Dernière mise à jour le 01/06/2026
BOTOX 200 UNITÉS ALLERGAN, poudre pour solution injectable
Indications thérapeutiques
Classe pharmacothérapeutique - code ATC : M03AX01
BOTOX est un relaxant musculaire utilisé pour traiter un certain nombre d’affections de l’organisme. Il contient de la toxine botulinique de type A, substance active qui est injectée aussi bien dans les muscles que dans la paroi de la vessie ou profondément dans la peau. Il agit en bloquant partiellement les impulsions nerveuses de tout muscle qui a été injecté et, réduit les contractions excessives de ces muscles.
Lorsqu’il est injecté sous la peau des aisselles, BOTOX agit sur les glandes sudoripares pour réduire la quantité de sueur produite. Lorsqu'il est injecté dans la paroi de la vessie, BOTOX agit sur le muscle de la vessie pour réduire les fuites urinaires (incontinence urinaire).
Lorsqu’il est injecté dans les muscles de la tête et du cou, BOTOX bloque les signaux de la douleur pour prévenir et traiter la migraine chronique.
Indications chez l’adulte :
Troubles de la vessie :
Lorsqu’il est injecté dans la paroi de la vessie, BOTOX agit sur le muscle de la vessie pour réduire les fuites urinaires (incontinence urinaire) et contrôler les situations suivantes :
· Hyperactivité vésicale idiopathique avec fuites urinaires (3 épisodes sur trois jours) : envie soudaine d’uriner et d’aller aux toilettes plus souvent que d’habitude (d’au moins 8 ou plus épisodes par jour) lorsqu’un traitement par un autre médicament (appelé anticholinergique) n’a pas agi.
· Hyperactivité vésicale due à des problèmes associés à une lésion de la moelle épinière ou à une sclérose en plaques, conduisant à des fuites urinaires (émission involontaire d’urine pendant la journée ou la nuit).
Troubles neurologiques :
BOTOX est utilisé pour prévenir la migraine chronique chez les adultes qui ont eu des maux de tête pendant 15 jours ou plus par mois, dont au moins 8 jours de migraine et qui n'ont pas bien répondu aux autres médicaments préventifs contre la migraine.
La migraine chronique est une maladie qui affecte le système nerveux. Les patients présentent généralement des maux de tête souvent associés à une sensibilité excessive à la lumière, aux sons forts ou aux odeurs, ainsi que de nausées et/ou de vomissements. Ces maux de tête surviennent au moins 15 jours par mois.
Indications chez l'adulte et l'enfant de plus de 12 ans :
BOTOX peut être injecté directement dans les muscles pour contrôler les situations suivantes :
· spasmes musculaires persistants dans les muscles autour des yeux, des paupières et du visage, provoquant un strabisme ou des yeux qui louchent ;
· spasmes musculaires persistants des paupières et du visage ;
· spasmes musculaires persistants dans le cou et les épaules.
BOTOX peut être injecté profondément dans la peau et ainsi agir sur les glandes sudoripares pour réduire la transpiration excessive des aisselles ; symptômes pouvant perturber les activités quotidiennes lorsque les autres traitements locaux n’agissent pas.
Indication chez l'adulte et l'enfant de 2 ans et plus :
BOTOX peut être injecté directement dans les muscles et ainsi contrôler les spasmes musculaires persistants dans les bras et/ou les jambes.
Présentations
> 1 flacon(s) en verre
Code CIP : 370 832-0 ou 34009 370 832 0 1
Déclaration de commercialisation : 01/10/2009
Cette présentation est agréée aux collectivités
Service médical rendu (SMR)
Les libellés affichés ci-dessous ne sont que des résumés ou extraits issus des avis rendus par la Commission de la Transparence. Seul l'avis complet de la Commission de la Transparence fait référence.
Cet avis est consultable à partir du lien `Avis du jj/mm/aaaa` ou encore sur demande auprès de la HAS (plus d'informations dans l'aide). Les avis et synthèses d'avis contiennent un paragraphe sur la place du médicament dans la stratégie thérapeutique.
| Valeur du SMR | Avis | Motif de l'évaluation | Résumé de l'avis |
|---|---|---|---|
| Modéré | Avis du 17/11/2021 | Extension d'indication | Le service médical rendu par BOTOX (toxine botulinique A) est modéré dans l’indication de traitement prophylactique de la migraine chronique (présence de céphalées au moins 15 jours par mois dont au moins 8 jours de migraine par mois) chez des patients adultes qui n’ont pas répondu ou sont intolérants aux autres traitements prophylactiques de la migraine. |
| Important | Avis du 05/06/2019 | Extension d'indication | Le service médical rendu par BOTOX est important dans cette indication de l’AMM. |
| Important | Avis du 19/11/2014 | Extension d'indication | Le service médical rendu par BOTOX est important dans cette extension d’indication au traitement de l’hyperactivité vésicale idiopathique. |
| Important | Avis du 18/07/2012 | Extension d'indication | Le service médical rendu par cette spécialité est important dans l’extension d’indication : traitement de l’hyperactivité détrusorienne neurologique conduisant à une incontinence urinaire non contrôlée par un traitement anticholinergique chez les patients blessés médullaires et les patients atteints de sclérose en plaques et utilisant l’autosondage comme mode mictionnel. |
| Important | Avis du 13/01/2010 | Extension d'indication | Le service médical rendu par ces spécialités est important dans l’extension d’indication thérapeutique : traitement symptomatique local de la spasticité (hyperactivité musculaire) des membres supérieurs et/ou inférieurs chez l’enfant de plus de 2 ans. |
| Important | Avis du 06/09/2006 | Inscription (CT) | Le service médical rendu de ces spécialités est important dans les indications : « troubles de l'oculomotricité : strabisme, paralysies oculomotrices récentes, myopathie thyroïdienne récente, blépharospasme, spasme hémifacial, torticolis spasmodique », « traitement symptomatique local de la spasticité (hyperactivité musculaire) du membre supérieur et/ou inférieur chez l'adulte » et « Traitement de la déformation dynamique du pied en équin chez les enfants (de plus de 2 ans) présentant une spasticité due à une infirmité motrice cérébrale ». |
| Modéré | Avis du 06/09/2006 | Inscription (CT) | Le service médical rendu de ces spécialités est modéré dans l'indication : « Hyperhidrose axillaire sévère ayant résisté aux traitements locaux et entraînant un retentissement psychologique et social important ». |
Amélioration du service médical rendu (ASMR)
Les libellés affichés ci-dessous ne sont que des résumés ou extraits issus des avis rendus par la Commission de la Transparence. Seul l'avis complet de la Commission de la Transparence fait référence.
Cet avis est consultable à partir du lien `Avis du jj/mm/aaaa` ou encore sur demande auprés de la HAS (plus d'informations dans l'aide). Les avis et synthéses d'avis contiennent un paragraphe sur la place du médicament dans la stratégie thérapeutique.
| Valeur de l'ASMR | Avis | Motif de l'évaluation | Résumé de l'avis |
|---|---|---|---|
| V (Inexistant) | Avis du 17/11/2021 | Extension d'indication | Compte-tenu : • de la démonstration de la supériorité de la toxine botulinique A, seulement à court terme, par rapport au placebo au cours d’une seule étude sur deux, randomisées en double-aveugle dans la migraine chronique sur : o la réduction du nombre de jours avec céphalées par mois à 24 semaines (critère de jugement principal) avec une quantité d’effet toutefois modérée (différence de -2,3 jours chez des patients présentant en moyenne 20 jours de céphalées par mois à l’inclusion) o les critères de jugement secondaires d’efficacité hiérarchisés suivants évalués à 24 semaines avec une quantité d’effet modérée : variation du nombre de jours de migraine/migraine probable par mois, variation du nombre de jours avec céphalée d’intensité modérée/sévère par mois, variation du nombre cumulé total d’heures de céphalées pendant les jours avec céphalée par mois et variation de la fréquence des épisodes avec céphalée par mois • de la démonstration de la supériorité par rapport au placebo au cours de cette même étude sur le critère de jugement secondaire hiérarchisé de qualité de vie de pourcentage de patients ayant obtenu un score d’impact des céphalées HIT-6 sévère (i.e., = 60), mais au regard : • de l’absence de comparaison par rapport aux traitements de recours dont les anticorps anti-CGRP, la Commission considère que BOTOX (toxine botulinique A) n’apporte pas d’amélioration du service médical rendu (ASMR V) dans la stratégie thérapeutique actuelle en traitement prophylactique de la migraine chronique chez des patients adultes qui n’ont pas répondu ou sont intolérants aux autres traitements prophylactiques de la migraine |
| III (Modéré) | Avis du 05/06/2019 | Extension d'indication | Compte tenu des données disponibles, la Commission estime que l’amélioration du service médical rendu par BOTOX n’est pas modifié dans cette indication (ASMR modéré, niveau III), y compris lorsque les patients atteints de SEP ont un mode mictionnel spontané. |
| IV (Mineur) | Avis du 19/11/2014 | Extension d'indication | BOTOX apporte une amélioration du service médical rendu mineure (ASMR IV) dans le traitement de l’hyperactivité vésicale chez les patients en échec des traitements médicamenteux et non médicamenteux (traitements comportementaux et de la rééducation périnéo-sphinctérienne). |
| III (Modéré) | Avis du 18/07/2012 | Extension d'indication | BOTOX apporte une amélioration du service médical rendu modérée (ASMR III) dans la prise en charge de l'incontinence urinaire par hyperactivité détrusorienne neurologique non contrôlée par un traitement anticholinergique chez les patients blessés médullaires et chez les patients ayant une sclérose en plaques et utilisant l'autosondage comme mode mictionnel. |
| IV (Mineur) | Avis du 13/01/2010 | Extension d'indication | Les spécialités BOTOX (toxine botulinique de type A) apportent une amélioration du service médical rendu mineure (ASMR IV) en termes d'efficacité dans la prise en charge de la spasticité (hyperactivité musculaire) des membres supérieurs et/ou inférieurs chez l'enfant de 2 ans et plus. |
| V (Inexistant) | Avis du 06/09/2006 | Inscription (CT) | Ces spécialités sont un complément de gamme qui n'apporte pas d'amélioration du service médical rendu. |
Autres informations
- Titulaire de l'autorisation : ABBVIE
- Conditions de prescription et de délivrance :
- liste I
- réservé à l'usage HOSPITALIER
- Statut de l'autorisation : Valide
- Type de procédure : Procédure nationale
- Code CIS : 6 731 295 0
ANSM - Mis à jour le : 06/02/2026
BOTOX 200 UNITÉS ALLERGAN, poudre pour solution injectable
2. COMPOSITION QUALITATIVE ET QUANTITATIVE
Toxine botulinique de type A*1...................................................................... 200 unités*2 ALLERGAN
pour un flacon.
*1 (de Clostridium botulinum)
*2 Une unité correspond à la dose létale 50 (DL50) du produit reconstitué et injecté par voie intrapéritonéale chez la souris.
Les unités de toxine botulinique ne sont pas interchangeables d’un produit à l’autre.
Pour la liste complète des excipients, voir rubrique 6.1.
Poudre pour solution injectable.
Poudre blanche.
Le produit BOTOX apparaît sous la forme d'un fin dépôt blanc qui peut être difficile à voir dans le fond du flacon.
4.1. Indications thérapeutiques
Dysfonctions vésicales
Traitement de l’hyperactivité vésicale idiopathique associée à des symptômes incluant :
· 3 épisodes d’incontinence urinaire avec urgenturie sur 3 jours,
et
· fréquence urinaire définie par un nombre de mictions ≥ 8 par jour et ne répondant pas de manière adéquate aux anticholinergiques (après 3 mois de traitement) ou intolérants au traitement anticholinergique et ne répondant pas à une kinésithérapie bien conduite.
Traitement de l’hyperactivité détrusorienne neurologique conduisant à une incontinence urinaire non contrôlée par un traitement anticholinergique chez :
· les patients blessés médullaires,
· les patients atteints de sclérose en plaques.
Troubles neurologiques
Traitement prophylactique de la migraine chronique (présence de céphalées au moins 15 jours par mois dont au moins 8 jours de migraine par mois) chez des patients adultes qui n’ont pas répondu ou sont intolérants aux autres traitements prophylactiques de la migraine.
Adultes et enfants de plus de 12 ans
· Troubles de l'oculomotricité : strabisme, paralysies oculomotrices récentes, myopathie thyroïdienne récente.
· Blépharospasme.
· Spasme hémifacial.
· Torticolis spasmodique.
· Hyperhidrose axillaire sévère ayant résisté aux traitements locaux et entraînants un retentissement psychologique et social important.
Adultes et enfants de 2 ans et plus
· Traitement symptomatique local de la spasticité (hyperactivité musculaire) des membres supérieurs et/ou inférieurs.
4.2. Posologie et mode d'administration
BOTOX doit être administré dans le cadre d’une prise en charge globale multidisciplinaire par des médecins spécialistes ayant déjà une bonne expérience de l’utilisation de la toxine dans ces indications et avec un plateau technique adapté.
Les doses recommandées de BOTOX ne sont pas interchangeables avec les autres préparations de toxines botuliniques. Elles sont exprimées en UNITÉS ALLERGAN (voir rubrique 4.4) et sont différentes des autres préparations de toxine botulinique.
Si différentes présentations de BOTOX sont utilisées dans le cadre d’une procédure d’injection, une attention doit être apportée à l’utilisation de la bonne quantité de solvant en reconstituant le nombre d’unités par 0,1 ml déterminé. La quantité de solvant varie entre BOTOX 50 UNITÉS ALLERGAN, BOTOX 100 UNITÉS ALLERGAN et BOTOX 200 UNITÉS ALLERGAN. Chaque seringue sera étiquetée en conséquence.
En cas d'antécédents d'atteinte neurogène de la face, il est recommandé de réduire la posologie lors de la première séance d'injections (voir rubrique 4.4).
Patients âgés
Le traitement initial doit être débuté avec la plus faible dose recommandée pour l’indication considérée. Les patients âgés ayant des antécédents médicaux significatifs et des traitements concomitants doivent être traités avec prudence.
Population pédiatrique
La sécurité et l’efficacité de BOTOX dans d’autres indications que celles décrites dans la rubrique 4.1 du Résumé des Caractéristiques du Produit pour la population pédiatrique n’ont pas été établies. Dans la population pédiatrique, aucune recommandation posologique ne peut être faite pour d’autres indications que le traitement symptomatique local de la spasticité. Pour cette indication, BOTOX ne doit être administré que par des médecins expérimentés dans l’évaluation et le traitement symptomatique local de la spasticité pédiatrique et dans le cadre d’un programme structuré de réadaptation.
Les données actuellement disponibles dans la population pédiatrique sont décrites dans les rubriques 4.2, 4.4, 4.8, 5.1 du Résumé des Caractéristiques du Produit comme indiqué dans le tableau ci-dessous.
|
Troubles de l’oculomotricité |
12 ans (voir rubriques 4.4 et 4.8) |
|
Blépharospasme/Spasme hémifacial |
12 ans (voir rubriques 4.4 et 4.8) |
|
Dystonie cervicale |
12 ans (voir rubriques 4.4 et 4.8) |
|
Traitement symptomatique local de la spasticité |
2 ans (voir rubriques 4.2, 4.4 et 4.8) |
|
Hyperhidrose axillaire sévère |
12 ans (expérience limitée chez les adolescents entre 12 et 17 ans, voir rubriques 4.4, 4.8 et 5.1) |
|
Hyperactivité détrusorienne neurologique chez l’enfant |
5-17 ans (voir rubriques 4.8 et 5.1) |
|
Hyperactivité vésicale chez l’enfant |
12-17 ans (voir rubriques 4.8 et 5.1) |
Intervalle minimum entre 2 séances d'injection
La présence d’anticorps dirigés contre la toxine botulinique de type A peut réduire l’efficacité du traitement par BOTOX. En conséquence, par mesure de prudence, un intervalle minimum entre 2 séances d'injection doit être respecté :
· Pour l'indication dans l'hyperhidrose axillaire sévère ayant résisté aux traitements locaux : 4 mois.
· Pour l'indication traitement symptomatique local de la spasticité (hyperactivité musculaire) des membres supérieurs et/ou inférieurs chez l'adulte et l'enfant de 2 ans et plus : 3 mois.
· Pour les autres indications chez l'adulte et l'enfant de plus de 12 ans : 2 mois et 10 semaines pour le traitement du torticolis spasmodique.
· Pour l’indication dans l’hyperactivité vésicale chez l’adulte : lorsque les bénéfices de l’injection précédente s’estompent (en général, 6 mois) et en respectant un intervalle minimum de 3 mois.
· Pour l’indication dans l’hyperactivité détrusorienne neurologique chez l’adulte : lorsque les bénéfices de l’injection précédente s’estompent (en général, 9 mois) et en respectant un intervalle minimum de 3 mois.
· Pour l’indication dans la migraine chronique chez l’adulte : 3 mois.
Technique d'injection
Voie intramusculaire stricte ou intradermique stricte suivant l'indication. Se reporter aux recommandations spécifiques à chaque indication, décrites plus bas.
De façon générale, la dose optimale comme le nombre de sites d’injection par muscle n’ont pas été établis pour toutes les indications. Dans ce cas, les schémas posologiques devront être établis individuellement par le praticien. Les doses optimales doivent toujours être établies par titration et ne doivent pas dépasser la dose maximale recommandée.
Après désinfection de la peau, la dose de BOTOX diluée est injectée à l'aide d'une aiguille stérile de calibre adapté au muscle à injecter.
Le guidage électromyographique peut augmenter la précision de l'injection : l'activité électrique enregistrée par la pointe de l'aiguille d'injection est utilisée comme guide pour le positionnement dans le muscle cible.
Ce guidage est impératif dans le traitement du strabisme.
Chez l’enfant, les injections de toxine botulinique doivent être administrées par des médecins spécialisés et ayant une forte expérience de ce traitement chez l’enfant. Ce traitement médicamenteux doit être inclus dans une prise en charge globale multidisciplinaire (associant neurologue, pédiatre, médecin de médecine physique et de réadaptation, chirurgien orthopédiste ) et associé à une prise en charge réadaptative.
Chez les patients adultes atteints d’hyperactivité vésicale, ce traitement médicamenteux par injection dans le détrusor doit être inclus dans une prise en charge globale multidisciplinaire associant médecin urologue et gynécologue-obstétricien ayant reçu une formation spécifique d’utilisation de la toxine botulinique dans cette indication sous la supervision d’un urologue. Les injections de toxine botulinique doivent être réalisées sous visualisation cystoscopique, via un cystoscope flexible ou rigide, en évitant le trigone.
Chez les patients adultes atteints d’hyperactivité détrusorienne neurologique, ce traitement médicamenteux par injection dans le détrusor doit être inclus dans une prise en charge globale multidisciplinaire associant médecin urologue et médecin de médecine physique et de réadaptation ayant reçu une formation spécifique d’utilisation de la toxine botulinique dans cette indication sous la supervision d’un urologue. Les injections de toxine botulinique doivent être réalisées sous visualisation cystoscopique, via un cystoscope flexible ou rigide, en évitant le trigone.
Pour les instructions concernant la reconstitution du médicament avant administration, la manipulation et l’élimination, voir la rubrique 6.6.
Après reconstitution, BOTOX ne doit être utilisé que pour une seule séance d'injections pour un seul patient.
BLÉPHAROSPASME
1) Préparation du produit
Préparer une solution contenant 2,5 unités pour 0,1 ml.
2) Posologie et mode d'administration
Utiliser une aiguille de 27 ou 30 gauges (0,40 ou 0,30 mm).
Le guidage électromyographique n'est pas nécessaire.
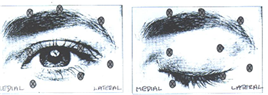
La dose initiale recommandée pour le traitement du blépharospasme bilatéral est de 17,5 unités par œil (0,70 ml) avec la répartition suivante :
· 7,5 unités (0,30 ml) répartis entre trois sites (2,5 unités par site) : partie interne et externe du muscle orbiculaire de la paupière supérieure et partie externe prétarsienne du muscle orbiculaire de la paupière inférieure.
· 5 unités (0,20 ml) dans l'arcade sourcilière répartis en 2 sites (2,5 unités par site).
· 5 unités (0,20 ml) dans la zone faciale supérieure.
La dose initiale ne doit pas dépasser 25 unités (1 ml) par orbiculaire.
Le fait d’éviter d'injecter près du muscle releveur de la paupière supérieure permet de réduire la complication par un ptosis.
Le fait d’éviter d’injecter dans la paupière inférieure médiane, et donc d’atténuer la diffusion dans le petit oblique, permet de réduire la complication par une diplopie.
Les schémas suivants indiquent les sites possibles d'injection :
|
|
En général, l'effet des injections apparaît dans les trois jours et atteint un pic une ou deux semaines après le traitement. Chaque traitement dure environ trois mois, à la suite desquels la procédure peut être répétée indéfiniment. Lors des réinjections, la dose peut être augmentée jusqu'à deux fois si le résultat du traitement initial est considéré insuffisant. Toutefois, il semble n'y avoir qu'un faible bénéfice à injecter plus de 5 Unités par site.
La dose totale ne doit pas dépasser 100 Unités (4 ml) tous les 3 mois.
Normalement, un traitement plus fréquent que tous les 3 mois n'apporte aucun bénéfice supplémentaire.
SPASME HÉMIFACIAL
Les doses et la technique d'injection sont identiques à celles décrites pour le traitement du blépharospasme unilatéral.
Un contrôle électromyographique peut être nécessaire pour identifier les petits muscles circulaires.
Les patients présentant un spasme hémifacial ou des troubles du VIIe nerf crânien seront traités comme pour un blépharospasme unilatéral ; les autres muscles affectés du visage étant injectés si besoin.
TORTICOLIS SPASMODIQUE
1) Préparation du produit
Préparer une solution contenant 10 unités pour 0,1 ml.
2) Posologie et mode d'administration
Utiliser une aiguille de 25, 27 ou 30 gauges (0,50, 0,40 ou 0,30 mm).
Lors des essais cliniques, le traitement du torticolis spasmodique consistait typiquement en l'injection de BOTOX dans le(s) muscle(s) sterno-cléido-mastoïdien(s), releveur(s) de l'omoplate, scalène, splénius de la tête, grand complexius, transversaire du cou, et/ou trapèze. Cette liste n'est pas exhaustive ; tout muscle responsable du contrôle de la position de la tête peut être impliqué et donc nécessiter un traitement.
La masse musculaire et le degré d'hypertrophie ou d'atrophie sont des facteurs à prendre en considération lors de la sélection de la dose appropriée. Les schémas d'activation musculaire peuvent changer spontanément en cas de torticolis spasmodique, sans changement de la présentation clinique de la dystonie.
En cas de difficulté pour isoler les muscles à injecter, les injections doivent être faites avec assistance électromyographique.
· Les doses initiales recommandées sont les suivantes :
· sterno-cleido-mastoïdien : 40 à 75 Unités (0,40 à 0,75 ml), splénius : 75 Unités - 50 à 150 Unités (0,75 ml) et trapèze : 75 Unités - 50 à 100 Unités (0,75 ml). Pour chaque muscle, la dose sera répartie en 3 sites.
· élévateur de l'omoplate : 50 Unités (0,5 ml) répartis en 2 sites ;
· scalène : 25 Unités (0,25 ml).
Lors des essais cliniques contrôlés initiaux visant à établir la tolérance et l'efficacité dans le torticolis spasmodique (dystonie cervicale), les doses de solution reconstituée de BOTOX allaient de 140 à 280 Unités.
Lors d'études plus récentes, les doses allaient de 95 à 360 Unités (avec une moyenne approximative de 240 Unités). Comme pour tout traitement médicamenteux, la dose initiale chez un patient naïf sera la dose minimale efficace.
Ne pas administrer plus de 50 unités (0,50 ml) par site.
Afin de minimiser le risque de dysphagie, le sterno-cleido-mastoïdien ne doit pas être injecté de façon bilatérale, ni recevoir plus de 100 unités (1 ml) par séance.
La dose totale ne doit jamais excéder 200 unités (2 ml) lors de la première séance.
Des ajustements pourront être faits lors des séances suivantes en fonction de la réponse initiale, sans dépasser une dose totale de 300 unités (3 ml) par séance.
Le nombre optimal de sites d’injection dépend de la taille du muscle.
L'amélioration clinique apparaît généralement au cours des deux semaines suivant l'injection. Le bénéfice clinique apparaît généralement vers la sixième semaine après l'injection. Une nouvelle injection peut être faite lorsque l'effet clinique de l'injection précédente a diminué.
La durée de l'effet bénéfique rapportée dans les essais cliniques a montré des variations importantes (de 2 à 33 semaines), avec une durée plus souvent rencontrée de 3 mois, le tout dépendant des symptômes et des réponses individuelles des patients. Le schéma posologique doit donc être adapté aux besoins de chaque patient.
Les séances d'injection doivent être répétées en fonction de la durée de l'effet clinique. Elles seront toujours espacées d'au moins 10 semaines.
STRABISME
1) Préparation du produit
Préparer une solution contenant 2,5 unités pour 0,1 ml.
2) Posologie et mode d'administration
Utiliser une aiguille de 27 gauges longue de 40 mm.
BOTOX est destiné à être injecté dans les muscles extra-oculaires en utilisant impérativement un guidage électromyographique (voir Technique d'injection).
Afin de préparer l'œil à l'injection de BOTOX, il est recommandé d'administrer quelques minutes avant l'injection, quelques gouttes d'anesthésique local et de décongestionnant oculaire.
Doses initiales : utiliser les plus petites doses pour le traitement de faibles déviations et les doses plus fortes pour les déviations importantes.
Pour les muscles verticaux et pour le strabisme horizontal de moins de 20 dioptries prismatiques : 1,25 unités à 2,5 unités (de 0,05 ml à 0,10 ml) quel que soit le muscle.
Pour le strabisme horizontal de 20 à 50 dioptries prismatiques : 2,5 unités à 5 unités (de 0,10 ml à 0,20 ml) quel que soit le muscle.
Pour une paralysie persistante du nerf moteur externe d'un mois ou plus : 1,25 unités à 2,5 unités (0,05 ml à 0,10 ml) dans le droit interne.
Les doses initiales de BOTOX dilué provoquent habituellement la paralysie des muscles injectés un à deux jours après l'injection : l'intensité de cette paralysie augmente pendant la première semaine. La paralysie dure 2 à 6 semaines et se résorbe progressivement pendant une période à peu près équivalente. Les sur-corrections durant plus de 6 mois sont rares.
La moitié des patients environ aura besoin de doses supplémentaires, du fait d'une paralysie insuffisante du muscle après la dose initiale, ou à cause de facteurs mécaniques tels que d'importantes déviations ou restrictions, ou encore à cause du manque de fusion motrice binoculaire pour stabiliser l'alignement.
ADULTE : TRAITEMENT SYMPTOMATIQUE LOCAL DE LA SPASTICITÉ (HYPERACTIVITÉ MUSCULAIRE) DES MEMBRES SUPÉRIEURS ET/OU INFÉRIEURS
1) Préparation du produit
Préparer une solution dont la dilution est adaptée à la posologie.
2) Posologie et mode d'administration
Posologie
La posologie exacte et le nombre de sites d'injection doivent être adaptés à chaque individu d'après la taille, le nombre et l'emplacement des muscles impliqués, la sévérité de la spasticité, la présence d'une faiblesse musculaire localisée et la réponse du patient au traitement précédent.
Posologies moyennes par muscle injecté :
|
Muscle |
Dose totale ; Nombre de sites |
|
Biceps brachial (Biceps brachii) |
100 à 200 Unités; jusqu'à 4 sites |
|
Fléchisseur commun profond des doigts (Flexor digitorum profundus) |
15 à 50 Unités; 1 à 2 sites |
|
Fléchisseur commun superficiel des doigts (Flexor digitorum sublimis) |
15 à 50 Unités; 1 à 2 sites |
|
Grand palmaire (Flexor carpi radialis) |
15 à 60 Unités; 1 à 2 sites |
|
Cubital antérieur (Flexor carpi ulnaris) |
10 à 50 Unités; 1 à 2 sites |
|
Adducteur du pouce (Adductor pollicis) |
20 Unités; 1 à 2 sites |
|
Long fléchisseur propre du pouce (Flexor pollicis longus) |
20 Unités; 1 à 2 sites |
|
Tibial postérieur (Posterior tibialis) |
50 à 150 Unités; 2 à 4 sites |
|
Tibial antérieur (Anterior tibialis) |
70 à 100 Unités; 2 sites |
|
Soléaire (Soleus) |
50 à 200 Unités; 2 à 4 sites |
|
Long fléchisseur commun et court fléchisseur des orteils (Flexor digitorum longus/brevis) |
50 à 150 Unités; 2 à 4 sites |
|
Gastrocnémien chefs médial et latéral (Gastrocnemius medial/lateral) |
50 à 200 Unités; 2 à 4 sites |
|
Long extenseur de l'hallux (extensor hallucis longus) |
50 à 100 Unités; 2 à 4 sites |
|
Adducteurs (adductor) |
50 à 200 Unités; 2 à 4 sites |
|
Ischiojambiers et sartorius (biceps femoris, semitendinosus, semimembranosus, sartorius) |
50 à 200 Unités; 2 à 4 sites |
Dans les essais cliniques, la dose totale administrée par séance d'injection n'a pas dépassé 360 Unités. La dose totale doit être répartie entre les différents muscles sélectionnés.
En général, la dose totale maximale est fixée à 6 Unités/kg.
Mode d’administration
Utiliser une aiguille de 25, 27 ou 30 gauges pour les muscles superficiels et une aiguille plus longue pour les muscles plus profonds.
Afin d'isoler les muscles concernés, le recours à un guidage électromyographique ou à des techniques de stimulation nerveuse peut être utile. Le choix de plusieurs sites d'injection par muscle permet de répartir plus uniformément BOTOX et s'avère particulièrement utile pour les gros muscles.
L'amélioration clinique de l'hypertonie musculaire s'observe généralement au cours des deux semaines qui suivent la séance d'injection. L'effet clinique maximal apparaît généralement quatre à six semaines après le traitement. Les séances d'injection pourront être répétées si besoin, mais seront toujours espacées d'au moins 3 mois.
Dans les essais cliniques, l'intervalle entre 2 séances d'injection était compris entre 12 et 16 semaines.
Lors d'une nouvelle séance d'injection, l'intensité et le type de spasticité musculaire peuvent conduire à modifier la dose de BOTOX administrée et le choix des muscles à injecter.
ENFANT DE 2 ANS ET PLUS : TRAITEMENT SYMPTOMATIQUE LOCAL DE LA SPASTICITÉ (HYPERACTIVITÉ MUSCULAIRE) DES MEMBRES SUPÉRIEURS ET/OU INFÉRIEURS
1) Préparation du produit
Préparer une solution dont la dilution est adaptée à la posologie.
2) Posologie et mode d'administration
Après reconstitution, BOTOX est injecté avec une aiguille stérile de 27 ou 30 gauges et de longueur adaptée aux muscles ciblés.
Pour localiser les muscles concernés, le recours à un guidage électromyographique (EMG) ou à des techniques de stimulation nerveuse peut être utile.
La posologie exacte et le nombre de sites d'injection doivent être adaptés à chaque individu d'après la taille, le nombre et l'emplacement des muscles impliqués, la sévérité de la spasticité, la présence d'une faiblesse musculaire locale et la réponse du patient au traitement précédent. Dans les essais cliniques, des doses par muscle comprises entre 0,5 et 2,0 Unités/kg de masse corporelle pour le membre supérieur et de 2,0 à 4,0 Unités/kg de masse corporelle pour le membre inférieur ont été administrées à chaque séance de traitement.
Ne pas dépasser la dose initiale recommandée à l'initiation du traitement.
Dans le traitement du pied en équin, deux injections sont faites dans chacun des chefs médial (interne) et latéral (externe) du muscle gastrocnémius (jumeau) atteint.
En cas d'hémiplégie, la dose initiale recommandée est de 4 Unités par kg injectées dans le membre concerné.
En cas de diplégie, la dose initiale recommandée est de 6 Unités par kg, à répartir entre les deux membres concernés.
Il conviendra de strictement respecter la dose initiale recommandée chez les enfants, en particulier, pour ceux :
· qui présentent des comorbidités associées notamment celles avec troubles de déglutition ou respiratoire préexistant,
· dont les muscles à traiter sont peu développés,
· qui nécessitent une injection multisite,
· qui bénéficient d'injections sous anesthésie générale.
A titre indicatif, le tableau ci-dessous fournit des directives pour l'injection de BOTOX dans le traitement de la spasticité locale chez les enfants âgés de 2 ans et plus.
Dans tous les cas, lors du choix de la dose, une évaluation individuelle du rapport bénéfice/risque devra être envisagée, afin de réduire le risque des effets indésirables notamment le risque de diffusion de la toxine à distance du site d'administration (voir rubriques 4.4 et 4.8). En fonction de la réponse au traitement précédent, la dose peut être augmentée au-dessus de la dose initiale recommandée avec une extrême précaution, sans toutefois dépasser la dose maximale par session indiquée ci-dessous. La dose et le plan de traitement devront être réévalués en cas de survenue d'effets indésirables.
|
Enfants âgés de 2 ans et plus (muscles cités à titre indicatif) |
Dose par muscle (Unités/kg) |
Dose maximale par session |
|
Muscles du membre supérieur |
15 Unités/kg ou 350 Unités ou 50 Unités par site |
|
|
Biceps brachii, brachialis, brachioradialis |
1-2 |
|
|
Pronator quadratus |
0,5-1 |
|
|
Pronator teres |
1-2 |
|
|
Flexor carpi ulnaris/radialis |
1-2 |
|
|
Flexor pollicis longus/brevis/opponens |
0,5-1 |
|
|
Adductor pollicis |
0,5-1 |
|
|
Flexor digitorum profundis/superficialis |
1-2 |
|
|
Muscles du membre inférieur |
||
|
Adductor longus/brevis/magnus |
2-4 |
|
|
Biceps femoris, semitendinosus, semimembranosus, sartorius |
2-4 |
|
|
Gastrocnemius |
2-4 |
|
|
Soleus |
2-3 |
L'amélioration clinique survient généralement au cours des deux semaines qui suivent la séance d'injection. Les séances d'injection doivent être répétées en fonction de la durée de l'effet clinique.
Elles seront toujours espacées d'au moins 3 mois. Une posologie adaptée devrait permettre d'obtenir un intervalle d'au moins 6 mois entre deux séances.
HYPERHIDROSE AXILLAIRE
1) Préparation du produit
Préparer une solution contenant 100 Unités pour 4 ml ou 2,5 Unités pour 0,1 ml.
2) Posologie et mode d'administration
Utiliser une aiguille de 30 gauges.
Injecter 50 Unités de BOTOX en injection intradermique stricte, réparties uniformément en plusieurs sites de la zone d'hyperhidrose de chaque aisselle distants les uns des autres d'environ 1 à 2 cm. La zone d'hyperhidrose peut être déterminée en utilisant des méthodes standardisées, comme la méthode de Minor (test à l'iode). Des doses différentes de 50 Unités par aisselle n'ont pas été étudiées et ne peuvent donc pas être recommandées.
L'amélioration clinique survient en général au cours de la première semaine suivant la séance d'injection.
La réponse au traitement est supérieure à 4 mois et peut durer 1 an ou plus. Des injections supplémentaires peuvent être faites lorsque l'effet clinique des injections précédentes diminue, mais il est nécessaire de respecter un délai minimum de 4 mois entre 2 séances d'injection.
ADULTES : DYSFONCTIONS VÉSICALES
1) Préparation du produit
Préparer une solution dont la dilution est adaptée à la posologie (voir rubrique 6.6).
2) Posologie et mode d'administration
Au moment du traitement, les patients ne doivent pas présenter d’infection urinaire (voir rubrique 4.3).
Un examen cyto-bactériologique des urines doit être systématiquement réalisé 5 jours avant le traitement. En cas de stérilité, une antibiothérapie prophylactique doit être administrée au patient 1 à 3 jours avant le traitement, le jour du traitement et 1 à 3 jours après le traitement. En cas de colonisation bactérienne asymptomatique, une antibiothérapie adaptée doit être initiée au moins 2 jours avant, poursuivie le jour du traitement et au moins 2 jours après.
Une interruption du traitement par antiagrégant plaquettaire est recommandée au moins 3 jours avant la procédure d’injection. Les patients sous anticoagulants doivent être pris en charge de façon appropriée pour réduire le risque de saignement.
ADULTES : HYPERACTIVITÉ VÉSICALE
Les patients doivent être informés que des sondages intermittents propres pour vider leur vessie pourront être nécessaires. Ils doivent eux-mêmes ou leur entourage être capables de les réaliser (voir rubrique 4.4).
Une instillation intra-vésicale d’une solution anesthésique diluée avec ou sans sédation associée peut être pratiquée avant l’injection selon les pratiques locales. En cas d’instillation d’anesthésique local, la vessie doit être drainée et rincée par une solution de chlorure de sodium stérile avant de poursuivre la procédure d’injection.
Débuter le traitement avec une dose de 50 Unités de BOTOX. Si la réponse est insuffisante, la dose étudiée de 100 Unités de BOTOX pourrait être utilisée lors des injections suivantes.
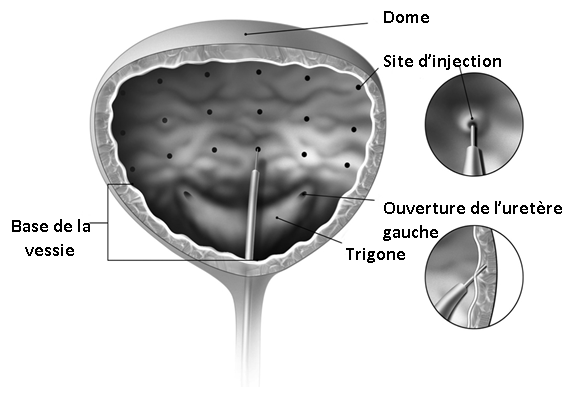
La solution reconstituée de BOTOX (voir rubrique 6.6) est injectée dans le muscle détrusorien via un cystoscope flexible ou rigide, en évitant le trigone. La vessie doit être instillée avec une quantité suffisante de solution de chlorure de sodium pour permettre une visualisation adéquate des injections, tout en évitant une distension excessive.
L’aiguille doit être remplie avec approximativement 1 ml de solution reconstituée de BOTOX (selon la longueur de l’aiguille) avant le début des injections afin de vider l’air du volume mort.
L’aiguille doit être introduite environ 2 mm à l’intérieur du détrusor, et les injections de 0,5 ml doivent être réparties en 20 sites espacés d’environ 1 cm (voir schéma ci-dessous). La dernière injection doit être réalisée avec 1 ml de solution de chlorure de sodium stérile afin de s’assurer que la dose totale aura été injectée. Une fois les injections réalisées, la solution de chlorure de sodium utilisée pour la visualisation des parois de la vessie ne doit pas être drainée afin que le patient puisse démontrer sa capacité à uriner avant de quitter l’établissement de santé. Le patient doit rester en observation au moins 30 minutes après la séance d’injection et jusqu’au retour à une miction spontanée.
L’amélioration clinique est généralement observée dans les 2 premières semaines suivant l’injection. Une nouvelle injection peut être considérée quand le bénéfice clinique de la précédente s’estompe (durée moyenne de l’effet observée, basée sur une demande de retraitement exprimée par le patient, dans les études de phase III avec 100 Unités de BOTOX : 166 jours (environ 24 semaines)), et en respectant un intervalle minimum de 3 mois. Pour les patients ayant reçu 100 Unités de BOTOX dans les études de phase III puis dans l’étude d’extension en ouvert (N=438), la durée moyenne de l’effet observée, basée sur une demande de retraitement exprimée par le patient, était de 212 jours (environ 30 semaines).
ADULTES : HYPERACTIVITÉ DÉTRUSORIENNE NEUROLOGIQUE
Les patients doivent être informés que, s’ils n’avaient pas recours à l’autosondage avant le traitement par BOTOX, des sondages intermittents propres pourront être nécessaires pour vider leur vessie au cours du traitement et après le traitement. Ils doivent eux-mêmes ou leur entourage être capables de les réaliser (voir rubrique 4.4).
Une instillation intravésicale d’une solution anesthésique diluée avec ou sans sédation associée, ou une anesthésie générale peut être pratiquée avant l’injection selon les pratiques locales. En cas d’instillation d’anesthésique local, la vessie doit être drainée et rincée par une solution de chlorure de sodium stérile avant de poursuivre la procédure d’injection.
Chez les blessés médullaires, la dose recommandée est de 200 Unités de BOTOX.
Chez les patients atteints de sclérose en plaques, la dose initiale recommandée est de 100 Unités de BOTOX. Chez les patients utilisant l’autosondage comme mode mictionnel, une dose de 200 Unités de BOTOX pourra être envisagée. L’injection est réalisée sous visualisation cystoscopique, via un cystoscope flexible ou rigide, en évitant le trigone. La vessie doit être instillée avec une quantité suffisante de solution de chlorure de sodium pour permettre une visualisation adéquate des injections, tout en évitant une distension excessive.
L’aiguille doit être remplie avec approximativement 1 ml de solution reconstituée de BOTOX (selon la longueur de l’aiguille) avant le début des injections afin de vider l’air du volume mort.
L’aiguille doit être introduite environ 2 mm à l’intérieur du détrusor, et les injections de 1 ml doivent être réparties en 30 sites espacés d’environ 1 cm (voir schéma). La dernière injection doit être faite avec 1 ml de solution de chlorure de sodium stérile afin de s’assurer que la dose totale aura été injectée. Une fois les injections réalisées, la solution de chlorure de sodium utilisée pour la visualisation des parois de la vessie doit être drainée. Le patient doit rester en observation au moins 30 minutes après la séance d’injection.
L’amélioration clinique est généralement observée dans les 2 premières semaines suivant l’injection. Une nouvelle injection peut être considérée quand le bénéfice clinique de la précédente s’estompe, environ 9 mois après (durée moyenne de l’effet observée, basée sur une demande de retraitement exprimée par le patient, dans les études de phase III : 256 à 295 jours avec 200 Unités de BOTOX (environ 36-42 semaines)), et en respectant un intervalle minimum de 3 mois. Pour les patients ayant reçu 200 Unités de BOTOX dans les études de phase III puis dans l’étude d’extension en ouvert (N=174), la durée moyenne de l’effet observée, basée sur une demande de retraitement exprimée par le patient, était de 253 jours (environ 36 semaines).
|
|
ADULTES : MIGRAINE CHRONIQUE
1) Préparation du produit
Préparer une solution dont la dilution est adaptée à la posologie.
2) Posologie et mode d'administration
Utiliser une aiguille de 30 gauges longue de 13 mm.
Les injections doivent être réparties sur 7 zones musculaires spécifiques de la tête / du cou, comme indiqué dans le tableau ci-dessous. Une aiguille de 25 mm peut être nécessaire dans la région du cou pour les patients dont les muscles du cou sont extrêmement épais.
À l'exception du muscle procerus, qui doit être injecté en 1 site (ligne médiane), tous les muscles doivent être injectés bilatéralement avec la moitié des sites d'injection administrés du côté gauche et l’autre moitié du côté droit de la tête et du cou. Si une ou plusieurs localisations sont plus douloureuses, des injections supplémentaires peuvent être administrées unilatéralement ou bilatéralement dans un maximum de 3 groupes musculaires spécifiques (occipitalis, temporalis et trapezius), sans toutefois dépasser la dose maximale par muscle indiquée dans le tableau ci-dessous.
L’intervalle recommandé entre les traitements est de 3 mois. A l’issue des deux premiers cycles de traitement, la pertinence de poursuivre le traitement en l’absence d’efficacité doit être réévaluée avant chaque nouvelle séance de traitement.
Les sites d’injection sont répertoriés sur les schémas ci-dessous : 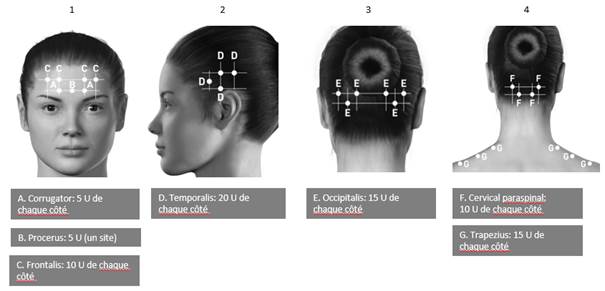
Les groupes de muscles recommandés pour les injections supplémentaires facultatives sont indiqués sur les schémas ci-après :
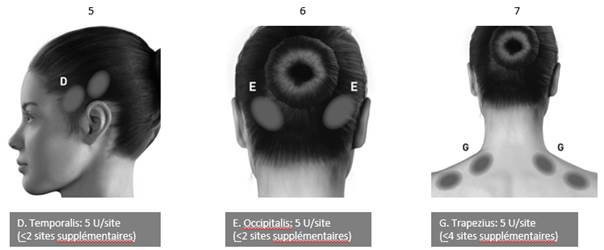
La dose recommandée est comprise entre 155 unités et 195 unités administrées par voie intramusculaire sous forme d'injections de 0,1 ml (5 unités) pratiquées dans 31 à 39 sites d’injection.
Posologies par muscle injecté :
|
|
Dose recommandée |
|
Zones de la tête/du cou |
Dose totale (nombre de sites d’injectiona) |
|
Corrugatorb |
10 Unités (2 sites) |
|
Procerus |
5 Unités (1 site) |
|
Frontalisb |
20 Unités (4 sites) |
|
Temporalisb |
40 Unités (8 sites) à 50 Unités (jusqu’à 10 sites) |
|
Occipitalisb |
30 Unités (6 sites) à 40 Unités (jusqu’à 8 sites) |
|
Muscles cervicaux paraspinauxb |
20 Unités (4 sites) |
|
Trapeziusb |
30 Unités (6 sites) à 50 Unités (jusqu’à 10 sites) |
|
Dose totale comprise entre: |
155 Unités à 195 Unités (31 à 39 sites) |
a1 site d’injection intramusculaire = 0.1 ml = 5 Unités de BOTOX
bDoses réparties de manière bilatérale
Toutes indications thérapeutiques :
En cas d'échec après une première séance de traitement, c'est-à-dire en l'absence, un mois après la séance, d'amélioration fonctionnelle significative par rapport au bilan initial, il y a lieu de :
· vérifier cliniquement, et au mieux par un examen électromyographique en milieu spécialisé, l'action de la toxine sur le(s) muscle(s) injecté(s),
· analyser les causes de l'échec :
o mauvaise sélection des muscles injectés,
o dose insuffisante,
o technique d'injection inadaptée,
o apparition d'une rétraction fixée,
o muscles antagonistes trop faibles,
o formation d’anticorps neutralisants,
· réévaluer la pertinence du traitement par la toxine botulinique de type A,
· en l'absence d'effet indésirable suite à la première séance de traitement, pratiquer une deuxième injection comme suit :
o ajuster la dose en prenant en compte les données de l'analyse de l'échec du traitement précédent,
o utiliser un guidage électromyographique,
o respecter l'intervalle de 3 mois entre la première et la deuxième injection.
En cas d’échec du traitement ou de diminution de l’effet après des injections répétées, des alternatives thérapeutiques devront être employées.
BOTOX est contre-indiqué dans les situations suivantes :
· hypersensibilité à la substance active ou à l’un des excipients mentionnés à la rubrique 6.1,
· myasthénie grave,
· en présence d’une infection au(x) site(s) d’injection.
Dans le traitement des patients atteints de dysfonctions vésicales associées à une incontinence urinaire, BOTOX est également contre-indiqué :
· chez les patients présentant une infection de l’appareil urinaire au moment du traitement,
· chez les patients présentant une rétention urinaire aigüe ou chronique chez lesquels le sondage intermittent est contre-indiqué ou refusé par le patient.
4.4. Mises en garde spéciales et précautions d'emploi
LA CONCENTRATION DE LA SOLUTION RECONSTITUÉE DE BOTOX EST EXPRIMÉE EN UNITÉS ALLERGAN.
ÉTANT DONNÉ L'ABSENCE D'HARMONISATION DES SYSTÈMES D'UNITÉS POUR LES DIFFÉRENTES TOXINES BOTULINIQUES COMMERCIALISÉES, IL EST NÉCESSAIRE DE FAIRE PREUVE D'UNE EXTRÊME PRUDENCE AU CAS OÙ LE PASSAGE D'UNE TOXINE BOTULINIQUE D'UN LABORATOIRE PHARMACEUTIQUE À LA TOXINE BOTULINIQUE D'UN AUTRE LABORATOIRE PHARMACEUTIQUE S'AVÉRERAIT NÉCESSAIRE.
RECOMMANDATIONS POUR LA RECONSTITUTION DU PRODUIT
La préparation du produit doit être réalisée dans un local approprié et par du personnel expérimenté (voir rubrique 6.6).
RECOMMANDATIONS EN CAS D'INCIDENT LORS DE LA MANIPULATION DE LA TOXINE
En cas d'incident lors de la manipulation de la toxine, des mesures appropriées doivent être prises (voir rubrique 6.6).
L'INJECTION DOIT ÊTRE REALISÉE PAR UN MÉDECIN SPÉCIALISTE AYANT UNE BONNE EXPÉRIENCE DE L'UTILISATION DE LA TOXINE BOTULINIQUE DANS LES INDICATIONS PRÉVUES PAR L'AUTORISATION DE MISE SUR LE MARCHÉ.
Les doses recommandées et les fréquences d’administration ne doivent pas être dépassées en raison du risque de surdosage, de faiblesse musculaire excessive, de diffusion de la toxine à distance du site d’administration et de formation d’anticorps neutralisants (voir rubrique 4.2). La dose initiale pour le traitement de patients naïfs doit correspondre à la plus faible dose recommandée pour l’indication considérée.
L'existence d'antécédents d'atteinte neurogène de la face (paralysie faciale, polyradiculonévrite...) nécessite lors de la première injection, d'utiliser des doses égales au quart de la dose recommandée.
Les professionnels de santé comme les patients doivent savoir que des effets indésirables peuvent survenir même si les injections précédentes ont été bien tolérées. Toutes les précautions doivent être prises lors de chaque administration.
Des effets indésirables, liés à la diffusion de la toxine à distance du site d'administration, ont été rapportés (voir rubrique 4.8), ayant parfois conduit à des décès suite à une dysphagie, une pneumonie et/ou une asthénie significative. Les symptômes sont cohérents avec le mécanisme d’action de la toxine botulinique et ont été rapportés quelques heures à quelques semaines après l’injection. Le risque d’apparition de ces symptômes est probablement plus important chez les patients présentant des pathologies sous-jacentes ou des comorbidités qui les prédisposent à ces symptômes, notamment les enfants et les patients adultes traités pour spasticité, et recevant de fortes doses.
Les patients traités à dose thérapeutique peuvent aussi présenter une faiblesse musculaire excessive.
Les patients âgés et les patients affaiblis doivent être traités avec prudence. Généralement, les études cliniques menées avec BOTOX n’ont pas identifié de différences dans la réponse au traitement entre les patients âgés et les patients plus jeunes. La sélection de la dose pour un patient âgé doit se faire avec prudence en commençant généralement par la dose la plus faible de l’intervalle de dose recommandé.
Le rapport bénéfice/risque doit être évalué pour chaque patient avant tout traitement par BOTOX.
Une dysphagie a également été rapportée après injection dans des sites autres que les muscles cervicaux (voir rubrique 4.4. "Torticolis spasmodique").
BOTOX ne sera utilisé qu'avec d'extrêmes précautions et sous surveillance étroite chez les patients ayant des manifestations infracliniques ou cliniques d'anomalie de la transmission neuromusculaire, par exemple une myasthénie grave ou un syndrome de Lambert-Eaton, chez les patients présentant des neuropathies périphériques motrices (telles que sclérose latérale amyotrophique ou neuropathie motrice) et chez les patients avec des troubles neurologiques sous-jacents.
Ces patients peuvent présenter une sensibilité accrue aux médicaments tels que BOTOX, même à des doses thérapeutiques, avec pour résultat une faiblesse musculaire excessive et un risque élevé d'effets systémiques cliniquement significatifs, y compris une dysphagie sévère et une atteinte de la fonction respiratoire.
La toxine botulinique doit être utilisée par des médecins spécialistes et uniquement si le bénéfice du traitement est supérieur aux risques.
Les patients ayant des antécédents de dysphagie et d’aspiration pulmonaire doivent être traités avec la plus grande prudence.
Les patients et leur entourage doivent être avertis de ces risques et de la nécessité d'une prise en charge médicale immédiate en cas de troubles de la déglutition, de troubles du langage ou de troubles respiratoires.
Comme pour tout traitement permettant à des patients précédemment sédentarisés de reprendre des activités, il est nécessaire de leur conseiller une reprise d'activité progressive.
Une connaissance de l'anatomie et de toute altération de l'anatomie due à des interventions chirurgicales antérieures, est nécessaire avant l'administration de BOTOX, et l'injection dans des structures anatomiques fragilisées doit être évitée.
Un pneumothorax lié à la procédure d’injection a été rapporté suite à l’administration de BOTOX au niveau du thorax. La prudence est recommandée lors d’injections proches des poumons, particulièrement au niveau des apex pulmonaires ou d’autres structures anatomiques sensibles.
Des effets indésirables graves d’évolutions fatales ont été rapportés chez des patients ayant reçu des injections hors AMM de BOTOX directement dans les glandes salivaires, la sphère oro-linguo-pharyngée, l’œsophage et l’estomac. Certains patients présentaient une dysphagie ou une faiblesse significative.
Des réactions d'hypersensibilité grave et/ou immédiate, incluant anaphylaxie, maladie sérique, urticaire, œdème des tissus mous et dyspnée, ont été rarement rapportées. Certaines de ces réactions ont été rapportées après utilisation de BOTOX seul ou en association à d'autres produits impliqués dans des réactions similaires. En cas de survenue d'une telle réaction, les injections de BOTOX doivent être arrêtées et un traitement médical approprié, tel que l’adrénaline, doit être immédiatement instauré. Un cas d'anaphylaxie a été rapporté dans lequel le patient est décédé, après avoir reçu une injection de BOTOX inadéquatement dilué dans 5 ml de lidocaïne à 1%.
Comme pour toute injection, des traumatismes liés à la procédure sont possibles. Une injection peut entraîner localement une infection, une douleur, une inflammation, des paresthésies, une hypoesthésie, une sensibilité, un gonflement, un érythème, et/ou un saignement/une ecchymose. La douleur et/ou l'anxiété liée à l'aiguille peuvent entraîner des réactions vasovagales telles que syncope, hypotension etc.
Des précautions sont nécessaires en cas d'utilisation de BOTOX en présence d'une inflammation au(x) site(s) proposé(s) d'injection, ou d'une faiblesse excessive ou d'une atrophie du muscle cible. Des précautions sont également nécessaires en cas d'utilisation de BOTOX pour traiter des patients ayant une neuropathie motrice périphérique (telle qu’une sclérose latérale amyotrophique ou une neuropathie motrice).
Des effets indésirables impliquant le système cardiovasculaire, dont arythmie et infarctus du myocarde, pouvant parfois être fatals, ont été également rapportés. Certains de ces patients présentaient des facteurs de risque, dont une maladie cardiovasculaire.
De nouvelles crises convulsives ou des convulsions récurrentes ont été rapportées, typiquement chez des patients prédisposés à de tels événements. La relation exacte de ces événements avec l'injection de toxine botulinique n'a pas été établie. Les cas recueillis chez l'enfant concernaient principalement des patients atteints de paralysie cérébrale traités pour une spasticité.
La formation d'anticorps neutralisants contre la toxine botulinique de type A est susceptible de réduire l'efficacité du traitement par BOTOX en inactivant l'action biologique de la toxine. Les résultats de certaines études suggèrent que des injections de BOTOX à des intervalles plus courts ou à des doses plus élevées pourraient conduire à une incidence plus élevée de formation d'anticorps. Le cas échéant, le potentiel de formation d'anticorps peut être minimisé en injectant la dose minimale efficace en respectant les intervalles les plus longs cliniquement recommandés entre les injections.
Les variations dans la réponse clinique constatée lors d’injections répétées de BOTOX (comme avec les autres toxines botuliniques) peuvent résulter des différences entre les procédures de reconstitution, les intervalles entre les injections, les muscles injectés et les faibles variations des valeurs d’activité de la toxine en fonction du test biologique utilisé.
Ce médicament contient moins de 1 mmol (23 mg) de sodium par flacon, c.-à-d. qu’il est essentiellement « sans sodium ».
Population pédiatrique
La sécurité et l’efficacité de BOTOX dans des indications autres que celles décrites dans la rubrique 4.1 pour la population pédiatrique n’ont pas été établies. De très rares cas de pharmacovigilance de possible diffusion de la toxine à distance du site d’injection ont été rapportés après la commercialisation de BOTOX chez des patients pédiatriques présentant des comorbidités, principalement une paralysie cérébrale. Généralement, la dose utilisée dans ce cas était au-dessus de celle recommandée (voir rubrique 4.8).
De très rares cas spontanés de décès parfois associés à une pneumonie d’inhalation ont concerné des enfants atteints d'infirmité motrice cérébrale sévère, après traitement par la toxine botulinique, dont des cas d’utilisation hors AMM (par exemple dans la région du cou). Une précaution extrême est nécessaire lors du traitement de patients pédiatriques ayant un déficit neurologique significatif, une dysphagie, ou des antécédents récents de pneumonie par inhalation ou de pneumopathie.
Le traitement de patients présentant un mauvais état général ne doit se faire que si le bénéfice potentiel est supérieur aux risques.
Dans la déformation dynamique du pied en équin chez les enfants présentant une infirmité motrice cérébrale, une évaluation fonctionnelle initiale précise doit être effectuée en milieu spécialisé. Elle permet :
· d'évaluer la pertinence de l'indication :
o spasticité prédominante,
o absence de faiblesse musculaire parfois masquée par l'hypertonie. Cette faiblesse pourrait être aggravée par une injection de toxine botulinique,
o absence de rétraction fixée importante ou de cicatrice post-chirurgicale rendant inutile une injection de toxine botulinique,
· de déterminer les différentes composantes du traitement (kinésithérapie, port d'attelles...),
· d'adapter le traitement en fonction de l'évolution du résultat de l'évaluation.
Blépharospasme
La diminution du clignement à la suite de l'injection de la toxine botulinique dans le muscle orbiculaire peut conduire à une exposition prolongée de la cornée, à une lésion épithéliale persistante et à une ulcération de la cornée en particulier chez les patients ayant présenté une paralysie faciale. Dans ce cas, des mesures préventives et curatives doivent être prises.
Un examen attentif de la sensibilité cornéenne des yeux ayant été opérés précédemment doit être réalisé, il ne faut pas effectuer d’injection dans la région de la paupière inférieure afin d'éviter un ectropion et un traitement efficace de toute lésion épithéliale est requis. Ceci peut nécessiter l’utilisation de gouttes oculaires protectrices, d’une pommade, de lentilles de contact thérapeutiques souples, ou la fermeture de l'œil par un patch occlusif ou d'autres moyens.
Des ecchymoses surviennent aisément dans les tissus mous palpébraux. Ceci peut être minimisé en appliquant une légère pression au site d’injection immédiatement après celle-ci.
En raison de l'action anticholinergique de la toxine botulinique, des précautions sont nécessaires lors du traitement de patients à risque de glaucome à angle fermé, y compris les patients ayant des angles anatomiquement étroits.
Torticolis spasmodique
Les patients atteints de torticolis spasmodique (dystonie cervicale) doivent être informés de la possibilité de survenue d'une dysphagie, laquelle peut être très légère mais peut également être sévère. La dysphagie peut persister durant deux à trois semaines après l'injection, mais a été rapportée jusqu'à cinq mois post-injection. Du fait de la dysphagie, il existe un risque potentiel d’aspiration pulmonaire, de dyspnée et occasionnellement de nécessité d’alimentation par sonde gastrique. De très rares cas de dysphagie ayant entraîné une pneumopathie d’inhalation et un décès ont été rapportés.
La limitation de la dose injectée dans le muscle sterno-cléido-mastoïdien à moins de 100 Unités pourrait diminuer la survenue de dysphagie. Il a été rapporté que les patients ayant une masse musculaire cervicale plus faible, ou que les patients recevant des injections sterno-cléido-mastoïdiennes bilatérales, présentaient un risque plus élevé de dysphagie.
La dysphagie est attribuée à la diffusion de la toxine aux muscles œsophagiens. Des injections dans le releveur de l'omoplate pourraient être associées à un risque accru d'infection des voies respiratoires supérieures et de dysphagie.
Une dysphagie pourrait contribuer à une diminution de la prise alimentaire et hydrique, avec pour conséquence une perte de poids et une déshydratation. Les patients ayant une dysphagie infraclinique pourraient encourir un risque accru de dysphagie plus sévère après une injection de BOTOX.
Spasticité des membres supérieurs/inférieurs chez l’enfant de plus de 2 ans et chez l’adulte
Dans le traitement symptomatique local de la spasticité (hyperactivité musculaire) des membres supérieurs et/ou inférieurs, BOTOX n'a été étudié qu'en association avec les traitements habituels, et ne vise pas à les remplacer. Il est peu probable que BOTOX puisse améliorer la mobilité d'une articulation bloquée par une contracture fixée.
Des cas de décès post-commercialisation ont été rapportés (parfois associés à des pneumopathies d’inhalation) et de diffusion de la toxine à distance du site d’injection chez des enfants présentant des co-morbidités, principalement une infirmité motrice cérébrale après traitement par toxine botulinique (voir les mises en garde de la rubrique 4.4 « Population pédiatrique »).
Hyperhidrose axillaire
Le recueil des antécédents médicaux et un examen clinique, ainsi que des explorations spécifiques supplémentaires le cas échéant, sont nécessaires pour éliminer des causes potentielles d'hyperhidrose secondaire (telles qu’une hyperthyroïdie ou un phéochromocytome). Ceci évitera le traitement symptomatique de l'hyperhidrose sans diagnostic et/ou le traitement de la pathologie sous-jacente.
Dans le traitement des patients atteints de dysfonctions vésicales :
Les précautions médicales d’usage doivent être mises en œuvre lors de la cystoscopie.
Chez les patients atteints d’hyperactivité vésicale :
Le volume résiduel d’urine post-mictionnel doit être évalué pendant les 2 semaines suivant le traitement puis de façon périodique selon avis médical jusqu’à 12 semaines. Les patients doivent être avertis de contacter leur médecin s’ils présentent des difficultés mictionnelles car les sondages intermittents seront nécessaires.
Les hommes atteints d’hyperactivité vésicale et présentant des signes ou symptômes d’obstruction urinaire ne doivent pas être traités par BOTOX.
Chez les patients atteints d’hyperactivité détrusorienne neurologique :
Le traitement de l’hyperactivité détrusorienne neurologique par injection intradétrusorienne de BOTOX peut conduire à une rétention urinaire et nécessiter que le patient utilise un sondage intermittent propre pour vider la vessie.
Par conséquent, les patients doivent être informés et doivent accepter que des sondages intermittents propres pour vider leur vessie seront nécessaires. Ils doivent, eux-mêmes ou leur entourage, être capables de les réaliser.
Chez les patients conservant des mictions spontanées au moins partielles, le volume résiduel d’urine post-mictionnel doit être évalué pendant les 2 semaines suivant le traitement puis de façon périodique selon avis médical jusqu’à 12 semaines. Les patients doivent être avertis de contacter leur médecin s’ils présentent des difficultés mictionnelles car les sondages intermittents seront nécessaires.
Une hyper-réflexie autonome associée à la procédure de cystoscopie et d’injection dans le détrusor peut survenir chez les patients traités pour hyperactivité détrusorienne neurologique. Une prise en charge médicale appropriée et rapide peut alors être nécessaire.
Chez les patients atteints de migraine chronique :
La sécurité et l’efficacité de BOTOX n’ont pas été établies dans la prophylaxie des céphalées chez les patients atteints de migraine épisodique (céphalées < 15 jours par mois) ou sur les céphalées de tension chroniques.
La cause la plus fréquente de symptômes évocateurs d’une migraine chronique est l’abus médicamenteux. Environ la moitié des patients ayant apparemment une migraine chronique reviennent à une migraine épisodique après le sevrage médicamenteux.
4.5. Interactions avec d'autres médicaments et autres formes d'interactions
L’utilisation de tels médicaments doit se faire avec prudence chez les patients traités par toxine botulinique.
L'effet de l'administration, concomitante ou séparée de plusieurs mois, de différents sérotypes de neurotoxine botulinique, n'est pas connu. Une faiblesse neuromusculaire excessive peut être exacerbée par l'administration d'une autre toxine botulinique avant la disparition totale des effets de la toxine botulinique administrée précédemment.
Aucune étude d'interaction n'a été réalisée. Aucune interaction cliniquement significative n'a été rapportée.
Population pédiatrique
Les études d’interaction n’ont été réalisées que chez l’adulte.
4.6. Fertilité, grossesse et allaitement
Grossesse
On ne dispose pas de données pertinentes concernant l'utilisation de la toxine botulinique de type A chez la femme enceinte. Les études chez l’animal ont montré une toxicité de la reproduction (voir rubrique 5.3). En clinique, il n'existe pas actuellement de données suffisamment pertinentes pour évaluer un éventuel effet malformatif ou fœtotoxique de la toxine botulinique lorsqu'elle est administrée pendant la grossesse dans l’espèce humaine.
En conséquence, la toxine botulinique ne doit pas être utilisée pendant la grossesse et chez les femmes en âge de procréer n’utilisant pas de moyens de contraception, sauf nécessité majeure.
Allaitement
L'innocuité de l'utilisation de la toxine botulinique chez la femme allaitante n'a pas été démontrée. En conséquence, l'allaitement n’est pas recommandé pendant le traitement.
Fertilité
Il n’existe pas de données suffisantes sur les effets de l’utilisation de la toxine botulinique de type A sur la fertilité des femmes en âge de procréer. Les études conduites chez des rats mâles et femelles ont montré des diminutions de la fertilité (voir rubrique 5.3).
4.7. Effets sur l'aptitude à conduire des véhicules et à utiliser des machines
Généralités
Dans les études cliniques contrôlées, les événements indésirables considérés comme reliés à BOTOX par les investigateurs ont été rapportés chez 35 % des patients dans le blépharospasme, 28 % des patients dans la dystonie cervicale, 17 % des patients dans la paralysie cérébrale chez l’enfant, 11 % des patients dans l’hyperhidrose axillaire et 16% des patients dans la spasticité focale du membre supérieur associée à un accident vasculaire cérébral. Dans les études cliniques conduites dans l’hyperactivité vésicale idiopathique, l’incidence des événements indésirables étaient de 26% après le premier traitement et de 22% après le second traitement. Dans les études cliniques conduites dans l'hyperactivité détrusorienne neurologique chez l'adulte, l'incidence des évènements indésirables était de 32 % après le premier traitement et diminuait à 18 % après le second traitement. Pour l'hyperactivité détrusorienne neurologique chez l’enfant, l'incidence des évènements indésirables était de 6,2% après le premier traitement. Dans les études cliniques conduites dans la migraine chronique, l’incidence des événements indésirables était de 26% après le premier traitement et diminuait à 11% après le deuxième traitement.
En général, les effets indésirables surviennent dans les tous premiers jours suivant l'injection, et, bien que généralement transitoires, ils peuvent durer plusieurs mois voire plus longtemps dans de rares cas.
Une faiblesse musculaire locale correspond à l'action pharmacologique attendue de la toxine botulinique sur les tissus musculaires. Cependant, une faiblesse musculaire des muscles adjacents et/ou à distance du site d’injection a été rapporté.
Comme on peut s'y attendre lors de toute procédure d'injection, la survenue d'une douleur localisée, d'une inflammation, de paresthésie, d'une hypoesthésie, d'une sensibilité douloureuse, d'un gonflement / œdème, d'un érythème, d'une infection locale, d'un saignement et/ou d'une ecchymose a été associée à l'injection. La douleur liée à l’injection et/ou l'anxiété ont entraîné des réactions vasovagales, incluant une hypotension symptomatique transitoire et une syncope. Des cas de fièvre et de syndrome pseudo-grippal ont également été rapportés après des injections de toxine botulinique.
Effets indésirables : fréquence par indication
Les effets indésirables rapportés pendant les essais cliniques sont classés par indication, classe de systèmes d’organes et fréquence selon la définition suivante : Très fréquent (≥ 1/10) ; fréquent (≥ 1/100 à < 1/10) ; peu fréquent (≥ 1/1 000 à < 1/100) ; rare (≥ 1/10 000 à < 1/1 000) ; très rare (< 1/10 000).
Effets indésirables indépendants du site d'injection
· Des effets indésirables liés à la diffusion de la toxine, à distance du site d'injection ont été très rarement rapportés (faiblesse musculaire excessive, dysphagie, pneumopathie d'inhalation, pouvant être fatales) (voir rubrique 4.4).
· Rares réactions allergiques générales (rash, érythème, prurit, réaction anaphylactique).
· Douleurs/brûlures au point d'injection, possibles quels que soient le site d'injection ou l'indication.
Blépharospasme et Spasme hémifacial
Affections du système nerveux
Peu fréquents : Vertiges, parésie faciale et paralysie faciale.
Affections oculaires
Très fréquent : Ptosis palpébral.
Fréquents : Kératite ponctuée, lagophtalmie, œil sec, photophobie, irritation oculaire et augmentation des larmoiements.
Peu fréquents : Kératite, ectropion, diplopie, entropion, troubles visuels et vision floue.
Rare : Œdème palpébral.
Très rares : Kératite ulcérative, anomalie de l'épithélium cornéen, perforation de la cornée.
Affections de la peau et du tissu sous-cutané
Fréquent : Ecchymose.
Peu fréquent : Eruption cutanée / Dermatite.
Troubles généraux et anomalies au site d'administration
Fréquents : Irritation et œdème facial.
Peu fréquent : Fatigue.
Dystonie cervicale (Torticolis spasmodique)
Infections et infestations
Fréquents : Rhinite et infection des voies respiratoires supérieures.
Affections du système nerveux
Fréquents : Vertiges, hypertonie, hypoesthésie, somnolence et céphalée.
Affections oculaires
Peu fréquents : Diplopie et ptosis palpébral.
Affections respiratoires, thoraciques et médiastinales
Peu fréquents : Dyspnée et dysphonie.
Affections gastro-intestinales
Très fréquent : Dysphagie
Fréquents : Sécheresse buccale et nausées.
Affections musculo-squelettiques et systémiques :
Très fréquent : Faiblesse musculaire.
Fréquents : Rigidité et douleur musculo-squelettiques.
Troubles généraux et anomalies au site d'administration
Très fréquent : Douleur.
Fréquents : Asthénie, syndrome pseudo-grippal et malaise.
Peu fréquent : Pyrexie.
Spasticité (hyperactivité musculaire) des membres supérieurs et/ou inférieurs chez l'adulte
Affections psychiatriques
Peu fréquents : Dépression, insomnie.
Affections du système nerveux
Fréquent : Hypertonie.
Peu fréquents : Hyperesthésie, hypoesthésie, céphalée, paresthésie, perte de la coordination et amnésie.
Affections de l’oreille et du labyrinthe
Peu fréquent : Vertiges.
Affections vasculaires
Peu fréquent : Hypotension orthostatique.
Affections gastro-intestinales
Peu fréquents : Nausées, paresthésie orale.
Affections de la peau et du tissu sous-cutané
Fréquents : Ecchymoses, purpura.
Peu fréquents : Dermatite, prurit, éruption cutanée.
Affections musculo-squelettiques et systémiques
Fréquents : Douleur dans les extrémités, faiblesse musculaire, douleurs des membres injectés.
Peu fréquents : Arthralgies et bursite.
Troubles généraux et anomalies au site d'administration
Fréquents : Douleur aux sites d’injection, fièvre, syndrome pseudo-grippal, hémorragie aux points d'injection et irritation aux sites d’injection.
Peu fréquents : Asthénie, douleur, hypersensibilité aux points d’injection, malaise, hémorragie et œdème périphérique.
Spasticité (hyperactivité musculaire) des membres supérieurs et/ou inférieurs chez l'enfant de 2 ans et plus
Spasticité du membre supérieur chez l’enfant de 2 ans et plus :
Infections et infestations
Fréquents : Syndrome grippal, pneumonie.
Affections du système nerveux
Fréquents : Maladresse, hypokinésie.
Affections de la peau et du tissu sous-cutané
Fréquents : Ecchymoses, purpura.
Peu fréquents : Dermatite, prurit, éruption cutanée.
Affections musculo-squelettiques et systémiques
Fréquents : Faiblesse musculaire, spasmes musculaires, doigt à ressaut.
Affections du rein et des voies urinaires
Fréquent : Pollakiurie.
Affections gastro-intestinales
Fréquent : Vomissement.
Lésions, intoxications et complications liées aux procédures
Fréquents : Luxation, chute, contusion.
Troubles généraux et anomalies au site d'administration
Très fréquent : Réaction aux points d'injection.
Fréquents : Ecchymose, douleurs aux points d'injection
Spasticité du membre inférieur chez l’enfant de 2 ans et plus :
Infections et infestations
Très fréquents : Infection virale, infection auriculaire.
Affections du système nerveux
Fréquents : Somnolence, démarche anormale, paresthésie.
Affections de la peau et du tissu sous-cutané
Fréquent : Eruption cutanée.
Affections musculo-squelettiques et systémiques
Fréquents : Myalgie, faiblesse musculaire, douleur aux extrémités.
Affections du rein et des voies urinaires
Fréquent : Incontinence urinaire.
Lésions, intoxications et complications liées aux procédures
Fréquent : Chute.
Troubles généraux et anomalies au site d'administration
Fréquents : Malaise, douleur aux points d'injection, asthénie.
Depuis la mise sur le marché, des cas possibles de diffusion de la toxine ont très rarement été rapportés chez les enfants ayant surtout des troubles associés à une infirmité motrice cérébrale. Généralement, la dose utilisée dans ces cas était au-dessus de celle recommandée (voir rubrique 4.2).
Hyperhidrose axillaire
Affections du système nerveux
Fréquents : Céphalées, paresthésies.
Affections vasculaires
Fréquent : Bouffées de chaleur.
Affections gastro-intestinales
Peu fréquent : Nausées.
Affections de la peau et du tissu sous-cutané
Fréquents : Hyperhidrose (transpiration non-axillaire), odeur cutanée anormale, prurit, nodule sous-cutané et alopécie.
Affections musculo-squelettiques et systémiques
Fréquent : Douleur des extrémités.
Peu fréquents : Faiblesse musculaire, myalgie et arthropathie.
Troubles généraux et anomalies au site d'administration
Très fréquent : Douleur au site d'injection.
Fréquents : Douleur, œdème au site d'injection, hémorragie au site d'injection, hypersensibilité au site d'injection, irritation au site d'injection, asthénie, et réactions au site d'injection.
Lors de la prise en charge de l’hyperhidrose axillaire, une augmentation de la transpiration autre qu'axillaire a été rapportée chez 4,5 % des patients dans le mois suivant l'injection, sans corrélation avec les sites anatomiques affectés. On a observé que ces effets disparaissent chez approximativement 30 % des patients dans les 4 mois suivant l'injection.
Une faiblesse du bras a également été rapportée. Cet effet indésirable peu fréquent (0,7 %), transitoire et d’intensité légère, n’a pas nécessité de traitement, et s’est résolu sans séquelles. Cet effet indésirable pourrait être lié au traitement et/ou à la technique d'injection. Dans le cas peu fréquent où une faiblesse musculaire serait rapportée, un examen neurologique pourra être envisagé. En outre, une réévaluation de la technique d'injection avant toute injection ultérieure est conseillée pour garantir l’administration intradermique des injections.
Lors d'une étude non contrôlée de la tolérance de BOTOX (50 Unités par aisselle) chez des patients pédiatriques âgés de 12 à 17 ans (N=144), les effets indésirables survenus chez plus d'un patient (2 patients dans chacun des cas) comprenaient une douleur au site d'injection et une hyperhidrose (transpiration non-axillaire).
Dysfonctions vésicales
Hyperactivité vésicale idiopathique chez l’adulte
Infections et infestations
Très fréquent : Infection du tractus urinaire.
Fréquent : Bactériurie.
Affections du rein et des voies urinaires
Très fréquent : Dysurie
Fréquents : Rétention urinaire, pollakiurie, leucocyturie.
Investigations
Fréquent : Augmentation du volume post-mictionnel*.
*résidu post-mictionnel augmenté ne nécessitant pas le recours à l’autosondage intermittent propre.
Les effets indésirables fréquents liés à la procédure sont la dysurie et l’hématurie.
L’autosondage intermittent propre a été initié chez 6,5% des patients traités par BOTOX 100 Unités versus 0,4% du groupe placebo.
Les données disponibles sur l’administration répétée de BOTOX rapportent les mêmes types d’effets indésirables.
Parmi les 1242 patients inclus dans les études cliniques contrôlées versus placebo, 41,4% (n=514) étaient de 65 ans et plus et 14,7% (n=182) étaient âgés de 75 ans et plus. Il n’y avait pas de différence en termes de profil de tolérance entre les patients âgés de moins de 65 ans et ceux de 65 ans et plus, à l’exception de l’incidence des infections urinaires qui était supérieure chez les patients âgés de 65 ans et plus dans les groupes placebo et BOTOX en comparaison aux patients plus jeunes.
Hyperactivité vésicale chez l’enfant
Infections et infestations
Fréquent : Infection du tractus urinaire
Affections du rein et des voies urinaires
Fréquent : Dysurie*, douleur urétrale
Affections gastro-intestinales
Fréquent : Douleur abdominale, douleur abdominale basse
*liés à la procédure
Lors d’une étude clinique multicentrique, randomisée, en double aveugle, en groupes parallèles menée chez 55 patients de 12 à 17 ans, les effets indésirables étaient généralement comparables au profil de sécurité connu en cas d’hyperactivité vésicale idiopathique chez l’adulte. Toutefois, des événements de douleur abdominale et urétrale ont également été notés lors de cette petite étude pédiatrique sur l’hyperactivité vésicale.
Voir rubriques 4.2 et 5.1
Hyperactivité détrusorienne neurologique chez l’adulte
Effets indésirables issus des études cliniques pivotales de phase III
Les données sur la tolérance de BOTOX ont été évaluées sur 809 patients ayant participé à 4 études : 537 patients dans le groupe BOTOX (dont 262 par 200 UNITÉS ALLERGAN de BOTOX) et 272 dans le groupe placebo.
Les effets indésirables les plus fréquemment rapportés étaient l’infection du tractus urinaire et la rétention urinaire.
Infections et infestations
Fréquent : Infection du tractus urinaire.
Affections musculo-squelettiques et systémiques
Fréquent : Faiblesse musculaire.
Affections du rein et des voies urinaires
Très fréquent : Rétention urinaire.
Fréquents : Hématurie*, dysurie*.
Lésions, intoxications et complications liées aux procédures
Fréquent : Hyper-réflexie autonome*.
Troubles généraux et anomalies au site d’administration
Fréquent : Fatigue.
Investigations
Fréquent : Volume d’urine résiduelle augmenté (voir rubrique 4.4).
* liés à la procédure
Des cas de poussées de la sclérose en plaques ont été observés chez les patients atteints de sclérose en plaques dans les études cliniques dans le groupe 300 Unités de BOTOX (5,1%, dose non recommandée), 200 Unités de BOTOX (1,9%) et le groupe placebo (2,2%). Aucune différence significative entre la dose 200 Unités de BOTOX et le placebo n’a été mise en évidence.
Les données disponibles sur l’administration répétée de BOTOX rapportent les mêmes types d’effets indésirables.
Effets indésirables issus d’une étude post-AMM (phase III) conduite avec 100 Unités de BOTOX chez des patients atteints de sclérose en plaques dont le mode mictionnel à l’inclusion était spontané.
Infections et infestations
Très Fréquent : Infection du tractus urinaire, bactériurie.
Investigations
Très fréquent : Volume d’urine résiduelle augmenté*
Affections du rein et des voies urinaires
Très fréquent : Rétention urinaire.
Fréquents : Hématurie**, dysurie**.
* Volume d’urine résiduel augmenté sans nécessité d’autosondage
** liés à la procédure
Aucune différence du taux annualisé d’exacerbation de la sclérose en plaques (c’est-à-dire le nombre d’exacerbation de la sclérose en plaques par patient-année) n’a été observée (BOTOX 100 U = 0 ; placebo = 0,07).
Dans l’étude post-AMM de phase III, l’autosondage a été initié chez 15,2% des patients après traitement par 100 Unités de BOTOX versus 2,6% des patients du groupe placebo alors que dans les études cliniques pivotales, l’autosondage a été initié chez 31,4% des patients après traitement par 200 Unités de BOTOX versus 4,5% des patients du groupe placebo.
Hyperactivité détrusorienne neurologique chez l’enfant
Les données sur la tolérance de BOTOX ont été évaluées sur 113 patients âgés de 5 à 17 ans souffrant d'incontinence urinaire due à une hyperactivité détrusorienne associée à une affection neurologique et utilisant un sondage intermittent propre (voir rubrique 5.1)
Infections et infestations
Très fréquent : Bactériurie
Fréquents : Infection du tractus urinaire, leucocyturie
Affections du rein et des voies urinaires
Fréquent : Hématurie, douleurs vésicales*
*Effet indésirable liée à la procédure
Aucun changement n'a été observé dans le profil de sécurité global lors de l'administration de doses répétées.
Voir rubriques 4.2 et 5.1
Migraine chronique
Les données sur la tolérance de BOTOX ont été évaluées dans deux essais cliniques en double aveugle, contrôlées versus placebo, impliquant 687 patients traités avec 155 à 195 UNITÉS ALLERGAN de BOTOX. Les effets indésirables suivants ont été rapportés :
Affections du système nerveux
Fréquents : Céphalées, migraine, parésie faciale.
Affections oculaires
Fréquent : Ptôse palpébrale.
Affections de la peau et du tissu sous-cutané
Fréquents : Prurit, éruption cutanée.
Peu fréquent : Douleur cutanée.
Affections musculo-squelettiques et systémiques
Fréquents : Douleur cervicale, myalgie, douleur musculo-squelettique, raideur musculo-squelettique, spasme musculaire, contracture musculaire, faiblesse musculaire.
Peu fréquent : Douleur dans la mâchoire.
Fréquence indéterminée : effet Méphisto (élévation latérale des sourcils).
Troubles généraux et anomalies au site d'administration
Fréquent : Douleur au site d'injection.
Affections gastro-intestinales
Peu fréquent : Dysphagie.
Une migraine, incluant une aggravation de la migraine, a été rapportée dans 3,8% des patients sous BOTOX et 2,6% des patients sous placebo, survenant généralement dans le premier mois du traitement. Ces effets ne se sont pas reproduits systématiquement au cours des cycles suivants de traitement, et l'incidence globale a diminué lors de traitements répétés.
Le taux d'arrêt du traitement en raison d'événements indésirables dans ces essais cliniques de phase III était de 3,8% pour le groupe BOTOX contre 1,2% pour le groupe placebo.
Informations supplémentaires
La liste suivante inclut les effets indésirables ou autres événements indésirables cliniquement pertinents rapportés depuis la mise sur le marché du médicament quelle que soit l’indication et pouvant être redondants avec ceux cités à la rubrique 4.4 (Mises en garde spéciales et précautions d’emploi) ou à la rubrique 4.8 (Effets indésirables).
Affections du système immunitaire
Anaphylaxie, angio-œdème, maladie sérique et urticaire.
Troubles du métabolisme et de la nutrition
Anorexie.
Affections du système nerveux
Atteinte du plexus brachial, dysphonie, dysarthrie, parésie faciale, hypoesthésie, faiblesse musculaire, myasthénie grave, neuropathie périphérique, paresthésie, radiculopathie, convulsions, syncope et paralysie faciale.
Affections de l’oreille et du labyrinthe
Hypoacousie, acouphènes et vertiges.
Affections oculaires
Glaucome à angle fermé (dans le traitement du blépharospasme), strabisme, vison floue, troubles visuels, sécheresse oculaire (notamment dans les indications impliquant des injections au niveau du visage) et œdème palpébral.
Affections cardiaques
Arythmie, infarctus du myocarde.
Affections respiratoires, thoraciques et médiastinales
Pneumonie d’inhalation (d’issue parfois fatale), dyspnée, détresse respiratoire et insuffisance respiratoire.
Affections gastro-intestinales
Douleur abdominale, diarrhée, constipation, bouche sèche, dysphagie, nausée et vomissements.
Affections de la peau et du tissu sous-cutané
Alopécie, dermatite psoriasiforme, érythème polymorphe, hyperhidrose, madarose, prurit et éruption cutanée.
Affections musculo-squelettiques et systémiques
Atrophie musculaire, myalgie et contractions musculaires localisées / contractions musculaires involontaires.
Troubles généraux et anomalies au site d’administration
Atrophie de dénervation, malaise, fièvre.
Déclaration des effets indésirables suspectés
La déclaration des effets indésirables suspectés après autorisation du médicament est importante. Elle permet une surveillance continue du rapport bénéfice/risque du médicament. Les professionnels de santé déclarent tout effet indésirable suspecté via le système national de déclaration : Agence nationale de sécurité du médicament et des produits de santé (ANSM) et réseau des Centres Régionaux de Pharmacovigilance - Site internet : https://signalement.social-sante.gouv.fr/
Un surdosage de BOTOX est un terme relatif et dépend de la dose, du site d'injection, et des propriétés tissulaires sous-jacentes.
Aucun cas de toxicité systémique résultant d'une injection accidentelle de BOTOX n'a été observé. Des doses excessives peuvent entraîner une paralysie neuromusculaire généralisée et profonde, locale ou à distance du site d’injection. Aucun cas d'ingestion de BOTOX n'a été rapporté.
Les signes et symptômes d'un surdosage ne s’observent pas immédiatement après l'injection. En cas d'injection ou d'ingestion accidentelle ou de suspicion de surdosage, le patient sera surveillé médicalement sur une période pouvant aller jusqu'à plusieurs semaines, à la recherche de signes et symptômes progressifs de faiblesse musculaire locaux ou à distance du site d'injection, pouvant inclure un ptosis, une diplopie, une dysphagie, des dysarthries, une faiblesse généralisée ou une défaillance respiratoire. Chez ces patients, des examens médicaux complémentaires sont à envisager, et un traitement médical approprié sera immédiatement instauré, pouvant inclure une hospitalisation.
Si la musculature de l'oropharynx et de l'œsophage est affectée, une fausse route peut survenir, avec développement possible d'une pneumonie d’inhalation. En cas d'apparition d'une paralysie des muscles respiratoires ou d’une faiblesse excessive de ces muscles, une intubation et une ventilation assistée seront requises jusqu'à la guérison pouvant nécessiter une trachéotomie et une ventilation mécanique prolongée en plus des autres traitements de soutien.
Il n'existe pas d'antidote. Il conviendra d'avoir recours à un traitement symptomatique si nécessaire.
5. PROPRIETES PHARMACOLOGIQUES
5.1. Propriétés pharmacodynamiques
Classe pharmacothérapeutique : AUTRES MYORELAXANTS À ACTION PÉRIPHÉRIQUE, code ATC : M03AX01 (M : Muscle et squelette).
La toxine botulinique de type A bloque la libération périphérique d'acétylcholine au niveau des terminaisons nerveuses cholinergiques présynaptiques, en clivant la SNAP-25, une protéine impliquée dans le stockage et la libération de l'acétylcholine à partir des vésicules situées dans les terminaisons nerveuses.
Après un certain temps, de nouvelles terminaisons nerveuses se forment et les connexions se rétablissent.
Des preuves cliniques suggèrent que BOTOX réduit les seuils de douleur, d’inflammation neurogène et de douleur cutanée liée à la chaleur dans un modèle de sensibilisation du trijumeau induite par capsaïcine. Dans les modèles précliniques, au niveau des neurones sensoriels, BOTOX inhibe la libération des neurotransmetteurs sensoriels (exemple : Substance P, CGRP) et diminue l’expression des récepteurs cellulaires de surface (exemple : TRPV1). De plus, BOTOX prévient et inverse la sensibilisation des neurones sensoriels nociceptifs dans ces modèles précliniques, ce qui pourrait également inhiber la sensibilisation centrale.
La récupération après une injection intramusculaire a normalement lieu dans les 12 semaines qui suivent l'injection, du fait du bourgeonnement des terminaisons nerveuses et de leur reconnexion avec les plaques motrices.
Après injection dans le détrusor, BOTOX inhibe le message efférent contrôlant l’activité motrice du détrusor en bloquant la libération d’acétylcholine (voie parasympathique).
ÉTUDES CLINIQUES
Hyperhidrose axillaire
Un essai clinique multicentrique en double aveugle a été réalisé chez des patients présentant une hyperhidrose axillaire bilatérale persistante (définie à l'inclusion par une mesure gravimétrique d'une quantité de sueur par aisselle d'au moins 50 mg sur une période de 5 minutes à température ambiante et au repos). 320 patients ont été randomisés pour recevoir soit 50 Unités de BOTOX (N=242) soit le placebo (N=78).
Les répondeurs étaient définis comme les patients montrant une réduction de production de sueur axillaire d'au moins 50 % par rapport à la valeur initiale de la mesure de la production de sueur. Le critère principal défini comme étant le taux de répondeurs à quatre semaines après l'injection était de 93,8 % dans le groupe BOTOX et de 35,9 % dans le groupe placebo (p< 0,001). Le taux de répondeurs est resté significativement plus élevé (p<0,001) dans le groupe BOTOX que dans le groupe placebo, à tous les points de mesure jusqu'à 16 semaines suivant l'injection.
Une étude de suivi en ouvert a été réalisée sur 12 mois, incluant 207 patients ayant reçu jusqu'à 3 injections de BOTOX.
Au total, sur les 2 études 174 patients ont terminé la période de 16 mois (4 mois en double aveugle et 12 mois de suivi en ouvert). Le taux de répondeurs à la 16ème semaine suivant la première (N=287), la deuxième (N=123) et la troisième (N=30) injection était respectivement de 85,0 %, 86,2 % et 80,0 %. La durée moyenne de l'effet après la première injection (basée sur l'ensemble des 2 études) était de sept mois et demi, cependant, pour 27,5 % des patients, la durée de l'effet était de 1 an ou plus.
Hyperactivité vésicale chez l’adulte
Deux études cliniques de phase III randomisées en double aveugle, multicentriques, contrôlées versus placebo sur 24 semaines ont été réalisées chez des patients présentant une hyperactivité vésicale avec des symptômes incluant incontinence urinaire, urgenturie et pollakiurie. Mille cent cinq (1105) patients dont les symptômes étaient insuffisamment contrôlés par un traitement anticholinergique (réponse insuffisante ou intolérance) ont été randomisés pour recevoir soit 100 Unités Allergan de BOTOX (n = 557), soit du placebo (n = 548).
Dans les deux études de phase III, des améliorations significatives, comparées au placebo, ont été observées en faveur de BOTOX, sur la variation par rapport à l’inclusion, de la fréquence quotidienne des épisodes d’incontinence urinaire à 12 semaines incluant également le pourcentage de patients continents. Sur l’échelle d’évaluation du bénéfice du traitement (Treatment Benefit Scale), la proportion de patients rapportant une réponse positive au traitement (« très améliorée » ou « améliorée ») était significativement plus importante dans le groupe BOTOX comparé au groupe placebo dans les deux études. Des améliorations significatives comparées au placebo ont été également observées pour la fréquence quotidienne des mictions (pollakiurie), de l’urgenturie, et de la nycturie. Le volume mictionnel était aussi significativement plus élevé. Des améliorations significatives ont été observées sur tous les symptômes de l’hyperactivité vésicale dès la deuxième semaine.
Aucune différence d’efficacité n’a été observée entre les patients de 65 ans et plus et ceux de moins de 65 ans.
Le traitement par BOTOX était associé à des améliorations significatives, comparées au placebo, sur la qualité de vie mesurée par le questionnaire I-QOL (Incontinence Quality of Life) (incluant les comportements d’évitement et de limitation, l’impact psychosocial, la gêne sociale) et le questionnaire KHQ (King’s Health Questionnaire) incluant l’impact de l’incontinence, le manque de confiance en soi, la gêne sociale, la gêne physique, les relations avec les autres, les émotions, le sommeil/énergie et la capacité d’adaptation.
Résultats poolés des critères principaux et secondaires des études pivots
|
Temps d’évaluation |
BOTOX 100 UNITÉS ALLERGAN (N=557) |
Placebo (N=548) |
p |
|
Fréquence quotidienne des épisodes d’incontinence urinaire* Moyenne à l’inclusion Variation moyenne à la semaine 12a |
5,49 - 2,80 |
5,39 -0,95 |
<0,001 |
|
Proportion de patients avec une réponse positive au traitement selon l’échelle d’évaluation au traitement TBS (%) Semaine 12a |
61,8 |
28,0 |
<0,001 |
|
Fréquence quotidienne des mictions Moyenne à l’inclusion Variation moyenne à la semaine 12b |
11,99 -2,35 |
11,48 -0,87 |
<0,001 |
|
Fréquence quotidienne des épisodes d’urgenturie Moyenne à l’inclusion Variation moyenne à la semaine 12b |
8,82 -3,30 |
8,31 -1,23 |
<0,001 |
|
Score total I-QOL Moyenne à l’inclusion Variation moyenne à la semaine 12b,c |
34,1 +22,5 |
34,7 +6,6 |
<0,001 |
|
KHQ (King’s Health Questionnaire) : Manque de confiance en soi Moyenne à l’inclusion Variation moyenne à la semaine 12b,c |
65,4 -25,4 |
61,2 -3,7 |
<0,001 |
|
KHQ (King’s Health Questionnaire) : gène sociale Moyenne à l’inclusion Variation moyenne à la semaine 12b,c |
44,8 -16,8 |
42,4 -2,5 |
<0,001 |
a Co-critères principaux
b Critères secondaires
c Les variations minimales prédéfinies comme importantes par rapport à l’inclusion étaient de +10 points pour le score I-QOL et de -5 points pour le score KHQ
La durée médiane de l’effet du traitement dans les deux études pivots, basée sur une demande de re-traitement exprimée par le patient, était de 166 jours (environ 24 semaines) (l’éligibilité au re-traitement nécessitait au moins 2 épisodes d’incontinence urinaire en 3 jours). Pour les patients ayant reçu 100 Unités de BOTOX dans les études de phase III puis dans l’étude d’extension en ouvert (N=438), la durée médiane de l’effet observée, basée sur une demande de retraitement exprimée par le patient, était de 212 jours (environ 30 semaines).
Huit cent trente-quatre (834) patients ont été suivis à long terme. L’efficacité du traitement a été maintenue lors des traitements successifs.
Dans les études pivots, aucun des 615 patients dont les échantillons sanguins ont été analysés n’ont développé d’anticorps neutralisants. Chez les patients dont les échantillons sanguins ont été analysés dans les études de phase III puis dans l’étude d’extension en ouvert, des anticorps neutralisants ont été retrouvés chez aucun des 954 patients (0,0%) ayant reçu des doses de 100 Unités de BOTOX et chez 3 des 260 patients (1,2%) ayant reçu au moins une dose de 150 Unités de BOTOX. Un de ces trois patients continuait à présenter un bénéfice clinique. En comparaison à l’ensemble de la population traitée avec BOTOX, les patients ayant développé des anticorps neutralisants présentaient généralement une durée d’effet plus courte et, en conséquence, recevaient des traitements plus fréquents (voir rubrique 4.4).
Hyperactivité vésicale chez l’enfant
Des données d’efficacité limitées sont issues d’une étude clinique multicentrique, randomisée, en double aveugle, en groupes parallèles (191622-137) menée chez des patients de 12 à 17 ans atteints d’hyperactivité vésicale avec symptômes d’incontinence urinaire. Au total, 55 patients (parmi les 108 prévus) ayant présenté une réponse inadéquate ou une intolérance à au moins un médicament anticholinergique ont été recrutés, entraînant une taille d’échantillon insuffisante pour conclure à l’efficacité dans cette population. Les patients ont été randomisés dans des groupes recevant 25 Unités, 50 Unités ou 100 Unités, sans dépasser 6 Unités/kg de poids corporel ; N=18, N=17, N=20 pour BOTOX 25 Unités, BOTOX 50 Unités et BOTOX 100 Unités, respectivement. Avant l’administration du traitement, les patients ont reçu une anesthésie selon les pratiques locales. Tous les patients ont reçu une anesthésie générale ou une sédation consciente.
Résultats des critères principaux et secondaires à l’inclusion et variation par rapport à l’inclusion dans une étude clinique en double aveugle, en groupes parallèles
|
|
BOTOX 100 Unités N=20 |
BOTOX 50 Unités N=17 |
BOTOX 25 Unités N=18 |
Valeur p
BOTOX 100 vs. 25 Unités |
Valeur p
BOTOX 50 vs. 25 Unités |
|
Fréquence quotidienne des épisodes d’incontinence urinaire diurnea Moyenne à l’inclusion Variation moyenne* à la semaine 12** (IC à 95 %) |
3,6
-2,3 (-3,8 ; -0,9) |
3,5
-1,0 (-2,6 ; 0,7) |
5,3
|
0,3802 |
0,7330 |
|
-1,4 (-3,0 ; 0,2) |
|||||
|
Proportion de patients présentant une réduction d’au moins 50 % par rapport à l’inclusion de la fréquence quotidienne des épisodes d’IU diurneb (%) Semaine 12c (IC à 95 %)
|
80,0 (56,3 ; 94,3) |
47,1 (23,0 ; 72,2) |
50,0 (26,0 ; 74,0) |
0,0472 |
0,9924 |
|
Réponse positive au traitement (« très améliorée » ou « améliorée »)b (%) Semaine 12c (IC à 95 %) |
68,4 (43,5 ; 87,4)
|
70,6 (44,0 ; 89,7)
|
52,9 (27,8 ; 77,0) |
0,6092 |
0,4824 |
|
Fréquence quotidienne des épisodes de miction diurneb Moyenne à l’inclusion |
8,1 |
8,5 |
11,2 |
0,5743 |
0,1451 |
|
Variation moyenne* à la semaine 12** (IC à 95 %) |
-1,0 (-3,0 ; 1,0)
|
0,3 (-1,7 ; 2,4)
|
-1,8 (-3,9 ; 0,2) |
||
|
Fréquence quotidienne des épisodes d’urgenturie diurneb Moyenne à l’inclusion Variation moyenne* à la semaine 12** (IC à 95 %) |
4,4 -2,2 (-4,1 ; -0,3) |
5,4 -1,8 (-3,8 ; 0,2) |
7,5 -1,9 (-3,9 ; 0,2) |
0,8206 |
0,9604 |
IC = Intervalle de confiance
* La variation moyenne aux moindres carrés (MC) par rapport à l’inclusion, la différence de traitement, l’IC à 95 % et la valeur p sont basés sur un modèle ANCOVA avec la valeur d’inclusion comme covariable et le groupe de traitement comme facteur. Les valeurs de dernière observation reportée ont été utilisées pour analyser la variable d’efficacité principale.
** Temps principal d’évaluation
a. Variable principale.
b. Variable secondaire.
c. Les valeurs p sont obtenues à l’aide d’un test de Cochran– Mantel– Haenszel, stratifié en fonction des épisodes d’incontinence d’urgence urinaire diurne à l’inclusion (≤ 6 ou > 6). L’IC à 95 % exact (Clopper-Pearson) est calculé à l’aide de la distribution binomiale.
Au cours d’une étude randomisée en double aveugle, sur les 55 enfants qui avaient un résultat négatif d’anticorps de liaison ou neutralisants à l’inclusion et qui avaient au moins une valeur évaluable, aucun n’a développé d’anticorps neutralisants après avoir reçu 25 à 100 Unités de BOTOX.
Hyperactivité détrusorienne neurologique chez l’adulte
Etudes cliniques de phase III
Deux études cliniques de phase III randomisées en double aveugle, contrôlées versus placebo ont été réalisées chez des patients présentant une incontinence urinaire due à une hyperactivité détrusorienne neurologique soit en miction spontanée soit sondés. Six cent quatre-vingt-onze (691) patients atteints de sclérose en plaques (SEP) ou blessés médullaires ont été inclus dans ces études. Les patients ont été randomisés pour recevoir soit 200 Unités de BOTOX (n = 227), soit 300 Unités de BOTOX (n = 223), soit du placebo (n = 241).
Dans les deux études de phase III, des améliorations significatives, comparées au placebo, ont été observées en faveur de BOTOX (200 UNITÉS ALLERGAN et 300 UNITÉS ALLERGAN) sur le critère principal d’efficacité de diminution, par rapport à l’inclusion, de la fréquence hebdomadaire des épisodes d’incontinence urinaire à 6 semaines (temps principal d’évaluation) incluant également le pourcentage de patients continents.
Des améliorations significatives des paramètres urodynamiques ont également été observées : augmentation de la capacité cystométrique maximale et diminution du pic de la pression maximale détrusorienne durant la première contraction involontaire du détrusor.
Aucun bénéfice additionnel n’a été observé avec la dose de 300 Unités de BOTOX par rapport à la dose de 200 UNITÉS ALLERGAN. L’incidence des événements indésirables était plus élevée avec la dose de 300 Unités de BOTOX par rapport à la dose de 200 UNITÉS ALLERGAN.
Les résultats poolés des études pivots sont présentés ci-dessous :
Résultats « poolés » des 2 études pivots sur le critère d’évaluation principal
|
|
BOTOX 200 UNITÉS ALLERGAN (N=227) |
Placebo (N=241) |
|
Fréquence hebdomadaire de l’incontinence urinaire |
|
|
|
Valeur moyenne à l’inclusion |
32,4 |
31,5 |
|
Variation moyenne à la semaine 2 |
-17,7* |
-9,0 |
|
Variation moyenne à la semaine 6a |
-21,3* |
-10,5 |
|
Variation moyenne à la semaine 12 |
-20,6* |
-9,9 |
a critère principal (à partir du calendrier mictionnel sur 7 jours)
La durée de l’effet du traitement dans les deux études pivots, basée sur une demande de re-traitement (2 injections au total) exprimée par le patient, était de 256-295 jours (36-42 semaines) pour le groupe ayant reçu 200 Unités de BOTOX comparé à 92 jours (13 semaines) pour le groupe placebo. Pour les patients ayant reçu 200 Unités de BOTOX dans les études de phase III puis dans l’étude d’extension en ouvert (N=174), la durée moyenne de l’effet observée, basée sur une demande de retraitement exprimée par le patient, était de 253 jours (environ 36 semaines).
L’efficacité du traitement a été observée également chez les patients ayant reçu une deuxième injection.
Etude post-AMM (phase III)
Une étude post-AMM de phase III en double-aveugle contrôlée par placebo a été conduite chez des patients atteints de sclérose en plaques et présentant une incontinence urinaire due à une hyperactivité détrusorienne neurologique ne répondant pas de manière adéquate aux traitements anticholinergiques et dont le mode mictionnel à l’inclusion était spontané. Les patients étaient randomisés pour recevoir 100 Unités de BOTOX (n=66) ou un placebo (n=78).
Des améliorations significatives du critère principal de variation par rapport à l’inclusion de la fréquence des épisodes d’incontinence urinaire a été observé par rapport au placebo au premier temps de mesure à 6 semaines.
Des améliorations significatives des paramètres urodynamiques, et du questionnaire de qualité de vie lié à l’incontinence (I-QoL) pour notamment les comportements d’évitement, l’impact psychologique et l’embarras social ont également été observées.
Les résultats de l’étude post-AMM (phase III) sont présentés ci-dessous.
|
Critère d’évaluation Temps d’évaluation |
BOTOX 100 UNITÉS ALLERGAN (N=66) |
Placebo (N=78) |
p |
|
Fréquence quotidienne des épisodes d’incontinence urinaire* |
|
|
|
|
Moyenne à l’inclusion |
4,2 |
4,3 |
|
|
Variation moyenne à la semaine 2 |
-2,9 |
-1,2 |
p<0,001 |
|
Variation moyenne à la semaine 6a |
-3,3 |
-1,1 |
p<0,001 |
|
Variation moyenne à la semaine 12 |
-2,8 |
-1,1 |
p<0,001 |
|
Capacité cystométrique maximale (ml) |
|
|
|
|
Moyenne à l’inclusion |
246,4 |
245,7 |
|
|
Variation moyenne à la semaine 6b |
+127,2 |
-1,8 |
<0,001 |
|
Pression maximale du détrusor lors de sa première contraction involontaire (cmH2O) |
|
|
|
|
Moyenne à l’inclusion |
35,9 |
36,1 |
|
|
Variation moyenne à la semaine 6b |
-19,6 |
+3,7 |
= 0,007 |
|
Score total I-QOLc,d |
|
|
|
|
Moyenne à l’inclusion |
32,4 |
34,2 |
|
|
Variation moyenne à la semaine 6b |
+40,4 |
+9,9 |
<0,001 |
|
Variation moyenne à la semaine 12 |
+38,8 |
+7,6 |
<0,001 |
* Le pourcentage de patients continents (sans incontinence urinaire) durant les 6 premières semaines était de 53,0% pour le groupe BOTOX et de 10,3% pour le groupe placebo
a critère principal (à partir du calendrier mictionnel sur 3 jours)
b critères secondaires
c le score total I-QoL varie de 0 (difficultés maximales) à 100 (aucunes difficultés)
d la différence significative minimale (MID) pré-déterminée pour le score I-QoL total était de 11 points en se basant sur des estimations de MID allant de 4 à 11 points obtenues chez des patients atteints d’hyperactivité vésicale neurologique
La durée de l’effet du traitement dans cette étude, basée sur une demande de re-traitement exprimée par le patient, était de 362 jours (~ 52 semaines) pour le groupe ayant reçu 100 Unités de BOTOX comparé à 88 jours (~ 13 semaines) pour le groupe placebo.
Dans l’étude post-AMM de phase III, l’autosondage a été initié chez 15,2% des patients après traitement par 100 Unités de BOTOX versus 2,6% des patients du groupe placebo alors que dans les études cliniques pivotales, l’autosondage a été initié chez 31,4% des patients après traitement par 200 Unités de BOTOX versus 4,5% des patients du groupe placebo (voir rubrique 4.8).
Hyperactivité détrusorienne neurologique chez l’enfant et l’adolescent
Une étude clinique multicentrique, randomisée, en double aveugle et en groupes parallèles a été menée chez des patients âgés de 5 à 17 ans souffrant d'incontinence urinaire due à une hyperactivité détrusorienne associée à une affection neurologique et utilisant un sondage intermittent propre. 113 enfants et adolescents (dont 99 souffrant d'un dysraphisme spinal tel que le spina bifida, 13 blessés médullaires et 1 d'une myélite transverse) n’avaient pas répondu de manière adéquate ou ont été intolérants à au moins un traitement anticholinergique. Ces patients ont été randomisés pour recevoir 50 unités, 100 unités ou 200 unités, ne devant pas dépasser 6 Unités/kg de poids corporel. Les patients recevant une dose inférieure à la dose randomisée en raison de la limite de 6 Unités/kg de poids corporel ont été affectés au groupe de dose le plus proche pour l'analyse ; N= 38, N=45 et N=30 pour BOTOX 50 unités, BOTOX 100 unités et BOTOX 200 unités, respectivement.
Avant l'administration du traitement, les patients ont reçu une anesthésie locale ou générale (requise pour les enfants de moins de 12 ans) selon leur âge et les pratiques locales. 109 patients (97,3 %) ont reçu une anesthésie générale ou une sédation consciente et 3 patients (2,7 %) ont reçu une anesthésie locale (autorisée pour les enfants de plus de 12 ans).
Valeurs moyennes à l’inclusion et variation par rapport à l’inclusion de la fréquence quotidienne des épisodes d'incontinence urinaire diurne, du volume d'urine lors du premier sondage matinal et de la pression maximale du détrusor pendant la phase de stockage (cmH2O) dans une étude clinique en double aveugle et en groupes parallèles.
|
|
BOTOX 200 Unités (N=30) |
BOTOX 100 Unités (N=45) |
BOTOX 50 Unités (N=38) |
Valeur p* BOTOX 200 vs. 50 Unités
|
Valeur p* BOTOX 100 vs. 50 Unités
|
|
Fréquence quotidienne des épisodes d'incontinence urinaire diurnea Valeur moyenne à l’inclusion (Ecart type)
Variation moyenne* à la semaine 2
Variation moyenne* à la semaine 6** (95%IC)
Variation moyenne* à la semaine 12 |
3.7 (5.06)
-1.1
-1.3 (-1.8, -0.9)
-0.9 |
3.0 (1.07)
-1.0
-1.3 (-1.7, -0.9)
-1.4 |
2.8 (1.04)
-1.2
-1.3 (-1.7, -0.9)
-1.2 |
0.9123 |
0.9949 |
|
Volume d'urine au premier sondage matinal (mL)b Valeur moyenne à l’inclusion (Ecart type)
Variation moyenne* à la semaine 2
Variation moyenne* à la semaine 6** (95%IC)
Variation moyenne* à la semaine 12 |
187.7 (135.70)
63.2
87.5 (52.1, 122.8)
45.2 |
164.2 (114.48)
29.4
34.9 (7.9, 61.9)
55.8 |
203.5 (167.48)
31.6
21.9 (-7.2, 51.1)
12.9 |
0.0055 |
0.5117 |
|
Pression maximale du détrusor pendant la phase de stockage (cmH2O)b
Valeur moyenne à l’inclusion (Ecart type)
Variation moyenne* à la semaine 6** (95% IC) |
56.7 (33.89)
-27.3 (-36.4, -18.2) |
56.5 (26.86)
-20.1 (-27.3, -12.8) |
58.2 (29.45)
-12.9 (-20.4, -5.3) |
0.0157 |
0.1737 |
IC = Intervalle de Confiance
*La variation moyenne aux moindres carrés et l'IC à 95 % et les valeurs p sont basés sur un modèle ANCOVA avec la valeur d’inclusion comme covariable, et le groupe de traitement, l'âge (< 12 ans ou ≥ 12 ans), les épisodes d'incontinence urinaire diurne à l’inclusion (≤ 6 ou > 6), et le traitement anticholinergique (oui/non) à l’inclusion comme facteurs.
** Temps principal d’évaluation
aCritère principal
bCritère secondaire
La durée médiane de réponse dans cette étude, basée sur la demande de re-traitement exprimée par le patient, était de 214,0 (31 semaines), 169,0 (24 semaines) et 207 jours (30 semaines) pour BOTOX 50 unités, BOTOX 100 unités et BOTOX 200 unités, respectivement.
Chez 99 patients pédiatriques qui avaient un résultat négatif à l’inclusion pour les anticorps de liaison ou les anticorps neutralisants et qui avaient au moins une valeur évaluable après inclusion d'une étude randomisée en double aveugle et d'une étude d'extension en double aveugle, aucun patient n'a développé d'anticorps neutralisants après avoir reçu 50 unités à 200 unités de BOTOX.
BOTOX a été évalué dans deux études (étude 1 et étude 2) multinationales et multicentriques de 56 semaines comprenant une phase en double aveugle de 24 semaines, avec deux cycles d'injection, comparant BOTOX au placebo, suivie d'une phase en ouvert de 32 semaines, avec trois cycles d'injection. Au total, 1 384 adultes atteints de migraine chronique, n’ayant jamais reçu ou pris de prophylaxie concomitante pour leurs céphalées au cours d'une période de 28 jours avant inclusion, et qui ont plus de 15 jours de céphalées, dont 50 % de migraines ou de migraines probables, et plus de 4 épisodes de céphalées ont été étudiés dans le cadre des deux essais cliniques de phase III. Les patients ont été randomisés pour recevoir du placebo ou BOTOX à une posologie comprise entre 155 et 195 unités toutes les 12 semaines pendant la phase de double aveugle sur 2 cycles de traitement. Ils ont pu bénéficier de 3 cycles supplémentaires de traitement en ouvert avec un total de 5 cycles d'injection maximum. Les patients ont été autorisés à prendre des traitements contre les crises de céphalées (65,5 % étaient en abus médicamenteux de ces traitements pendant la période initiale).
Dans l’étude 1, sur la période des 28 derniers jours de la phase en double aveugle de 24 semaines, les patients traités par Botox n’ont pas présenté de réduction statistiquement significative comparativement aux patients recevant un placebo sur le critère principal (fréquence des épisodes de céphalées). Néanmoins, des réductions statistiquement significatives ont été observées sur les critères secondaires suivants: fréquence des jours de céphalées, fréquence des jours de migraine/migraine probable.
Dans l’étude 2, sur la période des 28 derniers jours de la phase en double aveugle de 24 semaines, les patients traités par Botox ont présenté des réductions statistiquement significatives comparativement aux patients recevant un placebo sur le critère principal et les critères secondaires suivants: fréquence des jours de céphalées (critère principal), fréquence des jours de migraine/migraine probable, fréquence des jours de céphalées modérées à sévères, nombre total d’heures cumulées de céphalées les jours de céphalées, fréquence des épisodes de céphalées, proportion de patients dont les céphalées ont un impact majeur (score HIT-6).
Lors de l’analyse post hoc des données combinées des deux études, les patients traités par Botox ont présenté des réductions statistiquement significatives comparativement aux patients recevant un placebo sur les critères suivants: fréquence des jours de céphalées, fréquence des jours de migraine/migraine probable, fréquence des jours de céphalées modérées à sévères, nombre total d’heures cumulées de céphalées les jours de céphalées, nombre total d’heures cumulées de céphalées les jours de céphalées, fréquence des épisodes de céphalées.
Résultats combinés des critères principaux et secondaires d’efficacité dans les deux études pivots à la semaine 24 (temps principal d’évaluation)
|
Critère d’efficacité (variation sur une période de 28 jours) |
Etude 1 |
Etude 2 |
Résultats combinés des deux études |
||||||
|
BOTOX (N=341) |
Placebo (N=338) |
p |
BOTOX (N=347) |
Placebo (N=358) |
p |
BOTOX (N=688) |
Placebo (N=696) |
p |
|
|
Fréquence des jours de céphaléesa |
-7.8 |
-6.4 |
0.006 |
-9.0 |
-6.7 |
<0.001 |
-8.4 |
-6.6 |
<0.001 |
|
Fréquence des jours de migraine/migraine probableb |
-7.6 |
-6.1 |
0.002 |
-8.7 |
-6.3 |
<0.001 |
-8.2 |
-6.2 |
<0.001 |
|
Fréquence des jours de céphalées modérées à sévèresc |
-7.2 |
-5.8 |
0.004 |
-8.3 |
-5.8 |
<0.001 |
-7.7 |
-5.8 |
<0.001 |
|
Nombre total d’heures cumulées de céphalées les jours de céphaléesc |
-106.70 |
-70.40 |
0.003 |
-132.41 |
-90.01 |
<0.001 |
-119.67 |
-80.49 |
<0.001 |
|
Fréquence des épisodes de céphaléesd |
-5.2 |
-5.3 |
0.344 |
-5.3 |
-4.6 |
0.003 |
-5.2 |
-4.9 |
0.009 |
|
Proportion de patients dont les céphalées ont un impact majeur (scores HIT6>60)c |
68.9% |
79.9% |
0.001 |
66.3% |
76.5% |
0.003 |
67.6% |
78.2% |
<0.001 |
a critère principal d’évaluation de l’efficacité dans l’étude 2 et critère secondaire d’évaluation de l’efficacité dans l’étude 1
b critère secondaire d’évaluation de l’efficacité dans les études 1 et 2
c critère secondaire d’évaluation de l’efficacité dans l’étude 2 et critère exploratoire (analyse post hoc) d’évaluation de l’efficacité dans l’étude 1
d critère principal d’évaluation de l’efficacité dans l’étude 1 et critère secondaire d’évaluation de l’efficacité dans l’étude 2.
5.2. Propriétés pharmacocinétiques
Des études cinétiques ont été réalisées en marquant la toxine avec l'iode 125.
Lorsque le produit est injecté dans le muscle jumeau de rat, la radioactivité locale décline rapidement de telle façon que seulement 5 % de la radioactivité persiste après 24 heures. La radioactivité n'apparaît pas au-delà de 10 millimètres du chemin de l'aiguille. Des observations comparables ont été faites lors d'injections réalisées dans la partie supérieure de la paupière de lapin. On ne retrouve dans les urines que 7 % du produit intact.
5.3. Données de sécurité préclinique
La toxicité aiguë chez le rat par voie intramusculaire se situe autour de 39 Unités/Kg.
Des administrations répétées à raison d'une injection par mois chez le rat (6 injections) et le singe adulte (7 injections) et d'une injection toutes les 8 semaines chez le singe juvénile (3 injections) provoquent une atrophie et une dégénérescence du muscle et une paralysie respiratoire. Les doses sans effet toxique ou NOAEL exprimées en Unités/Kg sont estimées pour ces 3 études à 16 (rat), 4 (singe adulte) et 8 (singe juvénile).
Etudes de toxicité sur la reproduction
Suite à des injections intramusculaires de BOTOX à des souris, des rates ou des lapines gravides durant la période d'organogenèse, le NOAEL ("No Observed Adverse Effect Level" ; dose sans effet toxique observé) concernant le développement était de 4,1 et 0,25 Unités/kg respectivement.
Des doses plus élevées étaient associées à des réductions du poids corporel fœtal et/ou à un retard d'ossification, et, chez les lapines, des avortements spontanés ont été notés.
Etude de toxicité sur la fertilité
Les doses sans effet toxique sur la fertilité ou NOAEL après injection intramusculaire de BOTOX chez le rat étaient de 4 Unités/Kg pour le mâle et 8 Unités/Kg pour la femelle. Des doses plus élevées ont été associées à une diminution dose-dépendante de la fertilité probablement liée à une paralysie de l'arrière-train du mâle et à une altération du cycle ovarien. Sous réserve qu’une imprégnation ait eu lieu, aucun effet indésirable n’a été observé sur le nombre ou la viabilité des embryons engendrés ou conçus par les rats mâles ou femelles traités.
Aucune toxicité systémique n’a été observée après une injection unique dans le détrusor < 50 Unités/Kg de BOTOX chez le rat. Afin de simuler une injection accidentelle, une dose unique de BOTOX (~ 7 Unités/Kg) a été administrée dans l’urètre prostatique et le rectum proximal, la vésicule séminale et la paroi vésicale ainsi que dans l’utérus (~ 3 Unités/Kg) de singes sans qu’aucun effet indésirable n’ait été observé. Dans une étude d’administration répétée dans le détrusor pendant 9 mois (4 injections), un ptosis a été observé à la dose de 24 Unités/kg et des doses ≥ 24 Unités/Kg ont été mortelles. Aucun effet indésirable n’a été observé chez le singe à la dose de 12 Unités/Kg qui correspond à une exposition 3 fois plus importante que celle attendue avec la dose clinique recommandée de 200 UNITÉS ALLERGAN dans le traitement des patients atteints d’hyperactivité détrusorienne neurologique (basée sur une personne de 50 Kg).
Il n'y a aucun potentiel mutagène ni clastogène.
Albumine humaine, chlorure de sodium.
3 ans.
Après reconstitution dans son flacon, d’un point de vue microbiologique, une utilisation immédiate de la solution est recommandée. Toutefois, la stabilité physico-chimique a été démontrée pendant 24 heures entre + 2 °C et + 8 °C.
6.4. Précautions particulières de conservation
A conserver entre + 2 °C et + 8 °C (au réfrigérateur).
Pour les conditions de conservation du médicament après reconstitution, voir la rubrique 6.3.
6.5. Nature et contenu de l'emballage extérieur
Poudre en flacon (verre de type I) muni d'un bouchon (caoutchouc) et d'une bague (aluminium), boîte de 1.
6.6. Précautions particulières d’élimination et de manipulation
Préparation du produit
La préparation du produit doit être réalisée dans un local approprié et par du personnel expérimenté afin de minimiser le risque d'incident lors de la manipulation.
Pour reconstituer BOTOX, utiliser uniquement une solution stérile sans conservateur de sérum physiologique (solution injectable de chlorure de sodium à 0,9 pour cent).
Aspirer une quantité de solvant adaptée à la dilution souhaitée dans une seringue de taille adéquate (cf. tableau de dilution).
Nettoyer à l'alcool la partie centrale du bouchon en caoutchouc. Afin d'éviter la dénaturation du produit, injecter délicatement le solvant dans le flacon et agiter doucement en évitant la formation de bulles.
Ne pas utiliser le flacon si la dépression n'entraîne pas l'aspiration du solvant à l'intérieur du flacon.
Une fois reconstituée, la solution obtenue doit être contrôlée visuellement avant utilisation, afin de vérifier qu'elle est limpide, incolore ou jaune très pâle et qu'elle ne contient pas de particules.
Après reconstitution dans son flacon, d’un point de vue microbiologique, une utilisation immédiate de la solution est recommandée. Toutefois, la solution peut être conservée au réfrigérateur et utilisée dans les 24 heures (voir rubrique 6.4).
Si différentes présentations de BOTOX sont utilisées dans le cadre d’une procédure d’injection, une attention doit être apportée à l’utilisation de la bonne quantité de solvant en reconstituant le nombre d’unités par 0,1 ml déterminé. La quantité de solvant varie entre BOTOX 50 UNITÉS ALLERGAN, BOTOX 100 UNITÉS ALLERGAN et BOTOX 200 UNITÉS ALLERGAN. Chaque seringue sera étiquetée en conséquence.
Instructions pour la reconstitution dans le traitement des dysfonctions vésicales
Lorsque BOTOX est dilué dans une seringue pour le traitement des dysfonctions vésicales, il doit être utilisé immédiatement.
Instructions pour la reconstitution de BOTOX 200 UNITÉS ALLERGAN afin d’obtenir une dose de 100 UNITÉS ALLERGAN :
Il est recommandé d’utiliser préférentiellement des flacons de BOTOX 100 UNITÉS ALLERGAN afin de faciliter la reconstitution dans cette indication.
· Reconstituer 1 flacon de BOTOX 200 UNITÉS ALLERGAN, avec 8 ml de solution stérile sans conservateur de sérum physiologique (solution injectable de chlorure de sodium à 0,9 pour cent) puis mélanger le flacon doucement.
· Aspirer 4 ml du flacon dans une seringue de 10 ml.
· Terminer la reconstitution en ajoutant 6 ml de solution stérile sans conservateur de sérum physiologique (solution injectable de chlorure de sodium à 0,9 pour cent) dans la seringue de 10 ml et mélanger doucement.
Vous obtiendrez ainsi une seringue de 10 ml contenant un total de 100 Unités de solution reconstituée de BOTOX.
Instructions pour la reconstitution de BOTOX 200 UNITÉS ALLERGAN afin d’obtenir une dose de 200 UNITÉS ALLERGAN :
· Reconstituer 1 flacon de BOTOX 200 UNITÉS ALLERGAN avec 6 ml de solution stérile sans conservateur de sérum physiologique (solution injectable de chlorure de sodium à 0,9 pour cent) puis mélanger le flacon doucement.
· Aspirer 2 ml du flacon dans chacune des trois seringues de 10 ml.
· Terminer la reconstitution en ajoutant 8 ml de solution stérile sans conservateur de sérum physiologique (solution injectable de chlorure de sodium à 0,9 pour cent) dans chacune des seringues de 10 ml puis mélanger doucement.
Vous obtiendrez ainsi trois seringues de 10 ml contenant un total de 200 Unités de solution reconstituée de BOTOX.
Tableau de dilution – Autres indications
|
|
Flacon de 50 UNITÉS ALLERGAN |
Flacon de 100 UNITÉS ALLERGAN |
Flacon de 200 UNITÉS ALLERGAN |
|
Concentration en UNITÉS ALLERGAN / 0,1 ml |
Solvant ajouté (solution stérile sans conservateur de sérum physiologique (solution injectable de chlorure de sodium à 0,9 pour cent)) |
Solvant ajouté (solution stérile sans conservateur de sérum physiologique (solution injectable de chlorure de sodium à 0,9 pour cent)) |
Solvant ajouté (solution stérile sans conservateur de sérum physiologique (solution injectable de chlorure de sodium à 0,9 pour cent)) |
|
20 |
0,25 ml |
0,5 ml |
1 ml |
|
10 |
0,5 ml |
1 ml |
2 ml |
|
5 |
1 ml |
2 ml |
4 ml |
|
2,5 |
2 ml |
4 ml |
8 ml |
|
1,25 |
4 ml |
8 ml |
N/A |
Recommandations en cas d'incident lors de la manipulation de la toxine botulinique
En cas d'incident lors d'une manipulation du produit qu'il soit à l'état de poudre ou reconstitué, les mesures appropriées décrites ci-dessous doivent être mises en route immédiatement.
La toxine botulinique est très sensible à la chaleur et à certains agents chimiques.
Toute projection doit être essuyée :
· soit avec un matériel absorbant imbibé d'une solution d'hypochlorite de sodium (eau de Javel) en cas de produit sec ;
· soit avec un matériel absorbant sec en cas de produit reconstitué.
Les surfaces contaminées seront nettoyées avec un matériel absorbant imbibé d'une solution d'hypochlorite de sodium (eau de Javel), puis séchées.
En cas de bris de flacon, procéder comme indiqué ci-dessus au ramassage méticuleux des particules de verre et à l'essuyage du produit, en évitant les coupures cutanées.
En cas de projection, laver avec une solution d'hypochlorite de sodium (eau de Javel), puis rincer abondamment à l'eau.
En cas de projection oculaire, rincer abondamment avec de l'eau ou avec une solution ophtalmique de rinçage oculaire.
En cas de blessure du manipulateur (coupure, autopiqûre), procéder comme ci-dessus et prendre les mesures médicales appropriées en fonction de la dose injectée.
Recommandations pour l'élimination du matériel utilisé
Les aiguilles, les seringues et les flacons, qui ne doivent pas être vidés, seront placés, après usage, dans des récipients adaptés qui devront être incinérés.
Le matériel contaminé (tissu absorbant, gants, débris d'ampoule) doit être placé dans un sac intraversable et éliminé par incinération.
Tout médicament non utilisé ou déchet doit être éliminé conformément à la réglementation en vigueur.
7. TITULAIRE DE L’AUTORISATION DE MISE SUR LE MARCHE
ABBVIE
5/13 BOULEVARD DE LA REPUBLIQUE
92100 BOULOGNE-BILLANCOURT
8. NUMERO(S) D’AUTORISATION DE MISE SUR LE MARCHE
· 34009 370 832 0 1 : Poudre en flacon (verre de type I) muni d'un bouchon (caoutchouc) et d'une bague (aluminium), boîte de 1.
9. DATE DE PREMIERE AUTORISATION/DE RENOUVELLEMENT DE L’AUTORISATION
[à compléter ultérieurement par le titulaire]
10. DATE DE MISE A JOUR DU TEXTE
[à compléter ultérieurement par le titulaire]
Sans objet.
12. INSTRUCTIONS POUR LA PREPARATION DES RADIOPHARMACEUTIQUES
Liste I.
Médicament réservé à l’usage hospitalier.
ANSM - Mis à jour le : 06/02/2026
BOTOX 200 UNITÉS ALLERGAN, poudre pour solution injectable
Toxine botulinique de type A
· Gardez cette notice. Vous pourriez avoir besoin de la relire.
· Si vous avez d’autres questions, interrogez votre médecin ou votre pharmacien.
· Ce médicament vous a été personnellement prescrit. Ne le donnez pas à d’autres personnes. Il pourrait leur être nocif, même si les signes de leur maladie sont identiques aux vôtres.
· Si vous ressentez un quelconque effet indésirable, parlez-en à votre médecin ou votre pharmacien. Ceci s’applique aussi à tout effet indésirable qui ne serait pas mentionné dans cette notice. Voir rubrique 4.
1. Qu'est-ce que BOTOX 200 UNITÉS ALLERGAN, poudre pour solution injectable et dans quels cas est-il utilisé ?
2. Quelles sont les informations à connaître avant d'utiliser BOTOX 200 UNITÉS ALLERGAN, poudre pour solution injectable ?
3. Comment utiliser BOTOX 200 UNITÉS ALLERGAN, poudre pour solution injectable ?
4. Quels sont les effets indésirables éventuels ?
5. Comment conserver BOTOX 200 UNITÉS ALLERGAN, poudre pour solution injectable ?
6. Contenu de l’emballage et autres informations.
1. QU’EST-CE QUE BOTOX 200 UNITÉS ALLERGAN, poudre pour solution injectable ET DANS QUELS CAS EST-IL UTILISE ?
Classe pharmacothérapeutique - code ATC : M03AX01
BOTOX est un relaxant musculaire utilisé pour traiter un certain nombre d’affections de l’organisme. Il contient de la toxine botulinique de type A, substance active qui est injectée aussi bien dans les muscles que dans la paroi de la vessie ou profondément dans la peau. Il agit en bloquant partiellement les impulsions nerveuses de tout muscle qui a été injecté et, réduit les contractions excessives de ces muscles.
Lorsqu’il est injecté sous la peau des aisselles, BOTOX agit sur les glandes sudoripares pour réduire la quantité de sueur produite. Lorsqu'il est injecté dans la paroi de la vessie, BOTOX agit sur le muscle de la vessie pour réduire les fuites urinaires (incontinence urinaire).
Lorsqu’il est injecté dans les muscles de la tête et du cou, BOTOX bloque les signaux de la douleur pour prévenir et traiter la migraine chronique.
Indications chez l’adulte :
Troubles de la vessie :
Lorsqu’il est injecté dans la paroi de la vessie, BOTOX agit sur le muscle de la vessie pour réduire les fuites urinaires (incontinence urinaire) et contrôler les situations suivantes :
· Hyperactivité vésicale idiopathique avec fuites urinaires (3 épisodes sur trois jours) : envie soudaine d’uriner et d’aller aux toilettes plus souvent que d’habitude (d’au moins 8 ou plus épisodes par jour) lorsqu’un traitement par un autre médicament (appelé anticholinergique) n’a pas agi.
· Hyperactivité vésicale due à des problèmes associés à une lésion de la moelle épinière ou à une sclérose en plaques, conduisant à des fuites urinaires (émission involontaire d’urine pendant la journée ou la nuit).
Troubles neurologiques :
BOTOX est utilisé pour prévenir la migraine chronique chez les adultes qui ont eu des maux de tête pendant 15 jours ou plus par mois, dont au moins 8 jours de migraine et qui n'ont pas bien répondu aux autres médicaments préventifs contre la migraine.
La migraine chronique est une maladie qui affecte le système nerveux. Les patients présentent généralement des maux de tête souvent associés à une sensibilité excessive à la lumière, aux sons forts ou aux odeurs, ainsi que de nausées et/ou de vomissements. Ces maux de tête surviennent au moins 15 jours par mois.
Indications chez l'adulte et l'enfant de plus de 12 ans :
BOTOX peut être injecté directement dans les muscles pour contrôler les situations suivantes :
· spasmes musculaires persistants dans les muscles autour des yeux, des paupières et du visage, provoquant un strabisme ou des yeux qui louchent ;
· spasmes musculaires persistants des paupières et du visage ;
· spasmes musculaires persistants dans le cou et les épaules.
BOTOX peut être injecté profondément dans la peau et ainsi agir sur les glandes sudoripares pour réduire la transpiration excessive des aisselles ; symptômes pouvant perturber les activités quotidiennes lorsque les autres traitements locaux n’agissent pas.
Indication chez l'adulte et l'enfant de 2 ans et plus :
BOTOX peut être injecté directement dans les muscles et ainsi contrôler les spasmes musculaires persistants dans les bras et/ou les jambes.
2. QUELLES SONT LES INFORMATIONS A CONNAITRE AVANT D’UTILISER BOTOX 200 UNITÉS ALLERGAN, poudre pour solution injectable ?
N’utilisez jamais BOTOX 200 UNITÉS ALLERGAN, poudre pour solution injectable :
· si vous êtes allergique à la toxine botulinique de type A ou à l’un des autres composants contenus dans ce médicament, mentionnés dans la rubrique 6,
· si vous souffrez de myasthénie grave (maladie chronique affectant vos muscles),
· si vous présentez une infection au niveau du site d’injection,
· chez les patients atteints d’hyperactivité vésicale sans cause connue ou associée à une lésion de la moelle épinière ou à une sclérose en plaques, en cas d’infection urinaire au moment du traitement ou en cas d’impossibilité de vider votre vessie sans être habitué à utiliser une sonde urinaire.
Avertissements et précautions
Adressez-vous à votre médecin avant d’utiliser BOTOX 200 UNITÉS ALLERGAN, poudre pour solution injectable :
· si vous avez déjà eu des problèmes pour avaler ou si des aliments ou tout liquide sont déjà passés accidentellement dans vos poumons, en particulier si vous êtes traité pour des spasmes musculaires persistants du cou et des épaules ;
· si vous êtes âgé de plus de 65 ans et que vous souffrez d’autres maladies graves ;
· si vous souffrez de tout autre problème musculaire ou de maladies chroniques affectant vos muscles (telles que myasthénie grave ou syndrome de Lambert-Eaton) ;
· si vous souffrez de certaines maladies affectant le système nerveux (telles que sclérose latérale amyotrophique (Maladie de Charcot) ou neuropathie motrice) ;
· si vous souffrez de faiblesse importante ou de perte musculaire dans les muscles que votre médecin prévoit d’injecter ;
· si vous avez déjà subi une intervention chirurgicale ou avez eu une blessure qui a pu modifier d’une façon ou d’une autre, le muscle qui doit recevoir l’injection ;
· si vous avez déjà ressenti des troubles au cours de précédentes injections (tel qu’un évanouissement) ;
· si vous souffrez d’une inflammation des muscles ou de la surface de la peau où votre médecin prévoit d’injecter ;
· si vous souffrez de maladies cardiovasculaires (maladie du cœur ou des vaisseaux sanguins) ;
· si vous souffrez ou avez souffert de convulsions ;
· si vous souffrez d’une maladie oculaire appelée glaucome à angle fermé (pression élevée dans l’œil) ou si vous avez été informé qu’il y avait un risque que vous développiez ce type de glaucome ;
· si vous allez être traité pour une hyperactivité de la vessie avec fuites urinaires et que vous êtes un homme présentant des signes et des symptômes de rétention ou d’obstruction urinaire, tels que des difficultés à uriner ou un flux faible ou interrompu.
Après avoir reçu une injection de BOTOX 200 UNITÉS ALLERGAN, poudre pour solution injectable
Vous ou toute personne qui vous aide devra contacter votre médecin afin de recevoir une assistance médicale immédiate si vous ressentez un des effets suivants :
· difficulté à respirer, avaler ou parler ;
· urticaire, œdème incluant gonflement du visage ou de la gorge, respiration sifflante, étourdissement et essoufflement (symptômes possibles d’une réaction allergique sévère).
Précautions générales
· Ce produit est réservé à l'usage hospitalier en raison de ses caractéristiques : vous ne pouvez en détenir. La préparation du produit doit être réalisée par un personnel expérimenté, dans un local approprié.
· L'injection doit être réalisée par un médecin ayant une bonne formation et de l’expérience sur l'utilisation de la toxine botulinique dans les indications prévues par l'autorisation de mise sur le marché.
· Comme pour toute injection, il se peut que la procédure entraîne une infection, une douleur, un œdème, des brûlures et picotements, une sensibilité accrue, une douleur au toucher, une rougeur et/ou des saignements/hématomes au site d’injection.
· Des effets indésirables liés à la diffusion de la toxine à distance du site d'injection ont été rapportés (par exemple faiblesse musculaire, difficultés à avaler, fausse route d’aliments ou de liquide dans les voies respiratoires) avec des conséquences pouvant être graves, voire fatales dans de très rares cas. Le risque de survenue de tels effets est plus important chez les patients présentant une maladie neurologique avec difficultés à avaler.
· Si vous recevez des injections de BOTOX trop fréquemment ou si la dose injectée est trop élevée, il est possible que vous ressentiez une faiblesse musculaire ainsi que des effets indésirables liés à la diffusion de la toxine ou que votre corps produise des anticorps, ce qui peut réduire l’effet de BOTOX. Pour limiter ce risque, un intervalle de temps minimum doit être respecté entre 2 séances d'injection (voir rubrique 3. Comment utiliser BOTOX 200 UNITÉS ALLERGAN, poudre pour solution injectable ?).
· Lorsque BOTOX est utilisé dans le traitement d'une maladie qui n’est pas mentionnée dans cette notice, cela pourrait entraîner des réactions graves, en particulier chez les patients qui rencontrent déjà des difficultés à avaler ou qui ressentent une extrême faiblesse générale.
· Si vous n’avez pas eu d’activités physiques régulières pendant une longue période avant de recevoir un traitement par BOTOX, vous devrez alors débuter progressivement toute activité après les injections.
· Il est peu probable que ce médicament améliore l'amplitude de mouvement des articulations dont le muscle environnant a perdu son élasticité.
· Lorsque BOTOX est utilisé dans le traitement des spasmes musculaires persistants de la paupière, une diminution du clignement des yeux peut se produire, ce qui peut endommager la surface de vos yeux. Pour éviter cela, vous aurez besoin d’un traitement par gouttes oculaires, pommades, lentilles de contact souples ou même d’une protection qui couvre et maintient l’œil fermé. Votre médecin vous informera si cela est nécessaire.
· En cas de traitement de l’hyperactivité vésicale idiopathique :
o Vous devez être capable d’utiliser des sondes urinaires stériles pour vider votre vessie.
o Vous serez revu par votre médecin 2 semaines après l’injection. Vous devrez uriner et le volume d’urine restant dans votre vessie sera mesuré par échographie. Votre médecin décidera alors si vous devez revenir pour répéter cet examen pendant les 12 semaines suivantes. Vous devez contacter votre médecin si vous êtes dans l’impossibilité d’uriner.
o Une interruption du traitement par antiagrégants plaquettaires (type aspirine) et/ou anticoagulants (fluidifiants sanguins) est recommandée 3 jours avant la procédure d’injection.
· En cas de traitement de l’hyperactivité du détrusor associée à une lésion de la moelle épinière ou à une sclérose en plaques :
o Vous ou quelqu’un de votre entourage devez être capable d’utiliser des sondes urinaires stériles pour vider votre vessie.
o Vous serez revu par votre médecin 2 semaines après l’injection, si vous aviez des mictions spontanées ou des mictions spontanées partielles avant l’injection. Vous devrez uriner et le volume d’urine restant dans votre vessie sera mesuré par échographie. Votre médecin décidera alors si vous devez revenir pour répéter cet examen pendant les 12 semaines suivantes. Vous devez contacter votre médecin si vous êtes dans l’impossibilité d’uriner.
Une interruption du traitement par antiagrégants plaquettaires (type aspirine) et/ou anticoagulants (fluidifiants sanguins) est recommandée 3 jours avant la procédure d’injection.
Enfants et adolescents
Sans objet.
Autres médicaments et BOTOX 200 UNITÉS ALLERGAN, poudre pour solution injectable
Informez votre médecin si :
· vous prenez des antibiotiques (utilisés dans le traitement des infections), des anticholinestérasiques ou des relaxants musculaires. Certains de ces médicaments peuvent augmenter l’effet de BOTOX,
· vous avez été récemment injecté avec un médicament contenant de la toxine botulinique (la substance active contenue dans BOTOX) car l’effet de BOTOX pourrait être augmenté considérablement,
· vous prenez des antiagrégants plaquettaires (type aspirine) et/ou des anticoagulants (fluidifiants sanguins).
Informez votre médecin ou pharmacien si vous prenez, avez récemment pris ou pourriez prendre tout autre médicament.
BOTOX 200 UNITÉS ALLERGAN, poudre pour solution injectable avec des aliments, des boissons et de l’alcool
Sans objet.
L'utilisation de BOTOX n’est pas recommandée pendant la grossesse et chez les femmes en âge de procréer ne prenant pas de contraception, sauf en cas de nécessité. BOTOX n’est pas recommandé chez les femmes qui allaitent.
Si vous êtes enceinte ou que vous allaitez, si vous pensez être enceinte ou planifiez une grossesse, demandez conseil à votre médecin ou pharmacien avant de prendre ce médicament.
Demandez conseil à votre médecin ou à votre pharmacien avant de prendre tout médicament.
Conduite de véhicules et utilisation de machines
L'attention est attirée, notamment chez les conducteurs de véhicules et les utilisateurs de machines, sur les risques de vertiges, de somnolence, de fatigue, de faiblesse musculaire ou de problèmes de vue liés à l’utilisation de ce médicament, pouvant rendre dangereuse la conduite de véhicules ou l'utilisation de machines.
BOTOX contient du sodium
Ce médicament contient moins de 1 mmol (23 mg) de sodium par flacon, c.-à-d. qu’il est essentiellement « sans sodium ».
3. COMMENT UTILISER BOTOX 200 UNITÉS ALLERGAN, poudre pour solution injectable ?
|
La préparation et l'injection de BOTOX sont réservées à un personnel de santé expérimenté (voir rubrique 2. Prendre des précautions particulières avec BOTOX 200 UNITÉS ALLERGAN, poudre pour solution injectable). |
Posologie
La posologie varie en fonction de l'indication et de l'âge. Il revient à votre médecin de déterminer la dose qui vous convient, conformément à la rubrique Posologie du Résumé des Caractéristiques du Produit.
A titre indicatif, la posologie peut varier de 17,5 UNITÉS ALLERGAN par injection à une dose maximale de 360 UNITÉS ALLERGAN par séance d'injection dans certaines indications.
Mode d'administration
Injection dans les muscles (voie intramusculaire), dans la paroi de la vessie en utilisant un instrument spécifique (cystoscope) ou sous la peau (voie intradermique) suivant l'indication. BOTOX est injecté directement dans la zone à traiter. Votre médecin injectera généralement BOTOX dans plusieurs sites de chaque zone traitée.
La préparation du produit doit être réalisée dans un local approprié et par du personnel expérimenté afin de minimiser le risque d'accident lors de la manipulation.
La reconstitution de BOTOX, poudre pour solution injectable est réalisée à l'aide d'une solution injectable de chlorure de sodium à 0,9 pour cent.
Informations générales sur la posologie
Le nombre d’injections par muscle ainsi que la dose varient selon les indications. C’est pourquoi, votre médecin décidera de la quantité, de la fréquence et dans quel(s) muscle(s) BOTOX vous sera injecté. Il est recommandé à votre médecin d’utiliser la dose minimale effective.
La posologie et la durée d’action de BOTOX varient selon la maladie pour laquelle vous êtes traité. Vous trouverez ci-dessous les détails correspondants à chaque maladie.
La sécurité et l'efficacité de BOTOX ont été établies chez les enfants et les adolescents âgés de plus de deux ans dans le traitement symptomatique local de la spasticité.
|
Spasmes musculaires persistants des bras et/ou des jambes |
2 ans |
Des informations limitées sont disponibles sur l’utilisation de BOTOX chez les enfants et les adolescents dont les âges sont indiqués dans le tableau ci-dessous dans les indications suivantes.
Aucune recommandation posologique ne peut être faite pour ces indications.
|
Spasmes musculaires persistants dans les muscles autour des yeux, des paupières et du visage, provoquant un strabisme ou des yeux qui louchent |
12 ans |
|
Spasmes musculaires persistants des paupières et du visage |
12 ans |
|
Spasmes musculaires persistants du cou et des épaules |
12 ans |
|
Transpiration excessive des aisselles |
12 ans |
|
Hyperactivité détrusorienne neurologique chez l’enfant |
5-17 ans |
|
Hyperactivité vésicale chez l’enfant |
12-17 ans |
Posologie
|
Indication |
Dose maximale (UNITÉS ALLERGAN par zone traitée) |
Intervalle minimal entre 2 séances d’injection |
|
|
|
1ère séance d’injection |
Séances d’injection suivantes |
|
|
Spasmes musculaires persistants des bras et/ou des jambes chez les enfants de 2 ans et plus |
Votre médecin pourra effectuer plusieurs injections dans les muscles à traiter. La posologie exacte et le nombre de sites d’injection sont adaptés aux besoins individuels. La dose totale est de 350 UNITÉS ALLERGAN réparties entre les différents sites ou 50 UNITÉS ALLERGAN par site. |
La posologie exacte et le nombre de sites d’injection sont adaptés aux besoins individuels. La dose totale est de 350 UNITÉS ALLERGAN réparties entre les différents sites injectés ou 50 UNITÉS ALLERGAN par site. |
3 mois* |
|
Spasmes musculaires persistants des bras et/ou des jambes chez l’adulte |
Votre médecin pourra effectuer plusieurs injections dans les muscles à traiter. La posologie exacte et le nombre de sites d’injection sont adaptés aux besoins individuels. En général, la dose totale est de 6 UNITÉS ALLERGAN/kg réparties entre les différents sites. |
La posologie exacte et le nombre de sites d’injection sont adaptés aux besoins individuels. En général, la dose totale est de 6 UNITÉS ALLERGAN/kg réparties entre les différents sites. |
3 mois |
|
Spasmes musculaires persistants dans les muscles autour des yeux, des paupières et du visage, provoquant un strabisme ou des yeux qui louchent |
Votre médecin pourra effectuer plusieurs injections dans les muscles à traiter. La posologie exacte est adaptée aux besoins individuels. |
La posologie exacte est adaptée aux besoins individuels. |
Jusqu’à ce que les effets de la dose précédente ne cessent. |
|
Spasmes musculaires persistants des paupières et du visage |
25 UNITÉS ALLERGAN par œil |
Jusqu’à 100 UNITÉS ALLERGAN |
3 mois |
|
Spasmes musculaires persistants du cou et des épaules |
200 UNITÉS ALLERGAN |
Jusqu’à 300 UNITÉS ALLERGAN |
10 semaines |
|
Transpiration excessive des aisselles |
50 UNITÉS ALLERGAN par aisselle |
50 UNITÉS ALLERGAN par aisselle |
4 mois |
|
Hyperactivité vésicale idiopathique avec fuites urinaires |
50 UNITÉS ALLERGAN |
Jusqu’à 100 UNITÉS ALLERGAN |
3 mois |
|
Hyperactivité vésicale neurologique associée à une sclérose en plaques chez l’adulte |
100 ou 200** UNITÉS ALLERGAN |
100 ou 200** UNITÉS ALLERGAN |
3 mois |
|
Hyperactivité vésicale neurologique associée à une lésion de la moelle épinière chez l’adulte |
200 UNITÉS ALLERGAN |
200 UNITÉS ALLERGAN |
3 mois |
|
Maux de tête chez l’adulte atteint de migraine chronique |
155 à 195 UNITÉS ALLERGAN Il convient de ne pas injecter plus de 5 unités sur un site d’injection |
155 à 195 UNITÉS ALLERGAN
|
3 mois |
* Selon la dose choisie par le médecin, l’intervalle entre 2 séances d’injection peut aller jusqu’à 6 mois.
** Si vous utilisez des sondes urinaires stériles pour vider votre vessie.
L’intervalle doit être adapté à l’état de santé du patient et dépendra de la réapparition des symptômes.
Temps nécessaire à l’amélioration et durée de traitement
Toujours se conformer à la prescription médicale.
Spasmes musculaires persistants des bras et/ou des jambes chez les enfants de 2 ans et plus : l'amélioration apparaît généralement dans les 2 premières semaines après injection.
Spasmes musculaires persistants des bras et/ou des jambes chez l’adulte : l'amélioration apparaît généralement dans les 2 premières semaines après injection. L'effet maximal est habituellement observé environ 4 à 6 semaines après injection.
Spasmes musculaires persistants dans les muscles autour des yeux, des paupières et du visage, provoquant un strabisme ou des yeux qui louchent : l'amélioration apparaît généralement dans les 2 jours après injection. Habituellement, l’effet dure 2 à 6 semaines après injection.
Spasmes musculaires persistants des paupières et du visage : l'amélioration apparaît généralement dans les 3 premiers jours après injection. L’effet maximal est habituellement observé après 1 à 2 semaines.
Spasmes musculaires persistants du cou et des épaules : l'amélioration apparaît généralement dans les 2 semaines après injection. L'effet maximal est habituellement observé environ 6 semaines après injection.
Hyperactivité vésicale idiopathique avec fuites urinaires : l'amélioration apparaît généralement dans les 2 semaines après injection. Habituellement, l'effet dure approximativement 6-7 mois après injection.
Hyperactivité vésicale associée à une lésion de la moelle épinière ou à une sclérose en plaques chez l’adulte : l'amélioration apparaît généralement dans les 2 semaines après injection. Habituellement, l'effet dure approximativement 8-9 mois après injection.
Transpiration excessive des aisselles : l'amélioration apparaît généralement dans la première semaine après injection. L'effet dure habituellement plus de 4 mois et peut persister un an voire plus.
Si vous avez reçu plus de BOTOX 200 UNITÉS ALLERGAN, poudre pour solution injectable que vous n’auriez dû :
Consultez immédiatement votre médecin ou votre pharmacien.
Les signes d’une dose excessive de BOTOX peuvent ne pas apparaître pendant plusieurs jours après injection. Si vous avalez BOTOX ou qu’il vous a été injecté accidentellement, vous devez consulter votre médecin qui pourra vous garder en observation pendant plusieurs semaines.
Si vous avez reçu une dose excessive de BOTOX, l’un des symptômes mentionnés ci-après peut apparaître. Vous devez contacter votre médecin immédiatement qui décidera si vous devez aller à l'hôpital :
· faiblesse musculaire locale ou à distance du site d'injection ;
· difficulté à respirer, avaler ou parler en raison d’une paralysie des muscles ;
· pneumonie (infection des poumons) en raison d’une paralysie des muscles, provoquée par ingestion accidentelle d’aliments ou liquide dans les poumons ;
· affaissement des paupières, vision double ;
· faiblesse généralisée.
Si vous oubliez d’utiliser BOTOX 200 UNITÉS ALLERGAN, poudre pour solution injectable
Sans objet.
Si vous arrêtez d’utiliser BOTOX 200 UNITÉS ALLERGAN, poudre pour solution injectable
Sans objet.
Si vous avez d’autres questions sur l’utilisation de ce médicament, demandez plus d’informations à votre médecin ou à votre pharmacien.
4. QUELS SONT LES EFFETS INDESIRABLES EVENTUELS ?
En général, les effets indésirables apparaissent au cours des premiers jours suivant l'injection. Habituellement, ils durent seulement pendant une courte période, mais ils peuvent durer plusieurs mois et dans de rares cas, plus longtemps.
Si vous avez des difficultés à respirer, avaler ou parler après avoir reçu une injection de BOTOX, CONTACTEZ IMMÉDIATEMENT VOTRE MÉDECIN.
En cas d’apparition d’urticaire, d’œdème incluant gonflement du visage ou de la gorge, d’une respiration sifflante, d’un étourdissement et d’un essoufflement, CONTACTEZ IMMÉDIATEMENT VOTRE MÉDECIN.
Effets indésirables indépendants du site injecté ou de l'indication
· Des effets indésirables liés à la diffusion de la toxine à distance du site d'injection ont été très rarement rapportés : faiblesse musculaire excessive, difficultés à avaler, pneumonie due à une fausse route des aliments ou de liquide dans les voies respiratoires, pouvant être fatals dans certains cas.
· Rares réactions allergiques générales (éruption cutanée, érythème, prurit, réaction anaphylactique).
· Douleurs/ brûlures au site d'injection, possibles quel que soit le site d'injection ou l'indication.
Les effets secondaires sont classés dans les catégories suivantes, en fonction de leur fréquence :
|
Très fréquents |
peuvent affecter plus de 1 personne sur 10 |
|
Fréquents |
peuvent affecter jusqu’à 1 personne sur 10 |
|
Peu fréquents |
peuvent affecter jusqu’à 1 personne sur 100 |
|
Rares |
peuvent affecter jusqu’à 1 personne sur 1 000 |
|
Très rares |
peuvent affecter jusqu’à 1 personne sur 10 000 |
Les listes d’effets indésirables ci-après, varient selon la partie du corps où BOTOX est injecté. Si l’un des effets indésirables devient grave ou si vous remarquez des effets indésirables non mentionnés dans cette notice, contactez votre médecin ou votre pharmacien.
Traitement des spasmes musculaires persistants de la paupière et du visage
|
Très fréquent |
Affaissement de la paupière |
|
Fréquents |
Lésion localisée de la cornée (membrane transparente recouvrant la surface de l’œil), difficulté à fermer l’œil complètement, sécheresse oculaire, sensibilité à la lumière, irritation oculaire, larmoiement excessif, hématome sous la peau, irritation de la peau, gonflement du visage |
|
Peu fréquents |
Vertiges, faiblesse des muscles du visage, affaissement des muscles d'un côté du visage, inflammation de la cornée (membrane transparente recouvrant la surface de l'œil), enroulement anormal des paupières vers l'extérieur ou vers l'intérieur, vision double, difficultés à voir clairement, vision floue, éruption cutanée, fatigue et paralysie du visage |
|
Rare |
Gonflement de la paupière |
|
Très rares |
Inflammation ulcérative, lésion ou perforation de la cornée (membrane transparente recouvrant la surface de l’œil) |
Traitement des spasmes musculaires persistants du cou et des épaules
|
Très fréquents |
Difficultés à avaler, faiblesse musculaire, douleurs |
|
Fréquents |
Gonflement et irritation à l’intérieur du nez (rhinite), nez bouché ou qui coule, toux, mal de gorge, chatouillement ou irritation de la gorge, vertiges, tension musculaire accrue (crampes), sensibilité de la peau réduite, somnolence, mal de tête, sécheresse buccale, nausées, muscles raidis ou endoloris, fatigue, syndrome grippal, malaise |
|
Peu fréquents |
Vision double, fièvre, affaissement de la paupière, essoufflement, changement dans votre voix |
Traitement de la transpiration excessive des aisselles
|
Très fréquent |
Douleurs au site d’injection |
|
Fréquents |
Mal de tête, engourdissement, bouffées de chaleur, transpiration accrue sur les sites autres que les aisselles, odeur anormale de la peau, démangeaisons, grosseur sous la peau, perte de cheveux, douleurs aux extrémités (mains et doigts), douleurs, gonflement, saignements ou brûlures au site d’injection, sensibilité accrue au site d’injection, fatigue |
|
Peu fréquents |
Nausées, faiblesse musculaire, douleurs musculaires, problèmes aux articulations |
Traitement des spasmes musculaires persistants des bras et/ou des jambes chez l’adulte
|
Fréquents |
Douleurs au site d’injection, membre injecté douloureux, hématomes et saignements sous la peau provoquant des plaques rouges, tension musculaire accrue, faiblesse musculaire, douleurs aux extrémités (mains et doigts), fièvre, syndrome grippal, irritation ou saignements au site d’injection |
|
Peu fréquents |
Douleurs articulaires, sensation de faiblesse, hémorragie, sensibilité de la peau accrue ou diminuée, sensations cutanées anormales (par exemple picotements ou engourdissements), manque de coordination des mouvements, sensation vertigineuse ou de « tête qui tourne » (vertiges), chute de la pression artérielle en position debout qui provoquent des étourdissements, malaise ou évanouissement, engourdissement autour de la bouche, douleurs, dépression, insomnie, mal de tête, amnésie, faiblesse momentanée, nausées, réactions cutanées, douleur ou inflammation des articulations, hypersensibilité au site d’injection, saignements, gonflement des extrémités (mains et pieds) |
Traitement des spasmes musculaires persistants des bras et/ou des jambes chez les enfants de 2 ans et plus
Chez l’enfant, les effets indésirables suivants ont été observés pendant le traitement des spasmes musculaires persistants des bras :
|
Très fréquent |
Douleurs aux sites d’injection |
|
Fréquents |
Manque de coordination et amplitude réduite des mouvements, vomissements, envie fréquente d’uriner (pollakiurie), faiblesse musculaire, spasmes musculaires, syndrome du doigt à gâchette (doigt à ressaut), syndrome pseudo-grippal, pneumonie, luxation, chutes, hématomes et saignements sous la peau provoquant des plaques rouges |
|
Peu fréquents |
Dermatite, prurit, éruption cutanée |
Chez l’enfant, les effets indésirables suivants ont été observés pendant le traitement des spasmes musculaires persistants des jambes :
|
Très fréquents |
Infection virale, infection de l’oreille |
|
Fréquents |
Somnolence, troubles de la marche, sensations cutanées anormales (par exemple picotements ou engourdissements), miction involontaire (incontinence), réactions cutanées, douleurs musculaires, faiblesse musculaire, douleurs dans les mains et les pieds, chutes, sensation de malaise général, douleurs aux sites d’injection, fatigue |
Traitement de l’hyperactivité vésicale idiopathique avec fuites urinaires
|
Très fréquents |
Infections urinaires, douleurs en urinant après injection* |
|
Fréquents |
Bactéries dans les urines, impossibilité d’uriner (rétention urinaire), vidange incomplète de la vessie, envie fréquente d’uriner dans la journée, présence de sang dans les urines après injection**, globules blancs dans les urines |
* Cet effet indésirable peut être lié à la procédure d’injection
** Cet effet indésirable est lié à la procédure d’injection
Traitement de l’hyperactivité vésicale avec fuites urinaires chez l’enfant
|
Fréquents |
Infections urinaires, douleurs en urinant après injection*, douleur dans l’urètre (le tube qui transporte l’urine de la vessie en dehors du corps)*, douleur abdominale, douleur abdominale basse |
*Cet effet indésirable est uniquement lié à la procédure d’injection
Traitement de l’hyperactivité vésicale chez l’adulte associée à une lésion de la moelle épinière ou à une sclérose en plaques conduisant à des fuites urinaires
|
Très fréquent |
Impossibilité d’uriner (rétention urinaire) |
|
Fréquents |
Infection urinaire, faiblesse musculaire, présence de sang dans les urines après injection*, douleurs en urinant après injection*, fatigue, possible réaction réflexe non contrôlée du corps (c'est-à-dire transpiration importante, maux de tête lancinants ou augmentation du pouls) dans les jours suivant l’injection (hyperréflexie autonome)* |
* Certains de ces effets indésirables fréquents peuvent être liés à la procédure d’injection
Traitement de l’hyperactivité vésicale chez l’enfant associée à un spina-bifida, une lésion de la moelle épinière ou une myélite transverse conduisant à des fuites urinaires.
|
Très fréquent |
Bactéries dans les urines |
|
Fréquents |
Infection urinaire, globules blancs dans les urines, présence de sang dans les urines après injection, douleurs au niveau de la vessie (liée à l’administration du produit) |
Migraine chronique
|
Fréquents |
Maux de tête, migraine, faiblesse des muscles du visage, affaissement de la paupière, éruption cutanée, démangeaisons, douleurs au cou, douleurs musculaires, spasme musculaire, raideur musculaire, contracture musculaire, faiblesse musculaire, douleur au point d’injection |
|
Peu fréquents |
Difficulté à avaler, douleurs cutanées, douleur à la mâchoire |
|
Fréquence indéterminée |
Effet Méphisto (élévation de la partie externe des sourcils) |
Depuis sa commercialisation, des effets indésirables supplémentaires ont été rapportés avec BOTOX, communs à toutes situations :
· Réactions allergiques comprenant des réactions aux protéines ou aux sérums injectés ;
· Gonflement des couches profondes de la peau ;
· Urticaire ;
· Troubles de l’alimentation, perte d’appétit ;
· Lésions nerveuses (brachialgie) ;
· Troubles de la voix et de la parole ;
· Affaissement des muscles d’un côté du visage ;
· Faiblesse des muscles du visage ;
· Sensibilité de la peau réduite ;
· Faiblesse musculaire ;
· Maladie chronique affectant les muscles (myasthénie grave) ;
· Difficultés à bouger le bras et l’épaule ;
· Engourdissement ;
· Douleurs/engourdissement/faiblesse, partant de la colonne vertébrale ;
· Convulsions et évanouissement ;
· Augmentation de la pression oculaire ;
· Strabisme ;
· Vision floue ;
· Difficultés à voir clairement ;
· Diminution de l’ouïe ;
· Impression de bruits dans l’oreille ;
· Sensation d’étourdissements ou de « tête qui tourne » (vertiges) ;
· Problèmes cardiaques y compris crise cardiaque ;
· Pneumonies d’inhalation (inflammation des poumons provoquée par aspiration accidentelle d’aliments, de boissons, de salive, ou de vomissures) ;
· Troubles respiratoires, dépression respiratoire et/ou insuffisance respiratoire ;
· Douleurs abdominales ;
· Diarrhée, constipation ;
· Sécheresse de la bouche ;
· Difficultés à avaler ;
· Nausées, vomissements ;
· Perte de cheveux ;
· Démangeaisons ;
· Différents types d’éruptions cutanées de plaques rouges ;
· Transpiration excessive ;
· Perte des cils/des sourcils ;
· Douleurs musculaires, perte de l’innervation et atrophie du muscle injecté ;
· Sensation de malaise généralisé ;
· Fièvre ;
· Œil sec ;
· Contractions musculaires localisées / contractions musculaires involontaires ;
· Gonflement de la paupière.
Déclaration des effets secondaires
Si vous ressentez un quelconque effet indésirable, parlez-en à votre médecin ou votre pharmacien. Ceci s’applique aussi à tout effet indésirable qui ne serait pas mentionné dans cette notice. Vous pouvez également déclarer les effets indésirables directement via le système national de déclaration : Agence nationale de sécurité du médicament et des produits de santé (ANSM) et réseau des Centres Régionaux de Pharmacovigilance - Site internet : https://signalement.social-sante.gouv.fr/
En signalant les effets indésirables, vous contribuez à fournir davantage d’informations sur la sécurité du médicament.
5. COMMENT CONSERVER BOTOX 200 UNITÉS ALLERGAN, poudre pour solution injectable ?
Tenir ce médicament hors de la vue et de la portée des enfants.
N’utilisez pas ce médicament après la date de péremption indiquée sur le flacon après EXP. La date de péremption fait référence au dernier jour de ce mois.
A conserver à une température comprise entre +2°C et +8°C (au réfrigérateur).
Après reconstitution dans son flacon, une utilisation immédiate de la solution est recommandée, bien que la stabilité ait été démontrée pendant 24 heures entre +2°C et +8°C.
Ne jetez aucun médicament au tout-à-l’égout ou avec les ordures ménagères. Demandez à votre pharmacien d’éliminer les médicaments que vous n’utilisez plus. Ces mesures contribueront à protéger l’environnement.
6. CONTENU DE L’EMBALLAGE ET AUTRES INFORMATIONS
Ce que contient BOTOX 200 UNITÉS ALLERGAN, poudre pour solution injectable
· La substance active est :
Toxine botulinique de type A*1................................................................ 200 unités*2 ALLERGAN
pour un flacon.
*1 (de Clostridium botulinum)
*2 Une unité correspond à la dose létale 50 (DL50) du produit reconstitué et injecté par voie intrapéritonéale chez la souris.
· Les autres composants sont : albumine humaine, chlorure de sodium.
Ce médicament se présente sous forme d'une fine poudre blanche pour solution injectable qui peut être difficile à voir dans le fond du flacon (en flacon de 200 UNITÉS ALLERGAN de toxine botulinique de type A).
Titulaire de l’autorisation de mise sur le marché
5/13 BOULEVARD DE LA REPUBLIQUE
92100 BOULOGNE-BILLANCOURT
Exploitant de l’autorisation de mise sur le marché
5/13 BOULEVARD DE LA REPUBLIQUE
92100 BOULOGNE-BILLANCOURT
ALLERGAN PHARMACEUTICALS IRELAND
CASTLEBAR ROAD
WESTPORT
COUNTY MAYO
IRLANDE
Ou
ABBVIE DEUTSCHLAND GMBH & CO. KG
KNOLLSTRASSE
67061 LUDWIGSHAFEN
ALLEMAGNE
Noms du médicament dans les Etats membres de l'Espace Economique Européen
Sans objet.
La dernière date à laquelle cette notice a été révisée est :
[à compléter ultérieurement par le titulaire]
{mois AAAA}.
Des informations détaillées sur ce médicament sont disponibles sur le site Internet de l’ANSM (France).
Les informations suivantes sont destinées exclusivement aux professionnels de santé :
A L'INTENTION DES PROFESSIONNELS DE SANTÉ
Veuillez vous référer au Résumé des Caractéristiques du Produit pour une information complète sur la prescription de BOTOX.
L’injection de BOTOX doit être réalisée par des médecins ayant les qualifications nécessaires et l’expérience dans le traitement et l’utilisation du matériel requis.
Les unités de toxine botulinique ne sont pas interchangeables d’un produit à un autre. Les doses recommandées en UNITÉS ALLERGAN sont différentes de celles recommandées pour les autres préparations à base de toxine botulinique.
BOTOX est indiqué dans le traitement symptomatique local de la spasticité (hyperactivité musculaire) des membres supérieurs et / ou inférieurs chez les patients âgés de 2 ans et plus, des troubles de l’oculomotricité, du blépharospasme, du spasme hémifacial, du torticolis spasmodique, de l’hyperhidrose axillaire sévère persistante chez les patients âgés de 12 ans et plus, de l’hyperactivité vésicale idiopathique et de l’hyperactivité détrusorienne neurologique chez les adultes.
La sécurité et l'efficacité de BOTOX dans d’autres indications que celles décrites dans la rubrique 4.1 du Résumé des Caractéristiques du Produit (RCP) pour la population pédiatrique n'ont pas été établies. Dans la population pédiatrique, aucune recommandation posologique ne peut être faite pour d’autres indications que le traitement symptomatique local de la spasticité. Les données actuellement disponibles par indication sont décrites dans les rubriques 4.2, 4.4, 4.8, 5.1 du Résumé des Caractéristiques du Produit (RCP) comme indiqué dans le tableau ci-dessous.
|
Troubles de l’oculomotricité |
12 ans (voir rubriques 4.4 et 4.8 du RCP) |
|
Blépharospasme/Spasme hémifacial |
12 ans (voir rubriques 4.4 et 4.8 du RCP) |
|
Dystonie cervicale |
12 ans (voir rubriques 4.4 et 4.8 du RCP) |
|
Traitement symptomatique local de la spasticité |
2 ans (voir rubriques 4.2, 4.4 et 4.8 du RCP) |
|
Hyperhidrose axillaire sévère |
12 ans (expérience limitée chez les adolescents entre 12 et 17 ans, voir rubriques 4.4, 4.8 et 5.1 du RCP) |
|
Hyperactivité détrusorienne neurologique chez l’enfant |
5-17 ans (voir rubriques 4.8 et 5.1) |
|
Hyperactivité vésicale chez l’enfant |
12-17 ans (voir rubriques 4.8 et 5.1) |
Aucun ajustement posologique spécifique n’est requis pour une utilisation chez les sujets âgés.
La posologie initiale doit commencer à la dose minimale recommandée pour chaque indication.
En cas d’injections répétées, la dose efficace la plus faible associée au plus long intervalle cliniquement nécessaire entre deux injections, est recommandée.
Les sujets âgés présentant des antécédents médicaux importants et recevant un traitement concomitant, doivent être traités avec prudence.
Les doses optimales pouvant être injectées ainsi que le nombre de sites d’injection par muscle n’ont pas été établis pour toutes les indications. Un schéma thérapeutique individuel devra alors être défini par le médecin. Les doses optimales doivent être déterminées par titration sans injecter au-delà de la dose maximale recommandée. Comme pour tout traitement médicamenteux, la posologie initiale chez un patient naïf doit commencer à la dose efficace la plus faible.
Posologie et mode d’administration (voir rubriques 4.2 et 4.4 du RCP)
Toutes indications :
Des effets indésirables liés à la diffusion de la toxine à distance du site d’administration ont été rapportés. Ces effets ont parfois été fatals, qui dans certains cas étaient associés à une dysphagie, une pneumonie et/ou un affaiblissement important.
Les symptômes sont conformes au mécanisme d’action de la toxine botulinique et ont été rapportés quelques heures à quelques jours après injection. Le risque de symptômes est probablement plus élevé chez les patients ayant une maladie sous-jacente et présentant des comorbidités, qui les prédisposent à ces symptômes, comme les adultes et les enfants traités à doses élevées pour une spasticité.
Les patients traités à doses thérapeutiques peuvent également ressentir une faiblesse musculaire excessive. Un pneumothorax lié à la procédure d’injection a été rapporté à la suite de l’administration de BOTOX près du thorax. Une prudence s’impose lors d’une injection à proximité du poumon, en particulier la région apicale ou toutes autres structures anatomiques vulnérables.
Des événements indésirables pouvant être fatals, ont été rapportés chez des patients qui avaient reçu des injections de BOTOX dans des indications non listées dans l’AMM, telles que directement dans les glandes salivaires, la région oro-pharyngo-linguale, l’œsophage et l’estomac. Certains patients souffraient déjà de dysphagie ou d’une fatigue générale importante.
De rares cas de décès ont été spontanément rapportés. Ils étaient parfois associés à une pneumonie d’inhalation chez les enfants atteints de paralysie cérébrale grave à la suite d’injection avec de la toxine botulinique, y compris lors d’une utilisation hors AMM comme dans la région du cou. Une extrême prudence est recommandée lors du traitement des sujets pédiatriques souffrant de faiblesse neurologique importante, de dysphagie ou qui ont des antécédents récents de pneumonie d’inhalation ou de maladie des poumons.
Une réaction anaphylactique peut très rarement se produire après injection de toxine botulinique. De l’épinéphrine (adrénaline) et d’autres mesures anti-anaphylactiques doivent par conséquent être à disposition.
Pour une information complète sur les produits BOTOX, veuillez vous référer aux Résumés des Caractéristiques du Produit respectifs.
En cas d’échec du traitement après la première séance d’injection, c’est-à-dire en l’absence, un mois après l’injection, d’amélioration clinique significative par rapport à l’état initial du patient, les mesures suivantes doivent être prises :
· Vérification clinique, qui peut comprendre un examen électromyographique dans un service spécialisé, de l’action de la toxine sur le(s) muscle(s) injecté(s) ;
· Analyse des causes de l’échec comme une mauvaise sélection des muscles à injecter, une dose insuffisante, une mauvaise technique d’injection, l’apparition d’une contracture fixe, des muscles antagonistes trop faibles, une formation d’anticorps neutralisant la toxine ;
· Réévaluation de la pertinence du traitement par la toxine botulinique de type A.
En l’absence de tout effet indésirable secondaire à la première séance d’injection, initier une seconde séance d’injection en i) ajustant la dose en tenant compte de l’analyse de l’échec de la séance d’injection précédente ; ii) réalisant un électromyogramme ; iii) maintenant un intervalle de 3 mois entre les deux séances d’injection.
En cas d’échec du traitement ou d’un effet diminué à la suite de séances répétées d’injection, un traitement alternatif devra être envisagé.
Préparation du produit
Si différentes présentations de BOTOX sont utilisées dans le cadre d’une procédure d’injection, un soin doit être apporté à l’utilisation de la bonne quantité de solvant en reconstituant le nombre d’unités par 0,1 ml déterminé. La quantité de solvant varie entre BOTOX 50 UNITÉS ALLERGAN, BOTOX 100 UNITÉS ALLERGAN et BOTOX 200 UNITÉS ALLERGAN. Chaque seringue sera étiquetée en conséquence.
La préparation du produit doit être réalisée dans un local approprié et par du personnel expérimenté afin de minimiser le risque d'accident lors de la manipulation.
Pour reconstituer BOTOX, utiliser uniquement une solution stérile sans conservateur de sérum physiologique (solution injectable de chlorure de sodium à 0,9 pour cent).
Aspirer une quantité de solvant adaptée à la dilution souhaitée dans une seringue de taille adéquate (cf. tableau de dilution ci-après).
Nettoyer à l'alcool la partie centrale du bouchon en caoutchouc. Afin d'éviter la dénaturation du produit, injecter délicatement le solvant dans le flacon et agiter doucement en évitant la formation de bulles.
Ne pas utiliser le flacon si la dépression n'entraîne pas l'aspiration du solvant à l'intérieur du flacon.
Une fois reconstituée, la solution obtenue doit être contrôlée visuellement avant utilisation, afin de vérifier qu'elle est limpide, incolore ou jaune très pâle et qu'elle ne contient pas de particules.
Après reconstitution dans son flacon, d’un point de vue microbiologique, une utilisation immédiate de la solution est recommandée. Toutefois, la solution peut être conservée au réfrigérateur et utilisée dans les 24 heures (voir rubrique 6.4 du RCP – « Précautions particulières de conservation »).
Instructions pour la reconstitution dans le traitement des dysfonctions vésicales
Lorsque BOTOX est dilué dans une seringue pour le traitement des dysfonctions vésicales, il doit être utilisé immédiatement.
Instructions pour la reconstitution de BOTOX 100 UNITÉS ALLERGAN afin d’obtenir une dose de 50 UNITÉS ALLERGAN
Il est recommandé d’utiliser préférentiellement des flacons de BOTOX 100 UNITÉS ALLERGAN afin de faciliter la reconstitution dans cette indication.
· Reconstituer 1 flacon de BOTOX 100 UNITÉS ALLERGAN, avec 8 ml de solution stérile sans conservateur de sérum physiologique (solution injectable de chlorure de sodium à 0,9 pour cent) puis mélanger le flacon doucement.
· Aspirer 4 ml du flacon dans une seringue de 10 ml.
· Finir la reconstitution en ajoutant 6 ml de solution stérile sans conservateur de sérum physiologique (solution injectable de chlorure de sodium à 0,9 pour cent) dans la seringue de 10 ml et agiter doucement.
Vous obtiendrez ainsi une seringue de 10 ml contenant un total de 50 Unités de solution reconstituée de BOTOX.
Instructions pour la reconstitution de BOTOX 50 UNITÉS ALLERGAN afin d’obtenir une dose de 100 UNITÉS ALLERGAN
· Reconstituer 2 flacons de BOTOX 50 UNITÉS ALLERGAN, chacun avec 5 ml de solution stérile sans conservateur de sérum physiologique (solution injectable de chlorure de sodium à 0,9 pour cent) puis mélanger le flacon doucement.
· Aspirer 5 ml de chaque flacon dans une seringue de 10 ml.
Vous obtiendrez ainsi une seringue de 10 ml contenant un total de 100 Unités de solution reconstituée de BOTOX.
Instructions pour la reconstitution de BOTOX 100 UNITÉS ALLERGAN afin d’obtenir une dose de 100 UNITÉS ALLERGAN
· Reconstituer 1 flacon de BOTOX 100 UNITÉS ALLERGAN avec 10 ml de solution stérile sans conservateur de sérum physiologique (solution injectable de chlorure de sodium à 0,9 pour cent) et agiter le flacon doucement.
· Aspirer 10 ml de la solution reconstituée à partir du flacon dans une seringue de 10 ml.
Vous obtiendrez ainsi une seringue de 10 ml contenant un total de 100 Unités de solution reconstituée de BOTOX.
Instructions pour la reconstitution de BOTOX 200 UNITÉS ALLERGAN afin d’obtenir une dose de 100 UNITÉS ALLERGAN
Il est recommandé d’utiliser préférentiellement des flacons de BOTOX 100 UNITÉS ALLERGAN afin de faciliter la reconstitution dans cette indication.
· Reconstituer 1 flacon de BOTOX 200 UNITÉS ALLERGAN, avec 8 ml de solution stérile sans conservateur de sérum physiologique (solution injectable de chlorure de sodium à 0,9 pour cent) puis mélanger le flacon doucement.
· Aspirer 4 ml du flacon dans une seringue de 10 ml.
· Terminer la reconstitution en ajoutant 6 ml de solution stérile sans conservateur de sérum physiologique (solution injectable de chlorure de sodium à 0,9 pour cent) dans la seringue de 10 ml et mélanger doucement.
Vous obtiendrez ainsi une seringue de 10 ml contenant un total de 100 Unités de solution reconstituée de BOTOX.
Instructions pour la reconstitution de BOTOX 50 UNITÉS ALLERGAN afin d’obtenir une dose de 200 UNITÉS ALLERGAN
Il est recommandé d’utiliser préférentiellement des flacons de BOTOX 100 UNITÉS ALLERGAN ou BOTOX 200 UNITÉS ALLERGAN afin de faciliter la reconstitution dans cette indication.
· Reconstituer 4 flacons de BOTOX 50 UNITÉS ALLERGAN, chacun avec 3 ml de solution stérile sans conservateur de sérum physiologique (solution injectable de chlorure de sodium à 0,9 pour cent) puis mélanger les flacons doucement.
· Aspirer 3 ml du premier flacon et 1 ml du second flacon avec une seringue de 10 ml.
· Aspirer 3 ml du troisième flacon et 1 ml du quatrième flacon avec une seringue de 10 ml.
· Aspirer les 2 ml restant du second et du quatrième flacon avec une troisième seringue de 10 ml.
· Terminer la reconstitution en ajoutant 6 ml de solution stérile sans conservateur de sérum physiologique (solution injectable de chlorure de sodium à 0,9 pour cent) dans chacune des seringues de 10 ml puis mélanger doucement.
Vous obtiendrez ainsi trois seringues de 10 ml contenant un total de 200 Unités de solution reconstituée de BOTOX.
Instructions pour la reconstitution de BOTOX 100 UNITÉS ALLERGAN afin d’obtenir une dose de 200 UNITÉS ALLERGAN
· Reconstituer 2 flacons de BOTOX 100 UNITÉS ALLERGAN, chacun avec 6 ml de solution stérile sans conservateur de sérum physiologique (solution injectable de chlorure de sodium à 0,9 pour cent) puis mélanger les flacons doucement.
· Aspirer 4 ml de chaque flacon avec 2 seringues de 10 ml.
· Aspirer les 2 ml restant dans chaque flacon avec une troisième seringue de 10 ml.
· Terminer la reconstitution en ajoutant 6 ml de solution stérile sans conservateur de sérum physiologique (solution injectable de chlorure de sodium à 0,9 pour cent) dans chacune des seringues de 10 ml puis mélanger doucement.
Vous obtiendrez ainsi trois seringues de 10 ml contenant un total de 200 Unités de solution reconstituée de BOTOX.
Instructions pour la reconstitution de BOTOX 200 UNITÉS ALLERGAN afin d’obtenir une dose de 200 UNITÉS ALLERGAN
· Reconstituer 1 flacon de BOTOX 200 UNITÉS ALLERGAN avec 6 ml de solution stérile sans conservateur de sérum physiologique (solution injectable de chlorure de sodium à 0,9 pour cent) puis mélanger le flacon doucement.
· Aspirer 2 ml du flacon dans chacune des 3 seringues de 10 ml.
· Terminer la reconstitution en ajoutant 8 ml de solution stérile sans conservateur de sérum physiologique (solution injectable de chlorure de sodium à 0,9 pour cent) dans chacune des seringues de 10 ml puis mélanger doucement.
Vous obtiendrez ainsi trois seringues de 10 ml contenant un total de 200 Unités Allergan de solution reconstituée de BOTOX.
Tableau de dilution – Autres indications
|
|
Flacon de 50 UNITÉS ALLERGAN |
Flacon de 100 UNITÉS ALLERGAN |
Flacon de 200 UNITÉS ALLERGAN |
|
Concentration en UNITÉS ALLERGAN / 0,1 ml |
Solvant ajouté (solution stérile sans conservateur de sérum physiologique (solution injectable de chlorure de sodium à 0,9 pour cent)) |
Solvant ajouté (solution stérile sans conservateur de sérum physiologique (solution injectable de chlorure de sodium à 0,9 pour cent)) |
Solvant ajouté (solution stérile sans conservateur de sérum physiologique (solution injectable de chlorure de sodium à 0,9 pour cent)) |
|
20 |
0,25 ml |
0,5 ml |
1 ml |
|
10 |
0,5 ml |
1 ml |
2 ml |
|
5 |
1 ml |
2 ml |
4 ml |
|
2,5 |
2 ml |
4 ml |
8 ml |
|
1,25 |
4 ml |
8 ml |
N/A |
Recommandations en cas d'accident lors de la manipulation de la toxine botulinique
En cas d'accident lors d'une manipulation du produit qu'il soit à l'état de poudre ou reconstitué, les mesures appropriées décrites ci-dessous doivent être mises en route immédiatement.
La toxine botulinique est très sensible à la chaleur et à certains agents chimiques.
Toute projection doit être essuyée :
· soit avec un matériel absorbant imbibé d'une solution d'hypochlorite de sodium (eau de Javel) en cas de produit sec ;
· soit avec un matériel absorbant sec en cas de produit reconstitué.
Les surfaces contaminées seront nettoyées avec un matériel absorbant imbibé d'une solution d'hypochlorite de sodium (eau de Javel), puis séchées.
En cas de bris de flacon, procéder comme indiqué ci-dessus au ramassage méticuleux des particules de verre et à l'essuyage du produit, en évitant les coupures cutanées.
En cas de projection, laver avec une solution d'hypochlorite de sodium (eau de Javel), puis rincer abondamment à l'eau.
En cas de projection oculaire, rincer abondamment avec de l'eau ou avec une solution ophtalmique de rinçage oculaire.
En cas de blessure du manipulateur (coupure, autopiqûre), procéder comme ci-dessus et prendre les mesures médicales appropriées en fonction de la dose injectée.
Recommandations pour l'élimination du matériel utilisé
Les aiguilles, les seringues et les flacons, qui ne doivent pas être vidés, seront placés, après usage, dans des récipients adaptés qui devront être incinérés.
Le matériel contaminé (tissu absorbant, gants, débris d'ampoule) doit être placé dans un sac intraversable et éliminé par incinération.
Identification du produit
Afin de vérifier que le produit que vous avez reçu sous le nom de BOTOX est bien un authentique produit AbbVie, recherchez la présence de perforations intactes (au niveau de la pré-découpe d'ouverture) sur les boîtes BOTOX correspondant aux signes d’inviolabilité et un pelliculage holographique sur l’étiquette du flacon. Afin de visualiser ce pelliculage, examinez le flacon sous une lampe de bureau ou une source de lumière fluorescente. Faites tourner le flacon d’avant en arrière entre vos doigts pour visualiser des lignes horizontales multicolores et vérifier la présence de l’inscription « abbvie » entre ces lignes horizontales multicolores.
N’utilisez pas le produit et contactez AbbVie pour plus d’informations si les perforations sont endommagées, si la boîte semble avoir été ouverte ou si les lignes horizontales multicolores ou l’inscription « abbvie » ne sont pas présentes sur l’étiquette du flacon.
De plus, des étiquettes détachables sont incluses sur l’étiquette des flacons de BOTOX, indiquant le numéro du lot et la date d’expiration du produit. Ces étiquettes peuvent être décollées et placées dans le dossier de votre patient pour assurer la traçabilité du produit. L’inscription « UTILISÉ » apparaîtra une fois l’étiquette de traçabilité décollée de l’étiquette du flacon, ce qui confirmera que vous utilisez un authentique flacon de BOTOX.