilisé chez les patients adultes pour traiter :
· le cancer du sein avancé avec récepteurs hormonaux positifs chez les femmes ménopausées chez qui d'autres traitements (appelés « inhibiteurs non-stéroïdiens de l'aromatase ») ne permettent plus de contrôler la maladie. Il est administré avec un médicament appelé exémestane, un inhibiteur stéroïdien de l'aromatase, qui est utilisé dans le traitement hormonal du cancer.
· des tumeurs avancées appelées tumeurs neuroendocrines d’origine gastrique, intestinale, pulmonaire ou pancréatique. Il est administré si les tumeurs sont inopérables et si elles ne produisent pas en excès certaines hormones spécifiques ou d’autres substances naturelles apparentées.
2. QUELLES SONT LES INFORMATIONS A CONNAITRE AVANT DE PRENDRE EVEROLIMUS EG 10 mg, comprimé ?
EVEROLIMUS EG ne vous sera prescrit que par un médecin ayant l’expérience des traitements anticancéreux. Respectez attentivement les instructions du médecin. Elles peuvent être différentes des informations générales présentées dans cette notice. Si vous avez des questions sur EVEROLIMUS EG ou concernant la raison pour laquelle ce médicament vous a été prescrit, veuillez consulter votre médecin.
Ne prenez jamais EVEROLIMUS EG 10 mg, comprimé :
· si vous êtes allergique à l’évérolimus, à des substances apparentées telles que le sirolimus ou le temsirolimus, ou à l’un des autres composants contenus dans ce médicament, mentionnés dans la rubrique 6.
Si vous pensez que vous pouvez être allergique, demandez conseil à votre médecin.
Avertissements et précautions
Adressez-vous à votre médecin avant de prendre EVEROLIMUS EG 10 mg, comprimé :
· si vous avez des problèmes de foie ou si vous avez eu dans le passé une maladie qui aurait pu atteindre votre foie. Dans ce cas, il sera peut-être nécessaire de vous prescrire une dose différente d’EVEROLIMUS EG ;
· si vous êtes diabétique (taux élevé de sucre dans le sang). EVEROLIMUS EG peut augmenter le taux sanguin de glucose et aggraver le diabète. Cela peut nécessiter un traitement par insuline et/ou par antidiabétiques oraux. Parlez-en à votre médecin si vous constatez que vous avez une sensation de soif excessive ou que vous urinez de façon plus fréquente ;
· si vous devez être vacciné(e) pendant le traitement par EVEROLIMUS EG ;
· si vous avez un taux élevé de cholestérol. EVEROLIMUS EG peut augmenter le cholestérol et/ou les autres graisses dans le sang ;
· si vous avez eu récemment une opération chirurgicale majeure ou si la plaie n’est pas encore cicatrisée après la chirurgie. EVEROLIMUS EG peut augmenter le risque de problèmes liés à la cicatrisation des plaies ;
· si vous avez une infection. Cela peut nécessiter le traitement de l’infection avant l’instauration du traitement par EVEROLIMUS EG ;
· si vous avez eu précédemment une hépatite B, car cette infection peut se réactiver au cours d’un traitement par EVEROLIMUS EG (voir la rubrique 4 « Quels sont les effets indésirables éventuels ? ») ;
· si vous avez reçu ou allez recevoir une radiothérapie.
EVEROLIMUS EG peut également :
· affaiblir votre système immunitaire. Ainsi, vous pouvez vous trouver exposé(e) au risque de contracter une infection pendant que vous prenez votre traitement par EVEROLIMUS EG. Si vous avez de la fièvre, ou tout autre signe d’infection, vous devez consulter votre médecin. Certaines infections peuvent être graves et peuvent avoir des conséquences fatales ;
· avoir un effet sur votre fonction rénale. En conséquence, votre médecin surveillera votre fonction rénale au cours de votre traitement par EVEROLIMUS EG ;
· provoquer essoufflement, toux et fièvre ;
· provoquer des complications de la radiothérapie. Des complications sévères de la radiothérapie (tels que essoufflement, nausée, diarrhée, éruptions cutanées et douleur dans la bouche, les gencives et la gorge), y compris des cas mortels, ont été observés chez certains patients qui prenaient de l’évérolimus en même temps que la radiothérapie ou qui prenaient de l’évérolimus peu de temps après avoir reçu une radiothérapie. De plus, des phénomènes de rappel (comprenant rougeur cutanée ou inflammation pulmonaire au niveau d’un site précédemment irradié) ont été rapportés chez des patients ayant préalablement reçu une radiothérapie ;
· Informez votre médecin si vous prévoyez de recevoir une radiothérapie prochainement, ou si vous avez déjà reçu une radiothérapie.
Si vous ressentez ces symptômes, veuillez-en informer votre médecin.
Vous effectuerez régulièrement des analyses de sang pendant le traitement. Elles permettront de contrôler la quantité de cellules sanguines (globules blancs, globules rouges et plaquettes) dans votre organisme pour savoir si EVEROLIMUS EG a des effets indésirables sur ces cellules. Des analyses de sang seront également pratiquées pour surveiller vos fonctions rénale (taux de créatinine) et hépatique (taux de transaminases) et votre taux de sucre (glycémie) et de cholestérol dans le sang parce qu’ils peuvent eux aussi être modifiés par EVEROLIMUS EG.
Enfants et adolescents
EVEROLIMUS EG ne doit PAS être utilisé chez l’enfant ou l’adolescent (âgé de moins de 18 ans).
Autres médicaments et EVEROLIMUS EG 10 mg, comprimé
EVEROLIMUS EG peut interférer avec certains autres médicaments. Si vous prenez d’autres médicaments en même temps qu’EVEROLIMUS EG, il sera peut-être nécessaire que votre médecin modifie la dose d’EVEROLIMUS EG ou des autres médicaments.
Informez votre médecin ou pharmacien si vous prenez, avez récemment pris ou pourriez prendre tout autre médicament.
Les médicaments suivants peuvent augmenter le risque d’effets indésirables avec EVEROLIMUS EG :
· le kétoconazole, l’itraconazole, le voriconazole ou le fluconazole et les autres antifongiques utilisés pour traiter les infections fongiques ;
· la clarithromycine, la télithromycine ou l’érythromycine, antibiotiques utilisés pour traiter des infections bactériennes ;
· le ritonavir et d’autres médicaments utilisés dans le traitement de l’infection au VIH/SIDA ;
· le vérapamil ou le diltiazem utilisés pour traiter les affections cardiaques ou l’hypertension ;
· la dronédarone, un médicament utilisé pour aider à réguler les battements de votre cœur ;
· la ciclosporine, un médicament utilisé pour empêcher l’organisme de rejeter les organes transplantés ;
· l’imatinib, utilisé pour inhiber la croissance de cellules anormales ;
· les inhibiteurs de l’enzyme de conversion de l’angiotensine (ECA) (tels que le ramipril) utilisés pour traiter l’hypertension ou d’autres problèmes cardiovasculaires ;
· la néfazodone, utilisée pour traiter la dépression ;
· le cannabidiol (utilisé entre autres pour le traitement des crises d'épilepsie).
Les médicaments suivants peuvent réduire l’efficacité d’EVEROLIMUS EG :
· la rifampicine, utilisée pour traiter la tuberculose (TB) ;
· l’éfavirenz ou la névirapine, utilisés dans le traitement de l’infection au VIH/SIDA ;
· le millepertuis (Hypericum perforatum), un produit à base de plante utilisé pour traiter la dépression et d’autres affections ;
· la dexaméthasone, un corticoïde utilisé pour traiter de nombreux types d’affections, incluant les troubles inflammatoires ou immunitaires ;
· la phénytoïne, la carbamazépine ou le phénobarbital et les autres antiépileptiques utilisés pour faire cesser les convulsions ou les crises d’épilepsie.
Ces médicaments doivent être évités pendant le traitement par EVEROLIMUS EG. Si vous prenez l’un de ces médicaments, votre médecin le remplacera peut-être par un autre ou pourra modifier la dose d’EVEROLIMUS EG.
EVEROLIMUS EG 10 mg, comprimé avec des aliments et boissons
Vous devez prendre EVEROLIMUS EG à la même heure chaque jour avec ou sans aliment. Évitez de consommer du pamplemousse ou du jus de pamplemousse pendant le traitement par EVEROLIMUS EG. Cela peut augmenter la quantité d'EVEROLIMUS EG dans le sang, potentiellement à un niveau dangereux.
Grossesse, allaitement et fertilité
Grossesse
EVEROLIMUS EG peut nuire au fœtus et son utilisation pendant la grossesse n’est PAS recommandée.
Si vous êtes enceinte ou que vous allaitez, si vous pensez être enceinte ou planifiez une grossesse, demandez conseil à votre médecin ou pharmacien avant de prendre ce médicament. Votre médecin décidera si vous devez prendre ce médicament pendant votre grossesse.
Les femmes qui peuvent débuter une grossesse doivent utiliser une méthode contraceptive hautement efficace pendant le traitement et jusqu’à 8 semaines après l’arrêt du traitement. Si, malgré ces mesures, vous pensez que vous pouvez être enceinte, demandez conseil à votre médecin avant de reprendre EVEROLIMUS EG.
Allaitement
EVEROLIMUS EG peut nuire au nourrisson allaité. Vous ne devez pas allaiter pendant le traitement et pendant les 2 semaines qui suivent la prise de la dernière dose d’EVEROLIMUS EG. Si vous allaitez, informez votre médecin.
Fertilité féminine
Une absence des règles (aménorrhée) a été observée chez certaines patientes recevant EVEROLIMUS EG.
EVEROLIMUS EG peut avoir un effet sur la fertilité féminine. Prévenez votre médecin si vous souhaitez avoir des enfants.
Fertilité masculine
EVEROLIMUS EG peut avoir un effet sur la fertilité masculine. Prévenez votre médecin si vous souhaitez avoir des enfants.
Conduite de véhicules et utilisation de machines
Si vous vous sentez fatigué(e) de façon inhabituelle (la fatigue est un effet indésirable très fréquent), faites attention quand vous conduisez des véhicules et utilisez des machines.
EVEROLIMUS EG 10 mg, comprimé contient du lactose
EVEROLIMUS EG contient du lactose. Si votre médecin vous a informé(e) d’une intolérance à certains sucres, contactez-le avant de prendre ce médicament.
3. COMMENT PRENDRE EVEROLIMUS EG 10 mg, comprimé ?
La dose recommandée est de 10 mg en une prise par jour. Votre médecin vous dira combien de comprimés d’EVEROLIMUS EG vous devez prendre.
Si vous avez des problèmes au foie, votre médecin pourra vous faire débuter le traitement à une dose plus faible d’EVEROLIMUS EG (2,5, 5 ou 7,5 mg par jour).
Si vous présentez certains effets indésirables au cours de votre traitement par EVEROLIMUS EG (voir rubrique 4), votre médecin pourra réduire la dose de votre traitement ou arrêter votre traitement pendant une courte durée ou définitivement.
Prenez EVEROLIMUS EG une fois par jour, au même moment de la journée tous les jours, avec ou sans aliment de façon constante.
Avalez le(s) comprimé(s) entier(s) avec un verre d’eau. Les comprimés ne doivent pas être mâchés ou écrasés.
Si vous avez pris plus d’EVEROLIMUS EG 10 mg, comprimé que vous n’auriez dû
· Si vous avez pris trop d’EVEROLIMUS EG, ou si une autre personne a pris accidentellement vos comprimés, consultez immédiatement votre médecin ou un hôpital. Un traitement médical d’urgence pourra être nécessaire.
· Emportez la boîte de comprimés et cette notice pour que le médecin sache ce qui a été pris.
Si vous oubliez de prendre EVEROLIMUS EG 10 mg, comprimé
Si vous oubliez de prendre une dose, prenez la dose suivante comme prévu. Ne prenez pas de dose double pour compenser les comprimés que vous avez oublié de prendre.
Si vous arrêtez de prendre EVEROLIMUS EG 10 mg, comprimé
Vous ne devez pas arrêter de prendre EVEROLIMUS EG sans l’avis de votre médecin.
Si vous avez d’autres questions sur l’utilisation de ce médicament, demandez plus d’informations à votre médecin ou à votre pharmacien.
4. QUELS SONT LES EFFETS INDESIRABLES EVENTUELS ?
ARRÊTEZ de prendre EVEROLIMUS EG et demandez immédiatement l’aide d’un médecin si vous présentez l’un des signes de réaction allergique suivants :
· difficulté à respirer ou à avaler ;
· gonflement du visage, des lèvres, de la langue ou de la gorge ;
· démangeaisons sévères de la peau, avec une éruption rouge ou des bosses rouges.
Les effets indésirables graves d’EVEROLIMUS EG incluent :
Très fréquent (pouvant affecter plus de 1 personne sur 10)
· Augmentation de la température, frissons (signe d’une infection).
· Fièvre, toux, difficulté à respirer, respiration sifflante (signes d’une inflammation des poumons, également connue sous le nom de pneumopathie).
Fréquent (pouvant affecter jusqu’à 1 personne sur 10)
· Soif excessive, émission d’urine importante, augmentation de l’appétit avec une perte de poids, fatigue (signes d’un diabète).
· Saignement (hémorragie), par exemple dans la paroi intestinale.
· Diminution importante de l’émission d’urine (signe d’une défaillance rénale).
Peu fréquent (pouvant affecter jusqu’à 1 personne sur 100)
· Fièvre, éruption cutanée, douleur et inflammation des articulations, ainsi que fatigue, perte d’appétit, nausée, jaunisse (jaunissement de la peau), douleur dans la partie haute de l’abdomen du côté droit, selles pâles, urine foncée (peuvent être des signes de réactivation d’une hépatite B).
· Essoufflement, difficulté à respirer en position allongée, gonflement des pieds ou des jambes (signes d’une défaillance cardiaque).
· Gonflement et/ou douleur dans une des jambes, souvent dans le mollet, rougeur ou peau chaude dans la zone affectée (signes d’un blocage d’un vaisseau sanguin (veine) dans les jambes dû à un caillot de sang).
· Survenue soudaine d’un essoufflement, douleur de la poitrine ou crachat de sang (signes potentiels d’une embolie pulmonaire, une maladie qui survient lorsqu’une ou plusieurs artères de vos poumons sont bloquées).
· Diminution importante de l’émission d’urine, gonflement des jambes, sensation de confusion, douleur dans le dos (signes d’une défaillance soudaine de vos reins).
· Éruption, démangeaison, urticaire, difficulté à respirer ou à avaler, étourdissement (signes d’une réaction allergique grave, également connue sous le nom d’hypersensibilité).
Rare (pouvant affecter jusqu’à 1 personne sur 1 000)
· Essoufflement ou respiration rapide (signes d’un syndrome de détresse respiratoire aiguë).
Si vous présentez l’un de ces effets indésirables, prévenez immédiatement votre médecin car ils peuvent mettre votre vie en danger.
Les autres effets indésirables possibles d’EVEROLIMUS EG incluent :
Très fréquent (pouvant affecter plus d’1 personne sur 10)
· Taux élevé de sucre dans le sang (hyperglycémie).
· Perte d’appétit.
· Sens du goût perturbé (dysgueusie).
· Maux de tête.
· Saignement du nez (épistaxis).
· Toux.
· Ulcérations de la bouche.
· Estomac dérangé incluant une sensation de mal au cœur (nausées) ou diarrhée.
· Éruption cutanée.
· Démangeaisons (prurit).
· Sensation de faiblesse ou fatigue.
· Fatigue, essoufflement, étourdissement, pâleur cutanée, signes d’un taux faible de globules rouges (anémie).
· Gonflement des bras, mains, pieds, chevilles ou d’une autre partie du corps (signes d’un œdème).
· Perte de poids.
· Taux élevé de lipides (graisses) dans le sang (hypercholestérolémie).
Fréquent (pouvant affecter jusqu’à 1 personne sur 10)
· Saignements spontanés ou bleus (signes d’un taux faible de plaquettes, également connu sous le nom de thrombopénie).
· Essoufflement (dyspnée).
· Soif, faible émission d’urine, urine foncée, peau rouge et sèche, irritabilité (signes de déshydratation).
· Troubles du sommeil (insomnies).
· Maux de tête, étourdissement (signe d’une tension artérielle élevée, également connue sous le nom d’hypertension).
· Fièvre, mal de gorge ou ulcération de la bouche dus à des infections (signes d’un taux faible de globules blancs, leucopénie, lymphopénie et/ou neutropénie).
· Fièvre.
· Inflammation de la paroi intérieure de la bouche, de l’estomac, de l’intestin.
· Sècheresse buccale.
· Brûlure à l’estomac (dyspepsie).
· Avoir mal au cœur (vomissements).
· Difficulté à avaler (dysphagie).
· Douleur abdominale.
· Acné.
· Éruption et douleur sur les paumes des mains et les plantes des pieds (syndrome mains-pieds).
· Rougeur de la peau (érythème).
· Douleur articulaire.
· Douleur buccale.
· Troubles menstruels tels que règles irrégulières.
· Taux élevé de lipides (graisses) dans le sang (hyperlipémie, triglycérides augmentés).
· Taux faible de potassium dans le sang (hypokaliémie).
· Taux faible de phosphate dans le sang (hypophosphatémie).
· Taux faible de calcium dans le sang (hypocalcémie).
· Sécheresse cutanée, exfoliation cutanée, lésions cutanées.
· Anomalies des ongles, ongles cassants.
· Légère perte des cheveux.
· Résultats anormaux des tests sanguins de la fonction hépatique (augmentation de l’alanine et l’aspartate aminotransférase).
· Résultats anormaux des tests sanguins de la fonction rénale (augmentation de la créatinine).
· Écoulement oculaire avec démangeaisons, rougeurs et gonflement.
· Protéines dans les urines.
Peu fréquent (pouvant affecter jusqu’à 1 personne sur 100)
· Faiblesse, saignements spontanés ou bleus et infections fréquentes avec des signes tels que fièvre, frissons, mal de gorge ou ulcérations dans la bouche (signes d’un taux faible de cellules dans le sang, également connu sous le nom de pancytopénie).
· Perte des sensations du goût (agueusie).
· Crachat de sang (hémoptysie).
· Troubles menstruels tels qu'absence de règles (aménorrhée).
· Émission d’urine plus fréquente pendant la journée.
· Douleur dans la poitrine.
· Cicatrisation des plaies anormale.
· Bouffées de chaleur.
· Ecoulement oculaire avec démangeaisons et rougeurs, yeux roses ou rouges (conjonctivites).
Rare (pouvant affecter jusqu’à 1 personne sur 1 000)
· Fatigue, essoufflement, étourdissements, pâleur cutanée (signes d’un taux faible de globules rouges dans le sang, éventuellement dû à un type d’anémie appelée érythroblastopénie).
· Gonflement du visage, autour des yeux, de la bouche et à l’intérieur de la bouche et/ou de la gorge, ainsi que de la langue et difficulté à respirer ou avaler (également connu sous le nom d’angiœdème ou œdème de Quincke), pouvant être des signes d’une réaction allergique.
Fréquence indéterminée (ne peut être estimée sur la base des données disponibles)
· Réaction au niveau d’un site précédemment irradié, par exemple rougeur cutanée ou inflammation pulmonaire (phénomène de rappel).
Aggravation des effets secondaires de la radiothérapie.
Si ces effets indésirables deviennent sévères, informez votre médecin et/ou pharmacien. La plupart des effets indésirables sont légers à modérés et disparaissent généralement si votre traitement est interrompu pendant quelques jours.
Déclaration des effets secondaires
Si vous ressentez un quelconque effet indésirable, parlez-en à votre médecin ou votre pharmacien. Ceci s’applique aussi à tout effet indésirable qui ne serait pas mentionné dans cette notice. Vous pouvez également déclarer les effets indésirables directement via le système national de déclaration : Agence nationale de sécurité du médicament et des produits de santé (ANSM) et réseau des Centres Régionaux de Pharmacovigilance - Site internet : https://signalement.social-sante.gouv.fr/.
En signalant les effets indésirables, vous contribuez à fournir davantage d’informations sur la sécurité du médicament.
5. COMMENT CONSERVER EVEROLIMUS EG 10 mg, comprimé ?
Tenir ce médicament hors de la vue et de la portée des enfants.
N’utilisez pas ce médicament après la date de péremption indiquée sur la boîte et la plaquette. La date de péremption fait référence au dernier jour de ce mois.
Ce médicament ne nécessite pas de précautions particulières de conservation concernant la température. A conserver dans l’emballage d’origine, à l’abri de la lumière.
Ouvrir la plaquette juste avant de prendre les comprimés.
N’utilisez pas ce médicament si vous remarquez que l’emballage est endommagé ou a été ouvert.
Ne jetez aucun médicament au tout-à-l’égout ou avec les ordures ménagères. Demandez à votre pharmacien d’éliminer les médicaments que vous n’utilisez plus. Ces mesures contribueront à protéger l’environnement.
6. CONTENU DE L’EMBALLAGE ET AUTRES INFORMATIONS
Ce que contient EVEROLIMUS EG 10 mg, comprimé
· La substance active est :
Évérolimus....................................................................................................................... 10 mg
Pour un comprimé
· Les autres composants sont :
Butylhydroxytoluène (E321), hypromellose type 2910 (E464), lactose, crospovidone type A (E1202), stéarate de magnésium.
Qu’est-ce que EVEROLIMUS EG 10 mg, comprimé et contenu de l’emballage extérieur
EVEROLIMUS EG 10 mg, comprimé se présente sous forme de comprimé ovale, plat, blanc à blanc cassé, d’environ 15 mm de long et 6 mm de large, portant la mention « EVR » sur une face et « NAT » sur l’autre face.
EVEROLIMUS EG est disponible en boîtes de 10, 30 ou 90 comprimés ou 10x1, 30x1 ou 90x1 comprimés (dose unitaire).
Toutes les présentations ou dosages peuvent ne pas être commercialisés.
Titulaire de l’autorisation de mise sur le marché
EG LABO - LABORATOIRES EUROGENERICS
CENTRAL PARK
9 – 15 RUE MAURICE MALLET
92130 ISSY LES MOULINEAUX
Exploitant de l’autorisation de mise sur le marché
EG LABO - LABORATOIRES EUROGENERICS
CENTRAL PARK
9 – 15 RUE MAURICE MALLET
92130 ISSY LES MOULINEAUX
18 KM MARATHONOS AVENUE
153 51 PALLINI ATTIKI
GRECE
PHARMADOX HEALTHCARE LTD
KW20A KORDIN INDUSTRIAL PARK
PAOLA, PLA 3000
MALTE
STADA ARZNEIMITTEL AG
STADASTRASSE 2-18
BAD VILBEL 61118
ALLEMAGNE
STADA ARZNEIMITTEL GmBH
MUTHGASSE 36/2
WIEN 1190
AUTRICHE
PHARMACARE PREMIUM LTD
HHF003 HAL FAR INDUSTRIAL ESTATE
BIRZEBBUGIA, BBG3000
MALTE
Noms du médicament dans les Etats membres de l'Espace Economique Européen
Ce médicament est autorisé dans les États membres de l'Espace Économique Européen sous les noms suivants : Conformément à la réglementation en vigueur.
[À compléter ultérieurement par le titulaire]
La dernière date à laquelle cette notice a été révisée est :
[à compléter ultérieurement par le titulaire]
Des informations détaillées sur ce médicament sont disponibles sur le site Internet de l’ANSM (France).