Dernière mise à jour le 01/06/2026
EMLAPATCH 5 POUR CENT, pansement adhésif cutané
Indications thérapeutiques
Classe pharmacothérapeutique : ANESTHESIQUES LOCAUX - (N : système nerveux central), Code ATC N01B B20.
EMLAPATCH 5 POUR CENT contient deux substances actives appelées lidocaïne et prilocaïne. Ces dernières appartiennent à un groupe de médicaments appelés anesthésiques locaux.
EMLAPATCH 5 POUR CENT, pansement adhésif cutané fonctionne en provoquant une anesthésie de la surface de la peau pendant une courte durée. Il est appliqué sur la peau avant certains actes médicaux. Cela permet d’arrêter la douleur au niveau de la peau ; cependant, vous pouvez encore avoir les sensations de pression et de toucher.
Adultes, adolescents et enfants
Il peut être utilisé pour anesthésier la peau avant :
· l’insertion d’aiguilles (par exemple, si vous devez avoir une injection ou faire une prise de sang).
· des opérations mineures de la peau.
Présentations
> 1 boite(s) de 1 pansement(s)
Code CIP : 340 007-1 ou 34009 340 007 1 3
Déclaration de commercialisation : 19/01/1998
Cette présentation est agréée aux collectivités
- Prix hors honoraire de dispensation : 2,13 €
- Honoraire de dispensation : 1,02 €
- Prix honoraire compris : 3,15 €
- Taux de remboursement :65%
> 1 boîte(s) de 20 pansement(s)
Code CIP : 34009 302 740 5 7
Déclaration de commercialisation : 15/01/2025
Cette présentation est agréée aux collectivités
Service médical rendu (SMR)
Les libellés affichés ci-dessous ne sont que des résumés ou extraits issus des avis rendus par la Commission de la Transparence. Seul l'avis complet de la Commission de la Transparence fait référence.
Cet avis est consultable à partir du lien `Avis du jj/mm/aaaa` ou encore sur demande auprès de la HAS (plus d'informations dans l'aide). Les avis et synthèses d'avis contiennent un paragraphe sur la place du médicament dans la stratégie thérapeutique.
| Valeur du SMR | Avis | Motif de l'évaluation | Résumé de l'avis |
|---|---|---|---|
| Important | Avis du 30/08/2023 | Inscription (CT) | Le service médical rendu par EMLAPATCH 5 POUR CENT (lidocaïne/prilocaïne), pansement adhésif cutané est important dans l’indication de l’AMM. |
| Important | Avis du 06/07/2016 | Renouvellement d'inscription (CT) | Le service médical rendu par EMLA et EMLAPATCH reste important dans les indications de l’AMM. |
Amélioration du service médical rendu (ASMR)
Les libellés affichés ci-dessous ne sont que des résumés ou extraits issus des avis rendus par la Commission de la Transparence. Seul l'avis complet de la Commission de la Transparence fait référence.
Cet avis est consultable à partir du lien `Avis du jj/mm/aaaa` ou encore sur demande auprés de la HAS (plus d'informations dans l'aide). Les avis et synthéses d'avis contiennent un paragraphe sur la place du médicament dans la stratégie thérapeutique.
| Valeur de l'ASMR | Avis | Motif de l'évaluation | Résumé de l'avis |
|---|---|---|---|
| V (Inexistant) | Avis du 30/08/2023 | Inscription (CT) | Cette spécialité est un complément de gamme qui n’apporte pas d’amélioration du service médical rendu (ASMR V) par rapport aux présentations déjà inscrites. |
ANSM - Mis à jour le : 22/03/2024
EMLAPATCH 5 POUR CENT, pansement adhésif cutané
2. COMPOSITION QUALITATIVE ET QUANTITATIVE
Lidocaïne ..........................................................................................................................25 mg
Prilocaïne ........................................................................................................................ 25 mg
1 g d'émulsion pour un pansement adhésif de 10 cm2
Pour la liste complète des excipients, voir rubrique 6.1.
4.1. Indications thérapeutiques
· Anesthésie topique de la peau saine lors de :
o l’insertion d’aiguilles, par exemple insertion de cathéters intraveineux ou prélèvements sanguins
o procédures chirurgicales superficielles.
chez les adultes et dans la population pédiatrique.
4.2. Posologie et mode d'administration
Adultes et adolescents
Les détails des indications ou des procédures d’utilisation, ainsi que la dose et la durée d’application sont fournis dans les Tableaux 1 et 2.
Pour plus de renseignements sur l’utilisation appropriée du produit selon de telles procédures, veuillez-vous référer au Mode d’administration.
Tableau 1 Adultes et adolescents de 12 ans et plus
|
Indication/Procédure |
Dose et durée d’application |
|
Peau |
|
|
Petites interventions, par exemple insertion d’aiguilles et traitement chirurgical de lésions localisées. |
1 ou plusieurs patch(s) sont appliqués sur la (les) zone(s) de la peau sélectionnée(s) pendant 1 à 5 heures1 |
1) Après une période d’application plus longue l’anesthésie diminue.
Population pédiatrique
Tableau 2 Patients pédiatriques âgés de 0 à 11 ans
|
Groupe d’âge |
Procédure |
Dosage et temps d’application |
|
|
Petites interventions, par exemple insertion d’aiguilles et traitement chirurgical de lésions localisées. |
1 ou plusieurs patch(s) pendant 1 heure (voir détails ci-dessous) |
|
Nouveau‑nés et nourrissons 0‑2 mois 1) 2) 3) 7) |
|
Ne pas dépasser 1 patch pendant 1 heure. 4) |
|
Nourrissons 3‑11 mois 1) 2) 7) |
|
Jusqu’à 2 patchs pendant une heure5) |
|
Jeunes enfants et enfants |
|
Jusqu’à 10 patchs pendant 1‑5 heures6) |
|
Enfants |
|
Jusqu’à 20 patchs pendant 1‑5 heures6) |
|
Patients pédiatriques avec dermatite atopique |
Avant curetage de molluscum |
Temps d’application : 30 minutes |
1) Chez les nouveau‑nés nés à terme et les nourrissons de moins de 3 mois, une unique dose devra seulement être appliquée sur une période de 24 heures. Pour les enfants âgés de 3 mois et plus, un maximum de 2 doses, séparées d’au moins 12 heures, peut être donné sur une période de 24 heures, voir rubriques 4.4 et 4.8.
2) EMLA ne doit pas être utilisé chez les enfants de moins de 12 mois qui sont traités par des médicaments inducteurs de méthémoglobinémie pour des raisons de sécurité, voir rubriques 4.4 et 4.8.
3) EMLA ne doit pas être utilisé chez les prématurés de moins de 37 semaines d’âge gestationnel pour des raisons de sécurité, voir rubrique 4.4.
4) Un temps d’application > 1 heure n’a pas été étudié.
5) Aucune augmentation cliniquement significative des taux de méthémoglobine n’a été observée après application de 2g d’EMLA crème pendant un maximum de 4 heures sur 16 cm2.
6) Après une période d’application plus longue, l’anesthésie diminue.
7) La taille du patch le rend moins adapté pour l’utilisation sur certaines parties du corps chez les nouveaux nés et les nourrissons.
Patients âgés
Aucune réduction de dose n’est nécessaire chez les patients âgés (voir rubriques 5.1 et 5.2).
Atteinte de la fonction hépatique
Une réduction de dose unique n’est pas nécessaire chez les patients présentant une atteinte de la fonction hépatique (voir rubrique 5.2).
Atteinte de la fonction rénale
Une réduction de dose n’est pas nécessaire chez les patients présentant une fonction rénale diminuée.
Mode d’administration
Voie cutanée.
Après séparation de la couche protectrice et application du patch sur la peau, une pression doit être appliquée uniquement aux bords extérieurs. Ne pas appuyer sur le centre du patch. Cela peut provoquer la propagation de l’émulsion d’EMLA sous l'adhésif et diminuer l'adhérence.
EMLAPATCH doit être appliqué au moins 1 heure avant l'intervention. Si nécessaire, enlever les poils dans la zone avant l'application. Le patch ne doit pas être coupé ou divisé en parties plus petites.
4.4. Mises en garde spéciales et précautions d'emploi
Les patients souffrant d’un déficit congénital en glucose-6-phosphate-déhydrogénase ou de méthémoglobinémie idiopathique sont plus susceptibles de présenter des signes de méthémoglobinémie induite par la substance active. Chez les patients présentant un déficit en glucose‑6‑phosphate déshydrogénase, l’antidote bleu de méthylène est inefficace pour réduire le taux de méthémoglobine et peut oxyder l’hémoglobine elle-même. Par conséquent, une thérapie par le bleu de méthylène ne peut pas être effectuée.
En raison de données insuffisantes sur l’absorption, EMLA ne doit pas être appliqué sur les blessures ouvertes.
Il faut faire attention lors de l’application d'EMLA chez les patients atteints de dermatite atopique. Un temps d’application réduit à 15 à 30 minutes peut être suffisant (voir rubrique 5.1). Des temps d’application supérieurs à 30 minutes, chez des patients atteints de dermatite atopique peuvent conduire à une augmentation de l’incidence des réactions vasculaires locales, en particulier une rougeur au site d’application, et dans certains cas des pétéchies et un purpura (voir rubrique 4.8.). Avant curetage de molluscum chez les enfants atteints de dermatites atopiques un temps d’application de 30 minutes est recommandé.
Lors d’une application à proximité des yeux, EMLA doit être utilisé avec précaution car cela peut provoquer des irritations au niveau des yeux. La perte des réflexes de protection des yeux peut également conduire à une irritation de la cornée et à une potentielle abrasion. En cas de contact oculaire, les yeux doivent être immédiatement rincés avec de l’eau ou une solution saline et protégés jusqu’au retour de la sensibilité.
La lidocaïne et la prilocaïne ont des propriétés bactéricides et antivirales pour des concentrations supérieures à 0.5 – 2 %. C’est pourquoi, bien que les résultats d’une étude clinique suggèrent que l’utilisation d’EMLA avant une vaccination contre la tuberculose (BCG) n’influence pas la réponse immunitaire, évaluée par la formation d’une papule locale, il est nécessaire de suivre les résultats d’injections intradermiques concernant les vaccins vivants.
EMLA contient de l’hydroxystéarate de macrogolglycérol qui peut causer des réactions cutanées.
Population pédiatrique
Lors d’études, l’efficacité d’EMLA lors des prélèvements capillaires au talon chez les nouveau-nés n’a pas été démontrée.
Chez les nouveau-nés et les nourrissons de moins de 3 mois, une augmentation transitoire dépourvue de signification clinique des taux de méthémoglobine est couramment observée, jusqu’à 12 heures après application de la dose recommandée d’EMLA.
Si la dose recommandée est dépassée, le patient doit être surveillé en cas de survenue d’effets indésirables systémiques secondaires à une méthémoglobinémie (voir rubriques 4.2, 4.8 et 4.9).
EMLA ne doit pas être utilisé :
· chez les nouveau-nés/nourrissons jusqu’à 12 mois traités par des médicaments inducteurs de méthémoglobine ;
· chez les nouveau‑nés prématurés de moins de 37 semaines d’âge gestationnel car ils risquent de développer des taux élevés de méthémoglobine.
4.5. Interactions avec d'autres médicaments et autres formes d'interactions
De fortes doses de prilocaïne peuvent augmenter les taux de méthémoglobine, en particulier chez des patients traités par des médicaments inducteurs de méthémoglobine (ex : les sulfamides, nitrofurantoïne, phénytoïne, phénobarbital). Cette liste n’est pas exhaustive.
Avec des doses élevées d'EMLA, le risque de toxicité systémique supplémentaire doit être envisagé chez les patients recevant d'autres anesthésiques locaux ou des médicaments de structure apparentée aux anesthésiques locaux, puisque les effets toxiques sont additifs.
Les médicaments qui réduisent la clairance de la lidocaïne (par exemple la cimétidine ou les bétabloquants) peuvent causer des concentrations plasmatiques potentiellement toxiques lorsque la lidocaïne est administrée à fortes doses répétées sur une longue période. De telles interactions devraient cependant être sans conséquence clinique lors d’un traitement de courte durée avec la lidocaïne aux doses recommandées (par exemple : EMLAPATCH).
Population pédiatrique
Des études d’interaction spécifiques chez les enfants n’ont pas été réalisées. Les interactions semblent similaires à celles de la population adulte.
4.6. Fertilité, grossesse et allaitement
Grossesse
Bien qu’une application topique soit uniquement associée à un faible taux d’absorption systémique, l’utilisation d’EMLA chez les femmes enceintes doit être considérée avec précaution car les données disponibles concernant l’utilisation d’EMLA chez les femmes enceintes sont insuffisantes. Cependant, des études chez l’animal n’ont pas montré d’effets nocifs directs ou indirects sur la grossesse, le développement embryonnaire/fœtal, l’accouchement ou le développement postnatal. Une toxicité de reproduction a été montrée lors d’une administration sous cutanée/intramusculaire de fortes doses de lidocaïne ou de prilocaïne excédant considérablement l’exposition lors d’une application topique (voir rubrique 5.3).
La lidocaïne et la prilocaïne traversent la barrière placentaire, et peuvent être absorbées par les tissus fœtaux. Il est raisonnable de penser que la lidocaïne et la prilocaïne ont été utilisées chez de nombreuses femmes enceintes ou en âge de procréer. Jusqu’à présent, aucun trouble spécifique de la fonction reproductrice n’a été rapporté, par exemple : une incidence accrue de malformations ou autres effets directement ou indirectement nocifs pour le fœtus.
La lidocaïne et en toute probabilité la prilocaïne sont excrétées dans le lait, mais en quantités si faibles qu’il n’y a généralement pas de risque que l’enfant soit affecté à des doses thérapeutiques. EMLA peut être utilisé pendant l’allaitement en cas de nécessité clinique.
Fertilité
Des études chez l’animal n’ont montré aucune altération de la fertilité des rats mâles ou femelles (voir rubrique 5.3).
4.7. Effets sur l'aptitude à conduire des véhicules et à utiliser des machines
Résumé du profil de sécurité
Les effets indésirables les plus fréquemment observés sont liés au site d’administration (réactions locales transitoires au site d’application) et sont rapportés comme fréquents.
Liste des réactions indésirables sous forme de tableau
Les incidences des effets indésirables (EIs) associés à un traitement par EMLA sont présentées dans le tableau ci-dessous. Le tableau est basé sur les évènements indésirables rapportés pendant les essais cliniques et/ou l’utilisation post-commercialisation. Les effets indésirables sont listés par ordre de fréquence, selon la terminologie MedDRA par Classe de Système d’Organe (SOC), au niveau des termes préférés.
Dans chaque Classe de Système d’Organe, les fréquences des effets indésirables sont répertoriées comme suit : très fréquent (≥ 1/10), fréquent (≥ 1/100, < 1/10), peu fréquent (≥ 1/1000, < 1/100), rare (≥ 1/10 000, <1/1000) et très rare (< 1/10 000). Dans chaque groupe de fréquence, les effets indésirables sont présentés dans un ordre de sévérité décroissante.
Tableau 3 Effets indésirables
|
Classe de Système d’Organe |
Fréquent |
Peu fréquent |
Rare |
|
Affections hématologiques et du système lymphatique |
|
|
Méthémoglobinémie |
|
Affections du système immunitaire |
|
|
Hypersensibilité |
|
Affections oculaires |
|
|
Irritation de la cornée |
|
Affections de la peau et du tissu sous-cutané |
|
|
Purpura, pétéchies (en particulier après des durées d’application plus longues chez les enfants souffrant de dermatite atopique ou de molluscum contagiosum) |
|
Troubles généraux et anomalies au site d’administration |
Erythème au site d’application Œdème au site d’application Pâleur au site d’application |
Sensation de brûlure Prurit au site d’application Chaleur au site d’application |
|
Population pédiatrique
La fréquence, le type et la sévérité des effets indésirables sont similaires dans les groupes d’âges pédiatriques et adultes, excepté en ce qui concerne la méthémoglobinémie qui est observée plus fréquemment, souvent en relation avec un surdosage (voir la rubrique 4.9), chez les nouveau‑nés et les nourrissons âgés de 0 à 12 mois.
Déclaration des effets indésirables suspectés
La déclaration des effets indésirables suspectés après autorisation du médicament est importante. Elle permet une surveillance continue du rapport bénéfice/risque du médicament. Les professionnels de santé déclarent tout effet indésirable suspecté via le système national de déclaration : Agence nationale de sécurité du médicament et des produits de santé (ANSM) et réseau des Centres Régionaux de Pharmacovigilance - Site internet : https://signalement.social-sante.gouv.fr/.
De rares cas de méthémoglobinémie cliniquement significative ont été rapportés.
La prilocaïne à fortes doses peut entrainer une augmentation du taux de méthémoglobine en particulier chez les individus sensibles (voir rubrique 4.4), avec une administration trop fréquente chez les nouveau‑nés et les nourrissons de moins de 12 mois (voir rubrique 4.2) et traités par des médicaments inducteurs de méthémoglobinémie (ex. sulfamides, nitrofurantoïne, phénytoïne et phénobarbital). Il convient de tenir compte du fait que les valeurs de saturation à l’oxymètre de pouls peuvent surestimer la saturation réelle en oxygène dans le cas d’une augmentation de la fraction de méthémoglobine ; par conséquent, si une méthémoglobinémie est suspectée, il peut être plus utile de surveiller la saturation d’oxygène par co‑oxymétrie.
Une méthémoglobinémie cliniquement significative doit être traitée par une injection intraveineuse lente de bleu de méthylène dilué (voir également rubrique 4.4).
Si d’autres symptômes de toxicité systémique survenaient, les signes devraient être similaires à ceux qui se produiraient suite à l’administration d’anesthésiques locaux par d’autres voies d’administration. La toxicité d’un anesthésique local se manifeste par des symptômes nerveux centraux : excitation et, dans les cas sévères, dépression du système nerveux central et cardiovasculaire.
Des symptômes neurologiques graves (convulsions, dépression du système nerveux central) doivent être traités symptomatiquement par assistance respiratoire et par administration de médicaments anticonvulsivants, les signes circulatoires sont traités suivant les recommandations de réanimation.
Le taux d’absorption du produit sur peau intacte étant lent, un patient montrant des signes de toxicité doit être maintenu sous observation pendant plusieurs heures après le traitement d’urgence.
5. PROPRIETES PHARMACOLOGIQUES
5.1. Propriétés pharmacodynamiques
Classe pharmacothérapeutique : anesthésiques, local ; amides. Code ATC N01B B20.
Mécanisme d’action
EMLA produit une anesthésie de la peau par la libération de lidocaïne et de prilocaïne dans les couches de la peau et à proximité de récepteurs dermiques de la douleur et des terminaisons nerveuses.
La lidocaïne et la prilocaïne sont des anesthésiques locaux de type amide. Ces deux composants stabilisent les membranes neuronales en inhibant les flux ioniques nécessaires à l’initiation et à la conduction des influx, produisant ainsi une anesthésie locale. La qualité de l’anesthésie dépend du temps d’application et de la dose utilisée.
Peau
EMLA Patch est appliqué sur la peau intacte Le temps nécessaire pour obtenir une anesthésie fiable de la peau intacte est de 1 à 2 heures, en fonction du type d’intervention. L’effet anesthésique local est renforcé avec des durées d’application plus longues de 1 à 2 heures sur la plupart des parties du corps, à l’exception de la peau du visage. Compte tenu de la finesse de la peau du visage et d’une irrigation sanguine élevée des tissus, un effet anesthésique local maximal est obtenu après 30 à 60 minutes sur le front et les joues. La durée de l’anesthésie après l’application d’EMLA pendant 1 à 2 heures est d’au moins 2 heures après le retrait du pansement, excepté sur le visage où ce délai est plus court. EMLA est tout aussi efficace et a le même temps de déclenchement de l’effet anesthésiant quel que soit le niveau de pigmentation de la peau de claire à foncée (types de peau de I à VI).
Au cours des études cliniques avec application d’EMLA sur la peau intacte, aucune différence en termes de sécurité d’emploi ou d'efficacité (y compris pour le délai d’installation de l’anesthésie) n’a été mise en évidence entre les patients âgés (65-96 ans) et les patients plus jeunes.
EMLA produit une réponse vasculaire biphasique impliquant une vasoconstriction initiale suivie d’une vasodilatation au site d'application (voir rubrique 4.8). Indépendamment de la réponse vasculaire, EMLA facilite l’insertion de l'aiguille par rapport au placebo.
Chez les patients atteints de dermatite atopique, une réaction vasculaire similaire mais plus courte a été observée, avec survenue d’un érythème dans les 30-60 minutes, ce qui indique une absorption plus rapide par la peau (voir rubrique 4.4). EMLA peut causer une augmentation transitoire de l’épaisseur cutanée, provoquée en partie par l’hydratation de la peau sous le pansement occlusif. L’épaisseur de la peau se réduit au cours des 15 minutes suivant l’exposition à l’air.
La profondeur de l'anesthésie cutanée augmente avec le temps d'application. Chez 90 % des patients, l'anesthésie est suffisante pour l'insertion d'un poinçon de biopsie (de 4 mm de diamètre) à une profondeur de 2 mm après 60 minutes et 3 mm après 120 minutes de traitement par EMLA.
Population pédiatrique
Des études cliniques impliquant plus de 2300 patients pédiatriques de tout âge ont démontré une efficacité pour la douleur liée à l’aiguille (prélèvement sanguin veineux, cathétérisme, vaccination sous‑cutanée et intramusculaire, ponction lombaire), pour le traitement des lésions vasculaires au laser, et le curetage des molluscum contagiosum. EMLA a diminué la douleur lors de l’insertion d’aiguilles et lors de l’injection de vaccins. L’efficacité analgésique a augmenté d’une application de 15 minutes à une application de 90 minutes sur une peau normale ; mais sur des lésions vasculaires, une application de 90 minutes n’a pas fourni de bénéfice supérieur à une application de 60 minutes. EMLA ne montre pas de bénéfice versus placebo concernant la cryothérapie à l’azote liquide des verrues communes.
Onze études cliniques réalisées chez des nouveau‑nés et des nourrissons ont montré que les pics de concentrations de méthémoglobine qui survenaient environ 8 heures après l’administration épicutanée d’EMLA, étaient cliniquement non significatifs au dosage recommandé et reviennent à des valeurs normales après environ 12‑13 heures. La formation de méthémoglobine est associée à la quantité cumulée de prilocaïne absorbée par voie percutanée et peut donc augmenter en fonction de l’allongement de la durée d’application d’EMLA.
L’utilisation d’EMLA avant une vaccination rougeole‑oreillons‑rubéole ou avant l’administration intramusculaire d’un vaccin inactivé diphtérie‑coqueluche‑tétanos‑poliovirus ‑Haemophilus influenzae b ou d’un vaccin contre l’hépatite B n’a pas affecté la moyenne des titres en anticorps, le taux de séroconversion ou la proportion de patients obtenant des titres d’anticorps protecteurs ou positifs après immunisation, par rapport aux patients traités par un placebo.
5.2. Propriétés pharmacocinétiques
Absorption, distribution, biotransformation et élimination
L'absorption systémique de la lidocaïne et de la prilocaïne à partir d’EMLA est fonction de la dose, de la surface d'application et du temps d’application. Des facteurs additionnels incluent l’épaisseur de la peau (qui varie selon les parties du corps) ainsi que d’autres facteurs tels que les maladies de la peau, et le rasage. Les concentrations plasmatiques après traitement par EMLA sont de 20 à 60 % inférieures pour la prilocaïne par rapport à la lidocaïne en raison d’un volume de distribution supérieur et d’une clairance plus rapide. La voie d’élimination principale de la lidocaïne et de la prilocaïne est le métabolisme hépatique et les métabolites sont éliminés par voie rénale. Toutefois, les taux de métabolisation et d’élimination des anesthésiques locaux après une application topique d’EMLA dépendent de la vitesse d’absorption. Par conséquent, une réduction de la clairance, notamment chez les patients présentant une atteinte sévère de la fonction hépatique, a des effets limités sur les concentrations plasmatiques systémiques après administration d’une dose unique d’EMLA et après administrations réitérées une fois par jour de doses uniques à court terme (jusqu’à 10 jours).
Les symptômes de toxicité anesthésique locale deviennent de plus en plus marqués lorsque la concentration plasmatique de l’une ou l’autre des substances actives augmente de 5 à 10 μg/ml. Il convient de considérer que la toxicité de la lidocaïne et celle de la prilocaïne sont additives.
Peau intacte
Les données pharmacocinétiques disponibles se réfèrent à l’application d’EMLA crème 5% sur la peau saine.
Après l'application sur les cuisses chez les adultes (60 g de crème /400 cm2 pendant 3 heures), le taux d'absorption était d'environ 5 % de la lidocaïne et de la prilocaïne. Les concentrations plasmatiques maximales (en moyenne de 0,12 et 0,07 µg / ml) ont été atteintes environ 2-6 heures après l'application.
Le degré d'absorption systémique était d'environ 10% après l'application sur le visage (10 g/100 cm2 pendant 2 heures). Les concentrations plasmatiques maximales (en moyenne de 0,16 et 0,06 µg / ml) ont été atteintes après environ 1,5 à 3 heures.
Populations spéciales
Patients âgés
Après application d’EMLA sur la peau intacte de patients gériatriques ou sur d’autres patients non gériatriques, les taux plasmatiques de lidocaïne et de prilocaïne sont très faibles et nettement inférieurs aux niveaux toxiques potentiels.
Après application d’EMLA répétée quotidiennement pendant 10 jours consécutifs, les concentrations plasmatiques maximales ne dépendent pas de l’âge du patient, mais sont significativement (p<0,01) liées à la surface de la zone traitée.
Population pédiatrique
Les concentrations plasmatiques maximales de lidocaïne et de prilocaïne après application d’EMLA chez les patients pédiatriques de différents âges étaient également en dessous des niveaux potentiellement toxiques. Voir tableau 4.
Tableau 4. Concentrations plasmatiques de lidocaïne et de prilocaïne dans les groupes pédiatriques âgés de 0 mois à 8 ans
|
Age |
Quantité de crème appliquée |
Temps d’application de la crème sur la peau |
Concentration plasmatique [ng/ml] Lidocaïne Prilocaïne |
|
0 ‑ 3 mois |
1 g/10 cm2 |
1 heure |
135 107 |
|
3 ‑ 12 mois |
2 g/16 cm2 |
4 heures |
155 131 |
|
2 ‑ 3 ans |
10 g/100 cm2 |
2 heures |
315 215 |
|
6 ‑ 8 ans |
10 – 16 g/100‑160 cm2 (1 g/ 10 cm2) |
2 heures |
299 110 |
5.3. Données de sécurité préclinique
Lorsque la lidocaïne et la prilocaïne étaient administrés en association, seuls des effets additifs ont été observés, sans effet synergique et sans toxicité inattendue.
Après ingestion involontaire d’EMLA, les deux substances actives présentaient une faible toxicité orale aiguë avec une bonne marge de sécurité.
Dans des études de toxicité sur la reproduction, des effets embryotoxiques ou fœto‑toxiques ont été détectés à des doses de 25 mg/kg de lidocaïne par voie sous‑cutanée chez le lapin ; et à partir des doses de 100 mg/kg de prilocaïne par voie intramusculaire chez le rat. A des doses inférieures à la dose maternelle toxique chez le rat, la lidocaïne n’a pas d’effet sur le développement postnatal des petits. La fertilité chez les rats mâles ou femelles traités par la lidocaïne ou la prilocaïne n’a pas été altérée. La lidocaïne passe la barrière placentaire par diffusion simple. Le rapport entre la dose embryo‑fœtale et les concentrations sériques de la mère est de 0,4 à 1,3.
Aucun des deux anesthésiques locaux n’a montré un potentiel génotoxique lors des tests in vitro ou in vivo. Etant donné l’indication et la durée d’utilisation thérapeutique de ces substances actives, aucune étude de carcinogénicité n’a été réalisée avec la lidocaïne ou la prilocaïne, seules ou en association.
Un métabolite de la lidocaïne, la 2,6-diméthylaniline, et un métabolite de la prilocaïne, l’σ-toluidine, ont fait preuve d’une activité génotoxique. Au cours d’études précliniques toxicologiques évaluant l’exposition chronique, ces métabolites ont montré un potentiel carcinogène. Les évaluations du risque comparant l’exposition humaine maximale calculée lors de l’utilisation intermittente de lidocaïne et de prilocaïne à l’exposition au cours des études précliniques, indique une marge importante de sécurité lors de l’utilisation clinique.
Les études de tolérance locale utilisant un mélange de lidocaïne et de prilocaïne 1:1 (m/m) sous forme d'émulsion, de crème ou de gel ont indiqué que ces formulations sont bien tolérées sur la peau intacte et endommagée et sur les membranes des muqueuses.
Lors d’une étude chez l’animal, une réaction irritative prononcée a été observée après application unique dans l’œil d’une émulsion contenant 50 mg/g de lidocaïne + prilocaïne 1:1 (m/m). La concentration en anesthésiques locaux était identique et de la formulation était similaire à celle d’EMLA.
Cette réaction oculaire peut avoir été influencée par le pH élevé (environ 9) de la formulation de l’émulsion mais c’est probablement aussi en partie le résultat du potentiel d'irritation des anesthésiques locaux eux-mêmes.
Les études précliniques sur l’adhésif utilisé dans le patch n’ont pas mis en évidence de problème.
Composition du pansement adhésif cutané : feuille stratifiée (aluminium/plastique), disque absorbant (cellulose), bande circulaire de mousse adhésive (PE), adhésif (acrylate).
2 ans
6.4. Précautions particulières de conservation
A conserver à une température ne dépassant pas 25° C.
Ne pas mettre au réfrigérateur. Ne pas congeler.
6.5. Nature et contenu de l'emballage extérieur
Le pansement adhésif est composé d'un pansement occlusif contenant 1 g d'émulsion et d'un revêtement protecteur. Le pansement est constitué d'une feuille stratifiée aluminium/plastique, d'un disque absorbant en cellulose contenant les anesthésiques locaux et d'une bande circulaire de mousse adhésive. Cette bande est du polyéthylène recouverte d'un adhésif en acrylate. Boîte de 1, 2 ou 20.
Toutes les présentations peuvent ne pas être commercialisées.
6.6. Précautions particulières d’élimination et de manipulation
Le pansement adhésif doit être appliqué au moins une heure avant l'intervention.
S'assurer que la surface cutanée à anesthésier est propre et sèche.
Pour ouvrir le pansement adhésif, prendre le coin aluminium prévu à cet effet.
La partie blanche du pansement adhésif contient la crème anesthésique, ne pas la toucher.
Appliquer le pansement adhésif de manière à ce que la pastille blanche recouvre la surface à anesthésier.
Ne pas appuyer sur la partie centrale du pansement adhésif, pour éviter une fuite sous l'adhésif.
Assurer la bonne adhésion du pansement adhésif en appuyant fermement sur le pourtour.
Indiquer l'heure d'application à même le pansement adhésif. Celui-ci doit rester en place au minimum 60 minutes chez l'adulte et l'enfant de plus de 3 mois.
Chez le nourrisson de 0 à 2 mois, la durée d'application maximale est de 1 heure pour une surface de 10 cm2 maximum.
Tout médicament non utilisé ou déchet doit être éliminé conformément à la réglementation en vigueur.
7. TITULAIRE DE L’AUTORISATION DE MISE SUR LE MARCHE
3016 Lake Drive
Citywest Business Campus
DUBLIN 24
IRLANDE
8. NUMERO(S) D’AUTORISATION DE MISE SUR LE MARCHE
· 34009 340 007 1 3 : boîte de 1 pansement adhésif cutané.
· 34009 339 110 7 2 : boîte de 2 pansements adhésifs cutanés.
· 34009 302 740 5 7 : boîte de 20 pansements adhésifs cutanés.
9. DATE DE PREMIERE AUTORISATION/DE RENOUVELLEMENT DE L’AUTORISATION
[à compléter ultérieurement par le titulaire]
10. DATE DE MISE A JOUR DU TEXTE
[à compléter ultérieurement par le titulaire]
Sans objet.
12. INSTRUCTIONS POUR LA PREPARATION DES RADIOPHARMACEUTIQUES
Liste II
ANSM - Mis à jour le : 22/03/2024
EMLAPATCH 5 POUR CENT, pansement adhésif cutané
· Gardez cette notice. Vous pourriez avoir besoin de la relire.
· Si vous avez d’autres questions, interrogez votre médecin, ou votre pharmacien.
· Ce médicament vous a été personnellement prescrit. Ne le donnez pas à d’autres personnes. Il pourrait leur être nocif, même si les signes de leur maladie sont identiques aux vôtres.
· Si vous ressentez un quelconque effet indésirable, parlez-en à votre médecin, ou votre pharmacien. Ceci s’applique aussi à tout effet indésirable qui ne serait pas mentionné dans cette notice. Voir rubrique 4.
1. Qu'est-ce que EMLAPATCH 5 POUR CENT, pansement adhésif cutané et dans quels cas est-il utilisé ?
2. Quelles sont les informations à connaître avant d'utiliser EMLAPATCH 5 POUR CENT, pansement adhésif cutané ?
3. Comment utiliser EMLAPATCH 5 POUR CENT, pansement adhésif cutané ?
4. Quels sont les effets indésirables éventuels ?
5. Comment conserver EMLAPATCH 5 POUR CENT, pansement adhésif cutané ?
6. Contenu de l’emballage et autres informations.
1. QU’EST-CE QUE EMLAPATCH 5 POUR CENT, pansement adhésif cutané ET DANS QUELS CAS EST-IL UTILISE ?
Classe pharmacothérapeutique : ANESTHESIQUES LOCAUX - (N : système nerveux central), Code ATC N01B B20.
EMLAPATCH 5 POUR CENT contient deux substances actives appelées lidocaïne et prilocaïne. Ces dernières appartiennent à un groupe de médicaments appelés anesthésiques locaux.
EMLAPATCH 5 POUR CENT, pansement adhésif cutané fonctionne en provoquant une anesthésie de la surface de la peau pendant une courte durée. Il est appliqué sur la peau avant certains actes médicaux. Cela permet d’arrêter la douleur au niveau de la peau ; cependant, vous pouvez encore avoir les sensations de pression et de toucher.
Adultes, adolescents et enfants
Il peut être utilisé pour anesthésier la peau avant :
· l’insertion d’aiguilles (par exemple, si vous devez avoir une injection ou faire une prise de sang).
· des opérations mineures de la peau.
2. QUELLES SONT LES INFORMATIONS A CONNAITRE AVANT D’UTILISER EMLAPATCH 5 POUR CENT, pansement adhésif cutané ?
N’utilisez jamais EMLAPATCH 5 POUR CENT, pansement adhésif cutané :
· Si vous êtes allergique à la lidocaïne ou à la prilocaïne, à d’autres anesthésiques locaux similaires ou à l’un des autres composants contenus dans ce médicament (mentionnés dans la rubrique 6).
Avertissements et précautions
Mise en garde et précautions d’emploi
Adressez‑vous à votre médecin ou votre pharmacien avant d’utiliser EMLAPATCH 5%, pansement adhésif cutané.
· Si vous ou votre enfant avez une maladie héréditaire rare qui affecte le sang appelée « déficit en glucose‑6‑phosphate‑déshydrogénase ».
· Si vous ou votre enfant avez un problème au niveau de la quantité de pigment sanguin appelé « méthémoglobinémie »
· N’utilisez pas EMLAPATCH 5%, pansement adhésif cutané sur les zones présentant des éruptions cutanées, coupures, écorchures ou autres plaies ouvertes. Si l’un de ces problèmes est présent, vérifiez avec votre médecin ou votre pharmacien avant d’utiliser le patch.
· Si vous ou votre enfant souffrez de démangeaisons cutanées appelée « dermatite atopique », un temps d’application réduit peut être suffisant. Un temps d’application de plus de 30 minutes peut conduire à une augmentation de l’incidence des réactions cutanées locales (voir aussi rubrique 4 « Effets indésirables éventuels »).
· Evitez le contact d’EMLAPATCH 5%, pansement adhésif cutané avec les yeux car il peut provoquer une irritation. Si vous recevez accidentellement EMLAPATCH 5 %, pansement adhésif cutané dans votre œil, vous devez le rincer immédiatement à l’eau tiède ou avec une solution saline (chlorure de sodium). Veillez à ne rien recevoir d’autre dans les yeux jusqu’au retour de la sensibilité.
Si vous utilisez EMLAPATCH 5% pansement adhésif cutané avant d’être vacciné par des vaccins vivants (par exemple le vaccin contre la tuberculose), vous devez consulter à nouveau votre médecin ou votre infirmier/ère après la période de délai demandée dans le cadre du suivi de la vaccination.
Enfants et adolescents
Chez les nourrissons et les nouveau-nés de moins de 3 mois, une augmentation transitoire, cliniquement non significative, de la quantité de pigments sanguins appelés « méthémoglobinémie » est fréquemment observée jusqu’à 12 heures après l’application d’EMLAPATCH 5 POUR CENT, pansement adhésif cutané.
L’efficacité d’EMLA 5 POUR CENT, pansement adhésif cutané lors du prélèvement sanguin au talon chez les nouveau-nés n’a pas pu être confirmée dans les études cliniques.
EMLA 5 POUR CENT, pansement adhésif cutané ne doit pas être utilisé chez les enfants âgés de moins de 12 mois qui reçoivent simultanément d’autres traitements affectant la quantité de pigments sanguins « méthémoglobinémie » (par exemple les sulfamides, voir aussi rubrique 2 « Autres médicaments et EMLA 5 POUR CENT, pansement adhésif cutané »).
EMLA 5 POUR CENT, pansement adhésif cutané ne doit pas être utilisé chez les nourrissons prématurés.
Autres médicaments et EMLAPATCH 5%, pansement adhésif cutané
Informez votre médecin ou pharmacien si vous utilisez/prenez ou avez récemment utilisé/pris ou pourriez utiliser/prendre tout autre médicament incluant des médicaments obtenus sans ordonnance et les médicaments à base de plantes. Cela est dû au fait qu’EMLAPATCH 5 % peut affecter le mécanisme d’action de certains médicaments et certains médicaments peuvent avoir un effet sur EMLAPATCH 5%, pansement adhésif cutané.
Informez en particulier votre médecin ou votre pharmacien, si vous ou votre enfant avez récemment utilisé ou reçu l’un des médicaments suivants :
· Des médicaments utilisés pour traiter les infections, appelés « sulfamides » et nitrofurantoïne.
· Des médicaments utilisés pour traiter l’épilepsie, appelés phénytoïne et phénobarbital.
· D’autres anesthésiques locaux.
· La cimétidine ou les bêtabloquants qui peuvent augmenter les concentrations de lidocaïne dans le sang. Cette interaction n’a pas d’impact clinique si EMLA est utilisé aux doses recommandées pour une courte durée de traitement.
EMLAPATCH 5%, pansement adhésif cutané avec des aliments et boissons
Sans objet.
Grossesse, allaitement et fertilité
Si vous êtes enceinte ou que vous allaitez, si vous pensez être enceinte ou planifiez une grossesse, demandez conseil à votre médecin ou pharmacien avant de prendre ce médicament.
Il est peu probable qu’EMLA 5%, pansement adhésif cutané, utilisé occasionnellement pendant la grossesse, provoque des effets indésirables sur le fœtus.
Les substances actives d’EMLAPATCH 5%, pansement adhésif cutané (la lidocaïne et la prilocaïne) sont excrétées dans le lait maternel. Cependant, les quantités sont si faibles qu’il n’y a généralement pas de risque pour l’enfant.
Des études chez l’animal ont montré une absence d’altération de la fertilité chez le mâle ou la femelle.
Conduite de véhicules et utilisation de machines
EMLAPATCH 5%, pansement adhésif cutané n’a aucune ou qu’une influence négligeable sur l’aptitude à conduire et à utiliser des machines quand il est utilisé aux doses recommandées.
EMLAPATCH 5%, pansement adhésif cutané contient de l’hydroxystéarate de macrogolglycérol.
L’hydroxystéarate de macrogolglycérol peut provoquer des réactions cutanées.
3. COMMENT UTILISER EMLAPATCH 5 POUR CENT, pansement adhésif cutané ?
Utilisation d’EMLA 5 %, pansement adhésif cutané
· Le site d’application du patch, le nombre de patchs à utiliser ainsi que le temps d’application dépendront de la raison pour laquelle EMLA 5%, pansement adhésif cutané est utilisé.
· Votre médecin, votre pharmacien ou votre infirmier/ère vous appliqueront le patch ou vous monteront comment le faire vous-même.
Ne pas utiliser EMLA 5%, pansement adhésif cutané sur les zones suivantes :
· Coupures, écorchures ou plaies.
· Zone avec éruptions cutanées ou eczéma.
· Près des yeux.
· A l’intérieur de la bouche.
Utilisation sur la peau avant de petits actes médicaux (comme l’insertion d’aiguille ou des opérations mineures de la peau) :
· Le patch est appliqué sur la peau. Votre médecin, pharmacien ou votre infirmier/ère vous diront où vous devrez l’appliquer.
· Le patch est retiré juste avant le début de l’intervention.
· La dose usuelle chez les adultes et les adolescents âgés de plus de 12 ans est de 1 patch ou plus.
· Chez les adultes et adolescents âgés de plus de 12 ans, appliquer le patch au moins 60 minutes avant le début de l’intervention. Cependant, ne pas l’appliquer plus de 5 heures avant le début de l’intervention.
· Chez les enfants, le nombre de patchs EMLA utilisés et le temps d’application dépendent de l’âge. Votre médecin, pharmacien ou votre infirmier/ère vous diront combien de patchs utiliser et quand ils doivent être appliqués.
Utilisation chez les enfants
Utilisation sur la peau avant de petits actes médicaux (comme l’insertion d’aiguille ou des opérations mineures de la peau) : le temps d’application approximatif est d’une heure.
Nouveau-nés et nourrissons de 0-2 mois : un patch est appliqué sur la zone de peau choisie. La durée d’application ne doit pas excéder 1 heure. Sur une période de 24 heures on n’appliquera pas plus qu’une seule dose. La taille du patch le rend moins adapté pour une utilisation sur certaines parties du corps du nouveau-né ou du nourrisson.
Nourrissons âgés de 3-11 mois : jusqu’à deux patchs sont appliqués sur la zone de peau choisie. La durée d’application est approximativement 1 heure.
Enfants âgés de de 1-5 ans : jusqu’à 10 patchs sont appliqués sur la zone de peau choisie. La durée d’application est approximativement 1 heure, maximum 5 heures.
Enfants âgés de 6-11 ans : jusqu’à 20 patchs sont appliqués sur la zone de peau choisie. La durée d’application est de 1 heure, maximum 5 heures.
Un maximum de 2 doses (comme spécifié ci-dessus), à au moins 12 heures d’intervalles, peut être donnés aux nourrissons et enfants de plus de 3 mois sur une période de 24 heures.
EMLAPATCH peut être utilisé chez les enfants présentant une maladie de peau appelé « dermatite atopique » mais la durée d’application est alors de 30 minutes, pas plus.
Lors de l’application du patch, il est très important de suivre exactement les instructions suivantes :
EMLAPATCH doit être appliqué au moins 1 heure avant l’intervention (sauf chez les patients avec une dermatite atopique, voir rubrique avertissement et précaution). Si nécessaire, enlever les poils de la zone avant l’application. Le patch ne peut pas être coupé ou divisé en morceaux plus petits.
|
|
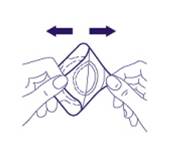
1/ Assurez-vous que la surface de la peau à anesthésier est propre et sèche. Saisissez le rebord en aluminium dans le coin du patch et tirez vers l’extérieur. Ensuite, saisissez le coin du patch de la couleur de la peau. Assurez-vous que les deux couches dans le coin soient bien séparées avant de continuer. |
|
|
2. Détachez les deux couches, en séparant la surface adhésive de la feuille protectrice, comme indiqué. Assurez-vous de ne pas toucher le disque rond blanc, qui contient EMLA. |
|
|
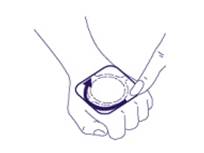
3. N’appuyez pas au centre du patch. EMLA pourrait se répandre en-dessous de l’adhésif et empêcher que le patch adhère bien à la peau. Appuyez fermement sur les bords afin de garantir une bonne adhérence de la peau. |
|
|
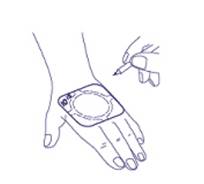
4. Le temps d’application peut facilement être indiqué directement sur le patch. (Un stylo à bille peut être utilisé à cette fin.) |
|
|
5. Laissez le patch en place pendant au moins une heure (excepté chez les patients atteints de dermatite atopique, voir rubrique Avertissements et précautions). Chez les enfants de moins de 3 mois, le patch ne doit pas être laissé en place pendant plus d’une heure. |
|
|
6. Lorsque le temps d’application est passé, le patch doit être retiré de la peau. |
Utilisation sur la peau avant l’enlèvement de taches ressemblant à des verrues appelées « molluscum »
· EMLAPATCH 5%, pansement adhésif cutané peut être utilisé chez les enfants et adolescents présentant une maladie de peau appelée « dermatite atopique ».
· La dose usuelle dépend de l’âge de l’enfant et est appliquée pendant 30 à 60 minutes (30 minutes si le patient a une dermatite atopique). Votre médecin, infirmier/ère ou pharmacien vous diront combien de patchs utiliser.
Si vous avez utilisé plus de EMLAPATCH 5%, pansement adhésif cutané que vous n’auriez dû :
Si vous avez utilisé plus d’EMLAPATCH 5%, pansement adhésif cutané que votre médecin, pharmacien ou infirmier/ère vous a dit, parlez-en à l’un d’entre eux immédiatement même si vous ne ressentez aucun symptôme.
Les symptômes d’un surdosage d’EMLAPATCH 5%, pansement adhésif cutané sont listés ci-dessous. Il est peu probable que de tels symptômes surviennent si EMLAPATCH 5%, pansement adhésif cutané est utilisé aux doses recommandées.
· Sensation d’étourdissement ou sensation vertigineuse.
· Picotements de la peau autour de la bouche et engourdissement de la langue.
· Goût anormal.
· Vision floue.
· Bourdonnement dans les oreilles.
· Un risque de « méthémoglobinémie aiguë » existe aussi (un problème avec les quantités de pigments sanguins). Ceci est plus probable quand certains médicaments ont été pris simultanément. Si cela se produit, la peau prend une coloration bleu gris en raison d’un manque d’oxygène.
Dans les cas graves de surdosage, les symptômes peuvent inclure des convulsions, une baisse de la pression artérielle, un ralentissement de la respiration, un arrêt respiratoire et une altération du rythme cardiaque. Ces effets peuvent engager le pronostic vital.
Si vous oubliez d’utiliser EMLAPATCH 5%, pansement adhésif cutané
Si vous arrêtez d’utiliser EMLAPATCH 5%, pansement adhésif cutané
Sans objet.
Si vous avez d’autres questions sur l’utilisation de ce médicament, demandez plus d’informations à votre médecin, à votre pharmacien ou à votre infirmier/ère.
4. QUELS SONT LES EFFETS INDESIRABLES EVENTUELS ?
Contactez votre médecin ou votre pharmacien si l’un des effets indésirables suivants vous dérange ou persiste. Informez votre médecin de tout événement qui entraîne une sensation de mal être lorsque vous utilisez EMLAPATCH 5%, pansement adhésif cutané.
Une légère réaction (pâleur ou rougeur de la peau, légère enflure, brûlure initiale ou démangeaisons) peut survenir au niveau de la zone sur laquelle EMLAPATCH 5%, pansement adhésif cutané a été appliqué. Ce sont des réactions normales liées au patch et à la présence des anesthésiques et elles disparaîtront rapidement sans que des mesures spécifiques ne soient nécessaires.
Si vous ressentez des effets gênants ou inhabituels pendant que vous utilisez EMLAPATCH 5%, pansement adhésif cutané, cessez son utilisation et consultez votre médecin, ou votre pharmacien aussi vite que possible.
Fréquents (peut affecter jusqu’à 1 personne sur 10)
· Réactions locales transitoires de la peau (pâleur, rougeur, gonflement) au site d’application sur la zone traitée.
Peu fréquents (peut affecter jusqu’à 1 personne sur 100)
· Sensation initiale légère de brûlure, de démangeaison ou de chaleur au site d’application sur la zone traitée.
Rares (peut affecter jusqu’à 1 personne sur 1000)
· Réactions allergiques, qui dans de rares cas peuvent conduire à un choc anaphylactique (éruption cutanée, gonflement, fièvre, difficultés respiratoires et évanouissement).
· Méthémoglobinémie (trouble sanguin).
· Petits saignements en forme de points sur la zone traitée (particulièrement chez les enfants présentant de l’eczéma en cas d’application prolongée).
· Irritations des yeux lorsqu’EMLAPATCH 5%, pansement adhésif cutané a été accidentellement en contact avec les yeux pendant le traitement de la peau.
Autres effets indésirables chez les enfants
Méthémoglobinémie, une maladie du sang, qui est plus souvent observée, dans le cas de surdosage chez les nouveau-nés et les nourrissons âgés de 0 à 12 mois.
Déclaration des effets secondaires
Si vous ressentez un quelconque effet indésirable, parlez-en à votre médecin, votre pharmacien ou à votre infirmier/ère. Ceci s’applique aussi à tout effet indésirable qui ne serait pas mentionné dans cette notice. Vous pouvez également déclarer les effets indésirables directement via le système national de déclaration : Agence nationale de sécurité du médicament et des produits de santé (ANSM) et réseau des Centres Régionaux de Pharmacovigilance - Site internet : https://signalement.social-sante.gouv.fr/.
En signalant les effets indésirables, vous contribuez à fournir davantage d’informations sur la sécurité du médicament.
5. COMMENT CONSERVER EMLAPATCH 5 POUR CENT, pansement adhésif cutané ?
Tenir ce médicament hors de la vue et de la portée des enfants.
N’utilisez pas ce médicament après la date de péremption indiquée sur l’emballage. La date de péremption fait référence au dernier jour de ce mois.
A conserver à une température ne dépassant pas 25°C.
Ne pas mettre au réfrigérateur. Ne pas congeler.
Ne jetez aucun médicament au tout-à-l’égout ou avec les ordures ménagères. Demandez à votre pharmacien d’éliminer les médicaments que vous n’utilisez plus. Ces mesures contribueront à protéger l’environnement.
6. CONTENU DE L’EMBALLAGE ET AUTRES INFORMATIONS
Ce que contient EMLAPATCH 5 POUR CENT, pansement adhésif cutané
· Les substances actives sont :
Lidocaïne......................................................................................................................... . 0,0250 g
Prilocaïne ......................................................................................................................... 0,0250 g
1 g d'émulsion pour un pansement adhésif* de 10 cm2
· Les autres composants excipients sont :
Huile de ricin hydrogénée polyoxyéthylénée (Arlatone 289), carbomère (carbopol 974P), hydroxyde de sodium, eau purifiée.
*Composition du pansement adhésif cutané : feuille stratifiée (aluminium/plastique), disque absorbant (cellulose), bande circulaire de mousse adhésive (PE), adhésif (acrylate).
Qu’est-ce que EMLAPATCH 5 POUR CENT, pansement adhésif cutané et contenu de l’emballage extérieur
Ce médicament se présente sous forme de pansement adhésif cutané.
Boîtes de 1,2 ou 20.
Toutes les présentations peuvent ne pas être commercialisées.
Titulaire de l’autorisation de mise sur le marché
3016 Lake Drive
Citywest Business Campus
DUBLIN 24
IRLANDE
Exploitant de l’autorisation de mise sur le marché
Aspen France
21 Avenue Edouard Belin
92500 Rueil Malmaison
BJORKBORNSVAGEN 5
691 33 KARLSKOGA
SUEDE
Noms du médicament dans les Etats membres de l'Espace Economique Européen
Sans objet.
La dernière date à laquelle cette notice a été révisée est :
[à compléter ultérieurement par le titulaire]
{MM/AAAA} {mois AAAA}
Sans objet.
Des informations détaillées sur ce médicament sont disponibles sur le site Internet de l’ANSM (France).