Dernière mise à jour le 01/06/2026
UMULINE NPH 100 UI/mL, suspension injectable en flacon
Indications thérapeutiques
Classe pharmacothérapeutique - code ATC : A10AC01.
UMULINE NPH contient l’insuline humaine comme substance active et est utilisée dans le traitement du diabète. Vous êtes diabétique lorsque votre pancréas ne produit pas suffisamment d'insuline pour réguler le niveau de glucose dans votre sang (glycémie). UMULINE NPH assure un contrôle du glucose à long terme. Son action est prolongée par la présence du sulfate de protamine dans la suspension.
Votre médecin peut vous prescrire UMULINE NPH ainsi qu’une insuline d'action rapide. Une notice spécifique accompagne cette autre insuline. Ne changez pas d'insuline sauf sur prescription de votre médecin. Observez la plus grande prudence si vous changez d'insuline. Chaque type d’insuline a une couleur et un symbole différents sur la boîte et le flacon ce qui vous permettra de les différencier facilement.
Présentations
> 1 flacon(s) en verre de 10 ml
Code CIP : 335 234-3 ou 34009 335 234 3 5
Déclaration de commercialisation : 01/03/2000
Cette présentation n'est pas agréée aux collectivités
- Prix hors honoraire de dispensation : 15,82 €
- Honoraire de dispensation : 1,02 €
- Prix honoraire compris : 16,84 €
- Taux de remboursement :65%
Documents de bon usage du médicament
- Stratégie médicamenteuse du contrôle glycémique du diabète de type 2
Auteur : Haute autorité de santé
Type : Recommandation de bonne pratique
Date de mise à jour :Janvier 2013
Service médical rendu (SMR)
Les libellés affichés ci-dessous ne sont que des résumés ou extraits issus des avis rendus par la Commission de la Transparence. Seul l'avis complet de la Commission de la Transparence fait référence.
Cet avis est consultable à partir du lien `Avis du jj/mm/aaaa` ou encore sur demande auprès de la HAS (plus d'informations dans l'aide). Les avis et synthèses d'avis contiennent un paragraphe sur la place du médicament dans la stratégie thérapeutique.
| Valeur du SMR | Avis | Motif de l'évaluation | Résumé de l'avis |
|---|---|---|---|
| Important | Avis du 03/05/2017 | Renouvellement d'inscription (CT) | Le service médical rendu par les spécialités UMULINE reste important dans l’indication de leur AMM. |
Amélioration du service médical rendu (ASMR)
Pas d'ASMR disponible pour ce médicament (plus d'informations dans l'aide )
ANSM - Mis à jour le : 17/08/2020
UMULINE NPH 100 UI/mL, suspension injectable en flacon
2. COMPOSITION QUALITATIVE ET QUANTITATIVE
1 mL contient : 100 UI d’insuline humaine (produite dans Escherichia coli par la technique de l’ADN recombinant).
Un flacon contient 10 mL correspondant à 1000 UI d’insuline isophane.
Pour la liste complète des excipients, voir rubrique 6.1.
Suspension injectable en flacon de 10 mL.
UMULINE NPH est une suspension stérile formée d’un précipité blanc cristallisé, d’insuline humaine isophane dans un tampon phosphate isotonique.
4.1. Indications thérapeutiques
4.2. Posologie et mode d'administration
La posologie est déterminée par le médecin, selon les besoins du patient.
Population pédiatrique
Aucune donnée n’est disponible.
Mode d’administration
UMULINE NPH doit être injectée par voie sous-cutanée, mais peut également, bien que cela ne soit pas recommandé, être injectée par voie intramusculaire. Cette spécialité ne doit pas être administrée par voie intraveineuse.
L’administration sous-cutanée doit être effectuée dans les bras, les cuisses, les fesses ou l’abdomen. Il est nécessaire de varier les points d’injection afin de ne pas utiliser le même point d’injection plus d’une fois par mois environ afin de diminuer le risque de développer une lipodystrophie ou une amyloïdose cutanée (voir rubriques 4.4 et 4.8).
Lors de l’injection de toute préparation d’insuline UMULINE, s’assurer que l’aiguille n’a pas pénétré dans un vaisseau sanguin. Après l’injection, ne pas masser le point d’injection. Les patients doivent être éduqués pour utiliser une technique d’injection correcte.
UMULINE NPH (insuline isophane) peut être administré en association avec UMULINE RAPIDE (insuline soluble) (Voir Instructions pour l’utilisation et la manipulation pour le mélange d’insulines).
Chaque boîte contient une notice d’information contenant des instructions pour pratiquer l’injection d’insuline.
Hypersensibilité à la substance active ou à l’un des excipients mentionnés à la rubrique 6.1, sauf dans le cadre d’un programme de désensibilisation.
Les préparations d’UMULINE ne doivent en aucun cas être administrées par voie intraveineuse, à l’exception d’UMULINE RAPIDE (insuline soluble).
4.4. Mises en garde spéciales et précautions d'emploi
Les patients recevant de l’insuline humaine peuvent nécessiter un changement de posologie par rapport à leurs insulines d’origine animale. Si une adaptation posologique est nécessaire, elle peut être faite lors de la première administration ou au cours des premières semaines ou des premiers mois.
Quelques patients ayant présenté des réactions hypoglycémiques après transfert d’une insuline d’origine animale à une insuline humaine ont signalé que les symptômes précurseurs d’hypoglycémie étaient moins prononcés ou différents de ceux ressentis lors de leur traitement antérieur par l'insuline animale. Les patients dont la glycémie s’est améliorée de façon importante, par exemple grâce à l’insulinothérapie intensifiée, peuvent voir disparaître certains ou tous les symptômes précurseurs d’hypoglycémie et doivent donc en être informés. La modification ou la diminution des symptômes précurseurs d’hypoglycémie peut également survenir chez les patients présentant un diabète de longue durée, une neuropathie diabétique ou prenant simultanément d'autres médicaments tels que les bêta-bloquants. L'hypoglycémie non corrigée ou les réactions d'hyperglycémie peuvent entraîner une perte de connaissance, un coma ou le décès.
L’utilisation de posologies mal adaptées ou l’arrêt du traitement, en particulier chez les diabétiques insulino-dépendants, peuvent entraîner une hyperglycémie et une acido-cétose diabétique dont le pronostic peut être fatal.
Le traitement par insuline humaine peut provoquer la formation d’anticorps, mais leurs taux sont inférieurs à ceux observés avec l'insuline animale purifiée.
Les besoins en insuline peuvent changer de façon significative en cas de maladies surrénalienne, hypophysaire ou thyroïdienne et d’insuffisance rénale ou hépatique.
Les besoins en insuline peuvent augmenter au cours d’une maladie ou de perturbations affectives.
L’adaptation posologique peut être nécessaire si le patient modifie l’intensité de son activité physique ou modifie son régime alimentaire habituel.
Association d’insuline humaine avec la pioglitazone
Des cas d’insuffisance cardiaque ont été observés lorsque l’insuline était associée à la pioglitazone, en particulier chez des patients ayant des facteurs de risque de développement d’insuffisance cardiaque. Cela devra être pris en compte dans le cas où un traitement associant la pioglitazone et l’insuline humaine est envisagé. Dans ce cas, l’apparition de signes et symptômes d’insuffisance cardiaque, d’une prise de poids et d’œdèmes devra être surveillée chez ces patients. La pioglitazone doit être arrêtée en cas d'aggravation des symptômes cardiaques.
Afin d'améliorer la traçabilité des médicaments biologiques, le nom et le numéro de lot du produit administré doivent être clairement enregistrés.
Excipients
Ce médicament contient moins de 1 mmol (23 mg) de sodium par dose, c.-à-d. qu’il est essentiellement « sans sodium ».
4.5. Interactions avec d'autres médicaments et autres formes d'interactions
Un certain nombre de médicaments sont connus pour interagir avec le métabolisme du glucose, en conséquence, le médecin doit être consulté en cas de prise simultanée d’autres médicaments avec l’insuline humaine (voir rubrique 4.4). Le médecin doit donc considérer toute possibilité d’interaction et doit toujours demander à ses patients s’ils prennent d’autres médicaments.
Les besoins en insuline peuvent être augmentés par les substances à effet hyperglycémiant, telles que les glucocorticoïdes, les hormones thyroïdiennes, l’hormone de croissance, le danazol, les bêta-2 mimétiques (tels que ritodrine, salbutamol, terbutaline), les thiazidiques.
Les besoins en insuline peuvent être diminués en présence de substances à effet hypoglycémiant, telles que les hypoglycémiants oraux (ADO), les salicylés (par exemple l’acide acétylsalicylique), certains antidépresseurs (les inhibiteurs de la monoamine oxydase IMAO), certains inhibiteurs de l’enzyme de conversion de l’angiotensine (IEC) (captopril, enalapril), les antagonistes des récepteurs de l’angiotensine II, les agents bêta-bloquants non sélectifs et l'alcool.
Les analogues des somatostatines (octréotide, lanréotide) peuvent réduire ou accroître les besoins en insuline.
4.6. Fertilité, grossesse et allaitement
Grossesse
Il est essentiel de maintenir un bon équilibre glycémique chez la patiente traitée par l’insuline (diabète insulino-dépendant ou gestationnel) durant la grossesse. Les besoins en insuline chutent habituellement au cours du premier trimestre et augmentent au cours des deuxième et troisième trimestres. Les patientes diabétiques doivent informer leur médecin si elles sont enceintes ou si elles envisagent une grossesse.
Une surveillance attentive de la glycémie ainsi que de l’état de santé général est primordiale pendant la grossesse chez les patientes diabétiques.
Allaitement
Les patientes diabétiques allaitant peuvent nécessiter une adaptation de la dose d’insuline, de leur régime alimentaire ou des deux.
4.7. Effets sur l'aptitude à conduire des véhicules et à utiliser des machines
Les patients doivent être informés des précautions à prendre avant de conduire pour éviter une hypoglycémie, en particulier chez ceux chez qui les symptômes précurseurs d’hypoglycémie sont absents ou diminués ou chez ceux qui ont de fréquents épisodes d’hypoglycémie. La capacité à conduire un véhicule doit être réévaluée dans ces circonstances.
L’hypoglycémie est l’effet indésirable le plus fréquent lors du traitement insulinique chez le patient diabétique. L’hypoglycémie sévère peut entraîner la perte de connaissance allant dans des cas extrêmes jusqu'à la mort. L’hypoglycémie pouvant résulter à la fois d’un excès d’insuline et d’autres facteurs tels que l’apport alimentaire et la dépense énergétique, aucune fréquence de survenue des hypoglycémies ne peut être présentée.
L’allergie locale est fréquente (≥ 1/100 à < 1/10). Une rougeur, un œdème et des démangeaisons peuvent survenir au point d’injection. Cette réaction disparaît habituellement en quelques jours voire quelques semaines. Dans certains cas, ces réactions locales peuvent être liées à des facteurs autres que l’insuline, tels que des produits irritants contenus dans le désinfectant cutané ou une mauvaise technique d’injection.
L’allergie systémique, qui est très rare (< 1/10 000) mais potentiellement plus grave, correspond à une allergie généralisée à l’insuline. Elle peut entraîner une éruption généralisée sur tout le corps, une dyspnée, une respiration sifflante, une baisse de la pression artérielle, une accélération du pouls ou des sueurs. Les cas sévères d’allergie généralisée peuvent menacer le pronostic vital.
Dans les rares cas d’allergie sévère à UMULINE, un traitement doit être instauré immédiatement. Un changement d’insuline ou une désensibilisation peut être nécessaire.
Une lipodystrophie au site d'injection apparaît peu fréquemment (≥ 1/1 000 à < 1/100).
Affections de la peau et du tissu sous-cutané : fréquence « indéterminée » : Amyloïdose cutanée.
Affections de la peau et du tissu sous-cutané :
Une lipodystrophie et une amyloïdose cutanée peuvent survenir au site d’injection, ce qui peut retarder la résorption locale de l’insuline. Une rotation continue des sites d’injection dans une zone donnée peut aider à diminuer ou à éviter ces réactions (voir rubrique 4.4).
Des cas d’œdèmes ont été rapportés lors du traitement par insuline, en particulier si un mauvais contrôle métabolique précédent est amélioré par une insulinothérapie intensifiée.
Déclaration des effets indésirables suspectés
La déclaration des effets indésirables suspectés après autorisation du médicament est importante. Elle permet une surveillance continue du rapport bénéfice/risque du médicament. Les professionnels de santé déclarent tout effet indésirable suspecté via le système national de déclaration : Agence nationale de sécurité du médicament et des produits de santé (ANSM) et réseau des Centres Régionaux de Pharmacovigilance - Site internet : www.signalement-sante.gouv.fr..
Il n’existe aucune définition spécifique du surdosage insulinique. En effet, la glycémie résulte d’interactions complexes entre les concentrations d’insuline, la disponibilité du glucose et d’autres facteurs métaboliques. L’hypoglycémie peut résulter d’un excès d’insuline par rapport à l’apport alimentaire et à la dépense énergétique.
L’hypoglycémie peut être associée à une apathie, une confusion, des palpitations, des céphalées, des sueurs et des vomissements.
Les épisodes d’hypoglycémie légère seront compensés par l’administration orale de glucose ou d’autres produits sucrés.
Une hypoglycémie modérément sévère peut être corrigée par l’administration intramusculaire ou sous-cutanée de glucagon, suivie d’une prise orale d’hydrates de carbone lorsque le patient sera suffisamment rétabli. Les patients qui ne répondent pas au glucagon doivent recevoir une solution de sérum glucosé par voie intraveineuse.
En cas de coma hypoglycémique, le glucagon devra être administré par voie intramusculaire ou sous-cutanée. Cependant, s’il n’y a pas de glucagon disponible ou si le patient n’y répond pas, on injectera du sérum glucosé par voie intraveineuse. Dès que le patient aura repris connaissance, un repas lui sera donné.
La prise prolongée d’hydrates de carbone et une surveillance peuvent être nécessaires car une hypoglycémie peut survenir après un rétablissement clinique apparent.
5. PROPRIETES PHARMACOLOGIQUES
5.1. Propriétés pharmacodynamiques
UMULINE NPH est une préparation d’insuline d’action intermédiaire.
L’activité principale de l’insuline est la régulation du métabolisme glucidique.
D'autre part, l'insuline possède plusieurs actions anaboliques et anticataboliques dans différents tissus. Dans le muscle, ces effets comprennent une augmentation de la synthèse du glycogène, des acides gras, du glycérol, des protéines et une augmentation de la fixation des acides aminés, ainsi qu’une diminution de la glycogénolyse, de la néoglucogénèse, de la cétogénèse, de la lipolyse, du catabolisme protéique et de l’élimination des acides aminés.
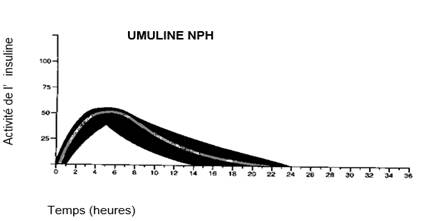
Le profil d’activité type (courbe d’utilisation du glucose) après injection sous-cutanée est représenté sur la courbe ci-dessous par la ligne épaisse. Les variations en durée et/ou intensité d’activité de l’insuline chez les patients sont représentées par la zone sombre. La variabilité individuelle dépend de facteurs tels que la dose, le site d’injection, la température et l’activité physique du patient.
5.2. Propriétés pharmacocinétiques
5.3. Données de sécurité préclinique
Glycérol,
Phénol,
Sulfate de protamine,
Phosphate disodique heptahydraté,
Oxyde de zinc,
Eau pour préparations injectables.
Peuvent être utilisés pour ajuster le pH : Acide chlorhydrique et/ou hydroxyde de sodium.
Flacons non ouverts : 3 ans.
Après première utilisation : 28 jours.
6.4. Précautions particulières de conservation
Ne pas congeler. Ne pas exposer à une chaleur excessive ou au soleil.
Flacons non ouverts
A conserver au réfrigérateur (entre 2°C et 8°C).
Après première utilisation
A conserver à une température ne dépassant pas 30°C.
6.5. Nature et contenu de l'emballage extérieur
10 mL de suspension injectable en flacon (verre de Type I) muni d’un bouchon (caoutchouc) et serti par une capsule (aluminium) et une capsule (plastique). Boîte de 1 flacon ou de 2 flacons ou emballage multiple de 5 boîtes de 1 flacon.
Toutes les présentations peuvent ne pas être commercialisées.
6.6. Précautions particulières d’élimination et de manipulation
Ne pas réutiliser les aiguilles. Eliminer l’aiguille de manière responsable. Les aiguilles ne doivent pas être partagées. Les flacons peuvent être utilisés jusqu’à ce qu’ils soient vides, puis doivent être convenablement jetés. Tout médicament non utilisé ou déchet doit être éliminé conformément à la réglementation en vigueur.
Instructions pour l’utilisation et la manipulation
Suspension injectable en flacon de 10 mL à utiliser avec une seringue appropriée marquée 100 UI/mL.
a) Préparation d’une dose
Immédiatement avant utilisation, faire rouler plusieurs fois entre la paume des mains le flacon contenant UMULINE NPH afin de remettre l’insuline en suspension, jusqu’à ce qu’elle prenne un aspect uniformément trouble ou laiteux. Si cet aspect n’est pas obtenu, répéter la procédure ci-dessus jusqu’à ce que le contenu du flacon soit bien mélangé. Ne pas agiter vigoureusement car la formation d’une mousse pourrait gêner la mesure exacte de la dose.
Examinez les flacons régulièrement et ne pas les utiliser si la suspension présente des floculations ou si des particules blanches restent collées au fond ou sur les parois du flacon, lui donnant un aspect givré.
Mélange d’insulines : commencer par aspirer l’insuline à action rapide dans la seringue, afin d’éviter la contamination du flacon par la préparation à durée d’action plus longue. Il est conseillé de réaliser l’injection immédiatement après le mélange. Toutefois, si un délai est nécessaire, procéder toujours de la même façon.
Il est également possible d’utiliser des seringues distinctes ou des cartouches distinctes d’UMULINE RAPIDE et d’UMULINE NPH pour administrer la quantité requise de chaque préparation.
Préparer votre seringue avant l’injection, en suivant les instructions de votre médecin ou infirmier/ère spécialiste du diabète.
Utiliser une seringue à insuline graduée, adaptée à la concentration de l’insuline à administrer.
b) Injection d’une dose
Injecter la dose correcte d’insuline en suivant les instructions de votre médecin ou infirmier/ère spécialiste du diabète.
Il est nécessaire de varier les points d’injection afin de ne pas utiliser le même point d’injection plus d’une fois par mois environ.
Chaque boîte contient une notice d’information contenant des instructions pour pratiquer l’injection d’insuline.
7. TITULAIRE DE L’AUTORISATION DE MISE SUR LE MARCHE
24, BOULEVARD VITAL BOUHOT
CS 50004
92521 NEUILLY-SUR-SEINE CEDEX
8. NUMERO(S) D’AUTORISATION DE MISE SUR LE MARCHE
· 34009 335 234 3 5 : 10 mL de suspension en flacon (verre de Type I) muni d’un bouchon (caoutchouc) ; boîte de 1.
· 34009 389 901 8 8 : 10 mL de suspension en flacon (verre de Type I) muni d’un bouchon (caoutchouc) ; boîte de 2.
· 34009 301 483 1 0 : 10 mL de suspension en flacon (verre de Type I) muni d’un bouchon (caoutchouc) ; boîte de 5.
9. DATE DE PREMIERE AUTORISATION/DE RENOUVELLEMENT DE L’AUTORISATION
[À compléter ultérieurement par le titulaire]
10. DATE DE MISE A JOUR DU TEXTE
[À compléter ultérieurement par le titulaire]
Sans objet.
12. INSTRUCTIONS POUR LA PREPARATION DES RADIOPHARMACEUTIQUES
Liste II.
ANSM - Mis à jour le : 17/08/2020
UMULINE NPH 100 UI/mL, suspension injectable en flacon
Insuline humaine isophane
· Gardez cette notice. Vous pourriez avoir besoin de la relire.
· Si vous avez d’autres questions, interrogez votre médecin, votre pharmacien ou votre infirmier/ère.
· Ce médicament vous a été personnellement prescrit. Ne le donnez pas à d’autres personnes. Il pourrait leur être nocif, même si les signes de leur maladie sont identiques aux vôtres.
· Si vous ressentez un quelconque effet indésirable, parlez-en à votre médecin, votre pharmacien ou votre infirmier/ère. Ceci s’applique aussi à tout effet indésirable qui ne serait pas mentionné dans cette notice. Voir rubrique 4.
1. Qu'est-ce que UMULINE NPH 100 UI/mL, suspension injectable en flacon et dans quels cas est-il utilisé ?
2. Quelles sont les informations à connaître avant d'utiliser UMULINE NPH 100 UI/mL, suspension injectable en flacon ?
3. Comment utiliser UMULINE NPH 100 UI/mL, suspension injectable en flacon ?
4. Quels sont les effets indésirables éventuels ?
5. Comment conserver UMULINE NPH 100 UI/mL, suspension injectable en flacon ?
6. Contenu de l’emballage et autres informations.
1. QU’EST-CE QUE UMULINE NPH 100 UI/mL, suspension injectable en flacon ET DANS QUELS CAS EST-IL UTILISE ?
Classe pharmacothérapeutique - code ATC : A10AC01.
UMULINE NPH contient l’insuline humaine comme substance active et est utilisée dans le traitement du diabète. Vous êtes diabétique lorsque votre pancréas ne produit pas suffisamment d'insuline pour réguler le niveau de glucose dans votre sang (glycémie). UMULINE NPH assure un contrôle du glucose à long terme. Son action est prolongée par la présence du sulfate de protamine dans la suspension.
Votre médecin peut vous prescrire UMULINE NPH ainsi qu’une insuline d'action rapide. Une notice spécifique accompagne cette autre insuline. Ne changez pas d'insuline sauf sur prescription de votre médecin. Observez la plus grande prudence si vous changez d'insuline. Chaque type d’insuline a une couleur et un symbole différents sur la boîte et le flacon ce qui vous permettra de les différencier facilement.
2. QUELLES SONT LES INFORMATIONS A CONNAITRE AVANT D’UTILISER UMULINE NPH 100 UI/mL, suspension injectable en flacon ?
N’utilisez jamais UMULINE NPH 100 UI/mL, suspension injectable en flacon :
· si vous estimez qu’une hypoglycémie (abaissement du niveau de sucre dans le sang) est imminente. Il est indiqué plus loin dans cette notice comment prendre en charge une hypoglycémie légère (voir paragraphe A à la rubrique 4).
· si vous êtes allergique à l’insuline humaine ou à l’un des autres composants contenus dans ce médicament, mentionnés dans la rubrique 6.
Avertissements et précautions
Adressez-vous à votre médecin, pharmacien ou votre infirmier/ère avant d’utiliser UMULINE NPH.
· Si votre glycémie est bien contrôlée par votre insuline actuelle, vous pourriez ne pas ressentir les symptômes annonciateurs de l'hypoglycémie. Ces symptômes sont énumérés ultérieurement dans cette notice. Réfléchissez soigneusement à l'heure à laquelle vous prendrez vos repas, à la fréquence et à l'intensité de vos exercices physiques. Vous devrez également étroitement surveiller les niveaux de sucre dans votre sang grâce à des tests fréquents de votre glycémie.
· Quelques patients ayant présenté des hypoglycémies (abaissement du niveau de sucre dans le sang) après passage d'une insuline animale à une insuline humaine ont rapporté que les symptômes annonciateurs d'hypoglycémie étaient moins prononcés ou différents de ceux provoqués par une insuline animale. En cas d'hypoglycémies fréquentes ou si vous avez des difficultés à les reconnaître, parlez-en avec votre médecin.
· Si vous répondez par l'AFFIRMATIVE à l'une des questions suivantes, informez-en votre médecin, pharmacien ou infirmière.
o Etes-vous récemment tombé malade ?
o Avez-vous des problèmes rénaux ou hépatiques ?
o Faites-vous davantage d'exercice que d'habitude ?
· Vos besoins en insuline pourront aussi varier si vous prenez de l'alcool.
· Si vous envisagez de vous rendre à l'étranger, pensez à en informer votre médecin, pharmacien ou infirmière. Le décalage horaire pourrait vous contraindre à modifier vos heures d'injection et de repas par rapport à vos heures habituelles.
· Certains patients avec un diabète de type II ancien et une maladie cardiaque ou un antécédent d’accident vasculaire cérébral, qui étaient traités avec la pioglitazone et l’insuline ont développé une insuffisance cardiaque. Informez votre médecin le plus rapidement possible si vous avez des signes d’insuffisance cardiaque tels qu’un essoufflement inhabituel ou une augmentation rapide de poids ou un gonflement localisé (œdème).
Modifications cutanées au site d’injection :
Il faut effectuer une rotation des sites d’injection pour prévenir des modifications cutanées telles que des grosseurs sous la peau. L’insuline risque de ne pas agir correctement si vous l’injectez dans une zone présentant des grosseurs (voir Comment utiliser UMULINE NPH 100 UI/mL, suspension injectable en flacon). Contactez votre médecin si vous injectez actuellement dans une zone présentant des grosseurs avant de commencer à injecter dans une autre zone. Votre médecin peut vous demander de contrôler votre glycémie de plus près et d’ajuster votre dose d’insuline ou celle de vos autres médicaments antidiabétiques.
Autres médicaments et UMULINE NPH 100 UI/mL, suspension injectable en flacon
Informez votre médecin ou pharmacien si vous prenez, avez récemment utilisé ou pourriez prendre tout autre médicament.
Vos besoins en insuline peuvent être différents si vous prenez un des traitements ci-dessous :
· corticostéroïdes,
· traitement substitutif par hormones thyroïdiennes,
· hypoglycémiants oraux (médicaments traitant le diabète),
· acide acétylsalicylique (aspirine),
· hormone de croissance,
· octréotide, lanréotide,
· bêta 2 stimulants (par exemple, ritodrine, salbutamol ou terbutaline),
· bêtabloquants,
· thiazidiques ou certains antidépresseurs (inhibiteurs de la monoamine oxydase),
· danazol,
· certains inhibiteurs de l’enzyme de conversion de l’angiotensine (IEC) (par exemple, captopril, enalapril) ou antagonistes des récepteurs de l’angiotensine II.
Grossesse, allaitement et fertilité
Les besoins en insuline diminuent généralement durant les trois premiers mois de grossesse et augmentent au cours des six autres mois. Si vous allaitez, l'administration de votre insuline devra peut-être être adaptée ainsi que votre régime alimentaire.
Si vous êtes enceinte ou que vous allaitez, si vous pensez être enceinte ou planifiez une grossesse, demandez conseil à votre médecin ou pharmacien avant de prendre ce médicament.
Conduite de véhicules et utilisation de machines
Vos capacités de concentration et vos réflexes peuvent être diminués en cas d'hypoglycémie (abaissement du niveau de sucre dans le sang). Vous devez y penser dans toutes les situations où vous pourriez mettre votre vie en danger ou celles d'autres personnes, comme la conduite automobile ou l'utilisation de machines. Vous devez interroger votre médecin ou votre infirmière sur vos capacités à conduire si vous avez :
· de fréquents épisodes d'hypoglycémie,
· des symptômes annonciateurs d'hypoglycémie diminués ou absents.
UMULINE NPH 100 UI/mL, suspension injectable en flacon contient du sodium :
Ce médicament contient moins de 1 mmol (23 mg) de sodium par dose, c.-à-d. qu’il est essentiellement « sans sodium ».
3. COMMENT UTILISER UMULINE NPH 100 UI/mL, suspension injectable en flacon ?
Veillez à toujours utiliser ce médicament en suivant exactement les indications de votre médecin ou pharmacien. Vérifiez auprès de votre médecin ou pharmacien en cas de doute.
Posologie
· Vous devez normalement injecter UMULINE NPH comme votre insuline basale. Votre médecin vous aura avisé du type d’insuline à utiliser, de la dose exacte de l'injection, de son heure et de sa fréquence. Ces consignes vous sont exclusivement réservées. Observez-les scrupuleusement et rendez-vous régulièrement chez votre médecin.
· Si vous changez de type d'insuline (passage, par exemple, d'une insuline animale à une insuline humaine), vous pouvez nécessiter un changement de dose. Ce changement peut être effectué lors de la première injection ou progressivement au cours de plusieurs semaines ou plusieurs mois.
· Injectez UMULINE NPH sous la peau. Ne l’administrez jamais par une autre voie d’administration. N’injectez jamais UMULINE NPH par voie intraveineuse.
Préparation d'UMULINE NPH :
Immédiatement avant utilisation, faites rouler le flacon d’UMULINE NPH plusieurs fois entre la paume des mains afin de remettre l’insuline en suspension, jusqu’à ce qu’elle prenne un aspect uniformément trouble ou laiteux. Si cet aspect n’est pas obtenu, répétez la procédure ci-dessus jusqu’à ce que le contenu du flacon soit bien mélangé. Ne pas agiter vigoureusement car la formation d’une mousse pourrait gêner la mesure exacte de la dose. Examinez les flacons régulièrement et ne pas les utiliser si la suspension présente des floculations ou si des particules blanches restent collées au fond ou sur les parois du flacon, lui donnant un aspect givré. Vérifiez-le avant chaque injection.
Injection d'UMULINE NPH :
· Lavez-vous d'abord les mains.
· Avant de procéder à l'injection, nettoyez la peau selon les instructions que l’on vous a données. Nettoyez le bouchon en caoutchouc du flacon sans toutefois le retirer.
· Prenez une seringue et une nouvelle aiguille stériles pour percer le bouchon en caoutchouc et prélevez le volume d’UMULINE NPH voulu. Suivez les instructions de votre médecin ou de votre infirmière. Ne partagez pas vos aiguilles et seringues.
· Injectez sous la peau comme on vous l'a appris. N’injectez pas directement dans une veine. Après l’injection, laissez l’aiguille sous la peau pendant 5 secondes afin de vous assurer d’avoir injecté votre dose en totalité. Ne frottez pas l’endroit où vous venez de faire l’injection. Assurez-vous que vous injectez à une distance d’au moins 1 cm de la dernière injection et que vous observez une « rotation » des emplacements d’injection, comme on vous l’a appris.
· Votre médecin vous dira si vous devez mélanger UMULINE RAPIDE avec UMULINE NPH. Si, par exemple, vous devez injecter un mélange, prélevez l’UMULINE RAPIDE en premier lieu dans la seringue avant l’insuline d’action longue durée. Procédez à l'injection immédiatement après le mélange. Observez la même procédure à chaque fois. Normalement, vous ne devez pas mélanger UMULINE NPH avec des mélanges d'insuline humaine. UMULINE NPH ne doit jamais être mélangé avec des insulines produites par d’autres fabricants ou avec des insulines d’origine animale.
· Vous ne devez pas injecter UMULINE NPH par voie intraveineuse. Injectez UMULINE NPH comme votre médecin ou votre infirmière vous l’a appris.
Si vous avez utilisé plus d’UMULINE NPH 100 UI/mL, suspension injectable en flacon que vous n’auriez dû :
Si vous avez pris plus d’UMULINE NPH que vous n’auriez dû, un abaissement du niveau de sucre dans le sang peut survenir. Vérifiez votre niveau de sucre (voir paragraphe A à la rubrique 4).
Si vous oubliez d’utiliser UMULINE NPH 100 UI/mL, suspension injectable en flacon :
Si vous prenez moins d’UMULINE NPH que vous n’auriez dû, une augmentation du niveau de sucre dans le sang peut survenir. Vérifiez votre niveau de sucre. Ne vous injectez pas de double dose pour compenser la dose que vous avez oublié de prendre.
Si vous arrêtez d’utiliser UMULINE NPH 100 UI/mL, suspension injectable en flacon :
Si vous prenez moins d’UMULINE NPH que vous n’auriez dû, une augmentation du niveau de sucre dans le sang peut survenir. Ne changez pas d’insuline sauf sur prescription de votre médecin.
Si vous avez d’autres questions sur l’utilisation de ce médicament, demandez plus d’informations à votre médecin, à votre pharmacien ou à votre infirmier/ère.
4. QUELS SONT LES EFFETS INDESIRABLES EVENTUELS ?
L’insuline humaine peut provoquer une hypoglycémie (abaissement du niveau de sucre dans le sang). Reportez-vous au paragraphe ci-dessous « Problèmes courants du diabète » pour plus d’information sur l’hypoglycémie.
Effets indésirables possibles
L’allergie générale est très rare (affecte moins d’un utilisateur sur 10 000). En voici les symptômes :
|
· baisse de la tension artérielle, |
· éruption généralisée sur tout le corps, |
|
· difficultés de respiration, |
· respiration sifflante de type asthmatique, |
|
· accélération du pouls, |
· transpiration. |
Si vous pensez avoir ce type d'allergie à l'insuline avec UMULINE NPH, consultez votre médecin immédiatement.
L’allergie locale est fréquente (affecte moins d’un utilisateur sur 10). Elle peut se manifester par une rougeur, un œdème ou des démangeaisons au point d'injection. Cette réaction disparaît habituellement en quelques jours voire quelques semaines. Consultez votre médecin si ceci vous arrive.
Modifications cutanées au site d’injection :
Si vous injectez trop souvent votre insuline au même endroit, le tissu adipeux peut devenir plus mince (lipoatrophie) ou plus épais (lipohypertrophie) (pouvant affecter jusqu’à 1 personne sur 100). Des grosseurs sous la peau peuvent également être causées par l’accumulation d’une protéine appelée amyloïde (amyloïdose cutanée, la fréquence de survenue est indéterminée). L’insuline risque de ne pas agir correctement si vous l’injectez dans une zone présentant des grosseurs. Changez de site d’injection à chaque fois pour éviter de telles modifications cutanées.
Un œdème (par exemple, gonflement au niveau des bras, des chevilles ; rétention d’eau) a été rapporté en particulier au début du traitement par insuline ou lors d’un changement du traitement pour améliorer le contrôle de votre glycémie.
Problèmes courants du diabète
A. Hypoglycémie
Hypoglycémie (abaissement du niveau de sucre dans le sang) signifie qu’il n’y a pas suffisamment de sucre dans le sang et peut être provoquée par :
· une dose trop élevée d'UMULINE NPH ou d’une autre insuline ;
· le fait que vous sautiez ou retardiez vos repas ou changiez de régime alimentaire ;
· des exercices ou des activités intenses juste avant ou après un repas ;
· une infection ou une maladie (en particulier des diarrhées ou des vomissements) ;
· des changements de vos besoins en insuline ou
· une insuffisance rénale ou hépatique en voie d'aggravation.
Les boissons alcoolisées ainsi que certains médicaments sont susceptibles d'influencer votre glycémie.
Les premiers symptômes de l'hypoglycémie apparaissent habituellement rapidement et comprennent :
· fatigue, · accélération du pouls,
· nervosité ou tremblements, · malaise,
· maux de tête, · sueurs froides.
Lorsque vous n'êtes pas certain de votre capacité à en reconnaître les symptômes précurseurs, évitez toute situation, comme la conduite de véhicule, dans laquelle vous ou d’autres personnes pourraient être exposés à un risque du fait de votre hypoglycémie.
N’utilisez pas UMULINE NPH si vous estimez qu’une hypoglycémie (abaissement du niveau de sucre dans le sang) est imminente.
Si votre glycémie est basse, consommez des préparations glucosées, du sucre ou des boissons sucrées. Puis, consommez des fruits, des biscuits ou un sandwich selon les conseils de votre médecin et reposez-vous. Ceci vous permettra fréquemment de maîtriser une hypoglycémie légère ou un surdosage mineur d'insuline. En cas d'aggravation, si votre respiration est superficielle et si votre peau devient pâle, avisez-en immédiatement votre médecin. Une injection de glucagon peut traiter une hypoglycémie assez grave. Prenez du glucose ou du sucre après l'injection de glucagon. Si vous n'obtenez aucune réaction au glucagon, un traitement hospitalier sera nécessaire. Demandez à votre médecin des renseignements concernant le glucagon.
B. Hyperglycémie et acidocétose diabétique
Hyperglycémie (trop de sucre dans le sang) signifie que vous n'avez pas suffisamment d'insuline. L’hyperglycémie peut être provoquée par le fait que :
· vous n'avez pas pris votre dose d'UMULINE NPH ou d’une autre insuline ;
· vous prenez moins d'insuline que la dose prescrite par votre médecin ;
· votre alimentation dépasse de beaucoup les limites permises par votre régime alimentaire ; ou
· vous avez de la fièvre, une infection ou êtes en état de stress.
L'hyperglycémie peut entraîner une acidocétose diabétique. Les premiers symptômes se manifestent lentement sur plusieurs heures, voire plusieurs jours. Parmi ceux-ci, citons les suivants :
· somnolence, · absence d'appétit,
· rougeur au visage, · odeur acétonique de l'haleine,
· soif, · sensation de malaise.
Les symptômes graves sont une respiration difficile et un pouls rapide. Consultez un médecin immédiatement.
Si une hypoglycémie (abaissement du niveau de sucre dans le sang) ou une hyperglycémie (trop de sucre dans le sang) n’est pas traitée, il peut s’ensuivre des complications sérieuses telles que maux de tête, nausées, vomissements, déshydratation, évanouissement, coma voire même décès.
Trois étapes simples pour éviter une hypoglycémie ou une hyperglycémie :
· Ayez toujours avec vous des seringues et un flacon d’UMULINE NPH de remplacement.
· Ayez toujours avec vous un document indiquant que vous êtes diabétique.
· Ayez toujours du sucre avec vous.
C. En cas de maladie
Si vous êtes malade et plus particulièrement, si vous vous sentez malade ou si vous avez des nausées, vos besoins en insuline pourront être différents. Même si vous ne vous alimentez pas normalement, vous avez néanmoins toujours besoin d'insuline. Testez vos urines ou votre sang, suivez les instructions qui vous ont été données dans ce cas et consultez votre médecin ou votre infirmière.
Déclaration des effets secondaires
Si vous ressentez un quelconque effet indésirable, parlez-en à votre médecin, votre pharmacien ou à votre infirmier/ère. Ceci s’applique aussi à tout effet indésirable qui ne serait pas mentionné dans cette notice. Vous pouvez également déclarer les effets indésirables directement via le système national de déclaration : Agence nationale de sécurité du médicament et des produits de santé (ANSM) et réseau des Centres Régionaux de Pharmacovigilance - Site internet : www.signalement-sante.gouv.fr.
En signalant les effets indésirables, vous contribuez à fournir davantage d’informations sur la sécurité du médicament.
5. COMMENT CONSERVER UMULINE NPH 100 UI/mL, suspension injectable en flacon ?
Tenir ce médicament hors de la vue et de la portée des enfants.
Avant la première utilisation, conservez votre UMULINE NPH au réfrigérateur (entre 2°C et 8°C). Ne pas congeler. Vous pouvez conserver votre flacon en cours d’utilisation à température ambiante (inférieure à 30°C) pendant 28 jours maximum. Ne pas exposer à la chaleur ou au soleil.
N’utilisez pas ce médicament après la date de péremption indiquée sur l’étiquette et l’emballage. La date de péremption fait référence au dernier jour de ce mois.
N’utilisez pas ce médicament si la suspension présente des floculations ou si des particules blanches restent collées au fond ou sur les parois du flacon, lui donnant un aspect givré. Vérifiez cela avant chaque injection.
Ne jetez aucun médicament au tout-à-l’égout ou avec les ordures ménagères. Demandez à votre pharmacien d’éliminer les médicaments que vous n’utilisez plus. Ces mesures contribueront à protéger l’environnement.
6. CONTENU DE L’EMBALLAGE ET AUTRES INFORMATIONS
· La substance active est l’insuline humaine. L’insuline humaine est produite en laboratoire par la « technique de l’ADN recombinant ». Elle a la même structure que l’hormone naturelle produite par le pancréas. Elle est par conséquent différente des insulines animales. L’insuline humaine contenue dans UMULINE NPH est présente dans une suspension mélangée avec du sulfate de protamine.
· Les autres composants sont le métacrésol, le glycérol, le phénol, le sulfate de protamine, le phosphate disodique heptahydraté, l’oxyde de zinc et l'eau pour préparations injectables. L’hydroxyde de sodium ou l'acide chlorhydrique peuvent avoir été utilisés pendant la fabrication pour ajuster le pH.
Qu’est-ce que UMULINE NPH et contenu de l’emballage extérieur
UMULINE NPH (isophane) 100 UI/mL, suspension injectable est une suspension stérile de couleur blanche et contient 100 unités internationales d’insuline par millilitre (100 UI/mL). Chaque flacon contient 1000 unités internationales (10 millilitres).
UMULINE NPH 100 UI/mL est disponible en boîte de 1 flacon ou de 2 flacons ou en emballage multiple de 5 boîtes de 1 flacon.
Toutes les présentations peuvent ne pas être commercialisées.
Titulaire de l’autorisation de mise sur le marché
24, BOULEVARD VITAL BOUHOT
CS 50004
92521 NEUILLY-SUR-SEINE CEDEX
Exploitant de l’autorisation de mise sur le marché
LILLY FRANCE
24, BOULEVARD VITAL BOUHOT
CS 50004
92521 NEUILLY-SUR-SEINE CEDEX
AVDA. DE LA INDUSTRIA 30,
28108 ALCOBENDAS MADRID,
ESPAGNE
LILLY FRANCE S.A.S.
RUE DU COLONEL LILLY
67640 FEGERSHEIM
Noms du médicament dans les Etats membres de l'Espace Economique Européen
Ce médicament est autorisé dans les Etats membres de l'Espace Economique Européen sous les noms suivants : Conformément à la réglementation en vigueur.
[À compléter ultérieurement par le titulaire]
La dernière date à laquelle cette notice a été révisée est :
[à compléter ultérieurement par le titulaire]
< {MM/AAAA}>< {mois AAAA}.>
Des informations détaillées sur ce médicament sont disponibles sur le site Internet de l’ANSM (France).