Dernière mise à jour le 28/04/2026
XEOMIN 50 unités, poudre pour solution injectable
Indications thérapeutiques
Classe pharmacothérapeutique : M03AX01
XEOMIN 50 unités, poudre pour solution injectable est un médicament qui contient comme principe actif la Neurotoxine Botulique de Type A qui détend les muscles injectés ou qui diminue le flux salivaire en fonction du site d’administration.
XEOMIN 50 unités, poudre pour solution injectable est utilisé chez l'adulte pour le traitement :
· du spasme des paupières (blépharospasme) et du spasme touchant un côté du visage (spasme hémifacial),
· du torticolis spasmodique,
· de l’hyperactivité musculaire ou de la raideur incontrôlable des muscles de l’épaule, du bras et/ou de la main. (spasticité des membres supérieurs)
· de l’excès de salive (sialorrhée) en raison de troubles neurologiques.
XEOMIN est indiqué dans le traitement symptomatique chez les enfants et adolescents âgés de 2 à 17 ans et pesant ≥ 12kg de
· l’excès de salive (sialorrhée) en raison de troubles neurologiques / troubles neurologiques du développement.
Présentations
> 1 flacon(s) en verre de 50 unité(s)
Code CIP : 580 884-7 ou 34009 580 884 7 6
Déclaration de commercialisation : 16/01/2012
Cette présentation est agréée aux collectivités
Service médical rendu (SMR)
Les libellés affichés ci-dessous ne sont que des résumés ou extraits issus des avis rendus par la Commission de la Transparence. Seul l'avis complet de la Commission de la Transparence fait référence.
Cet avis est consultable à partir du lien `Avis du jj/mm/aaaa` ou encore sur demande auprès de la HAS (plus d'informations dans l'aide). Les avis et synthèses d'avis contiennent un paragraphe sur la place du médicament dans la stratégie thérapeutique.
| Valeur du SMR | Avis | Motif de l'évaluation | Résumé de l'avis |
|---|---|---|---|
| Important | Avis du 07/06/2023 | Extension d'indication | Le service médical rendu par les spécialités XEOMIN (Neurotoxine de Clostridium botulinum de type A) est important pour le traitement symptomatique chez l’adulte de la spasticité des membres supérieurs. |
| Insuffisant | Avis du 30/03/2022 | Extension d'indication | Le service médical rendu par XEOMIN est insuffisant dans les autres situations cliniques de l’AMM. |
| Modéré | Avis du 30/03/2022 | Extension d'indication | Le service médical rendu par XEOMIN est modéré dans l’indication du traitement symptomatique de la sialorrhée chronique chez l’adulte atteint de troubles neurologiques uniquement dus à la maladie de Parkinson, un traumatisme crânien ou un AVC, en cas d’intolérance ou d’échec aux anticholinergiques. |
| Important | Avis du 23/05/2012 | Inscription (CT) | Le service médical rendu par cette spécialité est important dans le traitement symptomatique du blépharospasme, le traitement symptomatique de la dystonie cervicale, le traitement de la spasticité des membres supérieurs avec flexion de poignet et fermeture de la main à la suite d’un accident vasculaire cérébral. |
Amélioration du service médical rendu (ASMR)
Les libellés affichés ci-dessous ne sont que des résumés ou extraits issus des avis rendus par la Commission de la Transparence. Seul l'avis complet de la Commission de la Transparence fait référence.
Cet avis est consultable à partir du lien `Avis du jj/mm/aaaa` ou encore sur demande auprés de la HAS (plus d'informations dans l'aide). Les avis et synthéses d'avis contiennent un paragraphe sur la place du médicament dans la stratégie thérapeutique.
| Valeur de l'ASMR | Avis | Motif de l'évaluation | Résumé de l'avis |
|---|---|---|---|
| V (Inexistant) | Avis du 07/06/2023 | Extension d'indication | Les spécialités XEOMIN (Neurotoxine de Clostridium botulinum de type A) n’apportent pas d’amélioration du service médical rendu (ASMR V), comme les autres spécialités à base de toxine botulinique A actuellement disponibles, dans la stratégie thérapeutique pour le traitement symptomatique chez l’adulte de la spasticité des membres supérieurs. |
| V (Inexistant) | Avis du 30/03/2022 | Extension d'indication | XEOMIN n’apporte pas d’amélioration du service médical rendu (ASMR V) dans la stratégie de prise en charge des patients adultes atteint de sialorrhée |
| V (Inexistant) | Avis du 23/05/2012 | Inscription (CT) | XEOMIN 50U est un complément de gamme de XEOMIN 100U qui n'apporte pas d'amélioration du service médical rendu (ASMR V) par rapport aux autres présentations disponibles. |
Autres informations
- Titulaire de l'autorisation : MERZ THERAPEUTICS GmbH
- Conditions de prescription et de délivrance :
- liste I
- réservé à l'usage HOSPITALIER
- Statut de l'autorisation : Valide
- Type de procédure : Procédure décentralisée
- Code CIS : 6 741 166 7
ANSM - Mis à jour le : 10/03/2026
XEOMIN 50 unités, poudre pour solution injectable
2. COMPOSITION QUALITATIVE ET QUANTITATIVE
XEOMIN 50 unités poudre pour solution injectable
1 flacon contient 50 unités de neurotoxine de Clostridium botulinum de type A (150 kD), sans protéines complexantes*.
* Neurotoxine botulinique de type A purifiée obtenue par culture de Clostridium botulinum (souche Hall).
Pour la liste complète des excipients, voir rubrique 6.1.
Poudre pour solution injectable.
Poudre blanche.
4.1. Indications thérapeutiques
XEOMIN est indiqué pour le traitement symptomatique chez l’adulte
· du blépharospasme et du spasme hémifacial,
· de la dystonie cervicale à prédominance rotationnelle (torticolis spasmodique),
· de la spasticité des membres supérieurs
· de la sialorrhée chronique due à des troubles neurologiques.
XEOMIN est indiqué pour le traitement symptomatique chez les enfants et adolescents âgés de 2 à 17 ans et pesant ≥ 12 kg de
· la sialorrhée chronique due à des troubles neurologiques / troubles neurologiques du développement.
4.2. Posologie et mode d'administration
Les tests évaluant l’activité biologique de la neurotoxine sont différents. Aussi, les unités de XEOMIN ne sont pas interchangeables avec les autres préparations de toxine botulinique de type A.
Pour une information plus complète sur les études cliniques comparant XEOMIN à la toxine botulinique de type A conventionnelle (900kD), voir rubrique 5.1.
XEOMIN doit être administré uniquement par des médecins ayant les qualifications adéquates et une bonne expérience de l'utilisation de la toxine botulinique de type A.
Les doses optimales, la fréquence et le nombre de sites d'injection doivent être déterminés par le médecin, individuellement pour chaque patient. La dose optimale doit être déterminée par augmentation progressive des doses.
Ne pas dépasser les doses recommandées de XEOMIN.
Posologie
Blépharospasme et spasme hémifacial. La dose initiale recommandée est de 1,25 à 2,5 unités par site d’injection. La dose initiale ne doit pas dépasser 25 unités par œil. La dose totale ne doit pas dépasser 50 unités par œil et par séance de traitement. Les réinjections ne doivent généralement pas être espacées de moins de 12 semaines. Les intervalles de traitement par XEOMIN doivent être adaptés aux besoins cliniques réels et individuels du patient.
L'effet du traitement apparaît dans un délai médian de quatre jours suivant l'injection. Généralement, l’effet du traitement par XEOMIN dure environ 3 à 5 mois ; il peut toutefois durer plus ou moins longtemps.
Si le résultat du traitement initial est considéré comme insuffisant, la dose peut être au maximum doublée à la session suivante. Cependant, l'injection de plus de 5 unités par site ne semble apporter aucun bénéfice supplémentaire.
Les patients atteints d’un spasme hémifacial doivent être traités de la même manière que pour un blépharospasme unilatéral.
Torticolis spasmodique
Pour le traitement du torticolis spasmodique, la dose de XEOMIN doit être adaptée individuellement pour chaque patient selon l'orientation de la tête et du cou du patient, la localisation de la douleur éventuelle, l'hypertrophie du muscle, le poids du patient et sa réponse à l’injection.
Au cours de la première session de traitement, la dose maximale de 200 unités doit être respectée. En fonction de la réponse, des ajustements posologiques peuvent être réalisés au cours des sessions suivantes. Ne pas administrer plus de 300 unités au cours d’une séance. Ne pas administrer plus de 50 unités par site d’injection.
L'effet du traitement apparaît dans un délai médian de sept jours suivant l'injection. Généralement, l'effet de XEOMIN dure environ 3 à 4 mois ; il peut toutefois durer plus ou moins longtemps. Des intervalles de traitement de moins de 10 semaines ne sont pas recommandés. Les intervalles de traitement par XEOMIN doivent être adaptés aux besoins cliniques réels et individuels du patient.
Spasticité des membres supérieurs
La posologie exacte et le nombre de sites d’injection doivent être adaptés à chaque patient d'après la taille, le nombre et l'emplacement des muscles impliqués, la sévérité de la spasticité et la présence d'une faiblesse musculaire locale.
Doses recommandées par muscle :
|
Tableau clinique Muscle |
Unités (Gamme) |
Nombre de sites d’injection par muscle |
|
Fléchisseurs du poignet |
||
|
Flexor carpi radialis |
25-100 |
1-2 |
|
Flexor carpi ulnaris |
20-100 |
1-2 |
|
Fléchisseurs des doigts |
||
|
Flexor digitorum superficialis |
25-100 |
2 |
|
Flexor digitorum profundus |
25-100 |
2 |
|
Fléchisseurs du coude |
||
|
Brachioradialis |
25-100 |
1-3 |
|
Biceps |
50-200 |
1-4 |
|
Brachialis |
25-100 |
1-2 |
|
Pronateurs de l’avant-bras |
||
|
Pronator quadratus |
10-50 |
1 |
|
Pronator teres |
25-75 |
1-2 |
|
Fléchisseurs propres du pouce |
||
|
Flexor pollicis longus |
10-50 |
1 |
|
Adductor pollicis |
5-30 |
1 |
|
Flexor pollicis brevis/Opponens pollicis |
5-30 |
1 |
|
Rotateurs internes, extenseurs et adducteurs de l’épaule |
|
|
|
Deltoideus, pars clavicularis |
20-150 |
1-3 |
|
Latissimus dorsi |
25-150 |
1-4 |
|
Pectoralis major |
20-200 |
1-6 |
|
Subscapularis |
15-100 |
1-4 |
|
Teres major |
20-100 |
1-2 |
La dose totale maximale par session de traitement de la spasticité des membres supérieurs ne doit pas excéder 500 unités et pas plus de 250 unités dans les muscles de l’épaule.
Les patients ont noté une efficacité clinique dans les 4 jours avec une amélioration maximale du tonus musculaire dans les 4 semaines qui suivent l’injection. Comme l’effet du traitement persiste, en général12 semaines ; les réinjections peuvent être espacées d’au moins 12 semaines. Il peut cependant être plus court ou plus long, et de ce fait, les intervalles de traitement par XEOMIN doivent être adaptés aux besoins cliniques réels et individuels du patient.
Sialorrhée chronique (adultes)
Une solution reconstituée de concentration de 5 unités/0,1 mL doit être utilisée.
XEOMIN est injecté dans les glandes parotides et sous-maxillaires de chaque côté (quatre injections au total par traitement). La dose est répartie en un ratio de 3:2 entre les glandes parotides et sous-maxillaires comme suit:
|
Glandes |
Unités |
Volume |
|
Glandes parotides |
30 par côté |
0.6 mL par injection |
|
Glandes sous-maxillaires |
20 par côté |
0.4 mL par injection |
Le site d’injection doit être proche du centre de la glande.
La dose recommandée est de 100 unités par session de traitement. Cette dose maximale ne doit pas être dépassée.
Bien que les intervalles de traitement de moins de 16 semaines ne soient pas recommandés, les intervalles de traitement doivent être adaptés aux besoins cliniques réels et individuels des patients.
Sialorrhée chronique (enfants/adolescents)
Une solution reconstituée de concentration de 2,5 unités/0,1 ml doit être utilisée.
XEOMIN est injecté dans les glandes parotides et sous-maxillaires de chaque côté (quatre injections au total par traitement). La dose ajustée au poids corporel est répartie en un ratio de 3:2 entre les glandes parotides et sous-maxillaires comme indiqué dans le tableau ci-dessous. Aucune recommandation posologique ne peut être faite pour les enfants pesant moins de 12 kg.
|
Poids corporel |
Glande parotide, |
Glande sous-maxillaire, |
Dose totale, des deux glandes,des deux côtés |
||
|
Dose par glande |
Volume par injection |
Dose par glande |
Volume par injection |
||
|
[kg] |
[Unités] |
[ml] |
[Unités] |
[ml] |
[Unités] |
|
≥ 12 et < 15 |
6 |
0.24 |
4 |
0.16 |
20 |
|
≥ 15 et < 19 |
9 |
0.36 |
6 |
0.24 |
30 |
|
≥ 19 et < 23 |
12 |
0.48 |
8 |
0.32 |
40 |
|
≥ 23 et < 27 |
15 |
0.60 |
10 |
0.40 |
50 |
|
≥ 27 et < 30 |
18 |
0.72 |
12 |
0.48 |
60 |
|
≥ 30 |
22.5 |
0.90 |
15 |
0.60 |
75 |
Le site d’injection doit être proche du centre de la glande.
Les intervalles de traitement doivent être adaptés aux besoins cliniques réels et individuels de chaque patient, ces intervalles ne devraient pas être inférieurs à 16 semaines.
Toutes les indications
En l'absence d'amélioration dans le mois qui suit la première séance d'injections, il y a lieu de :
· Vérifier cliniquement l’effet de la toxine sur le muscle injecté (ex : un examen électromyographique en milieu spécialisé).
· Analyser les causes de non-réponse telles que : une mauvaise isolation des muscles injectés, une dose insuffisante, une technique d'injection inadaptée, une contracture fixe, des muscles antagonistes trop faibles, la formation éventuelle d'anticorps.
· Réévaluer la pertinence d’un traitement par neurotoxine botulinique de type A.
· En l'absence d'effets indésirables lors du traitement initial, une deuxième session d’injections peut être réalisée dans les conditions suivantes :
1) ajustement de la dose, en tenant compte de l'échec précédent ;
2) localisation du muscle à injecter à l’aide de techniques telles que le guidage électromyographique ;
3) respect des intervalles de temps minimum entre les injections.
La tolérance et l’efficacité de XEOMIN dans des indications autres que celle décrite pour la population pédiatrique dans la rubrique 4.1 n’ont pas été établies. Aucune recommandation sur la posologie ne peut être faite pour des indications autres que la sialorrhée chronique chez les enfants et les adolescents âgés de 2 à 17 ans et pesant ≥ 12kg.
Les données cliniques pédiatriques actuellement disponibles avec XEOMIN sont détaillées dans la rubrique 5.1.
Mode d’administration
Toutes les indications
Pour les instructions sur la reconstitution des flacons avant administration, voir rubrique 6.6. Après reconstitution, XEOMIN doit être utilisé pour une seule session d'injection et pour un seul patient.
XEOMIN est destiné à la voie intramusculaire et intraglandulaire (glandes salivaires).
Blépharospasme et spasme hémifacial
La solution reconstituée de XEOMIN est injectée par voie intramusculaire au moyen d'une aiguille stérile appropriée (diamètre de 27-30 Gauge/0,30-0,40 mm et longueur de 12,5 mm). Le guidage électromyographique n'est pas nécessaire. Un volume d'injection d'environ 0,05 à 0,1 ml est recommandé.
XEOMIN est injecté dans la partie interne et externe du muscle orbiculaire de la paupière supérieure et dans la partie externe latérale du muscle orbiculaire de la paupière inférieure. Si des spasmes gênent la vision, le produit peut être injecté dans l'arcade sourcilière, la partie latérale du muscle orbiculaire et la zone supérieure du visage.
En cas de blépharospasme unilatéral, les injections doivent être limitées à l’œil atteint.
Les patients atteints d’un spasme hémifacial doivent être traités de la même manière que pour un blépharospasme unilatéral.
Il n’existe pas d’information d’études cliniques concernant des injections de XEOMIN dans la zone faciale inférieure. Les muscles de la zone faciale inférieure ne doivent pas être injectés en raison du risque prononcé de faiblesse locale rapporté dans la littérature faisant suite à des injections de toxine botulinique dans cette zone chez des patients atteints de spasme hémifacial.
Torticolis spasmodique
Une aiguille stérile appropriée (diamètre de 25-30 Gauge/0,30-0,50 mm et longueur de 37 mm) doit être utilisée pour l'injection dans les muscles superficiels et par exemple, une aiguille de diamètre de 22 Gauge/0,70 mm et de longueur de 75 mm pour des injections dans les muscles plus profonds. Un volume d'injection d'environ 0,1 à 0,5 ml par site d’injection est recommandé.
Pour le traitement du torticolis spasmodique, XEOMIN est injecté dans le sterno-cléido-mastoïdien, le muscle élévateur de l'omoplate, les scalènes, le splénius et/ou les trapèzes. Cette liste n'est pas exhaustive car tous les muscles contrôlant la position de la tête peuvent être concernés et nécessiter un traitement. En cas de difficulté pour isoler les muscles, les injections peuvent être réalisées à l’aide de techniques telles que le guidage électromyographique ou échographique. La masse musculaire et le degré d'hypertrophie ou d'atrophie sont des facteurs à prendre en considération dans le choix de la dose.
La multiplicité des sites d'injection permet une diffusion plus uniforme de XEOMIN dans les zones innervées du muscle dystonique et s'avère particulièrement utile dans les grands muscles. Le nombre optimal de sites d’injection dépend de la taille des muscles à dénerver chimiquement.
Le sterno-cléido-mastoïdien ne doit pas être injecté de façon bilatérale ni recevoir des doses supérieures à 100 U, en raison d’un risque accru d'événements indésirables (en particulier de dysphagie).
Spasticité des membres supérieurs
La solution reconstituée de XEOMIN est injectée au moyen d'une aiguille stérile appropriée (ex : 26 Gauge/0,45 mm de diamètre/37 mm de longueur, pour les muscles superficiels et une aiguille plus longue ex : 22 Gauge/0,7 mm de diamètre/75 mm de longueur, pour les muscles plus profonds).
La localisation des muscles à l’aide de techniques telles que le guidage électromyographique ou l’échographique est recommandée en cas de difficulté pour isoler un muscle. La multiplicité des sites d’injection permet un contact plus uniforme de XEOMIN avec les zones innervées des muscles et s'avère particulièrement utile pour les grands muscles.
Sialorrhée chronique (adultes/enfants/adolescents)
La solution reconstituée de XEOMIN est injectée dans les glandes au moyen d’une aiguille stérile appropriée (par exemple de diamètre 27-30 Gauge/0,30-0,40 mm et de longueur 12,5 mm).
Chez l’adulte, un repérage anatomique ou un guidage échographique sont tous deux possibles pour la localisation des glandes salivaires impliquées, toutefois la méthode de guidage échographique devrait être privilégiée, car elle pourrait aboutir à un meilleur résultat thérapeutique (voir section 5.1).
Pour le traitement chez les enfants et les adolescents, un guidage échographique doit être utilisé. Une anesthésie locale (telle qu’une crème anesthésiante), une sédation ou une anesthésie combinée à une sédation peuvent être proposées aux enfants et aux adolescents avant l’injection après une évaluation minutieuse des bénéfices et des risques et selon la pratique locale du site.
· Hypersensibilité à la substance active ou à l'un des excipients mentionnés à la rubrique 6.1.
· Troubles généralisés de l'activité musculaire (ex: myasthénie grave, syndrome de Lambert-Eaton).
· Infection ou inflammation au site d'injection concerné.
4.4. Mises en garde spéciales et précautions d'emploi
Dans le but d'améliorer la traçabilité des médicaments biologiques, le nom et le numéro de lot du produit administré doivent être clairement enregistrés.
Généralités :
Avant toute administration de XEOMIN, le médecin doit se familiariser à l’anatomie du patient et à toute modification anatomique consécutive à des traitements chirurgicaux antérieurs.
Des précautions doivent être prises afin de s’assurer que XEOMIN n’est pas injecté dans un vaisseau sanguin.
XEOMIN doit être utilisé avec précaution :
· en cas de troubles hémorragiques
· chez les patients sous traitement anticoagulant ou sous un traitement susceptible d’avoir un effet anticoagulant.
Les effets cliniques de la neurotoxine botulinique de type A peuvent varier aux cours des sessions d’injections. Les raisons de ces changements peuvent être en relation avec la technique de reconstitution, les intervalles de traitement choisis, les sites injectés ou l’activité de la toxine qui peut légèrement différer selon la procédure de dosage de l’activité biologique choisie ou une non-réponse secondaire.
Diffusion locale ou à distance de la toxine
Des effets indésirables, liés à une injection de toxine botulinique de type A mal ciblée peut temporairement paralyser les muscles situés à proximité. De fortes doses peuvent causer une paralysie des muscles situés à distance du site d’injection.
Des effets indésirables liés à la diffusion de la toxine botulinique de type A à distance du site d'administration, ont été rapportés (voir rubrique 4.8). Des cas de mise en jeu du pronostic vital et de décès lesquels dans certains cas, étaient associés à une dysphagie, une pneumonie et/ou une faiblesse extrême, ont été rapportés.
Les patients traités par des doses thérapeutiques peuvent présenter une faiblesse musculaire excessive.
Des cas de botulisme iatrogène ont été rapportés après injection de produits contenant de la toxine botulinique.
Les patients et leur entourage doivent être avertis de la nécessité d'une prise en charge médicale immédiate, s’ils présentent des signes ou symptômes compatibles avec la diffusion de l’effet de la toxine botulinique ou en cas de troubles de la déglutition, de troubles de l’élocution ou de troubles respiratoires. (Voir rubrique 4.9).
Des cas de dysphagie ont également été rapportés à la suite d’injections dans d’autres sites que les muscles du cou.
Troubles musculaires préexistants
Les patients souffrant de troubles neuromusculaires sont plus à risque de présenter une faiblesse musculaire, en particulier lorsqu’ils sont traités en intramusculaire. Chez ces patients, la toxine botulinique de type A devra être utilisée sous le contrôle d'un médecin spécialiste et uniquement si le bénéfice du traitement l'emporte sur le risque.
Généralement, les patients ayant des antécédents de pneumopathie d’inhalation et de dysphagie doivent être traités avec précaution, et particulièrement dans le cadre d’une dystonie cervicale.
XEOMIN doit être utilisé avec précaution :
· Chez les patients souffrant de sclérose latérale amyotrophique
· Chez les patients présentant d'autres troubles entraînant un dysfonctionnement neuromusculaire périphérique,
· Dans les muscles cibles qui présentent une faiblesse ou une atrophie prononcée.
Réactions d’hypersensibilité
Des réactions d’hypersensibilité ont été rapportées avec la neurotoxine botulinique de type A. En cas de réaction d’hypersensibilité grave (ex : réaction anaphylactique) et/ou immédiate, un traitement médical approprié doit être instauré.
Formation d’anticorps
Des doses trop fréquentes peuvent augmenter le risque de formation d'anticorps et conduire à un échec thérapeutique (voir rubrique 4.2).
La formation potentielle d’anticorps peut être minimisée par l’administration de la plus petite dose efficace indiquée et ce, dans le respect des intervalles d’injection minimum entre 2 sessions.
Population pédiatrique
Chez les patients pédiatriques présentant des comorbidités, principalement des personnes atteintes de paralysie cérébrale, des notifications spontanées de diffusion à distance de la toxine ont été très rarement rapportées avec d’autres préparations de toxine botulinique de type A. En général, la dose utilisée dans ces cas était supérieure à celle recommandée pour ces produits.
De rares notifications spontanées de décès parfois associés à une pneumopathie d’inhalation chez les enfants atteints de paralysie cérébrale grave après traitement avec des produits à base de toxine botulinique, y compris après une utilisation hors indication (région du cou) ont été signalés.
Le risque est considéré comme particulièrement élevé chez les patients pédiatriques dont l'état de santé sous-jacent est médiocre ou chez les patients présentant une faiblesse neurologique importante, une dysphagie, ou chez les patients ayant des antécédents récents de pneumopathie d’inhalation ou de maladie pulmonaire.
Indications - Mises en garde spéciales
Blépharospasme et spasme hémifacial
Des injections à proximité du muscle releveur de la paupière supérieure doivent être évitées afin de réduire la survenue d’une ptose. Une diplopie peut survenir en cas de diffusion de la neurotoxine botulinique dans le muscle oblique inférieur. Eviter d’injecter dans la partie médiane de la paupière inférieure pour réduire ce risque.
Du fait de l'activité anticholinergique de la neurotoxine botulinique de type A, XEOMIN doit être utilisé avec précaution chez les patients à risque de développer un glaucome par fermeture de l'angle.
Afin de prévenir l'apparition d'un ectropion, des injections dans la paupière inférieure doivent être évitées et, si besoin, un traitement vigoureux en cas de lésion épithéliale doit être initié à base de collyre protecteur, de pommade, de lentilles souples thérapeutiques ou par occlusion de l'œil à l’aide d’un bandeau ou de tout autre moyen similaire.
La diminution du clignement due à l'injection de XEOMIN dans le muscle orbiculaire peut conduire à une exposition prolongée de la cornée, à une lésion épithéliale persistante et à une ulcération de la cornée, en particulier chez les patients ayant présenté des troubles du nerf crânien (nerf facial). Des tests de sensibilité cornéenne doivent être effectués chez les patients ayant été opérés des yeux.
Des ecchymoses se produisent facilement dans les tissus mous de la paupière. L'application d'une légère pression immédiatement après l'injection peut limiter ce risque.
Torticolis spasmodique
XEOMIN doit être injecté avec précaution lors de l’injection au niveau des sites situés à proximité de zones sensibles telles que la carotide, l’apex des poumons et l’œsophage.
Il convient de rappeler aux patients précédemment akinétiques ou sédentaires que la reprise d’activités après l’injection de XEOMIN doit être progressive.
Les patients doivent être informés que les injections de XEOMIN pour le traitement du torticolis spasmodique peuvent provoquer une dysphagie légère à sévère avec un risque de fausses routes et de dyspnée. Des mesures peuvent s'avérer nécessaires comme par exemple la pose d’une sonde d'alimentation gastrique (voir rubrique 4.8). Le risque de survenue d’une dysphagie est minimisé en limitant la dose d’injection à 100 unités maximum dans le muscle sterno-cléido-mastoïdien. Les patients ayant une faible masse musculaire au niveau du cou ou qui nécessitent des injections bilatérales dans le muscle sterno-cléido-mastoïdien présentent des risques plus élevés.
L'apparition d'une dysphagie est liée à l'effet pharmacologique de XEOMIN suite à la diffusion de la neurotoxine dans la musculature œsophagienne.
Spasticité des membres supérieurs
XEOMIN doit être injecté avec précaution lors de l’injection au niveau des sites situés à proximité de zones sensibles telles que la carotide, l’apex des poumons et l’œsophage.
Il convient de rappeler aux patients précédemment akinétiques ou sédentaires que la reprise d’activités après l’injection de XEOMIN doit être progressive.
XEOMIN comme traitement local de la spasticité, a été étudié en association avec les traitements habituels et ne vise pas à les remplacer. XEOMIN n'est pas efficace dans l'amélioration des mouvements au niveau d'une articulation touchée par une contracture musculaire fixe.
L’apparition ou la récurrence de crises convulsives ont été rapportées chez les patients prédisposés. L’imputabilité à la toxine botulinique de type A n’a pas été établie.
Sialorrhée chronique (adultes/enfants/adolescents)
En cas de sialorrhée induite par un médicament (par exemple par aripiprazole, clozapine, pyridostigmine), il faut tout d’abord envisager la possibilité de remplacer, de réduire ou même d’arrêter le médicament concerné avant d’utiliser XEOMIN pour le traitement de la sialorrhée.
L’efficacité et la sécurité de XEOMIN chez les patients atteints de sialorrhée induite par un médicament n’ont pas été étudiées.
Si des cas de sécheresse buccale se développent en association avec l’administration de XEOMIN, une réduction de dose doit être considérée.
Une visite dentaire au début du traitement est recommandée. Le dentiste doit être informé du traitement de la sialorrhée avec XEOMIN pour être capable de prendre les mesures appropriées pour la prophylaxie des caries.
4.5. Interactions avec d'autres médicaments et autres formes d'interactions
Aucune étude d’interactions n’a été réalisée.
Théoriquement, l'effet de la neurotoxine botulinique peut être potentialisé par les aminosides ou par d'autres médicaments qui interfèrent avec la transmission neuromusculaire, tels que les myorelaxants de type tubocurarine.
Par conséquent, l'utilisation concomitante de XEOMIN avec des aminosides ou la spectinomycine nécessite une vigilance particulière. Les myorelaxants périphériques sont à utiliser avec précaution ; au besoin en réduisant la dose de départ du myorelaxant ou en utilisant une substance à action intermédiaire comme le vécuronium ou l'atracurium plutôt que des substances ayant des effets plus durables.
Lors de l’irradiation de la tête et du cou y compris des glandes salivaires, et/ou de la co-administration d’anticholinergiques (par exemple atropine, glycopyrronium, scopolamine) l’effet de XEOMIN peut être augmenté dans le cadre du traitement de la sialorrhée chronique. Le traitement de la sialorrhée avec XEOMIN pendant la radiothérapie n’est pas recommandé.
Les amino-4-quinoléines peuvent réduire l'effet de XEOMIN.
4.6. Fertilité, grossesse et allaitement
Grossesse
Il n'existe pas actuellement de données appropriées sur l'utilisation de la neurotoxine botulinique de type A pendant la grossesse. Les études effectuées chez l'animal ont mis en évidence une toxicité sur la reproduction (voir rubrique 5.3). Le risque potentiel chez la femme n'est pas connu.
Par conséquent, XEOMIN ne doit pas être utilisé pendant la grossesse à moins d'une nécessité absolue et si le bénéfice potentiel justifie le risque.
Il n’existe pas de données quant à l’excrétion de la neurotoxine botulinique de type A dans le lait maternel. Par conséquent, XEOMIN ne doit pas être utilisé pendant l'allaitement.
Fertilité
Il n’existe pas de données cliniques sur l’utilisation de la neurotoxine botulinique de type A. Aucun effet sur la fertilité du mâle ou de la femelle n’a été mis en évidence au cours des études précliniques (voir rubrique 5.3).
4.7. Effets sur l'aptitude à conduire des véhicules et à utiliser des machines
Généralement, les effets indésirables se manifestent au cours de la première semaine suivant le traitement et sont de nature transitoire. Ils sont dus à la substance active, à la technique d’injection ou aux deux.
Effets indésirables indépendants de l’indication.
Effets indésirables liés à l’injection
Des douleurs localisées, une inflammation, des paresthésies, une hypoesthésie, une sensibilité, un gonflement, un œdème, un érythème, un prurit, une infection localisée, un hématome, un saignement et/ou des ecchymoses peuvent apparaître. La douleur liée à la piqûre et/ou à l'anxiété peut induire une réponse vagale, avec hypotension transitoire, nausée, acouphène et syncope.
Effets indésirables liés à la classe
Une faiblesse musculaire localisée est un effet pharmacologique attendu de la toxine botulinique de type A
Diffusion de la toxine
Des effets indésirables liés à une réelle diffusion de la toxine de type A à distance du site d’injection ont été très rarement rapportés (faiblesse musculaire excessive, dysphagie, pneumopathie d’inhalation avec issue fatale dans certains cas) (voir section 4.4).
Réactions d’hypersensibilité
Des réactions d’hypersensibilité graves et/ou immédiates incluant des chocs anaphylactiques, une maladie sérique, une urticaire, un œdème des tissus mous et une dyspnée ont été rarement rapportés. Certaines de ces réactions ont été décrites à la suite d’injection de toxine botulinique de type A conventionnelle soit seule soit en association avec d’autres agents connus pour causer des réactions similaires.
Expérience clinique des effets indésirables
Sur la base des études cliniques, la fréquence des effets indésirables est donnée pour chaque indication. La fréquence est définie comme suit : très fréquent (≥1/10) ; fréquent (≥1/100 à <1/10) ; peu fréquent (≥1/1 000 à <1/100) ; rare (≥1/10 000 à <1/1 000) ; très rare (<1/10 000) ; inconnu (ne peut être estimé sur les données disponibles).
Blépharospasme
|
Réactions indésirables |
Fréquence |
|
|
Affections du système nerveux |
Céphalées, parésie faciale |
Peu fréquent |
|
Affections oculaires |
Ptosis |
Très fréquent |
|
Sécheresse oculaire, vision trouble, trouble de la vision |
Fréquent |
|
|
Diplopie, larmoiement important |
Peu fréquent |
|
|
Affections gastro-intestinales |
Sécheresse buccale |
Fréquent |
|
Dysphagie |
Peu fréquent |
|
|
Affections de la peau et du tissu sous-cutané |
Rash |
Peu fréquent |
|
Affections musculosquelettiques et du tissu conjonctif |
Faiblesse musculaire |
Peu fréquent |
|
Troubles généraux et réactions au site d’administration |
Douleur au site d’injection |
Fréquent |
|
Fatigue |
Peu fréquent |
Spasme hémifacial
Des effets indésirables similaires à ceux du blépharospasme peuvent être attendus avec le spasme hémifacial.
Torticolis spasmodique
|
Réactions indésirables |
Fréquence |
|
|
Infections et infestations |
Infection des voies aériennes supérieures |
Fréquent |
|
Affections du système nerveux |
Céphalées, présyncope, vertiges |
Fréquent |
|
Troubles de l’élocution |
Peu fréquent |
|
|
Affections respiratoires, thoraciques et médiastinales |
Dysphonie, dyspnée |
Peu fréquent |
|
Affections gastro-intestinales |
Dysphagie |
Très fréquent |
|
Sécheresse buccale, nausées |
Fréquent |
|
|
Affections de la peau et du tissu sous-cutané |
Hyperhidrose |
Fréquent |
|
Rash |
Peu fréquent |
|
|
Affections musculosquelettiques et du tissu conjonctif |
Douleur du cou, faiblesse musculaire, myalgie, spasmes musculaires, raideurs musculaires |
Fréquent |
|
Troubles généraux et réactions au site d’administration |
Douleur au site d’injection, asthénie |
Fréquent |
Le traitement du torticolis spasmodique peut causer une dysphagie d’intensité variable avec un risque de fausses routes nécessitant une intervention médicale. La dysphagie peut persister 2 à 3 semaines après l’injection. Un cas de dysphagie persistant jusqu’à cinq mois a été rapporté.
Spasticité des membres supérieurs
|
Réactions indésirables |
Fréquence |
|
|
Affections du système nerveux |
Céphalées, hypoesthésie |
Peu fréquent |
|
Affections gastro-intestinales |
Bouche sèche |
Fréquent |
|
Dysphagie, nausée |
Peu fréquent |
|
|
Affections musculosquelettiques et du tissu conjonctif |
Faiblesse musculaire, douleur des extrémités, myalgie |
Peu fréquent |
|
Troubles généraux et réactions au site d’administration |
Asthénie |
Peu fréquent |
|
Douleur au site d’injection |
Inconnu |
Sialorrhée chronique (adultes)
|
Réactions indésirables |
Fréquence |
|
|
Affections du système nerveux |
Paresthésie |
Fréquent |
|
Trouble de la parole |
Peu fréquent |
|
|
Affections gastro-intestinales |
Sécheresse buccale, dysphagie |
Fréquent |
|
Altération (épaississement) de la salive, dysgueusie |
Peu fréquent |
Des cas de sécheresse buccale persistante (> 110 jours) d’intensité sévère ont été rapportés, ce qui pourrait entraîner d’autres complications comme une gingivite, une dysphagie et des caries.
Sialorrhée chronique (enfants/adolescents)
|
Classes de systèmes d’organes MedDRA |
Réactions indésirables |
Fréquence |
|
Affections gastro-intestinales |
Dysphagie |
Peu fréquent |
|
Altération (épaississement) de la salive, dysgueusie, douleurs buccales, caries dentaires |
Inconnu |
Données après commercialisation
Depuis la mise sur le marché de XEOMIN, les effets indésirables suivants ont été rapportés à une fréquence inconnue :
|
Réactions indésirables |
|
|
Affection du système immunitaire |
Réactions d’hypersensibilité telles que gonflement, œdème (y compris à distance du site d’injection), érythème, prurit, rash (localisé ou généralisé) et des difficultés respiratoires |
|
Affections musculosquelettiques et du tissu conjonctif |
Atrophie musculaire |
|
Troubles généraux et réactions au site d’administration |
Syndromes pseudo-grippaux |
Déclaration des effets indésirables suspectés
La déclaration des effets indésirables suspectés après autorisation du médicament est importante. Elle permet une surveillance continue du rapport bénéfice/risque du médicament. Les professionnels de santé déclarent tout effet indésirable suspecté via le système national de déclaration : Agence nationale de sécurité du médicament et des produits de santé (ANSM) et réseau des Centres Régionaux de Pharmacovigilance - Site internet : https://signalement.social-sante.gouv.fr/.
Veuillez consulter les informations sur les risques associés à une diffusion locale ou à distance de la toxine dans le paragraphe 4.4.
Des doses importantes de toxine botulinique peuvent provoquer, par diffusion à distance du site d’injection, des paralysies neuromusculaires prononcées avec une variété de symptômes. Les symptômes peuvent se traduire par une faiblesse généralisée, un ptosis, une diplopie, des troubles de la respiration ou d’élocution, une paralysie des muscles respiratoires ou des troubles de la déglutition pouvant entraîner une pneumopathie d'inhalation.
Mesures en cas de surdosage
En cas de surdosage ou de diffusion de la toxine, une surveillance médicale des symptômes évocateurs de faiblesse musculaire excessive ou de paralysie musculaire doit être mise en place. Un traitement symptomatique peut être nécessaire et une assistance respiratoire peut être requise en cas de paralysie des muscles respiratoires.
5. PROPRIETES PHARMACOLOGIQUES
5.1. Propriétés pharmacodynamiques
Classe pharmacothérapeutique : autres myorelaxants, agentS à action périphérique, Code ATC : M03AX01.
La neurotoxine botulinique de type A bloque la transmission cholinergique dans la jonction neuromusculaire en inhibant la libération d'acétylcholine. Les terminaisons nerveuses de la plaque motrice ne répondent plus aux impulsions nerveuses empêchant la sécrétion des neurotransmetteurs au niveau de cette plaque motrice (dénervation chimique). La formation de nouvelles terminaisons nerveuses et de plaques motrices permet le rétablissement d'une transmission par impulsion au niveau de la plaque motrice.
Mécanisme d’action
Le mécanisme d'action de la toxine botulinique de type A sur les terminaisons nerveuses cholinergiques peut être décrit par un procédé séquentiel en quatre temps faisant intervenir les étapes suivantes:
· Liaison : la chaîne lourde de la neurotoxine botulinique de type A se fixe de manière hautement sélective et avec une très forte affinité aux récepteurs cholinergiques.
· Internalisation : invagination de la membrane nerveuse terminale et encapsulation de la toxine dans la terminaison nerveuse (endocytose).
· Translocation : la fonction amine terminale de la chaîne lourde de la neurotoxine forme un pore dans la membrane de la vésicule ; le pont disulfure est rompu et la chaîne légère de la neurotoxine passe à travers ce pore dans le cytosol.
· Inhibition : lorsque la chaîne légère est libérée, elle clive de façon très sélective la protéine cible spécifique (SNAP 25) qui est indispensable à la libération d'acétylcholine.
Le rétablissement complet de la fonction motrice/conduction nerveuse intervient normalement dans les 3 à 4 mois qui suivent l’injection intramusculaire, après régénération des terminaisons nerveuses et rétablissement des connexions avec la plaque motrice.
Efficacité et sécurité clinique
L’équivalence thérapeutique d’une dose unique de XEOMIN a été démontrée contre le comparateur Botox (complexe d`onabotulinumtoxin A ou toxine botulinique de type A 900kD) dans deux études comparatives de phase 3, une réalisée chez des patients atteints de blépharospasme (étude MRZ 60201-0003, n=300) et une autre réalisée chez des patients atteints de torticolis spasmodique (étude MRZ 60201-0013, n=463). Les résultats de ces études suggèrent également que XEOMIN et ce comparateur ont des profils d’efficacité et de tolérance comparables chez les patients présentant un blépharospasme ou un torticolis spasmodique pour un ratio de conversion de dose de 1:1 (voir rubrique 4.2)
Blépharospasme
Une étude multicentrique de phase III, randomisée, en double-aveugle contre placebo a été réalisée chez des patients présentant un blépharospasme essentiel bénin (N=109). Les patients présentaient à l’inclusion un sous-score de sévérité ≥ 2 sur l’échelle Jankovic (JRS) avec une réponse clinique satisfaisante et stable à un traitement par Botox(onabotulinumtoxin A) .
Les patients randomisés (2:1) étaient traités avec une dose unique de XEOMIN (n=75) ou de placebo (n=34) à la même posologie (± 10%) que les 2 sessions d’injection par le produit comparateur qui ont précédé l’inclusion. La dose maximale autorisée dans cette étude était de 50 unités par œil avec une moyenne de 32 unités par œil pour XEOMIN.
Le critère principal d’efficacité, défini par la modification du sous-score de sévérité sur l’échelle JRS à 6 semaines par comparaison à la baseline (ITT), a été évalué en intention de traiter (ITT) sachant que les données manquantes étaient remplacées par la dernière donnée disponible du patient. La différence de -1,0 point (IC95%:-1,4;-0,5) du sous-score de sévérité sur l’échelle JRS entre la baseline et à 6 semaines retrouvée entre le groupe XEOMIN et le groupe placebo, est statistiquement significative (p<0,001).
Les patients pouvaient être inclus dans la période d’extension si une nouvelle injection était nécessaire. Les patients ont reçu jusqu’à cinq injections de XEOMIN. Un intervalle minimum d’au moins 6 semaines était requis entre 2 sessions d’injection. La durée totale de l’étude était de 48 à 69 semaines. Au cours de cette étude, les intervalles médians des sujets traités par XEOMIN était compris entre 10,14 (1er intervalle) et 12,00 semaines (du 2nd au 5ème intervalle).
Une autre étude clinique contrôlée de phase III, en double aveugle versus placebo, comportant une phase d’extension ouverte, a été réalisée afin d’évaluer l’efficacité de XEOMIN sur un total de 61 patients, présentant un diagnostic clinique de blépharospasme essentiel bénin avec un sous-score de sévérité ≥ 2 sur la base de l’échelle d’évaluation Jankovic Rating Scale (JRS) et naïfs de traitement par la toxine botulique, c’est-à-dire n’ayant reçu aucun traitement de toxine botulique pour leur blépharospasme depuis au moins 12 mois avant l'administration de XEOMIN. Au cours de la phase principale (6-20 semaines), les patients randomisés étaient traités respectivement avec une dose unique de XEOMIN 12,5 unités par œil (n-22), XEOMIN 25 unités par œil (n-19) ou placebo (n=20). Les patients nécessitant une nouvelle injection pouvaient poursuivre la phase d'extension et recevoir une nouvelle injection de XEOMIN.
Pendant la phase principale, la durée médiane d'intervalle de traitement était de 6 semaines dans le groupe placebo, de 11 semaines dans le groupe traité avec 12,5 unités par œil, et de 20 semaines dans le groupe traité avec 25 unités par œil. La variation du LS moyen d’ANCOVA entre la valeur initiale et la semaine 6 par rapport au placebo (IC 95%) au niveau de l’évolution du sous-score de sévérité de JRS était statistiquement significative, -1,2 (-1,9 ;-0,6) dans le groupe XEOMIN 25 unités par œil alors qu’elle n’était pas statistiquement significative, -0,5 (1,1 ; 0,2) dans le groupe XEOMIN 12,5 unités par œil.
Pendant la phase d'extension, les patients ont reçu une injection de XEOMIN (n-39) à une dose moyenne proche de 25 unités (intervalle : 15-30 unités) par œil et la durée médiane d’'intervalle de traitement était de 19,9 semaines.
Torticolis spasmodique
Une étude multicentrique de phase III, randomisée, en double-aveugle contre placebo a été réalisée chez des patients présentant une dystonie cervicale à prédominance rotationnelle (N=233). Les patients présentaient à l’inclusion un score total ≥ 20 sur l’échelle TWSTRS (Toronto Western Spasmodic Torticolis Rating Score). Les patients randomisés (1:1:1) étaient traités avec une dose unique de XEOMIN 240 unités (n=81), XEOMIN 120 unités (n=78) ou de placebo (n=74). Le nombre et les sites d’injection étaient déterminés par l’investigateur.
Le critère principal d’efficacité, défini par la modification du LS moyen à 4 semaines par comparaison à la baseline (ITT), a été évalué en intention de traiter (ITT) sachant que les données manquantes étaient remplacées par la valeur à la baseline du patient. La modification du score total TWSTRS entre la baseline et la semaine 4 était significativement plus importante dans le groupe XEOMIN que dans le groupe placebo (p<0,001 pour tous les modèles statistiques). Ces différences étaient également significatives sur le plan clinique : -9,0 points entre XEOMIN 240U et le placebo ; -7,5 points entre XEOMIN 120U et le placebo dans ce modèle statistique.
Les patients pouvaient être inclus dans la période d’extension si une nouvelle injection était nécessaire. Les patients ont reçu jusqu’à cinq injections de XEOMIN 120U ou 240U. Un intervalle minimum d’au moins 6 semaines était requis entre 2 sessions d’injection. La durée totale de l’étude était de 48 à 69 semaines.
Basée sur la demande du patient à se faire retraiter, la durée médiane de réponse après le traitement par XEOMIN dans cette étude (à la fois dans la phase en double-aveugle et dans la phase d’extension en ouvert) était de 12 semaines (intervalle interquartile : 9 à 15 semaines). Dans la majorité des cycles d'injection (96.3%), le délai avant une réinjection était compris entre 6 et 22 semaines et dans certains cas pouvait aller jusqu'à 28 semaines
Spasticité des membres supérieurs (adultes)
Dans l'étude pivot multicentrique réalisée en double-aveugle, contre placebo chez des patients présentant une spasticité des membres supérieurs en post-AVC, 148 patients ont été randomisés : XEOMIN (n=73) ou Placebo (n=75). La dose cumulée après traitements répétés jusqu'à 6 fois dans l'étude clinique était en moyenne de 1333 unités (maximum 2395 unités) et ce, pour une période de traitement jusqu’à 89 semaines.
Le critère primaire d'efficacité (taux de réponse des fléchisseurs du poignet au score d'Ashworth à la semaine 4) était défini comme une amélioration d'au moins 1 point sur l'échelle d'Ashworth en 5 points. Les patients traités par XEOMIN (taux de réponse: 68,5%) avaient 3,97 fois plus de chances d'être répondeurs en comparaison aux patients sous placebo (taux de réponse: 37,3% ; IC95% : 1,90-8,30 ; p < 0,001, population en ITT).
Cette étude à dose fixe n'a pas été conçue pour distinguer les patients de sexe féminin et masculin. Néanmoins, dans une analyse post-hoc, les taux de réponse étaient plus élevés chez la femme (89,3%) par rapport à l'homme (55,6%), la différence étant statistiquement significative pour les femmes seulement. Toutefois, les taux de réponse des hommes sur l'échelle d'Ashworth après 4 semaines de traitement par XEOMIN, étaient plus élevés dans tous les groupes musculaires traités comparativement au placebo. Basée sur la demande du patient de se faire retraiter, la durée médiane de l’effet dans cette étude pivot suivie par la période d’extension en ouvert était de 14 semaines (intervalles interquartiles : 13 à 17 semaines) avec, pour la majorité des cycles d’injection (95,9%), un intervalle compris entre 12 et 28 semaines.
Les taux de réponse étaient comparables chez l'homme et chez la femme au cours de la période de prolongation en ouvert de l'étude pivot (doses variables autorisées au cours de cette période), pour laquelle 145 patients ont été inclus et ont reçu jusqu'à 5 sessions d'injection, ainsi que dans l'étude en simple aveugle (EudraCT N°2006-003036-30) où l'efficacité et la sécurité de XEOMIN, à deux dilutions différentes, ont été évaluées chez 192 patients présentant une spasticité des membres supérieurs d'étiologies diverses.
Une autre étude clinique contrôlée de phase III, réalisée en double aveugle contre placebo, a inclus 317 patients, naïfs de tout traitement, présentant une spasticité du membre supérieur post AVC depuis au moins 3 mois. Au cours de la phase principale (PP), une dose totale de 400 unités de XEOMIN a été injectée par voie intramusculaire en fonction de l’objectif clinique principal choisi entre la flexion du coude, la flexion du poignet ou le poing serré, ainsi que dans tout autre groupe musculaire atteint (n=210). L’analyse du critère principal et de l’ensemble des critères d’efficacité à 4 semaines ont démontré une amélioration significative du taux de répondeurs (score d’Ashworth), du score d’Ashworth versus baseline et de l’impression globale de l’investigateur.
296 patients ayant finalisé la PP ont été inclus dans la phase d’extension en ouvert (OLEX) et ont reçu jusqu’à 3 injections à chaque cycle. Les patients étaient traités avec une seule dose totale de 400 unités de XEOMIN répartie à l’ensemble des muscles affectés puis suivis 12 semaines. La durée totale de l’étude était de 48 semaines.
Une étude de phase III en ouvert, évaluant, entre autres, le traitement des muscles de l’épaule, a inclus 155 patients présentant une spasticité des membres supérieurs et des membres inférieurs. Des doses jusqu’à 600 unités de XEOMIN dans le membre supérieur pouvaient être administrées. Cette étude a montré une corrélation positive entre l’augmentation des doses de XEOMIN et l’amélioration des patients ; amélioration objectivée par le score d’Ashworth et par d’autres variables d’efficacité ; sans modification de la sécurité des patients ou du profil de tolérance de XEOMIN.
Spasticité des membres inférieurs et supérieurs due à une paralysie cérébrale (enfants / adolescents)
Membres inférieurs
Dans une étude clinique de phase III en double aveugle, dose-effet en groupes parallèles, incluant 311 enfants (âgés de 2 à 17 ans) présentant une spasticité uni ou bilatérale des membres inférieurs due à une paralysie cérébrale. Pour le traitement de la spasticité des membres inférieurs, XEOMIN a été administré dans les trois groupes de traitement suivants (4 unités / Kg poids corporel avec un maximum de 100 unités, 12 unités / kg de poids corporel avec un maximum de 300 unités ou 16 unités / kg de poids corporel avec un maximum de de 400 unités) pour le traitement des deux schémas cliniques sélectionnés des membres inférieurs (pied équin, genou fléchi, adduction de cuisse).
Dans cette étude, le groupe ayant reçu une dose faible devait servir de groupe témoin. Aucune différence statistiquement significative n’a été démontrée entre la dose faible et la dose forte, ni sur le critère principal ni sur le co-critère principal d’efficacité. Le changement du LS-Mean (SE, IC 95%) entre la valeur initiale et la valeur à la semaine 4 après injection du score de l’échelle d'Ashworth du fléchisseur plantaire était pour la dose forte de -0,70 (0,061, IC 95% ; -0,82 ; -0,58) et pour la dose faible -0,66 (0.084, IC 95% : -0,82 ; -0,50) avec une valeur de p de 0.650. L’amélioration du tonus musculaire ne se traduisait pas par un effet sur la fonction ou sur l’échelle d’impression globale des investigateurs. La posologie adéquate de XEOMIN pour le traitement de la spasticité des membres inférieurs chez les enfants et les adolescents ne peut pas être déterminée. Aucun événement indésirable inattendu n'a été observé lors du traitement en double aveugle et du traitement en ouvert à long terme avec XEOMIN sur quatre cycles d'injection.
Membres supérieurs
Une deuxième étude clinique de phase III en double aveugle, dose-effet en groupe parallèle, incluant 350 enfants et adolescents (âgés de 2 à 17 ans) atteints de spasticité des membres supérieurs seuls ou associés à une spasticité des membres supérieurs et inférieurs, due à une paralysie cérébrale traités par XEOMIN.
Concernant le traitement du membre supérieur (fléchissement du coude, du poignet, du poing, de l'avant-bras en pronation, du pouce dans la paume) ou de la spasticité combinée des membres supérieurs et des membres inférieurs (pied équin, genou fléchi, adduction de cuisse), lors de la phase principale (PP), XEOMIN a été administré en trois groupes avec un cycle d'injection: 2 à 5 unités / kg de poids corporel avec un maximum de 50 à 125 unités, 6 à 15 unités / kg de poids corporel avec un maximum de 150 à 375 unités et 8 à 20 unités / kg poids corporel avec un maximum de 200 à 500 unités. Les patients ont poursuivi lors de la phase d’extension avec 3 cycles d’injection à la dose la plus élevée.
Une différence statistiquement significative entre les doses faible et élevée a été observée dans le changement par rapport à la valeur initiale dans l'échelle d'Ashworth pour le fléchisseur du coude ou du poignet à la semaine 4 après l'injection -0,22 (0,091; IC à 95%: -0,40; -0,04) p = 0,017. Les améliorations du tonus musculaire ne se sont pas traduites par un effet sur la fonction et sur l'échelle de l'impression globale du changement des investigateurs. La posologie adéquate de XEOMIN pour le traitement de la spasticité des membres supérieurs chez les patients pédiatriques ne peut donc pas être déterminée à partir de cette étude.
Aucun problème de tolérance inattendue n'a été signalé dans le traitement de la spasticité des membres supérieurs et inférieurs avec XEOMIN jusqu'à quatre cycles d'injection (14 ± 2 semaines chacun).
Sialorrhée chronique (adultes)
L’étude pivot de phase III en double aveugle, contre placebo a été réalisée chez des patients (n=184) présentant une sialorrhée de plus de trois mois résultant d’une maladie de Parkinson, d’un parkinsonisme atypique, d’un accident vasculaire cérébral ou d’un traumatisme crânien.
Au cours de la phase principale (PP), une dose totale de XEOMIN (100 ou 75 unités) ou de placebo a été injectée en intraglandulaire à un ratio de dose défini de 3:2 dans les glandes salivaires parotides et sous-maxillaires respectivement.
|
|
|
uSFR (g/min) |
GICS (score points) |
||
|
Traitement |
Instant |
n obs |
LS mean (SE) |
n obs |
LS mean (SE) |
|
Placebo |
Semaine 4 |
36 |
-0.04 (0.033) |
36 |
0.67 (0.186) |
|
100 unités |
Semaine 4 |
73 |
-0.13 (0.026) |
74 |
1.25 (0.144) |
|
100 unités |
Semaine 8 |
73 |
-0.13 (0.026) |
74 |
1.30 (0.148) |
|
100 unités |
Semaine 12 |
73 |
-0.12 (0.026) |
74 |
1.21 (0.152) |
|
100 unités |
Semaine 16 |
73 |
-0.11 (0.027) |
74 |
0.93 (0.152) |
|
uSFR : Unstimulated Salivary Flow Rate ; GICS : Global Impression of Change Scale ; n obs : nombre observé ; LS (Least Squares): Moyenne des moindres carrés par rapport à la baseline ; SE : Standard Error |
|||||
À la quatrième semaine, une amélioration d’au moins 1 point sur le GICS (co-critère principal d’évaluation) a été observée chez 73% des patients traités par 100 unités de XEOMIN comparativement à 44% des patients du groupe placebo. L’analyse de suivi d’efficacité des deux co-critères principaux (uSFR et GICS à la semaine 4 post injection) a démontré des améliorations statistiquement significatives du groupe traité par des unités de 100 en comparaison avec le placebo. Les améliorations des paramètres d’efficacité aux semaines 8 et 12 après l’injection ont pu être démontrées et ce jusqu’à la fin de la PP à la semaine 16. Les variables des co-critères principaux d’efficacité à la semaine 4 ont montré des résultats supérieurs pour le guide échographique par ultrasons en comparaison avec la méthode de repérage anatomique (pour uSFR valeur de p 0.019 vs 0,099 et pour GICS valeur de p 0,003 vs 0,171).
173 patients traités ont terminé la PP et ont été inclus dans la phase d’extension (PE). La PE consistait en trois cycles de doses administrées en aveugle, chacun avec une seule séance de traitement (dose totale de 100 ou 75 unités de XEOMIN, avec le même ratio de dose que dans la PP) suivis d’une période d’observation de 16 semaines. 151 patients ont terminé la PE.
Les résultats de la PE confirment les conclusions de la PP en montrant les améliorations continues du traitement de 100 unités XEOMIN.
Sialorrhée chronique (enfants/adolescents)
Dans un essai clinique de phase III en double aveugle, contrôlé versus placebo, un total de 255 enfants et adolescents (âgés de 2 à 17 ans) ayant un poids corporel (PC) d'au moins 12 kg et souffrant de sialorrhée chronique associée à des troubles neurologiques et/ou à une déficience intellectuelle ont été traités. Au cours de la phase principale (PP), 220 patients âgés de 6 à 17 ans ont reçu un traitement par XEOMIN selon la classe de poids corporel et ce jusqu'à 75 U, ou un placebo. Le traitement a été administré par voie intraglandulaire sous repérage échographique à un ratio de dose défini de 3:2 dans les glandes salivaires parotides et sous-maxillaires, respectivement.
|
|
|
uSFR (g/min) |
GICS (score points) |
||
|
Traitement |
Instant |
n obs |
LS mean (SE) |
n obs |
LS mean (SE) |
|
Placebo |
Semaine 4 |
72 |
-0.07 (0.015) |
72 |
0.63 (0.104) |
|
XEOMIN selon la classe BW |
Semaine 4 |
148 |
-0.14 (0.012) |
148 |
0.91 (0.075) |
|
Semaine 8 |
146 |
-0.16 (0.012) |
146 |
0.94 (0.068) |
|
|
Semaine 12 |
147 |
-0.16 (0.013) |
147 |
0.87 (0.073) |
|
|
Semaine 16 |
145 |
-0.15 (0.013) |
146 |
0.77 (0.070) |
|
|
uSFR: Unstimulated Salivary Flow Rate; GICS: Global Impression of Change Scale; BW: Body Weight; n obs: Nombre observé; LS (Least square): Moyenne des moindres carrés par rapport à la baseline; SE: Standard Error |
|||||
L’analyse des deux co-critères d’efficacité (uSFR et GICS à la semaine 4 post injection) a démontré des améliorations statistiquement significatives et cliniquement pertinentes du groupe XEOMIN par rapport au placebo. Pour ces deux critères d’efficacité, des différences statistiquement significatives entre les groupes de traitement ont été observées jusqu'à la fin de la PP à la semaine 16.
Les 35 enfants âgés de 2 à 5 ans ont été traités avec XEOMIN selon leur classe BW, aucun bras placebo n'a été utilisé comme contrôle afin de montrer une amélioration des variables d'efficacité étudiées similaire à celle observée dans le groupe de traitement XEOMIN 6-17 ans.
247 patients ont participé au premier cycle de la phase d'extension en ouvert (OLEX). L'OLEX consistait en trois cycles supplémentaires, chacun avec une seule séance de traitement suivie d'une période d'observation de 16 semaines. Tous les patients ont reçu XEOMIN selon le même schéma posologique prédéterminé et le même ratio de dose que celui utilisé dans la PP. Au total, 222 patients ont participé à l’OLEX. Les résultats de la phase OLEX ont confirmé les conclusions de la PP, montrant des améliorations continues du traitement. Aucun effet indésirable nouveau ou inattendu n’a été identifié.
Population pédiatrique
L’agence européenne des médicaments a accordé une dérogation à l’obligation de soumettre les résultats d’études réalisées avec XEOMIN :
· dans tous les sous-groupes de la population pédiatrique dans le traitement de la dystonie
· dans la population pédiatrique entre 0 et 24 mois dans le traitement de la spasticité des muscles et de la sialorrhée chronique.
Voir la rubrique 4.2 pour des informations sur l’utilisation pédiatrique.
5.2. Propriétés pharmacocinétiques
Caractéristiques générales de la substance active
Les études de distribution et de cinétique classiques ne peuvent pas être effectuées avec la neurotoxine botulinique de type A car la substance active est administrée en très petites quantités (picogrammes par injection) et se lie rapidement et de manière irréversible aux terminaisons nerveuses cholinergiques.
La toxine botulinique naturelle de type A est un complexe de haut poids moléculaire qui, en plus de la neurotoxine (150 kD), contient d'autres protéines non toxiques hémagglutinantes et non hémagglutinantes. Contrairement à la toxine botulinique de type A conventionnelle, XEOMIN contient la neurotoxine pure (150 kD) puisque celle-ci est dénuée de protéines complexantes et de ce fait, contient peu de protéines étrangères. La teneur en protéines étrangères administrée est considérée comme l’un des facteurs d’échec secondaire au traitement.
La neurotoxine botulinique de type A a montré qu'elle subissait un transport axonal rétrograde après injection intramusculaire. Le passage transsynaptique rétrograde de la neurotoxine botulinique active de type A dans le système nerveux central n'a cependant pas été observé aux doses thérapeutiques préconisées.
La neurotoxine botulinique de type A liée aux récepteurs membranaires est endocytosée dans la terminaison nerveuse avant d'atteindre sa cible (SNAP 25) puis est dégradée par voie intracellulaire. Les molécules de la neurotoxine botulinique de type A circulant librement, qui n'ont pas été liées aux récepteurs des terminaisons nerveuses cholinergiques présynaptiques, subissent un processus de phagocytose ou de pinocytose et sont dégradées comme toute autre protéine circulant librement.
Distribution
En raison de la nature du produit, aucune étude cinétique n'a été conduite chez l'homme.
5.3. Données de sécurité préclinique
Les conclusions des études de toxicité à doses répétées sur la toxicité systémique de XEOMIN après injection intramusculaire chez l’animal telles qu’atonie, parésie et atrophie du muscle injecté, étaient essentiellement liées à l'activité pharmacodynamique.
De même, le poids de la glande salivaire sous-maxillaire injectée a été réduit à toutes les doses, et une atrophie acineuse des glandes salivaires a été observée à la dose la plus élevée de 40 unités/kg après quatre injections répétées de XEOMIN à 8 semaines d’intervalle chez le rat.
Aucun signe d'intolérance locale n'a été noté. Les études de toxicité sur la reproduction réalisées avec XEOMIN chez le lapin n’ont pas montré de toxicité sur la fertilité du mâle ou de la femelle ni d’effet direct sur le développement embryo-fœtal ou sur le développement pre- et post-natal chez le rat et/ou le lapin. Cependant des études d’embryotoxicité, après administration quotidienne, hebdomadaire ou bihebdomadaire de XEOMIN à une femelle, à des doses exposant la femelle à une perte de poids ; une augmentation du nombre d'avortements chez le lapin et une légère diminution du poids du fœtus chez le rat ont été observées. Une exposition systémique en continu de femelles gravides pendant la phase sensible (inconnue) de l'organogenèse comme pré-requis pour l'induction d'effets tératogènes ne peut pas nécessairement être envisagée dans ces études.
Dans une étude de toxicité juvénile post-sevrage chez le rat, une atrophie de l'épithélium germinal testiculaire et une hypospermie ont été observées à la dose la plus élevée testée (30 unités / kg / adm) sans aucune incidence sur la fertilité masculine. Lorsque les mâles et les femelles ont été appariés à l'âge de 14 semaines, la performance d'accouplement a été réduite chez les mâles exposés à la dose élevée, probablement en raison de la faiblesse du membre ou du poids corporel nettement inférieur. En l'absence de tout effet sur le nombre moyen de corps jaunes, la perte préimplantatoire était augmentée à 10 unités / kg / adm et plus. Que cette constatation soit un effet induit pour l'homme ou pour la femme n'a pas pu être clarifié de manière concluante.
En conséquence, les marges de sécurité cliniques étaient généralement étroites à fortes doses.
Aucune étude de génotoxicité ou de carcinogénicité n'a été conduite avec XEOMIN.
Saccharose.
Flacon fermé: 3 ans
Solution reconstituée : la stabilité physique et chimique en cours d’utilisation a été démontrée pendant 24 heures entre 2°C et 8°C. D'un point de vue microbiologique, le produit doit être utilisé immédiatement. Dans le cas contraire, la durée et les conditions de conservation de la solution reconstituée relèvent de la responsabilité de l’utilisateur et ne devraient pas excéder 24 heures à une température comprise entre +2°C et +8°C, à moins que la reconstitution n’ait été effectuée dans des conditions d’asepsie maitrisées et validées.
6.4. Précautions particulières de conservation
A conserver à une température ne dépassant pas 25°C.
Pour les conditions de conservation après reconstitution du médicament, voir rubrique 6.3.
6.5. Nature et contenu de l'emballage extérieur
Flacon (verre de type 1), muni d’un bouchon (caoutchouc bromobutyle) et d’une bague de sécurité (aluminium).
Boîtes de 1, 2, 3 ou 6 flacons, chacun contenant 50 unités.
Toutes les présentations peuvent ne pas être commercialisées.
6.6. Précautions particulières d’élimination et de manipulation
Reconstitution
XEOMIN est reconstitué avant utilisation avec une solution injectable de chlorure de sodium à 9 mg/ml (0,9 %). La reconstitution et la dilution doivent être réalisées selon les bonnes pratiques cliniques, particulièrement dans le respect de l'asepsie.
Il est de bonne pratique de reconstituer le produit et préparer les seringues sur des supports papier doublés de plastique pour recevoir tout déversement accidentel. Un volume approprié de solution de chlorure de sodium est aspiré dans une seringue (voir tableau de dilution). Une aiguille courte en biseau de 20-27 Gauge est recommandée pour la reconstitution. Après insertion verticale de l’aiguille à travers le bouchon en caoutchouc, le solvant est injecté délicatement dans le flacon afin d’éviter la formation de mousse. Si la dépression n'entraîne pas l'aspiration du solvant à l'intérieur du flacon, le flacon doit être jeté. La seringue doit être retirée du flacon et XEOMIN doit être mélangé délicatement au solvant par des mouvements circulaires et par retournement du flacon. La solution ne doit pas être mélangée vigoureusement.
Si besoin, l’aiguille ayant servi à la reconstitution de la solution peut rester dans le flacon. Le volume de produit nécessaire est aspiré avec une nouvelle seringue stérile adaptée à l’injection.
|
|
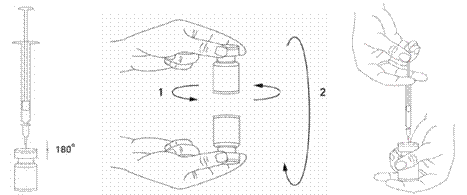
La solution reconstituée de XEOMIN est limpide et incolore.
XEOMIN ne doit pas être utilisé si la solution reconstituée présente un aspect trouble ou si elle contient des particules ou des matières floconneuses.
Afin de prévenir tout surdosage accidentel, il convient de bien s’assurer lors de la reconstitution du produit que le volume de solvant est adapté à la présentation choisie. Si différentes présentations sont utilisées au cours d’une même séance d’injection, il convient de s’assurer que le volume de solvant utilisé lors de la reconstitution du produit donne le bon nombre d’unités par 0,1ml. Le volume de solvant à ajouter diffère entre XEOMIN 50 unités, XEOMIN 100 unités et XEOMIN 200 unités. Chaque seringue utilisée doit être identifiée en conséquence.
Les concentrations possibles pour XEOMIN 50 unités sont indiquées dans le tableau suivant :
|
Dose obtenue (unités par 0,1 ml) |
Solvant ajouté (solution injectable de chlorure de sodium à 9 mg/ml (0,9%)) |
|
|
20 unités |
0,25ml |
|
|
10 unités |
0,5 ml |
|
|
8 unités |
0,625 ml |
|
|
5 unités |
1 ml |
|
|
4 unités |
1,25 ml |
|
|
2,5 unités |
2 ml |
|
|
2 unités |
2,5 ml |
|
|
1,25 unités |
4 ml |
|
Toute solution injectable qui a été conservée pendant plus de 24 heures et toute fraction de solution injectable restante ou non utilisée doivent être jetées.
Procédure à suivre pour une élimination en toute sécurité des flacons, des seringues et matériels utilisés.
Tous flacons non utilisés ou contenant des solutions reconstituées résiduelles et/ou les seringues peuvent être autoclavés.
Il est également possible d’inactiver toute fraction restante de XEOMIN par addition d’éthanol à 70%, d’isopropanol à 50%, de lauryl sulfate de sodium à 0,1% (détergent anionique), d’une solution diluée d'hydroxyde de sodium (NaOH 0,1 N) ou d’une solution diluée d'hypochlorite de sodium (NaOCl à au moins 0,1%).
Les flacons utilisés, seringues et matériels ne doivent pas être vidés mais doivent être jetés dans des récipients adaptés et éliminés conformément à la règlementation locale en vigueur.
Recommandations en cas d'incident lors de la manipulation de la toxine botulinique de type A
· Toute projection doit être essuyée avec un matériel absorbant imbibé d'une des solutions listées ci-dessus pour la poudre ou bien avec un matériel absorbant sec pour le produit reconstitué.
· Les surfaces contaminées sont nettoyées avec un matériel absorbant, imbibé d'une des solutions listées ci-dessus puis séchées.
· Si le flacon est brisé, il faut procéder comme indiqué ci-dessus au ramassage méticuleux des particules de verre et essuyer le produit, en évitant toute coupure cutanée.
· En cas de contact avec la peau, la zone touchée doit être rincée abondamment à l'eau.
En cas de contact avec les yeux, ils doivent être abondamment rincés avec de l'eau ou avec une solution pour lavage ophtalmique.
En cas de contact du produit avec une blessure, une coupure ou une piqûre, la peau doit être abondamment rincée avec de l'eau. Les mesures médicales appropriées en fonction de la dose injectée doivent être prises.
Ces instructions d'utilisation, de manipulation et d'élimination doivent être scrupuleusement suivies.
7. TITULAIRE DE L’AUTORISATION DE MISE SUR LE MARCHE
ECKENHEIMER LANDSTRASSE 100
60318 FRANKFURT / MAIN
ALLEMAGNE
8. NUMERO(S) D’AUTORISATION DE MISE SUR LE MARCHE
· 34009 580 884 7 6 : 50 unités de neurotoxine de Clostridium botulinum de type A en flacon (verre de type I), muni d'un bouchon (caoutchouc bromobutyle) et d'une bague de sécurité (aluminium), boîte de 1 flacon.
· 34009 580 885 3 7 : 50 unités de neurotoxine de Clostridium botulinum de type A en flacon (verre de type I), muni d'un bouchon (caoutchouc bromobutyle) et d'une bague de sécurité (aluminium), boîte de 2 flacons.
· 34009 580 887 6 6 : 50 unités de neurotoxine de Clostridium botulinum de type A en flacon (verre de type I), muni d'un bouchon (caoutchouc bromobutyle) et d'une bague de sécurité (aluminium), boîte de 3 flacons.
· 34009 580 888 2 7 : 50 unités de neurotoxine de Clostridium botulinum de type A en flacon (verre de type I), muni d'un bouchon (caoutchouc bromobutyle) et d'une bague de sécurité (aluminium), boîte de 6 flacons.
9. DATE DE PREMIERE AUTORISATION/DE RENOUVELLEMENT DE L’AUTORISATION
[à compléter ultérieurement par le titulaire]
10. DATE DE MISE A JOUR DU TEXTE
[à compléter ultérieurement par le titulaire]
Sans objet.
12. INSTRUCTIONS POUR LA PREPARATION DES RADIOPHARMACEUTIQUES
Liste I.
Médicament réservé à l'usage hospitalier.
ANSM - Mis à jour le : 10/03/2026
XEOMIN 50 unités, poudre pour solution injectable
Neurotoxine de Clostridium botulinum de type A (150 kD), sans protéines complexantes
· Gardez cette notice. Vous pourriez avoir besoin de la relire.
· Si vous avez d’autres questions, interrogez votre médecin ou votre pharmacien.
· Ce médicament vous a été personnellement prescrit. Ne le donnez pas à d’autres personnes. Il pourrait leur être nocif, même si les signes de leur maladie sont identiques aux vôtres.
· Si vous ressentez un quelconque effet indésirable, parlez-en à votre médecin ou votre pharmacien. Ceci s’applique aussi à tout effet indésirable qui ne serait pas mentionné dans cette notice. Voir rubrique 4.
1. Qu'est-ce que XEOMIN 50 unités, poudre pour solution injectable et dans quels cas est-il utilisé ?
2. Quelles sont les informations à connaître avant d'utiliser XEOMIN 50 unités, poudre pour solution injectable?
3. Comment utiliser XEOMIN 50 unités, poudre pour solution injectable ?
4. Quels sont les effets indésirables éventuels ?
5. Comment conserver XEOMIN 50 unités, poudre pour solution injectable ?
6. Contenu de l’emballage et autres informations.
1. QU’EST-CE QUE XEOMIN 50 unités, poudre pour solution injectable ET DANS QUELS CAS EST-IL UTILISE ?
Classe pharmacothérapeutique : M03AX01
XEOMIN 50 unités, poudre pour solution injectable est un médicament qui contient comme principe actif la Neurotoxine Botulique de Type A qui détend les muscles injectés ou qui diminue le flux salivaire en fonction du site d’administration.
XEOMIN 50 unités, poudre pour solution injectable est utilisé chez l'adulte pour le traitement :
· du spasme des paupières (blépharospasme) et du spasme touchant un côté du visage (spasme hémifacial),
· du torticolis spasmodique,
· de l’hyperactivité musculaire ou de la raideur incontrôlable des muscles de l’épaule, du bras et/ou de la main. (spasticité des membres supérieurs)
· de l’excès de salive (sialorrhée) en raison de troubles neurologiques.
XEOMIN est indiqué dans le traitement symptomatique chez les enfants et adolescents âgés de 2 à 17 ans et pesant ≥ 12kg de
· l’excès de salive (sialorrhée) en raison de troubles neurologiques / troubles neurologiques du développement.
2. QUELLES SONT LES INFORMATIONS A CONNAITRE AVANT D’UTILISER XEOMIN 50 unités, poudre pour solution injectable ?
N’utilisez jamais XEOMIN 50 unités, poudre pour solution injectable :
· si vous êtes allergique à la neurotoxine botulinique de type A ou à l'un des autres composants contenus dans ce médicament mentionnés dans la rubrique 6 ;
· si vous souffrez de troubles généralisés de l'activité musculaire (par ex: myasthénie grave, syndrome de Lambert-Eaton);
· si vous présentez une infection ou une inflammation au niveau du site d'injection concerné.
Avertissements et précautions
En cas d’injection mal ciblée de neurotoxine botulinique de type A, des effets indésirables paralysant temporairement les muscles à proximité du site d’injection et à un botulisme peuvent être observés. Ces effets indésirables en relation avec une réelle diffusion de la toxine à distance du site d’injection ont été très rarement rapportés (par exemple : vision double, vision floue et/ou paupières tombantes, troubles de la parole ou de la respiration faiblesse musculaire excessive, difficultés ou troubles de la déglutition à l’origine de fausses routes de boissons ou de nourriture dans les voies aériennes). Les patients traités aux doses recommandées peuvent présenter une faiblesse musculaire excessive.
Si la dose est trop forte ou si les injections trop fréquentes, le risque de formation d’anticorps peut être augmenté. La formation d’anticorps peut être à l’origine d’un échec au traitement par toxine botulinique de type A, et ce, quelle que soit son indication.
Adressez-vous à votre médecin ou votre pharmacien avant tout traitement par XEOMIN 50 unités, poudre pour solution injectable:
· si vous souffrez de tout type de trouble hémorragique ;
· si vous recevez des substances qui empêchent la coagulation du sang telles que coumariniques, héparine, aspirine ou clopidogrel ;
· si vous souffrez de faiblesse prononcée ou d'une diminution de volume du muscle dans lequel les injections sont effectuées ;
· si vous souffrez de sclérose latérale amyotrophique (SLA) pouvant entraîner une fonte musculaire généralisée ;
· si vous souffrez d'autres maladies qui atteignent les nerfs et les muscles squelettiques (dysfonctionnement neuromusculaire périphérique) ;
· si vous avez ou avez eu des difficultés pour avaler ;
· si vous souffrez ou avez souffert de convulsions
· si vous avez rencontré des problèmes dans le passé avec des injections de toxine botulinique de type A ;
· si vous allez être opéré.
Contactez immédiatement votre médecin pour une surveillance médicale si vous présentez l’un des symptômes suivants :
· des difficultés à respirer, à déglutir, ou à parler.
· en cas d’urticaire, de gonflement, y compris de la face ou de la gorge, de sifflement, de sensation d’évanouissement, d’essoufflement (symptômes possibles d’une réaction allergique sévère).
Injections répétées de XEOMIN 50 unités, poudre pour solution injectable
En cas d'injections répétées, l'effet thérapeutique de XEOMIN 50 unités, poudre pour solution injectable peut varier. Les raisons possibles d'une augmentation ou d’une diminution de l’efficacité peuvent résulter :
· de différences dans la technique de reconstitution de la solution par votre médecin ;
· de différences dans les intervalles de traitement ;
· d’injections dans d'autres muscles ;
· de très faibles différences d'efficacité de la substance active de XEOMIN 50 unités, poudre pour solution injectable;
· de l'absence de réponse ou d’un échec thérapeutique.
Spasme des paupières (blépharospasme) et spasme touchant un côté du visage (spasme hémifacial)
Veuillez informer votre médecin avant tout traitement par XEOMIN 50 unités, poudre pour solution injectable, si vous :
· avez subi une opération chirurgicale des yeux. Votre médecin prendra les précautions nécessaires.
· présentez un risque de développer une maladie appelée glaucome par fermeture de l'angle. Cette maladie peut causer une augmentation de la pression intraoculaire et détériorer le nerf optique. Votre médecin saura si vous présentez ce risque.
Au cours du traitement, des petits points hémorragiques peuvent se produire au niveau de la paupière. Votre médecin peut les limiter en exerçant immédiatement une légère pression sur le site d'injection.
Après injection de XEOMIN 50 unités, poudre pour solution injectable dans les muscles autour de votre œil, le clignement de la paupière peut être ralenti. Ceci peut conduire à une exposition prolongée d’une partie de l'œil (la cornée). Cette exposition peut abimer la surface de l’œil et créer une inflammation (ulcération de la cornée).
Torticolis spasmodique
Vous pouvez ressentir des difficultés à avaler de sévérité variable. Ceci peut provoquer des difficultés pour respirer et des risques plus élevés de fausses routes. Des substances étrangères inhalées dans vos poumons peuvent créer une inflammation ou une infection (pneumonie). Votre médecin vous donnera un traitement médical spécial si cela s'avère nécessaire (par ex sous la forme d'une alimentation artificielle).
Les difficultés à avaler peuvent durer jusqu'à deux à trois semaines après l'injection (jusqu'à cinq mois pour un patient).
Si vous avez été inactif pendant longtemps, toute activité doit être reprise progressivement après l'injection de XEOMIN 50 unités, poudre pour solution injectable.
Hyperactivité musculaire ou raideur incontrôlable des muscles
XEOMIN 50 unités, poudre pour solution injectable peut être utilisé pour traiter une hyperactivité musculaire ou une raideur incontrôlable de certaines parties des membres supérieurs tels que le bras ou la main. XEOMIN 50 unités, poudre pour solution injectable est efficace en association avec des traitements standards. XEOMIN 50 unités, poudre pour solution injectable devrait être utilisé en association à ces autres traitements.
Il est improbable que ce médicament améliore les mouvements des articulations si le muscle les entourant a perdu sa capacité d’élongation.
Si vous avez été inactif pendant longtemps, toute activité doit être reprise progressivement après l'injection de XEOMIN 50 unités, poudre pour solution injectable.
Excès de salive (Sialorrhée)
Certains médicaments (clozapine, aripiprazole, pyridostigmine) peuvent entraîner une production excessive de salive. Pour commencer, la possibilité de remplacement, de réduction ou même d’arrêt du médicament responsable doit être envisagée avant l’utilisation de XEOMIN comme traitement de l’excès de salive. L’utilisation de XEOMIN pour réduire la salivation induite par les médicaments n’a pas été étudiée.
Si des cas de sécheresse buccale se développent en association avec l’administration de XEOMIN, votre médecin considérera une réduction de la dose.
Lorsque votre débit de salive est réduit par XEOMIN, des problèmes de santé bucco-dentaire tels que les caries dentaires peuvent se développer ou des problèmes existants peuvent progresser. Contactez un dentiste lorsque vous commencez à utiliser XEOMIN pour le traitement de l’excès de salive. Si nécessaire, votre dentiste peut décider de prendre des mesures pour la prévention des caries.
Enfants et adolescents
Ne pas administrer ce médicament aux enfants de moins de 2 ans, aux enfants pesant moins de 12kg et aux enfants et adolescents pour des traitements autres que l’excès de salive car l’utilisation de XEOMIN n’a pas été établie dans cette population et n’est donc pas recommandée.
Autres médicaments et XEOMIN 50 unités, poudre pour solution injectable
Si vous prenez, avez pris récemment ou pourriez prendre tout autre médicament, informez votre médecin ou votre pharmacien.
L’effet de XEOMIN 50 unités, poudre pour solution injectable peut être augmenté:
· par des médicaments indiqués pour traiter certaines infections tels que la spectinomycine ou les antibiotiques du groupe des aminosides (ex : néomycine, kanamycine, tobramycine) ;
· par d’autres médicaments qui relâchent les muscles, tels que les myorelaxants de type tubocurarine. Ces produits sont, par exemple, utilisés lors d’une anesthésie. Avant toute intervention chirurgicale, informez votre anesthésiste de votre traitement par XEOMIN 50 unités, poudre pour solution injectable.
· par d’autres médicaments qui eux-mêmes réduisent le débit salivaire (par exemple, les anticholinergiques comme l’atropine, le glycopyrronium ou la scopolamine) ou par irradiation thérapeutique de la tête et du cou, y compris des glandes salivaires lorsqu’il est utilisé pour le traitement de l’excès de salive. Informez votre médecin si vous recevez une radiothérapie ou si une radiothérapie est prévue.
Dans ces cas, XEOMIN 50 unités, poudre pour solution injectable doit être utilisé avec précaution.
L’effet de XEOMIN 50 unités, poudre pour solution injectable peut être réduit par :
· certains médicaments antipaludéens/antirhumatismaux (amino-4-quinoléines).
XEOMIN 50 unités, poudre pour solution injectable avec des aliments et boissons
Sans objet.
Grossesse et allaitement et fertilité
Si vous êtes enceinte, ou que vous allaitez, si vous pensez être enceinte ou planifiez une grossesse, demandez conseil à votre médecin ou votre pharmacien avant de prendre ce médicament.
XEOMIN 50 unités, poudre pour solution injectable ne doit pas être utilisé au cours de la grossesse sauf si votre médecin considère que ce traitement est nécessaire et que le bénéfice potentiel justifie ce possible risque pour le fœtus.
XEOMIN 50 unités, poudre pour solution injectable ne doit pas être utilisé pendant l’allaitement.
Conduite de véhicules et utilisation de machines
Vous ne devez pas conduire ou avoir des activités potentiellement dangereuses en cas d’affaissement des paupières, de faiblesse (asthénie), de faiblesse musculaire ou de troubles de la vision. En cas de doute, demandez conseil à votre médecin.
XEOMIN 50 unités, poudre pour solution injectable contient du saccharose et de l’albumine humaine
3. COMMENT UTILISER XEOMIN 50 unités, poudre pour solution injectable?
XEOMIN 50 unités, poudre pour solution injectable doit être administré uniquement par des médecins ayant l’expérience de l’utilisation de la toxine botulinique de type A.
La dose optimale, la fréquence et le nombre de sites d'injection seront spécifiquement déterminés par votre médecin. Les résultats d’un premier traitement par XEOMIN 50 unités, poudre pour solution injectable doivent être évalués et peuvent conduire à un ajustement des doses jusqu'à obtention de l'effet thérapeutique souhaité. Les intervalles de traitement seront déterminés par votre médecin en fonction de vos besoins cliniques réels.
Si vous avez l'impression que l'effet de XEOMIN 50 unités, poudre pour solution injectable est trop fort ou trop faible, informez votre médecin. Dans le cas où aucun effet thérapeutique n'apparaît, un autre traitement doit être envisagé.
Spasme des paupières (blépharospasme) et spasme touchant un côté du visage (spasme hémifacial)
La dose initiale recommandée est au maximum de 25 unités par œil et la dose totale recommandée pour les sessions suivantes de traitement est au maximum de 50 unités par œil. En général, les premiers effets s'observent dans les quatre jours qui suivent l'injection. L'effet de chaque traitement dure environ 3 à 5 mois ; il peut toutefois durer plus ou moins longtemps. Un intervalle de traitement de moins de 12 semaines n’est pas recommandé.
Normalement, il n'y a aucun bénéfice à traiter plus souvent que tous les 3 mois.
Si vous souffrez de spasmes touchant un côté de votre visage (spasme hémifacial), votre médecin suivra les recommandations de traitement pour le spasme des paupières (blépharospasme) limité à un côté du visage. Le spasme touchant un côté de votre visage (spasme hémifacial) ne sera traité que dans la partie supérieure du visage, car les injections de XEOMIN dans la partie inférieure du visage peuvent accroître le risque d'effets indésirables, tels qu'un risque prononcé de faiblesse locale.
Torticolis spasmodique
La dose recommandée par site d’injection est au maximum de 50 unités et la dose maximale recommandée pour la première session de traitement est de 200 unités. Des doses jusqu’à 300 unités peuvent être indiquées par votre médecin pour les sessions suivantes selon la réponse au traitement. Généralement, les premiers effets s'observent dans les sept jours qui suivent l'injection. L'effet de chaque traitement dure environ 3 à 4 mois; il peut toutefois durer plus ou moins longtemps. Bien que des intervalles de traitement de moins de 10 semaines ne soient pas recommandés, les intervalles de traitement seront adaptés individuellement par votre médecin à vos besoins réels.
Hyperactivité musculaire ou raideur incontrôlable des muscles de l’épaule, du bras ou de la main (spasticité des membres supérieurs).
La dose maximale recommandée est de 500 unités par traitement et ne doit pas dépasser 250 unités dans les muscles de l’épaule. Les patients ont noté une efficacité clinique dans les 4 jours avec une amélioration du tonus musculaire dans les 4 semaines qui suivent l’injection. Comme l’effet du traitement persiste, en général 12 semaines, les réinjections peuvent être espacées d’au moins 12 semaines. Il peut cependant être plus court ou plus long, de ce fait les intervalles de traitement seront adaptés individuellement par votre médecin selon vos besoins réels.
Excès de salive (Sialorrhée, adultes)
La dose recommandée est de 100 unités par session de traitement. Cette dose maximale ne doit pas être dépassée. Bien que les intervalles de traitement de moins de 16 semaines ne soient pas recommandés, les intervalles de traitement doivent être adaptés à vos besoins cliniques réels et individuels.
Excès de salive (Sialorrhée, enfants/adolescents)
La dose recommandée par session de traitement dépend du poids corporel. La dose maximale ne doit pas dépasser 75 unités. L'intervalle entre chaque session de traitement doit être d'au moins 16 semaines.
Mode d’administration
La solution de XEOMIN 50 unités, poudre pour solution injectable est destinée à être injectée dans le muscle (voie intramusculaire), et dans les glandes salivaires (utilisation intraglandulaire) (voir les informations destinées aux professionnels de santé à la fin de cette notice). En ce qui concerne la localisation des glandes chez l’adulte, un repérage anatomique ou un guidage échographique sont tous deux possibles, toutefois la méthode de guidage échographique devrait être privilégiée pour des raisons d’efficacité. Chez les enfants et les adolescents, la méthode de guidage échographique doit être utilisée.
Avant l’injection, les enfants et les adolescents peuvent recevoir une anesthésie locale (telle qu’une crème anesthésiante), un sédatif ou un anesthésique associé à un sédatif.
Si vous avez utilisé plus de XEOMIN 50 unités, poudre pour solution injectable que vous n’auriez dû :
Consultez immédiatement votre médecin ou votre pharmacien.
Symptômes de surdosage
Les symptômes de surdosage n'apparaissent pas immédiatement après l'injection et peuvent se traduire par une faiblesse généralisée, un affaissement des paupières, une vision double, des difficultés à respirer ou à parler, une paralysie des muscles respiratoires ou des difficultés à avaler pouvant conduire à une pneumonie.
Mesures en cas de surdosage
En cas de symptômes de surdosage, veuillez contacter immédiatement les services médicaux d'urgence ou demandez à votre entourage de le faire pour vous et faites-vous hospitaliser. Une surveillance médicale pendant plusieurs jours et une assistance respiratoire s'avèreront peut-être nécessaires.
Si vous avez d'autres questions sur l'utilisation de ce médicament, demandez à votre médecin ou à votre pharmacien.
Si vous oubliez d’utiliser XEOMIN 50 unités, poudre pour solution injectable :
Si vous arrêtez d’utiliser XEOMIN 50 unités, poudre pour solution injectable :
Sans objet.
4. QUELS SONT LES EFFETS INDESIRABLES EVENTUELS ?
Comme tous les médicaments, ce médicament peut provoquer des effets indésirables, mais ils ne surviennent pas systématiquement chez tout le monde.
Généralement, les effets indésirables se manifestent au cours de la première semaine suivant le traitement et sont de nature transitoire. Ils peuvent être dûs au médicament, à la technique d’injection ou aux deux. Les effets peuvent être limités à la zone autour du site d'injection (ex : faiblesse musculaire localisée, douleur locale, inflammation, picotements et sensation d’aiguilles (paresthésie), diminution de la sensibilité au toucher (hypoesthésie), sensibilité, gonflement (général), gonflement des tissus mous (œdème), rougeur de la peau (érythème), prurit, infection localisée, hématome, hémorragie, saignement ou bleus).
La piqûre peut être douloureuse. La douleur ou l’anxiété due à la piqûre peuvent provoquer un étourdissement, des nausées, des acouphènes (bourdonnements d’oreilles) ou une baisse de la pression artérielle.
Des effets indésirables tels qu’une faiblesse musculaire excessive ou des difficultés à avaler peuvent être causés par un relâchement des muscles à distance du site d'injection de XEOMIN 50 unités, poudre pour solution injectable. Les difficultés à avaler peuvent être à l’origine de fausses routes pouvant provoquer une inflammation des poumons voire, dans certains cas, un décès.
Des réactions allergiques avec XEOMIN 50 unités, poudre pour solution injectable peuvent être observées. Des réactions allergiques graves et/ou immédiates (anaphylaxie) ou des réactions allergiques à l’albumine contenue dans ce médicament (maladie sérique) peuvent par exemple être à l’origine de difficultés respiratoires (dyspnée), d’urticaire ou de gonflement des tissus mous (œdème), ont rarement été décrites. Certaines de ces réactions ont été observées à la suite d’un traitement par toxine botulinique de type A conventionnelle seule ou en association à d’autres médicaments connus pour causer des réactions similaires. Une réaction allergique peut causer l’un des symptômes suivants :
· difficultés à respirer, à avaler ou à parler en raison du gonflement du visage, des lèvres, de la bouche ou de la gorge
· gonflement des mains, des pieds ou des chevilles.
Si vous présentez un de ces effets indésirables, contactez immédiatement un service d'urgence ou demandez à votre entourage de le faire.
Les effets secondaires suivants ont été notés avec XEOMIN 50 unités, poudre pour solution injectable :
Spasme de la paupière (blépharospasme)
Très fréquent (peut concerner plus de 1 patient sur 10) : chute de la paupière (ptosis)
Fréquent (peut concerner jusqu’à 1 patient sur 10) : sécheresse oculaire, vision floue, troubles de la vision, sécheresse buccale, douleur au site d’injection
Peu fréquent (peut concerner jusqu’à 1 patient sur 10) : Maux de tête, faiblesse des muscles du visage (parésie faciale), vision double (diplopie), larmoiement important, difficulté à déglutir (dysphagie), fatigue, faiblesse musculaire, rash
Spasme touchant un côté du visage (spasme hémifacial)
Des effets secondaires similaires à ceux du spasme de la paupière peuvent être attendus lors du traitement des spasmes affectant un côté du visage.
Torsion du cou (torticolis spasmodique)
Très fréquent (peut concerner plus de 1 patient sur 10) : difficultés à avaler (dysphagie)
Fréquent (peut concerner jusqu’à 1 patient sur 10) : douleur du cou, faiblesse musculaire, douleurs musculaires (myalgies), raideur musculaire, spasmes musculaires, céphalées, vertiges, douleur au site d’injection, faiblesse (asthénie), sécheresse buccale, nausée, transpiration excessive (hyperhidrose), infection des voies aériennes supérieures, sensation de malaise (présyncope).
Peu fréquent (peut concerner jusqu’à 1 patient sur 100) : troubles du langage, dysphonie, respiration ralentie (dyspnée), rash.
Le traitement du torticolis spasmodique peut provoquer des difficultés à avaler de sévérité variable pouvant conduire à des fausses routes et nécessiter une intervention médicale. Les difficultés à avaler peuvent persister jusqu’à deux à trois semaines après l'injection (cinq mois rapportés pour un cas). Ces difficultés à avaler semblent être en relation avec la dose.
Hyperactivité musculaire ou raideur incontrôlable des muscles de l’épaule, du bras ou de la main (spasticité des membres supérieurs).
Fréquent (peut concerner jusqu’à 1 patient sur 10) : bouche sèche.
Peu fréquent (peut concerner jusqu’à 1 patient sur 100) : maux de tête, sensation diminuée au toucher (hypoesthésie), faiblesse musculaire, douleurs des extrémités, faiblesse (asthénie), douleur musculo-squelettique (myalgie), difficultés à avaler (dysphagie), nausées.
De fréquence inconnue (impossible à estimer compte-tenu des données actuelles) : douleur au site d’injection
Certains de ces évènements indésirables peuvent être dus à la maladie.
Excès de salive (sialorrhée) chez l’adulte
Fréquent (peut concerner jusqu’à 1 patient sur 10): sécheresse buccale, difficultés à avaler (dysphagie), sensation d’épingles et d’aiguilles (paresthésie)
Peu fréquent (peut concerner jusqu’à 1 patient sur 100): salive épaissie, trouble de la parole, trouble du goût (dysgueusie)
Des cas de sécheresse buccale persistante (> 110 jours) d’intensité sévère ont été signalés, ce qui pourrait entraîner d’autres complications telles qu’une inflammation des gencives (gingivite), des difficultés à avaler (dysphagie) et des caries.
Excès de salive (sialorrhée) chez les enfants/adolescents
Peu fréquent (peut concerner jusqu’à 1 patient sur 100): difficultés à avaler (dysphagie)
Inconnu (ne peut être estimé à partir des données disponibles): sécheresse buccale, salive épaissie, douleurs buccales, caries dentaires
Expérience après commercialisation
Depuis la mise sur le marché de XEOMIN, les effets indésirables suivants ont été rapportés à une fréquence inconnue : syndromes pseudo-grippaux, diminution du volume du muscle injecté et des réactions d’hypersensibilité, telles que gonflement, œdèmes des tissus mous (également à distance du site d’injection), rougeurs, démangeaisons, rash (localisé ou généralisé) et des difficultés respiratoires.
Déclaration des effets secondaires
Si vous ressentez un quelconque effet indésirable, parlez-en à votre médecin ou votre pharmacien. Ceci s’applique aussi à tout effet indésirable qui ne serait pas mentionné dans cette notice. Vous pouvez également déclarer les effets indésirables directement via le système national de déclaration : Agence nationale de sécurité du médicament et des produits de santé (ANSM) et réseau des Centres Régionaux de Pharmacovigilance - Site internet: https://signalement.social-sante.gouv.fr/.
En signalant les effets indésirables, vous contribuez à fournir davantage d’informations sur la sécurité du médicament.
5. COMMENT CONSERVER XEOMIN 50 unités, poudre pour solution injectable ?
Tenir ce médicament hors de la vue et de la portée des enfants.
N’utilisez pas ce médicament après la date de péremption mentionnée sur la boîte et le flacon après la mention « EXP ». La date d'expiration fait référence au dernier jour du mois.
Flacon fermé: à conserver à une température ne dépassant pas 25°C
Solution reconstituée: la stabilité physique et chimique en cours d’utilisation a été démontrée pendant 24 heures entre 2°C et 8°C.
D’un point de vue microbiologique, le produit doit être utilisé immédiatement Dans le cas contraire, la durée et les conditions de conservation de la solution reconstituée relèvent de la responsabilité de l’utilisateur et ne devraient pas excéder 24 heures à une température comprise entre +2°C et +8°C, à moins que la reconstitution ait été effectuée dans des conditions d’asepsie maitrisées et validées.
Votre médecin ne devra pas utiliser XEOMIN 50 unités, poudre pour solution injectable si la solution reconstituée est trouble ou contient des particules visibles.
La procédure pour l’élimination est donnée à la fin de cette notice, dans la partie réservée aux professionnels de santé.
6. CONTENU DE L’EMBALLAGE ET AUTRES INFORMATIONS
Ce que contient XEOMIN 50 unités, poudre pour solution injectable
· La substance active est :
La neurotoxine de Clostridium botulinum de type A (150 kD), sans protéines complexantes.
XEOMIN 50 unités poudre pour solution injectable
Un flacon contient 50 unités de neurotoxine de Clostridium botulinum de type A (150 kD), sans protéines complexantes*.
*Neurotoxine botulique de type A, purifiée à partir de cultures de Clostridium botulinum (souche de Hall)
· Les autres composants sont : albumine humaine, saccharose.
Qu’est-ce que XEOMIN 50 unités, poudre pour solution injectable et contenu de l’emballage extérieur
XEOMIN 50 unités, poudre pour solution injectable est présenté sous forme de poudre pour solution injectable. La poudre est blanche.
La reconstitution de la poudre donne une solution limpide et incolore.
Boîtes de 1, 2, 3 et 6 flacons.
Toutes les présentations peuvent ne pas être commercialisées.
Titulaire de l’autorisation de mise sur le marché
ECKENHEIMER LANDSTRASSE 100
60318 FRANKFURT / MAIN
ALLEMAGNE
Exploitant de l’autorisation de mise sur le marché
MERZ PHARMA FRANCE
TOUR EQHO
2, AVENUE GAMBETTA
92400 COURBEVOIE
ECKENHEIMER LANDSTRASSE 100
60318 FRANCFORT/MAIN
ALLEMAGNE
Noms du médicament dans les Etats membres de l'Espace Economique Européen
Ce médicament est autorisé dans les Etats membres de l'Espace Economique Européen sous les noms suivants : Conformément à la réglementation en vigueur.
La dernière date à laquelle cette notice a été révisée est
[à compléter ultérieurement par le titulaire]
Des informations détaillées sur ce médicament sont disponibles sur le site Internet de l’ANSM (France).
Les informations suivantes sont destinées exclusivement aux professionnels de santé :
Instructions pour la reconstitution de la solution
XEOMIN 50 unités, poudre pour solution injectable est reconstitué avant utilisation avec une solution injectable de chlorure de sodium à 9 mg/ml (0,9 %).
XEOMIN 50 unités, poudre pour solution injectable doit être utilisé pour traiter un seul patient au cours d’une seule session.
Il est de bonne pratique de reconstituer le produit et préparer les seringues sur des supports papier doublés de plastique pour recevoir tout déversement accidentel. Un volume de chlorure de sodium approprié est aspiré dans une seringue (voir tableau de dilution). Une aiguille courte en biseau de 20-27G est recommandée pour la reconstitution. Après insertion verticale de l’aiguille à travers le bouchon en caoutchouc, le solvant est injecté délicatement dans le flacon afin d’éviter la formation de mousse. Jeter le flacon si la dépression n'entraîne pas l'aspiration du solvant à l'intérieur du flacon. Retirer la seringue du flacon et mélanger délicatement XEOMIN 50 unités, poudre pour solution injectable au solvant par des mouvements circulaires et par retournement du flacon. Ne pas mélanger vigoureusement. Si besoin, l’aiguille ayant servi à la reconstitution de la solution peut rester dans le flacon. Le volume de produit nécessaire est aspiré avec une nouvelle seringue stérile adaptée à l’injection.
|
|
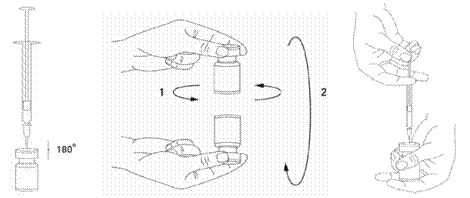
La solution reconstituée de XEOMIN 50 unités, poudre pour solution injectable est limpide et incolore.
XEOMIN 50 unités, poudre pour solution injectable ne doit pas être utilisé si la solution reconstituée (préparée selon les modalités décrites ci-dessus) présente un aspect trouble ou contient des particules ou des molécules floconneuses.
Afin de prévenir tout surdosage accidentel, il convient de bien s’assurer lors de la reconstitution du produit que le volume de solvant est adapté à la présentation choisie. Si différentes présentations sont utilisées au cours d’une même séance d’injection, il convient de s’assurer que le volume de solvant utilisé lors de la reconstitution du produit donne le bon nombre d’unités par 0,1ml. Le volume de solvant à ajouter diffère entre XEOMIN 50 unités, XEOMIN 100 unités et XEOMIN 200 unités. Chaque seringue utilisée doit être identifiée en conséquence.
Les concentrations possibles pour XEOMIN 50 unités sont indiquées dans le tableau suivant :
|
Dose obtenue (unités par 0,1 ml) |
Solvant ajouté (solution injectable de chlorure de sodium à 9 mg/ml (0,9%)) |
|
|
20 unités |
0,25 ml |
|
|
10 unités |
0,5 ml |
|
|
8 unités |
0,625 ml |
|
|
5 unités |
1 ml |
|
|
4 unités |
1,25 ml |
|
|
2,5 unités |
2 ml |
|
|
2 unités |
2,5 ml |
|
|
1,25 unités |
4 ml |
|
Précautions pour l’élimination
Toute solution reconstituée qui a été conservée pendant plus de 24 heures et toute fraction de solution injectable restante ou non utilisée doivent être jetées.
Procédure à suivre pour une élimination en toute sécurité des flacons, des seringues et matériels utilisés.
Tout flacon inutilisé ou contenant la solution reconstituée et/ou des seringues peuvent être autoclavés
Il est également possible d’inactiver toute fraction restante de XEOMIN 50 unités, poudre pour solution injectable par addition d’éthanol à 70%, d’isopropanol à 50%, de lauryl sulfate de sodium à 0,1% (détergent anionique), d’une solution diluée d'hydroxyde de sodium (NaOH 0,1 N) ou d’une solution diluée d'hypochlorite de sodium (NaOCl à au moins 0,1%).
Les flacons utilisés, seringues et matériels ne doivent pas être vidés mais doivent être jetés dans des récipients adaptés et éliminés conformément à la règlementation locale en vigueur.
Recommandations en cas d'incident lors de la manipulation de la toxine botulinique de type A
Toute projection doit être essuyée avec un matériel absorbant imbibé d'une des solutions listées ci-dessus pour la poudre ou bien avec un matériel absorbant sec pour le produit reconstitué.
Les surfaces contaminées sont nettoyées avec un matériel absorbant, imbibé d'une des solutions listées ci-dessus puis séchées.
Si le flacon est brisé, procéder comme indiqué ci-dessus au ramassage méticuleux des particules de verre et essuyer le produit, en évitant toute coupure cutanée.
En cas de contact avec la peau, rincer abondamment la zone touchée à l'eau.
En cas de contact avec les yeux, rincer abondamment avec de l'eau ou avec une solution pour lavage ophtalmique.
En cas de contact du produit avec une blessure, une coupure ou une piqûre, rincer abondamment avec de l'eau et prendre les mesures médicales appropriées en fonction de la dose injectée.
Ces instructions d'utilisation, de manipulation et d'élimination doivent être scrupuleusement suivies.