Dernière mise à jour le 03/08/2026
GADOVISTMANUEL 1,0 mmol/mL, solution injectable en seringue préremplie
Ce médicament n'est ou ne sera bientôt plus disponible sur le marché.
Si vous prenez actuellement ce médicament, il vous est recommandé d'en parler avec votre médecin
ou avec votre pharmacien qui pourra vous orienter vers un autre traitement.
Indications thérapeutiques
Classe pharmacothérapeutique : Produit de contraste paramagnétique - code ATC : V08C A09
GADOVIST peut également être utilisé pour l’IRM des anomalies d’autres régions du corps.
Il facilite la visualisation des structures ou des lésions anormales et aide à différencier les tissus sains des tissus malades.
Il est indiqué chez les adultes et les enfants de tout âge (y compris les nouveau-nés à terme).
Comment agit GADOVIST 1,0 mmol/mL, solution injectable en seringue préremplie
L’IRM est une technique d’imagerie médicale à visée diagnostique basée sur les différences de réaction des molécules d’eau entre les tissus normaux et les tissus anormaux. Cet examen est réalisé avec un système complexe utilisant un champ magnétique et des ondes radio. Un ordinateur enregistre l’activité et la traduit sous forme d’images.
GADOVIST est administré par injection dans une veine. Ce médicament est à usage diagnostique uniquement et ne vous sera administré que par des professionnels de santé ayant l’expérience de la pratique clinique de l’IRM.
Présentations
> 1 seringue(s) préremplie(s) en verre de 7,5 ml avec dispositif d'administration pour injection manuelle
Code CIP : 34009 301 817 3 7
Déclaration d'arrêt de commercialisation : 19/12/2025
Cette présentation n'est pas agréée aux collectivités
> 1 seringue(s) préremplie(s) en verre de 10 ml avec dispositif d'administration pour injection manuelle
Code CIP : 34009 301 817 4 4
Déclaration d'arrêt de commercialisation : 15/05/2025
Cette présentation n'est pas agréée aux collectivités
> 1 seringue(s) préremplie(s) en verre de 15 ml avec dispositif d'administration pour injection manuelle
Code CIP : 34009 301 817 5 1
Déclaration d'arrêt de commercialisation : 24/01/2025
Cette présentation n'est pas agréée aux collectivités
Service médical rendu (SMR)
Les libellés affichés ci-dessous ne sont que des résumés ou extraits issus des avis rendus par la Commission de la Transparence. Seul l'avis complet de la Commission de la Transparence fait référence.
Cet avis est consultable à partir du lien `Avis du jj/mm/aaaa` ou encore sur demande auprès de la HAS (plus d'informations dans l'aide). Les avis et synthèses d'avis contiennent un paragraphe sur la place du médicament dans la stratégie thérapeutique.
| Valeur du SMR | Avis | Motif de l'évaluation | Résumé de l'avis |
|---|---|---|---|
| Important | Avis du 10/07/2019 | Inscription (CT) | Le service médical rendu par GADOVISTMANUEL est important dans les indications de l’AMM. |
Amélioration du service médical rendu (ASMR)
Les libellés affichés ci-dessous ne sont que des résumés ou extraits issus des avis rendus par la Commission de la Transparence. Seul l'avis complet de la Commission de la Transparence fait référence.
Cet avis est consultable à partir du lien `Avis du jj/mm/aaaa` ou encore sur demande auprés de la HAS (plus d'informations dans l'aide). Les avis et synthéses d'avis contiennent un paragraphe sur la place du médicament dans la stratégie thérapeutique.
| Valeur de l'ASMR | Avis | Motif de l'évaluation | Résumé de l'avis |
|---|---|---|---|
| V (Inexistant) | Avis du 10/07/2019 | Inscription (CT) | Ces présentations sont des compléments de gamme qui n’apportent pas d’amélioration du service médical rendu (ASMR V) par rapport aux présentations déjà inscrites. |
ANSM - Mis à jour le : 11/07/2025
GADOVISTMANUEL* 1,0 mmol/mL, solution injectable en seringue préremplie
* GADOVISTMANUEL est identique au médicament GADOVIST et ne diffère que par les dispositifs d’administration ajoutés dans l’emballage. Etant donné que toute l’expérience clinique a été acquise avec GADOVIST, cette dénomination est utilisée dans tout le texte.
2. COMPOSITION QUALITATIVE ET QUANTITATIVE
1 mL de solution injectable contient 604,72 mg de gadobutrol (correspondant à 1,0 mmol de gadobutrol contenant 157,25 mg de gadolinium).
1 seringue préremplie de 7,5 mL de solution contient 4535,4 mg de gadobutrol.
1 seringue préremplie de 10 mL de solution contient 6047,2 mg de gadobutrol.
1 seringue préremplie de 15 mL de solution contient 9070,8 mg de gadobutrol.
Excipient à effet notoire :
1 mL de solution injectable contient 0,00056 mmol (correspondant à 0,013 mg) de sodium (voir rubrique 4.4).
Pour la liste complète des excipients, voir rubrique 6.1.
Solution injectable en seringue préremplie.
Solution limpide, incolore à jaune pâle.
Propriétés physico-chimiques :
Osmolalité à 37 °C : 1603 mOsm/kg H2O
Viscosité à 37 °C : 4,96 mPa.s
4.1. Indications thérapeutiques
Ce médicament est à usage diagnostique uniquement.
GADOVIST est indiqué chez l’adulte et chez l’enfant de tout âge (y compris le nouveau-né à terme), pour le :
· Rehaussement du contraste en Imagerie par Résonance Magnétique (IRM) des territoires crâniens et rachidiens.
· Rehaussement du contraste en IRM du foie ou des reins chez les patients avec une forte suspicion ou une présence évidente de lésions focalisées, afin de classer ces lésions comme bénignes ou malignes.
· Rehaussement du contraste en Angiographie par Résonance Magnétique (ARM).
GADOVIST peut également être utilisé pour l’Imagerie par Résonance Magnétique des pathologies du corps entier.
Il facilite la visualisation des structures ou des lésions anormales et aide à la différenciation entre les tissus sains et pathologiques.
GADOVIST ne doit être utilisé que lorsque le diagnostic est nécessaire et que ce diagnostic ne peut pas être obtenu par imagerie par résonance magnétique (IRM) sans rehaussement de contraste.
4.2. Posologie et mode d'administration
Mode d’administration
Ce médicament est administré exclusivement par voie intraveineuse.
La dose nécessaire est administrée en bolus par voie intraveineuse. L’examen IRM avec rehaussement du contraste peut débuter immédiatement après l’injection (dans un délai dépendant des séquences d’IRM utilisées et du protocole d’examen).
Le rehaussement optimal du signal est observé pendant le premier passage artériel en ARM et dans les 15 minutes suivant l’injection de GADOVIST pour les indications du SNC (ce délai dépendant du type de lésion ou de tissu).
Les séquences pondérées en T1 sont particulièrement adaptées aux examens avec injection d’un produit de contraste gadoliné.
Le patient doit être, si possible, allongé lors de l’injection intravasculaire du produit de contraste et doit être surveillé pendant au moins une demi-heure après celle-ci, la majorité des effets indésirables survenant au cours de cette période (voir rubrique 4.4).
Instructions d’utilisation :
Ce produit n'est destiné qu'à un usage unique.
La solution doit être contrôlée visuellement juste avant utilisation.
GADOVIST ne doit pas être utilisé en cas de changement important de coloration, de présence de particules ou d’emballage défectueux.
Seringues préremplies
La seringue préremplie doit être sortie de son emballage et préparée pour l’injection juste avant son administration.
L’opercule de protection doit être retiré de la seringue préremplie juste avant utilisation.
Dispositifs médicaux pour injection intraveineuse
Pour plus d’informations sur l’assemblage et le bon usage des dispositifs médicaux fournis dans l’emballage secondaire, veuillez-vous reporter aux pictogrammes figurant sur la notice et suivre les instructions d’utilisation de chacun des dispositifs médicaux.
Posologie
La dose la plus faible permettant un rehaussement de contraste suffisant à des fins diagnostiques doit être utilisée. La dose doit être calculée en fonction de la masse corporelle du patient et ne doit pas dépasser la dose recommandée par kilogramme de masse corporelle, détaillée dans cette rubrique.
Adultes
Indications dans le SNC :
La dose recommandée chez l’adulte est de 0,1 mmol/kg de masse corporelle, ce qui équivaut à 0,1 mL/kg de la solution à 1,0 M.
En cas de forte suspicion clinique d’une lésion non confirmée à l’IRM ou si des informations plus précises peuvent modifier la prise en charge thérapeutique du patient, une seconde injection pouvant aller jusqu’à 0,2 mL/kg au maximum peut être effectuée dans les 30 minutes suivant la première injection.
Une dose de 0,075 mmol de gadobutrol par kg de masse corporelle (équivalent à 0,075 mL de Gadovist par kg de masse corporelle) peut être administrée au minimum pour l’imagerie du SNC (voir rubrique 5.1).
IRM du corps entier (à l’exception de l’Angiographie par Résonance Magnétique) :
De manière générale, l’administration de 0,1 mL de GADOVIST par kg de masse corporelle est suffisante pour apporter une réponse à la question clinique.
Angiographie par Résonance Magnétique :
Image d'un seul champ d'acquisition : 7,5 mL pour un patient de moins de 75 kg ; 10 mL pour un patient de 75 kg et plus (équivalent à 0,1-0,15 mmol/kg).
Image de plusieurs champs d’acquisition : 15 mL pour un patient de moins de 75 kg ; 20 mL pour un patient de 75 kg ou plus (équivalent à 0,2-0,3 mmol/kg).
Populations particulières
Insuffisants rénaux
GADOVIST ne doit être administré aux patients présentant une insuffisance rénale sévère (DFG < 30 mL/min/1,73 m²) et aux patients en période périopératoire de transplantation hépatique qu’après une évaluation approfondie du rapport bénéfice/risque et que si les informations diagnostiques sont indispensables et ne peuvent être obtenues au moyen d'une IRM sans rehaussement du contraste (voir rubrique 4.4).
S’il est nécessaire d’administrer GADOVIST, la dose ne doit pas excéder 0,1 mmol/kg de masse corporelle. Ne pas administrer plus d’une dose au cours de l’examen IRM.
En raison du manque d’information sur les administrations répétées, les injections de GADOVIST ne doivent pas être réitérées sauf si l’intervalle entre les injections est d’au moins 7 jours.
Population pédiatrique
Pour les enfants de tout âge (y compris les nouveau-nés à terme), la dose recommandée est de 0,1 mmol de gadobutrol par kg de masse corporelle (équivalent à 0,1 mL de GADOVIST par kg de masse corporelle) pour toutes les indications (voir rubrique 4.1).
Nouveau-nés jusqu’à l’âge de 4 semaines et nourrissons jusqu’à l’âge d’un an
En raison de l’immaturité de la fonction rénale chez le nouveau-né jusqu’à l’âge de 4 semaines et chez le nourrisson jusqu’à l’âge d’un an, GADOVIST ne doit être utilisé chez ces patients qu’après une évaluation attentive et à une dose n’excédant pas 0,1 mmol/kg de masse corporelle. Ne pas administrer plus d’une dose au cours de l'examen. En raison du manque d’information sur les administrations répétées, les injections de GADOVIST ne doivent pas être réitérées sauf si l’intervalle entre les injections est d’au moins 7 jours.
Sujets âgés (à partir de 65 ans)
Aucune adaptation posologique n’est nécessaire. Utiliser avec prudence chez les sujets âgés (voir rubrique 4.4).
Hypersensibilité à la substance active ou à l’un des excipients mentionnés à la rubrique 6.1.
4.4. Mises en garde spéciales et précautions d'emploi
Le gadobutrol ne doit pas être administré par voie intrathécale. Des réactions graves, pouvant engager le pronostic vital et ayant entraîné la mort dans certains cas, principalement des réactions neurologiques (coma, encéphalopathie, convulsions, par exemple), ont été signalées en cas d’administration intrathécale.
L’injection de GADOVIST dans des veines de petit calibre peut provoquer des effets indésirables tels que rougeur et gonflement.
Les mesures de sécurité habituelles en IRM, en particulier l’éviction de tout matériel ferromagnétique s’appliquent à l’emploi de GADOVIST.
Réactions d’hypersensibilité ou autres réactions idiosyncrasiques
Comme pour les autres agents de contraste administrés par voie intraveineuse, l’utilisation de GADOVIST peut s’accompagner de réactions anaphylactoïdes/d’hypersensibilité ou d’autres réactions idiosyncrasiques (par exemple, syndrome de détresse respiratoire aiguë / œdème pulmonaire avec ou sans contexte de réactions d'hypersensibilité), se traduisant par des manifestations cardiovasculaires, respiratoires ou cutanées, potentiellement sévères, allant même jusqu’au choc.
En général, les patients souffrant de maladies cardio-vasculaires sont plus susceptibles que les autres de présenter une évolution grave ou fatale lors d’une réaction d’hypersensibilité sévère.
Le risque de développer des réactions d’hypersensibilité peut être augmenté en cas :
· de précédente réaction aux produits de contraste,
· d’antécédents d’asthme bronchique,
· d’antécédents de troubles allergiques.
Chez les patients présentant un terrain allergique, la décision d’utiliser GADOVIST ne doit être prise qu’après une évaluation particulièrement attentive du rapport bénéfice/risque.
La plupart de ces réactions surviennent dans la demi-heure suivant l’administration. Aussi, il est recommandé de garder le patient en observation après examen.
Il est impératif d’avoir à disposition le traitement médicamenteux nécessaire à la prise en charge de réactions d’hypersensibilité ou d’autres réactions idiosyncrasiques ; il faut également être prêt pour la mise en place immédiate de mesures d’urgence (voir rubrique 4.2).
Des réactions retardées (de quelques heures à plusieurs jours) ont été rarement observées (voir rubrique 4.8).
Insuffisance rénale
Avant l’administration de GADOVIST, il est recommandé de procéder à des examens de laboratoire chez tous les patients afin de rechercher une altération de la fonction rénale.
Des cas de fibrose systémique néphrogénique (FSN) ont été rapportés après injection de certains produits de contraste contenant du gadolinium chez des patients ayant une insuffisance rénale sévère aiguë ou chronique (clairance de la créatinine < 30 mL/min/1,73m²). Les patients devant bénéficier d’une transplantation hépatique sont particulièrement à risque, car l’incidence de l’insuffisance rénale aiguë est élevée dans ce groupe.
Etant donné qu’il est possible que des cas de FSN surviennent avec GADOVIST, ce produit ne doit être administré aux patients présentant une insuffisance rénale sévère ou durant la période pré ou post-opératoire d’une transplantation hépatique qu’après une évaluation approfondie du rapport bénéfice/risque et que si le diagnostic est indispensable et ne peut être obtenu par d’autres moyens que l’IRM avec injection de gadolinium.
La réalisation d’une hémodialyse peu de temps après l’administration de GADOVIST pourrait faciliter l’élimination de ce produit de l’organisme. Il n’est pas établi que l’instauration d’une hémodialyse puisse prévenir ou traiter la FSN chez les patients qui ne sont pas déjà hémodialysés.
Nouveau-nés et nourrissons
En raison de l’immaturité de la fonction rénale des nouveau-nés jusqu’à l’âge de 4 semaines et des nourrissons jusqu’à l’âge d’un an, GADOVIST ne doit être administré à ces patients qu’après un examen approfondi de la situation.
Sujets âgés
L’élimination rénale de gadobutrol pouvant être altérée chez les sujets âgés, il est particulièrement important de rechercher un dysfonctionnement rénal chez les sujets âgés de 65 ans et plus.
Troubles épileptiques
Comme avec les autres produits de contraste contenant du gadolinium, des précautions particulières s'imposent en cas de seuil épileptogène bas.
Excipients
Ce médicament contient moins de 1 mmol de sodium (23 mg) par dose (calculé pour une dose moyenne administrée à une personne de 70 kg) c’est-à-dire qu’il est essentiellement « sans sodium ».
4.5. Interactions avec d'autres médicaments et autres formes d'interactions
Aucune étude d’interaction n’a été réalisée.
4.6. Fertilité, grossesse et allaitement
Grossesse
Les données sur l’utilisation de produits de contraste à base de gadolinium y compris du gadobutrol chez la femme enceinte sont limitées. Le gadolinium peut traverser la barrière placentaire. On ignore si l’exposition au gadolinium est associée à des effets indésirables chez le fœtus. Les études effectuées chez l’animal ont mis en évidence une toxicité sur la reproduction à des doses répétées élevées (voir rubrique 5.3).
GADOVIST ne doit pas être utilisé pendant la grossesse à moins que la situation clinique de la patiente ne nécessite l’administration de gadobutrol.
Les produits de contraste contenant du gadolinium sont excrétés dans le lait maternel en très petites quantités (voir rubrique 5.3).
Aux doses cliniques, aucun effet n’est attendu chez le nourrisson allaité en raison de la faible quantité excrétée dans le lait et de la faible absorption intestinale. Le médecin et la mère allaitante doivent décider s’il faut poursuivre l’allaitement ou le suspendre pendant les 24 heures suivant l’administration de GADOVIST.
Fertilité
Les études réalisées chez l’animal n’indiquent pas d’effet sur la fertilité.
4.7. Effets sur l'aptitude à conduire des véhicules et à utiliser des machines
Le profil général de tolérance de GADOVIST est basé sur des données issues des essais cliniques sur plus de 6300 patients, et rapportées en période post-commercialisation.
Les effets indésirables les plus fréquemment observés (³ 0,5%) chez les patients recevant GADOVIST sont : céphalées, nausées et sensations vertigineuses.
Les effets indésirables les plus graves observés chez les patients recevant GADOVIST sont : arrêt cardiaque, syndrome de détresse respiratoire aiguë / œdème pulmonaire et réactions anaphylactoïdes sévères (incluant arrêt respiratoire et choc anaphylactique).
Des réactions anaphylactoïdes retardées ou autres réactions idiosyncrasiques (de quelques heures à quelques jours après l’administration) ont été rarement observées (voir rubrique 4.4).
La plupart des effets indésirables ont été d’intensité légère à modérée.
Les effets indésirables observés sous GADOVIST sont représentés dans le tableau ci-dessous. Ils sont classés selon la classification par système-organe (MedDRA). La terminologie MedDRA la plus appropriée est utilisée pour décrire une certaine réaction et ses synonymes ainsi que les conditions qui y sont associées.
Les effets indésirables du médicament issus des données des essais cliniques sont classés en fonction de leur fréquence.
Les groupes de fréquence sont définis selon la convention suivante : fréquent : ³ 1/100 à < 1/10 ; peu fréquent : ³ 1/1000 à < 1/100 ; rare : ³ 1/10 000 à < 1/1000. Les effets indésirables du médicament identifiés uniquement en période post-commercialisation, pour lesquels la fréquence n’a pas pu être évaluée, sont listés sous la fréquence « indéterminée ».
Au sein de chaque groupe de fréquence, les effets indésirables sont présentés suivant un ordre décroissant de gravité.
Tableau 1 : Effets indésirables issus des données des essais cliniques ou rapportés en période post-commercialisation chez les patients traités par GADOVIST
|
|
Fréquence |
|||
|
Classe de systèmes d’organes |
Fréquent |
Peu fréquent |
Rare |
Indéterminée |
|
Affections du système immunitaire |
|
Hypersensibilité / réaction anaphylactoïde*# (p.ex. choc anaphylactoïde§*, collapsus circulatoire§*, arrêt respiratoire§*, bronchospasme§, cyanose§, gonflement oropharyngé§*, œdème laryngé§, hypotension*, augmentation de la pression artérielle§, douleur dans la poitrine§, urticaire, œdème facial, angio-œdème§, conjonctivite§, œdème palpébral, bouffées de chaleur, hyperhidrose§, toux§, éternuements§, sensation de brûlure§, pâleur§) |
|
|
|
Affections du système nerveux |
Céphalées |
Sensations vertigineuses, Dysgueusie, Paresthésie |
Perte de conscience*, Convulsions, Parosmie |
|
|
Affections cardiaques |
|
|
Tachycardie, Palpitations |
Arrêt cardiaque* |
|
Affections respiratoires, thoraciques et médiastinales |
|
Dyspnée* |
|
Syndrome de détresse respiratoire aiguë (SDRA)*1 Œdème pulmonaire*1 |
|
Affections gastro-intestinales |
Nausées |
Vomissements |
Bouche sèche |
|
|
Affections de la peau et du tissu sous-cutané |
|
Erythème, prurit (dont prurit généralisé), éruptions cutanées (dont éruption généralisée, maculaire, papuleuse, rash pruritique) |
|
Fibrose Systémique Néphrogénique (FSN) |
|
Troubles généraux et anomalies au site d’administration |
|
Réaction au point d’injection0, Sensation de chaleur |
Malaise, Sensation de froid |
|
¹ Ces effets indésirables ont été rapportés avec et sans contexte de réactions d'hypersensibilité.
* Des cas ayant mis en jeu le pronostic vital et/ou mortels ont été rapportés pour cet effet indésirable.
# Aucun des effets indésirables listés individuellement sous l’item « Hypersensibilité/réactions anaphylactoïdes » n’a été identifié, lors des études cliniques, à une fréquence de survenue supérieure à « rare » (excepté l’urticaire)
§ Hypersensibilité / réactions anaphylactoïdes identifiées uniquement en période post-commercialisation (fréquence indéterminée)
0 Réactions au point d’injection (différents types) incluant les manifestations suivantes : extravasation au point d’injection, brûlure au point d’injection, sensation de froid au point d’injection, sensation de chaleur au point d’injection, érythème ou éruption cutanée au point d’injection, douleur au point d’injection, hématome au point d’injection.
Les réactions d’hypersensibilité sont plus fréquentes chez les patients présentant un terrain allergique.
Des cas isolés de fibrose systémique néphrogénique (FSN) ont été rapportés avec GADOVIST (voir rubrique 4.4).
Des fluctuations des paramètres de la fonction rénale, y compris une augmentation de la créatinine sérique ont été observées après l'administration de GADOVIST.
Population pédiatrique
Sur la base de deux études de Phase I/III, à dose unique, chez 138 sujets âgés de 2 à 17 ans et chez 44 sujets âgés de 0 à moins de 2 ans (voir rubrique 5.1), il a été démontré que la fréquence, le type et la gravité des effets indésirables chez l’enfant de tout âge (y compris le nouveau-né à terme) concordent avec le profil connu d’effets indésirables chez l’adulte. Ceci a été confirmé lors d’une étude de phase IV portant sur plus de 1100 patients pédiatriques et par l’expérience en période post‑commercialisation.
Déclaration des effets indésirables suspectés
La déclaration des effets indésirables suspectés après autorisation du médicament est importante. Elle permet une surveillance continue du rapport bénéfice/risque du médicament. Les professionnels de santé déclarent tout effet indésirable suspecté via le système national de déclaration : Agence nationale de sécurité du médicament et des produits de santé (ANSM) et réseau des Centres Régionaux de Pharmacovigilance - Site internet : https://signalement.social-sante.gouv.fr/
La dose journalière maximale testée chez l’homme est d’1,5 mmol de gadobutrol par kg de masse corporelle.
Aucun signe d’intoxication par surdosage n’a été rapporté à ce jour en clinique.
En cas de surdosage accidentel, une surveillance cardiovasculaire (incluant un ECG) et un contrôle de la fonction rénale sont recommandés à titre de précaution.
En cas de surdosage chez les patients présentant une insuffisance rénale, GADOVIST peut être éliminé par hémodialyse. Après 3 séances d’hémodialyse, environ 98 % de l’agent de contraste est éliminé du corps. Toutefois, il n’est pas démontré que l’hémodialyse soit appropriée dans la prévention de la fibrose systémique néphrogénique (FSN).
5. PROPRIETES PHARMACOLOGIQUES
5.1. Propriétés pharmacodynamiques
Classe pharmacothérapeutique : Produit de contraste paramagnétique, code ATC : V08C A09.
Mécanisme d’action
L’effet de rehaussement du contraste est dû au gadobutrol, un complexe non ionique associant du gadolinium (III) et un ligand macrocyclique, l’acide dihydroxy-hydroxyméthylpropyl-tétraazacyclododécane-triacétique (butrol).
Effets pharmacodynamiques
La relaxivité du gadobutrol, mesurée in vitro dans du sang / plasma humain dans des conditions physiologiques et à des intensités de champ cliniquement pertinentes (1,5 et 3,0 T), est comprise entre 3,47 et 4,97 L/mmol/s.
Aux doses cliniques, la relaxivité prononcée du gadobutrol entraîne un raccourcissement des temps de relaxation des protons dans l'eau tissulaire.
La stabilité du complexe de gadobutrol a été étudiée in vitro dans des conditions physiologiques (dans du sérum humain natif, à pH 7,4 et 37°C) sur une période de 15 jours.
Les quantités d'ions gadolinium libérés par le gadobutrol étaient inférieures à la limite de quantification de 0,1 mol % du gadolinium total, ce qui démontre la stabilité élevée du complexe de gadobutrol dans les conditions testées.
Efficacité clinique
Dans une étude pivot de phase III sur le foie, la sensibilité moyenne combinée pré et post contraste en IRM, pour les patients traités par GADOVIST, a été de 79 % et la spécificité a été de 81 % pour la détection des lésions et la classification des lésions hépatiques malignes suspectes (analyse basée sur les patients).
Dans une étude pivot de phase III sur le rein, la sensibilité moyenne a été de 91 % (analyse par patients) et de 85 % (analyse par lésions) pour la classification des lésions rénales malignes ou bénignes. La spécificité moyenne a été de 52 % dans l’analyse basée sur les patients et de 82 % dans l’analyse basée sur les lésions.
L’augmentation de la sensibilité de l’IRM sans contraste à l’IRM combinée pré et post contraste, pour les patients traités par GADOVIST, a été de 33 % dans l’étude sur le foie (analyse basée sur les patients) et 18 % dans l’étude sur le rein (analyse basée sur les patients et analyse basée sur les lésions). L’augmentation de la spécificité de l’IRM sans contraste à l’IRM combinée pré et post contraste, a été de 9 % dans l’étude sur le foie (analyse basée sur les patients) alors qu’il n’y a pas eu d’augmentation de la spécificité dans l’étude sur le rein (analyse par patients et analyse par lésions).
L’ensemble des résultats sont des résultats moyens obtenus par lecture en aveugle lors d’études cliniques.
Lors d’une étude conçue sous la forme d’une comparaison intra-individuelle croisée, et menée chez 132 patients, GADOVIST a été comparé au gadotérate de méglumine (tous deux administrés à 0,1 mmol/kg) dans le rehaussement du contraste de tumeurs cérébrales.
Le critère principal d’évaluation était la préférence générale (valeur médiane des lectures en aveugle) pour les images réalisées soit avec GADOVIST, soit avec le gadotérate de méglumine. Pour ce critère, la supériorité de GADOVIST a été démontrée (p = 0,0004). Plus particulièrement, la préférence a été donnée à GADOVIST chez 42 patients (32 %), alors qu’elle a été généralement donnée pour le gadotérate de méglumine chez 16 patients (12 %). Chez 74 patients (56 %), aucune préférence n’a été donnée entre l’un ou l’autre des produits de contraste.
En ce qui concerne les critères secondaires d’évaluation, le rapport lésion-cerveau a été statistiquement significatif en faveur de GADOVIST (p < 0,0003). Le taux de rehaussement des lésions a été plus important avec le GADOVIST qu’avec le gadotérate de méglumine, avec une différence statistiquement significative chez le lecteur en aveugle (p < 0,0003).
Le rapport contraste/bruit a montré une valeur moyenne supérieure pour GADOVIST (129) comparé au gadotérate de méglumine (98), avec une différence statistiquement non significative.
Lors d’une étude conçue sous la forme d’une comparaison croisée intra-individuelle, le gadobutrol à une dose réduite de 0,075 mmol/kg a été comparé au gadotérate de méglumine à sa dose standard de 0,1 mmol/kg dans le rehaussement de contraste en IRM du SNC chez 141 patients ayant déjà présenté un rehaussement de lésions du SNC à l’IRM avec le gadotérate de méglumine. Les variables principales étaient l’amélioration du contraste des lésions, la morphologie des lésions et la délimitation des bordures des lésions. Les images ont été analysées par trois lecteurs indépendants en aveugle. La non-infériorité au gadotérate de méglumine du degré d’amélioration de rehaussement par rapport à l’imagerie non rehaussée a été démontrée pour les trois variables principales (au moins 80% de l’effet préservé) basé sur la moyenne des lecteurs. Le nombre moyen de lésions détectées par le gadobutrol (2,14) et le gadotérate (2,06) était similaire.
Population pédiatrique
Deux études de phase I/III à dose unique ont été menées, l’une chez 138 sujets pédiatriques pour lesquels une IRM du système nerveux central, du foie et des reins ou une ARM était prévue et l’autre chez 44 sujets âgés de 0 à moins de 2 ans (y compris des nouveau-nés à terme) pour lesquels une IRM de routine de n’importe quelle région du corps était prévue. L’efficacité du diagnostic et l’amélioration de la confiance du diagnostic ont été démontrées pour tous les paramètres évalués dans ces études et il n’y avait pas de différence entre les groupes d’âge pédiatrique, ni par rapport aux adultes. GADOVIST a été bien toléré dans ces études avec un profil de sécurité du gadobutrol identique à celui chez l’adulte.
Tolérance clinique
Le type et la fréquence des effets indésirables survenus suite à l'administration de GADOVIST dans diverses indications ont été évalués dans le cadre d'un vaste essai international prospectif non interventionnel (GARDIAN). La population de l’étude comprenait 23 708 patients de tous les groupes d'âge, y compris les enfants (n = 1142; 4,8%) et les personnes âgées (n = 4330; 18,3% entre 65 et <80 ans et n = 526; 2,2% ≥ 80 ans). L'âge médian était de 51,9 ans.
Deux cent deux patients (0,9%) ont rapporté au total 251 événements indésirables (EIs) et 170 (0,7%) ont rapporté 215 événements classés comme effets indésirables médicamenteux (EIM), dont la majorité (97,7%) étaient d'intensité légère ou modérée.
Les effets indésirables médicamenteux les plus fréquemment documentés étaient les nausées (0,3%), les vomissements (0,1%) et les sensations vertigineuses (0,1%). La fréquence de survenue des EIM étaient de 0,9% chez les femmes et de 0,6% chez les hommes.
Il n'y a pas eu de différence de fréquence dans la survenue des EIM en fonction des doses de gadobutrol administrées. Quatre des 170 patients ayant présenté des effets indésirables médicamenteux (0,02%) ont présenté un EIM grave, dont un EIM (choc anaphylactique) ayant entraîné le décès.
Dans la population pédiatrique, des évènements indésirables ont été signalés chez 8 des 1 142 enfants (0,7%). Chez six enfants, ces évènements indésirables ont été classés comme effets indésirables médicamenteux (0,5%).
Insuffisance rénale
Dans une étude pharmaco-épidémiologique prospective (GRIP) visant à évaluer l'ampleur du risque potentiel de développement d’une fibrose systémique néphrogénique (FSN) chez les patients insuffisants rénaux, 908 patients présentant divers stades d'insuffisance rénale ont reçu GADOVIST à la dose standard approuvée pour l'IRM.
Tous les patients, dont 234 atteints d'insuffisance rénale sévère (DFG estimé <30 mL/min/1,73 m²) qui n'avaient pas reçu d'autres produits de contraste gadolinés ont été suivis pendant deux ans pour détecter les signes et symptômes de NSF. Aucun patient participant à l'étude n'a développé de FSN.
5.2. Propriétés pharmacocinétiques
Le gadobutrol injecté par voie intraveineuse se distribue rapidement dans le compartiment extracellulaire. Son taux de liaison aux protéines plasmatiques est négligeable. Chez l’homme, les paramètres pharmacocinétiques du gadobutrol sont proportionnels à la dose. Après des doses de gadobutrol jusqu’à 0,4 mmol/kg de masse corporelle, la concentration plasmatique diminue de manière biphasique. Les concentrations plasmatiques moyennes de gadobutrol mesurées 2 et 60 minutes après l’injection d’une dose de 0,1 mmol/kg ont été respectivement de 0,59 mmol/L et 0,3 mmol/L.
Biotransformation
Aucun métabolite n’est détecté dans le plasma ni dans les urines.
Élimination
Plus de 50 % de la dose est éliminé dans les urines en l’espace de 2 heures et plus de 90 % en l’espace de 12 heures, avec une demi-vie terminale moyenne de 1,8 heures (entre 1,3 et 2,1 heures), correspondant à la vitesse d’élimination rénale. A la dose de 0,1 mmol de gadobutrol par kg de masse corporelle, 100,3 ± 2,6 % de la dose (en moyenne) sont excrétés dans les 72 heures suivant l’administration. Chez les sujets sains, la clairance rénale du gadobutrol est de 1,1 à 1,7 mL.min-1.kg-1, c’est-à-dire comparable à la clairance rénale de l’inuline, ce qui indique que le gadobutrol est essentiellement éliminé par filtration glomérulaire. L’excrétion fécale représente moins de 0,1 % de la dose.
Caractéristiques des populations de patients spécifiques
Population pédiatrique
Le profil pharmacocinétique du gadobutrol chez l’enfant âgé de moins de 18 ans est similaire à celui chez l’adulte (voir rubrique 4.2).
Deux études de phase I/III à dose unique ont été menées chez l’enfant de moins de 18 ans. Les paramètres pharmacocinétiques ont été évalués chez 130 enfants âgés de 2 à moins de 18 ans et chez 43 enfants âgés de moins de 2 ans (y compris des nouveau-nés à terme).
Il a été démontré que le profil pharmacocinétique du gadobutrol chez les enfants de tout âge est similaire à celui chez les adultes, avec des valeurs similaires pour l’aire sous la courbe (ASC), la clairance plasmatique normalisée par kg de masse corporelle (CLtot) et le volume de distribution (Vss), ainsi que pour la demi-vie d’élimination et la vitesse d’excrétion.
Environ 99 % (valeur médiane) de la dose administrée a été éliminée dans les urines dans les 6 heures (cette information provient du groupe d’enfants âgés de 2 à moins de 18 ans).
Sujets âgés (à partir de 65 ans)
En raison des modifications physiologiques de la fonction rénale liées à l’âge, chez des volontaires âgés sains (âgés de 65 ans ou plus), l’exposition systémique a été augmentée d’environ 33 % (chez l’homme) et 54 % (chez la femme) et la demi-vie terminale d’environ 33 % (chez l’homme) et 58 % (chez la femme). La clairance plasmatique est réduite d’environ 25 % (chez l’homme) et de 35 % (chez la femme), respectivement. L’élimination dans les urines de la dose administrée a été complète chez tous les volontaires après 24 heures et il n’y avait aucune différence entre les volontaires sains âgés et non âgés.
Insuffisance rénale
Chez les patients atteints d’insuffisance rénale, la demi-vie sérique de gadobutrol est prolongée en raison de la filtration glomérulaire réduite. La demi-vie terminale moyenne a été prolongée à 5,8 heures chez les patients en insuffisance rénale modérée (30 < CLCR < 80 mL/min) et prolongée à 17,6 heures chez les patients en insuffisance rénale sévère non dialysés (CLCR < 30 mL/min). La clairance sérique moyenne a été réduite à 0,49 mL/min/kg chez les patients atteints d’une insuffisance rénale légère à modérée (30 < CLCR < 80 mL/min) et à 0,16 mL/min/kg chez les patients insuffisants sévères non dialysés (CLCR < 30 mL/min). L’élimination complète dans les urines a été observée chez les patients atteints d’une insuffisance légère à modérée dans les 72 heures. Chez les patients en insuffisance rénale sévère, environ 80 % de la dose administrée a été éliminée dans les urines dans les 5 jours (voir également rubriques 4.2 et 4.4).
Chez les patients sous dialyse, le gadobutrol a été presque entièrement éliminé après la troisième dialyse.
5.3. Données de sécurité préclinique
Les études animales de toxicité sur la reproduction à doses intraveineuses réitérées ont révélé un retard de développement embryonnaire chez le rat et le lapin et une augmentation de la mortalité embryonnaire chez le rat, le lapin et le singe à des doses allant de 8 à 16 fois (en fonction de la surface du corps) ou de 25 à 50 fois (en fonction du poids) au-delà de la dose à visée diagnostique chez l’homme. On ignore si ces effets peuvent également se produire après l’injection d’une dose unique.
Les études de toxicité à doses répétées et à dose unique chez le rat nouveau-né et juvénile n’ont mis en évidence aucun signe évocateur d’un risque spécifique quant à l’utilisation chez les enfants de tout âge, y compris chez les nouveau-nés à terme et les nourrissons.
Le gadobutrol radiomarqué administré par voie intraveineuse à des rates en période d'allaitement a été transmis aux nouveau-nés par le lait à une dose ne dépassant pas 0,1 % de la dose injectée.
Chez le rat, l’absorption après administration orale s’est avérée être très faible et elle atteint environ 5% en fonction de la dose excrétée dans les urines.
Les études pharmacologiques précliniques de tolérance cardiovasculaire ont montré, selon la dose administrée, une augmentation transitoire de la pression artérielle et de la contractilité du myocarde. Ces effets n’ont pas été observés chez l’homme.
Des études environnementales ont montré que la persistance et la mobilité des produits de contraste contenant du gadolinium indiquent un potentiel de distribution dans la colonne d’eau et éventuellement dans les eaux souterraines.
Calcobutrol sodique
Trométamol
Acide chlorhydrique 1N (ajustement du pH)
Eau pour préparations injectables.
En l’absence d’études de compatibilité, ce médicament ne doit pas être mélangé avec d’autres médicaments.
3 ans.
Durée de conservation après la première ouverture du récipient :
Il faut jeter toute solution injectable qui n’a pas été utilisée lors d’un examen. La stabilité physico‑chimique et microbiologique du produit après ouverture a été démontrée pendant 24 heures à 20‑25°C. Toutefois, d’un point de vue microbiologique, le produit doit être utilisé immédiatement. En cas d’utilisation non immédiate, les durées et les conditions de conservation en cours d’utilisation relèvent de la seule responsabilité de l’utilisateur.
6.4. Précautions particulières de conservation
Ce médicament ne nécessite pas de précautions particulières de conservation.
Pour les conditions de conservation du médicament après première ouverture, voir la rubrique 6.3.
6.5. Nature et contenu de l'emballage extérieur
Chaque boîte contient une seringue préremplie (contenant 7,5 mL, 10 mL ou 15 mL de solution injectable) conditionnée avec des dispositifs médicaux pour injection intraveineuse.
Seringues préremplies (verre)
Une seringue préremplie de 10 mL (verre de type I) avec un bouchon de piston (élastomère chlorobutylé) et un opercule de protection (élastomère chlorobutylé) contient 7,5 mL ou 10 mL de solution injectable.
Une seringue préremplie de 17 mL (verre de type I) avec un bouchon de piston (élastomère chlorobutylé) et un opercule de protection (élastomère chlorobutylé) contient 15 mL de solution injectable.
Seringues préremplies (plastique)
Une seringue préremplie de 10 mL (polymère cyclo-oléfine) avec un bouchon de piston (bromobutyle siliconé) et un embout d’étanchéité (élastomère thermoplastique) contient 7,5 mL de solution injectable.
Une seringue préremplie de 20 mL (polymère cyclo-oléfine) avec un bouchon de piston (bromobutyle siliconé) et un embout d’étanchéité (élastomère thermoplastique) contient 15 mL de solution injectable.
Dispositifs médicaux pour injection intraveineuse
Pour injection manuelle
· 1 raccord PVC incolore et transparent (50 cm)
· 1 seringue incolore PP/IR vide (20 mL)
· 1 cathéter de sécurité intraveineux PFEP/PUR avec une aiguille en acier inoxydable (22G)
Les dispositifs médicaux sont fournis dans leur propre emballage individuel, séparément de la seringue préremplie, le tout étant intégré dans la même boîte. Pour plus d’informations, veuillez-vous reporter aux instructions d’utilisation présentes dans la boîte.
Présentations
Boîte contenant 1 seringue préremplie de 7,5 mL, 10 mL ou 15 mL conditionnée avec des dispositifs d’administration pour injection manuelle.
Toutes les présentations peuvent ne pas être commercialisées.
6.6. Précautions particulières d’élimination et de manipulation
Toute solution non utilisée au cours d’un examen doit être éliminée conformément à la règlementation en vigueur.
Tout médicament non utilisé ou déchet doit être éliminé conformément à la réglementation en vigueur.
L’étiquette détachable de traçabilité placée sur les seringues préremplies doit être collée dans le dossier du patient afin de permettre un suivi précis du produit de contraste à base de gadolinium utilisé. La dose administrée doit également être enregistrée. En cas d’utilisation de dossier médical électronique, le nom du produit, le numéro de lot et la dose administrée doivent être enregistrés dans le dossier du patient.
7. TITULAIRE DE L’AUTORISATION DE MISE SUR LE MARCHE
1 RUE CLAUDE BERNARD
59000 LILLE
8. NUMERO(S) D’AUTORISATION DE MISE SUR LE MARCHE
· 34009 301 644 3 3 : 15 mL de solution en seringue préremplie (polymère cyclo-oléfine) avec dispositif d’administration pour injection manuelle, boîte de 1
· 34009 301 817 3 7 : 7.5 mL de solution en seringue préremplie (verre) avec dispositif d’administration pour injection manuelle, boîte de 1
· 34009 301 817 4 4 : 10 mL de solution en seringue préremplie (verre) avec dispositif d’administration pour injection manuelle, boîte de 1
· 34009 301 817 5 1 : 15 mL de solution en seringue préremplie (verre) avec dispositif d’administration pour injection manuelle, boîte de 1
9. DATE DE PREMIERE AUTORISATION/DE RENOUVELLEMENT DE L’AUTORISATION
[à compléter ultérieurement par le titulaire]
10. DATE DE MISE A JOUR DU TEXTE
[à compléter ultérieurement par le titulaire]
Sans objet.
12. INSTRUCTIONS POUR LA PREPARATION DES RADIOPHARMACEUTIQUES
Liste I
Ce médicament n'est ou ne sera bientôt plus disponible sur le marché.
Si vous prenez actuellement ce médicament, il vous est recommandé d'en parler avec votre médecin
ou avec votre pharmacien qui pourra vous orienter vers un autre traitement.
ANSM - Mis à jour le : 11/07/2025
GADOVISTMANUEL* 1,0 mmol/mL, solution injectable en seringue préremplie
Gadobutrol
* GADOVISTMANUEL est identique au médicament GADOVIST et ne diffère que par les dispositifs d’administration ajoutés dans l’emballage. Etant donné que toute l’expérience clinique a été acquise avec GADOVIST, cette dénomination est utilisée dans tout le texte.
Veuillez lire attentivement l’intégralité de cette notice avant d’utiliser ce médicament car elle contient des informations importantes pour vous.
· Gardez cette notice. Vous pourriez avoir besoin de la relire.
· Si vous avez d’autres questions, interrogez votre médecin ou la personne qui va vous administrer GADOVIST (le radiologue) ou le personnel de l’hôpital / du centre d’IRM.
· Si vous ressentez un quelconque effet indésirable, parlez-en à votre médecin ou votre radiologue. Ceci s’applique aussi à tout effet indésirable qui ne serait pas mentionné dans cette notice. Voir rubrique 4.
1. Qu'est-ce que GADOVIST 1,0 mmol/mL, solution injectable en seringue préremplie et dans quels cas est-il utilisé ?
2. Quelles sont les informations à connaître avant qu’on ne vous administre GADOVIST 1,0 mmol/mL, solution injectable en seringue préremplie?
3. Comment utiliser GADOVIST 1,0 mmol/mL, solution injectable en seringue préremplie?
4. Quels sont les effets indésirables éventuels ?
5. Comment conserver GADOVIST 1,0 mmol/mL, solution injectable en seringue préremplie?
6. Contenu de l’emballage et autres informations.
1. QU’EST-CE QUE GADOVIST 1,0 mmol/mL, solution injectable en seringue préremplie ET DANS QUELS CAS EST-IL UTILISE ?
Classe pharmacothérapeutique : Produit de contraste paramagnétique - code ATC : V08C A09
GADOVIST peut également être utilisé pour l’IRM des anomalies d’autres régions du corps.
Il facilite la visualisation des structures ou des lésions anormales et aide à différencier les tissus sains des tissus malades.
Il est indiqué chez les adultes et les enfants de tout âge (y compris les nouveau-nés à terme).
Comment agit GADOVIST 1,0 mmol/mL, solution injectable en seringue préremplie
L’IRM est une technique d’imagerie médicale à visée diagnostique basée sur les différences de réaction des molécules d’eau entre les tissus normaux et les tissus anormaux. Cet examen est réalisé avec un système complexe utilisant un champ magnétique et des ondes radio. Un ordinateur enregistre l’activité et la traduit sous forme d’images.
GADOVIST est administré par injection dans une veine. Ce médicament est à usage diagnostique uniquement et ne vous sera administré que par des professionnels de santé ayant l’expérience de la pratique clinique de l’IRM.
2. QUELLES SONT LES INFORMATIONS A CONNAITRE AVANT D’UTILISER GADOVIST 1,0 mmol/mL, solution injectable en seringue préremplie ?
N’utilisez jamais GADOVIST 1,0 mmol/mL, solution injectable en seringue préremplie
Si vous êtes allergique au gadobutrol ou à l’un des autres composants contenus dans ce médicament, mentionnés dans la rubrique 6.
Avertissements et précautions
Adressez-vous à votre médecin avant que GADOVIST ne vous soit administré, si :
· vous souffrez d’une allergie ou si vous avez des antécédents d’allergie (par exemple, rhume des foins, urticaire) ou d’asthme,
· vous avez des antécédents de réaction à un quelconque produit de contraste,
· vous présentez une fonction rénale altérée,
· vous souffrez d’une maladie cérébrale avec convulsions (troubles épileptiques) ou d’une autre maladie du système nerveux,
· vous portez un stimulateur cardiaque (pacemaker) ou si vous avez un implant ou des agrafes contenant du fer dans votre corps.
Votre médecin décidera si l’examen peut être réalisé ou non.
Des réactions de type allergique ou d’autres types de réactions pouvant entraîner des problèmes cardiaques, des difficultés à respirer ou des réactions au niveau de la peau peuvent survenir après l’utilisation de GADOVIST. La survenue de réactions graves est possible. La plupart de ces réactions surviennent dans la demi-heure suivant l’administration de GADOVIST, c’est pourquoi vous serez gardé(e) en observation après l’examen. Des cas de réactions retardées (survenant de quelques heures à plusieurs jours suivant l’administration) ont été rapportés (voir la rubrique 4).
Reins/Foie
Prévenez votre médecin si :
· vos reins ne fonctionnent pas correctement,
· vous avez récemment bénéficié ou allez prochainement bénéficier d’une greffe du foie.
· Votre médecin peut vous prescrire un examen sanguin afin de vérifier le fonctionnement de vos reins avant de prendre la décision de vous prescrire GADOVIST, particulièrement si vous êtes âgé de 65 ans ou plus.
Nouveau-nés et nourrissons
En raison de l’immaturité de la fonction rénale des nouveau-nés jusqu’à l’âge de 4 semaines et des nourrissons jusqu’à l’âge d’un an, GADOVIST ne doit être administré à ces patients qu’après un examen approfondi de la situation par le médecin.
Autres médicaments et GADOVIST 1,0 mmol/mL, solution injectable en seringue préremplie :
Informez votre médecin si vous prenez, avez récemment pris ou pourriez prendre tout autre médicament.
GADOVIST 1,0 mmol/mL, solution injectable en seringue préremplie avec des aliments et boissons
Sans objet
Grossesse et allaitement
Demandez conseil à votre médecin avant de prendre tout médicament.
Grossesse
Le gadobutrol peut traverser la barrière placentaire. On ignore si cela pourrait nuire à l’enfant à naître. Vous devez prévenir votre médecin si vous pensez être enceinte ou si vous envisagez de l’être, car GADOVIST ne doit pas être administré au cours de la grossesse, sauf en cas de stricte nécessité.
Allaitement
Prévenez votre médecin si vous allaitez ou prévoyez d’allaiter. Votre médecin déterminera avec vous si vous pouvez poursuivre l’allaitement ou si vous devez l’interrompre pendant une période de 24 heures après l’administration de GADOVIST.
Conduite de véhicules et utilisation de machines
Sans objet.
GADOVIST 1,0 mmol/mL, solution injectable en seringue préremplie contient du sodium.
Ce médicament contient moins de 23 mg de sodium par dose (calculé pour une dose moyenne administrée à une personne de 70 kg) c’est-à-dire qu’il est essentiellement « sans sodium ».
3. COMMENT UTILISER GADOVIST 1,0 mmol/mL, solution injectable en seringue préremplie ?
Après l’injection, vous serez gardé(e) en observation pendant au moins une demi-heure.
Posologie usuelle :
La dose de GADOVIST la mieux adaptée pour vous dépendra de votre poids ainsi que de la partie du corps examinée par IRM :
Chez l’adulte, une seule injection de 0,1 millilitre de GADOVIST par kilogramme de masse corporelle est recommandée (pour une personne de 70 kg, la dose est donc de 7 millilitres), cependant une injection supplémentaire pouvant aller jusqu’à 0,2 millilitre par kg de masse corporelle pourra vous être administrée dans les 30 minutes suivant la première injection. La dose totale maximale pouvant être administrée est de 0,3 millilitre de GADOVIST par kg de masse corporelle (cela signifie que pour une personne de 70 kg, la dose serait de 21 millilitres) pour l’imagerie du système nerveux central (SNC) et de l’angiographie par résonance magnétique (ARM) avec rehaussement. Une dose de 0,075 millilitre de GADOVIST par kg de masse corporelle peut être administrée au minimum (ce qui signifie que pour une personne de 70 kg, la dose serait de 5,25 millilitres) pour le SNC.
De plus amples renseignements concernant l'administration et la manipulation de GADOVIST sont donnés à la fin de la notice.
Utilisation dans des populations particulières
L'utilisation de GADOVIST n’est pas recommandée chez les patients qui ont des troubles rénaux sévères et chez les patients qui ont récemment eu ou doivent prochainement bénéficier d’une greffe du foie. Si l’administration de GADOVIST est cependant nécessaire, vous ne devrez recevoir qu’une seule dose au cours d’un examen et ne pas faire l’objet d’un second examen IRM avec injection de produit de contraste avant au moins sept jours.
Utilisation chez les nouveau-nés, nourrissons, enfants et adolescents
Chez les enfants de tout âge (y compris les nouveau-nés à terme), une seule dose de 0,1 millilitre de GADOVIST par kg de masse corporelle est recommandée pour tout examen (voir rubrique 1).
En raison de l’immaturité de la fonction rénale chez le nouveau-né jusqu’à l’âge de 4 semaines et chez le nourrisson jusqu’à l’âge d’un an, GADOVIST ne doit être administré qu’après un examen approfondi de la situation par le médecin. Les nouveau-nés et les nourrissons ne doivent recevoir qu’une seule dose au cours d’un examen et ne pas faire l’objet d’un second examen IRM avec injection de produit de contraste avant au moins sept jours.
Sujets âgés
Il n’est pas nécessaire d’adapter la dose si vous avez 65 ans ou plus, mais une analyse de sang pourra être faite afin de vérifier le fonctionnement de vos reins.
Si vous avez reçu plus de GADOVIST 1,0 mmol/mL, solution injectable en seringue préremplie que vous n’auriez dû
Le risque de surdosage est peu probable. En cas de surdosage, le médecin instaurera un traitement des symptômes associés et pourra recourir à une hémodialyse pour éliminer GADOVIST de votre organisme. Il n’a pas été établi que l’hémodialyse puisse prévenir le risque de Fibrose Systémique Néphrogénique (FSN ; voir rubrique 4) ni la traiter.
Dans certains cas, le médecin contrôlera votre cœur.
Si vous oubliez d’utiliser GADOVIST 1,0 mmol/mL, solution injectable en seringue préremplie:
Sans objet.
Si vous arrêtez d’utiliser GADOVIST 1,0 mmol/mL, solution injectable en seringue préremplie :
Sans objet
Si vous avez d'autres questions sur l'utilisation de ce médicament, demandez plus d’informations à votre médecin ou votre radiologue.
4. QUELS SONT LES EFFETS INDESIRABLES EVENTUELS ?
Les effets indésirables les plus graves (ayant mis en jeu le pronostic vital ou ayant eu une issue fatale dans certains cas) sont :
· le cœur qui s’arrête de battre (arrêt cardiaque), une maladie grave des poumons (syndrome de détresse respiratoire aiguë) / la présence de liquide dans les poumons (œdème pulmonaire) et les réactions sévères de type allergique (anaphylactoïdes) incluant arrêt respiratoire et choc anaphylactoïde.
De plus, pour les effets indésirables suivants, des évolutions fatales ou ayant mis en jeu le pronostic vital ont été observées dans certains cas :
· Essoufflement (dyspnée), perte de conscience, réactions sévères de type allergique, baisse importante de la pression artérielle pouvant aller jusqu’au collapsus, arrêt respiratoire, liquide dans les poumons, gonflement de la bouche et de la gorge, pression artérielle basse.
Dans de rares cas :
· des réactions de type allergique (hypersensibilité et anaphylaxie) peuvent se produire, y compris des réactions sévères (choc anaphylactique) nécessitant une prise en charge médicale immédiate.
Si vous remarquez:
· un gonflement du visage, des lèvres, de la langue ou de la gorge,
· une toux et des éternuements,
· des difficultés à respirer,
· des démangeaisons,
· un écoulement nasal,
· une urticaire (éruption cutanée ressemblant à celle d’une piqûre d’orties),
Veuillez prévenir immédiatement le personnel du service d’IRM. Ces symptômes peuvent être les premiers signes d’une réaction sévère. Votre examen pourra être interrompu et vous pourrez nécessiter un traitement supplémentaire.
Les effets indésirables les plus fréquemment observés (survenant chez 5 patients sur 1000 ou plus) sont :
· des maux de tête, une sensation de malaise (nausées) et des étourdissements.
· La plupart des effets indésirables sont légers à modérés.
Les effets indésirables éventuels qui ont été observés lors des études cliniques avant la commercialisation de GADOVIST sont listés ci-dessous, selon leur probabilité de survenue :
Fréquents : pouvant affecter jusqu’à 1 personne sur 10
· maux de tête
· sensation de malaise (nausées)
· Peu fréquents : pouvant affecter jusqu’à 1 personne sur 100
· réactions de type allergique, par exemple :
o pression artérielle basse,
o urticaire,
o gonflement du visage,
o gonflement (œdème) des paupières,
o bouffées de chaleur.
La fréquence des réactions de type allergique suivantes est inconnue :
o réaction de type allergique sévère (choc anaphylactoïde),
o baisse importante de la pression artérielle pouvant mener à un collapsus (choc),
o arrêt respiratoire,
o difficultés à respirer (bronchospasme),
o bleuissement des lèvres,
o gonflement de la bouche et de la gorge,
o gonflement de la gorge,
o augmentation de la pression artérielle,
o douleurs dans la poitrine,
o gonflement du visage, de la gorge, de la bouche, des lèvres et/ou de la langue (angio‑œdème),
o conjonctivite,
o augmentation de la transpiration,
o toux,
o éternuements,
o sensation de brûlures,
o pâleur de la peau (pâleur),
· étourdissements, troubles du goût, engourdissement et fourmillement,
· essoufflement (dyspnée),
· vomissements,
· rougeur de la peau (érythème),
· démangeaisons (prurit incluant un prurit généralisé),
· éruption cutanée (incluant éruption cutanée généralisée, taches rouges petites et plates [rash maculaire], petites lésions surélevées et limitées [rash papulaire], éruption qui démange [rash prurigineux]),
· réactions diverses au point d’injection (par exemple : débordement vers les tissus environnants, brûlure, froid, chaud, rougeur, rash, douleur ou bleus),
· sensation de chaleur.
· Rares : pouvant affecter jusqu’à 1 personne sur 1000
· évanouissement,
· convulsions,
· troubles de l’odorat,
· accélération du rythme cardiaque,
· palpitations,
· bouche sèche,
· sensation générale de malaise (malaise),
· sensation de froid.
· Autres effets indésirables rapportés après l’autorisation de mise sur le marché de GADOVIST avec une fréquence indéterminée (la fréquence ne peut pas être évaluée à partir des données disponibles) :
· cœur qui s’arrête de battre (arrêt cardiaque),
· maladie grave des poumons (syndrome de détresse respiratoire aiguë),
· présence de liquide dans les poumons (œdème pulmonaire),
· des cas de fibrose systémique néphrogénique - FSN (durcissement de la peau qui peut également affecter les tissus mous et les organes internes) ont été rapportés.
· Des variations des tests sanguins de la fonction rénale (par exemple, augmentation de la créatinine sérique) ont été observées après l'administration de GADOVIST.
Déclaration des effets secondaires
Si vous ressentez un quelconque effet indésirable, parlez-en à votre médecin ou votre radiologue. Ceci s’applique aussi à tout effet indésirable qui ne serait pas mentionné dans cette notice. Vous pouvez également déclarer les effets indésirables directement via le système national de déclaration : Agence nationale de sécurité du médicament et des produits de santé (ANSM) et réseau des Centres Régionaux de Pharmacovigilance - Site internet : https://signalement.social-sante.gouv.fr/
En signalant les effets indésirables, vous contribuez à fournir davantage d’informations sur la sécurité du médicament.
5. COMMENT CONSERVER GADOVIST 1,0 mmol/mL, solution injectable en seringue préremplie ?
Tenir ce médicament hors de la vue et de la portée des enfants.
N’utilisez pas ce médicament après la date de péremption indiquée sur l’étiquette et sur l'étui en carton après « EXP ». La date de péremption fait référence au dernier jour de ce mois. Ce médicament ne nécessite pas de conditions particulières de conservation.
La stabilité physico-chimique et microbiologique du produit après ouverture a été démontrée pendant 24 heures à 20-25°C. Toutefois, d’un point de vue microbiologique, il est préférable d’utiliser le produit immédiatement.
Ce médicament est une solution limpide, incolore à jaune pâle. Ne l'utilisez pas en cas de changement important de coloration, de présence de particules ou si l’emballage apparaît défectueux.
Ne jetez aucun médicament au tout-à-l’égout ou avec les ordures ménagères. Le professionnel de santé éliminera ce médicament lorsqu’il ne sera plus utilisé. Ces mesures contribueront à protéger l’environnement.
6. CONTENU DE L’EMBALLAGE ET AUTRES INFORMATIONS
Ce que contient GADOVIST 1,0 mmol/mL, solution injectable en seringue préremplie
· La substance active est : le gadobutrol.
1 mL de solution injectable contient 604,72 mg de gadobutrol (correspondant à 1,0 mmol de gadobutrol contenant 157,25 mg de gadolinium).
1 seringue préremplie de 7,5 mL de solution contient 4535,4 mg de gadobutrol.
1 seringue préremplie de 10 mL de solution contient 6047,2 mg de gadobutrol.
1 seringue préremplie de 15 mL de solution contient 9070,8 mg de gadobutrol.
· Les autres composants sont : le calcobutrol sodique (voir fin de la rubrique 2), le trométamol, l’acide chlorhydrique 1N et l’eau pour préparations injectables.
GADOVIST est une solution injectable limpide, incolore à jaune pâle.
Chaque boîte contient :
· 1 seringue préremplie (verre) contenant 7,5 mL, 10 mL ou 15 mL de solution injectable avec des dispositifs d’administration pour injection manuelle (une seringue vide de 20 mL, un raccord et un cathéter de sécurité I.V 22G).
· 1 seringue préremplie (plastique) contenant 7,5 mL ou 15 mL de solution injectable avec des dispositifs d’administration pour injection manuelle (une seringue vide de 20 mL, un raccord et un cathéter de sécurité I.V 22G).
· Toutes les présentations peuvent ne pas être commercialisées.
Titulaire de l’autorisation de mise sur le marché
1 RUE CLAUDE BERNARD
59000 LILLE
Exploitant de l’autorisation de mise sur le marché
BAYER HEALTHCARE SAS
1 RUE CLAUDE BERNARD
59000 LILLE
MULLERSTRASSE 178
13353 BERLIN
ALLEMAGNE
Noms du médicament dans les Etats membres de l'Espace Economique Européen
Ce médicament est autorisé dans les Etats membres de l'Espace Economique Européen sous les noms suivants : Conformément à la réglementation en vigueur.
[À compléter ultérieurement par le titulaire]
La dernière date à laquelle cette notice a été révisée est :
[à compléter ultérieurement par le titulaire]
{MM/AAAA}
Des informations détaillées sur ce médicament sont disponibles sur le site Internet de l’ANSM (France).
Les informations suivantes sont destinées exclusivement aux professionnels de santé :
· Insuffisance rénale
Avant l’administration de GADOVIST, il est recommandé de procéder à des examens de laboratoire chez tous les patients afin de rechercher une altération de la fonction rénale.
Des cas de fibrose systémique néphrogénique (FSN) ont été rapportés après injection de certains produits de contraste contenant du gadolinium chez des patients ayant une insuffisance rénale sévère aiguë ou chronique (clairance de la créatinine < 30 mL/min/1,73 m²). Les patients devant bénéficier d’une transplantation hépatique sont particulièrement à risque, car l’incidence de l’insuffisance rénale aiguë est élevée dans ce groupe. Etant donné qu’il est possible que des cas de FSN surviennent avec GADOVIST, ce produit ne doit être administré aux patients présentant une insuffisance rénale sévère ou aux patients durant la période pré ou post-opératoire d’une transplantation hépatique qu’après une évaluation approfondie du rapport bénéfice/risque et que si le diagnostic est indispensable et ne peut être obtenu par d’autres moyens que l’IRM avec injection de gadolinium. S’il est nécessaire d’administrer GADOVIST, la dose ne doit pas excéder 0,1 mmol/kg de masse corporelle. Ne pas administrer plus d’une dose au cours de l’examen IRM. En raison du manque d’information sur les administrations répétées, les injections de GADOVIST ne doivent pas être réitérées sauf si l’intervalle entre les injections est d’au moins 7 jours.
L’élimination rénale de gadobutrol pouvant être altérée chez les sujets âgés, il est particulièrement important de rechercher un dysfonctionnement rénal chez les sujets âgés de 65 ans et plus.
La réalisation d’une hémodialyse peu de temps après l’administration de GADOVIST pourrait faciliter l’élimination de ce produit de l’organisme. Il n’est pas établi que l’instauration d’une hémodialyse puisse prévenir ou traiter la FSN chez les patients qui ne sont pas déjà hémodialysés.
· Grossesse et allaitement
GADOVIST ne doit pas être utilisé pendant la grossesse à moins que la situation clinique de la patiente ne nécessite l’administration de GADOVIST.
Le médecin et la mère allaitante doivent décider s’il faut poursuivre l’allaitement ou le suspendre pendant les 24 heures suivant l’administration de GADOVIST.
· Réactions d’hypersensibilité
Comme pour les autres agents de contraste administrés par voie intraveineuse, l’utilisation de GADOVIST peut s’accompagner de réactions anaphylactoïdes/d’hypersensibilité ou d’autres réactions idiosyncrasiques, se traduisant par des manifestations cardiovasculaires, respiratoires ou cutanées, potentiellement sévères, allant même jusqu’au choc.
D’une manière générale, les patients souffrant de maladies cardio-vasculaires sont plus susceptibles que les autres de présenter une évolution grave ou fatale lors d’une réaction d’hypersensibilité sévère.
Le risque de développer des réactions d’hypersensibilité peut être augmenté en cas :
· de précédente réaction aux produits de contraste,
· d’antécédents d’asthme bronchique,
· d’antécédents de troubles allergiques.
Chez les patients présentant un terrain allergique, la décision d’utiliser GADOVIST ne doit être prise qu’après une évaluation particulièrement attentive du rapport bénéfice/risque.
La plupart de ces réactions surviennent dans la demi-heure suivant l’administration. Aussi, il est recommandé de garder le patient en observation après examen.
Il est impératif d’avoir à disposition le traitement médicamenteux nécessaire à la prise en charge de réactions d’hypersensibilité; il faut également être prêt pour la mise en place immédiate de mesures d’urgence.
Des réactions retardées (de quelques heures à plusieurs jours après l’administration) ont été rarement observées.
· Troubles épileptiques
Comme avec les autres produits de contraste contenant du gadolinium, des précautions particulières s'imposent en cas de seuil épileptogène bas.
· Surdosage
En cas de surdosage accidentel, une surveillance cardiovasculaire (incluant un ECG) et un contrôle de la fonction rénale sont recommandés à titre de précaution.
En cas de surdosage chez les patients présentant une insuffisance rénale, GADOVIST peut être éliminé par hémodialyse. Après 3 séances d’hémodialyse, environ 98 % de l’agent de contraste est éliminé du corps. Toutefois, il n’est pas démontré que l’hémodialyse soit appropriée dans la prévention de la fibrose systémique néphrogénique (FSN).
· Avant l’injection
Ce produit n'est destiné qu'à un usage unique.
Ce médicament est une solution limpide, incolore à jaune pâle. Elle doit être contrôlée visuellement avant utilisation.
GADOVIST ne doit pas être utilisé en cas de changement important de coloration, de présence de particules ou d’emballage défectueux.
· Instructions d’utilisation
Seringues préremplies
GADOVIST est prêt à l’emploi.
La seringue préremplie doit être préparée pour l’injection juste avant son administration.
L’opercule de protection doit être retiré de la seringue préremplie juste avant utilisation.
|
INJECTION MANUELLE |
|
|
Seringue en verre uniquement :
|
Seringue en plastique uniquement : |
|
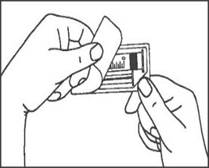
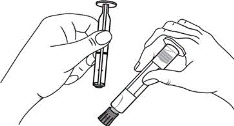
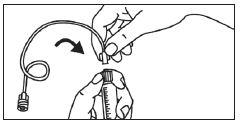
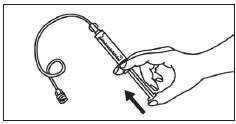
1. Ouvrir l’emballage. |
1. Prendre la seringue et le piston. |
|
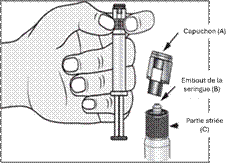
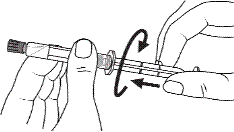
2. Tenir la seringue droite sur la partie striée (C) du système de fermeture. |
2. Tourner le piston dans le sens des aiguilles d'une montre pour le fixer sur la seringue. |
|
3. De l'autre main, saisir le capuchon (A) et le basculer doucement d'avant en arrière jusqu'à ce qu’il se détache et puisse être retiré (tous les joints seront rompus). |
3. Dévisser le capuchon en le tournant. |
|
4. Retirer le capuchon (A) en le dirigeant vers le haut. Ne pas toucher l'embout de la seringue (B) afin de préserver sa stérilité. |
4. Expulser l’air de la seringue. |
|
5. Expulser l’air de la seringue.
|
|
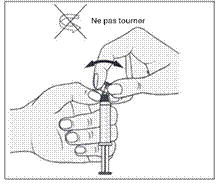
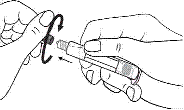
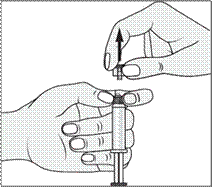

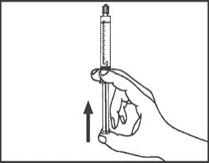
Toute solution restante non utilisée après un examen doit être jetée conformément aux réglementations nationales en vigueur.
Dispositifs médicaux pour injection intraveineuse
Pour plus d’informations sur l’assemblage et le bon usage, veuillez suivre les instructions d’utilisation de chacun des dispositifs médicaux.
Seringue en verre / seringue en plastique
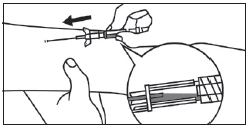
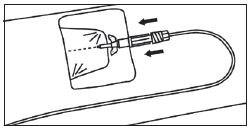
|
DISPOSITIFS POUR INJECTION MANUELLE |
|
|
|
1. Fixation du raccord basse pression à la seringue préremplie. |
|
|
|
2. Expulsion de l’air de la tubulure. |
|
|
|
3. Création d’un accès veineux périphérique avec le cathéter. |
|
|
|
4. Connexion de la tubulure au cathéter.
|
Durée de conservation après première ouverture de la seringue pré‑remplie
Il faut jeter toute solution injectable qui n’a pas été utilisée lors d’un examen. La stabilité physico chimique et microbiologique du produit après ouverture a été démontrée pendant 24 heures à 20- 25°C. Toutefois, d'un point de vue microbiologique, il est préférable d’utiliser le produit immédiatement. S'il n'est pas utilisé immédiatement, la durée et les conditions de conservation jusqu'à l'utilisation sont de la seule responsabilité de l'utilisateur.
L’étiquette détachable de traçabilité placée sur les seringues doit être collée dans le dossier du patient afin de permettre la traçabilité du produit de contraste à base de gadolinium administré. La dose administrée doit également être enregistrée. En cas d’utilisation de dossier médical électronique, le nom du produit, le numéro de lot et la dose administrée doivent être enregistrés dans le dossier du patient.
Posologie
La dose la plus faible permettant un rehaussement de contraste suffisant à des fins diagnostiques doit être utilisée. La dose doit être calculée en fonction de la masse corporelle du patient et ne doit pas dépasser la dose recommandée par kilogramme de masse corporelle, détaillée dans cette rubrique.
· Adultes
Indications dans le SNC :
La dose recommandée pour un adulte est de 0,1 mmol par kg de masse corporelle (mmol/kg), ce qui équivaut à 0,1 mL/kg de la solution à 1,0 M.
En cas de forte suspicion clinique d’une lésion non confirmée à l’IRM ou si des informations plus précises peuvent modifier la prise en charge thérapeutique du patient, une seconde injection pouvant aller jusqu’à 0,2 mL/kg au maximum peut être effectuée dans les 30 minutes suivant la première injection.
Une dose de 0,075 mmol de gadobutrol par kg de masse corporelle (équivalent à 0,075 mL de GADOVIST par kg de masse corporelle) peut être administrée au minimum pour l’imagerie du SNC.
IRM du corps entier (à l’exception de l’Angiographie par Résonance Magnétique) :
De manière générale, l’administration de 0,1 mL de GADOVIST par kg de masse corporelle est suffisante pour apporter une réponse à la question clinique.
Angiographie par Résonance Magnétique :
Image d'un seul champ d'acquisition : 7,5 mL pour un patient de moins de 75 kg, 10 mL pour un patient de 75 kg et plus (équivalent à 0,1-0,15 mmol/kg).
Image de plusieurs champs d’acquisition : 15 mL pour un patient de moins de 75 kg, 20 mL pour un patient de 75 kg ou plus (équivalent à 0,2-0,3 mmol/kg).
· Population pédiatrique
Pour les enfants de tout âge (y compris les nouveau-nés à terme), la dose recommandée est de 0,1 mmol de gadobutrol par kg de masse corporelle (équivalent à 0,1 mL de GADOVIST par kg de masse corporelle) pour toutes les indications (voir rubrique 1).
En raison de l’immaturité de la fonction rénale chez le nouveau-né jusqu’à l’âge de 4 semaines et chez le nourrisson jusqu’à l’âge d’un an, GADOVIST ne doit être administré à ces patients qu’après un examen approfondi de la situation et à une dose n’excédant pas 0,1 mmol/kg de masse corporelle. Ne pas administrer plus d’une dose au cours de l’examen IRM. En raison du manque d’information sur les administrations répétées, les injections de GADOVIST ne doivent pas être réitérées sauf si l’intervalle entre les injections est d’au moins 7 jours.
Imagerie
La dose nécessaire est administrée en bolus par voie intraveineuse. L’examen IRM avec rehaussement du contraste peut débuter immédiatement après l’injection (dans un délai dépendant des séquences d’IRM utilisées et du protocole d’examen).
Le rehaussement optimal du signal est observé pendant le premier passage artériel en angiographie par résonance magnétique et dans les 15 minutes suivant l’injection de GADOVIST pour les indications du SNC (ce délai dépendant du type de lésion ou de tissu).
Les séquences pondérées en T1 sont particulièrement adaptées aux examens avec injection d’un produit de contraste gadoliné.
De plus amples renseignements concernant l’utilisation de GADOVIST sont donnés à la rubrique 3 de la notice.